

Abstract
Background and Aims
Mutations in ATPase phospholipid transporting 8B1 (ATP8B1) can lead to familial intrahepatic cholestasis type 1 (FIC1) deficiency, or progressive familial intrahepatic cholestasis type 1. The rarity of FIC1 deficiency has largely prevented a detailed analysis of its natural history, effects of predicted protein truncating mutations (PPTMs), and possible associations of serum bile acid (sBA) concentrations and surgical biliary diversion (SBD) with long‐term outcome. We aimed to provide insights by using the largest genetically defined cohort of patients with FIC1 deficiency to date.
Approach and Results
This multicenter, combined retrospective and prospective study included 130 patients with compound heterozygous or homozygous predicted pathogenic ATP8B1 variants. Patients were categorized according to the number of PPTMs (i.e., splice site, frameshift due to deletion or insertion, nonsense, duplication), FIC1‐A (n = 67; no PPTMs), FIC1‐B (n = 29; one PPTM), or FIC1‐C (n = 34; two PPTMs). Survival analysis showed an overall native liver survival (NLS) of 44% at age 18 years. NLS was comparable among FIC1‐A, FIC1‐B, and FIC1‐C (% NLS at age 10 years: 67%, 41%, and 59%, respectively; P = 0.12), despite FIC1‐C undergoing SBD less often (% SBD at age 10 years: 65%, 57%, and 45%, respectively; P = 0.03). sBAs at presentation were negatively associated with NLS (NLS at age 10 years, sBAs < 194 µmol/L: 49% vs. sBAs ≥ 194 µmol/L: 15%; P = 0.03). SBD decreased sBAs (230 [125‐282] to 74 [11‐177] μmol/L; P = 0.005). SBD (HR 0.55, 95% CI 0.28‐1.03, P = 0.06) and post‐SBD sBA concentrations < 65 μmol/L (P = 0.05) tended to be associated with improved NLS.
Conclusions
Less than half of patients with FIC1 deficiency reach adulthood with native liver. The number of PPTMs did not associate with the natural history or prognosis of FIC1 deficiency. sBA concentrations at initial presentation and after SBD provide limited prognostic information on long‐term NLS.
Abbreviations
- ABCB11
ATP‐binding cassette, subfamily B, member 11
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- ATP8B1
ATPase phospholipid transporting 8B1
- AUC
area under the curve
- BRIC
benign recurrent intrahepatic cholestasis
- BSEP
bile salt export pump
- CI
confidence interval
- EHC
enterohepatic circulation
- FIC1
familial intrahepatic cholestasis type 1
- GGT
gamma‐glutamyltransferase
- IE
ileal exclusion
- LT
liver transplantation
- NAPPED
Natural course and Prognosis of PFIC and Effect of biliary Diversion
- NLS
native liver survival
- PEBD
partial external biliary diversion
- PFIC
progressive familial intrahepatic cholestasis
- PLT
platelet count
- PPTM
predicted protein truncating mutation
- REDCap
Research Electronic Data Capture
- ROC
receiver operating characteristic
- sBA
serum bile acid
- SBD
surgical biliary diversion
- TSB
total serum bilirubin
- UDCA
ursodeoxycholic acid
Familial intrahepatic cholestasis type 1 (FIC1) deficiency, or progressive familial intrahepatic cholestasis type 1 (PFIC), results from mutations in the adenosine triphosphate (ATP)ase phospholipid transporting 8B1 (ATP8B1) gene.( 1, 2, 3, 4 ) FIC1 is a member of ATP‐dependent phospholipid flippases, which maintain a phospholipid asymmetry across phospholipid bilayers, such as the canalicular membrane.( 5 ) Mutations in ATP8B1 are thought to disrupt the physiological membrane structure, leading to impaired biliary bile salt excretion,( 6 ) yet the exact mechanism by which this occurs remains elusive. Indirect inhibition of other canalicular transporters (e.g., bile salt export pump [BSEP]) by increased vulnerability to canalicular bile acids as well as down‐regulation of the farnesoid X receptor are among the proposed responsible mechanisms.( 5, 7, 8, 9, 10, 11, 12 ) Patients with FIC1 deficiency typically present with cholestasis and pruritus in early childhood, often progressing into end‐stage liver disease (ESLD).( 13, 14 ) FIC1 is expressed in tissues other than liver. Deficiency of FIC1 therefore results in a range of extrahepatic manifestations, such as diarrhea, pancreatitis, and hearing loss.( 14, 15, 16, 17, 18, 19, 20, 21, 22 ) FIC1 deficiency has been largely refractory to medical treatment, yet surgical biliary diversion (SBD) procedures may decrease pruritus and slow the development of liver fibrosis.( 13, 18, 23, 24, 25, 26 ) Patients in whom cholestasis does not (sufficiently) respond to medical treatment or SBD are potential candidates for liver transplantation (LT). Although LT may be indicated for severe cholestasis and/or ESLD, it cannot be considered as a total cure of the underlying genetic disease due to the extrahepatic expression of FIC1, and therefore the presence or even development of extrahepatic symptoms after LT. SBD can still be indicated after LT to prevent or treat graft steatofibrosis and secondary intestinal lymphangiectasia.( 27 )
FIC1 deficiency is a rare disease; thus, its natural history and genotype–phenotype relationships have not been characterized in large, genetically defined cohorts. We therefore aimed to provide detailed insights into the natural history and genotype–phenotype associations, and to assess the effect of SBD on native liver survival (NLS). Since 2017, we have been collecting patient data in a global consortium: NAPPED (NAtural course and Prognosis of PFIC and Effect of biliary Diversion). NAPPED has become a large, international initiative that aims to characterize the natural course of disease in severe FIC1 (and severe BSEP deficiency), to assess the effect of genetic, clinical, and therapeutic parameters on long‐term outcome.
Materials and Methods
Data Collection, Patient Selection, and Genetic Categorization
Since its start in 2017, NAPPED has collected retrospective data on patients with FIC1 deficiency (and severe BSEP deficiency caused by mutations in ATP‐binding cassette, subfamily B, member 11 [ABCB11], data that have recently been published( 28 )). The Childhood Liver Disease Research Network (ChiLDReN) collected data prospectively. NAPPED is currently comprised of 68 referral centers from Europe, North America, South America, Africa, Asia, and Australia. Data for the present study were collected, conforming to the guidelines of the 1975 Declaration of Helsinki. The study was reviewed by the institutional review committees of the participating centers, and was granted IRB approval or was exempted from IRB approval, which depended on the participating center‘s regulations. The requirement of approval by institutional review committees for participation to the study was institute‐dependent and, if required, was obtained. Data collection and management used a prespecified case‐record form and were captured using Research Electronic Data Capture (REDCap).( 29 ) Demographic, clinical, and outcome data were collected by investigators within each center, who identified all consecutive patients who had ever been under pediatric care (age 0‐18 years) since 1981. From ChiLDReN, all cases of FIC1 deficiency enrolled in the Longitudinal Study of Genetic Causes of Intrahepatic Cholestasis since 2007 were included. Data reported in the present manuscript were exported from REDCap on May 1, 2020. Patients with pathological compound heterozygous or homozygous ATP8B1 mutations were selected from the database. The pathogenicity of mutations was based on existing literature and databases, including in vitro studies.( 4, 30, 31, 32 ) Patients without available genetic reports or with mutations of no identifiable pathological significance were excluded. Included patients were further categorized based on their genotype vis‐à‐vis predicted protein truncating mutations (PPTMs) (i.e., splice site, frameshift due to deletion or insertion, nonsense, duplication): Patients with an FIC1‐A genotype harbored no PPTMs; patients with an FIC1‐B genotype harbored one PPTM; and patients with an FIC1‐C genotype harbored two PPTMs. We further categorized patients with homozygous mutations based on the location of their mutations in the FIC1 protein. Biochemistry at presentation in the tertiary center, as well as before SBD and between 2 months and 1 year after SBD, were analyzed. If such information was available from the medical file, pruritus was scored as “absent,” “mild to moderate,” or “severe” at the discretion of the participating center, which, for statistical purposes, was dichotomized later into “absent” or “present.” Effect of SBD on pruritus was noted as “no improvement in pruritus,” “transient (partial or complete) relief of pruritus,” or “sustained (partial or complete) relief of pruritus.” Analyses were performed with regard to important clinical events in the form of SBD, LT, or death. Extrahepatic features have not (yet) been documented in the NAPPED REDCap database, and therefore were not analyzed in the present paper.
