

Abstract
Two naphthalene‐diimide (NDI) bis‐imidazolium salts have been used as N‐heterocyclic carbene (NHC) precursors for the preparation of NDI‐functionalized complexes of rhodium and iridium of general formula [MCl(NDI‐NHC)(COD)] (M=Rh, Ir; NDI‐NHC=NDI‐functionalized NHC ligand). Comparison of the IR spectra of the complexes [IrCl(NDI‐NHC)(CO)2] and their related one‐ and two‐electron reduced forms, reveal that each one‐electron reduction produces a decrease of the average ν(CO) of 9–10 cm−1, indicating a significant enhancement of the electron‐richness of the metal. The [MCl(NDI‐NHC)(COD)] complexes were tested in the catalytic cycloisomerization of alkynoic acids. The one‐electron reduced forms showed greatly enhanced activities. For the cyclization of 5‐hexynoic acid, the two‐electron reduction of the ligand produced further enhancement of the catalytic activity, therefore showing that the catalyst can switch between three redox species with three distinct catalytic activities.
Keywords: homogeneous catalysis, iridium, naphthalenediimide (NDI), N-heterocyclic carbenes, redox-switchable, rhodium
We present a series of rhodium(I) and iridium(I) complexes with a naphthalene‐di‐imide N‐heterocyclic carbene ligand that behave as effective redox‐switchable catalysts for the cycloisomerization of alkynoic acids. The catalysts can adopt three electronically different stages with distinct catalytic activities. The activity of catalysts can toggle between the activated and inactivated forms for several cycles.
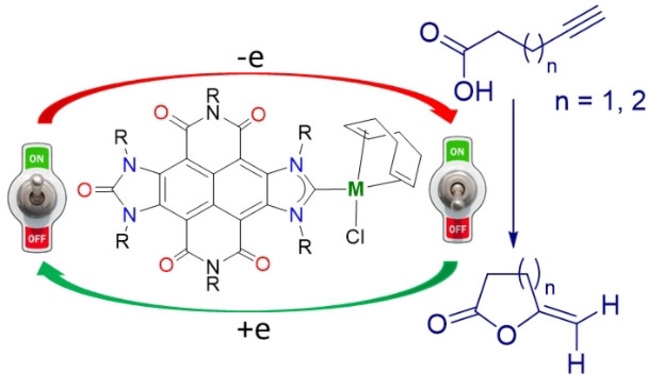
Introduction
Apart from the traditional role of spectator ligands that bind to the metal and provide electronic and steric‐defined pockets for binding the substrate in homogeneous catalysis, switchable ligands incorporate stimuli‐responsive units that can confer a biomimetic level of control over chemical transformations.[1] Such level of control potentially enables these systems to facilitate chemical transformations that are difficult to accomplish in other ways, and therefore provides a way for tuning the catalysts to specific needs. For reversible switchable catalysts, removal of the stimulus should cause the catalyst to revert to its initial form, and therefore to recover its original activity. Cases of reversible switchable catalysis can be found for redox,[1b, 1c, 1d, 2] pH‐,[3] or light‐driven[4] processes. Redox active ligands can participate in the catalytic cycle by only accepting/donating electrons that can be used for tuning the Lewis acidity of the metal, or in the formation/breaking of substrate covalent bonds.[1c] N‐heterocyclic carbene ligands (NHCs) have been highlighted recently for their ability to adapt their properties to the specific requirements of individual catalytic transformations, a reason for which they merited been qualified as “smart” ligands.[5] In this regard, NHCs have also demonstrated to be useful scaffolds for building redox switchable catalysts.[6] The first redox‐switchable catalyst based on a NHC ligand was described by Plenio and co‐workers,[7] who reported a Hoveyda–Grubbs catalyst bearing a NHC ligand with two pendant ferrocenyl groups that acted as redox‐responsive units. The strategy of using NHC ligands with ferrocenyl tags for electrochemically modifying the catalytic properties of the catalysts was also used by Bielawski,[8] Sarkar,[9] and us.[10] Ferrocenyl units were not the only ones used for designing redox‐active NHC ligands. For example, Bielawski and co‐workers reported a series of naphthoquinimidazolylidene complexes of NiII, PdII and PtII used in the Kumada cross‐coupling reaction.[11]
Although many ligand frameworks have been used for redox‐switchable purposes, fluorescent molecules that can promote electron transfer when illuminated have been used rarely.[12] Rylene dyes such as perylene‐diimides (PDI) and naphthalene‐diimides (NDI) are good examples of molecules that combine intense light absorption, high stability, electron accepting ability and high fluorescence quantum yields.[13] One remarkable example of the use of rylene‐dyes in transition metal catalysts, was reported by Crabtree, Brudvig and Wasielewski, when they described the photodriven intramolecular electron transfer from an iridium‐based water‐oxidation catalyst to a PDI unit.[14]
During the last few years, we focused most of our attention in using Janus‐type pyrene‐connected di‐NHC ligands for the preparation of catalysts[15] and supramolecular metallo‐assemblies.[16] Based on our previous results, we thought that preparing a NDI‐connected di‐NHC ligand would bring us the opportunity to prepare new materials exhibiting multifunctional structural, optical, electronic and magnetic properties, associated with the degree of through‐space electron sharing and accessible redox states of the NDI core. It is expected that a NDI‐bis‐imidazolylidene ligand displays similar structural features as those shown by the pyrene‐connected di‐NHC, but its electronic properties should significantly different, since the NDI core is highly electron‐deficient. Considering all the above mentioned, herein we describe the preparation of two NDI‐connected bis‐imidazolium salts and their use as NHC‐precursors in the preparation of rhodium and iridium complexes. The structural and electrochemical properties of the new complexes will be discussed. We will demonstrate how the redox‐active nature of the NDI dye can be used to generate catalysts that can be effectively toggled between their active and inactive forms, in the cyclization of acetylenic carboxylic acids for generating exocyclic enol lactones.
Results and Discussion
The NDI‐connected bis‐imidazolium salts [1 a](BF4)2 and [1 b](BF4)2 were obtained by bis‐annulation of the N,N′‐bis(n‐alkyl)‐2,3,6,7‐tetra(n‐alkyl)‐NDI compounds (alkyl=octyl, butyl)[17] with HBF4 in the presence of trimethyl orthoformate (Scheme 1). The resulting compounds were isolated as yellow products, which were characterized by NMR spectroscopy and Electrospray Ionization Mass spectrometry (ESI‐MS). The 1H NMR spectra of [1 a](BF4)2 and [1 b](BF4)2, showed the characteristic low field signals assigned to the two equivalent NCHN protons, at 9.68 and 9.62 ppm, respectively. The ESI‐mass spectrum of [1 a](BF4)2 showed peaks at m/z 1108.2 and 510.9, assigned to [M−BF4]+ and [M−2(BF4)]2+, respectively. In the case of [1 b](BF4)2, the ESI‐mass spectrum displayed peaks at m/z 771.9 and 342.5 for [M−BF4]+ and [M−2(BF4)]2+, respectively. The molecular structure of the bis‐imidazolium salt [1 a](BF4)2 (R=n‐octyl), was unambiguously determined by means of single crystal X‐ray diffraction. As can be seen in the structure shown in Figure 1, the molecule consists of a bis‐imidazolium salt connected by a central naphthalene‐tetracarboxylic‐diimide. The separation between the two carbon atoms located at the edges of the longer axis of the molecule is of 8.9 Å, very similar to the related distance shown by our previously described pyrene‐connected bis‐azolium salts (9.0 Å),[18] thus exemplifying the structural similarities between these two bis‐imidazolium salts connected by electronically dissimilar tetracyclic cores.
