

Abstract
Protein crystallography (PX) is widely used to drive advanced stages of drug optimization or to discover medicinal chemistry starting points by fragment soaking. However, recent progress in PX could allow for a more integrated role into early drug discovery. Here, we demonstrate for the first time the interplay of high throughput synthesis and high throughput PX. We describe a practical multicomponent reaction approach to acrylamides and ‐esters from diverse building blocks suitable for mmol scale synthesis on 96‐well format and on a high‐throughput nanoscale format in a highly automated fashion. High‐throughput PX of our libraries efficiently yielded potent covalent inhibitors of the main protease of the COVID‐19 causing agent, SARS‐CoV‐2. Our results demonstrate, that the marriage of in situ HT synthesis of (covalent) libraires and HT PX has the potential to accelerate hit finding and to provide meaningful strategies for medicinal chemistry projects.
Keywords: covalent inhibitors, high-throughput protein crystallography, multicomponent reactions, nanosynthesis, SARS-CoV-2
Parallel, automated, and practical multicomponent reaction synthesis of high diversity acrylamides/ester libraries and HT protein crystallography (PX) yielded potent covalent inhibitors of the main protease of the COVID‐19 causing agent SARS‐CoV‐2. More general, integration of HT PX and HT synthesis into the “design‐make‐test‐analyze” cycle will open unprecedented horizons in medicinal chemistry.

Introduction
Paul Ehrlich's Corpora non agunt, nisi fixata is the maxim of modern receptor pharmacology.[1] Protein crystallography (PX) like no other biophysical method contributed to the understanding of ligand receptor interactions.[2] Hydrogen bonds, charge–charge interactions, pi stacking interactions, halogen bonds, and shape complementarity of receptor and ligand can be “seen” at atomic resolution.[3] Over the last decades PX has changed the face of drug discovery. Many industrial and academic drug discovery projects are supported by PX with cocrystallized ligands. However, in most cases PX plays an auxiliary but not a leading role: hits from HTS are optimized to leads and sometimes crystal structures are performed and used retrospectively to explain SAR (Figure 1 A). However, novel technologies increased the pace of PX: bottlenecks of HT PX, such as improved synchrotron radiation,[4] improved detectors,[5] microcrystal delivery system for serial femtosecond crystallography,[6] parallel soaking of compound libraries,[7] crystal fishing and mounting,[8] data analysis of low occupancy compounds,[9] were developed over the last decade and a sizable library of compounds can be screened in only a couple of weeks.[10] HT soaking of static fragment libraries is nowadays routinely offered to clients at synchrotrons (Figure 1 B).[10, 11] These advances have been driven by the work of thousands of members of the structural biology community globally.[12] On the other hand there are tremendous developments in miniaturization and automation of synthetic chemistry.[13] Reaction optimization is performed in an automated fashion in 1536 well plates and large libraries of classical and novel chemistries can be rapidly produced nowadays using nano dispensing technologies.[14] Hence, its seems to be time to truly integrate HT synthesis with HT PX into the early drug discovery cycle (Figure 1 C).
Figure 1.

Protein crystallography and drug discovery. A) In many hit to lead projects protein crystallography is only sporadically used to confirm binding and enable faster SAR. B) Fragment soaking to find medicinal chemistry starting points. C) The high‐throughput synthesis performed herein combined with high throughput crystallography has the potential to accelerate early drug discovery.
Here, we show the synergy between HT synthesis and HT PX with the SARS‐CoV‐2 main protease 3CLpro, a cysteine protease that plays a critical role in CoV replication, displaying a very low mutation rate, and making it an attractive drug discovery target to combat COVID‐19.[15] The majority of described 3CLpro inhibitors bind covalently to the active site cysteine, as is the clinically tested PF‐07321332.[16] Here we focused on the synthesis of acrylamides and acrylic acid esters to create diverse libraries of electrophiles which we screened by HT PX. Owing to our longstanding experience in multicomponent reaction chemistry, we designed a one‐pot pathway to diversified libraries of highly substituted acrylamides and ‐esters.[17] We hypothesized that acrylic acid might be employed in Ugi (U‐4CR) and Passerini (P‐3CR) style reactions (Figure 1). However, acrylic acid is a very reactive molecule, readily undergoing polymerization reaction, as are acrylamides and ‐esters. Moreover, ammonia was often described in the literature as an inferior or non‐functional substrate in the Ugi reaction.[18] Thus, it was a priori, not clear if our reaction design will lead to a successful outcome.
Results and Discussion
We initiated our work with the 0.5 mmol reaction of isobutyric aldehyde A and equivalent amount of acrylic acid B, tert‐butyl isocyanide C, and aqueous ammonia D in 0.5 mL trifluoroethanol (Scheme 1). We chose trifluoroethanol as a non‐nucleophilic solvent, which seems to have beneficial properties in the suppression of side reactions.[19] Surprisingly, the expected product E1 precipitated and could be isolated by simple filtration at an acceptable 72 % yield and excellent purity.