Statistical Evaluation
Continuous variables are expressed as medians and interquartile range. Nonnormally distributed data were analyzed using the appropriate nonparametric methods, including the Mann‐Whitney U test and Kruskal‐Wallis test. Differences among categorical variables were assessed using χ2, McNemar, or Mantel’s test for trend, as appropriate. Unadjusted differences in disease‐free survival and NLS among patient subgroups were assessed using Kaplan‐Meier estimates and compared using the log‐rank test. The following factors were included in the analyses: sex, birth year, age at first visit at referral center, use of medical therapy before first visit (e.g., ursodeoxycholic acid [UDCA], rifampicin, phenobarbital, cholestyramine, antihistamines), occurrence of HCC, serum bile acids (sBAs) (not standardized for fasting/postprandial state), total serum bilirubin (TSB), alanine aminotransferase (ALT), aspartate aminotransferase (AST), gamma‐glutamyltransferase (GGT), and platelet count (PLT). Units of sBAs (μmol/L), TSB (μmol/L), AST (IU/L), ALT (IU/L), and GGT (IU/L) were converted, if needed. Time‐dependent Cox regression was applied for studying the association between SBD and NLS in all patients (HRs and 95% CIs). Sex, genotype, birth year, and SBD as a time‐dependent factor for NLS were included in the multivariate model. The model was extended with additional factors, one by one, to assess the association with the endpoint. Because data were collected from multiple sites, sensitivity analyses (i.e., stepwise exclusion of centers as well as stepwise exclusion of geographic regions from the analysis) were performed to control for the heterogeneity between sites. A clock‐reset approach was used to visualize the association of SBD with NLS. All patients started without SBD. Then, patients who underwent SBD during follow‐up were censored at the age of SBD and restarted with a new risk in the SBD curve. Receiver operating characteristic (ROC) curve analysis was used to determine the cutoff for biochemistry at presentation and post‐SBD biochemistry in relation to predicting NLS after these points in time. The number of patients for whom data were present for each variable is indicated. Imputation of missing data was not attempted.
A two‐sided P value < 0.05 was considered statistically significant. All analyses were performed using IBM SPSS Statistics (Armonk, NY). Figures were constructed using IBM SPSS Statistics and GraphPad Prism 7.02 (GraphPad Software, La Jolla, CA).
Results
Baseline Data
The present study included 130 patients with compound heterozygous or homozygous pathological ATP8B1 mutations and a low‐GGT cholestasis phenotype (Fig. 1; data exported from REDCap on May 1, 2020). Among the patients, 15 initially presented with a benign recurrent intrahepatic cholestasis (BRIC) phenotype that later evolved into a severe FIC1 deficiency (PFIC1) phenotype. Six patients fulfilling the genetic inclusion criteria were excluded because they persistently presented a BRIC phenotype and not a severe FIC1 deficiency (PFIC1) phenotype. Of the 130 patients, 48 (37%) had been described in the literature; 520 patients were excluded because mutations did not meet our inclusion criteria, and 103 were excluded because genetic reports were not available (Fig. 1). We identified two cohorts with a homozygous mutation (i.e., c.923G > T; p. Gly308Val mutation [n = 18] or c.1660G > A; p. Asp554Asn mutation [n = 8]). The included patients were followed in centers from Europe (n = 58), North America (n = 37), and Asia (n = 35). Supporting Table S1 provides the observed mutations per included patient, and Supporting Fig. S1 shows the locations of all observed mutations in the FIC1 protein.
Fig. 1.
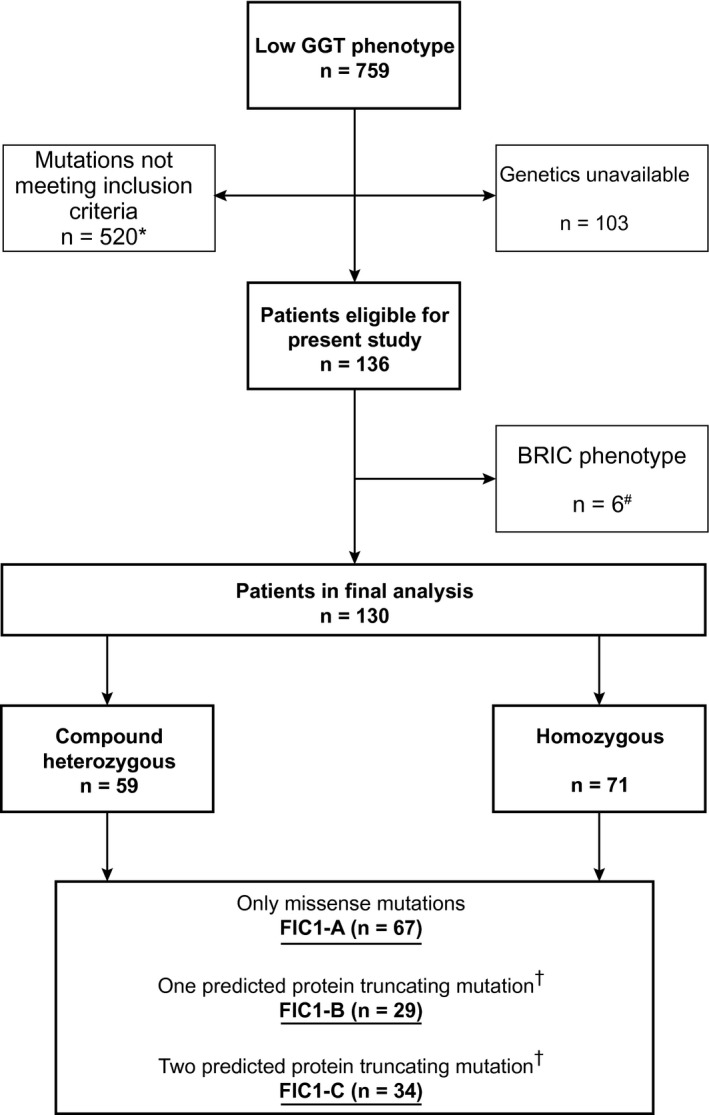
Flowchart of patient inclusion from the NAPPED database and subsequent categorization based on genotype. *Either only one mutation in ATP8B1, ATP8B1 mutations of no (known) clinical consequence, mutations in ABCB11/TJP2, or participation in investigational drug trials. sEpisodic cholestasis and/or pruritus and transient hepatocellular damage. †Splice site, frameshift due to deletion or insertion, nonsense, or duplication.
There were 71 (55%) males in this cohort of 130 patients with FIC1 deficiency (Table 1). The median birth year was 2007 (1999‐2012). Age at first presentation to the tertiary center was 0.6 (0.3‐2.2) years, illustrating the early onset of disease. Before or at presentation, 40% of the patients had used or were receiving UDCA. As expected, at presentation patients had elevated total sBAs and TSB with elevated aminotransferases. During follow‐up of a median of 4.2 (2.2‐9.8) years, 62 of 130 patients (48%) had undergone an SBD, and 38 of 130 patients (29%) had undergone LT (Supporting Table S2). HCC was not diagnosed in any of the patients, either before transplantation or in explant. A total of 8 patients (6%) died before LT, of whom 3 underwent SBD during follow‐up. Deaths were related to liver disease in 7 patients (age at death was 5.0 years [range 3.2‐10.7]) and unrelated to liver disease in 1 patient (death due to head trauma at age 13.2 years). Survival analysis showed that at 18 years of age, 44% of patients were alive with their native liver (Fig. 2). During adulthood (i.e., ≥ 18 years of age), 2 patients underwent LT (ages 20.0 and 20.2 years; indications for LT: pruritus [n = 1], unknown [n = 1]).
Table 1.
