

Abstract
The chemistry of stabilized α‐boryl carbanions shows remarkable diversity, and can enable many different synthetic routes towards efficient C−C bond formation. The electron‐deficient, trivalent boron center stabilizes the carbanion facilitating its generation and tuning its reactivity. Here, the electronic structure and the reactivity trends of a large dataset of α‐boryl carbanions are described. DFT‐derived parameters were used to capture their electronic and steric properties, computational reactivity towards model substrates, and crystallographic analysis within the Cambridge Structural Dataset. This study maps the reactivity space by systematically varying the nature of the boryl moiety, the substituents of the carbanionic center, the number of α‐boryl motifs, and the metal counterion. In general, the free carbanionic intermediates are described as borata‐alkene species with C−B π interactions polarized towards the carbon. Furthermore, it was possible to classify the α‐boryl alkylidene metal precursors into three classes directly related to their reactivity: 1) nucleophilic borata‐alkene salts with alkali and alkaline earth metals, 2) nucleophilic η 2‐(C−B) borata‐alkene complexes with early transition metals, Cu and Ag, and 3) α‐boryl alkyl complexes with late transition metals. This trend map aids selection of the appropriate reactive synthon depending on the reactivity sought.
Keywords: alpha-borylcarbanion, borata, descriptors, density functional calculations
Carbanion cartography: This study maps the stability/reactivity space for α‐boryl carbanions leading to the prediction of novel species. Free carbanionic intermediates can be described as borata‐alkene species with a C−B π‐interaction strongly polarized towards the carbon, whereas multi‐boryl species have very polar C−B bonds. Extensive analysis of α‐boryl alkylidene metal precursors identifies three structural patterns that can be related to reactivity.

Introduction
The access to primary, secondary, and tertiary alkylboronic esters, through the generation of α‐boryl carbanions and subsequent electrophilic trapping, is a new and powerful synthetic tool towards efficient C−C bond formation. The generation of α‐boryl carbanions can be conducted through four complementary pathways (Scheme 1), including a) deborylation of 1,1‐diboryl alkanes,[1, 2, 3, 4, 5, 6, 7, 8, 9, 10] b) deprotonation of the α‐hydrogen from an organoborane compound,[11, 12, 13, 14, 15, 16] c) metallation of α‐halo boronic esters[17] and d) transmetallation of α‐borylmethide metal salts with organometallic reagents.[18] α‐Boryl carbanions show a remarkable stability due to the valence deficiency of the adjacent three coordinate boron center, and they can be also described by their borata‐alkene resonance forms (Scheme 2).[19, 20]
Scheme 1.
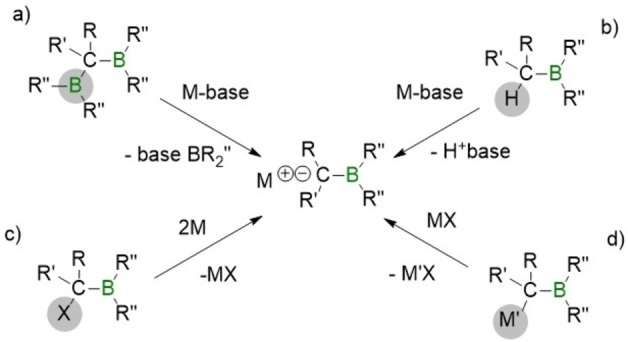
Strategic methods to access α‐boryl carbanions.
Scheme 2.

Resonance structures for α‐boryl carbanion and borata‐alkene.
The experimental outcomes are consistent with the delocalization of the electron density of the anion throughout the empty p orbital of the adjacent boron. This is demonstrated by the chemical shifts displacement on 11B (highfield) and 13C (downfield) NMR data for R2B‐CH2 − in comparison with the corresponding α‐boryl alkane.[12, 21] IR spectra of boron‐stabilized anions in the gas phase, in combination with DFT calculations, also suggest the double bond character of C=B bond in Me2BCH2 − α‐monoboryl anions.[22]
In the solid state, the shortened B−C bond lengths[23, 24, 25] of the borata‐alkene species provide an additional evidence of the their “boron ylide” character, which can be related to the analogs containing boron–carbon double bonds.[26] Computational studies have also supported the borata‐alkene character of α‐boryl carbanions by means of detailed analysis of their electronic structures.[25, 27, 28] The natural bond orbital (NBO) analysis on [Mes2B=CR2]− anion by Gabbaï et al. showed one σ‐ and one π‐interaction between the carbon and the boron atoms, where the π bond was polarized towards the carbon.[27] Erker et al. described a similar B−C interaction for related species, in which the HOMO formally corresponds to a C−B π‐orbital strongly polarized towards the carbanionic atom.[25]
The study by Erker et al.[25] compared three different α‐boryl moieties, B(C6F5)2, BMes2 and 9‐boryilbicyclo[3.3.1]nonane (Scheme 3). The authors concluded that the high degree of carbanion stabilization when the boryl moiety B(C6F5)2 is involved might be due to the presence of fluorine substituents on the aryl group. The mesityl substituents at boron led to a decrease of stabilization by about 16 kcal mol−1, followed by the borata‐alkenes containing the 9‐boryilbicyclo[3.3.1]nonane which showed a lower degree of carbanion stabilization (Scheme 3). The steric protection of the boron center seems to be necessary to ensure an appropriate proton abstraction of the α‐hydrogen from the organoborane (Scheme 1b), since there is a strong tendency to form a four‐coordinate boron”ate” salt upon addition of a base. For that reason, most of the borata‐alkenes reported so far bear bulky substituents on boron, such as B(C6F5)2, BMes2 or 9‐boryilbicyclo[3.3.1]nonane. Eventually, the pronounced α‐boryl carbanion stabilization represents an extra advantage to explore the significant reactivity of the borata‐alkenes.[25, 29]
Scheme 3.