Scheme 1.

Conditions and yield of the Ugi product (E1).
Next, we aimed to create a library of acrylamides using parallel synthesis techniques (Figure 2 A; S2‐S7). We performed the reactions using 96‐well glass plates (1.5 mL per well) equipped with small stirring bars. We used 16 different isocyanides and 24 different aldehydes (Figure 2 B; Supporting Information, Figure S1). The stock solutions of the building blocks (0.1 mL of each, 5 M in trifluoroethanol) were added by using single, 8‐, and 12‐channel pipettes. The order of addition was ammonia in water, followed by the aldehyde component, followed by acrylic acid and isocyanide. The 96‐well plate was sealed and stirred overnight at room temperature (Figure 2 C, Figure S2).
Figure 2.

96‐Well parallel set‐up of acrylamide syntheses on a 0.5 mmol scale. A) General reaction scheme. B) 2D structures of building blocks, and yields of isolated products. Wells with precipitated products during the reaction, precipitation after refrigeration, and wells without precipitations are shown in green, blue and white, respectively. C) Efficient, parallel reagent transfer using multi pipettes. D) Picture of the 96‐well reaction with partially precipitated products. E) Easy isolation of precipitated products by simple vacuum filtration in a 96‐well format manifold.
Then, 0.5 mL diethyl ether was added to all wells and stirred for another 15 min. A total of 45 wells (47 %) showed considerable precipitate (Figure 2 D). The non‐precipitated wells were stored in a−20 °C refrigerator, and four additional products precipitated after 4 h (4 %). Forty‐seven wells resulted in no precipitation (49 %). The precipitates were transferred into a 96‐well filter plate using a multichannel pipette. Using an in‐house constructed vacuum manifold, the filtrate was removed, and the precipitate was washed further with two times 0.5 mL diethyl ether (Figure 2 E). The filter plate with the precipitates was dried under vacuum in a desiccator. To our surprise, the quality of the compounds, as measured by NMR, was very good (S88‐S237). The yields varied from 22 % to quantitative, with an average of 62 %. Ten of the remaining forty‐seven unprecipitated reactions were purified by flash chromatography, and these products could be obtained in yields from 35 % to 80 % (average 58 %).
The diversity of the building blocks working in this reaction is noteworthy. Aliphatic, bulky (C16), linear (C1, C2) and cyclic (C10), aromatic and aliphatic heterocyclic isocyanides included thiophene (C7), tetrahydrofuran (C6), and benzodioxolane (C14) worked well. Tested compatible functional groups in the isocyanide included alkene (C8), ether (C2, C12), nitrile (C9) and halo substituted benzenes (C3, C5, C13). The diversity of the aldehyde component is also remarkable, ranging from simple aliphatic (A13, A14, A22, A24), to cyclic (A15), to aromatic (A1–3, A5–8, A10–A12, A19) and heteroaromatic (A9, A23). Tested compatible functional groups in the aldehyde building blocks included alkene (A4), halo (A5, A7, A11), nitrile (A6), and ether (A3, A10). Other compounds noteworthy from further plates are E2–6. E2 Is a phenylalanine derivative, E3 and E4 are phthalimide derivatives, and E5 has a flexible indoyl ethyl amide moiety (Scheme 2).
Scheme 2.

Examples of synthesized acrylamides with amino acid, heterocyclic, extended acrylic acid, and natural product motifs.
An advantage of our method is that a majority of the products precipitate from the crude reaction mixtures, thus providing high‐quality acrylamides using a straightforward filtration procedure.
Lipophilicity is the single most crucial descriptor affecting potency, distribution, and elimination of a compound in the body. Highly lipophilic compounds tend to be insoluble in water, have high plasma protein binding, and are unable to reach the target at a high enough concentration to affect a biological response.[20] With an average cLogP of 2.56 varying from −1.2 to 5.8 our 96‐well library represents a good range of lipophilicity. Also, the average molecular weight of 335 Dalton is not too high to create unnecessary problems in a future modification of any hits to increase potency. Even more hydrophilic compounds also incorporating heterocyclic and natural product moieties are shown in Scheme 2. Compound E7 is an excellent example of the superior functional group compatibility of the reaction. Unprotected sugars and amino sugar, α,β,γ,δ‐unsaturated ketone double bonds and a macrocyclic lactone structure present in the antibiotic starting material tylosin are not affecting the selectivity of the reaction and the product is formed in 62 % yield.