Characteristics and Biochemistry for All Patients According to FIC1 Genotype
| Parameter | All Patients n = 130) | Category of Mutations | P Value* | ||
|---|---|---|---|---|---|
| FIC1‐A (n = 67; no PPTM) | FIC1‐B (n = 29; 1 PPTM) | FIC1‐C (n = 34; 2 PPTMs) | |||
| Year of birth | 2007 (1999‐2012) | 2009 (2004‐2014) | 2006 (1995‐2011) | 2006 (1996‐2011) | 0.04 |
| Available n (%) | 130 (100) | 67 (100) | 29 (100) | 34 (100) | |
| Year of birth time frame | 1981‐2019 | 1981‐2019 | 1982‐2017 | 1984‐2017 | — |
| Male, n (%) | 71 (55) | 33 (49) | 17 (59) | 21 (62) | 0.09 |
| Available n (%) | 130 (100) | 67 (100) | 29 (100) | 34 (100) | |
| Age at first visit, years | 0.6 (0.3‐2.2) | 0.8 (0.4‐3.0) | 0.9 (0.4‐2.7) | 0.4 (0.2‐0.7) | 0.004 |
| Available n (%) | 130 (100) | 67 (100) | 29 (100) | 34 (100) | |
| Year of first visit | 2010 (2006‐2014) | 2011 (2008‐2014) | 2010 (1999‐2013) | 2007 (1996‐2013) | 0.01 |
| Available n (%) | 130 (100) | 67 (100) | 29 (100) | 34 (100) | |
| Year of first visit time frame | 1982‐2019 | 1982‐2019 | 1989‐2018 | 1985‐2017 | — |
| Before presentation, ever treated with: | |||||
| UDCA, n (%) | 41 of 103 (40) | 23 of 46 (50) | 10 of 26 (39) | 8 of 31 (26) | 0.01 |
| Rifampicin, n (%) | 16 of 103 (16) | 6 of 46 (9) | 3 of 26 (12) | 7 of 31 (23) | 0.48 |
| Phenobarbital, n (%) | 10 of 103 (10) | 2 of 46 (3) | 4 of 26 (15) | 4 of 31 (13) | 0.28 |
| Cholestyramine, n (%) | 12 of 103 (12) | 5 of 46 (8) | 2 of 26 (8) | 5 of 31 (16) | 0.74 |
| Antihistamines, n (%) | 9 of 103 (9) | 5 of 46 (8) | 2 of 26 (8) | 2 of 31 (7) | 0.35 |
| Laboratory data at presentation | |||||
| sBAs, μmol/L | 179 (122‐220) | 202 (138‐223) | 177 (129‐211) | 153 (117‐200) | 0.43 |
| Available n (%) | 69 (53) | 32 (48) | 14 (48) | 23 (68) | |
| TSB, μmol/L | 129 (64‐220) | 113 (61‐193) | 212 (123‐373) | 125 (65‐173) | 0.02 |
| Available n (%) | 103 (79) | 46 (69) | 24 (83) | 33 (97) | |
| ALT, IU/L | 48 (31‐82) | 55 (30‐86) | 41 (31‐67) | 49 (31‐73) | 0.67 |
| Available n (%) | 102 (78) | 45 (67) | 23 (79) | 34 (100) | |
| AST, IU/L | 66 (50‐86) | 64 (48‐84) | 67 (54‐107) | 69 (51‐86) | 0.64 |
| Available n (%) | 89 (68) | 40 (60) | 21 (72) | 28 (82) | |
| GGT, IU/L | 23 (17‐35) | 22 (15‐35) | 37 (21‐41) | 23 (15‐31) | 0.26 |
| Available n (%) | 90 (69) | 37 (55) | 24 (79) | 30 (88) | |
| PLT, 109/L | 461 (313‐569) | 365 (241‐538) | 399 (288‐484) | 530 (394‐625) | 0.02 |
| Available n (%) | 57 (44) | 24 (36) | 14 (48) | 19 (56) | |
Note: Data are presented as medians and interquartile ranges.
Mantel‐Haenszel test for trend or Kruskal‐Wallis tests, as appropriate, were used to test differences among FIC1‐A, FIC1‐B, and FIC1‐C.
Abbreviation: IQR, interquartile range.
Fig. 2.
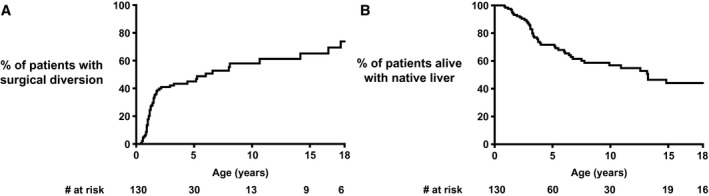
Proportion of all patients alive with native liver with a surgical diversion over time (A) and proportion of patients alive with native liver (B) in all patients.
Associations Between Genotype Category and Characteristics at Presentation
Table 1 presents the patient characteristics and biochemistry at presentation to the tertiary referral center for all patients with FIC1 genotypes according to the genotype categories (FIC1‐A, n = 67; FIC1‐B, n = 29; and FIC1‐C, n = 34). Patients with an FIC1‐C genotype presented to the referral center at 0.4 years (range, 0.2‐0.7) (i.e., 0.4 and 0.5 years earlier compared with FIC1‐A and FIC1‐B, respectively; P = 0.01). At presentation, levels of sBAs, aminotransferases, and GGT were comparable among the three genotype groups. Patients with an FIC1‐B genotype presented the highest TSB levels (FIC1‐A 113 [61‐193], FIC1‐B 212 [123‐373], and FIC1‐C 125 [65‐173] μmol/L; P = 0.02). Patients with an FIC1‐C genotype presented with higher PLTs (FIC1‐A 365 [241‐538], FIC1‐B 399 [288‐484], and FIC1‐C 530 [394‐625] 109/L; P = 0.02). Half of the patients with an FIC1‐A genotype had used or were using UDCA (50%) before or at presentation, which was a larger proportion of patients than in the FIC1‐B (39%) or FIC1‐C (26%) genotypes (P = 0.01). The difference in use of UDCA did not appear to result in markedly improved biochemistry in comparison with the other patient groups (Table 1). In patients with an FIC1‐A genotype, significant differences in biochemistry at presentation were not observed between patients who had used or were using UDCA and those who never used UDCA. A similar analysis could not reliably be performed for FIC1‐B and FIC1‐C due to lower numbers.
Associations Between Baseline Characteristics and SBD or NLS
Figure 2 shows that by the age of 18 years, 74% of the patients alive with their native liver had undergone SBD. A lower percentage of patients with an FIC1‐C genotype underwent SBD during follow‐up (% SBD at 10 years of age: FIC1‐A 65%, FIC1‐B 57%, and FIC1‐C 45%; P = 0.03; Fig. 3). NLS was comparable among the three genotype groups (% NLS at 10 years of age: FIC1‐A 67%, FIC1‐B 41%, and FIC1‐C 59%; P = 0.15; Fig. 3). In patients undergoing SBD during follow‐up, NLS was comparable (% NLS at 10 years of age: FIC1‐A 81%, FIC1‐B 67%, and FIC1‐C 77%; P = 0.19; Supporting Fig. S2), as well as in patients not undergoing SBD during follow‐up (% NLS at 10 years of age: FIC1‐A 39%, FIC1‐B 23%, and FIC1‐C 35%; P = 0.57; Supporting Fig. S2). It should be noted that the proportions of patients alive with native liver at the age of 10 years were lower in patients without SBD than in patients who had undergone an SBD during follow‐up (Supporting Fig. S2).
Fig. 3.
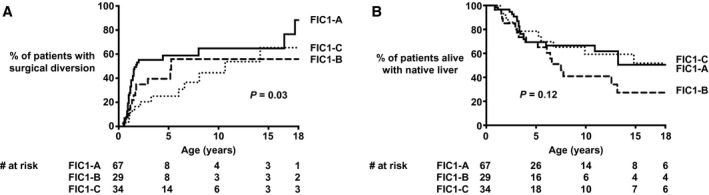
Proportion of patients with a surgical diversion (A) and proportion of patients alive with native liver (B) in patients without PPTMs (FIC1‐A), with one PPTM (FIC1‐B), or two PPTMs (FIC1‐C).