Scheme 1.

Synthesis of NDI‐based compounds.
Figure 1.

Two perspectives of the molecular structure of [1 a](BF4)2 obtained from single‐crystal X‐ray diffraction studies. Hydrogen atoms and counter‐anions (2 BF4 −) are omitted for clarity. The six n‐octyl chains are represented in the wireframe form. C gray, O red, N light blue.
In an attempt to use [1 a](BF4)2 and [1 b](BF4)2 as NHC precursors, we performed a reaction of these two salts with tBuOK in THF, in the presence of one equivalent of [MCl(COD)]2 (M=Rh, Ir). Although we expected to obtain the dimetallic complexes bound by a NDI‐connected di‐NHC ligand, all our attempts yielded NHC‐based monometallic complexes 2 and 3, in which one of the edges of the di‐imidazolium salt was oxidized to yield a NHC=O cyclic urea. Oxo‐substituted azoles may be produced from the reductive elimination of metal coordinated NHCs and oxygen‐containing anionic bases, a process that constitutes one of the main pathways of decomposition of NHC‐based metal catalysts in strong basic media.[19] We also observed the formation of these types of NHC=O adducts previously.[20] To avoid the formation of the cyclic urea, we tested some alternative metallation strategies, such as the in situ transmetallation from the silver‐NHC complexes, the use of weak bases like Cs2CO3 or NEt3, and different solvents (toluene, acetonitrile), but all our attempts resulted in the formation of complexes 2 and 3 in lower yields. Some of these attempts were accompanied by the concomitant formation of the bis‐urea product, which we isolated and characterized crystallographically (see SI for details). In any case, complexes 2 and 3 constitute unique examples of the introduction of the NDI moiety into the backbone of metal‐coordinated NHC ligands. In view of these findings, the synthesis of complexes 2 and 3 was modified and we used only 0.5 equivalents of [MCl(COD)]2 (M=Rh, Ir) with respect to the bis‐azolium salt (1 equivalent of metal/ligand, as detailed in the Supplementary Information). Complexes 2 and 3 were characterized by means of NMR spectroscopy. The 13C NMR spectra of the rhodium complexes 2 a and 2 b showed the diagnostic doublets assigned to the Rh‐Ccarbene carbons at 214.4 (1 J Rh‐C=51 Hz) and 214.5 ppm (1 J Rh‐C=51 Hz), respectively. The signals due to the Ir‐Ccarbene carbons of 3 a and 3 b were observed at 206.9 and 206.9 ppm, respectively. The infrared spectra of 2 and 3, show the characteristic band assigned to the urea C=O at ν(CO) in the range of 1736–1742 cm−1.
The molecular structure of 3 b was confirmed by means of single crystal X‐ray diffraction studies. The structure of 3 b (Figure 2) consists of a hexacyclic imidazolylidene ligand bound to an iridium atom, which completes its coordination sphere with one cyclooctadiene and a chloride ligand. The hexacyclic NHC ligand is formed by a NDI core flanked by an imidazolylidene ring and an oxo‐substituted azole. All six nitrogen atoms of the NDI‐decorated NHC ligand are functionalized with n‐butyl chains. The distance of the Ir‐Ccarbene bond is of 2.023 Å. The two imide‐functionalized six‐membered rings deviate from the plane formed by the naphthalene moiety by an angle of 23.8°, measured as the angle between the plane formed by the four oxygen atoms of the imide rings and the naphthalene plane. The crystal packing of the structure reveals the formation of extended π‐stacking interactions through the NDI part of the molecule.
Figure 2.

Two perspectives of the molecular structure of 3 b obtained from single‐crystal X‐ray diffraction studies. Hydrogen atoms are omitted for clarity. The n‐butyl chains are represented in the wireframe form. C gray, O red, N light blue, Cl green; Ir dark blue.
To attain some information about the electron‐donating character of the NDI‐functionalized NHC ligands, the carbonyl derivatives 4 a and 4 b were obtained by bubbling CO in a CH2Cl2 solution of the iridium complexes 3 a and 3 b at 0 °C, as depicted in Scheme 1. The resulting carbonyl‐substituted complexes were characterized by NMR spectroscopy. The IR spectrum (CH2Cl2) of the iridium complex 4 a shows the signals due to the C‐O stretching of the two carbonyl ligands at 2072 and 1991 cm−1. The IR spectrum of complex 4 b displays the two C−O stretching bands also at 2072 and 1991 cm−1. This indicates that both n‐butyl‐ and n‐octyl‐containing NDI‐NHC ligands exert the same electron‐donating character. By using the well‐accepted correlation,[21] and using the ν (C‐O) values obtained, we calculated that the Tolman Electronic Parameter (TEP) of the NDI‐NHC ligands is 2054 cm−1. This TEP value indicates that the electron‐donating character of these two NDI‐functionalized NHC ligands is very close to the ones shown by our previously reported pyrene‐functionalized NHC ligands.[18, 22]
Next, we turned our attention toward using cyclic voltammetry (CV) for evaluating the influence of the NHC ligand upon coordination to the metal complex. Figure 3 shows the cyclic voltammograms of the NDI‐functionalized bis‐imidazolium salt [1 a](BF4)2 and the NDI‐decorated NHC rhodium complex 2 a. The CV data for [1 a](BF4)2 reveals two well separated reversible one‐electron reduction processes. The first process (E 1/2=−0.23 V vs. Fc+/Fc) is the reduction of the dicationic salt to a radical monocation, while the second reduction (E 1/2=−0.70 V vs. Fc+/Fc) corresponds to the formation of a doubly reduced neutral species. For this data set, the peak potentials are independent of the scan rate, as expected for reversible processes. The reduction potential observed for [1 a](BF4)2 are significantly less negative than those obtained for the related neutral naphthalene diimides,[23] as a consequence of the doubly positive nature of the salt introduced by the presence of the two imidazolium rings. The CV diagram for complex 2 a, shows two reversible well separated reduction waves at −1.14 and −1.51 V (vs. Fc+/Fc), therefore showing that the reduction of the NDI core is produced at lower (more negative) potentials than those observed for the dicationic salt [1 a](BF4)2, and at similar values as those shown for neutral naphthalene diimides.[23] In this case, the first reduction produces a radical anion (2 a .−), while the second reduction produces a dianion (2 a 2−). The CV data for 2 a also shows another redox event produced at a value of E 1/2=+0.59 V (vs. Fc+/Fc), which corresponds to the quasi‐reversible RhI/RhII oxidation. The CV data for the rest of the complexes are depicted in Table 1. From the analysis of the data, we can conclude that: i) the substitution of the n‐octyl chains by n‐butyl groups, produces negligible changes (ca. 10 mV) in the redox potentials of the compounds, indicating that the change of the N‐alkyl group does not produce changes in the electron‐donating character of the NHC ligand; ii) the reduction potentials of the NDI core of the ligands present in the rhodium and iridium complexes are virtually identical; and iii) the reduction potentials of the COD‐substituted rhodium and iridium complexes 2 b and 3 b, are significantly more negative (110–120 mV) than the related values shown for the carbonyl complexes 4 a and 4 b, as a consequence of the reduction of the electron density in the metal upon introduction of the π‐accepting carbonyl ligands. The fact that the reduction potential of the NDI‐decorated NHC ligand is sensitive to changes in the nature of the metal centers bound to the NHC ligand (observation iii mentioned above) is an indication of the coupling between the metal center and the NDI core of the ligand.
