

Abstract
Darwinian evolution preferentially follows mutational pathways whose individual steps increase fitness. Alternative pathways with mutational steps that do not increase fitness are less accessible. Here, we show that mistranslation, the erroneous incorporation of amino acids into nascent proteins, can increase the accessibility of such alternative pathways and, ultimately, of high fitness genotypes. We subject populations of the beta‐lactamase TEM‐1 to directed evolution in Escherichia coli under both low‐ and high‐mistranslation rates, selecting for high activity on the antibiotic cefotaxime. Under low mistranslation rates, different evolving TEM‐1 populations ascend the same high cefotaxime‐resistance peak, which requires three canonical DNA mutations. In contrast, under high mistranslation rates they ascend three different high cefotaxime‐resistance genotypes, which leads to higher genotypic diversity among populations. We experimentally reconstruct the adaptive DNA mutations and the potential evolutionary paths to these high cefotaxime‐resistance genotypes. This reconstruction shows that some of the DNA mutations do not change fitness under low mistranslation, but cause a significant increase in fitness under high‐mistranslation, which helps increase the accessibility of different high cefotaxime‐resistance genotypes. In addition, these mutations form a network of pairwise epistatic interactions that leads to mutually exclusive evolutionary trajectories towards different high cefotaxime‐resistance genotypes. Our observations demonstrate that protein mistranslation and the phenotypic mutations it causes can alter the evolutionary exploration of fitness landscapes and reduce the predictability of evolution.
Keywords: epistasis, genetic diversification, mistranslation, molecular evolution, phenotypic mutations
High mistranslation can increase the fitness benefits of adaptive DNA mutations, and thus help open evolutionary paths to alternative high‐fitness genotypes (left panel) that would be inaccessible under low mistranslation (right panel).
![]()
1. INTRODUCTION
A fundamental question in evolutionary biology is how reproducible and predictable evolution is, and which factors affect this predictability (Dickinson et al., 2013; Lässig et al., 2017). Among these factors are epistatic (nonadditive) interactions between different mutations, where the fitness effect of one mutation depends on the presence of another mutation (Poelwijk et al., 2007). On the one hand, epistasis can increase the predictability of evolutionary pathways towards high fitness peaks by rendering only some pathways accessible (Weinreich et al., 2006). On the other hand, it may also help decrease predictability. This is evident from parallel evolution experiments, and from experimental reconstructions of evolutionary pathways, in which similar phenotypic evolution was accompanied by genetic diversification among populations (Bank et al., 2016; Dickinson et al., 2013; Kvitek et al., 2011; Salverda et al., 2011; Starr et al., 2017). This diversification resulted from a combination of the stochastic nature of DNA mutations and a kind of epistasis called sign epistasis. Sign epistasis occurs when the qualitative fitness effect of a mutation (positive, neutral or negative) depends on mutations at another site (De Visser & Krug, 2014). A special case is reciprocal sign epistasis, in which two mutations mutually depend on each other in this qualitative fitness effect (Kvitek et al., 2011). This kind of epistasis can create adaptive valleys in a fitness landscape, and thus help create a rugged landscape with multiple peaks (Kvitek et al., 2011; Poelwijk et al., 2007; Weinreich et al., 2005). Different populations evolving in such a rugged landscape may follow different evolutionary trajectories towards different fitness peaks, depending on the mutational history of a population. This history is determined by the order in which individual mutations arise, an order that is fundamentally stochastic. Because of such historical contingencies, once a mutation occurs, it may bias subsequent evolution towards few accessible mutational paths. In consequence, the mutant can render other evolutionary trajectories inaccessible, whose genotypes will not be seen in nature (Gould, 1989; Starr et al., 2017).
Previous work has demonstrated that the stochasticity of DNA mutations can create contingency, and thus reduce the reproducibility and predictability of evolution (Blount et al., 2018; Salverda et al., 2011). However, some nongenetic perturbations are much more frequent than DNA mutations, and previous experiments remain silent on their effect on contingency and reproducibility. An especially important such perturbation is mistranslation, the erroneous incorporation of amino acids into proteins by a ribosome. Mistranslation is an abundant source of amino acid variation because translational errors are up to ~105 times more frequent than DNA mutations (Kramer & Farabaugh, 2007; Ogle & Ramakrishnan, 2005; Parker, 1989; Willensdorfer et al., 2007). Such highly frequent phenotypic mutations might interact epistatically with DNA mutations and could thus change their fitness effects. As a result, the phenotypic mutations caused by mistranslation may affect the kind of DNA mutations that can spread through a population, and the order in which they do. If so, mistranslation may also alter how evolving populations explore fitness landscapes, and create evolutionary contingencies, especially for populations that evolve on a rugged fitness landscape. Previous pertinent work showed that elevated mistranslation can help proteins accumulate stabilizing mutations (Bratulic et al., 2015) and eliminate deleterious mutations (Bratulic et al., 2017). It provides a first hint that mistranslation may affect the exploration of fitness landscapes. In the present work, we study this effect of mistranslation in detail. Specifically, we show that mistranslation creates contingency in the exploration of a fitness landscape, and thus reduces the reproducibility and predictability of evolution.
For our work, we used directed evolution of the TEM‐1 beta‐lactamase enzyme in the bacterium E. coli. TEM‐1 beta‐lactamase, which can hydrolyse beta‐lactam antibiotics, is highly active against ampicillin but has negligible activity on extended‐spectrum cephalosporins such as cefotaxime (Palzkill & Botstein, 1992). However, TEM‐1 can evolve resistance against cefotaxime in a process that involves epistatically interacting amino acid changes (Salverda et al., 2011; Schenk et al., 2013, 2015; Weinreich et al., 2006). Some of these mutations show sign epistasis, help create a fitness landscape with multiple peaks (Salverda et al., 2011; Schenk et al., 2013), and render TEM‐1 a well‐suited model for testing our hypothesis.
2. MATERIALS AND METHODS
2.1. Strains and plasmids
We started from E. coli strain MG1655 to construct a strain with an elevated mistranslation rate. In short, we constructed this strain by transferring the mutated ribosomal protein S4 gene into the MG1655 genetic background to replace the wild‐type ribosomal protein S4 gene (Ballesteros et al., 2001), as reported in a previous study (Bratulic et al., 2015). The mutated ribosomal protein S4 gene substantially increases the rate of missense, read‐through, and frameshift errors during protein synthesis, including an 8.4‐fold increase in misreading the codon AAU (Kramer & Farabaugh, 2007). To ensure that our wild‐type (L for low mistranslation) and error‐prone strains (H for high mistranslation) have the same genetic background except for the ribosomal protein S4 gene, we used the same procedure for constructing the wild‐type strain, transferring the wild‐type ribosomal protein S4 gene into the MG1655 genetic background.
We used E. coli strain DH5α for cloning and preparing the TEM‐1 mutation library. We used the high‐copy number plasmid pHS13T, which harbours a chloramphenicol resistance marker, as the vector for TEM‐1 evolution(Bratulic et al., 2015). To facilitate isolation of the vector backbone for re‐cloning by gel extraction, we used the plasmid pHS13K, which we constructed by replacing the TEM‐1 gene in pHS13T with a KanR cassette from the pKD4 plasmid (Datsenko et al., 2000).
2.2. Preparation of electro‐competent cells for transformation
We used glycerol/mannitol step centrifugation to prepare electro‐competent cells, as described previously (Bratulic et al., 2015; Warren, 2011). Specifically, we grew E. coli strains in 5 ml SOB medium overnight (37°C, 250 rpm) and transferred 3 ml culture into 300 ml SOB medium for another 2–4 hr of incubation in a shaking incubator (INFORS HT, 37°C, 250 rpm) until the OD600 value reached 0.4–0.6 (optical path length: 1 cm). We then placed the culture on ice for 15 min and collected cells by centrifuging at 1,500 g for 15 min (4°C). We suspended the pellet in 60 ml ice‐cold ddH2O and distributed the resulting cell suspension into three 50 ml tubes. Then we used a 10 ml pipette to slowly add 10 ml ice‐cold glycerol/mannitol solution (consisting of 20% glycerol (w/v) and 1.5% mannitol (w/v)) to the bottom of each tube. We centrifuged the tubes at 1,500 g for 15 min (4°C, acceleration/deceleration set to zero). After aspiration of the supernatant, we suspended the pellets in 1.5 ml ice‐cold glycerol/mannitol solution. We divided the resulting suspensions into 1.5 ml precooling tubes, put them in a dry ice‐ethanol bath for about 1 min, and then stored them at −80°C.
2.3. Mutagenic PCR and preparation of the mutation library
We used mutagenic PCR (Zaccolo et al., 1996) to introduce mutations into the coding region of TEM‐1. This method is A → G and T → C biased, as has been characterized previously (Bershtein et al., 2006; Bratulic et al., 2015; Zaccolo et al., 1996). A 100 µl PCR reaction consisted of 10 ng template plasmid, 400 µM dNTPs (R0192, Thermo Scientific), 2.5 U Taq DNA polymerase (M0267L, NEB), 10 µl 10× ThermoPol buffer (M0267L, NEB), 3 µM 8‐oxo‐GTP/dPTP (Trilink Biotechnologies) and 400 nM primers (TEM‐1‐F6/TEM‐R6; Table S9). We executed 25 cycles of PCR using the following programme: 95°C/3 min, 25 cycles of 94°C/30 s, 47°C/30 s and 68°C/1 min, 68°C/5 min. We treated the PCR products with DpnI (R0176S, NEB) at 37°C for 2 hr to remove the template plasmid. We added 0.6 U of proteinase K (EO0491, Thermo Scientific) to each PCR reaction and incubated the mixture at 50°C for 1h and then at 80°C for 15 min to inactivate the Taq DNA polymerase. We digested the PCR products with 20 U SacI‐HF/HindIII‐HF (R3156S/R3104S, NEB) at 37°C overnight and then inactivated the restriction enzymes at 80°C for 20 min. We purified the digested products using the QIAquick PCR purification kit (Qiagen). To get the vector backbone, we digested plasmid pHS13K with 20 U SacI‐HF/HindIII‐HF overnight and purified the digested products using the QIAquick gel extraction kit (Qiagen). We dephosphorylated the purified vector backbone with 5 U Antarctic Phosphatase (M0289S, NEB), and re‐purified the dephosphorylated vector backbone using the QIAquick PCR purification kit.
