

Abstract
We report the synthesis of unprecedented tetra‐urea derivatives of calix[4]arene and calix[4]pyrrole containing four spiropyran (SP) units at their upper rim. We investigate the photo‐ and acid‐induced isomerization of the monomeric and homo‐dimeric tetra‐ureas derivatives using UV‐Vis and 1H NMR spectroscopies. At micromolar concentration, irradiation of the samples with 365 nm light induces changes in their absorption spectra that are consistent with SP→merocyanine (MC) isomerization. However, analogous experiments at millimolar concentration do not produce noticeable changes in the 1H NMR spectra. The addition of triflic acid to micromolar and millimolar solutions of the tetra‐ureas produces the quantitative isomerization of the SP units to the protonated merocyanine form (E‐MCH+) and the simultaneous disassembly of the capsular dimers to form ill‐defined aggregates. The neutralization of the acid solutions resets the SP form. Under these acid/base treatment conditions, the controlled release of the included guest and the reassembly of the all‐SP tetra‐urea dimers occurs at different extents depending on its calix[4]arene or calix[4]pyrrole scaffold.
Keywords: acid-isomerization, calix[4]pyrrole, calix[4]arene, photo-isomerization, spiropyran
Controlled uptake and release: We investigate the photo‐ and acid‐induced isomerization of monomeric and homo‐dimeric tetra‐spiropyran tetra‐urea calix[4]arene and calix[4]pyrrole derivatives. The acid‐induced isomerization of the switches was coupled to the homo‐dimeric calix[4]arene capsule's disassembly and the release of the cationic guest to the bulk solution. The neutralization of the acidified solution with Et3N completely restored the original encapsulation complex.
Introduction
The design of molecular and supramolecular containers incorporating responsive units (molecular switches) is relevant to different areas of chemistry. The switching of the responsive units, triggered by external stimuli (e. g. light, pH, temperature, redox), may impact on the thermodynamic stability of their inclusion and encapsulation complexes.[1, 2, 3] Ideally, and for practical uses, the modulation of the container's affinity should induce the quantitative release and uptake of molecular cargo to/from the bulk solution.[4]
Light has been widely employed as an external stimulus, owing to its non‐intrusive nature and high spatial and temporal resolution. The incorporation of photo‐switchable units in the scaffolds of molecular and supramolecular assemblies is demanded in applications such as controlled drug delivery[5, 6] and catalysis.[7, 8, 9]
Upon light irradiation, suitable photo‐switches reversibly interconvert between two isomeric states featuring significant structural and electronic changes. Examples of unimolecular containers having covalently attached photo‐switches and experiencing modulations of their binding properties in response to the isomerization state of the switch are well‐known.[10] Moreover, the incorporation of photo‐switches in molecular components of self‐assembled containers may allow coupling the isomerization of the switch to the assembly/disassembly process of the container.[11]
Well‐established families of photo‐switches include: azobenzene,[12] hemithioindigo,[13] acyl hydrazones,[14] spiropyrans (SPs),[15] diarylethenes (DAEs)[16] and donor‐acceptor Stenhouse adducts (DASAs).[17] Spiropyrans (SPs) are ideal switching units for incorporation into functional supramolecular containers due to their ease of synthetic manipulation and dual responsiveness to both light and chemical stimuli (i. e. pH, redox).[15, 18, 19] The closed‐SP neutral form of the switch is constituted by indoline and chromene moieties, which are perpendicularly oriented and connected through an sp3 spiro carbon atom. On the one hand, the irradiation of the SP‐form with UV‐light induces the electrocyclic ring‐opening of the furan, by cleavage of the Cspiro−O bond.[20] The subsequent isomerization of the of the metastable cisoid‐merocyanine (Z‐MC) affords the thermodynamically more stable transoid‐conjugated isomer (E‐MC). The isomerization process is accompanied by a significant change in colour of the solution, a.k.a. photochromism.[21] The E‐MC isomer displays quite different structural and electronic properties compared to the SP counterpart i. e. a planar structure and a larger dipole moment. The zwitterionic resonating structure of the E‐MC is characterized by a positively charged indolinium N+ atom and a negatively charged phenolate O− atom.
On the other hand, the addition of strong acids catalyses the cleavage of the Cspiro−O bond of the SP switches. The protonated cisoid‐merocyanine (Z‐MCH+), initially obtained, undergoes thermal isomerization of the double bond affording the protonated transoid‐merocyanine species (E‐MCH+). In this case, the observed change of colour is referred as acido‐chromism.
The switch can be fully reset to the initial SP‐form by simple thermal‐equilibration of the E‐MC isomer. The protonated counterpart E‐MCH+ requires addition of base (i. e. Et3N) to thermally restore the SP isomer. Noticeably, irradiation with visible light of E‐MC isomer produces a photo‐stationary state (PSS) with the SP‐form.
The incorporation of SP switches in guest's scaffolds is widely documented in literature.[22, 23, 24, 25] The main aim of this strategy resides in coupling the isomerization of the switch to the thermodynamic stability of the resulting supramolecular complexes. In contrast, the number of examples describing the incorporation of SP units into the structures of host molecules is less frequent.[26] Most likely, this difference is due to the greater synthetic challenge demanded by the latter approach.
Specifically, SP units were incorporated in macrocyclic receptors such as crown ethers,[27, 28] cyclodextrins[29] and calix[4]arenes.[30, 31, 32]
To the best of our knowledge, SP units have not been incorporated into the molecular components of self‐assembled containers. We envisaged that the SP→MC (or SP→MCH+) isomerization process of the switching units might be coupled to the assembly/disassembly of the container and the uptake and release of its cargo.
In this vein, we considered that hydrogen‐bonded dimeric capsules based on tetra‐urea calix[4]arene and calix[4]pyrrole scaffolds were privileged supramolecular containers to test our hypotheses.[33] These dimeric assemblies are known to encapsulate a variety of guests of different sizes and electronic nature.[34, 35, 36, 37] Consequently, the isomeric state of the switching unit could also be used to control the uptake and release of the cargo.
Recently, we described several photo‐responsive homo‐ and hetero‐dimeric tetra‐urea capsules equipped with four azobenzene units at their upper rims.[11, 38, 39, 40] Depending on the substitution of the terminal phenyl ring of the azobenzene units, the trans‐to‐cis photo‐isomerization reaction of the switches induced the dimer's disassembly at different extents. We discovered that the release of the cargo to the bulk solution was not directly related to the isomerization level. This result was attributed to the existence of cis‐enriched isomers that can still form capsular or non‐capsular aggregates displaying the guest binding affinity. The substitution of the azobenzene groups by SP units should produce larger structural changes in the isomerization process leading to a superior destabilization of dimeric aggregates. In addition, the use of SP units could allow controlling the uptake and release of molecular cargo using two different external stimuli: light and pH.
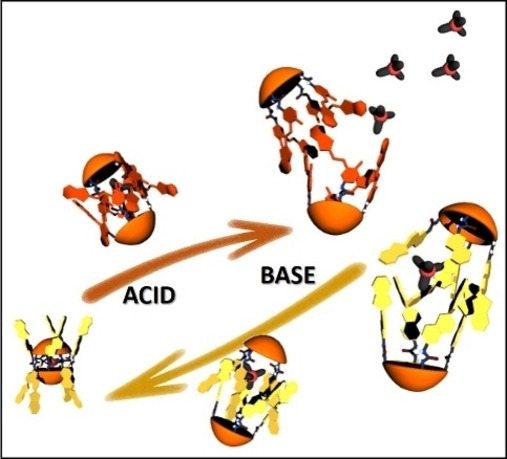
Herein, we report the synthesis of a tetra‐urea calix[4]arene and a tetra‐urea aryl‐extended calix[4]pyrrole, SP−C[4]A and SP−C[4]P, respectively, equipped with four spiropyran SP groups at their upper rims (Scheme 1). First, we describe the photo‐ and acid‐induced isomerization of the photo‐switches incorporated in the tetra‐ureas in (CH3)2SO solution. In this polar solvent, the tetra‐ureas, SP−C[4]A and SP−C[4]P, are present as monomers. We also report that in CH2Cl2 solution and in the presence of a suitable guest the two tetra‐ureas, SP−C[4]A and SP−C[4]P, self‐assemble into homo‐dimeric capsules. Remarkably, the self‐sorting of solutions containing equimolar amounts of the responsive tetra‐ureas (SP−C[4]A and SP−C[4]P) and their structural analogues lacking the SP groups (C[4]A and C[4]P) exclusively produced hetero‐dimeric capsules. We also study the photo‐ and acid‐induced isomerization of the SP units present in homo‐capsules by means of UV‐Vis and NMR spectroscopy. The acid‐promoted SP→E‐MCH+ isomerization significantly reduces the thermodynamic stability of the homo‐capsules. The coupled release of the encapsulated guest (cargo) to the bulk solution is evidenced in the case of the complex of the SP−C[4]A homo‐dimer.
Scheme 1.