Stabilizing order for selected borata‐alkene species.
To gain more insight into the electronic structure and the reactivity trends of α‐boryl carbanions, we conduct here a detailed computational study based on density functional calculations (ωB97X‐D functional) and wave function analysis. The study is performed on a varied dataset of compounds bearing commonly used boryl moieties, such as Bpin (pinacolboryl) and Bdan (naphthodiazaboryl). As illustrated in Scheme 4, we have gauged the influence of several structural features of the α‐boryl carbanions: a) the nature of the boryl moiety, b) the substituents on the carbanionic center, c) the comparison between α‐mono‐, α‐di‐ and α‐triboryl carbanions, and d) the nature of the metal involved. Ultimately, we aim to identify reliable descriptors derived from ground‐state structures that correlate with the stability and reactivity of α‐boryl carbanions and allow to build a map of trends for these species.
Scheme 4.

Analyzed structural features influencing the nature of the α‐boryl carbanions.
Results and Discussion
Influence of the nature of the boryl moiety
Using as starting point, the early work by Erker et al.,[25] we have initially gauged the electronic structure and reactivity of α‐monoboryl carbanions as a function of boryl fragment nature. We started with the comparison of the previously analyzed α‐monoboryl carbanions, containing the boryl moieties BMes2 and B(C6F5)2 (1ames and 1aPhF ), with those that include the commonly used Bpin and Bdan moieties in the structure, (1apin and 1adan respectively). Here, we have explored different geometric, electronic and energy descriptors aiming to rationalize the stability and reactivity trends along the carbanion series. Table 1 collects the values of the most meaningful descriptors for 1aPhF , 1ames , 1adan and 1apin species; and Tables S1 and S2 in the Supporting Information lists all the computed descriptors and their values.
Table 1.
Calculated protonation Gibbs energies (ΔG prot. [kcal mol−1]) and free‐energy barriers for nucleophilic substitution with bromoethane (Δ [kcal mol−1]), energy of the HOMO orbital (E HOMO [eV]), and Wiberg bond orders upon variation of boryl moiety nature (∑C−Bbo), species 1aPhF , 1ames , 1adan and 1apin .
|
| ||||
|---|---|---|---|---|
|
|
1aPhF |
1ames |
1adan |
1apin |
|
ΔG prot. |
+19.7 |
−0.2 |
−23.2 |
−33.6 |
|
Δ |
19.3 |
18.8 |
6.9 |
4.2 |
|
∑C−Bbo |
1.73 |
1.69 |
1.57 |
1.56 |
|
E HOMO |
−2.87 |
−2.20 |
−1.19 |
−0.58 |

In line with the analysis reported by Erker et al.,[25] the stabilization of α‐boryl carbanions can be assessed by calculating the protonation energies of the carbanions with respect to the cyclopentadienyl anion (ΔG prot) as illustrated in Scheme 5. Note that this calculation is equivalent to compute absolute proton affinities of carbanions but using cyclopenatadienyl as reference to set the zero, that is, subtracting the proton affinity of cyclopentadienyl (368.4 kcal mol−1) to each species. Moreover, we have determined the free‐energy barriers (Δ ) for the SN2 nucleophilic substitution reaction between the carbanions and bromoethane (Scheme 6) in order to quantify the nucleophilic reactivity of α‐boryl carbanions towards organic electrophiles. The values of energy barriers in Table 1 indicate the following trend on the reactivity 1apin >1adan >1ames >1aPhF .
Scheme 5.

Protonation of α‐boryl carbanion with cyclopentadiene.
Scheme 6.

Alkylation of α‐boryl carbanion with bromoethane.
The reactivity trend is inversely correlated to the stability of the α‐monoboryl carbanions. The least reactive species 1ames and 1aPhF show a marked stabilization as reflected in isoenergetic or endergonic, relative protonation free‐energies (ΔG prot=−0.2 and +19.7 kcal mol−1, respectively). Whereas the boron atom in 1aPhF and 1ames is protected by the steric bulkiness of Mes and C6F5 groups, the Bpin and Bdan boryl fragments depict the π‐donor ability from the O and N heteroatoms to the empty p orbital of the B atom. Consequently, in 1ames and 1aPhF the electron deficient boron center is fully available for delocalizing the carbanion negative charge. This also correlates with the Wiberg C−B bond order whose values increase from 1apin <1adan <1ames <1aPhF (Table 1). Moreover, the values are significantly larger than 1 (from 1.56 to 1.73), supporting the borata‐alkene character of these species.
The nucleophilic reactivity of organic reagents has been traditionally correlated with the energy level of HOMO orbital, or with the NBO atomic charges as we reported for related nucleophilic boryl species.[30, 31] Here, the energy of the HOMO, formally corresponding to a C−B π‐orbital strongly polarized towards the carbanionic atom (Figure 1), clearly correlates with the nucleophilic reactivity. The higher the energy is, the lower the free energy barrier (compare 2nd and 4th files in Table 1). Interestingly, the 1apin species is computed to be more reactive than 1adan (Δ =4.2 and 6.9 kcal mol−1, respectively).
Figure 1.