We mixed 50 ng plasmid insert (mutagenized TEM‐1 library), 70 ng digested and dephosphorylated vector backbone, 10 U T4 DNA ligase and 2 µl 10× Ligation buffer (M0202L, NEB) in a 20 µl ligation reaction. We incubated the ligation reaction at 20–22°C for ~16 hr and then inactivated the T4 DNA ligase at 65°C for 10 min. To precipitate the ligation product, we added 1 µl glycogen (R0551, Thermo Scientific), 50 µl 7.5 M ammonium acetate (A2706‐100Ml, Sigma), 375 µl ice‐cold absolute ethanol, and 80 µl ddH2O. After 20 min incubation at −20°C, we centrifuged the mixture at 18,000 g for 20 min. We washed the pellet in 800 µl of cold ethanol (70%) twice, dried the pellet using a concentrator 5,301 (Eppendorf), and then dissolved it in 10 µl ddH2O for transformation.
2.4. Preselection of TEM‐1 mutant libraries
To increase the transformation efficiency in wild‐type and error‐prone hosts, we first transformed the purified ligation product (TEM‐1 mutant pool) into electro‐competent DH5α cells to ensure methylation of plasmids. In short, we added 4 μl ligation product to 100 μl electro‐competent DH5α cells, and used a Micropulser electroporator (Bio‐Rad) at setting EC3 (15 kV/cm), and a 0.2 cm cuvette (EP202, Cell Projects) for electroporation. After that, we immediately added 1 ml prewarmed SOC medium and transferred the suspension into a 10 ml tube. After 1.5 hr incubation at 37°C with shaking at 200 rpm in a shaking incubator (INFORS HT), we added 10 ml LB medium with 34 μg/ml chloramphenicol (C1919, Sigma, to select for the presence of plasmids) to the transformed cells. We used 100 μl cell aliquots for estimating the library size by plating the serially diluted aliquot (in saline) on LB agar with 20 μg/ml chloramphenicol. (Our transformation protocol resulted in a library size of ~106 colony‐forming units for each population.). We incubated the rest of the culture at 37°C with shaking at 320 rpm overnight (in a Microtiter plate shaking incubator, FAUST Laborbedarf AG). Subsequently we isolated the plasmid from the overnight culture using the QIAprep spin miniprep kit (Qiagen).
2.5. Evolution under selection for ampicillin resistance
We transformed the plasmids from the preselection library into wild‐type and error‐prone electro‐competent cells and subjected them to ampicillin (A0166, Sigma) resistance selection, as also described previously (Bratulic et al., 2015). In short, we transformed 5 ng plasmid from the preselection library into 80 μl wild‐type or error‐prone electro‐competent cells. After 1.5 hr recovery in 1.0 ml SOC medium at 37°C and 200 rpm, we collected the recovered cells by centrifuging at 2,000 g for 15 min and then suspended them in 3 ml LB medium with 34 μg/ml of chloramphenicol and 250 μg/ml of ampicillin. We used 50 μl of recovered cells for estimating the library size by plating the serially diluted aliquot (in saline) on LB agar with 20 μg/ml chloramphenicol, resulting in ~107 colony‐forming units. After incubation for approximately 6 generations (2:07 hr for wild type host, 4:23 hr for error‐prone hosts), we sampled the culture for isolating plasmids using the QIAprep spin miniprep kit. We used the isolated plasmids as templates for the next mutation‐selection cycle. After eight mutation‐selection cycles, we used the isolated plasmids as template for evolution under selection for cefotaxime resistance.
2.6. Evolution under selection for cefotaxime resistance
We added 5 ng isolated plasmid from the preselection library to 100 μl error‐prone or wild‐type electro‐competent cells for electroporation under the same conditions as described above. After 1.5 hr of recovery in 1.0 ml SOC medium, we added 10 ml LB medium supplemented with 34 μg/ml chloramphenicol, and sampled 100 μl of the cell aliquot for estimating library size, using the same method as described above. We used 50 μl of the recovered cells for estimating the library size by plating the serially diluted aliquot (in saline) on LB agar with 20 μg/ml chloramphenicol, resulting in ~107 colony‐forming units. Subsequently, we incubated the recovered cells at 37°C with overnight shaking at 240 rpm. We transferred 1 ml overnight culture into 10 ml LB medium with 34 μg/ml chloramphenicol and incubated the cell culture until it reached an OD600 of 0.3–0.6. (We measured the OD600 in 200 μl of culture, using a TECAN Infinite F200 PRO micro‐plate reader.) We then diluted the culture using saline to an OD600 of 0.04. Then, we transferred 100 μl diluted culture (which contained ~107 cells, according to density estimation by plating of serially diluted cultures on LB agar) into 3 ml LB medium supplemented with 34 μg/ml chloramphenicol and a two‐fold dilution series of cefotaxime (C7039, Sigma). We recorded the minimal inhibitory concentrations (MIC) after 48 hr incubation for the populations evolved in wild‐type host, and after 60 hr of incubation for the populations evolved in error‐prone host. We used a longer incubation time for the populations evolved in the error‐prone host, since they grew more slowly, and since the MIC no longer changed in the wild‐type population after 48 hr incubation.
Following the above procedure, we subjected four replicate populations of TEM‐1, which had evolved for eight rounds of evolution on ampicillin in error‐prone (H) hosts, to evolution on cefotaxime in the same host. (We refer to this experimental treatment as the H → H treatment). We also subjected four replicate populations of TEM‐1 that had evolved for eight rounds in wild‐type (L) on ampicillin to evolution on cefotaxime (L → L treatment). In addition, we directly subjected four replicate populations of wild‐type (Anc) TEM‐1 which had not been subject to evolution on ampicillin, to evolution on cefotaxime in either the error‐prone host (Anc→H treatment) or the wild‐type host (Anc→L, see also Figure 1). After each round, we sampled cultures from the highest cefotaxime concentration on which all 8 error‐prone or wild‐type populations survived, and isolated plasmids from each sample. We used the isolated plasmids as starting points for the next mutation‐selection cycle. After the 3rd round, we sampled those populations that survived the highest concentration just below their MIC, and isolated plasmids from these samples for SMRT sequencing. For culture sampling, we appropriately extended the incubation time (6 ~ 12 hr) for those populations that grew more slowly, to ensure that they reached cell concentrations comparable to other populations. After each round, we also sampled 600 μl of each culture and mixed it with 400 μl 50% glycerol. We stored this glycerol stock at −80°C for MIC assays.
FIGURE 1.
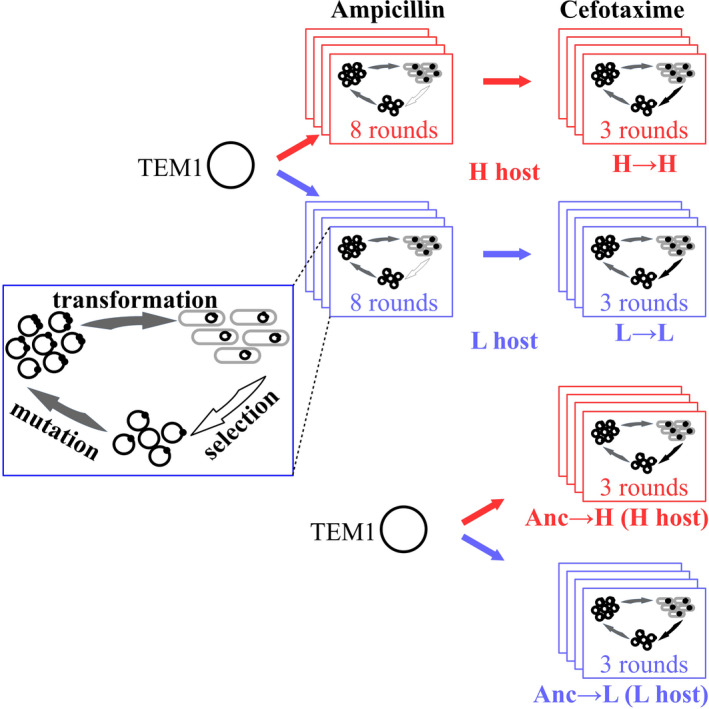
Experimental evolution of TEM‐1 under low and high mistranslation. We subjected four replicate populations of TEM‐1 to eight rounds (cycles, ‘generations’) of directed evolution in an Escherichia coli host with a high‐mistranslation rate (H), and under selection for ampicillin (250 μg/ml) resistance. Following that, we subjected these populations to three rounds of directed evolution for increased cefotaxime resistance in the same host (Figure S1). We refer to the combined experiment as the H → H experiment. We also evolved four replicate populations of TEM‐1 in low‐mistranslation (L, wild‐type) hosts by following the same procedure (L → L). Additionally, we directly subjected four replicate populations of wild‐type TEM‐1 to three rounds of directed evolution on cefotaxime in high‐mistranslation host (Anc→H) and low‐mistranslation host (Anc→L), respectively. During selection on ampicillin, we isolated TEM‐1 encoding plasmid libraries from each population after each round, and used the isolated plasmids as templates for the next mutation‐selection cycle. During directed evolution on cefotaxime, we determined the minimum inhibitory concentration of cefotaxime in each generation, and used TEM‐1 populations from the culture with the highest cefotaxime concentration on which all eight high‐ or low‐ mistranslating populations survived for the next round. In this way, we increased the cefotaxime concentration for selection in each round. In the third round, we isolated the plasmids from those populations that survived at the highest cefotaxime concentration. We subjected TEM‐1 populations isolated from the eighth round of evolution on ampicillin, and from each round of evolution on cefotaxime to SMRT sequencing (Figure S1)
2.7. Determination of mutation rates
We constructed two control libraries for each host strain per evolution cycle to estimate mutation rates. We prepared each control library with the same procedure as the libraries under selection on ampicillin and cefotaxime. After transformation, we plated the diluted culture (using saline) on LB agar which only contained 34 μg/ml chloramphenicol for plasmid maintenance, and neither ampicillin nor cefotaxime. We chose six clones randomly from the control library after each round of evolution, and subjected them to colony PCR and Sanger sequencing to estimate the mutation rate. Sanger sequencing indicated a mutation rate of 1.5 ± 0.6 mutations per TEM‐1 molecule and round of mutagenesis (mean ± standard deviation of mutation rates observed in all three rounds).