During follow‐up, relatively more females than males tended to undergo SBD during follow‐up (% SBD at 10 years of age: 67% vs. 53% in males; P = 0.06; Supporting Fig. S3). The NLS of females was comparable to that of males (% NLS at 10 years of age: 67% vs. 49%, respectively; P = 0.31; Supporting Fig. S3), as was NLS in females and males not undergoing SBD during follow‐up (% NLS at 10 years of age: 39% vs. 30%, respectively; P = 0.84; Supporting Fig. S4).
We performed ROC analyses for baseline biochemistry in relation to long‐term outcome (i.e., NLS). The areas under the curve (AUCs) were highest for sBAs and TSB: 0.71 (0.51‐0.92) and 0.62 (0.40‐0.83), respectively. Optimal cutoffs for prediction of NLS after first presentation were 194 µmol/L (sensitivity 85%, specificity 72%) for sBAs and 72 µmol/L (sensitivity 69%, specificity 50%) for TSB. These cutoffs were used in subsequent Kaplan‐Meier analyses, in which the time of presentation at the tertiary referral center was used as time point 0, because the biochemical parameters were recorded at that particular point in time. Patients with an sBA level < 194 µmol/L or TSB level < 72 µmol/L at presentation had a higher NLS compared to patients with levels above these thresholds: NLS at 15 years after presentation was 49% in patients with sBAs < 194 µmol/L and 15% in patients with sBAs ≥ 194 µmol/L (P = 0.03). NLS at 15 years after presentation was 47% in patients with a TSB < 72 µmol/L and 34% in patients with a TSB ≥ 72 µmol/L (P = 0.07). The long‐term outcome, compatible with the association between initial sBAs and outcome, could not be attributed to SBD or to either of the two subgroups (i.e., patients with and without a TSB < 72 µmol/L or ≥ 72 µmol/L at presentation); there were no significant differences observed in the proportion of patients undergoing SBD over time in these two groups (P values = 0.53 and 0.76, respectively). The proportion of patients undergoing SBD during follow‐up in patients with sBA levels < 194 µmol/L or ≥ 194 µmol/L at presentation was comparable; most patients (i.e., 78% and 90%, respectively) underwent SBD before the age of 2.5 years (P = 0.27). The distribution of genotype categories was comparable between the two groups (i.e., patients with sBA levels < 194 µmol/L or ≥ 194 µmol/L; P = 0.17).
SBD Baseline Data
A total of 62 patients underwent an SBD during follow‐up, at a median age of 5.9 years (Fig. 2). Based on the limited information available (n = 22), it appears that the main indication for SBD had been pruritus (21 of 22 [95%]). Of the 62 patients who underwent SBD, 49 underwent partial external biliary diversion (PEBD) (79%), 6 underwent gallbladder‐colic diversion (10%), 4 underwent ileal exclusion (5%), 1 underwent total biliary diversion (TBD) (2%), 1 underwent cholecystojejunostomy (2%), and 1 underwent an unknown procedure (2%) (Supporting Table S2). The estimated proportion of patients alive with native liver with an SBD at the ages of 5, 10, 15, and 18 years of age was 46%, 58%, 65%, and 74%, respectively (Fig. 2). Two patients underwent SBD during adulthood (at ages 18.8 and 20.0 years). Median follow‐up after SBD was 5.3 years (derived from Kaplan‐Meier curve). The initial diversion was converted to TBD in 3 patients: 2 within 1 month after initial SBD and 1 within 1.6 years after initial SBD. None of the initial diversions were surgically closed. LT was performed in a total of 15 of 62 patients (24%) who first underwent SBD (missing data in 1 patient). These patients underwent LT 2.5 (0.7‐6.2) years after initial SBD. Paired pruritus data, before and after SBD, were available in 29 patients. Before SBD, pruritus had been present in 28 of 29 patients (97%). Following SBD (i.e., at least 2 months and no more than 1 year after SBD), pruritus was present in 23 of 29 patients (79%) (P = 0.13). Retrospective analysis on pruritus data should be interpreted with caution. Nevertheless, some data could be derived from the patient files and could provide information of some relevance. In those patients for whom long‐term pruritus data were available (n = 23), half appeared to (partially) benefit from SBD: In 11 of 23 patients (48%), no improvement of pruritus was reported, whereas 6 of 23 patients (26%) had transient relief and 6 of 23 patients (26%) had sustained (partial or complete) relief of pruritus. A sustained partial or complete relief of pruritus was observed most often in FIC1‐C (n = 4 of 8 [50%]), compared with FIC1‐A (n = 1 of 9 [11%]) and FIC1‐B (n = 1 of 6 [17%]; P = 0.03).
Paired biochemistry data, before and after SBD, were analyzed. We noted a decrease in sBAs (230 [125‐282] to 74 [11‐177] μmol/L; median 49% decrease; P = 0.005; Fig. 4). There were no statistically significant differences in the post‐SBD sBA levels among patients with an FIC1‐A (81 [7‐191] μmol/L), FIC1‐B (74 [12‐296] μmol/L), and FIC1‐C (44 [4‐136] μmol/L) (P = 0.63) genotype or in TSB, ALT, or AST (P values: TSB, 0.89; ALT, 0.46; and AST, 0.42), respectively. Although numbers were small, the post‐SBD sBA levels associated with post‐SBD presence of pruritus were as follows: Patients with a post‐SBD sBA < 65 μmol/L were less likely to experience pruritus (n = 7 of 11 [63%]) compared to patients with a post‐SBD sBA ≥ 65 μmol/L (n = 9 of 9 [100%]) (P = 0.04).
Fig. 4.
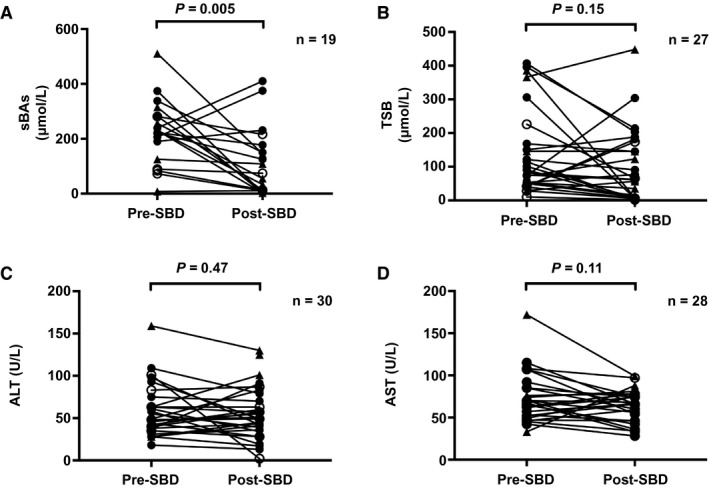
Pre‐SBD and post‐SBD biochemistry: sBAs (A), TSB (B), ALT (C), and AST (D) in patients with FIC1‐A (filled circles), FIC1‐B (open circles), and FIC1‐C (triangles) genotypes for whom paired data were available.
Association Between SBD and NLS
Time‐dependent Cox regression analysis (corrected for sex, genotype, and birth year) showed that SBD tended to be associated with NLS (overall HR, 0.55; 95% CI, 0.28‐1.03; P = 0.06; Fig. 5). However, the association between SBD and NLS was not similar across the three subgroups: An FIC1‐B genotype was associated with a significantly lower NLS (HR, 2.13; 95% CI, 1.09‐4.16; P = 0.03). We analyzed but could not detect a center bias to underlie this difference. We analyzed proportional hazards and observed that after 7 years of age, the proportional hazards appeared higher in FIC1‐B compared with FIC1‐A and FIC1‐C (log minus log plot; Supporting Fig. S5). This led us to use the landmark method (within the time‐dependent Cox regression, as stated previously) to scrutinize whether an FIC1‐B genotype was indeed associated with lower NLS, either before 7 years of age or after 7 years of age. The NLS did not significantly differ among the three genotypes of either age. However, in the latter period of time, a relatively high HR was observed in FIC1‐B (HR, 2.92; 95% CI, 0.77‐11.12; P = 0.11; 12 patients in analysis). To test the robustness of the overall regression model, sensitivity analyses were performed; we stratified for geographical region and caseload of the respective centers. HRs remained comparable when stratifying for geographic region (range, 0.52‐0.84). Excluding centers, one by one, yielded comparable results (range, 0.40‐0.79; analyses were performed for centers contributing more than 5 patients to the present cohort).