Representation of HOMO orbitals, formally corresponding to a C−B π‐orbital polarized towards the carbon, for 1aPhF , 1ames , 1adan and 1apin .
As illustrated in the HOMO representations of Figure 1, the aromatic fragment of Bdan 1adan moiety contributes to the delocalization of the carbanionic charge through the π‐channel, resulting in lower energy‐laying HOMO compared to 1apin . In fact, we had observed this effect in a previous study on boron‐stabilized carbanions generated from deborylation of 1,1‐diborylalkanes with alkoxides.[32] As the N atoms bound to B in Bdan are better π‐donors than O atoms in Bpin, one can envisage that in the absence of the aromatic fragment, the diazoboryl moieties should enhance carbanion nucleophilicity. To evaluate this effect, we computed the unprecedented compound 1aNpin (Scheme 7), in which the O substituents on 1apin were replaced by NH fragments. In line with previous reasoning, the 1aNpin is the most reactive species with a computed free energy barrier Δ of only 3.2 kcal mol−1. Finally, it is worth to mention that in this subset, the computed atomic charges at the carbanionic carbon (−0.88, −0.98, −1.14 and −1.20 a.u. for 1aPhF , 1ames , 1adan and 1apin , respectively) are consistent with the nucleophilicity of the α‐boryl methyl fragment, but this correlation is not observed for the other subsets in this work. A similar trend was suggested by Erker et al.[25] who attributed the observation to their borata‐alkane behavior. Therefore, HOMO energies will be used hereafter as a suitable descriptor of the reactivity of α‐boryl carbanions.
Scheme 7.

Schematic representation of the newly designed 4,4,5,5‐tetramethyl‐1,3,2‐diazaboryl methide anion 1aNpin .
Influence of the substituents on the carbanionic carbon
We explored next how the reactivity/stability and electronic structural properties can be affected by the influence of Me and Ph substituents on α‐monoboryl carbanions. The calculated ΔG prot energies for 1cpin (Table 2) demonstrates that the Ph group stabilizes the carbanion lone pair. This is also reflected in a larger sum of Wiberg bond orders for the three bonds of carbanion (3.68 for 1c pin vs. 3.55 and 3.62 for 1apin and 1bpin , respectively), and in the lower energy‐laying HOMO (−1.27 for 1c pin vs. −0.58 and −0.56 eV for 1a pin and 1b pin, respectively). Both descriptors capture the electron withdrawing effect of the Ph group. In line with the trend in stability, 1c pin shows lower reactivity for the SN2 nucleophilic substitution of bromoethane than for 1a pin (Δ =9.8 and 4.2 kcal mol−1 for 1c pin and 1a pin, respectively).
Table 2.
Calculated protonation Gibbs energies (ΔG prot. [kcal mol−1]), free‐energy barriers for nucleophilic substitution in bromoethane (Δ [kcal mol−1]) in, energy of the HOMO (E HOMO [eV]), and Wiberg bond orders upon variation of carbanion substituents, species 1cpin , 1apin and 1bpin .
|
|
|
|
|
|---|---|---|---|
|
Species |
1cpin |
1apin |
1bpin |
|
ΔG prot. |
−7.2 |
−33.6 |
−30.6 |
|
Δ [a] |
9.8 (13.0) |
4.2 (9.3) |
4.2 (8.5) |
|
∑C‐R3 bond order[b] |
3.68 |
3.55 |
3.62 |
|
E HOMO |
−1.27 |
−0.58 |
−0.56 |