2.8. SMRT sequencing
We performed two‐step PCRs to barcode TEM‐1 variants of each population for single molecule real‐time (SMRT) sequencing, as described previously(Bratulic et al., 2015). To this end, we used high‐fidelity Phusion DNA polymerase for the PCR amplification to reduce the mutation rate during this process. In short, we first amplified TEM‐1 variants from each population by a 12‐cycle PCR using primers TEM‐1FS‐F/TEM‐1FS‐R (Table S9). A 30 µl PCR reaction consisted of 1 ng template plasmid, 400 µM dNTPs, 1.5 U Phusion Hot Start II High‐Fidelity DNA Polymerase (F‐549L, Thermo Scientific), 6 µl 5× Phusion HF Buffer, 0.3 µl 100% DMSO, and 400 nM primers TEM‐1FS‐F/TEM‐1FS‐R. We used the following thermocycler programme for the PCR reaction: 98°C/30 s, 12 cycles of 98°C/15 s, 59°C/15 s and 72°C/30 s, 72°C/1 min. Then we used the resulting PCR product as a template in a barcoding PCR, using the different barcode‐tagged primers BCXX and ELP (Table S9). Each barcode‐tagged primer contained a unique 6 bp sequence(Chubiz et al., 2012). We prepared a 50 µl PCR reaction (consisting of 2 µl template, 400 µM dNTPs, 2.5 U Phusion Hot Start II High‐Fidelity DNA Polymerase, 10 µl 5× Phusion HF Buffer and 400 nM primers BCXX and ELP) and performed the PCR amplification with the following programme: 98°C/30 s, 28 cycles of 98°C/15 s, 63°C/15 s and 72°C/20 s, 72°C/1 min. We purified the barcode‐tagged PCR product and checked the quality and concentration of amplicons with a UV‐Vis spectrophotometer (NanoDrop, Thermo Fisher Scientific), as well as through agarose gel electrophoresis. We amplified the wild‐type TEM‐1 gene following the same procedure to detect potential errors that might occur during library preparation. At the end of these procedures, we combined 20 ng DNA of each population from the same round of evolution into one tube for sequencing. We used the DNA Template Prep Kit 2.0 (250 bp to 3 Kb, Pacific Biosciences) for constructing the SMRTbell library. We ligated blunt end adapters to amplicons and used them as SMRTbell templates. We created ready‐to‐sequence SMRTbell‐polymerase complexes using the DNA/Polymerase P4 binding kit (Pacific Biosciences). Subsequently, we sequenced each amplicon pool on one SMRT cell (v3.0) using the Pacific Biosciences RS2 instrument (Pacific Biosciences). We used P4/C2 chemistry and the magnetic bead loading method, and recorded two movies of 3‐hr duration for each cell.
2.9. Primary data analysis
We analysed SMRT sequencing data using a previously described pipeline (Bratulic et al., 2015). We assembled consensus reads from sub‐reads using the smrt Analysis v2.3 package (www.pacb.com/products‐and‐services/analytical‐software/devnet/). We filtered reads of TEM‐1 inserts by setting the full‐pass subread number to ≥4, the predicted consensus accuracy to ≥0.9 and the insert length to 850–1,200 bp. We mapped reads to the wild‐type TEM‐1 sequence (GenBank No.: KT391064) using BLASR (Chaisson & Tesler, 2012) by setting the mapped length to ≥850 bp and the mapping accuracy to ≥0.9. This generated 36,542–49,758 reads, depending on the sequencing reaction, with an average mapped read length of 963–967 bp and a mean mapped subread concordance (Kim et al., 2014) of 0.973–0.974. From this data, we identified mapped reads that span the entire TEM‐1 coding region and had an average Phred quality above 20. We used only these reads for further analysis, and de‐multiplexed the filtered, mapped reads according to each read's barcoding sequences using custom python scripts. We excluded those sequences from further analysis that lacked a stop codon or had an internal stop codon (~1% per library). At the end of these procedures, we were left with 20,421–29,863 sequences, depending on the sequencing reaction, for further analyses.
2.10. Identification of SNPs
Because more than 90% of sequencing errors that occur during SMRT sequencing are single‐nucleotide indels (Kim et al., 2014), and because more than 98% of indels render TEM‐1 nonfunctional (Firnberg et al., 2014), we ignored indels and only analysed point mutations. We considered a mismatch of a TEM‐1 variant sequence to the TEM‐1 reference sequence as a true SNP only if its Phred quality score was above 20. We wrote python scripts (python 2.7.12) to identify SNPs and calculated their frequencies in each population.
2.11. Engineering TEM‐1 variants
We engineered TEM‐1 variants using whole plasmid PCR (Liu & Naismith, 2008) by designing primers that carried the corresponding mutations. For introducing an amino acid mutation, we designed a pair of primers that carried mutated nucleotides and that complemented each other at the 5′‐terminus (Table S10). A 20 µl PCR reaction consisted of 10 ng template plasmid, 400 µM dNTPs, 1.0 U Phusion Hot Start II High‐Fidelity DNA Polymerase, 4 µl 5× Phusion HF Buffer and 400 nM primers (Table S10). We performed 25 cycles of PCR reaction using the following programme: 98°C/30 s, 28 cycles of 98°C/15 s, 63–72°C (Table S10) /20 s and 72°C/2 min, 72°C/5 min. We treated the PCR products with DpnI at 37°C for 2 hr to remove the template plasmid, and inactivated the enzyme at 80°C for 20 min. Then we added 2 µl precooled PCR products to electro‐competent DH5α cells for electroporation, using the same procedure described in the section Preselection of TEM‐1 mutant libraries. After 1.5 hr of recovery, we diluted the recovery culture 100 times using saline and plated 50 µl of the resulting dilution on LB agar with 34 μg/ml chloramphenicol. After incubation overnight, we chose three colonies from each library to check for the desired mutation by colony PCR and Sanger sequencing. We extracted the plasmids of correctly constructed mutants and used them as templates for the next round of whole plasmid PCR to introduce the second desired mutation. By repeating the above procedure, we engineered variants that harboured double, triple or quadruple mutations. For those mutations that were close to each other (N175D/D179G/M182T), we introduced two or three mutations into TEM‐1 at one time by designing pairs of primers harbouring two or three mutations, respectively (Table S10).
To reduce the incidence of random mutations in the plasmid backbone, we used High‐Fidelity DNA Polymerase (with an error rate >50‐fold lower than that of Taq DNA Polymerase (Frey & Suppmann, 1995)) for whole plasmid PCR. In addition, for those variants constructed by more than two rounds of whole plasmid PCR, we re‐inserted the coding region of TEM‐1 variants into the fresh plasmid backbones. In short, we isolated plasmids carrying the desired mutations from DH5α cells, digested them with 20 U SacI‐HF/HindIII‐HF, and re‐inserted the (mutated) coding region of TEM‐1 into the plasmid backbone as described in Mutagenic PCR and preparation of mutation libraries. We transformed the resulting plasmid into electro‐competent error prone and wild type hosts, respectively, as described in Preselection of TEM‐1 mutant libraries. After 1.5 hr recovery, we plated the cell culture (100 μl) on LB agar with 34 μg/ml chloramphenicol. After overnight growth at 37°C, we washed colonies from plates using LB medium and mixed the cultures with glycerol to a final glycerol concentration of 20% (w/v). We stored this glycerol stock at −80°C for further experiments.
2.12. Minimal inhibitory concentration assays
We placed the glycerol stock of each engineered TEM‐1 variant or evolved population (after each round of experimental evolution on cefotaxime) on ice until it had thawed, and transferred 10 μl glycerol stocks into 2 ml LB medium containing 34 μg/ml of chloramphenicol. After growth overnight at 37°C and 240 rpm, we transferred 200 μl of the culture into 2 ml LB medium containing 34 μg/ml of chloramphenicol. We then grew this culture until its OD600 reached a value of 0.3–0.6 (measured in 200 μl of culture using a micro‐plate reader). We diluted the culture using saline to an OD600 of 0.004. Then we transferred 5 μl of the diluted culture (which contained ~5 × 104 cells, as estimated by plating of a serial dilution on LB agar) into 200 μl of LB medium supplemented with 34 μg/ml chloramphenicol and a two‐fold dilution series of cefotaxime. After 60 hr of incubation at 37°C and 400 rpm in a micro‐plate incubator, we determined growth by visual inspection, and defined the MIC as the lowest concentration of antibiotic that completely prevented visible growth (Although different variants displayed different growth curves at the highest concentrations just below their MICs, their MICs no longer changed in both the wild‐type and error‐prone hosts after 60 hr of incubation). We replicated each assay three times (in one 96‐well microplate) or six times (in two 96‐well microplates) to determine the MICs of individual TEM‐1 variants. We replicated each assay eight times (in one 96‐well microplate) to determine the MICs of evolved populations. We calibrated data acquired on different days by comparing the MIC values of genetic variants to their ancestor TEM‐1 measured in the same host and on the same day.