Synthetic scheme for the preparation of tetra‐spiropyran tetra‐urea calix[4]arene SP−C[4]A and tetra‐spiropyran tetra‐urea calix[4]pyrrole SP−C[4]P, including their corresponding cartoon representations and proton assignment.
We demonstrate that the isomerization of the SP switches to the E‐MCH+ counterparts can be reversed by base addition (neutralization with triethylamine). Remarkably, the two responsive tetra‐ureas are not substantially degraded by the acid/base treatment. However, the thermodynamic stability of the hydrogen‐bonded ureas’ belt of the homo‐capsules is modified in the presence of the ‘in situ’ formed triethylammonium triflate following the acid/base treatment.
Results and Discussion
Synthesis of tetra‐spiropyran tetra‐urea calix receptors SP−C[4]A and SP−C[4]P
Tetra‐spiropyran tetra‐urea calix[4]arene SP−C[4]A and the analogous calix[4]pyrrole SP−C[4]P were prepared by the four‐fold condensation of spiropyran amino‐derivative 1 with the p‐nitrophenyl activated tetra‐carbamate derivatives 2 [38] and 3,[9] respectively (Scheme 1). The reactions were performed in DMF solution using Et3N as base. Tetra‐spiropyran tetra‐urea calix[4]arene SP−C[4]A was obtained in 67 % yield after trituration of the reaction crude with a CH2Cl2:MeOH (1 : 2) solvent mixture. Likewise, tetra‐spiropyran tetra‐urea calix[4]pyrrole SP−C[4]P was isolated in a 57 % yield (see Supporting Information for experimental details). The two responsive tetra‐ureas were characterized by a complete set of high‐resolution spectra (Figures S2–S11). In turn, 6‐aminospiro[2H‐1‐benzopyran‐2,2′‐indoline] 1 was prepared following a reported procedure.[41] After work‐up, the amino spiropyran 1 was isolated as an oil and used without further purification in the condensation reaction.
As mentioned above, spiropyrans (SP) are photochromic organic compounds known to isomerize to their open‐ring merocyanine (MC) isomer upon UV light‐irradiation and to the protonated merocyanine (MCH+) after treatment with a strong Brønsted acid.[42, 43] Therefore, the sequential isomerization of the four SP units at the upper rims of the tetra‐urea SP−C[4]A and SP−C[4]P, can lead up to a mixture of a six different stereoisomers: all‐SP and five MC‐ enriched isomers: SP−SP−SP−MC, SP−SP−MC−MC, SP−MC−SP−MC, SP−MC−MC−MC and all‐MC (MC will be MCH+ if acid is used instead of light irradiation to trigger the isomerization). We prepared 2 mM (CD3)2SO solutions of the isolated SP−C[4]A and SP−C[4]P solids in separate NMR tubes and we analysed both solutions using 1H NMR spectroscopy.
The 1H NMR spectra of both solutions displayed a single set of sharp and well‐resolved proton signals in agreement with C4v symmetry (Figure S2 and S8). These observations combined with the multiplicity and chemical shift values of the proton signals indicated that both tetra‐urea derivatives were exclusively present in solution as monomers of the thermodynamically more stable all‐SP‐isomer. Owing to the polar nature of (CD3)2SO and its high competition for hydrogen bonding interactions the all‐SP−C[4]A and all‐SP−C[4]P tetra‐urea isomers do not dimerize in this solvent.
An important point to be considered when moving from compounds containing a single SP chiral unit to molecular architectures with multiple SPs switches (i. e. all‐SP−C[4]A and all‐SP−C[4]P) has to do with the possible existence of diastereoisomers. For example, the chirality inversion (thermal or light‐induced) of the stereogenic carbon (spiro carbon) of a racemic SP mixture in a compound featuring a single SP unit involves two enantiomers (R‐SP and S‐SP) having identical 1H NMR spectra.[44] In contrast, the enantiomerization process of the SP units in all‐SP−C[4]A and all‐SP−C[4]P should render a mixture of racemic diastereoisomers, which might be distinguishable by 1H NMR spectroscopy. Remarkably and as mentioned above, we observed a single set of signals for the protons of all‐SP−C[4]A and all‐SP−C[4]P isomers. This result suggested that the thermal enantiomerization of their SP units, even in a crowded environment of the four switches installed in tetra‐urea scaffolds, involves low energy barriers. Most likely, the process occurs through the intermediacy of Z‐MC metastable state. In short, the chemical exchange between the diasteroisomers (R/S,R/S,R/S R/S) of all‐SP−C[4]A and all‐SP−C[4]P in (CD3)2SO solution displays fast kinetics on the proton chemical shift timescale. An alternative explanation would be that the chemical shifts of the protons of a SP unit are not affected by the absolute configurations of the adjacent ones. That is, all switches act as independent and identical units in the 1H NMR spectrum.
Spectral characterization of the photo‐ and acid‐induced isomerization of all‐SP−C[4]A and all‐SP−C[4]P
Initially, we investigated the photo‐isomerization process of the SP decorated tetra‐ureas, all‐SP−C[4]A and all‐SP−C[4]P, as monomeric species in (CH3)2SO solution by means of UV‐Vis and 1H NMR spectroscopy.
The UV‐Vis absorption spectrum of a thermally equilibrated (CH3)2SO solution of all‐SP−C[4]A showed a shoulder absorption band centred at λmax=345 nm. In agreement with literature, this band was assigned to the SP units (black solid line in Figure 1).[45] The irradiation of the solution with 365 nm light for 12 min produced noticeable changes in the absorption spectrum. Specifically, a new absorption band centred at 460 nm appeared (Figure S17). This new low‐frequency band is characteristic of the extended π‐conjugation in the open‐ring E‐MC photoisomer.[20] Taken together, these results indicated that the light irradiation induced the ring‐opening isomerization of terminal SP units of all‐SP−C[4]A to an unknown extent. This process would produce a mixture of C[4]A tetra‐urea isomers enriched with open‐ring E‐MC and Z‐MC units.
Figure 1.

Series of UV‐Vis absorption spectra of a 50 μM solution of SP−C[4]A in (CH3)2SO: after thermal equilibration in the dark (all‐SP isomer, black line); immediately after the addition of 200 μM of TfOH (grey line); 24 h after addition of 200 μM of TfOH (green line); irradiation with 365 nm light of the previous acidified solution (yellow line); standing in the dark for 24 h after irradiation of the acidified solution (orange line) and finally addition of 200 μM of Et3N to the equilibrated acidified solution in the dark (grey dashed line).
The reset of all switches to the SP state, was attempted by thermally equilibrating the previously irradiated solution at 60 °C in the dark for 24 h. The treatment produced the partial recovery of the original absorption spectrum (Figure S17). This result demonstrated that the photo‐isomerization process was reversible. It also indicated that the SP−C[4]A tetra‐ureas may be degraded (not photo‐stable) under the long irradiation conditions used to achieve the PSS.[46] We obtained similar results using the calix[4]pyrrole counterpart all‐SP−C[4]P (Figure S19).
In any case, the monitoring of the photo‐isomerization process using UV‐Vis spectroscopy did not allow the quantification of the level of SP→MC conversion. The determination of the extinction coefficients for the tetra‐ureas enriched with E‐MC and Z‐MC units was out of our reach and their absorption spectra displayed a significant overlap.
For this reason, we undertook the quantitative investigation of the photo‐isomerization process by performing light‐irradiation experiments using an NMR tube containing a 2 mM (CD3)2SO solution of all‐SP−C[4]A. We monitored the progress of the photo‐isomerization by analysing the ex‐situ light irradiated solution at regular intervals using 1H NMR spectroscopy (Figure S18).
As previously mentioned, the 1H NMR spectrum of the initial solution of all‐SP−C[4]A in (CD3)2SO displayed a single set of proton signals (Figure 2a). Specifically, the protons of the double bond, Hi and Hh, in the heterocyclic spiro−benzopyran moiety resonated as two doublets (J=10.0 Hz) centred at δ=6.9 and 5.7 ppm, respectively. The protons of the gem‐methyl groups in the indole unit, Hg and Hf, appear as two diastereotopic singlets at δ=1.1 and 1.2 ppm. Finally, the protons of the N‐methyl group of the indole (Ha) produced a sharp singlet centred at δ=2.6 ppm.
Figure 2.