Another critical descriptor of an acrylamide inhibitor is the substitution pattern around the double bond, which can be used to fine‐tune the reactivity towards nucleophiles. For example, different substituted acrylamides have been used to optimize the reactivity of the clinical RAS covalent inhibitor MRTX849.[21] Thus, we set out to synthesize exemplary substituted acrylamides to show the versatility of our method (Scheme 2). Once more, the reactions were performed on 0.5 mmol scales and resulted in satisfactory to excellent yields. Noteworthy, the acrylamide moiety in E12 was also used in the covalent kinase inhibitors afatinib, neratinib, and dacomitinib. Finally, we investigated the method scalability beyond the 0.5 mmol mark by preparing the acrylamide compound I‐H8 on a multigram scale following the procedure described above, obtaining 9 g of pure product, which corresponds to a 56 % yield (Figure 3).
Figure 3.
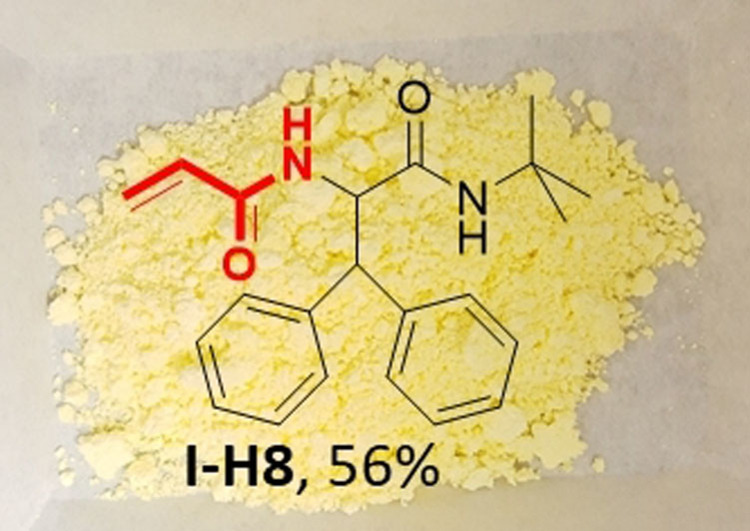
Structure of I‐H8 synthesized on a 9 g scale.
Similarly, we applied our HT synthesis to the creation of a Passerini reaction library using acrylic acid, aldehydes, and isocyanides (Figure 4, Figure S3). In contrast to Ugi reactions, the Passerini reaction favors aprotic, apolar solvents. Moreover, the products are less prone to precipitate from the reaction mixture due to one less amide group in the scaffold. After some experimentation, we found n‐pentane to be a suitable solvent where 13 % (12 wells) of the products of a 96‐well plate precipitated with excellent purity (Supporting Information). The 96‐well plate was based on the reaction of 71 different isocyanides (S9), 85 different aldehydes (S8) and acrylic acid, which were combined in a random fashion to avoid any bias. 1H NMR analysis of the full plate revealed that all reactions worked well and the purities in all the non‐precipitated cases were greater than 50 %, in most cases greater 80% and higher. Fast and efficient flash chromatography purification yielded the non‐precipitated products in high purity. Also noteworthy for the Passerini reaction is the great functional group compatibility, which can be seen in the products, for example the range of heterocycles, pyridine (II‐A11), indole (II‐F10), and benzopyran (II‐D2).
Figure 4.

High‐diversity parallel synthesis (96‐well plate) of acrylic acid ester library using 85 isocyanides and 88 aldehydes in the Passerini‐3CR (A). B) Heat plot with the yields as determined by 1H‐NMR. Explicit structures are compounds precipitated during the reactions (white boxed in the heat map).
Despite being a convenient one‐pot procedure, the above described 96‐well or multigram scales are not useful to create truly large acrylamide libraries (≫1000) with limited resources and in a given time. Therefore, we investigated our synthetic scheme under nanoscale synthesis conditions (Figure 5, Figure S4). For this, we used a positive‐pressure‐based low‐volume and contactless dispensing system (I‐DOT), as recently described by us.[22] Briefly, we used 384‐well polypropylene destination plates for our syntheses. Each destination plate was charged with 10 μL (excess amount) of acrylic acid/ammonium hydroxide mixture (0.5 M) using a multichannel pipette. Subsequently, oxo components (0.5 M, 125 nL) and then isocyanides (0.5 M, 125 nL) were transferred into the corresponding well using an I‐DOT nano dispenser, at a final concentration for the stoichiometric components of 6 mM. The 384‐well plates were filled using an algorithm developed in‐house that allows for random combinations of building blocks to avoid any synthesis bias (Figure S5).[22b] The total length of the autonomous dispensing procedure was ≈12 min per 384‐well plate. We produced a total of four 384‐well‐plates. The quality of the reactions on the nanomole scale was evaluated by direct injection into the mass spectrometer as recently described by us (Figure 5, S67).[14f] Accordingly, in 79 % of the reactions, product formation was observed, while for 21 % no product could be observed (Figure S6, S7, S8). To support our fast qualitative analysis of the nano synthesis, we reproduced 28 compounds on a 0.5 mmol scale, including full NMR and HRMS characterization (Figure 5, S88, S112). Gratifyingly, all 28 compounds precipitated during the upscaling reaction.
Figure 5.