2.13. Determination of specific growth rates
We grew L and H strains in 2 ml LB medium overnight (37°C, 240 rpm), and transferred 5 μl culture into 200 μl LB medium (96‐well micro‐plate) for another 20 hr of incubation in a TECAN Plate reader (37°C, 240 rpm). During incubation, we monitored OD600 values in 900s time intervals. We determined the specific growth rates of each host (cell mass increase per unit of time) according to the expression: μ = ln (N2/N1)/(t2 − t1). Here N1 and N2 indicate cell densities (as indicated by OD600 values) at time t1 (2.10 hr for L host and 1.31 hr for H host) and t2 (3.15 hr for L host and 2.36 hr for H host) during log phase, respectively. We grew 44 biological replicates to measure specific growth rates for each host.
To measure the effects of the mutation M182T on the growth of L and H hosts, we placed the glycerol stocks of the ancestor TEM‐1 and the variant M182T in L and H hosts on ice until they had thawed, and transferred 10 μl glycerol stocks into 2 ml LB medium containing 34 μg/ml of chloramphenicol. After growth overnight at 37°C and 220 rpm, we transferred 200 μl of the culture into 2 ml LB medium containing 34 μg/ml of chloramphenicol. We then grew this culture until its OD600 reached a value of 0.6–1.0 (measured in 200 μl of culture using a micro‐plate reader). We diluted the culture using saline to an OD600 of 0.004. Then we transferred 5 μl of the diluted culture into 200 μl of LB medium supplemented with 34 μg/ml chloramphenicol for 24 hr of incubation in a TECAN Plate reader (37°C, 240 rpm). We monitored the OD600 values in a 900 s interval. After 24 hr of incubation, we mixed 50 μl of the culture with 150 μl of saline, and measured the absorbance at 600nm. We grew 12 biological replicates to measure the growth of the ancestor TEM‐1 or the variant M182T in each host.
2.14. Statistical analysis
Unless specified otherwise, we used nonparametric two‐sided Wilcoxon–Mann–Whitney tests for statistical data analysis. MIC‐based measurements are semiquantitative, classifying antibiotic resistance into multiple discrete categories on a base 2 logarithmic scale. We thus transformed the MIC and MIC fold change values logarithmically (base 2) for statistical analyses. To test the statistical significance of the MIC fold change caused by every single mutation, we performed a one‐way ANOVA, using the two‐sided Dunnett test for multiple comparisons to one control (the wild‐type TEM‐1). For the analysis of epistatic interactions between paired mutations, we used a one‐way ANOVA with post hoc Bonferroni correction to conduct pairwise comparisons, that is, we compared MIC fold changes caused by double mutants and single mutants. We used R version 3.4.1 to perform all statistical analyses.
3. RESULTS
3.1. Phenotypic evolution under elevated mistranslation
As in previous work, we here use an E. coli host with a high (H) mistranslation rate that is caused by a mutation in the ribosomal protein S4 gene, as well as a matched, isogenic wild‐type host with a low (L) mistranslation rate (see Section 2). To find out whether mistranslation influences the evolutionary exploration of TEM‐1 populations in a multi‐peaked fitness landscape, we subjected four replicate TEM‐1 populations in an H host to eight rounds (‘generations’) of directed evolution on their native substrate ampicillin, which allows standing variation to accumulate that may facilitate adaptive evolution (Figure 1). Then we subjected these populations to three more rounds of directed evolution on the new substrate cefotaxime (Figure 1; Figure S1). We denote this experiment the H → H experiment. In parallel, we evolved four replicate TEM‐1 populations in the same way in the L host (the L → L experiment, Figure 1; Figure S1). Furthermore, we also subjected four replicate populations of the wild‐type (Anc) TEM‐1 molecule (without accumulating standing variation) to three rounds of directed evolution on their new substrate cefotaxime (Anc→H and Anc→L, Figure 1; Figure S1). In each round and replicate population of these experiments, we evolved a population of ~106 TEM‐1 variants subject to antibiotic selection and mutagenic PCR (1.5 ± 0.6 mutations per TEM‐1 gene per generation, see Materials and Methods). We quantified fitness through the minimal inhibitory concentrations (MIC), that is, the smallest concentration of cefotaxime that completely inhibits cell growth.
The H host grows significantly more slowly than the L host, even in the absence of antibiotics and TEM‐1 genes (Figure S2a; two‐sided Wilcoxon–Mann–Whitney test, W = 1,483.5, p = 1.723 × 10–05). In addition, the wild‐type TEM‐1 protein has a lower absolute MIC of cefotaxime when expressed in the H host than in the L host (Figure S2b; two‐sided Wilcoxon–Mann–Whitney test, W = 36, p = .001821). A likely explanation is that the greater numbers of mistranslation events that TEM‐1 is exposed to in the H host impair its function, at least on average. Furthermore, the generally lower fitness of the H host, regardless of TEM‐1’s presence, may contribute to the difference in MIC.
At the evolutionary endpoint, most of the populations that had evolved at low mistranslation rates (Figure 2a,b, Anc →L and L → L) reached higher MICs than those evolved under high mistranslation rates (Figure 2c,d, Anc →H and H → H). In addition, all the populations evolved at low mistranslation rates (Figure 2a,b, Anc →L and L → L) reached a similar MIC, but those evolved under high mistranslation rates (Figure 2c,d, Anc →L and L → L) reached quite different MICs. This increased variation in cefotaxime‐resistance provides a first hint—at the phenotypic level—of what our subsequent analyses showed on the genotypic level, namely that mistranslation can lead to evolutionary diversification.
FIGURE 2.
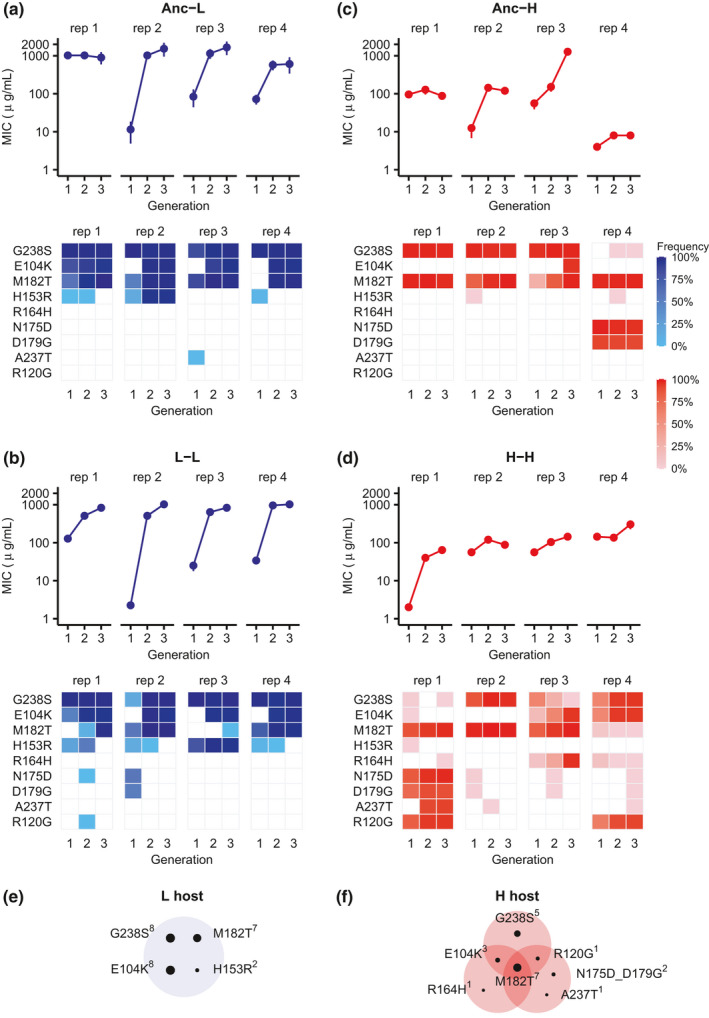
Evolution of resistance to cefotaxime under low and high mistranslation. (a–d) MICs of cefotaxime (upper panels, vertical axes) and frequency (lower panels, coloured squares) of SNPs (lower panels, vertical axes) known to be important for cefotaxime resistance during each round of evolution (horizontal axis) on cefotaxime for populations starting from wild‐type TEM‐1 in L (Anc→L, panel a) or H hosts (Anc→H, panel c), and for populations that first evolved on ampicillin and then on cefotaxime in L (L → L, panel b) or H (H → H, panel d) hosts. We considered a SNP in a replicate population only if it occurred in at least two TEM‐1 molecules in that replicate population. We performed an MIC assay (see Section 2) for each experimental population after each round of experimental evolution on cefotaxime. Error bars represent one standard deviation for eight biological replicates. (e and f) Different mutation combinations became fixed under low (L, panel e) and high (H, panel f) mistranslation. SNPs that were fixed together in the same replicate population are shown in the same shaded oval. The size of each small black circle is proportional to the number of replicate populations (superscript to SNP name) in which the corresponding SNP became fixed. ‘rep 1‐4’ indicate replicate populations 1‐4
3.2. Elevated mistranslation results in genetic diversification between populations
To characterize the genetic basis of phenotypic evolution in our TEM‐1 populations, we used single‐molecule real‐time (SMRT) sequencing to genotype more than 400 evolved variants per TEM‐1 replicate population after each generation of evolution (Table S1).