[a] Values in parenthesis correspond to calculations with continuum solvent mode (DMSO). [b] Sum of Wiberg bond orders for the three bonds of carbanion (C−B, C−H, and C−Ph/−H/−Me).
The presence of the Me group (1bpin ) does not alter significantly the nucleophilic character of the carbanion respect to hydrogen‐substituted 1apin . Although it is usually considered that alkyl groups destabilize carbanions due to their electron‐donating inductive effect, here calculations show that other subtle effects need to be considered since they do not provide a clear picture of stability/reactivity order between 1apin and 1bpin (Table 2). Besides the inductive effect one should considerer an electrostatic size effect, in which the negative charge in 1bpin is more stabilized by the larger molecular volume that reduces its charge density. Thus, calculations in vacuum pointed out that methyl‐substituted 1bpin is more stable (less negative ΔG prot) than 1apin , whereas its HOMO is higher in energy (−0.58 and −0.56 eV for 1apin and 1bpin , respectively). However, inclusion of the effect of polar solvent reduces the influence of electrostatic size effect, and the corresponding calculations predict that methyl‐substituted 1bpin is more reactive than 1apin (see values in parenthesis in Table 2).
Influence of the number of boryl substituents
We next explored the influence of the number of boryl fragments in the stability/reactivity of the corresponding carbanions. Table 3 compares the main computed parameters for α‐mono‐, di‐, and triboryl carbanions 1apin , 2a2pin and 33pin , respectively. Increasing the number of boryl moieties the stability of the carbanion is enhanced (1apin <2a2pin <33pin ) whereas the nucleophilicity is reduced (1apin >2a2pin >33pin ). Figure 2 shows the HOMO orbitals for 2a2pin and 33pin , where it can be clearly observed that each boryl moiety contributes to the stabilization of the carbanion through a strong delocalization of the carbanion p‐type electron density into the π channel. Consequently, the α‐triboryl carbanion 33pin has lower‐energy laying HOMO (−2.61 eV) than those for 2a2pin and 1apin (−1.85 and −0.58 eV, respectively) being the former the least prone to react with electrophiles and the most stable.
Table 3.
Calculated protonation Gibbs energies (ΔG prot. [kcal mol−1]) and free‐energy barriers for nucleophilic substitution in bromoethane (Δ [kcal mol−1]), energy of the HOMO orbital (E HOMO [eV]), Wiberg bond orders, and average C−B distances in Å upon variation of the number of boryl substituens, species 1apin , 2a2pin and 33pin .
|
|
|
|
|
|---|---|---|---|
|
Species |
1apin |
2a2pin |
33pin |
|
ΔG prot. |
−33.6 |
−10.0 |
−1.8 |
|
Δ |
4.2 |
10.3 |
14.0 |
|
∑C‐R3 bond order |
3.55 |
3.51 |
3.39 |
|
C−Bbo‐av. |
1.56 |
1.23 |
1.07 |
|
dC−B av. |
1.44 |
1.47 |
1.50 |
|
E HOMO |
−0.58 |
−1.85 |
−2.61 |



Figure 2.

Representation of HOMO orbitals, formally corresponding to a C−B π‐orbital polarized towards the carbon, for 2a2pin and 33pin .
Remarkably, the sums of Wiberg bond order of the carbanion for species 1apin , 2a2pin and 33pin (3.55, 3.51 and 3.39, respectively) do not seem to be consistent with their stability. In this case, the most stable 33pin species has the lowest overall bond order. This might be due to the loss of borata‐alkane character when the negative charge of carbanion has to be shared between several boryl moieties. In fact, the averaged individual C−B bond order in 33pin is low (1.07) and the averaged C−B bond distance (1.50 Å) is significantly longer than that for monoborylated species 1apin (1.44 Å). Thus, multi‐boryl carbanions can be viewed as carbanionic species with polar C−B bonds, in which the excess of negative charge is electrostatically stabilized by the boron substituents, as well as, by some amount of charge transfer to the empty perpendicular boron p orbitals. Figure 3 depicts the evolution of computed atomic charges from mono‐ to di‐, and to tri‐boryl carbanions, showing an increasing charge separation (polarization) at the C−B bonds. The estimated amount of charge transferred from the carbanion to the perpendicular boron p orbitals is still significant for 33pin (0.21 a.u.), contributing to the overall stabilization of this species (Table S3).
Figure 3.

NBO atomic charges for mono‐, di‐, and triboryl carbanions 1apin (Δq B‐C=2.10), 2a2pin (Δq B‐C=2.40), and 33pin (Δq B‐C=2.64).
Mapping the nucleophilicity of α‐boryl carbanions
Once it has been discussed in detail the influence of electronic and structural features on the stability/reactivity of several model α‐boryl carbanions, our next objective is the construction of a map from a full set of structures, in order to classify and identify certain trends in the chemical space. To this end, we additionally analyzed 14 α‐boryl carbanions depicted in Figure 4. Overall, the full dataset (set 1) comprises 22 anionic species, which were selected by varying systematically the number and the type of boryl moieties, as well as, the type of substituents on carbon (R=H, Me and Ph). The dataset also includes the α‐boryl vinyl system (1epin ), in order to compare the borata‐alkene character between [R2B=CH2]− and [R2B=CH=CH2]−. Interestingly, we found an inverse, linear correlation (correlation coefficient r 2=0.91) between the protonation energies of the carbanions and the HOMO energies, which can be consequently used as reliable descriptor to gauge the stability/reactivity trends in α‐boryl carbanions (Figure S1).
Figure 4.

Additional α‐boryl carbanion species forming dataset set1.
Figure 5 maps the full dataset using two descriptors, the energy of the HOMO (E HOMO) and the sum of Wiberg C−B bond order (∑C−Bbo). The E HOMO descriptor can be directly related to the stability/reactivity trends, while the ∑C−Bbo is a useful descriptor allowing to separate the mono‐, di‐ and triborylated species, and to differentiate between Me and Ph substituents on carbon. First, we identified a clear correlation between the number of boryl substituents and the carbanion nucleophilicity as reflected in the HOMO energy. α‐Triboryl carbanions are the least nucleophilic, with HOMO energies ranging from −3.3 to −2.6 eV, presumably due to the accumulation of the three stabilizing boryl substituents. The reactivity on these species follows the trend 33pin >32pindan >3pin2dan >33dan in agreement with the observation that Bdan moieties have an extra stabilization effect on the carbanion.[32] Within the α‐diboryl cabanions, the calculated HOMO energies are found between −2.6 to −1.7 eV, conforming a specific group where once again the species containing Bdan units become less reactive and more stabilized than the ones with Bpin moieties. In fact, the values for HOMO energy for 2a2dan and 2c2dan carbanions reach values close to the next group to the left, the α‐triboryl carbanions (Figure 5). The mixed α‐diboryl cabanions (2apindan , 2bpindan , and 2cpindan ) have intermediate energy HOMO values, and the 2b2pin might be the most reactive among the α‐diboryl carbanions.
Figure 5.