Figure 3.

Cyclic voltammograms of [1 a](BF4)2 (a) and 2 a (b) in dry CH2Cl2 with 1 mM analyte and 0.25 M [N(nBu)4][PF6]. Measurements performed at 100 mV s−1 and referenced vs. ferrocenium/ferrocene.
Table 1.
Electrochemical properties of compounds 1–4. Data shown from lower to higher potential values.[a]
|
Compound |
E1/2 [V] (ΔE [mV]) |
E′1/2 [V] (ΔE [mV]) |
E′′1/2 [V] |
|---|---|---|---|
|
[1 a](BF4)2 |
−0.70(69) |
−0.23(69) |
– |
|
[1 b](BF4)2 |
−0.71(65) |
−0.24(63) |
– |
|
2 a |
−1.51(73) |
−1.14(105) |
0.59 |
|
2 b |
−1.49(71) |
−1.16(69) |
0.56 |
|
3 a |
−1.50 (69) |
−1.16 (71) |
0.57 |
|
3 b |
−1.49(71) |
−1.15 (69) |
0.55 |
|
4 a |
−1.39 (69) |
−1.03 (69) |
– |
|
4 b |
−1.39 (60) |
−1.03 (69) |
– |
[a] Cyclic voltammograms performed in dry CH2Cl2 with 1 mM analyte and 0.25 M [N(nBu)4][PF6]. Measurements performed at 100 mV s−1 and referenced vs. ferrocenium/ferrocene.
To obtain information about the nature and stability of the species formed upon one‐ and two‐electron reduction of the salts [1](BF4)2, and the metal complexes obtained with the NDI‐functionalized NHC ligands, we performed a series of spectroelectrochemical (SEC) experiments. First, the bulk reduction of the NDI‐functionalized bis‐imidazolium salts [1 a](BF4)2 and [1 b](BF4)2 was performed in an Optically Transparent Thin Layer Electrochemical (OTTLE) cell in CH2Cl2, by progressively applying more negative potentials while recording the corresponding UV/Vis spectra. As can be seen in Figure 4 a, the absorption spectrum of [1 b]2+ shows one strong, structured absorption band below 500 nm. Upon application of stepwise electrolysis potential, the band associated to [1 b]2+ decreased, while a new intense band centered at 725 nm associated to [1 b. ]+ appeared. Further electrochemical reduction of this species gave rise to the appearance of a vibronically resolved band with peak maxima at 500 and 550 nm, attributed to the 2e‐reduced species [1 b]. Interestingly, the analysis of the UV/Vis spectra reveals that the transition between [1 b]2+ → [1 b. ]+, and then between [1 b. ]+ → [1 b] display clear isosbestic points at 450 and 525 nm, and 465 and 620 nm, for the transitions associated to the first and second reductions, respectively. This observation indicates that both transitions are produced without the appearance of further reaction intermediates and that the species involved in the two‐step reduction process are stable under the conditions used to carry out the SEC experiment.
Figure 4.

UV/Vis SEC monitoring reduction of a) [1 b]2+ in dry CH2Cl2 (0.1 M [N(nBu)4][PF6]), and b) 3 b in dry CH3CN (0.1 M [N(nBu)4][PF6]). The electrochemical reduction was performed applying progressively lower potentials with a Au working electrode, Pt counter‐electrode, and Ag wire pseudo‐reference electrode. The solid lines represent the spectra of the starting (red), singly reduced (blue) and doubly reduced (black) species. Arrows are used to signal the isosbestic points.
Similarly, SEC experiment was performed using the iridium complex 3 b. In this case, CH3CN was used as solvent. As can be seen from Figure 4 b, the UV/vis spectrum of 3 b shows a strong, structurally resolved band centered at 500 nm (solid red line in Figure 4 b), which is attributed to transitions centered in the NDI core. Upon progressive reduction, the intensity of this band decreases, while a new set of bands associated to [3 b. ]− appear (solid blue line in Figure 4 b). Further reduction produced the disappearance of the bands attributed to [3 b. ]−, and the appearance of new bands assigned to the doubly reduced di‐anionic species [3 b]2− (black solid line in Figure 4 b). Again, the transition between these three species occurred without the detection of reaction intermediates, as indicated by the presence of isosbestic points at 540 and 615 nm, for the first and second reduction steps, respectively. This observation also indicates that the three species involved in the reduction process are fairly stable under the conditions used for the experiments.
We expected that the reduction of the NDI‐containing NHCs in complexes 3 and 4 should increase significantly the electron‐donating abilities of the ligand. To quantify this effect, we performed IR‐SEC experiments to the iridium carbonyl complexes 4 a and 4 b. Figure 5 shows the set of IR spectra obtained for the progressive reduction of 4 b. The experiments were performed in CH2Cl2. The progressive reduction of 4 b is accompanied by a decrease of the intensity of the two CO stretching bands at 2072 and 1991 cm−1, while two new ones appear at 2061 and 1981 cm−1, which are assigned to the 1e‐reduced species [4 b. ]−. Further reduction to lower potentials results in the disappearance of the signals at 2061 and 1981 cm−1, with the concomitant appearance of two new C‐O stretching bands at 2052 and 1972 cm−1, assigned to the 2e‐reduced species [4 b]2−. This means that the first reduction produces an average Δν (CO) of −10.5 cm−1, while the second reduction shifts the ν (CO) by another −9 cm−1, thus showing that the ligand can enhance its donor properties in a two‐step tunable way.
Figure 5.