Previous studies had identified three DNA mutations that occur especially frequently during evolution of cefotaxime resistance and in clinical isolates (Bratulic et al., 2017; Dellus‐Gur et al., 2013; Orencia et al., 2001; Salverda et al., 2010, 2011, 2017). These are G238S, E104K, and M182T. The first two (G238S and E104K) improve the hydrolysis of cefotaxime, and show synergistic (positive) epistasis in doing so. That is, their combined beneficial effect is greater than the sum of the individual effects. However, they also destabilize TEM‐1. In contrast, M182T is a ‘global suppressor’ that stabilizes TEM‐1 and compensates for this destabilizing effect (Brown et al., 2010). At low mistranslation rates, these three mutations sweep through our populations (Figure 2e). Specifically, at the end of three rounds of evolution on cefotaxime, all three mutations (G238S, M182T and E104K) became fixed in all four Anc → L populations (Figure 2a), and three of four replicate L → L populations (Figure 2b). However, this was no longer the case at a high mistranslation rate, where the triple mutant was fixed in only one replicate Anc → H population (Figure 2c), and in none of four replicate H → H populations (Figure 2d). Random amino acid changes in proteins are on average weakly deleterious, and mistranslation introduces such changes into evolving proteins, in addition to those caused by genetic mutations (Bratulic et al., 2015; Drummond & Wilke, 2008). Such changes can reduce the benefits of strongly beneficial genetic mutations and slow down their ascent to fixation (Bratulic et al., 2017). However, the lack of fixation of the three canonical mutations may also have a second explanation (not mutually exclusive with the first), which is the focus of our present analysis. That is, mistranslation may enable the exploration of alternative evolutionary trajectories, by causing different DNA mutations (or combinations thereof) to have beneficial effects.
To find out whether this may be the case, we first examined our Anc → H and H → H populations for other fixed or high frequency DNA mutations that have been implicated in cefotaxime resistance (Figure 2c,d; Table S2). We found four distinct combinations of such high frequency mutants (Figure 2c,d,f). The first is the mutation triplet N175D, D179G and M182T, which became fixed in one Anc → H population (Figure 2c). Both N175D and D179G, which may improve the activity of TEM‐1 on cefotaxime, are not found in clinical isolates so far and rarely occur in laboratory evolution experiment (Salverda et al., 2010, 2011; Vakulenko et al., 1999). Another triplet (R164H, E104K and M182T) became fixed in one H → H population (Figure 2d). Its distinguishing mutation R164H has also been implicated in enhancing the activity of TEM‐1 (Salverda et al., 2010; Vakulenko et al., 1999). Thirdly, a combination of five mutations (N175D, D179G, A237T, M182T and R120G) dominated one H → H population (Figure 2d). In addition, the triplet (G238S, E104K and R120G) dominated another H → H population (Figure 2d). Previous work suggests that A237T is an activity‐improving mutation, and R120G is a stabilizing mutation (Salverda et al., 2010, 2011). Thus, the low incidence of activity‐improving mutation G238S or E104K may have been compensated by an increased incidence of R164H, N175D, D179G or A237T in some Anc → H and H → H populations. Similarly, the low incidence of stabilizing mutation M182T in one H → H population may have been compensated by R120G (Figure 2d).
We found that additional DNA mutations that reached a medium or high frequency in high‐mistranslating populations (Figure S3) affect the TEM‐1 signalling peptide, which directs TEM‐1 export into the periplasmic space (Figure S4). The efficient transportation of mature beta‐lactamase into the periplasmic space may enhance its ability to degrade antibiotics. In addition, the periplasm provides a better environment for protein folding where proteins can also escape degradation by proteases (Baneyx & Mujacic, 2004; Choi & Lee, 2004; Walker & Gilbert, 1994). Improving the secretion efficiency of beta‐lactamase into the periplasm can thus weaken any deleterious effects of mistranslation.
3.3. Elevated mistranslation affects the cefotaxime‐resistance benefit of multiple stability and activity enhancing mutations
The observation that different combinations of TEM‐1 mutations rose to high frequency under high mistranslation raises the possibility that mistranslation alters the cefotaxime‐resistance of TEM‐1 mutants. To find out, we first engineered multiple single nucleotide changes into wild‐type TEM‐1 (see Section 2). Specifically, we engineered each of the three stabilizing and each of the six activity‐improving mutations that had reached especially high frequency into wild‐type TEM‐1, measured the minimal inhibitory concentration of cefotaxime in these mutants, and did so under both high and low mistranslation (Table S3).
Every single one of these mutations conveyed higher cefotaxime‐resistance benefit under high mistranslation than under low mistranslation. Consider first the stability‐enhancing mutations. Whereas none of them conveyed increased MIC under low mistranslation, all three conferred increased MIC under high mistranslation, and the MIC increase relative to wild‐type TEM‐1 was statistically significant for two of these three mutations (Figure 3a; One‐way ANOVA: F 9,50 = 38.677, p = 1.7 × 10–19; post hoc two‐sided Dunnett tests, p = .011 for both M182T and R120G). Consistent with these observations, M182T or R120G achieved a higher frequency than all other mutations in six of eight populations evolved under high mistranslation, and they did so after merely one round of directed evolution on cefotaxime (Anc → H and H → H populations in Figure 2c,d). In addition, all three stability‐enhancing mutations were more beneficial under high mistranslation than under low mistranslation (Figure 3a; two‐sided Wilcoxon–Mann–Whitney test, W = 2, p = .008576 for M182T, W = 4, p = .02445 for R120G and W = 0, p = .003 for H153R). We suspect that these stabilizing mutations convey greater cefotaxime resistance under high mistranslation, because that is when they are especially important to buffer the deleterious effects of phenotypic mutations. In addition, their slower growth may enable high mistranslation populations (Figure S2) to persist longer in the presence of antibiotic, such that adaptive TEM‐1 variants have more time to degrade cefotaxime and thus convey greater fitness benefits. This possibility is also consistent with the cefotaxime‐resistance of the six activity‐enhancing mutations: Five of the six mutations convey greater cefotaxime‐resistance benefit under high mistranslation (Figure 3b; Text S1). In sum, mistranslation can increase the cefotaxime‐resistance benefit of multiple single mutations.
FIGURE 3.
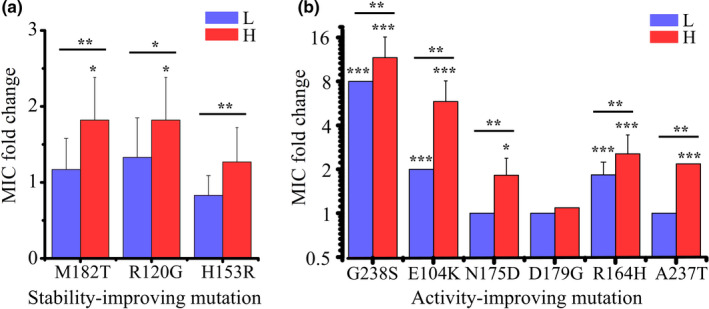
A greater incidence of beneficial mutations in TEM‐1 subject to high mistranslation. MIC fold change of TEM‐1 variants carrying (a) stabilizing mutations and (b) activity‐improving mutations in low‐mistranslation (L) and high‐mistranslation (H) hosts. We determined the MIC fold change by dividing the MIC value of the corresponding variant in the L or H host by the MIC value of wild‐type TEM‐1 in the corresponding host. Error bars represent one standard deviation. Absent error bars indicate that all replicate measurements had yielded identical MIC values. Note that MIC‐based measurements are semiquantitative, classifying antibiotic resistance into multiple discrete categories on a logarithmic scale. This is why multiple replicate measurements can yield the same MIC values. To test the statistical significance of the MIC fold change caused by single mutation in either L or H hosts compared to the wild‐type TEM‐1 in the corresponding host, we performed a one way ANOVA using the two‐sided Dunnett Test for multiple comparisons to one control (the wild‐type TEM‐1). To assess statistical significance of the difference in MIC fold change between L and H hosts caused by a single mutation, we performed a two‐sided Wilcoxon–Mann–Whitney test. We transformed the MIC and MIC fold change values logarithmically (base 2) for performing the above statistical analyses. *p < .05, **p < .01 and ***p < .001
3.4. Elevated mistranslation promotes the exploration of alternative evolutionary trajectories
We next engineered the three triple‐mutation genotypes that had attained high‐frequency in either low or high mistranslation populations and measured their cefotaxime MICs in both kinds of hosts (Table S4). Together with data from the double‐mutants (Table S4), this information allowed us to construct all possible evolutionary paths to the triple‐mutant genotypes, where each step in such a path involves a single point mutation. We distinguish three kinds of steps in such a path. An accessible mutational step is one that increases cefotaxime MIC significantly (Figure 4, thick solid arrows), an inaccessible step is one that does not increase cefotaxime MIC (dashed lines), and a conditionally accessible step is one that increases cefotaxime MIC significantly in the genetic background in which it occurs, but where wild‐type TEM‐1 would first have to experience one or more inaccessible steps to create this kind of genetic background (thin solid lines). The first of the triple‐mutants we analysed was the canonical or standard (S) combination G238S/E104K/M182T favoured under low mistranslation. The other two were the alternative (A) genotypes N175D/D179G/M182T and R164H/E104K/M182T favoured under high mistranslation (Figure 2). We will refer to them as the S, A1, and A2 triple‐mutant genotypes, respectively. The cefotaxime MIC of the standard genotype was significantly higher than those of both alternative genotypes under both high and low mistranslation (Table S5). Thus, if the alternative genotypes are fitness peaks in the TEM‐1 adaptive landscape, they are local rather than global peaks, consistent with the observation that replicate 4 of the Anc → H populations and replicates 1 and 3 of the H → H populations attain lower cefotaxime MICs (Figure 2). Among the total number of six paths to the standard high fitness genotype, three were accessible under low mistranslation and five were accessible under high mistranslation (Figure 4a). Thus, high mistranslation increases the number of accessible paths to this genotype.