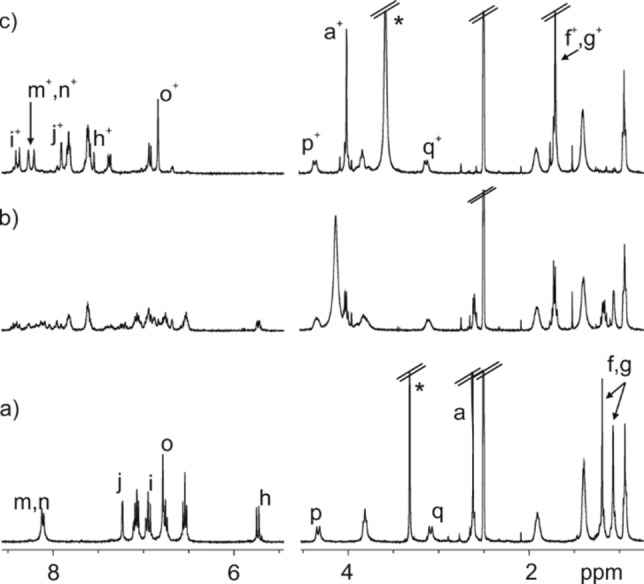
Selected region of the 1H NMR spectra (400 MHz, (CD3)2SO, 298 K) of a 2 mM solution of SP−C[4]A: a) thermally equilibrated sample; b) 24 h in the dark after the addition of 4 equiv. TfOH (8 mM). c) 72 h in the dark after the addition of 4 equiv. TfOH (8 mM). +Signals attributed to the E‐MCH+. *Residual solvent peak. See Scheme 1 for proton assignment.
The light‐irradiation (λex 365 nm) of the solution for 10 min did not produce noticeable changes in its 1H NMR spectrum (Figure S18).[47] Most likely, the amount of E‐MC and Z‐MC enriched isomers of the C[4]A present in solution is too low to be detected using 1H NMR spectroscopy. This can be due to a reduced photo‐isomerization yield to E‐MC and Z‐MC enriched isomers or/and to the existence of a fast and efficient thermal back‐isomerization to their SP form. Both effects derive from a reduced thermodynamic stability of the metastable MC isomer in DMSO solution. Previous work demonstrated that a nitro spiropyran carboxylic acid underwent reduced levels (∼3–10 %) of SP→MC photo‐isomerization in (CD3)2SO solution.[48] This study also emphasized the importance of the sample concentration and light fluxes. In the case at hand, the photo‐isomerization of the all‐SP decorated tetra‐ureas should produce up to five MC‐enriched stereoisomers. Considering a 3 % isomerization yield for each SP unit and that all of them are independently switched, we estimated a ratio of MC:SP enriched isomers of 3 : 97. This estimate serves to explain the lack of detection of the MC‐enriched isomers using 1H NMR spectroscopy owing to its low concentration in a 2 mM solution of the initial all‐SP tetra‐urea.
We already mentioned that the addition of acid catalyses the thermally induced conversion of the SP isomers to their protonated transoid‐merocyanine forms (E‐MCH+).[49] For this reason, we investigated the ring‐opening of the SP units of tetra‐urea monomeric derivatives induced by addition of triflic acid (TfOH). Firstly, we probed the isomerization process using UV‐Vis absorption spectroscopy. As depicted in Figure 1, the addition of 4 equiv. of TfOH to a 50 μM (CH3)2SO solution containing all‐SP−C[4]A did not result in immediate changes of the absorption spectrum (grey line in Figure 1). However, after standing in the dark for 24 h, the UV‐Vis spectrum of the acidic solution showed the emergence of three new bands with maxima at 355, 385 and 480 nm (green line in Figure 1). We attributed the new bands to the existence of a mixture of SP−C[4]A tetra‐urea isomers enriched with protonated merocyanines units (E/Z‐MCH+). Irradiation of the solution with 365 nm light produced an increase in the intensity of all absorption bands. Remarkably, the relative intensity of the absorption bands at 355 nm (attributed to Z‐MCH+) and those at 385 and 480 nm (attributed to E‐MCH+) changed in the PSS (yellow line in Figure 1).[18] After irradiation, the obtained solution was left standing in the dark for 24 h at r.t. The absorption spectrum of the thermally equilibrated solution was similar to that of the acidified but not light‐irradiated sample (putative mixture of E/Z‐MCH+, orange line in Figure 1). This observation suggested us that the thermal equilibration induced the isomerization of some of the upper‐rim Z‐MCH+ switches to the E‐MCH+ analogues. Finally, the addition of 4 equiv. of Et3N (200 μM) partially recovered the initial UV‐Vis spectrum corresponding to the all‐SP−C[4]A isomer (grey dashed line, Figure 1). In other words, the deprotonated E‐MC units rapidly and thermally isomerized to the Z‐MC form. In turn, the Z‐MCs appended at the upper rim of the tetra‐urea C[4]A underwent a fast thermal induced re‐cyclization reaction affording the closed SP isomers.
In order to quantify the level of SP→MCH+ conversion, we monitored the acid induced isomerization process using 1H NMR spectroscopy. The addition of 4 equiv. of TfOH acid (8 mM) to a 2 mM (CD3)2SO solution of all‐SP−C[4]A produced a slight broadening of the proton signals and the appearance of new signals of low intensity (Figure S21). The initial light‐brown coloured solution become red after the addition of the acid. The 1H NMR spectrum of the acidic solution after standing in the dark for 24 h was complex and showed multiple sets of proton signals (Figure 2b). However, after standing in the dark for a total of 72 h, the 1H NMR revealed the presence of a single set of proton signals different to that of the all‐SP−C[4]A isomer (Figure 2c).
The new set of proton signals featured a trans‐coupled pair of olefinic protons (Hi+ and Hh+, J=16.0 Hz), replacing the cis‐coupled protons of the double bond in the heterocyclic pyran unit of the SP (see above, J=10.0 Hz). In addition, the gem‐ dimethyl (Hf+, Hg+) protons of the new species resonated as a downfield shifted singlet at 1.7 ppm. The N‐methyl protons (Ha+) also moved downfield to 4.0 ppm. The deshielding of these signals agreed with the existence of an indolinium moiety in the new state of the switch.
Taken together, these observations supported the assignment of the new set of signals to the E‐MCH+ form of the upper rim switches. We concluded that the treatment of the all‐SP−C[4]A isomer with 4 equiv. of TfOH acid induced the almost quantitative formation of the all‐E‐MCH+−C[4]A counterpart. The complete acid catalysed isomerization process of the SP units seems to be slow on the human timescale (i. e. requires several days).[42]
Notably, some of the diagnostic signals of the E‐MCH+ substituents such as the gem‐dimethyl (Hf+, Hg+) and N‐methyl (Ha+) protons were visible immediately after the acid addition (low intensity signal in Figure S21b). We suggest, the existence of an equilibrium process between the spiropyran and the protonated cis‐merocyanine isomer of the switches, SP+H+⇌Z‐MCH+. This process displays intermediate dynamics on the chemical shift timescale. The Z‐MCH+ is metastable and might be formed in solution to a reduced extent. Subsequently, the protonated Z‐MCH+ unit undergoes a thermal isomerization process, which is slow on the human timescale, rendering the thermodynamically stable trans‐merocyanine form (E‐MCH+) of the switch. The C[4]A isomers enriched with Z‐MCH+ and E‐MCH+ switches do not experience a re‐cyclization reaction to the SP form owing to the protonation of the phenolate oxygen atom and the overall positive charge of the switch. The sequential acid‐induced isomerization of the four SP units initially renders a mixture of different isomers in solution which produce a complex 1H NMR spectrum (i. e. SP−SP−SP‐E/Z‐MCH+, SP−SP‐E/Z‐MCH+‐E/Z‐MCH+, etc.) (Figure 2b). With time, all isomers of the C[4]A enriched with MCH+ transform into the all‐E‐MCH+−C[4]A isomer featuring a nice and well‐defined 1H NMR spectrum (Figure 2c).
The addition of 4 equiv. of Et3N (8 mM) to the acidic deep red solution provoked an immediate colour change to deep blue. Blue coloured species are typically assigned to neutral MC species.[45] The deep blue colour of the solution quickly changed to light‐brown. This latter colour coincides with that of the original solution containing the all‐SP−C[4]A isomer. In agreement with the colour changes, the addition of the base restored the 1H NMR spectrum assigned to all‐SP−C[4]A isomer (Figure S21e). This result demonstrated that the base deprotonated the phenol groups inducing the formation of neutral E‐MC units possessing a zwitterionic resonating structure (Scheme 2). Rapidly, the zwitterion underwent thermal isomerization to Z‐MC and re‐cyclization reaction resetting the switches in the initial SP form.
Scheme 2.

Acid‐base and photo‐isomerization equilibria for the interconversion between SP, Z/E‐MC and Z/E‐MCH+.
In (CD3)2SO solution, the E‐ and Z‐forms of the merocyanine units (MC in Scheme 2) of the C[4]A isomers have a reduced lifetime. Our observations demonstrated the reversibility of the acid‐base isomerization process of the SP units and the chemical‐stability of the different C[4]A isomers under the used experimental conditions. We observed an almost identical behaviour in the acid‐base induced isomerization in (CD3)2SO solution of the all‐SP−C[4]P tetra‐urea (Figure S22 and S23).
Self‐assembly of homo‐dimeric capsules based on the calix[4]arene scaffold
We studied the dimerization process of all‐SP−C[4]A tetra‐urea into homo‐capsule (all‐SP−C[4]A)2 in CD2Cl2 solution using 1H NMR spectroscopy.