Fig. 5.
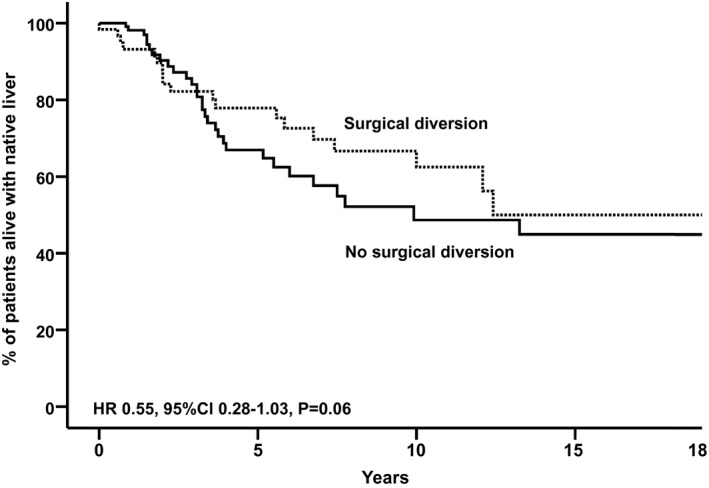
Observed native liver survival (x axis) in all patients with an FIC1 genotype, who underwent SBD (n = 62) or not (n = 68). The clock‐reset approach allows visualization of native liver survival up to SBD (solid line, all patients) and after SBD (dotted line, only patients who underwent SBD). The estimated HR is achieved by Cox regression, with SBD as a time‐dependent risk factor, adjusted for genotype, sex, and birth year.
Follow‐up After Diversion
The proportion of patients alive with native liver 10 years after SBD was 70% in FIC1‐A, 37% in FIC1‐B, and 66% in FIC1‐C (P = 0.18; Fig. 6). We compared NLS after SBD of patients who underwent PEBD to that of patients who underwent an SBD other than PEBD. NLS after SBD was comparable between the two groups (NLS at 10 years of age after SBD = 65% vs. 56%, respectively; P = 0.37). Because sBAs decreased significantly after SBD and had been associated with long‐term NLS in patients with severe BSEP deficiency,( 28 ) ROC analyses were performed for post‐SBD sBA levels in relation to NLS after SBD. A post‐SBD sBA level < 65 μmol/L tended to be associated with prolonged NLS after SBD (P = 0.05; AUC sBAs, 0.589; sensitivity, 80%; specificity, 61%; Fig. 7). A decrease of at least 76% (based on ROC) in sBAs was not associated with improved NLS after SBD (P = 0.21; AUC % change sBAs, 0.525; cutoff, 76%: sensitivity, 80%; specificity, 44%). The proportion of patients achieving a post‐SBD sBA level < 65 μmol/L was comparable among FIC1‐A (45%), FIC1‐B (50%), and FIC1‐C (63%) (P = 0.48), as was the proportion of patients achieving a decrease in sBA levels of at least 76% after SBD in FIC1‐A (22%), FIC1‐B (50%), and FIC1‐C (57%) (P = 0.16).
Fig. 6.
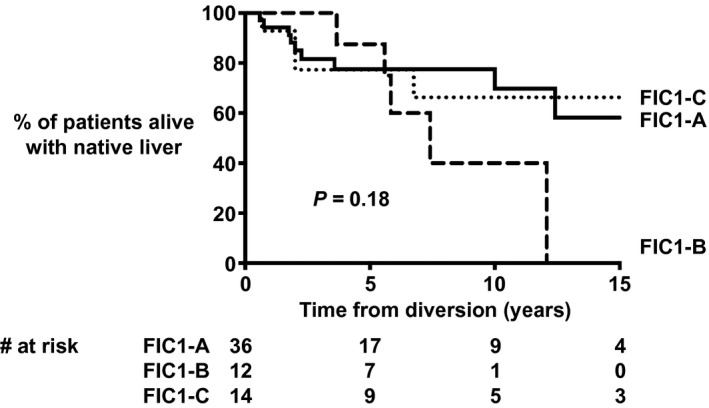
NLS after SBD in patients with an FIC1‐A (no truncating mutations), an FIC1‐B (one truncating mutation), or an FIC1‐C (two truncating mutations) genotype.
Fig. 7.
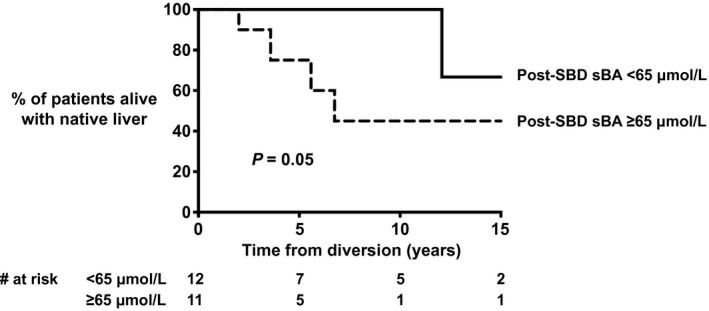
NLS after SBD in patients with a sBA concentration of < 65 umol/L versus ≥ 65 umol/L.
Discussion
This study aimed to define the natural history of FIC1 deficiency and assess the effect of the presence of PPTMs, sBA concentrations, and SBD on long‐term outcome. NAPPED herewith presents a cohort of genetically defined patients with FIC1 deficiency . The present data indicate that less than half of patients with FIC1 deficiency are alive with their native liver by the time they reach adulthood. Surprisingly, the genotype of patients with FIC1 deficiency, with respect to presence of PPTMs, did not appear to reliably predict the phenotype. sBA concentrations at baseline (i.e., < 194 µmol/L or ≥ 194 µmol/L) and after SBD (i.e., < 65 µmol/L or ≥ 65 µmol/L) may provide clinicians some prognostic information during counseling. For example, the proportion of patients who reach 15‐year survival with native liver is 3‐fold higher in patients with an sBA concentration at presentation below the threshold of 194 µmol/L than in those with sBA concentrations above the threshold. Additionally, SBD tended to be associated with improved NLS.
Our data indicate that most patients with FIC1 deficiency present to tertiary referral centers before the age of 1 year, but some patients present as late as in their second decade of life. Variability in age at presentation was observed in patients with identical genotypes. It has been described that, even within the same family, a variable expression of disease may be present,( 30, 33, 34 ) which supports the notion that the phenotype of FIC1 deficiency is only to a limited degree determined by the genotype. However, this might partially reflect the referral pathway specialist centers, rather than only disease presentation. Upon their presentation at the tertiary referral center, patients had elevated levels of total sBAs, TSB, and aminotransferases. As reported, aminotransferases were less markedly elevated compared with severe BSEP deficiency( 28 ) (up to an approximate 5‐fold lower median), which has been described.( 14 ) Alfa‐fetoprotein may also discriminate between severe BSEP deficiency and FIC1 deficiency,( 13 ) yet data unavailability precluded performing such an analysis.