Representation of the sum of C−B Wiberg bond orders versus the energy of the HOMO [eV] for α‐monoboryl (•), α‐diboryl (•), and α‐triboryl (•) carbanions.
The subset formed by the α‐monoborylated species with Bpin or Bdan boryl fragments shows higher HOMO energies (−1.3 to −0.2 eV). Among them, we predicted the vinyl carbanionic species 1epin , that contains a sp2 carbanion, as highly nucleophilic. Even more to the right, we found the newly designed species 1aNpin (Scheme 7). Whereas the sum of Wiberg C−B bond order for 1aNpin is similar to 1apin , the higher energy of the HOMO orbital of 1aNpin indicates that combining N‐substituted boron and non‐aromatic scaffold causes the largest nucleophilicity. On the other side, the species containing highly acidic BMes or BC6H5 moieties (1ames and 1aPhF ) have EHOMO values that lay in the range of di‐ and triborylated species (−2.2 and −2.9 eV, respectively). This clearly indicates that boryl substituents in 1ames and 1aPhF cause a strong stabilization on the carbanion because in the absence of heteroatom substituents on boron, its perpendicular p orbital is fully available for the overlap with the lone pair of the carbanion.
The structures with the phenyl substituents (1cpin , 1cdan , 2c2pin , 2cpindan and 2c2dan ) show the lowest C−B bond orders due to the electron‐releasing effect of the phenyl groups, which compete with electron delocalization through the borata‐alkene structure. On the other hand, the structures containing methyl substituents (1bpin , 1bdan , 2b2pin , 2bpindan and 2b2dan ) have slightly lower C−B bond orders than those species with hydrogen substituents. This latter trend can be correlated with a subtle effect of alkyl substituents, which induce some electrostatic delocalization.
Influence of the nature of the metal
The practical applications of borata‐alkenes as reactive synthons are achieved by different strategies which involve the preparation of boryl alkylidene metal salts and α‐boryl alkyl transition metal complexes. Here, we study the influence of those metals and transition metals in their stability/reactivity. Figure 6 depicts the selected structures including Li+, Cu+, Ag+ and Pd2+ α‐boryl methide metal salts, and Table 4 collects the most representative computed parameters. In this case, to evaluate the nucleophilic reactivity, we have determined the free‐energy barriers (Δ ) required to transfer the carbanions to the electrophilic carbon atom of the model substrate formaldehyde (Scheme 8). This substrate is a simple species that have been used to quantify the reactivity of related metal‐boryl compounds,[31, 33] and constitute a model of observed reactions such as the nucleophilic borylmethylation of aldehydes with α‐boryl alkyl copper[34] and silver[35] complexes, and with lithium dimesithylboron substituted carbanions [(Mes)2BC(H)RLi].[36] It is important to note that for Li, Cu and Ag species the nucleophilic additions to several organic electrophiles have been reported,[34, 35, 36, 37] while for transition metals such as Pd the observed reactivity involves mainly transmetalation processes.[38] Thus, we should expect low to moderate free‐energy barriers for Li, Cu and Ag salts, and larger barrier for Pd indicating a less favorable nucleophilic reactivity.
Figure 6.

Selected structures for the analysis of the effect of the metal cation (Li+, Cu+, Ag+, and Pd2+) on the stability/reactivity of α‐boryl carbanionic species.
Table 4.
Calculated free‐energy barriers for carbanion addition to formaldehyde (Δ [kcal mol−1]), overall charge of carbanion fragment (q[C]), Wiberg bond orders, C−B lengths (d C−B [Å]), and steric distance–weight volume (V W) upon variation the cationic fragment in 1apin‐Li, 1apin‐Cu, 1apin‐Ag and 1apin‐Pd.
|
Structure |
1apin‐Li |
1apin‐Cu |
1apin‐Ag |
1apin‐Pd |
|---|---|---|---|---|
|
Δ [a] |
2.2a |
15.0 |
18.4 |
52.7 |
|
q[C] |
−0.88 |
−0.66 |
−0.64 |
−0.21 |
|
C−B bond order |
1.36 |
1.17 |
1.17 |
0.97 |
|
d C−B |
1.48 |
1.51 |
1.51 |
1.55 |
|
V W |
0.0 |
40.6 |
38.1 |
46.3 |
[a] The free‐energy barrier is computed from a precursor complex in which the carbonyl oxygen of aldehyde is coordinated side‐on to lithium.
Scheme 8.