The 1H NMR spectrum of a 2 mM CD2Cl2 solution of all‐SP−C[4]A showed broad proton signals (Figure S24). This observation indicated the formation of ill‐defined aggregates deriving from the establishment of inter‐ and intra‐molecular hydrogen bonding interactions between the urea groups. We and others, have previously reported that dichloromethane is not a good fit for the cavity volume of C[4]A capsular dimers.[34]
For this reason, we investigated the assembly of dimeric capsules in CD2Cl2 solution in the presence of a guest suitable to fill 55 % of its interior volume (i. e. Me4P+ cation, 4).[50] The addition of 0.5 equiv. of tetrakis(3,5‐bis(trifluoromethyl)phenyl)borate (BArF) tetra‐methyl phosphonium salt (4⋅BArF) to a 4 mM solution of all‐SP−C[4]A in CD2Cl2 produced a 1H NMR spectrum with sharp proton signals that were diagnostic of the assembly of the encapsulation complex 4⊂(all‐SP−C[4]A)2 (Figure 3). Based on previously reported dimeric assemblies of tetra‐urea calix[4]arenes, the 4⊂(all‐SP−C[4]A)2 complex was expected to display S8 symmetry.[38] In agreement with this expectation, the aromatic protons ortho to the urea groups resonated as two separate meta coupled doublets (Ho1 ′ and Ho2 ′, Scheme 1 and Figure 3). This asymmetry is attributed to the unidirectional orientation of the eight urea groups forming a belt of 16 hydrogen bonds that stabilizes the dimeric capsule. Moreover, the observation of this asymmetry indicates that the exchange between the two senses of the unidirectional orientation of the urea groups is slow on the 1H NMR time scale.
Figure 3.
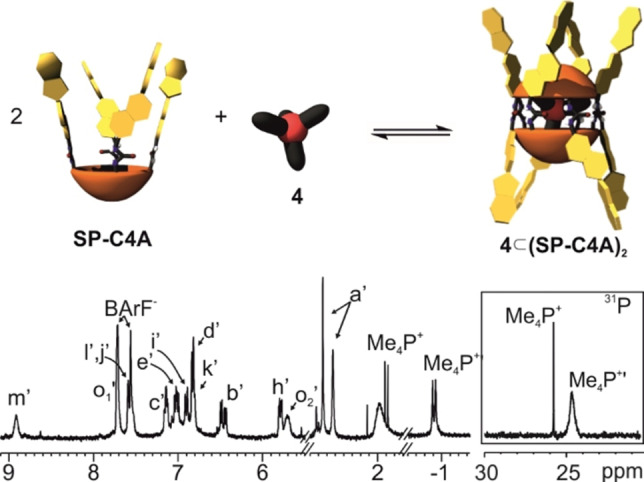
Top) Cartoon showing the equilibrium of the self‐assembly process of 4⊂(SP−C[4]A)2. Bottom) Selected regions of the 1H (400 MHz, 298 K) and 31P NMR spectra of a thermally equilibrated 4 mM CD2Cl2 solution of all‐SP−C[4]A containing 0.5 equiv. of the BArF salt of the tetra‐methyl phosphonium cation 4. Primed letters correspond to signals of the dimeric assembly. Non‐primed letters correspond to the signals of free guest in solution. See Scheme 1 for proton assignment and Figure S26 for full spectrum.
To our surprise, the protons of the N‐methyl group (Ha′) in the indole moiety of the SP units involved in the capsular assembly 4⊂(all‐SP−C[4]A)2 resonated as two separate singlets of similar intensity (δ=2.7 and 2.6 ppm) (Figure 3). Likewise, the diastereotopic gem‐methyl protons (Hf′, Hg′) split into four singlets (Figure S26).
To explain these observations, we considered that the dynamics of the enantiomerization process of the SP units in the dimeric capsule became slow on the 1H chemical shift timescale. Also, that all SP units experienced an independent racemization process. The origin of the observed multiplicity for the proton signals mentioned above could be assigned to the asymmetry provided by the unidirectional orientation of the urea groups. The unidirectional arrangement of the urea groups induces inherent chirality to the two hemispheres of the capsule. Thus, the protons of the R and S enantiomers of the SP switches become diastereotopic. Nevertheless, the 1H NMR spectrum of the encapsulation complex 4⊂(all‐SP−C[4]A)2 is not as complex as could be expected for a mixture of diastereomeric capsules having inherently chiral hemispheres with four chiral SP units. Both asymmetry elements are experiencing racemization processes at rates that are slow on the chemical shift timescale. Possibly, the simplification of the spectrum derives from a reduced transfer of chiral magnetic anisotropy between SP neighbours.
In the upfield region of the 1H NMR spectrum of 4⊂(all‐SP−C[4]A)2, we observed a broadened doublet (δ=−0.87 ppm, Δδ=2.8 ppm). This signal was ascribed to the methyl protons of the tetra‐methyl phosphonium cation 4 encapsulated in the capsular dimer. We assigned another doublet resonating at δ=1.9 ppm to the methyl protons of free 4 in solution. The existence of two separate signals corresponding to free and bound 4 indicated that their chemical exchange was slow on the chemical shift timescale. The integral values of the two doublets was used to calculate that ∼60 % of the Me4P+ cation 4 was encapsulated. This value is in good agreement with the ∼64 % yield assigned to the formation of the encapsulation complex, 4⊂(all‐SP−C[4]A)2, using integral values of its proton signals and those of an internal standard (i. e. 1,3,5‐trifluorobenzene). We assume that the remaining 37 % of the tetra‐urea all‐SP−C[4]A must be involved in ill‐defined aggregates producing proton signals that broaden beyond detection. The 31P NMR spectrum of the solution showed two signals resonating at δ=24.7 and 25.8 ppm. The upfield shifted signal is broad and is assigned to the encapsulated cation 4 in the multiple diastereomeric complexes. The free tetra‐methyl phosphonium 4 appears downfield as a sharp singlet (Figure 3‐bottom, right).
Self‐assembly of homo‐dimeric capsules based on the aryl‐extended calix[4]pyrrole scaffold
Next, we investigated the dimerization of the responsive calix[4]pyrrole tetra‐urea, all‐SP−C[4]P, using NMR spectroscopy. Similarly to the calix[4]arene counterpart, the tetra‐urea all‐SP−C[4]P did not assemble into capsular homo‐dimers in CD2Cl2 solution (Figure S25). We reported that tetra‐urea C[4]P dimerizes encapsulating one molecule of 4,4′‐bipyridine‐N,N′‐dioxide 5. The 1H NMR spectrum of a CD2Cl2 solution all‐SP−C[4]P with 0.5 equiv. of 5 showed the earmarks of capsule's formation, 5⊂(all‐SP−C[4]P)2 (Figure S32).[11, 51] The pyrrole NHs resonated as a singlet at δ=9.69 ppm. The downfield shift of the NHs is attributed to their involvement in hydrogen‐bonding interactions with the oxygen atom of 5. The aromatic protons of encapsulated 5, Hα′ and Hβ′, appeared at δ=4.89 (Δδ=−3.29) and δ=6.11 ppm (Δδ=−1.34), respectively. The upfield shift experienced by these protons, compared to the free guest, was attributed to the shielding effect exerted by the four meso‐aryl substituent of the calix[4]pyrrole. The magnitude of the complexation‐induced shifts located the pyridyl‐N‐oxide residues of 5 as deeply included in the capsule's hemispheres defined by the cone conformation of the aryl‐extended calix[4]pyrrole scaffolds. We also observed the splitting of the signal corresponding to the indole N‐methyl group (Ha′) in this capsular assembly, as observed for 4⊂(all‐SP−C[4]A)2.
A DOSY experiment showed a good fit of the decay of all proton signals for the 5⊂(all‐SP−C[4]P)2 encapsulation complex to the corresponding mono‐exponential function. The fit returned a diffusion constant value of D=4.48±0.06×10−10 m2/s (Figure S35) supporting that, in solution, 5 and the tetra‐urea all‐SP−C[4]P were involved in the same complex (Figure S36).
Self‐assembly of hetero‐dimeric capsules
Previously, we demonstrated that in CD2Cl2 solution a tolyl tetra‐urea calix[4]arene and a benzyl tetra‐urea calix[4]pyrrole exclusively self‐sorted into the hetero‐dimeric capsule. The dimeric assembly encapsulated one molecule of solvent and one molecule of trimethylamine N‐oxide 6 (Figure 4) rendering a four particles aggregate.[52] We were curious to investigate if the exclusive self‐sorting process was also operative with the newly synthesized tetra‐ureas, all‐SP−C[4]A and all‐SP−C[4]P. To facilitate the self‐assembly of hetero‐dimers, we used complementary tetra‐urea scaffolds not containing SP units, C[4]P 7 and C[4]A 8 (Figure 4).
Figure 4.

Line drawing and cartoon representation of the guests used for the self‐assembly of dimeric capsules and non‐responsive tetra‐urea calix[4]pyrrole (7) and calix[4]arene (8) used for the assembly of hetero‐dimeric capsules.