IR‐SEC reduction of 4 b in dry CH2Cl2 (0.1 M [N(nBu)4][PF6]). The electrochemical reduction was performed applying progressively lower potentials with a Au working electrode, Pt counter electrode, and Ag wire pseudo‐reference electrode. The solid lines represent the IR spectra of 4 b (red), [4 b. ]−(blue) and [4 b]2− (black) species. Arrows are used to indicate the isosbestic points.
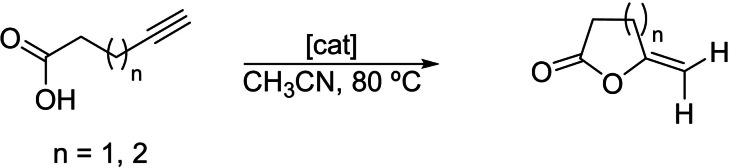
As we showed above, the reduction of the naphthalene‐diimide‐imidazolylidene ligand in complexes 2 and 3 increases significantly the electron density of the metal. We envisaged that this effect could be used for enhancing the activity of the catalysts, in processes where the rate‐determining step of the cycle is facilitated by increasing the electron‐richness of the metal. We decided to study the cyclization of acetylenic carboxylic acids, an atom‐economic process that constitutes a very convenient route for synthesizing exocyclic enol lactones. Exocyclic enol lactones are important reagents because they are prevalent in natural products with interesting biological activity.[24] In addition, lactones are useful and versatile intermediates in synthetic organic chemistry.[25] Some authors,[26] including us,[27] have devoted great attention to the development of catalysts for the synthesis of five‐ and six‐membered lactones via the cyclization of alkynoic acids. The intermolecular version of this process is the coupling of terminal alkynes with carboxylic acids to form vinyl esters.[28] Among the catalysts used to promote this reaction, those based on rhodium and iridium are particularly interesting,[29] and a significant number of investigations have been devoted to elucidating the mechanistic aspects of the process.[30]
We studied the cycloisomerization of 4‐pentynoic and 5‐hexynoic acids, using the cyclooctadiene‐containing complexes 2 and 3 (Scheme 1) The reactions were carried out in acetonitrile at 80 °C, using catalyst loadings of 0.25 and 0.01 mol % for the cyclization of 4‐pentynoic acid, and 1 mol % for the cyclization of 5‐hexynoic acid. The results that we obtained are shown in Table 2. For the cyclization of 4‐pentynoic acid (entries 1–8), the activities shown by 2 a (entry 1), 3 a (entry 3) and 3 b (entry 5) after 7 hours of reaction were very low. For the catalysts containing the n‐octyl‐substituted NHC ligand, we observed that the activity of the rhodium complex 2 a was higher than that of the iridium analogue 3 a (compare entries 1 and 3). This result is in accordance with previous studies that indicate that for this type of process, rhodium complexes normally outperform their iridium analogues.[26c, 30f] The activity shown by the n‐butyl‐substituted iridium complex 3 b, was higher compared to the one shown by n‐octyl‐containing complexes 2 a and 3 a, thus indicating a positive effect of the n‐butyl group in the process. To study the effect of the reduction of the NDI‐containing NHC ligand on the performance of the catalyst, we decided to carry out the reaction in the presence of cobaltocene. With a redox potential of −1.33 V,[31] we considered that cobaltocene is a suitable reducing agent for the selective one‐electron reduction of 2 and 3, since these complexes displayed redox couples at −1.14–(−1.16) V and −1.49–(1.51), for the one‐ and two‐electron reductions, respectively (vide supra). As can be seen from the data shown in Table 2, the addition of one equivalent of cobaltocene resulted in a clear improvement in the activity of the catalyst (entries 2, 4 and 6). The rhodium catalyst 2 a, afforded a 50 % yield, compared to the 17 % yield that was obtained without addition of cobaltocene. The differences in the activities shown for the iridium complexes were even more significant. For both 3 a and 3 b, the addition of cobaltocene allowed that the production of γ‐methylene‐γ‐butyrolactone became quantitative, an observation that is even more relevant for the case of 3 a, for which the reaction in the absence of cobaltocene produced negligible amounts of the lactone (compare entries 3 and 4). It is important to note that for the reaction carried out with 3 b in the presence of cobaltocene, the quantitative production of the lactone was achieved in just 2.5 hours (entry 6). A control experiment carried out using cobaltocene in the absence of catalyst did not yield any trace of product. In view of the good activity shown by catalyst 3 b in the presence of cobaltocene, we decided to see if we could enhance its activity by using a stronger reductant capable of producing the two‐electron reduction of the catalyst. Decamethylcobaltocene ([CoCp*2]) has a redox potential of −1.94 V, and thus we can use two equivalents of this reagent to transfer two electrons to 3 b. Taking this into account, we repeated the cycloisomerization of 4‐pentynoic acid in the presence of 3 b (0.25 mol %) with the addition of two equivalents of [CoCp*2], and observed that the reaction reached completion after 3 hours, therefore the activity of the catalyst did not improve with respect to the reaction carried out in the presence of [CoCp2] (compare entries 6 and 7). To check if the activity of the “reduced” catalyst was maintained after completion of the catalytic reaction, we added a second batch of starting material and allowed the reaction to proceed for 3 more hours. This experiment was repeated twice, and allowed us to confirm that the activity of the catalyst was maintained all over the three cycles (see Figure S46 in the Supplementary Information).
Table 2.
Catalytic cyclization of acetylenic carboxylic acids.[a]
|
Entry |
Alkynoic Acid |
Cat. |
Cat. Load. |
Additive |
t [h] |
Yield [%][a] |
|---|---|---|---|---|---|---|
|
1 |
4‐pentynoic |
2 a |
0.25 |
none |
7 |
17 |
|
2 |
4‐pentynoic |
2 a |
0.25 |
[CoCp2] |
7 |
50 |
|
3 |
4‐pentynoic |
3 a |
0.25 |
none |
7 |
7 |
|
4 |
4‐pentynoic |
3 a |
0.25 |
[CoCp2] |
7 |
>99 |
|
5 |
4‐pentynoic |
3 b |
0.25 |
none |
7 |
40 |
|
6 |
4‐pentynoic |
3 b |
0.25 |
[CoCp2] |
2.5 |
>99 |
|
7 |
4‐pentynoic |
3 b |
0.25 |
[CoCp*2] |
3 |
>99 |
|
8 |
4‐pentynoic |
3 b |
0.01 |
[CoCp2] |
75 |
>99 |
|
9 |
5‐hexynoic |
2 a |
1 |
none |
50 |
2 |
|
10 |
5‐hexynoic |
2 a |
1 |
[CoCp2] |
50 |
7 |
|
11 |
5‐hexynoic |
3 a |
1 |
none |
12.5 |
0 |
|
12 |
5‐hexynoic |
3 a |
1 |
[CoCp2] |
12.5 |
>99 |
|
13 |
5‐hexynoic |
3 b |
1 |
none |
7 |
0 |
|
14 |
5‐hexynoic |
3 b |
1 |
[CoCp2] |
7 |
>99 |
|
15 |
5‐hexynoic |
3 b |
1 |
[CoCp*2] |
5 |
>99 |
[a] Reactions carried out in 1.5 mL of CD3CN, at 80 °C. The cyclization of 4‐pentynoic acid was performed using a solution 0.33 M of the alkynoic acid. The cyclization of 5‐hexynoic acid was carried out with a solution 0.083 M of the alkynoic acid. The reactions performed in the presence of cobaltocene contained one equivalent of [CoCp2] (with respect to catalyst). The reactions performed in the presence of decamethylcobaltocene contained two equivalents of [CoCp*2] (with respect to catalyst). Yields were calculated by GC and 1H‐NMR spectroscopy, using 1,3,5‐trimethoxybenzene as integration standard.