Nucleophilic addition of α‐boryl methide metals to formaldehyde.
The computed Δ values (Table 4) predict an order of nucleophilic reactivity that is consistent with experimental background (Li>Cu>Ag >Pd, with Δ =2.2, 15.0, 18.4, and 52.7 kcal mol−1, respectively). Here, we identify the overall charge of carbanionic fragment (q[C]) as descriptor correlating with nucleophilicity; thus, the more negatively charged the carbanionic fragment, the lower is the energy barrier (Table 4, second row). The formation of coordination complexes changes the nature of the HOMO orbital which cannot be univocally assigned to the C−B π‐interaction, and consequently, we discarded it as descriptor in this case. Additionally, we evaluated the steric effects of the different metal fragments on the reactivity using the distance‐weighted volume (V W) parameter.[39, 40] The VW parameter measures the steric bulkiness of the metal fragment and its impact on carbanion center (see Computational Methods). Comparison of the closely related [(PPh3)2Cu(H2CBpin)] and [(PPh3)2Ag(H2CBpin)] complexes shows that the silver fragment induces less steric hindrance on the reactive carbanion due to the larger size of Ag+ ion, which move away the ligand substituents. However, the larger polarization of the metal‐carbon bond in Cu complex determines its higher nucleophilicity, indicating that steric effects are less influential than electronic ones when comparing different metals.
Along the series we observe substantial structural changes in the boron‐carbon moiety and in its interaction with the metal (Table 4, third and fourth rows). This indicates a continuous switch on the compound nature from borata‐alkene lithium salt to a α‐boryl alkyl palladium complex. In lithium species 1apin‐Li, both the Wiberg C−B bond order (1.36) and the C−B distance (1.48 Å) are indicative of borata‐alkene character. The Li cation interacts electrostatically with the three atoms of O−C−B moiety (Figure 7). Although Li+ does not change the nature of the species, it induces some C−B lengthening (+0.04 Å) and pyramidalization of the carbanionic carbon (−30°), with respect to the free carbanion 1apin . Note that introducing specific solvation molecules solvating Li+ cation[41] would diminish its effect on the electronic structure of borata‐alkene. On the other extreme, the computed C−B bond order of palladium complex 1apin‐Pd is close to 1 and the corresponding distance (1.55 Å) is significantly larger than for 1apin‐Li. Thus, this Pd compound can be better defined as an α‐boryl alkyl palladium complex. At an intermediate situation, the Bpin=CH2 − fragment in Cu and Ag compounds acts as an anionic η 2‐(C,B) ligand whose interaction with the transition metal is shifted toward the C atom (Figure 7). In line with bonding description, the HOMO orbital in 1apin‐Cu and 1apin‐Ag shows an overlap between transition metal d orbitals and the C=B π orbital (Figure S4). Note that borata‐alkenes acting as anionic ligands with η2 coordination to transition metals are known.[42]
Figure 7.

Three‐dimensional representation of computed structures for metal complexes 1apin‐Li, 1apin‐Cu, 1apin‐Ag and 1apin‐Pd. Selected bond lengths are given in Å.
Trend map for metal salts and complexes
To further assess the variation in the nature of α‐boryl carbanions with regard to the metal involved, we performed a systematic structural search within the Cambridge Structural Database (CSD; see Computational Methods), and constructed a histogram of the C=B bond lengths (Figure 8). The graph separates the crystallographic data for boryl methide salts into three main groups: red bars represents boryl methide Li salts, green bars covers boryl methide Cu salts, and other metals from group 4 and 5, and blue bars involve boryl methide salts of late transition metals including Pd2+. For each group, the d(C−B) values lie within a relatively wide range (0.1 Å), but their distributions are centered at different distances. To the far left we found the boryl alkylidene lithium salts, most of whose C−B distances range from 1.44 to 1.49 Å. Moving to the right, from 1.47 to 1.54 Å, there are several early transition metal complexes (Ti4+, Zr4+, Hf4+, and Ta5+), and within this distance range we also found the Cu+ complexes at 1.51 and 1.52 Å. At longer C−B distances (>1.52 Å), we find most of the late transition metal complexes such as Pd, Pt, Ru, Rh or Zn (see Table S4 for a complete list). Overall, our computed values fit quite well with the structural data available in CSD, allowing to classify the nature of α‐boryl carbanionic species into three main groups and to establish structure‐reactivity relationships.
Figure 8.

Histograms for B−C bond lengths of α‐boryl carbanionic species separated into 3 groups as a function of carbanion nature: 1) Li salts (▪), 2) Cu and groups 4 and 5 including Ti, Zr, Hf, and Ta complexes (▪), and 3) late transition metals including Fe, Ru, Rh, Ni, Pd, Pt, Au, Zn and Hg (▪). All bonds lengths are rounded to the nearest 0.01 Å. Data obtained from crystallographic data in the CSD.
The borata‐alkene lithium salts, and the η 2‐(C,B) borata‐alkene copper and silver complexes show highly polarized metal‐carbon interactions with a significant nucleophilic character. They are prone to generate carbanionic fragments stabilized by boryl moiety that are able to react with organic electrophiles as observed experimentally.[34, 35, 36, 37] Other promising species for nucleophilic additions are those based on early transition metals, although to the best of our knowledge their reactivity has not been tested yet.[37] Only few examples are currently reported on the use of silver salts for nucleophilic additions.[35] The α‐boryl alkyl palladium complexes have less polarized and stronger metal‐carbon bond. As consequence, the reported reactivity for Pd differs from Li, Cu and Ag. Although these palladium complexes do not serve as nucleophilic agents, they have been applied in C−C bond forming reactions, for example, by transmetalation.[38] Figure 8 shows that other late transition metals have similar structural features to Pd, suggesting that this reactivity can be extended to other complexes.
Finally, Figure 9 provides an extension of the electronic structure analysis for Li, Cu, Ag and Pd compounds with different type and number of boryl moieties and substituents on the carbanionic atom. The overall charge of the carbanionic fragment (q[C]) and the average C−B bond order (C−Bbo‐av.) can be used as descriptors to evaluate the nucleophilicity and the nature of the α‐boryl carbanionic species, respectively. Clearly, the extent of the nucleophilicity (increasing to the left of the graph) is mainly ruled by the type of metal, but it can be tuned by α‐boryl carbanionic fragment. We also observe that the decrease of C−B bond order, that is, the reduction of borata‐alkene character, has the following trend: Li>Cu≌Ag>Pd. The C−Bbo‐av. descriptor is more sensitive to the nature of the carbanionic fragment. Interestingly, among Cu species, calculations suggest that the vinyl carbanionic 1epin ‐Cu complex is located at a different area of the chemical space (Figure 9), and therefore, it could lead to new reactivity.
Figure 9.