The 1H NMR spectrum of an equimolar mixture of all‐SP−C[4]P and trimethyl‐N‐oxide 6 in CD2Cl2 solution revealed the diagnostic signals for the quantitative assembly of the homo‐dimeric encapsulation complex, 62 ⊂(all‐SP−C[4]P)2 (Figure 5 and Figure S37–S41).[39] The addition of 1 equiv. of tetra‐urea calix[4]arene 8 to the above solution produced the disappearance of all the proton signals corresponding to the homo‐capsule and the concomitant emergence of a complete new set of sharp and well‐defined proton signals.
Figure 5.
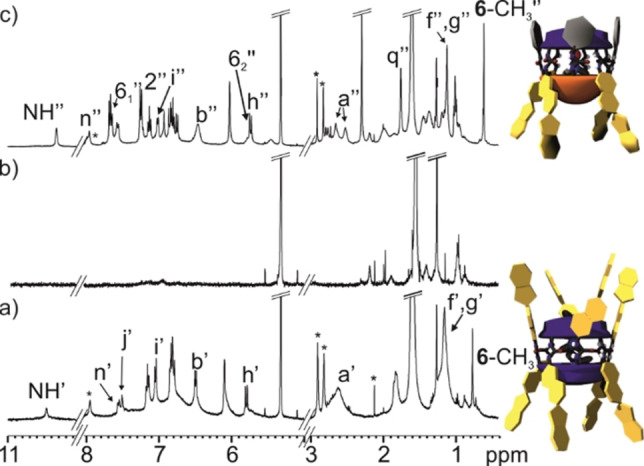
Selected regions of the 1H NMR (400 MHz, CD2Cl2, 298 K) spectra of a) 62 ⊂(SP−C[4]P)2 b) 8 and c) 6⊂(SP−C[4]P ⋅ 8). Primed letters correspond to proton signals of the homo‐dimeric assembly 62 ⊂(SP−C[4]P)2, double primed letters and numbers correspond to proton signals of the hetero‐dimeric assembly 6⊂(SP−C[4]P ⋅ 8). See Scheme 1 and Figure 4 for proton assignment. *Residual solvent peaks.
The new set of signals was assigned to the hetero‐dimeric encapsulation complex 6⊂(all‐SP−C[4]P ⋅ 8), which was quantitatively assembled in the solution (Figure 5c). The assembly of the hetero‐dimer benefits from the poor size of CD2Cl2 to fill 55 % of the internal volume of the homo‐dimer (8)2. In contrast, the combination of one molecule of CD2Cl2 and one of trimethyl‐N‐oxide 6 provides a close to optimal filling of the assembled hetero‐dimer. A DOSY experiment assigned a diffusion coefficient constants of D=4.22±0.02×10−10 m2/s to the hetero‐dimeric encapsulation complex 6⊂(all‐SP−C[4]P ⋅ 8) (Figure S51). The calculated diffusion constant value is in line with the ones determined for the homo‐dimeric assemblies 5⊂(all‐SP−C[4]P)2 and 62 ⊂(all‐SP−C[4]P)2 as expected owing to their similar sizes and shapes (Figure S35 and Figure S40, respectively).
We obtained analogous results in the self‐sorting experiment with the pair of tetra‐ureas all‐SP−C[4]A and tetra‐urea calix[4]arene 7. In this case, the encapsulation complex 6⊂(all‐SP−C[4]A ⋅ 7) was exclusively formed in solution (Figure S43–S47).
The obtained results demonstrated that hetero‐dimeric capsules based on tetra‐ureas C[4]A and C[4]P scaffolds containing four SP units in one hemisphere can be assembled in CD2Cl2 solution.
Studies of the photo‐ and acid‐ induced isomerization process of the SP units in homo‐dimeric capsules
We wanted to investigate the effect of light irradiation and acid/base treatment on the isomerization processes of the SP units installed in the tetra‐urea components of the homo‐dimeric capsules. We hypothesized that the isomerization process may reduce the thermodynamic stability of the resulting dimers enriched with MC/MCH+ forms of the switch. If this was the case, the disassembly of the dimers would affect the equilibrium distribution of the species (capsular dimer/oligomer) in solution. For suitable dimeric encapsulation complexes, this might also lead to the release (partial or total) of the encapsulated guest/s to the bulk solution (i. e. stimuli‐controlled cargo release).
In a similar fashion to the studies performed with the monomers in (CH3)2SO solution (see above), we first used UV‐Vis spectroscopy to monitor both light‐ and acid‐induced isomerization processes of the SP units in the homo‐dimeric encapsulation complexes. Please, recall that the tetra‐ureas dimerize in dichloromethane solution in the presence of suitable guests i. e. 4, 5 and 6 (Figure 4) producing the corresponding encapsulation complexes. The absorption spectrum of a 50 μM CH2Cl2 solution containing equimolar amounts of all‐SP−C[4]P and trimethyl‐N‐oxide 6 showed a low intensity band with a maximum at 340 nm. We attributed this band to absorption of the SP units in the 62 ⊂(all‐SP−C[4]P)2 encapsulation complex. The irradiation of the solution with 365 nm light for a period of 120 s produced an increase in the intensity of the band. Concomitantly, a new band with maximum at 490 nm emerged (Figure S53).[43, 53] We attributed the observed changes to the SP→MC isomerization of the switches installed at the upper‐rim of the tetra‐urea components in the 6 2⊂(all‐SP−C[4]P)2 encapsulation complex producing isomeric dimers and other aggregates enriched with Z/E‐MC units. The UV‐Vis experiment is not suitable for the quantification of the isomerization process owing to the unknown epsilons for the tetra‐urea enriched with Z/E‐MC units and the overlap of its absorption bands.
Next, we monitored the acid‐induced isomerization of the SP switches in the 62 ⊂(all‐SP−C[4]P)2 complex using UV‐Vis spectroscopy. The UV‐Vis spectrum of the solution acquired immediately after the acid addition (8 equiv. of TfOH) showed a small increase in the intensity of the bands centred at 340 nm and 490 nm (grey line in Figure 6). This observation was reminiscent to the effect produced by the light‐irradiation. Consequently, we attribute this change to the isomerization process of SP switches into the E/Z‐MCH+ counterparts. We allowed the acidic solution to equilibrate in the dark for 24 h. After this time, the absorption spectrum of the mixture showed the appearance of two intense bands centred at 395 and 490 nm (green line in Figure 6). The registered spectrum matches well with that reported for the Z‐MCH+ isomer of structurally related SP switches.[45]
Figure 6.
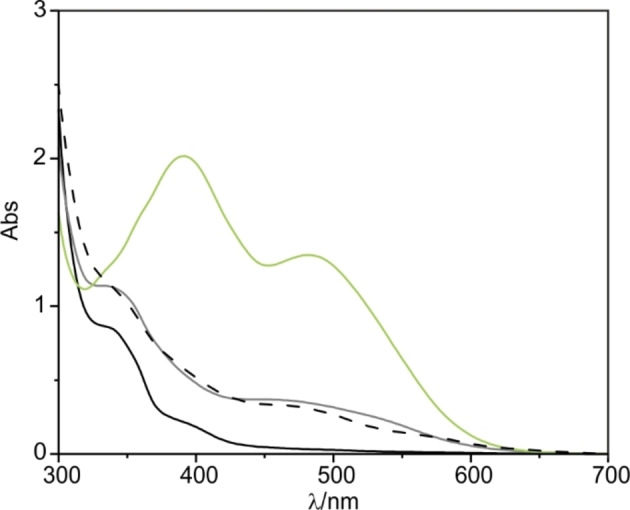
UV‐Vis spectra of a 50 μM equimolar solution of SP−C[4]P and 6 in CH2Cl2. Solid black line ‐ thermally equilibrated sample; solid grey line ‐ after the addition of 4 equiv. of TfOH (200 μM); green line ‐ 24 h after the addition of acid and dashed‐black line ‐ 24 h after addition of 4 equiv. of Et3N (200 μM).
After neutralization of the solution (addition of 8 equiv. of Et3N with respect to the dimeric assembly), the registered UV‐Vis spectrum almost coincided with that obtained immediately after the acid addition (dashed‐black line in Figure 6). Most likely, most of the switches were reset to the SP state. The presence of the triethylamine triflate salt (8 equiv.) could explain the increase of absorbance in comparison to the initial solution of 62 ⊂(all‐SP−C[4]P)2.
Unfortunately, the monitoring of the isomerization experiments (photo‐ and acid‐induced) by UV‐Vis spectroscopy do not provide direct structural information regarding the capsule's assembly/disassembly process and the extent of the reaction. For this reason, we re‐analysed the isomerization processes at higher concentration using 1H NMR spectroscopy.