FIGURE 4.
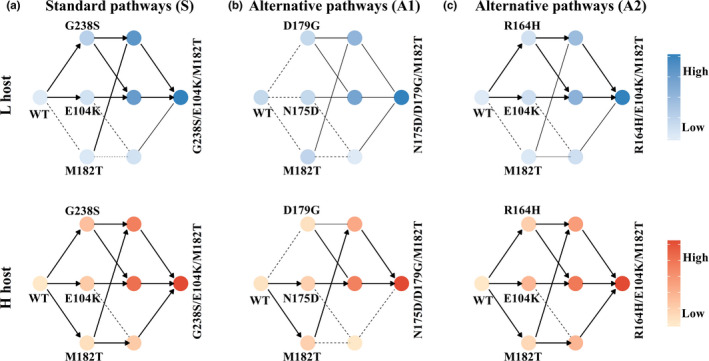
Elevated mistranslation increases the evolutionary accessibility of mutational pathways under directional selection. The accessible evolutionary trajectories to the highest cefotaxime‐resistance genotype G238S/E104K/M182T (a), to N175D/D179G/M182T (b) and to R164H/E104K/M182T (c) in low‐mistranslating (L) and high‐mistranslating (H) hosts. During directed evolution, an accessible mutational step is one that increases MIC significantly (thick solid lines with arrows), an inaccessible step is one that does not increase MIC (dashed lines), and a conditionally accessible step is one that increases MIC significantly in the genetic background in which it occurs, but where wild‐type TEM‐1 must experience one or more inaccessible mutations that create this kind of genetic background (thin solid lines). More saturated colours correspond to higher MIC (relative to wild‐type TEM‐1; see also Tables S3 and S4). We considered a difference in MIC between two genotypes significant only when p < .05 (One‐way ANOVA with post hoc Bonferroni correction)
Just as for the standard triple‐mutant genotype, high mistranslation increased the number of paths to the alternative genotypes. For the A1 genotype, the difference is most extreme. Specifically, under low mistranslation, not a single path to this genotype is accessible, unless the mutations N175D, D179G or M182T are fixed in advance (Figure 4b). In contrast, under high mistranslation, TEM‐1 can follow two evolutionary pathways towards this genotype. What is more, an additional mutation A237T, which became fixed together with the A1 genotype in one high‐mistranslating population, and causes an increase in cefotaxime MIC specifically under high mistranslation, also increases the accessibility of this genotype (Figure S5). For the A2 genotype, there are three accessible paths under low mistranslation, and two additional paths become accessible under high mistranslation (Figure 4c). In sum, high mistranslation increases the accessibility of all three high cefotaxime‐resistance genotypes, and can thus help diversify the evolutionary trajectories of populations.
3.5. Elevated mistranslation does not qualitatively change pairwise epistasis between mutations
Changing epistatic relationships between pairs of mutations can affect whether a population reaches a high fitness genotype from the same wild‐type genotype. For example, in positive (synergistic) epistasis of beneficial mutations, the joint occurrence of two beneficial mutations conveys a greater fitness benefit than the sum of their individual benefits. In negative (antagonistic) epistasis, their joint benefit is smaller than that of their individual effects. Epistatic interactions like these might differ between low‐ and high‐mistranslating strains, which may change the evolutionary pathways that such strains follow. To find out whether this may be the case, we engineered multiple pairs of individual mutations into wild‐type TEM‐1, and measured their MIC under both low and high mistranslation (Table S4). We found strong positive epistatic interactions in the following mutation pairs: G238S/E104K, G238S/M182T, R164H/E104K, R164H/M182T, N175D/D179G, D179G/M182T and N175D_D179G/A237T (Figure S6). (Because N175D and D179G always appeared together in our replicate populations (Table S5), and because they convey a cefotaxime‐resistance increase jointly but not individually under low mistranslation, we considered the double mutant N175D_D179G as a single mutant in our pairwise epistasis analysis.) Likewise, we found strong negative sign epistatic interactions in several other paired mutations: G238S/R164H, G238S/N175D_D179G and R164H/N175D_D179G (Figure S7 and Table S6). However, these interactions did not change qualitatively between low‐ and high‐mistranslating strains. They can therefore not be the sole cause of altered evolutionary trajectories.
3.6. An epistatic network can help explain genetic diversification between populations under elevated mistranslation
The observation that high mistranslation increases the accessibility of all three genotypes raises the question why the standard triple‐mutant genotype is less prevalent under high mistranslation, especially since it has higher cefotaxime MIC than the alternatives. To resolve this apparent paradox, we turned again to an analysis of pairwise epistasis, but in the context of the triple‐mutant high cefotaxime resistance genotypes (Figure 5).
FIGURE 5.
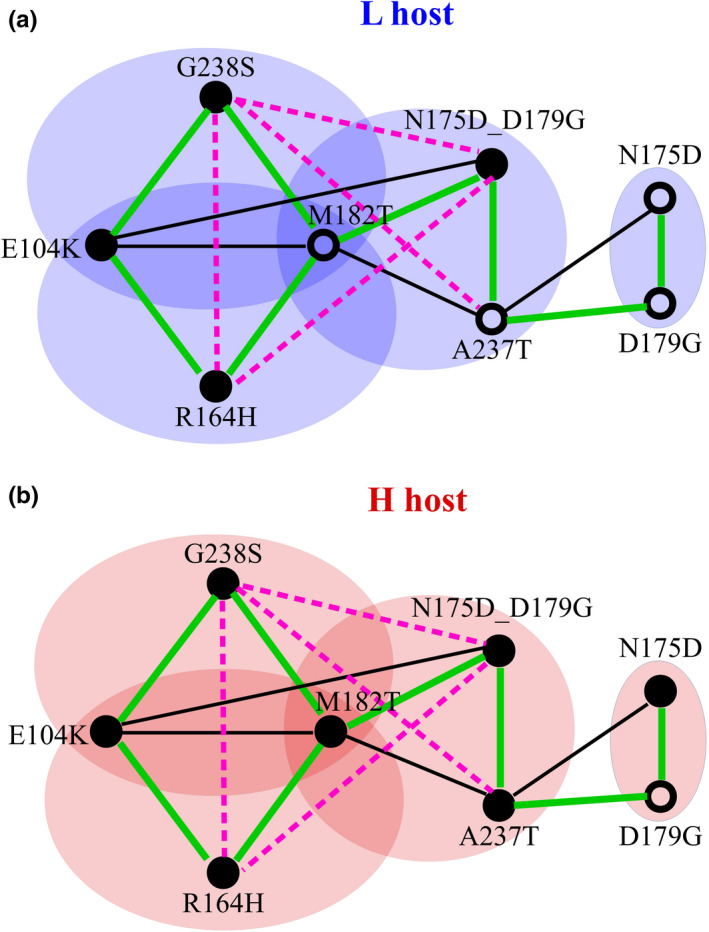
Epistatic interactions between mutations under high and low mistranslation rates. Lines connecting mutations indicate positive epistasis (green), weak negative epistasis (dashed black) and strong negative sign epistasis (pink) in relative MIC of the indicated variants (see also Figures S6 and S7). Filled circles indicate an increase in MIC of the labelled single mutation relative to the wild‐type, and open circles indicate no increase in MIC. We determined the relative MIC by dividing the MIC of any one variant by the MIC of wild‐type TEM‐1 in the same host (L or H). The schematic is based on data in Tables S3, S4 and S6. The strong negative sign epistasis between G238S and A237T was reported in a previous study (Salverda et al., 2011)
Two crucial observations can help resolve this paradox. The first comes from epistatic interactions between the mutations that are unique to each of the triple‐mutant genotypes, and that distinguish these genotypes from each another. These are G238S (for the S genotype), N175D_D179G (for A1) and R164H (for A2). All three of these mutations show strong negative pairwise sign epistasis with one another (Figure S7). That is, whenever one of these mutations rises to high frequency, negative sign epistasis prevents any of the other two mutations from co‐occurring with it at high frequency. In other words, these mutations mutually exclude each other. This is not only predicted by our cefotaxime‐resistance measurements, it is also demonstrated by the co‐occurrence frequencies of paired mutations in our evolving populations. Specifically, any two of these mutations occurred together in less than 0.1% of a population's genotypes (Note that SMRT sequencing allows us to sequence each full‐length TEM‐1 variant; Figure S8 and Table S5).
The second observation regards the suppressor mutation M182T, which does not increase the cefotaxime MIC by itself under low mistranslation, but significantly increases the cefotaxime MIC under high mistranslation (Figure 3a). Even in the absence of cefotaxime, the mutation conveys a significantly greater growth benefit on the H host (47.7% increase in cell density) than on the L host (13.0% increase; two‐sided Wilcoxon–Mann–Whitney test, W = 0, p = 3.602 × 10–5; Figure S9a,b). Consistent with these observations, sequencing the high‐mistranslating and low‐mistranslating populations after eight generations of selection on ampicillin (and before even selecting on cefotaxime) showed that M182T reached a 1.94‐fold higher frequency on average in high‐mistranslating populations, a difference that is statistically significant (two‐sided Wilcoxon–Mann–Whitney test, W = 16, p = .02857; Figure S9c). Importantly, M182T also shows strong epistatic interactions with the three mutations above, but these interactions are positive (Figure 5; Figure S7). Any one of these mutations, if it co‐occurs with M182T, has an increased chance to outcompete the others over time (Figure S10). The net result is multiple, mutually exclusive paths to a high cefotaxime‐resistance genotype (Text S2).