We also tested the cyclization of 4‐pentynoic acid using a catalyst loading of 0.01 mol %, in the presence of cobaltocene, and observed that the reaction was complete after 75 h (entry 8). This result is interesting because it gives a record TON value of 10 000 for this reaction, but also because it demonstrates that the activity of the catalyst is maintained for long reaction times. It is also important to mention that the comparison of the normalized time‐dependent profiles of the reactions carried out with catalyst 3 b at these two concentrations (0.25 and 0.01 mol %) in the presence of cobaltocene is consistent with a reaction order of 1 in catalyst (details about determination of reaction order can be found in the SI).[32]
Since the cyclization of 5‐hexynoic acid is known to be much less efficient,[26c, 27a, 27d, 29e] we performed the reaction using a larger catalyst loading than that used for the cyclization of 4‐pentynoic acid (1 mol % vs. 0.25 mol %). Under the reaction conditions indicated in Table 2, all three catalysts were completely inefficient in the process (entries 9, 11 and 13). Only traces of the product were observed for the reaction catalyzed with the rhodium complex 2 a, after 50 hours of reaction (entry 9). Again, addition of cobaltocene resulted in a clear enhancement of the catalytic activity. For the reaction catalyzed with the rhodium complex 2 a, the product yield was increased to 7 % after 50 hours (entry 10). The results observed for the two iridium complexes were much more dramatic. Complex 3 a afforded quantitative production of the lactone in 12.5 hours in the presence of cobaltocene, while 3 b needed only 7 hours to complete the process (see entries 12 and 14). When the reaction was carried out using 3 b in the presence of two equivalents of [CoCp*2], the reaction reached completion after only 5 hours (entry 15), thus indicating that, for this substrate, the addition of the stronger reductant had a positive effect in the activity of the catalyst.
To provide further insights on the effect of the addition of cobaltocene on the performance of the activity of complexes 2 and 3, we decided to study some time‐dependent reaction profiles. Figure 6 shows the profile for the cyclization of 4‐pentynoic acid using catalysts 2 a and 3 a, with and without the addition of cobaltocene. The visual analysis of the plots shown in Figure 6, indicates a zeroth order dependence on the substrate. The addition of cobaltocene produces a threefold increase in the kinetic constant of the reaction catalyzed by the rhodium complex 2 a (from 3.2×10−6 to 9.5×10−6 M s−1). This increase is even more pronounced in the case of the iridium‐catalyzed process, for which the kinetic constant experiences an increase of more than one order of magnitude (from 5.3×10−7 to 1.9×10−5 M s−1).
Figure 6.

Time‐dependent reaction profiles for the cyclization of 4‐pentynoic acid using catalysts 2 a and 3 a with and without addition of cobaltocene. The reactions were carried out in acetonitrile, with an initial concentration of 4‐pentynoic acid of 0.33 M, and a catalyst loading of 0.25 mol %. Cobaltocene was added in a 0.25 mol % with respect to the substrate. Yields were determined by GC, using 1,3,5‐trimethoxybenzene as integration standard. Final yields were also corroborated by 1H NMR spectroscopy. The Figure also shows the kinetic constants for each reaction.
The results found are in agreement with the reaction mechanism shown in Scheme 2, which is also supported by previous mechanistic studies.[26d, 26g, 29e, 30a, 30e, 30f] The zeroth order dependence with respect to the substrate, also observed in other systems,[30a] strongly suggests that the substrate is coordinated to the coordination sphere of the metal in the resting state of the catalytic cycle. Then the fact that the one‐electron reduction of the ligand produces an improvement in the catalytic performance of the catalyst, indicates that the rate‐determining step of this reaction very likely involves a process in which the metal is oxidized, since increasing the electrondonicity of the ligand facilitates the oxidative process. With this data in hand, we believe that the first step of the reaction is the coordination of the alkynoic substrate to the metal via the formation of an alkyne–metal bond, to form I. The following step would be an intramolecular oxidative addition of the O−H bond of the carboxylic acid, to form the MIII species II, or the generation of the MIII species IV through a reaction intermediate III in which the proton of the carboxylic acid electrophilically attacks the terminal carbon of the alkyne bound to the metal. A rhodium‐based metallo‐lactone related to IV was isolated and characterized by Marder and co‐workers in 1988.[30e] While most of the reports on the mechanism of this process suggest that the reaction proceeds through the oxidative addition of the O−H bond through II,[26d, 26g, 29e, 30e, 30f] the detailed mechanistic studies performed by Breit and co‐workers indicate that the process involves an intermediate such as III.[30a] In either case, the rate‐determining step would involve an oxidation of the metal, in agreement with our experimental observation. It is interesting to recall that the addition of a stronger reductant such a [CoCp*2] has a different effect, depending on whether the substrate is 4‐pentynoic acid or 5‐hexynoic acid. In the case of the cyclization of 4‐pentynoic acid, the activity of the catalyst seems to be slightly reduced, compared to the situation in which cobaltocene is added. For the reaction with 5‐hexynoic acid, the addition of [CoCp*2] produces an acceleration of the reaction with respect to the reaction performed in the presence of cobaltocene, although the effect is not as pronounced as when comparing the activities of the catalyst in the absence and in the presence of one equivalent of cobaltocene. One plausible explanation for this effect is that, while the reduction of the catalyst facilitates the oxidative addition in the catalytic cycle, it also makes the reductive elimination step more difficult. The catalytic reaction will be favored by the addition of a reductant as long as the rate‐determining step of the process is the oxidative addition, but a very strong reductant can make that the rate‐determining step switches to the reductive elimination, and therefore can be detrimental for the catalytic process.
Scheme 2.

Representation of the mechanism proposed for the cyclization of alkynoic acids.