Not surprisingly to us, the 1H NMR spectrum of the photo‐induced isomerization (365 nm, 5 min) of 4⊂(all‐SP−C[4]A)2 (Figure S55) did not reveal noticeable changes in relation to the one initially acquired (t=0 sec). We hypothesized that in dichloromethane solution, as observed in DMSO, the isomerization process producing the MC‐enriched ureas occurred to a reduced extent. This behaviour makes 1H NMR spectroscopy not suitable for the detection of MC‐enriched isomers of the C[4]A scaffold both in capsular dimers and other aggregates.
We moved to monitoring the acid‐induced isomerization process of the SP units in dimeric capsules by 1H NMR spectroscopy. The addition of 8 equiv. of TfOH acid to a 2 mM CD2Cl2 solution of the encapsulation complex 4⊂(all‐SP−C[4]A)2 produced an immediate change in the solution's colour, from light brown to deep red. The 1H NMR spectrum of the solution, registered after few minutes of the acid addition, showed broadening of most proton signals. Nevertheless, the proton signals corresponding to the encapsulation complex 4⊂(all‐SP−C[4]A)2 were still visible (Figure S57b). We left the solution equilibrating in the dark at r.t. for 24 h and re‐analysed using 1H NMR spectroscopy. The registered spectrum revealed the complete disappearance of the proton signals for the 4⊂(all‐SP−C[4]A)2 encapsulation complex (Figure 7b). We also observed the total fading of the most upfield shifted doublet (δ=−0.87 ppm), assigned to the encapsulated methyl protons of 4, and a noticeable increase in intensity for the doublet of the free cation 4 in solution (δ=1.9 ppm). In the same vein, the 31P NMR spectrum of the acidic solution revealed the presence of only one singlet corresponding to the free cation 4 in solution (Figure 7b, right). Taken together, these observations suggested that the acid treatment induced the isomerization of the SP units of the C[4]A to the positively charged indolinium isomer MCH+. The new species did not form capsular dimers or other aggregates suitable for the encapsulation of the tetra‐methyl phosphonium cation 4. Consequently, the acid treatment produced the complete release of 4 to the bulk solution. The observation of broad signals for the all‐MCH+−C[4]A isomers suggested the formation of ill‐defined aggregates that were stabilized by intermolecular hydrogen‐bonding interactions established between its urea groups. We consider that the aggregation driven by stacking of the MCH+ units is less likely owing to repulsive coulombic interactions.
Figure 7.
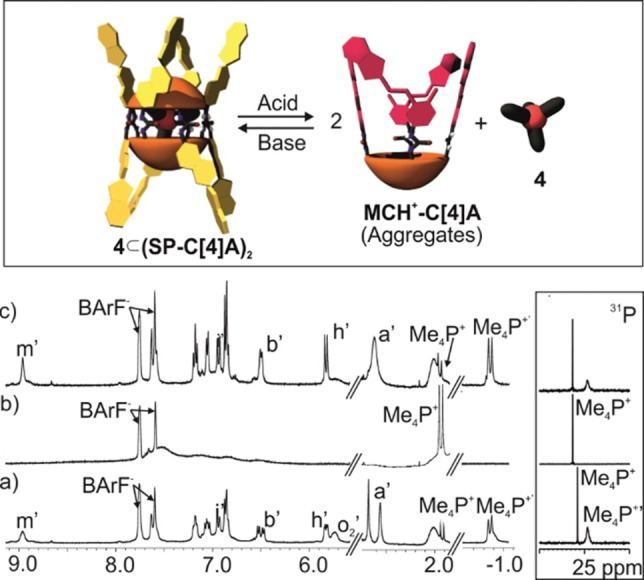
Top) Cartoon of the acid/base‐regulated disassembly/assembly equilibrium of the 4⊂(SP−C[4]A)2 homo‐dimeric encapsulation complex. Selected regions of the 1H NMR (400 MHz, CD2Cl2, 298 K) spectra of a 2 mM solution of a) 4⊂(SP−C[4]A)2; b) 4⊂(SP−C[4]A)2 24 h after the addition of 8 equiv. of TfOH (16 mM) and c) 24 h after the addition of Et3N. Primed letters and numbers correspond to proton signals of 4⊂(SP−C[4]A)2. See Scheme 1 for proton assignment.
The addition of 50 μL of DMSO‐d 6 to the acidified solution produced the sharpening of most proton signals (Figure S58c). The polar and competitive hydrogen‐bonding nature of DMSO eliminated the formation of aggregates in solution. The 1H NMR spectrum of acid‐solution containing DMSO‐d 6 showed the diagnostic signals of all‐E‐MCH+ isomer of the monomeric tetra‐urea. The doublets of protons Hi+ and Hh+ moved downfield (δ=8.5 and 7.4 ppm, respectively), compared to the neutral SP form, and featured a J coupling of 16.0 Hz in agreement with the double bond trans configuration. The gem‐methyl protons (Hg+ and Hf+) of the indole moiety appeared downfield shifted (δ=1.7 ppm) resonating as a broad singlet, in comparison to the two pairs of singlets observed for the same protons in the SP units in the encapsulation complex 4⊂(all‐SP−C[4]A)2. The N‐methyl protons also moved downfield and resonated as a singlet (Ha+, δ=4.0 ppm) compared to the two singlets (δ=2.5 and 2.7 ppm) observed for the SP units in the complex.
The addition of 8 equiv. of dry Et3N (16 mM) to an acidified solution thermally equilibrated for 24 h but not containing DMSO produced an immediate change in colour from red to deep blue. As mentioned above, a blue‐colour is attributed to the presence of neutral MC species.[45] The blue colour faded away in few seconds restoring the original light brown colour of the 4⊂(all‐SP−C[4]A)2 encapsulation complex. The 1H NMR spectrum of the solution obtained after the addition of the base testified the presence of the encapsulation complex 4⊂(all‐SP−C[4]A)2 in solution (Figure 7c). Altogether, these observations demonstrated that the assembly/disassembly process of the dimeric encapsulation complex 4⊂(all‐SP−C[4]A)2 was reversible and its molecular components were stable under the used acid/base conditions.
It is worthy to note that the acid/base treatment provoked some changes in the proton signals of the encapsulation complex 4⊂(all‐SP−C[4]A)2 in comparison to those registered for the untreated counterpart. For example, the aromatic protons of the calix[4]arene scaffold ortho to the urea groups (Ho′) broaden beyond detection and the protons of gem‐methyl groups (Ha′) in the SP units resonated as a broad singlet. The multiplicity of other protons signals (e. g. Hb′ and Hh′) was also simplified. Most likely, the ‘in situ’ formation of triethylammonium triflate following the acid/base treatment reduces the energy barrier of the chemical exchange between the two senses of the belt of unidirectionally orientated and hydrogen‐bonded urea groups stabilizing the capsular dimer.
We performed an analogous acid‐base treatment using a solution of the encapsulation complex 5⊂(all‐SP−C[4]P)2 (Figure S59). This complex involves two tetra‐urea calix[4]pyrroles, with appended SP units, encapsulating one molecule of 4,4′‐bipyridine‐N,N′‐dioxide 5. Upon addition of 8 equiv. of TfOH acid to a 2 mM solution of 5⊂(all‐SP−C[4]P)2 complex we observed an immediate colour change from light brown to deep red that was accompanied by the precipitation of a red solid. The 1H NMR spectrum of the acidified solution showed broad signals and a significant decrease in the signal‐to‐noise ratio. These results are in agreement with protonation and reduction of the concentration of the tetra‐urea calix[4]pyrrole in solution (Figure S59b). We did not detect the proton signals corresponding to free 4,4′‐bipyridine‐N,N′‐dioxide 5 in solution. Hence, we hypothesized that the acid‐treatment induced the isomerization of the SP switches to the MCH+ counterparts provoking the formation of aggregates insoluble in dichloromethane, which are also able to bind bis‐N‐oxide 5. The equilibration of the suspension (48 h) did not induce changes in the 1H NMR spectrum.
The addition of 8 equiv. of Et3N to the equilibrated suspension produced the solubilization of the precipitate and a colour change of the solution from deep red to deep blue. The blue colour rapidly faded away rendering a light brown solution. This behaviour parallels the findings described previously for the C[4]A analogue. It also supports the neutralization and isomerization equilibria: all‐E‐MCH+⇌E‐MC+H+ and E‐MC⇌SP experienced by the upper rim switches of the C[4]P scaffold. Remarkably, the 1H NMR spectrum of the neutralized solution showed two separate sets of signals of almost equal intensity for the protons of all‐SP−C[4]P (Figure S59c). One of them was assigned to the 5⊂(all‐SP−C[4]P)2 capsular assembly. The other must correspond to a different complex involving all‐SP−C[4]P and bis‐N‐oxide 5. As observed for the acid/base treatment of 4⊂(SP−C[4]A)2, the presence of 8 equiv. of triethylammonium triflate modified the energy barrier for the interconversion between the two senses of the belt of hydrogen‐bonded and unidirectionally oriented ureas. This effect may also reduce the thermodynamic stability of the dimeric encapsulation complex. In the case at hand, the presence of a small excess of bis‐N‐oxide 5 (i. e. the original mixture contained 0.7 equiv. of 5) may also assist the dissociation of the weakened capsular assembly. Based on these considerations, we assigned the second set of the tetra‐urea proton signals to the formation of a 1 : 1 complex 5⊂all‐SP−C[4]P.[54] These results evidenced that the assembly/disassembly process of the tetra‐urea C[4]P encapsulation complex is reversible to a reduced extent under the used acid/base conditions.