Representation of the average of C−B Wiberg bond orders (C−Bbo‐av.) versus the overall charge of the carbanionic fragment (q[C]) for the α‐boryl carbanionic Li (•), Cu (•), Ag (•) and Pd (•) species.
Conclusion
We have systematically studied several types of α‐boryl carbanion, analyzing their electronic structures and reactivity trends as a function of the nature of boryl moieties, the type of carbanionic substituents, the number of boryl motifs, and the metal interaction with the carbanion. In general, the free carbanionic intermediates are better described as borata‐alkene species with a C−B π‐interaction strongly polarized towards the carbanionic atom. By taking into consideration the energy of the HOMO and Wiberg bond order, we were able to establish a gradient of stability and nucleophilic reactivity for these intermediates. π‐Acidic boron atoms (i. e., BMes2 or B(C5F5)2), aromatic substituents on boron (i. e., Bdan), or electron withdrawing substituents on carbon (i. e., Ph) induce a larger delocalization of the carbanionic charge through the π‐channel that results in more stable and less reactive intermediates. The multi‐boryl carbanionic species lose part of their borata‐alkene character but enhance their stability electrostatically through the additive effect of several polar C−B bonds. This map of the reactivity landscape has predicted a novel α‐boryl carbanion, the newly designed 4,4,5,5‐tetramethyl‐1,3,2‐diazaboryl methide anion 1aNpin [H2CB(NH)2R2]−, which should show enhanced nucleophilicity.
For α‐boryl alkylidene metal precursors, both computational and crystallographic analysis of a large dataset identify three different types of carbanionic species that can be directly related to the observed reactivity: 1) borata‐alkene salts with alkali and alkaline earth metals such as Li, 2) η 2‐(C−B) borata‐alkene complexes with early transition metals, Cu and Ag, and 3) α‐boryl alkyl complexes with late transition metals such as Pd. The two first groups show highly polarized metal‐carbon interactions with a significant nucleophilic character that make them suitable synthons for reacting with organic electrophiles. The third group has a less polarized and stronger metal‐carbon bond, and they are prone to undergoing other types of C−C bond forming reactions such as cross coupling through transmetalation strategies. We hope that this map and the underlying dataset will facilitate the optimization of novel α‐boryl carbanion reagents and assist their selection along with the desired reactivity parameters, contributing to the developing of this emerging area in chemistry.
Computational Methods
Geometry optimizations and transition state searches were performed with Gaussian16[43] package. The quantum mechanics calculations were performed within the framework of density functional theory (DFT)[44] by using the ωB97X−D functional.[45] The basis set employed effective core potentials (ECPs) with double‐ζ valence basis set (LANDL2DZ)[46] for Cu, Ag, Pd, Cl, Br and P, and were supplemented with polarized shells with the following exponents: Cu (f=3.525), Ag (f=1.611), Pd (f=1.472), Cl (d=0.650), Br (d=0.428) and P (d=0.387).[47] For all other electrons of all other atoms 6–31G(d) basis set was used.[48] The solvent effects of DMSO were included by means of SMD model[49] as implemented in Gaussian16.[43] The bonding of the molecules as well as the fragment charges were analyzed by using the NBO method,[50] from which we derived the Wiberg bond order and carbanion fragment charge (q[C]) descriptors. The NBO method analyses the resultant wave function in terms of optimally chosen localized orbitals, corresponding to a Lewis structure representation of chemical bonding. For computing orbital populations consistently, we had defined the carbanion carbon bonded to 3 substituents with single bonds.