Since we observed such great difference in the catalytic behavior of the iridium catalysts between their neutral and reduced forms, we decided to investigate whether we would be able to toggle between an active and inactive catalyst during the course of a catalytic reaction. To do this, we chose to study the cyclization of 4‐pentynoic acid using catalyst 3 a (Figure 7). We chose 3 a because according to the data shown in Table 2, and to the reaction profiles shown in Figure 6, catalyst 3 a is the one that shows larger differences in the activities of the neutral and the one‐electron reduced forms. First, the reaction was allowed to proceed at 80 °C in CD3CN in the presence of 3 a (0.25 mol %) for 3 hours. After this time, we did not observe the formation of any trace of the lactone product. Then, we added 1 equivalent of [CoCp2] and observed the activation of the catalyst, which produced 29 % of the lactone in the following 3 hours. The sequential addition of [FeCp2](BF4) and [CoCp2] separated by periods of 3 hours, produced a total of three cycles of activations and deactivations of the catalyst until the quantitative formation of the lactone was obtained after 19 hours (Figure 4). These results clearly show how the activity of catalyst 3 a can be “switched on” upon addition of [CoCp2], and then “switched off” back again by adding [FeCp2](BF4). Interestingly, the activity of the catalyst is restored in almost 100 % its capacity after every “switch on” process, as can be observed by comparing the three quasi‐identical kinetic constants found for the time gaps where the catalyst was in its active form. These studies demonstrate that the catalyst is stable both in the active/reduced and inactive/oxidized states, and that extended durations in the inactive form do still allow reactivation upon subsequent reduction.
Figure 7.

Plot showing the cycloisomerization of 4‐pentynoic acid with 0.25 mol % of 3 a and sequential additions of [Cp2Co] and [FeCp2](BF4). The reaction was carried out in CD3CN at 80 °C, with an initial concentration of 4‐pentynoic acid of 0.33 M. Product yields determined by GC, using 1,3,5‐trimethoxybenzene as integration standard. Final yields were also corroborated by 1H‐NMR spectroscopy.
Conclusion
We prepared a naphthalene‐diimide‐functionalized NHC ligand that was coordinated to rhodium and iridium. The CV studies revealed that the resulting coordination complexes show two reversible reduction waves associated to two successive one‐electron reductions of the NDI core of the NHC ligand. This reduction is accompanied by a significant increase of the electron‐richness of the metal, as observed by comparing the C‐O stretching frequencies of the infrared spectra of the neutral, singly reduced and doubly reduced metal carbonyl complexes, thus affording a rare example in which a switchable ligand is capable of adopting three levels of electronic control. The iridium and rhodium complexes obtained were tested in the cycloisomerization of alkynoic acids, where the one‐electron reduction of the ligand produces a great enhancement of the catalytic activity of the process. This enhancement can be further improved in the case of the cyclization of 5‐hexynoic acid if the ligand is doubly reduced. The stability of the neutral and reduced forms of the catalysts is evident from the plots that represent the concentration time‐dependent profiles of the catalytic reactions, where it is observed that the reaction rates are maintained all along the process, regardless its duration. Furthermore, the activity of catalysts can toggle between the activated and inactivated form for several cycles by adding subsequently a reductant and an oxidant. The use of these redox‐switchable catalysts allowed us to achieve TON values of up to 10 000, for the cycloisomerization of 4‐pentynoic acid, which, to our knowledge, is the highest reported for this specific reaction. Finally, the redox control of this particular reaction enables an enhanced understanding of the reaction mechanism.
Collectively, these results underscore the potential of NDI‐functionalized NHCs to impart redox‐switchable properties to metal catalysts. Given the wide applicability of NHCs in homogeneously catalyzed processes, we expect that this new ligand will be useful for expanding the use of redox‐switchable catalysts for improving the efficiency of a large number of synthetically appealing organic transformations. We think that this improvement will be most likely observed in catalytic cycles whose rate‐determining steps are facilitated by electron‐rich metals, such as oxidative additions, or electrophilic attacks to the coordinated substrate.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We gratefully acknowledge financial support from the Ministerio de Ciencia y Universidades (PGC2018‐093382‐B‐I00), Generalitat Valenciana (AICO/2019/149), and the Universitat Jaume I (UJI‐B2017‐07 and UJI‐B2018‐46). S.G. thanks the Juan de la Cierva Incorporación program (IJC2019‐039982‐I). We are grateful to the Serveis Centrals d'Instrumentació Científica (SCIC‐UJI) for providing with spectroscopic facilities.
C. Ruiz-Zambrana, A. Gutiérrez-Blanco, S. Gonell, M. Poyatos, E. Peris, Angew. Chem. Int. Ed. 2021, 60, 20003.
Contributor Information
Macarena Poyatos, Email: poyatosd@uji.es.
Eduardo Peris, Email: eperis@uji.es.
References
- 1.
- 1a.Crabtree R. H., New J. Chem. 2011, 35, 18–23; [Google Scholar]
- 1b.Berben L. A., de Bruin B., Heyduk A. F., Chem. Commun. 2015, 51, 1553–1554; [DOI] [PubMed] [Google Scholar]
- 1c.Lyaskovskyy V., de Bruin B., ACS Catal. 2012, 2, 270–279; [Google Scholar]
- 1d.de Bruin B., Eur. J. Inorg. Chem. 2012, 340–342; [Google Scholar]
- 1e.Lüning U., Angew. Chem. Int. Ed. 2012, 51, 8163–8165; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 8285–8287; [Google Scholar]
- 1f.Blanco V., Leigh D. A., Marcos V., Chem. Soc. Rev. 2015, 44, 5341–5370; [DOI] [PubMed] [Google Scholar]
- 1g.Choudhury J., Tetrahedron Lett. 2018, 59, 487–495. [Google Scholar]
- 2.
- 2a.Allgeier A. M., Mirkin C. A., Angew. Chem. Int. Ed. 1998, 37, 894–908; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 936–952; [Google Scholar]
- 2b.Luca O. R., Huang D. L., Takase M. K., Crabtree R. H., New J. Chem. 2013, 37, 3402–3405. [Google Scholar]
- 3.
- 3a.Wu C. Y., Chen H. Q., Corrigan N., Jun K., Kan X. N., Li Z. B., Liu W. J., Xu J. T., Boyer C., J. Am. Chem. Soc. 2019, 141, 8207–8220; [DOI] [PubMed] [Google Scholar]
- 3b.Walczak A., Stefankiewicz A. R., Inorg. Chem. 2018, 57, 471–477; [DOI] [PubMed] [Google Scholar]
- 3c.Zhan P. F., Wang J. Y., Wang Z. G., Ding B. Q., Small 2014, 10, 399–406; [DOI] [PubMed] [Google Scholar]
- 3d.Dunbar M. A., Balof S. L., Roberts A. N., Valente E. J., Schanz H. J., Organometallics 2011, 30, 199–203; [Google Scholar]
- 3e.Balof S. L., P′Pool S. J., Berger N. J., Valente E. J., Shiller A. M., Schanz H. J., Dalton Trans. 2008, 5791–5799. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a.Neilson B. M., Bielawski C. W., ACS Catal. 2013, 3, 1874–1885; [Google Scholar]
- 4b.Freixa Z., Catal. Sci. Technol. 2020, 10, 3122–3139. [Google Scholar]
- 5.Peris E., Chem. Rev. 2018, 118, 9988–10031. [DOI] [PubMed] [Google Scholar]
- 6.Ryu Y., Ahumada G., Bielawski C. W., Chem. Commun. 2019, 55, 4451–4466. [DOI] [PubMed] [Google Scholar]
- 7.Süßner M., Plenio H., Angew. Chem. Int. Ed. 2005, 44, 6885–6888; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 7045–7048. [Google Scholar]
- 8.