We categorized patients according to the presence of PPTMs. Patients with a PFIC1 phenotype (i.e., continuous cholestasis and/or pruritus and continuous hepatocellular damage) were suggested to harbor truncating mutations more often than patients with a BRIC1 phenotype (i.e., episodic cholestasis and/or pruritus and transient hepatocellular damage).( 35 ) Quantification of FIC1 transport functionality has been difficult, partly because the exact transport mechanism remains elusive. Based on this limitation, we considered it reasonable to assume that the functionality of the FIC1 protein would be most severely affected in the case of a PPTM. We then hypothesized that a gene dose effect would be present. However, we could not observe a significant effect of the number of PPTMs on phenotype in contrast to, for example, patients with severe BSEP deficiency.( 28 ) The difference between FIC1 and BSEP deficiency in this respect could be related to the fact that mutations in ATP8B1 only indirectly affect bile acid transport across the canalicular membrane, in contrast to a direct effect on BSEP deficiency. The current hypothesis holds that the transport activity of BSEP is affected secondarily to alterations in the canalicular membrane composition, induced by FIC1 deficiency.( 6 ) Because the membrane composition is likely to be influenced by many more factors, genetic and environmental, the phenotype may be more influenced by variant compensatory influences. Support for this possibility can be derived from rather different phenotypic severities observed between individuals with identical FIC1 genotypes in the present study. The patients in our present study did not demonstrate clear phenotypic differences at baseline, nor were the genotype categories associated with NLS, biochemistry after SBD, or NLS after SBD. If anything, patients who would be expected to have a more severe genotype (i.e., FIC1‐C) presented at a significantly younger age than patients with predicted milder genotypes (i.e., FIC1‐A and FIC1‐B). On the contrary, patients with an FIC1‐C phenotype underwent SBD less often than patients with the FIC1‐A and FIC1‐B phenotypes, yet presented similar NLS. In addition, patients with the FIC1‐C phenotype were most likely to achieve a post‐SBD SBA concentration of less than 194 μmol/L. The fact that patients with identical genetic profiles may present different phenotypes, even within the same family,( 30, 34, 35 ) again indicates that the ATP8B1 genotype might not be the main determinant in the natural history of FIC1 deficiency. This is further supported by data from our study (which includes some patients described in the referenced papers). Patients with the homozygous c.2932‐3C > A mutation did not all express a BRIC phenotype; some already manifested a PFIC phenotype at presentation. Additionally, these patients underwent diversion at different ages (or did not undergo diversion at all). Notably, we excluded 1 patient with a homozygous c.2932‐3C > A mutation, due to the absence of a PFIC phenotype at last follow‐up. The case may be, therefore, that variations as described here result from factors other than the ATP8B1 genotype, as yet unknown. For example, environmental exposures (e.g., drugs, nutrition), local management strategies, or other genetic factors (such as modifiers) may exert a more significant effect than we currently anticipate.
This study identified two relative large patient subgroups with homozygous c.923G > T; p. Gly308Val or c.1660G > A; p. Asp554Asn mutations. The first mutation was originally found in the Amish kindred, first published by Clayton et al., who termed the disease “Byler disease.”( 36 ) Patients with homozygous p. Gly308Val mutations in the present study had mild disease in terms of NLS, provided they had undergone SBD at young age. Their NLS was significantly higher than patients with homozygous p. Asp554Asn, but it is unclear to what extent the lower fraction undergoing SBD in the latter group contributed to the observed difference. The p. Asp554Asn was originally identified in the Inuit and has subsequently been described in ethnically distinct individuals.( 30, 37 ) Both the p. Gly308Val and the p. Asp554Asn mutations are missense mutations, and both have been associated with a PFIC phenotype. Our study could not demonstrate clear differences in the phenotype of these two groups with homozygous mutations.
Although the number of nonfunctional protein‐type mutations could not reliably predict the phenotype of FIC1 deficiency, our data suggest that biochemistry at presentation in the tertiary center had prognostic value regarding the natural history of patients with FIC1 deficiency. An sBA level < 194 μmol/L was associated with long‐term NLS after presentation. This study provides a surrogate marker for long‐term outcome in FIC1 deficiency. The fact that an sBA level < 194 μmol/L at baseline is associated with improved NLS may suggest that it is beneficial for long‐term NLS to interrupt the enterohepatic circulation (EHC) and alleviate patients of bile acid accumulation. Our results should be interpreted with some caution, however. First, the sBA concentrations were not obtained in a standardized manner with respect to fasting or postprandial state. We cannot exclude that affected the results. Second, as stated previously, the results on sBA concentrations did not reach the predefined cutoff of statistical significance (P = 0.05 rather than P < 0.05).
The relationship between low sBAs and beneficial long‐term prognosis may be more complex than would be anticipated. SBD may protect the liver against systemic bile acid accumulation by interrupting the EHC. Our study demonstrates that SBD can successfully decrease sBAs, relatively soon after the procedure. Moreover, SBD tended to be associated with NLS in the multivariate, time‐dependent Cox regression model. Overall, an FIC1‐B genotype was associated with lower NLS, but NLS was not significantly different among the three groups if determined either before or after 7 years of NLS in this model. We could not identify why this difference existed. It should be noted that after 7 years of NLS, only 12 patients were still in the analysis. Although it is likely that these low numbers contributed (partly) to the observed trend toward lower NLS in patients with the FIC1‐B genotype, this outcome warrants further research. SBD appears to be associated with improved NLS in those patients who achieve a post‐SBD sBA level < 65 μmol/L. To characterize those patients who appeared to significantly benefit from SBD, we scrutinized patients with long‐term NLS and low sBAs after SBD. We could not, however, identify factors, such as genotype, sex, or age at diversion, that were associated with the response to SBD. In addition, at presentation, we could not identify differences in patient characteristics, biochemistry, or use of medication between patients who did and did not undergo SBD during follow‐up (data not shown). Our study lacked data regarding pre‐SBD fibrosis, which could have influenced post‐SBD outcomes. Nevertheless, our data do support one of our main findings, which is that genotype, with respect to the presence of PPTMs, does not appear to reliably predict the phenotype of FIC1 deficiency. The fact that FIC1 is not directly involved in bile acid transport may be of importance. Indeed, the molecular mechanisms by which FIC1 contributes to cholestasis still have to be elucidated in more detail. Cholestasis might result from the indirect inhibition of other canalicular transporters such as BSEP, resulting from increased vulnerability to canalicular bile acids.( 6, 11 ) Staining of these proteins does, however, appear to be unaffected in murine studies.( 26, 28 ) To elucidate these mechanisms, further (fundamental) research is required to gain more insight into this difficult disease.
In the present cohort, we did not observe any incidence of HCC before transplantation or following transplantation in explanted livers. Indeed, as far as we have found in the literature, there have been no reports of HCC in patients with FIC1 deficiency, which is in contrast to the high prevalence of HCC in severe BSEP deficiency.( 28, 38, 39 ) If indeed mutations in ATP8B1 affect bile acid transport indirectly, such as through BSEP, it is surprising that HCC does not develop in patients with FIC1 deficiency. Up until now, there is no proven explanation for this discrepancy between the two diseases. We speculate that the tumor‐promoting pathways described in severe BSEP deficiency( 40 ) are not induced in FIC1 deficiency. The zonal or subcellular accumulation of bile acids may differ between the two diseases, as well as the composition of the retained bile acids, which may lead to differences in the toxicity and/or carcinogenicity.
We realize that FIC1 deficiency extends beyond the liver, due to expression in tissues such as intestine, pancreas, lungs, and inner ear. Patients may present diarrhea, pancreatitis, or exocrine pancreatic insufficiency and sensorineural hearing loss.( 14, 16, 17, 18, 21 ) Currently, extrahepatic manifestations of FIC1 deficiency are not documented in our REDCap database; therefore, they could not be analyzed in the present paper. Some results in the present study should therefore be interpreted with caution, because we cannot exclude the possibility that factors other than the hepatic phenotype have contributed to decisions regarding SBD or LT. Future research initiatives of NAPPED will focus on elucidating the extrahepatic features and their impact on the disease, both before and after LT. For example, therapy‐resistant diarrhea may complicate the posttransplant disease course.( 27 )
Despite its large size, our study was limited by missing data. For example, data regarding the indication for LT were lacking in 10 patients. However, we feel that the numbers are sufficient to provide insights into the disease. Because patients in the present study have been included from regions all over the world, local treatment strategies may have interfered with outcomes. However, sensitivity analyses showed that neither the geographical region nor the center from which patients originated significantly affected our main results. Finally, follow‐up in this study stopped at the time of LT. Therefore, complications after LT, such as (aggravated) diarrhea, graft steatosis, and inflammation, have not been studied in this analysis.( 23, 27, 41, 42 )
In conclusion, our study shows that less than 50% of the patients with FIC1 deficiency reach adulthood with their native liver. The predicted genotypic severity with respect to PPTMs did not reliably predict the long‐term phenotype. Biochemical parameters, such as sBA concentrations at presentation and after SBD, can predict long‐term outcome and have potential to be used during counseling and treatment of patients. SBD tended to be associated with NLS. Our study further enhances the understanding of the difficult disease that FIC1 deficiency is, which allows for better care during childhood and adulthood, better prognostication of the disease, and better means to assess the effect of interruption of the EHC.