Nanoscale, automated acrylamide synthesis. A 384‐well heat map indicating MS‐based analytics is shown. Shown below are the resynthesized compounds on a 0.5 mmol scale with isolated yields. III‐C6, III‐J24, and III‐N17 are shown in blue because of I‐DOT reagent transfer failure. However, their resynthesis showed medium product formation, indeed.
We tested our parallel synthesized electrophiles in high throughput crystallography to elaborate synergies. The SARS‐CoV‐2 3CLpro is an attractive workhorse, not only because it is a covalent drug discovery target to combat COVID‐19, but also because of the uncomplicated access to large amounts of crystallization‐quality enzyme.[15] Thus we established a high yielding 3CLpro expression/purification system which allowed us to soak highly concentrated solutions of our herein and previously described electrophile libraries into preformed crystals in a high throughput mode.[23] The compounds were selected to represent diversity in scaffolds and electrophiles. A total of 181 different compounds were soaked. The compounds were prepared as 100 mM stock solutions in DMSO. Large quantities of 3CLpro crystals were grown using the mosquito crystal liquid handler (SPT Labtech). After initial soak and diffraction tests to determine optimal DMSO concentrations (10 % v/v) and soak times (3 hours at room temperature), the crystals were grown for a day and selected for soaking using a software suite developed in‐house at the Swiss Light Source (SLS) as part of the internal Fast Fragment and Compound Screening (FFCS) platform. This software suite was used to design the experimental steps of the crystallographic compound screening as well as to keep track of each 3CLpro crystal from selection to soaking to harvesting. After crystal selection, compounds were dispensed accordingly and quickly (<3 min) with the Echo 550 Liquid Handler. Following a 3‐hour incubation time, crystals were harvested in a semi‐automated fashion with the Crystal Shifter, which is a motorized, x−y microscope stage allowing for rapid crystal mounting (≈100 crystals per hour), before cryopreservation and subsequent data collection.[8] A single X‐ray diffraction data set was successfully collected for 172 of the soaked crystals at the X06SA, X06DA, and X10S beamlines at SLS (Figure S9), with the remaining 9 soaking conditions yielding poorly diffracting crystals. In total, 245 soaking experiments were performed and one crystal was harvested from each experiment for subsequent data collection. Generally, multiple datasets were collected per crystal resulting in a total of 653 datasets collected for 245 mounted crystals. For each unique compound, either the diffraction data with the best properties or the only processable diffraction data was used for further analysis. High throughput, automated molecular replacement/refinement was performed on these datasets using the DIMPLE pipeline.[24] The resulting electron density maps and structures were then subjected to PanDDA analysis, a pan dataset analysis algorithm that can rapidly detect and identify weak or obscured signals caused by ligand binding or structural shifts versus a “ground‐state” apo structure.[25] Within 4 hours, all events were identified, with five datasets showing binding events in the 3CLpro active site (Figure 6 B). Three co‐crystal structures (II‐F10, II‐G11 and F3) were solved and refined using the regular electron density maps as they had interpretable density. For F1 and F2, which had weaker density in the active site, PanDDA ensemble modelling and refinement protocols were followed using the event maps to yield the refined co‐crystal structures. All structures were deposited in the Protein Data Bank (Table S1).
Figure 6.

High throughput crystallography of covalent library with SARS‐CoV‐2 3CLpro. A) Active site of 3CLpro (PDB ID 6Y2E).[26] The shape of the substrate pocket is indicated by the red dotted line. Cys145 is shown as yellow sticks. B) Examples of compounds giving a clear electron density: chloroacetamide F1 (PDB ID 7NUK); acrylic ester II‐F10 (PDB ID 7NT1); acrylic ester II‐G11 (PDB ID 7NT2); chloroacetamide F2 (PDB ID 7NTV); acrylamide F3 (PDB ID 7NT3); C) Table of inhibitions of cocrystallized compounds measured using a FRET assay. D) Representative FRET binding curve of compound F2.
In agreement with recent HT campaigns the structure release time of even larger screening campaigns can be as short as 2 weeks for over 90 different structures.[10] Four of the hits (II‐G11, II‐F10, F1, and F2) were found to be covalently bound to the active site Cys145 (Figure 6 B 1–4). They represent two classes of covalent inhibitors, the chloroacetamides and acrylic esters, both classes that have shown to be promising covalent inhibitors of multiple protein targets.[23, 27] They bind in a variety of modes, mainly interacting with the S1′, S1 and S2 subsites, primarily making π‐π interactions with His41 and hydrophobic interactions with the sidechains of the S1′ site (Figure 6 A). F3 was found to be non‐covalently bound to the 3Clpro, with its acrylamide warhead orientated away from the active Cys145. Instead, the phenol moiety forms a hydrogen bond with His163 and the benzodioxole forms π‐π interactions with His41. In order to verify whether the hits found by HT crystallography were capable of inhibiting the 3Clpro activity in vitro, a slightly modified, established FRET based IC50 assay was performed (S243).[28] The measured IC50 values ranged from 2.36 μM (F2) to exceeding 1 mM (F3) (Figures 6 C,D and Figure S10). The fact that F3 does not seem to inhibit the 3CLpro is not surprising considering the non‐covalent mode observed in the crystal structure combined with the concentration used in soaking.