Similarly, both G238S and R164H also showed strong positive epistatic interactions with E104K, and both compete with each other for co‐occurrence with E104K (Figure 5), resulting in mutually exclusive paths to a high cefotaxime‐resistance genotype and genetic diversification (Figure S10). Taken together, these observations suggest that epistatic interactions among adaptive mutations can reduce the predictability of adaptive evolution and increase the genetic divergence between populations. In addition, it can increase diversity within populations under high mistranslation (Text S3).
4. DISCUSSION
Elevated mistranslation increases the diversity of evolutionary trajectories towards high cefotaxime‐resistance genotypes, and the genetic mechanism behind this diversification involves two phenomena. First, high mistranslation promotes the accessibility of different high cefotaxime‐resistance genotypes by increasing the individual cefotaxime‐resistance benefit of adaptive DNA mutations (Figures 3 and 4). Second, the adaptive DNA mutations we observed form an epistatic network that can lead to mutually exclusive evolutionary trajectories towards different high cefotaxime‐resistance genotypes (Figure 5; Figure S10).
High mistranslation increased the cefotaxime‐resistance benefit of both stability‐ and activity‐enhancing mutations that are known to be important for cefotaxime resistance (Figure 3). To understand why stabilizing mutations are especially beneficial under high mistranslation, it is useful to keep in mind that 10%–50% of random amino‐acid mutations are deleterious to protein function, and most such deleterious mutations unfold and destabilize proteins (Bloom et al., 2006; Drummond & Wilke, 2008; Guo et al., 2004). Under high mistranslation, more such amino acid changes occur in the form of phenotypic mutations, which suggests that stabilizing genetic mutations may be more important to compensate their deleterious effects. This assertion is supported by the slower growth and lower antibiotic resistance of high mistranslation populations that harbour wild‐type TEM‐1 (Figure S2). It is also consistent with earlier observations that more stabilizing mutations and fewer destabilizing mutations accumulate during directed evolution of TEM‐1 under high mistranslation (Bratulic et al., 2015, 2017). Finally, it is consistent with our observation that all three individual stabilizing mutations did not change the MIC of cefotaxime under low mistranslation, but increased it significantly under high mistranslation (M182T, R120G and H153R; Figure 3). In addition, slower growth may cause adaptive TEM‐1 mutants to convey higher benefits in high mistranslation populations, because it allows cells to persist longer (Eng et al., 1991), such that these mutants have more time to degrade cefotaxime. Consistent with this hypothesis, we observed that all three stabilizing mutations and five of six activity‐improving mutations conveyed significantly higher fitness benefit under high mistranslation (Figure 3). Taken together, all these factors allowed TEM‐1 populations to evolve along more pathways to different high cefotaxime‐resistance genotypes, thus rendering diverse evolutionary trajectories accessible under high mistranslation (Figure 4).
Because epistasis can affect which mutations go to fixation and the order in which they do (Salverda et al., 2011; Weinreich et al., 2006), we suspected that our beneficial mutations interact epistatically, and that mistranslation alters these interactions. Pairs of such mutations did indeed show positive epistasis or negative sign epistasis, but for any one mutation pair, mistranslation did not alter the nature of this epistasis qualitatively (Figures S6 and S7). However, we observed a network of pairwise epistatic interactions that involves up to seven mutations (Figure 5), and that is brought to the fore by the increased cefotaxime‐resistance benefit of individual mutations under high mistranslation (Figure 3 and Figure 4). It results in reduced predictability of evolutionary trajectories and genetic diversification (Figure 2; Figure S10).
The adaptive evolution of TEM‐1 towards cefotaxime resistance involves the stabilizing mutation M182T, which enhances cefotaxime‐resistance under high mistranslation and displays strong positive epistasis with the activity‐enhancing mutations G238S, R164H and N175D_D179G. The latter three mutations, in contrast, display strong negative sign epistasis with each other (Figure 5), which reduces their individual cefotaxime‐resistance benefits (Figure S7). If any one of these mutations co‐occurs first with M182T, it has an increasing chance to outcompete the others over time (Figure S10). Because the epistatic network we observe involves both positive epistasis and negative sign epistasis, a specific high cefotaxime‐resistance mutant combination that arises first in a population may go to fixation by outcompeting other such combinations that arise later. In addition, in several of our populations that had accumulated standing variation before selection for cefotaxime‐resistance, multiple high‐fitness genotypes coexisted within the same population, at least at the initial stages of adaptive evolution (Figure 2d; Table S8 and Text S3). This within‐population diversification caused by the epistatic network may render evolutionary trajectories especially unpredictable, because the relative frequencies and fitness values of the high‐fitness genotypes may determine their probability of going to fixation. In our experiments, such unpredictability is especially evident from replicate populations that were initially polymorphic for high cefotaxime‐resistance genotypes, but experienced fixation of different such genotypes in different populations (Figure 2d,f; Table S8 and Text S3).
In sum, high mistranslation can help a population explore diverse evolutionary trajectories, and thus reduce the predictability of evolutionary pathways and the high fitness genotypes they attain. It can affect which genetic mutations are adaptive by altering the individual fitness benefit of such mutations. In combination with epistatic interactions among such mutations, it can affect the order in which combinations of such mutations achieve high frequency. The life of a cell is intrinsically noisy (Kærn et al., 2005; Wilkinson, 2009), and this stochasticity might have substantial impact on the trajectories of adaptive evolution.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
AUTHOR CONTRIBUTIONS
J.Z., S.B. and A.W. designed the experiments. J.Z. and S.B. performed the experiments. J.Z., S.B., H.L. and A.W. all contributed to data analysis. J.Z. and A.W. wrote the paper. All authors read and edited the paper.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/jeb.13892.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by ERC Advanced Grant 739874 and by Swiss National Science Foundation grant 31003A_172887. J.Z. and S.B. thank the Functional Genomics Center Zurich for sequencing the samples by SMRT sequencing. J.Z. thanks Fahad Khalid, Rzgar Hosseini, Maria Magdalena San Roman, Dr. Joshua Payne, Dr. Kathleen Sprouffske and Dr. Macarena Toll‐Riera for help in analysing the sequencing data and for useful discussion.
Zheng, J., Bratulic, S., Lischer, H. E. L., & Wagner, A. (2021). Mistranslation can promote the exploration of alternative evolutionary trajectories in enzyme evolution. Journal of Evolutionary Biology, 34, 1302–1315. 10.1111/jeb.13892
DATA AVAILABILITY STATEMENT
The SMRT sequencing project of TEM‐1 populations has been deposited at DDBJ/EMBL/GenBank under the accession KBRX00000000. The version described in this paper is the first version, KBRX01000000.
REFERENCES
- Ballesteros, M., Fredriksson, Å., Henriksson, J., & Nyström, T. (2001). Bacterial senescence: Protein oxidation in non‐proliferating cells is dictated by the accuracy of the ribosomes. EMBO Journal, 20, 5280–5289. 10.1093/emboj/20.18.5280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baneyx, F., & Mujacic, M. (2004). Recombinant protein folding and misfolding in Escherichia coli. Nature Biotechnology, 22, 1399–1408. 10.1038/nbt1029 [DOI] [PubMed] [Google Scholar]
- Bank, C., Matuszewski, S., Hietpas, R. T., & Jensen, J. D. (2016). On the (un)predictability of a large intragenic fitness landscape. Proceedings of the National Academy of Sciences of the United States of America, 113, 14085–14090. 10.1073/pnas.1612676113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bershtein, S., Segal, M., Bekerman, R., Tokuriki, N., & Tawfik, D. S. (2006). Robustness–epistasis link shapes the fitness landscape of a randomly drifting protein. Nature, 444, 929–932. 10.1038/nature05385 [DOI] [PubMed] [Google Scholar]
- Bloom, J. D., Labthavikul, S. T., Otey, C. R., & Arnold, F. H. (2006). Protein stability promotes evolvability. Proceedings of the National Academy of Sciences of the United States of America, 103, 5869–5874. 10.1073/pnas.0510098103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blount, Z. D., Lenski, R. E., & Losos, J. B. (2018). Contingency and determinism in evolution: Replaying life’s tape. Science, 362, eaam5979. 10.1126/science.aam5979 [DOI] [PubMed] [Google Scholar]
- Bratulic, S., Gerber, F., & Wagner, A. (2015). Mistranslation drives the evolution of robustness in TEM‐1 β‐lactamase. Proceedings of the National Academy of Sciences of the United States of America, 112, 12758–12763. 10.1073/pnas.1510071112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratulic, S., Toll‐Riera, M., & Wagner, A. (2017). Mistranslation can enhance fitness through purging of deleterious mutations. Nature Communications, 8, 15410. 10.1038/ncomms15410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, N. G., Pennington, J. M., Huang, W., Ayvaz, T., & Palzkill, T. (2010). Multiple global suppressors of protein stability defects facilitate the evolution of extended‐spectrum TEM β‐lactamases. Journal of Molecular Biology, 404, 832–846. 10.1016/j.jmb.2010.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson, M. J., & Tesler, G. (2012). Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): Application and theory. BMC Bioinformatics, 13, 238. 10.1186/1471-2105-13-238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, J. H., & Lee, S. Y. (2004). Secretory and extracellular production of recombinant proteins using Escherichia coli . Applied Microbiology and Biotechnology, 64, 625–635. 10.1007/s00253-004-1559-9 [DOI] [PubMed] [Google Scholar]