Conclusion
In summary, we report the synthesis and characterization of two tetra‐urea derivatives having calix[4]arene and calix[4]pyrrole scaffolds decorated with four SP groups appended at their upper rims. The tetra‐ureas exist as monomers in (CH3)2SO solution. In the monomers, the photo‐induced isomerization of the SP units occurs to a reduced extent. Isomers enriched with MC units cannot be detected by 1H NMR spectroscopy. In contrast, the addition of triflic acid induced the complete isomerization of the SP units into the protonated MCH+ forms. We detected the quantitative formation of all‐MCH+ tetra‐ureas by 1H NMR spectroscopy after thermal equilibration (48 h at r.t.) of the acidified solution in the dark. We demonstrated that in CH2Cl2 solution and in the presence of suitable guests, the responsive tetra‐ureas dimerize to produce homo‐ and hetero‐encapsulation complexes with all‐SP units. The SP units of the homo‐encapsulation dimers were photo‐isomerized to the MC forms to a reduced extent. We noticed the existence of tetra‐ureas enriched with MC units by UV‐Vis spectroscopy. These species were not observed using 1H NMR spectroscopy. Addition of TfOH acid to the homo‐encapsulation dimers evidenced the quantitative isomerization of the SP units to the MCH+ form and the existence of aggregation processes between the MCH+ enriched tetra‐ureas. In the particular case of the 4⊂(all‐SP−C[4]A)2 complex, the acid‐induced isomerization of the switches was coupled to the capsule's disassembly and the release of the cationic guest to the bulk solution. The acid‐induced isomerization of the SP units in the 5⊂(all‐SP−C[4]P)2 complex did not provoke the release of the bis‐N‐oxide to the bulk solution. Instead, the bis‐N‐oxide was bound by the insoluble aggregates produced by the all‐MCH+−C[4]P isomer. The neutralization of the acidified solution with Et3N completely restored the original 4⊂(all‐SP−C[4]A)2 encapsulation complex. In contrast, the addition of the base to the suspension of the all‐MCH+−C[4]P restored all the switches to the SP isomers but the re‐assembly of the 5⊂(all‐SP−C[4]P)2 complex was not quantitative. This partial re‐assembly of the encapsulation complex was attributed to a decrease in thermodynamic stability owing to the presence of an excess of triethylammonium triflate as by‐product of the acid/base treatment. We demonstrated that all the molecular components of the assemblies are stable under the used acid/base conditions. The use of non‐polar solvent demanded for the assembly of the hydrogen‐bonded dimeric capsules seems to be incompatible with efficient light‐induced SP to MC isomerization. On the contrary, the use of triflic acid produced the quantitative isomerization of the SP units into the MCH+ disrupting the formation of dimeric capsular aggregates. Unfortunately, the acidic treatment is too harsh to be investigated in more biologically relevant conditions.
Experimental Section
Synthesis of all‐SP−C[4]A: tetra‐carbamate calix[4]arene 2 [55] and 6‐aminospiro[2H‐1‐benzopyran‐2,2′‐indoline] 1 [41] were prepared in good yields following the reported procedures. Tetra‐carbamate calix[4]arene 2 (50 mg, 0.035 mmol, 1 equiv.) was added to an oven‐dried 10 mL Schlenk tube purged 3x with Ar, and dissolved in 2 mL anhydrous DMF under Ar atmosphere. Spiropyran amino derivative 1 (60 mg, 0.2 mmol, 5.8 equiv.) was dissolved in 2 mL anhydrous DMF and added dropwise to 2 under Ar. Finally, freshly distilled Et3N (28 μL, 0.2 mmol, 5.8 equiv.) was added dropwise to the solution mixture. The yellow solution was stirred overnight at r.t. protected from light and under Ar atmosphere. After 12 h, 10 mL of DCM were added to the reaction mixture. The solution was transferred to an extraction funnel and the organic phase was washed with 4 % aq. NaHCO3 solution (4×10 mL), and citric acid 1 M solution (1×10 mL). The organic phase was again washed with 4 % aq. NaHCO3 (1×10 mL) and finally with water (1×10 mL). The light brown organic phase was dried over sodium sulfate, filtered and concentrated under reduced pressure to leave around 0.5 mL DCM. 1 mL of MeOH was added dropwise to the brown solution to obtain a grey solid. The solid was filtered, collected and dried overnight under high vacuum (48 mg, 67 % yield). Rf=0.37 (CH2Cl2:CH3OH 99 : 1). m.p.=>230 °C (decompose). 1H NMR ((CD3)2SO, 500 MHz) δ (ppm): 8.11 (s, 4H); 8.09 (s, 4H); 7.22 (s, 4H); 7.08 (m, 4H); 7.06 (d, J=7.8 Hz, 4H); 6.95 (d, J=8.6 Hz, 4H); 6.93 (d, J=10.2 Hz, 4H); 6.78 (s, 8H); 6.75 (m, 4H); 6.55 (d, J=8.6 Hz, 4H); 6.52 (d, J=7.8 Hz, 4H); 5.73 (d, J=10.2 Hz, 4H); 4.32 (d, J=12.0 Hz, 4H); 3.80 (s, 8H); 3.08 (d, J=12.0 Hz, 4H); 2.61 (s, 12H); 1.90 (s, 8H); 1.38 (s, 16H); 1.18 (s, 8H); 1.06 (s, 8H); 0.93 (s, 12H). HRMS (MALDI/+) m/z: [M+H]+ Calcd. for C128H141N12O12: 2038.0786; found: 2038.0759.
Synthesis of all‐SP−C[4]P: tetra‐carbamate calix[4]pyrrole 3 [11] (87 mg, 0.062 mmol, 1 equiv.) and 6‐aminospiropyran 1 (91 mg, 0.091 mmol, 5 equiv.) were added to an oven‐dried 25 mL Schlenk tube purged 3x with Ar, and dissolved in 8 mL anhydrous DMF under Ar atmosphere. Then, freshly distilled Et3N (43 μL, 0.311 mmol, 5 equiv.) was added dropwise to the solution. The mixture was stirred overnight at r.t. protected from light and under Ar atmosphere. After 12 h, 10 mL of DCM were added to the mixture, followed by 10 mL of a 0.3 M aqueous solution of K2CO3. The reaction mixture was stirred for 10 min. The organic phase was washed with water (1×10 mL) and brine (1×10 mL). The organic phase was dried over sodium sulfate, filtered and concentrated under reduced pressure. The compound was purified by trituration in a DCM:MeOH mixture (1 : 2). The resulting solid was filtered and washed with MeOH and dried under high vacuum to obtain a grey solid (58 mg, 47 % yield). m.p.=>230 °C (decompose). 1H NMR (400 MHz, (CD3)2SO, 298 K): δ (ppm)= 9.56 (s, 4H); 8.50 (s, 4H); 8.347 (s, 4H); 7.30 (s, 12H); 7.05 (m, 8H); 6.99 (d, J=10.2 Hz, 4H); 6.84 (d, J=8.9 Hz, 8H); 6.73 (m, 4H); 6.54 (d, J=8.9 Hz, 4H); 6.50 (d, J=9.9 Hz, 4H); 5.94 (s, 8H); 5.67 (d, J=10.2 Hz, 4H); 2.58 (s, 12H); 1.77 (s, 12H); 1.61 (s, 12H); 1.03 (s, 12H). HRMS (ESI/+) m/z: [M+2H]2+ Calc for (C128H122N16O8): 1005.4810; found: 1005.4821. [M+3H]3+ Calc for (C128H123N16O8): 670.6565; found: 670.6587.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The authors thank Gobierno de España MICIN/AEI/FEDER (projects CTQ2017‐84319‐P, CEX2019‐000925‐S), the European Union (NOAH project H2020‐MSCA‐ITN project Ref. 765297) the CERCA Programme/Generalitat de Catalunya and AGAUR (2017 SGR 1123) for financial support. We thank D. Stares and Prof. C. Schalley from Freie Universität Berlin for the measurement of the HRMS of SP‐C4P. We also thank Dr. F. Arroyave for helpful discussions over the project.