Conclusion
X‐ray protein crystallography is an important tool in medicinal chemistry since it gives atomic resolution information about drugs binding to receptors which can be used to refine the compounds.[29] Our study provides several key conceptual advances in the context of HT synthesis and PX. This has important implications for an alternative early drug discovery cycle weighting PX. While the exponential increase in speed of structure determination is not reflected in the largely bystander role of PC, integration of HT PX with HT synthesis of more complex molecules than fragments could help to advance early drug discovery. We showed how HT synthesis can advantageously synergize with HT crystallography to yield single digit μM hits for the SARS‐CoV‐2 main protease with a variety of warheads involved in covalent bond formation, derived from newly synthesized libraries. This is conceptually different from the mostly performed approach of cocrystallizing or soaking existing static fragment libraries of low complexity.[10, 11] For this, we developed a novel, general, highly diverse multicomponent method for the one‐pot synthesis of acyl amides and esters, based on the building blocks acrylic acid, ammonia, aldehydes, and isocyanides. Noteworthy many substrates with different functional groups are compatible with the reaction and show good product formation. The reactions were performed on a 0.5 mmol scale in 96‐well plates, on a nanoscale in an automated fashion yielding 1.536 reactions, and also on a multigram scale. Thus, scalability has been established over 6 orders of magnitude. In the majority of 0.5 mmol and higher scale acrylamide reactions, the products precipitated, making this a valuable method for accessing high‐quality compound libraries. Furthermore, it highlights that, the 96‐well protocol is fast, and several plates can be produced per week, including NMR and SFC‐MS based quality control. A subset of 180 electrophiles were applied to high throughput crystallography by soaking of the main protease of the COVID‐19 causing agent SARS‐CoV‐2 yielding 5 cocrystal structures. The best compound featured a 2 μM inhibition of the protease in an enzymatic assay. While previous work describing screening of covalent libraries by high throughput crystallography was based on static covalent fragment libraries from commercial vendors, our herein exercised high throughput library synthesis is highly dynamic and can be easily adapted to the target of interest.[10] In another HT PX approach already approved drugs and drugs in clinical trials were cocrystallized with 3Clpro attempting a repurposing approach.[30] Due to the shear infinite scaffold and building block diversity applicable in MCRs, combined with the high throughput miniaturized synthesis format, many more structural opportunities can be realized.[31] With continuing technological advances in speed and scale of chemical synthesis[14a, 14b, 14c, 14d, 14f, 32] and macromolecular crystallography,[29a, 33] it is conceivable that a seamless process of in situ HT synthesis and HT PX can be performed in a cyclic fashion to truly guide compound optimization and complement the “design‐make‐test‐analyze” cycle (DMTA) in drug discovery. Assuming sufficient access to a synchrotron beamline and optimization of all working blocks, a cycle time of 1–2 weeks seems feasible which is comparable to current cycle times in the DMTA cycle but without crystallographic information. Clearly, the marriage of high throughput synthesis of small molecule (covalent) libraries and high throughput crystallography will help to accelerate hit finding and provide meaningful strategies for medicinal chemistry projects.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This research has been supported to (AD) by the National Institute of Health (NIH) (2R01GM097082‐05), the European Lead Factory (IMI) under grant agreement number 115489, the Qatar National Research Foundation (NPRP6‐065‐3‐012). Moreover, funding was received through ITN “Accelerated Early stage drug dIScovery” (AEGIS, grant agreement No 675555) and COFUND ALERT (grant agreement No 665250). Shabnam Shaabani was supported by a KWF Kankerbestrijding grant (grant agreement No 10504). Fandi Sutanto acknowledges Indonesian Endowment Fund for Education (Lembaga Pengelola Dana Pendidikan) for financial support. Deniz Eris is supported by the National Research Programme Covid‐19 (NRP78) from the Swiss National Science Foundation (grant number 4078P0_198290). We acknowledge the Paul Scherrer Institut, Villigen, Switzerland for provision of synchrotron radiation beamtime at X06SA, X06DA, and X10S beamlines of the SLS and would like to thank Justyna Wojdyla and Eric Plichta for assistance. We thank Silvia Mansueto, for helping with the purification of some compounds. We acknowledge academical licenses of the Instant Jchem and Scorpion software. We acknowledge Dispensix for borrowing us an I‐DOT instrument.
F. Sutanto, S. Shaabani, R. Oerlemans, D. Eris, P. Patil, M. Hadian, M. Wang, M. E. Sharpe, M. R. Groves, A. Dömling, Angew. Chem. Int. Ed. 2021, 60, 18231.