- 8a.Arumugam K., Varnado C. D., Sproules S., Lynch V. M., Bielawski C. W., Chem. Eur. J. 2013, 19, 10866–10875; [DOI] [PubMed] [Google Scholar]
- 8b.C. D.Varnado, Jr. , Rosen E. L., Collins M. S., Lynch V. M., Bielawski C. W., Dalton Trans. 2013, 42, 13251–13264. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a.Hettmanczyk L., Suntrup S. K., Hoyer C., Sarkar B., Chem. Eur. J. 2017, 23, 576–585; [DOI] [PubMed] [Google Scholar]
- 9b.Maity R., van der Meer M., Sarkar B., Dalton Trans. 2015, 44, 46–49; [DOI] [PubMed] [Google Scholar]
- 9c.Hettmanczyk L., Manck S., Hoyer C., Hohloch S., Sarkar B., Chem. Commun. 2015, 51, 10949–10952. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a.Ibáñez S., Poyatos M., Peris E., ChemCatChem 2016, 8, 3790–3795; [Google Scholar]
- 10b.Ibáñez S., Poyatos M., Dawe L. N., Gusev D., Peris E., Organometallics 2016, 35, 2747–2758. [Google Scholar]
- 11.Tennyson A. G., Lynch V. M., Bielawski C. W., J. Am. Chem. Soc. 2010, 132, 9420–9429. [DOI] [PubMed] [Google Scholar]
- 12.Thompson B. L., Simons C. R., Heiden Z. M., Chem. Commun. 2019, 55, 11430–11433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a.Feng J. J., Jiang W., Wang Z. H., Chem. Asian J. 2018, 13, 20–30; [DOI] [PubMed] [Google Scholar]
- 13b.Jiang W., Li Y., Wang Z. H., Acc. Chem. Res. 2014, 47, 3135–3147; [DOI] [PubMed] [Google Scholar]
- 13c.Tovar J. D., Acc. Chem. Res. 2013, 46, 1527–1537; [DOI] [PubMed] [Google Scholar]
- 13d.Maniam S., Higginbotham H. F., Bell T. D. M., Langford S. J., Chem. Eur. J. 2019, 25, 7044–7057; [DOI] [PubMed] [Google Scholar]
- 13e.Kumar S., Shukla J., Kumar Y., Mukhopadhyay P., Org. Chem. Front. 2018, 5, 2254–2276; [Google Scholar]
- 13f.Pan M., Lin X. M., Li G. B., Su C. Y., Coord. Chem. Rev. 2011, 255, 1921–1936; [Google Scholar]
- 13g.Bhosale S. V., Jani C. H., Langford S. J., Chem. Soc. Rev. 2008, 37, 331–342. [DOI] [PubMed] [Google Scholar]
- 14.Vagnini M. T., Smeigh A. L., Blakemore J. D., Eaton S. W., Schley N. D., D'Souza F., Crabtree R. H., Brudvig G. W., Co D. T., Wasielewski M. R., Proc. Natl. Acad. Sci. USA 2012, 109, 15651–15656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a.Nuevo D., Poyatos M., Peris E., Organometallics 2018, 37, 3407–3411; [Google Scholar]
- 15b.Gonell S., Peris E., ACS Catal. 2014, 4, 2811–2817; [Google Scholar]
- 15c.Gonell S., Peris E., Poyatos M., Eur. J. Inorg. Chem. 2019, 33, 3776–3781; [Google Scholar]
- 15d.Ibáñez S., Poyatos M., Peris E., Dalton Trans. 2016, 45, 14154–14159; [DOI] [PubMed] [Google Scholar]
- 15e.Peris E., Chem. Commun. 2016, 52, 5777–5787. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a.Martínez-Agramunt V., Peris E., Chem. Commun. 2019, 55, 14972–14975; [DOI] [PubMed] [Google Scholar]
- 16b.Martínez-Agramunt V., Peris E., Inorg. Chem. 2019, 58, 11836–11842; [DOI] [PubMed] [Google Scholar]
- 16c.Martínez-Agramunt V., Eder T., Darmandeh H., Guisado-Barrios G., Peris E., Angew. Chem. Int. Ed. 2019, 58, 5682–5686; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 5738–5742; [Google Scholar]
- 16d.Martínez-Agramunt V., Gusev D. G., Peris E., Chem. Eur. J. 2018, 24, 14802–14807; [DOI] [PubMed] [Google Scholar]
- 16e.Martínez-Agramunt V., Ruiz-Botella S., Peris E., Chem. Eur. J. 2017, 23, 6675–6681. [DOI] [PubMed] [Google Scholar]
- 17.Sasikumar M., Suseela Y. V., Govindaraju T., Asian J. Org. Chem. 2013, 2, 779–785. [Google Scholar]
- 18.Gonell S., Poyatos M., Peris E., Chem. Eur. J. 2014, 20, 9716–9724. [DOI] [PubMed] [Google Scholar]
- 19.Chernyshev V. M., Khazipov O. V., Shevchenko M. A., Chernenko A. Y., Astakhov A. V., Eremin D. B., Pasyukov D. V., Kashin A. S., Ananikov V. P., Chem. Sci. 2018, 9, 5564–5577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ibáñez S., Poyatos M., Peris E., Chem. Commun. 2017, 53, 3733–3736. [DOI] [PubMed] [Google Scholar]
- 21.R. A.Kelly III , Clavier H., Giudice S., Scott N. M., Stevens E. D., Bordner J., Samardjiev I., Hoff C. D., Cavallo L., Nolan S. P., Organometallics 2008, 27, 202–210. [Google Scholar]
- 22.Valdés H., Poyatos M., Peris E., Organometallics 2014, 33, 394–401. [Google Scholar]
- 23.Andric G., Boas J. F., Bond A. M., Fallon G. D., Ghiggino K. P., Hogan C. F., Hutchison J. A., Lee M. A. P., Langford S. J., Pilbrow J. R., Troup G. J., Woodward C. P., Aust. J. Chem. 2004, 57, 1011–1019. [Google Scholar]
- 24.