Author Contributions
D.B.E.v.W. contributed to the study concept and design, data acquisition, analysis and interpretation, statistical analysis, drafting of the manuscript, and obtainment of funding . R.J.T., E.G., I.J., B.L.S., and E. Sokal contributed to the study concept and design, data acquisition, and critical revision of the manuscript for important intellectual content. T.G., A.K., A.S., P.C., P.L., N.R., M.S., D.B., T.A., N.M., E.N., D.K., G.N., H.A., B.F., J.B.F.H., D.S., C.A., D.D., F.L., C.G., L.H., G.M.B., Y.M‐G., A.A., J.B., A.D., P.L.C., D.K‐S., E. Sturm, W.L.v.d.W., J‐S.W., L.L., Ö.D., N.K., M.H.J., R.F., C.J‐R., S.A., M.C., M. Ruiz, C.T.F., F.O., H.W., K.K., H‐L.C., E.C., A.F., J.Q.B., E.M.A., R.J.S., F.J.S., K.M.L., P.J.M., P.R., Y.T., G.S.R., S.H., B.K., M. Rogalidou, and W.W.K. contributed to the acquisition of data and critical revision of the manuscript for important intellectual content. E.J. contributed to the critical revision of the manuscript for important intellectual content. B.H. contributed to the study concept and design, data analysis and interpretation, statistical analysis, drafting of the manuscript, and obtainment of funding . H.V. contributed to the study concept and design, data acquisition, analysis and interpretation, drafting of the manuscript, and obtainment of funding.
Supporting information
Supplementary Material
Acknowledgment
The authors thank the patients and their families for the important data; all contributing centers and investigators who are currently or will soon be participating in NAPPED; and all of our individuals who provided care for the patients included in this study and/or have supported the present initiative.
Supported by an M.D./Ph.D. scholarship from the University of Groningen; European Society for Pediatric Gastroenterology, Hepatology, and Nutrition Networking Grant 2019; National Institutes of Health (U01DK062436, U01DK62453, UL1 TR002535, U01DK103149, U01DK062481, UL1TR000003, U01DK062466, U01DK062500, UL1TR001872, and U01DK084536); Seattle Children’s Hospital (DK084575); and unrestrictive research grants from Albireo and Mirum Pharmaceuticals.
Potential conflict of interest: Dr. Sokal consults, advises, owns stock, holds intellectual property rights, and is employed by Promethera. Dr. Thompson consults and owns stock in Generation Bio. He consults for Albireo, Mirum, EVOX, Alnylam, Rectify, Sana, Retrophin, and Qing. Dr. Hansen consults and received grants from CymaBay, Intercept, Albireo, and Mirum. She consults for Chemobay and Calliditas. Dr. Kamath consults and received grants from Mirum, Albireo, and Audentes. Dr. Kelly consults and received grants from Intercept, Mirum, and Albireo. Dr. Gonzalez consults for Mirum, Albireo, and CTRS. Dr. Verkade consults and received grants from Mirum and Albireo. Dr. Wang consults for Mirum and Albireo. Dr. Loomes consults and received grants from Mirum and Albireo. Dr. Rosenthal consults and received grants from Gilead, AbbVie, Merck, Retrophin, Albireo, and Mirum. He consults for Audentes and Dicerna. He received grants from Arrowhead. Dr. Czubkowski received grants from Albireo and Mirum. Dr. Sokol advises Retrophin, Mirum, and Albireo. Dr. Karnsakul received grants from Albireo and Gilead.
References
Author names in bold designate shared co‐first authorship.
- 1.Elferink RO, Groen AK. Genetic defects in hepatobiliary transport. Biochim Biophys Acta 2002;1586:129‐145. [DOI] [PubMed] [Google Scholar]
- 2.Bull LN, Thompson RJ. Progressive familial intrahepatic cholestasis. Clin Liver Dis 2018;22:657‐669. [DOI] [PubMed] [Google Scholar]
- 3.Noe J, Kullak‐Ublick GA, Jochum W, Stieger B, Kerb R, Haberl M, et al. Impaired expression and function of the bile salt export pump due to three novel ABCB11 mutations in intrahepatic cholestasis. J Hepatol 2005;43:536‐543. [DOI] [PubMed] [Google Scholar]
- 4.Bull LN, Van Eijk MJT, Pawlikowska L, DeYoung JA, Juijn JA, Liao M, et al. A gene encoding a P‐type ATPase mutated in two forms of hereditary cholestasis. Nat Genet 1998;18:219‐224. [DOI] [PubMed] [Google Scholar]
- 5.Andersen JP, Vestergaard AL, Mikkelsen SA, Mogensen LS, Chalat M, Molday RS. P4‐ATPases as phospholipid flippases: structure, function, and enigmas. Front Physiol 2016;7:275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paulusma CC, de Waart DR, Kunne C, Mok KS, Elferink RP. Activity of the bile salt export pump (ABCB11) is critically dependent on canalicular membrane cholesterol content. J Biol Chem 2009;284:9947‐9954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Folmer DE, van der Mark VA, Ho‐Mok KS, Oude Elferink RP, Paulusma CC. Differential effects of progressive familial intrahepatic cholestasis type 1 and benign recurrent intrahepatic cholestasis type 1 mutations on canalicular localization of ATP8B1. Hepatology 2009;50:1597‐1605. [DOI] [PubMed] [Google Scholar]
- 8.Cai SY, Gautam S, Nguyen T, Soroka CJ, Rahner C, Boyer JL. ATP8B1 deficiency disrupts the bile canalicular membrane bilayer structure in hepatocytes, but FXR expression and activity are maintained. Gastroenterology 2009;136:1060‐1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen F, Ananthanarayanan M, Emre S, Neimark E, Bull LN, Knisely AS, et al. Progressive familial intrahepatic cholestasis, type 1, is associated with decreased farnesoid X receptor activity. Gastroenterology 2004;126:756‐764. [DOI] [PubMed] [Google Scholar]
- 10.Frankenberg T, Miloh T, Chen FY, Ananthanarayanan M, Sun AQ, Balasubramaniyan N, et al. The membrane protein ATPase class I type 8B member 1 signals through protein kinase C zeta to activate the farnesoid X receptor. Hepatology 2008;48:1896‐1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Demeilliers C, Jacquemin E, Barbu V, Mergey M, Paye F, Fouassier L, et al. Altered hepatobiliary gene expressions in PFIC1: ATP8B1 gene defect is associated with CFTR downregulation. Hepatology 2006;43:1125‐1134. [DOI] [PubMed] [Google Scholar]
- 12.Koh S, Takada T, Kukuu I, Suzuki H. FIC1‐mediated stimulation of FXR activity is decreased with PFIC1 mutations in HepG2 cells. J Gastroenterol 2009;44:592‐600. [DOI] [PubMed] [Google Scholar]
- 13.Davit‐Spraul A, Fabre M, Branchereau S, Baussan C, Gonzales E, Stieger B, et al. ATP8B1 and ABCB11 analysis in 62 children with normal gamma‐glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatology 2010;51:1645‐1655. [DOI] [PubMed] [Google Scholar]
- 14.Pawlikowska L, Strautnieks S, Jankowska I, Czubkowski P, Emerick K, Antoniou A, et al. Differences in presentation and progression between severe FIC1 and BSEP deficiencies. J Hepatol 2010;53:170‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tygstrup N, Steig BA, Juijn JA, Bull LN, Houwen RH. Recurrent familial intrahepatic cholestasis in the Faeroe Islands: phenotypic heterogeneity but genetic homogeneity. Hepatology 1999;29:506‐508. [DOI] [PubMed] [Google Scholar]