References
- 1.Ehrlich P., Lancet 1913, 182, 445–451. [Google Scholar]
- 2.Goodsell D. S., Zardecki C., Di Costanzo L., Duarte J. M., Hudson B. P., Persikova I., Segura J., Shao C., Voigt M., Westbrook J. D., Young J. Y., Burley S. K., Protein Sci. 2020, 29, 52–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tickle I., Sharff A., Vinkovic M., Yon J., Jhoti H., Chem. Soc. Rev. 2004, 33, 558–565. [DOI] [PubMed] [Google Scholar]
- 4.Polyakova A., Bourenkov G., Karpics I., Fielder S., Liu F., Schneider T. R., Acta Crystallogr. Sect. A 2016, 72, s194. [Google Scholar]
- 5.Kraft P., Bergamaschi A., Broennimann C., Dinapoli R., Eikenberry E. F., Henrich B., Johnson I., Mozzanica A., Schlepütz C. M., Willmott P. R., Schmitt B., J. Synchrotron Radiat. 2009, 16, 368–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a.Lee D., Park S., Lee K., Kim J., Park G., Nam K. H., Baek S., Chung W. K., Lee J. L., Cho Y., Park J., J. Appl. Crystallogr. 2020, 53, 477–485; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b.Schlichting I., IUCrJ 2015, 2, 246–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collins P. M., Ng J. T., Talon R., Nekrosiute K., Krojer T., Douangamath A., Brandao-Neto J., Wright N., Pearce N. M., von Delft F., Acta Crystallogr. Sect. D 2017, 73, 246–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wright N. D., Collins P., Koekemoer L., Krojer T., Talon R., Nelson E., Ye M., Nowak R., Newman J., Ng J. T., Mitrovich N., Wiggers H., von Delft F., Acta Crystallogr. Sect. D 2021, 77, 62–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pearce N. M., Bradley A. R., Krojer T., Marsden B. D., Deane C. M., von Delft F., Struct. Dyn. 2017, 4, 032104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Douangamath A., Fearon D., Gehrtz P., Krojer T., Lukacik P., Owen C. D., Resnick E., Strain-Damerell C., Aimon A., Ábrányi-Balogh P., Brandão-Neto J., Carbery A., Davison G., Dias A., Downes T. D., Dunnett L., Fairhead M., Firth J. D., Jones S. P., Keeley A., Keserü G. M., Klein H. F., Martin M. P., Noble M. E. M., O'Brien P., Powell A., Reddi R. N., Skyner R., Snee M., Waring M. J., Wild C., London N., von Delft F., Walsh M. A., Nat. Commun. 2020, 11, 5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a.Nichols C., Ng J., Keshu A., Kelly G., Conte M. R., Marber M. S., Fraternali F., De Nicola G. F., J. Med. Chem. 2020, 63, 7559–7568; [DOI] [PubMed] [Google Scholar]
- 11b.Wollenhaupt J., Metz A., Barthel T., Lima G. M. A., Heine A., Mueller U., Klebe G., Weiss M. S., Structure 2020, 28, 694–706.e695; [DOI] [PubMed] [Google Scholar]
- 11c.Bentley M. R., Ilyichova O. V., Wang G., Williams M. L., Sharma G., Alwan W. S., Whitehouse R. L., Mohanty B., Scammells P. J., Heras B., Martin J. L., Totsika M., Capuano B., Doak B. C., Scanlon M. J., J. Med. Chem. 2020, 63, 6863–6875; [DOI] [PubMed] [Google Scholar]
- 11d.Schuller M., Correy G. J., Gahbauer S., Fearon D., Wu T., Díaz R. E., Young I. D., Carvalho Martins L., Smith D. H., Schulze-Gahmen U., Owens T. W., Deshpande I., Merz G. E., Thwin A. C., Biel J. T., Peters J. K., Moritz M., Herrera N., Kratochvil H. T., Aimon A., Bennett J. M., Brandao Neto J., Cohen A. E., Dias A., Douangamath A., Dunnett L., Fedorov O., Ferla M. P., Fuchs M. R., Gorrie-Stone T. J., Holton J. M., Johnson M. G., Krojer T., Meigs G., Powell A. J., Rack J. G. M., Rangel V. L., Russi S., Skyner R. E., Smith C. A., Soares A. S., Wierman J. L., Zhu K., O'Brien P., Jura N., Ashworth A., Irwin J. J., Thompson M. C., Gestwicki J. E., von Delft F., Shoichet B. K., Fraser J. S., Ahel I., Sci. Adv. 2021, 7, eabf8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For a broader overview of these developments the reader is referred to a book chapter: “High-Throughput Macromolecular Crystallography in Drug Discovery”: G. Bricogne in Structural Biology in Drug Discovery (Ed.: J.