- 24a.Kurume A., Kamata Y., Yamashita M., Wang Q. L., Matsuda H., Yoshikawa M., Kawasaki I., Ohta S., Chem. Pharm. Bull. 2008, 56, 1264–1269; [DOI] [PubMed] [Google Scholar]
- 24b.Pearce A. N., Chia E. W., Berridge M. V., Maas E. W., Page M. J., Webb V. L., Harper J. L., Copp B. R., J. Nat. Prod. 2007, 70, 111–113; [DOI] [PubMed] [Google Scholar]
- 24c.Beck J. J., Chou S. C., J. Nat. Prod. 2007, 70, 891–900; [DOI] [PubMed] [Google Scholar]
- 24d.Nomura T., Kushiro T., Yokota T., Kamiya Y., Bishop G. J., Yamaguchi S., J. Biol. Chem. 2005, 280, 17873–17879; [DOI] [PubMed] [Google Scholar]
- 24e.Richter S., Palumbo M., Mini-Rev. Med. Chem. 2003, 3, 37–49; [DOI] [PubMed] [Google Scholar]
- 24f.Hirota A., Nakagawa M., Hirota H., Agric. Biol. Chem. 1991, 55, 1187–1188; [Google Scholar]
- 24g.Yanagihara N., Lambert C., Iritani K., Utimoto K., Nozaki H., J. Am. Chem. Soc. 1986, 108, 2753–2754. [Google Scholar]
- 25.
- 25a.Quinn R. K., Konst Z. A., Michalak S. E., Schmidt Y., Szklarski A. R., Flores A. R., Nam S., Horne D. A., Vanderwal C. D., Alexanian E. J., J. Am. Chem. Soc. 2016, 138, 696–702; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25b.Valot G., Mailhol D., Regens C. S., O'Malley D. P., Godineau E., Takikawa H., Philipps P., Fürstner A., Chem. Eur. J. 2015, 21, 2398–2408. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a.Mancano G., Page M. J., Bhadbhade M., Messerle B. A., Inorg. Chem. 2014, 53, 10159–10170; [DOI] [PubMed] [Google Scholar]
- 26b.Man B. Y. W., Bhadbhade M., Messerle B. A., New J. Chem. 2011, 35, 1730–1739; [Google Scholar]
- 26c.Elgafi S., Field L. D., Messerle B. A., J. Organomet. Chem. 2000, 607, 97–104; [Google Scholar]
- 26d.Huang Y., Zhang X. H., Dong X. Q., Zhang X. M., Adv. Synth. Catal. 2020, 362, 782–788; [Google Scholar]
- 26e.Geier M. J., Vogels C. M., Decken A., Westcott S. A., Eur. J. Inorg. Chem. 2010, 4602–4610; [Google Scholar]
- 26f.Haydl A. M., Breit B., Chem. Eur. J. 2017, 23, 541–545; [DOI] [PubMed] [Google Scholar]
- 26g.Lumbroso A., Abermil N., Breit B., Chem. Sci. 2012, 3, 789–793; [Google Scholar]
- 26h.Man B. Y. W., Knuhtsen A., Page M. J., Messerle B. A., Polyhedron 2013, 61, 248–252. [Google Scholar]
- 27.
- 27a.Gutiérrez-Blanco A., Peris E., Poyatos M., Organometallics 2018, 37, 4070–4076; [Google Scholar]
- 27b.Mas-Marzá E., Peris E., Castro-Rodriguez I., Meyer K., Organometallics 2005, 24, 3158–3162; [Google Scholar]
- 27c.Viciano M., Mas-Marzá E., Sanau M., Peris E., Organometallics 2006, 25, 3063–3069; [Google Scholar]
- 27d.Mas-Marzá E., Sanau M., Peris E., Inorg. Chem. 2005, 44, 9961–9967. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a.Goossen L. J., Rodriguez N., Goossen K., Angew. Chem. Int. Ed. 2008, 47, 3100–3120; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 3144–3164; [Google Scholar]
- 28b.Goossen L. J., Goossen K., Rodriguez N., Blanchot M., Linder C., Zimmermann B., Pure Appl. Chem. 2008, 80, 1725–1733; [Google Scholar]
- 28c.Alonso F., Beletskaya I. P., Yus M., Chem. Rev. 2004, 104, 3079–3159; [DOI] [PubMed] [Google Scholar]
- 28d.Bruneau C., Neveux M., Kabouche Z., Ruppin C., Dixneuf P. H., Synlett 1991, 755–763; [Google Scholar]
- 28e.Haydl A. M., Breit B., Liang T., Krische M. J., Angew. Chem. Int. Ed. 2017, 56, 11312–11325; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 11466–11480. [Google Scholar]
- 29.
- 29a.Zeng L. W., Chen R. J., Zhang C., Xie H. J., Cui S. L., Chem. Commun. 2020, 56, 3093–3096; [DOI] [PubMed] [Google Scholar]
- 29b.Wei S. P., Pedroni J., Meissner A., Lumbroso A., Drexler H. J., Heller D., Breit B., Chem. Eur. J. 2013, 19, 12067–12076; [DOI] [PubMed] [Google Scholar]
- 29c.Nakagawa H., Okimoto Y., Sakaguchi S., Ishii Y., Tetrahedron Lett. 2003, 44, 103–106; [Google Scholar]
- 29d.Koschker P., Kahny M., Breit B., J. Am. Chem. Soc. 2015, 137, 3131–3137; [DOI] [PubMed] [Google Scholar]
- 29e.Chan D. M. T., Marder T. B., Milstein D., Taylor N. J., J. Am. Chem. Soc. 1987, 109, 6385–6388. [Google Scholar]
- 30.
- 30a.Gellrich U., Meissner A., Steffani A., Kahny M., Drexler H. J., Heller D., Plattner D. A., Breit B., J. Am. Chem. Soc. 2014, 136, 1097–1104; [DOI] [PubMed] [Google Scholar]
- 30b.Lumbroso A., Koschker P., Vautravers N. R., Breit B., J. Am. Chem. Soc. 2011, 133, 2386–2389; [DOI] [PubMed] [Google Scholar]
- 30c.Zlota A. A., Frolow F., Milstein D., Organometallics 1990, 9, 1300–1302; [Google Scholar]
- 30d.Bianchini C., Meli A., Peruzzini M., Zanobini F., Bruneau C., Dixneuf P. H., Organometallics 1990, 9, 1155–1160; [Google Scholar]
- 30e.Marder T. B., Chan D. M. T., Fultz W. C., Milstein D., J. Chem. Soc. Chem. Commun. 1988, 996–998; [Google Scholar]
- 30f.Marder T. B., Chan D. M. T., Fultz W. C., Calabrese J. C., Milstein D., J. Chem. Soc. Chem. Commun. 1987, 1885–1887; [Google Scholar]
- 30g.Lumbroso A., Vautravers N. R., Breit B., Org. Lett. 2010, 12, 5498–5501. [DOI] [PubMed] [Google Scholar]
- 31.Connelly N. G., Geiger W. E., Chem. Rev. 1996, 96, 877–910. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a.Burés J., Angew. Chem. Int. Ed. 2016, 55, 2028–2031; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2068–2071; [Google Scholar]
- 32b.Burés J., Angew. Chem. Int. Ed. 2016, 55, 16084–16087; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 16318–16321. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information