- 16.Stapelbroek JM, Peters TA, van Beurden DH, Curfs JH, Joosten A, Beynon AJ, et al. ATP8B1 is essential for maintaining normal hearing. Proc Natl Acad Sci U S A 2009;106:9709‐9714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ray NB, Durairaj L, Chen BB, McVerry BJ, Ryan AJ, Donahoe M, et al. Dynamic regulation of cardiolipin by the lipid pump Atp8b1 determines the severity of lung injury in experimental pneumonia. Nat Med 2010;16:1120‐1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Folvik G, Hilde O, Helge GO. Benign recurrent intrahepatic cholestasis: review and long‐term follow‐up of five cases. Scand J Gastroenterol 2012;47:482‐488. [DOI] [PubMed] [Google Scholar]
- 19.Nagasaka H, Yorifuji T, Kosugiyama K, Egawa H, Kawai M, Murayama K, et al. Resistance to parathyroid hormone in two patients with familial intrahepatic cholestasis: possible involvement of the ATP8B1 gene in calcium regulation via parathyroid hormone. J Pediatr Gastroenterol Nutr 2004;39:404‐409. [DOI] [PubMed] [Google Scholar]
- 20.Verhulst PM, van der Velden LM, Oorschot V, van Faassen EE, Klumperman J, Houwen RH, et al. A flippase‐independent function of ATP8B1, the protein affected in familial intrahepatic cholestasis type 1, is required for apical protein expression and microvillus formation in polarized epithelial cells. Hepatology 2010;51:2049‐2060. [DOI] [PubMed] [Google Scholar]
- 21.Walkowiak J, Jankowska I, Pawlowska J, Bull L, Herzig KH, Socha J. Normal pancreatic secretion in children with progressive familial intrahepatic cholestasis type 1. Scand J Gastroenterol 2006;41:1480‐1483. [DOI] [PubMed] [Google Scholar]
- 22.Li L, Deheragoda M, Lu Y, Gong J, Wang J. Hypothyroidism associated with ATP8B1 deficiency. J Pediatr 2015;167:1334‐1339.e1. [DOI] [PubMed] [Google Scholar]
- 23.Bull LN, Pawlikowska L, Strautnieks S, Jankowska I, Czubkowski P, Dodge JL, et al. Outcomes of surgical management of familial intrahepatic cholestasis 1 and bile salt export protein deficiencies. Hepatol Commun 2018;2:515‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ismail H, Kalicinski P, Markiewicz M, Jankowska I, Pawlowska J, Kluge P, et al. Treatment of progressive familial intrahepatic cholestasis: liver transplantation or partial external biliary diversion. Pediatr Transplant 1999;3:219‐224. [DOI] [PubMed] [Google Scholar]
- 25.Hollands CM, Rivera‐Pedrogo FJ, Gonzalez‐Vallina R, Loret‐de‐Mola O, Nahmad M, Burnweit CA. Ileal exclusion for Byler’s disease: an alternative surgical approach with promising early results for pruritus. J Pediatr Surg 1998;33:220‐224. [DOI] [PubMed] [Google Scholar]
- 26.Arnell H, Bergdahl S, Papadogiannakis N, Nemeth A, Fischler B. Preoperative observations and short‐term outcome after partial external biliary diversion in 13 patients with progressive familial intrahepatic cholestasis. J Pediatr Surg 2008;43:1312‐1320. [DOI] [PubMed] [Google Scholar]
- 27.Nicastro E, Stephenne X, Smets F, Fusaro F, de Magnee C, Reding R, et al. Recovery of graft steatosis and protein‐losing enteropathy after biliary diversion in a PFIC 1 liver transplanted child. Pediatr Transplant 2012;16:E177‐E182. [DOI] [PubMed] [Google Scholar]
- 28.van Wessel DBE, Thompson RJ, Gonzales E, Jankowska I, Sokal E, Grammatikopoulos T, et al. Genotype correlates with the natural history of severe bile salt export pump deficiency. J Hepatol 2020;73:84‐93. [DOI] [PubMed] [Google Scholar]
- 29.Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap): a metadata‐driven methodology and workflow process for providing translational research informatics support. J Biomed Inform 2009;42:377‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klomp LW, Vargas JC, van Mil SW, Pawlikowska L, Strautnieks SS, van Eijk MJ, et al. Characterization of mutations in ATP8B1 associated with hereditary cholestasis. Hepatology 2004;40:27‐38. [DOI] [PubMed] [Google Scholar]
- 31.Takatsu H, Tanaka G, Segawa K, Suzuki J, Nagata S, Nakayama K, et al. Phospholipid flippase activities and substrate specificities of human type IV P‐type ATPases localized to the plasma membrane. J Biol Chem 2014;289:33543‐33556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van der Woerd WL, Mulder J, Pagani F, Beuers U, Houwen RHJ, van de Graaf SFJ. Analysis of aberrant pre‐messenger RNA splicing resulting from mutations in ATP8B1 and efficient in vitro rescue by adapted U1 small nuclear RNA. Hepatology 2015;61:1382‐1391. [DOI] [PubMed] [Google Scholar]
- 33.Bourke B, Goggin N, Walsh D, Kennedy S, Setchell KD, Drumm B. Byler‐like familial cholestasis in an extended kindred. Arch Dis Child 1996;75:223‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bull LN, Juijn JA, Liao M, van Eijk MJ, Sinke RJ, Stricker NL, et al. Fine‐resolution mapping by haplotype evaluation: the examples of PFIC1 and BRIC. Hum Genet 1999;104:241‐248. [DOI] [PubMed] [Google Scholar]
- 35.van Ooteghem NA, Klomp LW, van Berge‐Henegouwen GP, Houwen RH. Benign recurrent intrahepatic cholestasis progressing to progressive familial intrahepatic cholestasis: low GGT cholestasis is a clinical continuum. J Hepatol 2002;36:439‐443. [DOI] [PubMed] [Google Scholar]
- 36.Clayton RJ, Iber FL, Ruebner BH, McKusick VA. Byler disease: fatal familial intrahepatic cholestasis in an Amish kindred. Am J Dis Child 1969;117:112‐124. [PubMed] [Google Scholar]
- 37.Klomp LW, Bull LN, Knisely AS, van Der Doelen MA, Juijn JA, Berger R, et al. A missense mutation in FIC1 is associated with Greenland familial cholestasis. Hepatology 2000;32:1337‐1341. [DOI] [PubMed] [Google Scholar]
- 38.Knisely AS, Strautnieks SS, Meier Y, Stieger B, Byrne JA, Portmann BC, et al. Hepatocellular carcinoma in ten children under five years of age with bile salt export pump deficiency. Hepatology 2006;44:478‐486. [DOI] [PubMed] [Google Scholar]
- 39.Strautnieks SS, Byrne JA, Pawlikowska L, Cebecauerova D, Rayner A, Dutton L, et al. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology 2008;134:1203‐1214. [DOI] [PubMed] [Google Scholar]
- 40.Iannelli F, Collino A, Sinha S, Radaelli E, Nicoli P, D’Antiga L, et al. Massive gene amplification drives paediatric hepatocellular carcinoma caused by bile salt export pump deficiency. Nat Commun 2014;5:3850. [DOI] [PubMed] [Google Scholar]
- 41.Miyagawa‐Hayashino A, Egawa H, Yorifuji T, Hasegawa M, Haga H, Tsuruyama T, et al. Allograft steatohepatitis in progressive familial intrahepatic cholestasis type 1 after living donor liver transplantation. Liver Transpl 2009;15:610‐618. [DOI] [PubMed] [Google Scholar]
- 42.Aydogdu S, Cakir M, Arikan C, Tumgor G, Yuksekkaya HA, Yilmaz F, et al. Liver transplantation for progressive familial intrahepatic cholestasis: clinical and histopathological findings, outcome and impact on growth. Pediatr Transplant 2007;11:634‐640. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material