-P. Renaud) 2020, 10.1002/9781118681121.ch10; as well as recent review article: [DOI]; Maveyraud L., Mourey L., Molecules 2020, 25, 1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krska S. W., DiRocco D. A., Dreher S. D., Shevlin M., Acc. Chem. Res. 2017, 50, 2976–2985. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a.Neochoritis C. G., Shaabani S., Ahmadianmoghaddam M., Zarganes-Tzitzikas T., Gao L., Novotna M., Mitrikova T., Romero A. R., Irianti M. I., Xu R. X., Olechno J., Ellson R., Helan V., Kossenjans M., Groves M. R., Domling A., Sci. Adv. 2019, 5, eaaw4607; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14b.Shaabani S., Xu R. X., Ahmadianmoghaddam M., Gao L., Stahorsky M., Olechno J., Ellson R., Kossenjans M., Helan V., Domling A., Green Chem. 2019, 21, 225–232; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14c.Wang Y. Z., Shaabani S., Ahmadianmoghaddam M., Gao L., Xu R. X., Kurpiewska K., Kalinowska-Tluscik J., Olechno J., Ellson R., Kossenjans M., Helan V., Groves M., Domling A., ACS Cent. Sci. 2019, 5, 451–457; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14d.Hadian M., Shaabani S., Patil P., Shishkina S. V., Boltz H., Domling A., Green Chem. 2020, 22, 2459–2467; [Google Scholar]
- 14e.Hadian M., Shaabani S., Patil P., Shishkina S. V., Böltz H., Dömling A., Green Chem. 2020, 22, 2459–2467; [Google Scholar]
- 14f.Osipyan A., Shaabani S., Warmerdam R., Shishkina S. V., Boltz H., Dömling A., Angew. Chem. Int. Ed. 2020, 59, 12423–12427; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 12523–12527. [Google Scholar]
- 15.
- 15a.Chen C.-C., Yu X., Kuo C.-J., Min J., Chen S., Ma L., Liu K., Guo R.-T., FEBS J. 2021, 10.1111/febs.15696; [DOI] [PubMed] [Google Scholar]
- 15b.Dömling A., Gao L., Chem 2020, 6, 1283–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.B. Boras, R. M. Jones, B. J. Anson, D. Arenson, L. Aschenbrenner, M. A. Bakowski, N. Beutler, J. Binder, E. Chen, H. Eng, H. Hammond, J. Hammond, R. E. Haupt, R. Hoffman, E. P. Kadar, R. Kania, E. Kimoto, M. G. Kirkpatrick, L. Lanyon, E. K. Lendy, J. R. Lillis, J. Logue, S. A. Luthra, C. Ma, S. W. Mason, M. E. McGrath, S. Noell, R. S. Obach, M. N. O'Brien, R. O'Connor, K. Ogilvie, D. Owen, M. Pettersson, M. R. Reese, T. F. Rogers, M. I. Rossulek, J. G. Sathish, N. Shirai, C. Steppan, M. Ticehurst, L. W. Updyke, S. Weston, Y. Zhu, J. Wang, A. K. Chatterjee, A. D. Mesecar, M. B. Frieman, A. S. Anderson, C. Allerton, bioRxiv 2021, 2020.2009.2012.293498.
- 17.
- 17a.Dömling A., Wang W., Wang K., Chem. Rev. 2012, 112, 3083–3135; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17b.Neochoritis C. G., Zhao T., Dömling A., Chem. Rev. 2019, 119, 1970–2042; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17c.Ref. [14c].
- 18.
- 18a.Zhao T., Boltjes A., Herdtweck E., Dömling A., Org. Lett. 2013, 15, 639–641; [DOI] [PubMed] [Google Scholar]
- 18b.Sung K., Chen F.-L., Huang P.-C., Synlett 2006, 2667–2669; [Google Scholar]
- 18c.Kazmaier U., Hebach C. J. S., Synlett 2003, 1591–1594. [Google Scholar]
- 19.Thompson M. J., Chen B., J. Org. Chem. 2009, 74, 7084–7093. [DOI] [PubMed] [Google Scholar]
- 20.Di L., Kerns E. H., Drug-like properties: concepts, structure design and methods from ADME to toxicity optimization, Academic Press, New York, 2015. [Google Scholar]
- 21.Fell J. B., Fischer J. P., Baer B. R., Blake J. F., Bouhana K., Briere D. M., Brown K. D., Burgess L. E., Burns A. C., Burkard M. R., J. Med. Chem. 2020, 63, 6679–6693. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a.Ref. [14d];
- 22b.Ref. [14f].
- 23.Sutanto F., Shaabani S., Neochoritis C. G., Zarganes-Tzitzikas T., Patil P., Ghonchepour E., Dömling A., Sci. Adv. 2021, 7, eabd9307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S., Acta Crystallogr. Sect. D 2011, 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pearce N. M., Krojer T., Bradley A. R., Collins P., Nowak R. P., Talon R., Marsden B. D., Kelm S., Shi J., Deane C. M., von Delft F., Nat. Commun. 2017, 8, 15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R., Science 2020, 368, 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.