- Chubiz, L. M., Lee, M.‐C., Delaney, N. F., Marx, C. J., & Haaland, P. (2012). FREQ‐Seq: A rapid, cost‐effective, sequencing‐based method to determine allele frequencies directly from mixed populations. PLoS One, 7, e47959. 10.1371/journal.pone.0047959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko, K. A., Wanner, B. L., & Beckwith, J. (2000). One‐step inactivation of chromosomal genes in Escherichia coli K‐12 using PCR products. Proceedings of the National Academy of Sciences of the United States of America, 97, 6640–6645. 10.1073/pnas.120163297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Visser, J. A. G., & Krug, J. (2014). Empirical fitness landscapes and the predictability of evolution. Nature Reviews Genetics, 15, 480–490. 10.1038/nrg3744 [DOI] [PubMed] [Google Scholar]
- Dellus‐Gur, E., Toth‐Petroczy, A., Elias, M., & Tawfik, D. S. (2013). What makes a protein fold amenable to functional innovation? Fold polarity and stability tradeoffs. Journal of Molecular Biology, 425, 2609–2621. 10.1016/j.jmb.2013.03.033 [DOI] [PubMed] [Google Scholar]
- Dickinson, B. C., Leconte, A. M., Allen, B., Esvelt, K. M., & Liu, D. R. (2013). Experimental interrogation of the path dependence and stochasticity of protein evolution using phage‐assisted continuous evolution. Proceedings of the National Academy of Sciences of the United States of America, 110, 9007–9012. 10.1073/pnas.1220670110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond, D. A., & Wilke, C. O. (2008). Mistranslation‐induced protein misfolding as a dominant constraint on coding‐sequence evolution. Cell, 134, 341–352. 10.1016/j.cell.2008.05.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng, R. H., Padberg, F. T., Smith, S. M., Tan, E. N., & Cherubin, C. E. (1991). Bactericidal effects of antibiotics on slowly growing and nongrowing bacteria. Antimicrobial Agents and Chemotherapy, 35(9), 1824–1828. 10.1128/AAC.35.9.1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firnberg, E., Labonte, J. W., Gray, J. J., & Ostermeier, M. (2014). A comprehensive, high‐resolution map of a gene’s fitness landscape. Molecular Biology and Evolution, 31, 1581–1592. 10.1093/molbev/msu081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey, B., & Suppmann, B. (1995). Demonstration of the Expand PCR system’s greater fidelity and higher yields with a lacI‐based PCR fidelity assay. Biochemica, 2, 8–9. [Google Scholar]
- Gould, S. J. (1989). Wonderful life: The burgess shale and the nature of history. WW Norton & Co. [Google Scholar]
- Guo, H. H., Choe, J., & Loeb, L. A. (2004). Protein tolerance to random amino acid change. Proceedings of the National Academy of Sciences of the United States of America, 101, 9205–9210. 10.1073/pnas.0403255101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kærn, M., Elston, T. C., Blake, W. J., & Collins, J. J. (2005). Stochasticity in gene expression: From theories to phenotypes. Nature Reviews Genetics, 6, 451–464. 10.1038/nrg1615 [DOI] [PubMed] [Google Scholar]
- Kim, K. E., Peluso, P., Babayan, P., Yeadon, P. J., Yu, C., Fisher, W. W., Chin, C.‐S., Rapicavoli, N. A., Rank, D. R., Li, J., Catcheside, D. E. A., Celniker, S. E., Phillippy, A. M., Bergman, C. M., & Landolin, J. M. (2014). Long‐read, whole‐genome shotgun sequence data for five model organisms. Scientific Data, 1, 140045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer, E. B., & Farabaugh, P. J. (2007). The frequency of translational misreading errors in E. coli is largely determined by tRNA competition. RNA, 13, 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvitek, D. J., Sherlock, G., Ricupero‐Hovasse, S., Dore, C., & Lepage, P. (2011). Reciprocal sign epistasis between frequently experimentally evolved adaptive mutations causes a rugged fitness landscape. PLoS Genetics, 7, e1002056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lässig, M., Mustonen, V., & Walczak, A. M. (2017). Predicting evolution. Nature Ecology & Evolution, 1, 0077. 10.1038/s41559-017-0077 [DOI] [PubMed] [Google Scholar]
- Liu, H., & Naismith, J. H. (2008). An efficient one‐step site‐directed deletion, insertion, single and multiple‐site plasmid mutagenesis protocol. BMC Biotechnology, 8, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogle, J. M., & Ramakrishnan, V. (2005). Structural insights into translational fidelity. Annual Review of Biochemistry, 74, 129–177. 10.1146/annurev.biochem.74.061903.155440 [DOI] [PubMed] [Google Scholar]
- Orencia, M. C., Yoon, J. S., Ness, J. E., Stemmer, W. P. C., & Stevens, R. C. (2001). Predicting the emergence of antibiotic resistance by directed evolution and structural analysis. Natural Structural Biology, 8, 238–242. [DOI] [PubMed] [Google Scholar]
- Palzkill, T., & Botstein, D. (1992). Identification of amino acid substitutions that alter the substrate specificity of TEM‐1 beta‐lactamase. Journal of Bacteriology, 174, 5237–5243. 10.1128/jb.174.16.5237-5243.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker, J. (1989). Errors and alternatives in reading the universal genetic code. Microbiological Reviews, 53, 273–298. 10.1128/mr.53.3.273-298.1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poelwijk, F. J., Kiviet, D. J., Weinreich, D. M., & Tans, S. J. (2007). Empirical fitness landscapes reveal accessible evolutionary paths. Nature, 445, 383–386. 10.1038/nature05451 [DOI] [PubMed] [Google Scholar]
- Salverda, M. L., De Visser, J. A., & Barlow, M. (2010). Natural evolution of TEM‐1 b‐lactamase: Experimental reconstruction and clinical relevance. FEMS Microbiology Reviews, 34, 1015–1036. [DOI] [PubMed] [Google Scholar]
- Salverda, M. L. M., Dellus, E., Gorter, F. A., Debets, A. J. M., Van Der Oost, J., Hoekstra, R. F., Tawfik, D. S., Arjan, J., & De Visser, G. M. (2011). Initial mutations direct alternative pathways of protein evolution. PLoS Genetics, 7, e1001321. 10.1371/journal.pgen.1001321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salverda, M. L. M., Koomen, J., Koopmanschap, B., Zwart, M. P., & de Visser, J. A. G. M. (2017). Adaptive benefits from small mutation supplies in an antibiotic resistance enzyme. Proceedings of the National Academy of Sciences, 114, 12773–12778. 10.1073/pnas.1712999114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk, M. F., Szendro, I. G., Salverda, M. L. M., Krug, J., & de Visser, J. A. G. M. (2013). Patterns of epistasis between beneficial mutations in an antibiotic resistance gene. Molecular Biology and Evolution, 30, 1779–1787. 10.1093/molbev/mst096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk, M. F., Witte, S., Salverda, M. L. M., Koopmanschap, B., Krug, J., & de Visser, J. A. G. M. (2015). Role of pleiotropy during adaptation of TEM‐1 β‐lactamase to two novel antibiotics. Evolutionary Applications, 8, 248–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr, T. N., Picton, L. K., & Thornton, J. W. (2017). Alternative evolutionary histories in the sequence space of an ancient protein. Nature, 549, 409–413. 10.1038/nature23902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakulenko, S. B., Taibi‐Tronche, P., Rta, M., Th, T., Massova, I., Lerner, S. A., & Mobashery, S. (1999). Effects on substrate profile by mutational substitutions at positions 164 and 179 of the class A TEMpUC19 β‐lactamase from Escherichia coli . Journal of Biological Chemistry, 274, 23052–23060. 10.1074/jbc.274.33.23052 [DOI] [PubMed] [Google Scholar]
- Walker, K. W., & Gilbert, H. F. (1994). Effect of redox environment on the in vitro and in vivo folding of RTEM‐1 beta‐lactamase and Escherichia coli alkaline phosphatase. Journal of Biological Chemistry, 269, 28487–28493. 10.1016/S0021-9258(18)46953-0 [DOI] [PubMed] [Google Scholar]
- Warren, D. J. (2011). Preparation of highly efficient electrocompetent Escherichia coli using glycerol/mannitol density step centrifugation. Analytical Biochemistry, 413, 206–207. 10.1016/j.ab.2011.02.036 [DOI] [PubMed] [Google Scholar]
- Weinreich, D. M., Delaney, N. F., DePristo, M. A., & Hartl, D. L. (2006). Darwinian evolution can follow only very few mutational paths to fitter proteins. Science, 312, 111–114. 10.1126/science.1123539 [DOI] [PubMed] [Google Scholar]
- Weinreich, D. M., Watson, R. A., & Chao, L. (2005). Perspective: Sign epistasis and genetic constraint on evolutionary trajectories. Evolution, 59, 1165–1174. [PubMed] [Google Scholar]
- Wilkinson, D. J. (2009). Stochastic modelling for quantitative description of heterogeneous biological systems. Nature Reviews Genetics, 10, 122–133. 10.1038/nrg2509 [DOI] [PubMed] [Google Scholar]
- Willensdorfer, M., Bürger, R., Nowak, M. A., Holtz, G., & Remaut, E. (2007). Phenotypic mutation rates and the abundance of abnormal proteins in yeast. PLoS Computational Biology, 3, e203. 10.1371/journal.pcbi.0030203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccolo, M., Williams, D. M., Brown, D. M., & Gherardi, E. (1996). An approach to random mutagenesis of DNA using mixtures of triphosphate derivatives of nucleoside analogues. Journal of Molecular Biology, 255, 589–603. 10.1006/jmbi.1996.0049 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The SMRT sequencing project of TEM‐1 populations has been deposited at DDBJ/EMBL/GenBank under the accession KBRX00000000. The version described in this paper is the first version, KBRX01000000.