P. Ferreira, G. Moncelsi, G. Aragay, P. Ballester, Chem. Eur. J. 2021, 27, 12675.
References
- 1.Clever G. H. in Switchable Host-Guest Interactions of Supramolecular Rings and Cages, Vol. Wiley-VCH, Weinheim, 2012, pp. 13–37. [Google Scholar]
- 2.Yan X., Zhang M., Wei P., Zheng B., Chi X., Ji X., Huang F., Chem. Commun. 2011, 47, 9840–9842. [DOI] [PubMed] [Google Scholar]
- 3.Wang Q., Cheng M., Zhao Y., Yang Z., Jiang J., Wang L., Pan Y., Chem. Commun. 2014, 50, 15585–15588. [DOI] [PubMed] [Google Scholar]
- 4.Chi X., Cen W., Queenan J. A., Long L., Lynch V. M., Khashab N. M., Sessler J. L., J. Am. Chem. Soc. 2019, 141, 6468–6472. [DOI] [PubMed] [Google Scholar]
- 5.Han M., Michel R., He B., Chen Y.-S., Stalke D., John M., Clever G. H., Angew. Chem. Int. Ed. 2013, 52, 1319–1323; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 1358–1362. [Google Scholar]
- 6.Catti L., Kishida N., Kai T., Akita M., Yoshizawa M., Nat. Commun. 2019, 10, 1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neilson B. M., Bielawski C. W., J. Am. Chem. Soc. 2012, 134, 12693–12699. [DOI] [PubMed] [Google Scholar]
- 8.Wang J., Feringa B. L., Science 2011, 331, 1429–1432. [DOI] [PubMed] [Google Scholar]
- 9.Vlatković M., Bernardi L., Otten E., Feringa B. L., Chem. Commun. 2014, 50, 7773–7775. [DOI] [PubMed] [Google Scholar]
- 10.Hua Y., Flood A. H., J. Am. Chem. Soc. 2010, 132, 12838–12840. [DOI] [PubMed] [Google Scholar]
- 11.Osorio-Planes L., Espelt M., Pericàs M. A., Ballester P., Chem. Sci. 2014, 5, 4260–4264. [Google Scholar]
- 12.Bandara H. M. D., Burdette S. C., Chem. Soc. Rev. 2012, 41, 1809–1825. [DOI] [PubMed] [Google Scholar]
- 13.Wiedbrauk S., Dube H., Tetrahedron Lett. 2015, 56, 4266–4274. [Google Scholar]
- 14.Aprahamian I., Chem. Commun. 2017, 53, 6674–6684. [DOI] [PubMed] [Google Scholar]
- 15.Kortekaas L., Browne W. R., Chem. Soc. Rev. 2019, 48, 3406–3424. [DOI] [PubMed] [Google Scholar]
- 16.Irie M., Fukaminato T., Matsuda K., Kobatake S., Chem. Rev. 2014, 114, 12174–12277. [DOI] [PubMed] [Google Scholar]
- 17.Lerch M. M., Wezenberg S. J., Szymanski W., Feringa B. L., J. Am. Chem. Soc. 2016, 138, 6344–6347. [DOI] [PubMed] [Google Scholar]
- 18.Kortekaas L., Chen J., Jacquemin D., Browne W. R., J. Phys. Chem. B 2018, 122, 6423–6430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin Fang Z., Baba R., Hashimoto K., Fujishima A., J. Photochem. Photobiol. A 1995, 92, 91–97. [Google Scholar]
- 20.Fleming C. L., Li S., Grøtli M., Andréasson J., J. Am. Chem. Soc. 2018, 140, 14069–14072. [DOI] [PubMed] [Google Scholar]
- 21.There are eight possible merocyanine conformers identified by the Z/E configuration about the α, β and γ bonds. Four of those correspond to the transoid merocyanine form: CTC, CTT, TTC and TTT. See reference [15] for details.
- 22.Hernández-Melo D., Tiburcio J., Chem. Commun. 2015, 51, 17564–17567. [DOI] [PubMed] [Google Scholar]
- 23.Samanta D., Galaktionova D., Gemen J., Shimon L. J. W., Diskin-Posner Y., Avram L., Král P., Klajn R., Nat. Commun. 2018, 9, 641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mohan Raj A., Raymo F. M., Ramamurthy V., Org. Lett. 2016, 18, 1566–1569. [DOI] [PubMed] [Google Scholar]
- 25.Howlader P., Mondal B., Purba P. C., Zangrando E., Mukherjee P. S., J. Am. Chem. Soc. 2018, 140, 7952–7960. [DOI] [PubMed] [Google Scholar]
- 26.Moncelsi G., Ballester P., ChemPhotoChem 2019, 3, 304–317. [Google Scholar]
- 27.Salhin A. M. A., Tanaka M., Kamada K., Ando H., Ikeda T., Shibutani Y., Yajima S., Nakamura M., Kimura K., Eur. J. Org. Chem. 2002, 2002, 655–662. [Google Scholar]
- 28.Khairutdinov R. F., Hurst J. K., Langmuir 2004, 20, 1781–1785. [Google Scholar]
- 29.Wu S., Luo Y., Zeng F., Chen J., Chen Y., Tong Z., Angew. Chem. Int. Ed. 2007, 46, 7015–7018; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 7145–7148. [Google Scholar]
- 30.Lee M., Cho D., Kim I., Lee J., Lee J. Y., Satheeshkumar C., Song C., ChemistrySelect 2017, 2, 3527–3533. [Google Scholar]
- 31.Liu Z., Jiang L., Liang Z., Gao Y., Tetrahedron 2006, 62, 3214–3220. [Google Scholar]
- 32.Nag R., Polepalli S., Althaf Hussain M., Rao C. P., ACS Omega 2019, 4, 13231–13240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neri P., Sessler J. L., Wang M.-X., Calixarenes and Beyond, Springer, Cham, 2016, p. [Google Scholar]
- 34.Mogck O., Pons M., Böhmer V., Vogt W., J. Am. Chem. Soc. 1997, 119, 5706–5712. [Google Scholar]
- 35.Castellano R. K., Kim B. H., Rebek J. J., J. Am. Chem. Soc. 1997, 119, 12671–12672. [Google Scholar]
- 36.Castellano R. K., Nuckolls C., Rebek J. J., J. Am. Chem. Soc. 1999, 121, 11156–11163. [Google Scholar]
- 37.Schalley C. A., Castellano R. K., Brody M. S., Rudkevich D. M., Siuzdak G., Rebek J. J., J. Am. Chem. Soc. 1999, 121, 4568–4579. [Google Scholar]
- 38.Arroyave F. A., Ballester P., J. Org. Chem. 2015, 80, 10866–10873. [DOI] [PubMed] [Google Scholar]
- 39.Díaz-Moscoso A., Arroyave F. A., Ballester P., Chem. Commun. 2016, 52, 3046–3049. [DOI] [PubMed] [Google Scholar]
- 40.Sekiya R., Díaz-Moscoso A., Ballester P., Chem. Eur. J. 2018, 24, 2182–2191. [DOI] [PubMed] [Google Scholar]
- 41.Zimmermann T., Brede O., J. Heterocycl. Chem. 2003, 40, 611–616. [Google Scholar]
- 42.Wojtyk J. T. C., Wasey A., Xiao N.-N., Kazmaier P. M., Hoz S., Yu C., Lemieux R. P., Buncel E., J. Phys. Chem. A 2007, 111, 2511–2516. [DOI] [PubMed] [Google Scholar]
- 43.Tian W., Tian J., Dyes Pigm. 2014, 105, 66–74. [Google Scholar]
- 44.Kießwetter R., Pustet N., Brandl F., Mannschreck A., Tetrahedron: Asymmetry 1999, 10, 4677–4687. [Google Scholar]
- 45.Klajn R., Chem. Soc. Rev. 2014, 43, 148–184. [DOI] [PubMed] [Google Scholar]
- 46.Zanoni M., Coleman S., Fraser K. J., Byrne R., Wagner K., Gambhir S., Officer D. L., Wallace G. G., Diamond D., Phys. Chem. Chem. Phys. 2012, 14, 9112–9120. [DOI] [PubMed] [Google Scholar]
- 47.Sample was not irradiated for more amount of time due to lack of changes observed upon 10 min of irradiation.
- 48.Wolff C., Kind J., Schenderlein H., Bartling H., Feldmeier C., Gschwind R. M., Biesalski M., Thiele C. M., Magn. Reson. Chem. 2016, 54, 485–491. [DOI] [PubMed] [Google Scholar]
- 49.Roxburgh C. J., Sammes P. G., Dyes Pigm. 1995, 27, 63–69. [Google Scholar]
- 50.Mogck O., Böhmer V., Vogt W., Tetrahedron 1996, 52, 8489–8496. [Google Scholar]
- 51.Ballester P., Gil-Ramírez G., Proc. Nat. Acad. Sci. 2009, 106, 10455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chas M., Gil-Ramírez G., Ballester P., Org. Lett. 2011, 13, 3402–3405. [DOI] [PubMed] [Google Scholar]
- 53.Please note that the UV-Vis spectrum of SP is highly dependent on the solvent.
- 54.If the original mixture of SP−C[4]P and 5 in CD2Cl2 contains a higher excess of bis-N-oxide guest 5 (∼1 equiv.) the 1H NMR spectrum of the resulting mixture after the acid/base treatment mainly shows the signals of the putative 1 : 1 complex.
- 55.Chas M., Ballester P., Chem. Sci. 2012, 3, 186–191. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information