- 27a.Zhao Z., Bourne P. E., Drug Discovery Today 2018, 23, 727–735; [DOI] [PubMed] [Google Scholar]
- 27b.Ghosh A. K., Samanta I., Mondal A., Liu W. R., ChemMedChem 2019, 14, 889–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.
- 28a.Oerlemans R., Ruiz-Moreno A. J., Cong Y., Dinesh Kumar N., Velasco-Velazquez M. A., Neochoritis C. G., Smith J., Reggiori F., Groves M. R., Dömling A., RSC Med. Chem. 2021, 12, 370-37; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28b.Blanchard J. E., Elowe N. H., Huitema C., Fortin P. D., Cechetto J. D., Eltis L. D., Brown E. D., Chem. Biol. 2004, 11, 1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.
- 29a.Blundell T. L., Jhoti H., Abell C., Nat. Rev. Drug Discovery 2002, 1, 45–54; [DOI] [PubMed] [Google Scholar]
- 29b.Thomas S. E., Collins P., James R. H., Mendes V., Charoensutthivarakul S., Radoux C., Abell C., Coyne A. G., Floto R. A., von Delft F., Blundell T. L., Philos. Trans. R. Soc. London Ser. A 2019, 377, 20180422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Günther S., Reinke P. Y. A., Fernández-García Y., Lieske J., Lane T. J., Ginn H. M., Koua F. H. M., Ehrt C., Ewert W., Oberthuer D., Yefanov O., Meier S., Lorenzen K., Krichel B., Kopicki J. D., Gelisio L., Brehm W., Dunkel I., Seychell B., Gieseler H., Norton-Baker B., Escudero-Pérez B., Domaracky M., Saouane S., Tolstikova A., White T. A., Hänle A., Groessler M., Fleckenstein H., Trost F., Galchenkova M., Gevorkov Y., Li C., Awel S., Peck A., Barthelmess M., Schluenzen F., Lourdu Xavier P., Werner N., Andaleeb H., Ullah N., Falke S., Srinivasan V., França B. A., Schwinzer M., Brognaro H., Rogers C., Melo D., Zaitseva-Doyle J. J., Knoska J., Peña-Murillo G. E., Mashhour A. R., Hennicke V., Fischer P., Hakanpää J., Meyer J., Gribbon P., Ellinger B., Kuzikov M., Wolf M., Beccari A. R., Bourenkov G., von Stetten D., Pompidor G., Bento I., Panneerselvam S., Karpics I., Schneider T. R., Garcia-Alai M. M., Niebling S., Günther C., Schmidt C., Schubert R., Han H., Boger J., Monteiro D. C. F., Zhang L., Sun X., Pletzer-Zelgert J., Wollenhaupt J., Feiler C. G., Weiss M. S., Schulz E. C., Mehrabi P., Karničar K., Usenik A., Loboda J., Tidow H., Chari A., Hilgenfeld R., Uetrecht C., Cox R., Zaliani A., Beck T., Rarey M., Günther S., Turk D., Hinrichs W., Chapman H. N., Pearson A. R., et al., Science 2021, 372, 642–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.
- 31a.Dömling A., Chem. Rev. 2006, 106, 17–89; [DOI] [PubMed] [Google Scholar]
- 31b.Dömling A., Wang W., Wang K., Chem. Rev. 2012, 112, 3083–3135; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31c.Ref. [17b].
- 32.
- 32a.Ref. [23];
- 32b.Perera D., Tucker J. W., Brahmbhatt S., Helal C. J., Chong A., Farrell W., Richardson P., Sach N. W., Science 2018, 359, 429–434; [DOI] [PubMed] [Google Scholar]
- 32c.Gesmundo N. J., Sauvagnat B., Curran P. J., Richards M. P., Andrews C. L., Dandliker P. J., Cernak T., Nature 2018, 557, 228–232; [DOI] [PubMed] [Google Scholar]
- 32d.Buitrago Santanilla A., Regalado E. L., Pereira T., Shevlin M., Bateman K., Campeau L.-C., Schneeweis J., Berritt S., Shi Z.-C., Nantermet P., Liu Y., Helmy R., Welch C. J., Vachal P., Davies I. W., Cernak T., Dreher S. D., Science 2015, 347, 49–53. [DOI] [PubMed] [Google Scholar]
- 33.
- 33a.Ref. [7];
- 33b.Ref. [8];
- 33c.Pearce N. M., Krojer T., Bradley A. R., Collins P., Nowak R. P., Talon R., Marsden B. D., Kelm S., Shi J., Deane C. M., von Delft F., Nat. Commun. 2017, 8, 15123; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33d.Lima G. M. A., Talibov V. O., Jagudin E., Sele C., Nyblom M., Knecht W., Logan D. T., Sjogren T., Mueller U., Acta Crystallogr. Sect. D 2020, 76, 771–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information