To quantify the steric hindrance of metal fragments, we used the distance‐weighed volume parameter (V W).[39, 40] which measures the steric bulkiness of the molecular environment and its impact on the carbanion center. The descriptor quantifies the bulk produced the metal fragment by considering three parameters: 1) The number of atoms, excluding the metal, 2) the size of the atom (r=van der Waals radii [Å]), and 3) the distance (d [Å]) from the atom to the boron center. The factor r 3 is divided by d for each atom, and the sum is extended to all of the atoms in the given fragment. Finally, the crystallographic structure search was carried out by using CSD software, using the February 2020 version. A data set collection of computational results is available in the ioChem‐BD repository and can be accessed at https://doi.org/10.19061/iochem‐bd‐2‐52.[51]
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We thank Ministerio de Economía y Competitividad y por el Fondo Europeo de Desarrollo Regional FEDER through projects PGC2018‐100780‐B‐l00 and PID2019‐109674GB−I00, and the Generalitat de Catalunya (grant no. 2017SGR629)
R. J. Maza, E. Fernández, J. J. Carbó, Chem. Eur. J. 2021, 27, 12352.
Contributor Information
Prof. Elena Fernández, Email: mariaelena.fernandez@urv.cat.
Dr. Jorge J. Carbó, Email: j.carbo@urv.cat.
References
- 1.Cainelli G., Dal Bello G., Zubiani G., Tetrahedron Lett. 1965, 6, 3429–3432. [Google Scholar]
- 2.Zweifel G., Steele R. B., Tetrahedron Lett. 1966, 48, 6021–6025. [Google Scholar]
- 3.Homer L., Hoffmann H., Wippel H. G., Chem. Ber. 1958, 91, 61–63. [Google Scholar]
- 4.Schmidbauer H., Tronich W., Chem. Ber. 1968, 101, 3556–3561. [Google Scholar]
- 5.Kauffmann T., Joussen R., Klas N., Vahrenhorst A., Top. Curr. Sci. 1980, 92, 109–147. [Google Scholar]
- 6.Kauffmann T., Angew. Chem. Int. Ed. 1982, 21, 410–429; [Google Scholar]; Angew. Chem. 1982, 94, 401–420. [Google Scholar]
- 7.Kauffmann T., Joussen R., Klas N., Vahrenhorst A., Tetrahedron Lett. 1978, 19, 4391–4395. [Google Scholar]
- 8.Bertini F., Grasselli P., Zubiani G., Cainelli G., Tetrahedron 1970, 26, 1281–1290. [Google Scholar]
- 9.Zweifel G., Fisher R. P., Horng A., Synthesis 1972, 557–558. [Google Scholar]
- 10.Zweifel G., Arzoumanian H., J. Am. Chem. Soc. 1967, 89, 291–295. [Google Scholar]
- 11.
- 11a.Rathke M. W., Kow R., J. Am. Chem. Soc. 1972, 94, 6854–6856; [Google Scholar]
- 11b.Kow R., Rathke M., J. Am. Chem. Soc. 1973, 95, 2715–2716. [Google Scholar]
- 12.Ashe A. J. III, Shu P., J. Am. Chem. Soc. 1971, 93, 1804–1805. [Google Scholar]
- 13.Pelter A., Singaram B., Warren L., Win J. W.Tetrahedron 1993, 49, 2965–2978. [Google Scholar]
- 14.Pelter A., Singaram B., Williams L., Wilson J. W., Tetrahedron Lett. 1983, 24, 623–626. [Google Scholar]
- 15.Pelter A., Williams L., Wilson J. W., Tetrahedron Lett. 1983, 24, 627–630. [Google Scholar]
- 16.Pelter A., Singaram B., Wilson J. W., Tetrahedron Lett. 1983, 24, 631–634. [Google Scholar]
- 17.Knochel P., J. Am. Chem. Soc. 1990, 112, 7431–7333. [Google Scholar]
- 18.
- 18a.Matteson D. S., Wilson J. W., Organometallics 1985, 4, 1690–1694; [Google Scholar]
- 18b.Tsai D. J. S., Matteson D. S., Organometallics 1983, 2, 236–241. [Google Scholar]
- 19.
- 19a.Brown H. C., Zweifel G., J. Am. Chem. Soc. 1961, 83, 3834–3840; [Google Scholar]
- 19b.Zweifel G., Arzoumanian H., Tetrahedron Lett. 1966, 7, 2535–2538; [Google Scholar]
- 19c.Matteson D. S., Synthesis 1975, 147–158; [Google Scholar]
- 19d.Paetzold P., Boeke B., Chem. Ber. 1976, 109, 1011–1016. [Google Scholar]
- 20.Maza R. J., Carbó J. J., Fernández E., Adv. Synth. Catal. 2021, 363, 2274–2289. [Google Scholar]
- 21.Hong K., Liu X., Morken J. P., J. Am. Chem. Soc. 2014, 136, 10581–10584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oomens J., Steill J. D., Morton Th. H., Inorg. Chem. 2010, 49, 6781–6783. [DOI] [PubMed] [Google Scholar]
- 23.Bartlett R. A., Power P. P., Organometallics 1986, 5, 1916–1917. [Google Scholar]
- 24.Olmstead M. M., Power P. P., Weese K. J., Doedens R. J., J. Am. Chem. Soc. 1987, 109, 2541–2542. [Google Scholar]
- 25.Moquist P., Chen G.-Q., Mück-Lichtenfeld Ch., Bussmann K., Daniliuc C. G., Kehr G., Erker G., Chem. Sci. 2015, 6, 816–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Power P. P., Chem. Rev. 1999, 99, 3463–3504. [DOI] [PubMed] [Google Scholar]
- 27.Chiu Ch.-W., Gabbaï F. P., Angew. Chem. Int. Ed. 2007, 46, 6878–6881; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 7002–7005. [Google Scholar]
- 28.Lafage M., Pujol A., Saffon-Merceron N., Mezailles N., ACS Catal. 2016, 6, 3030–3035. [Google Scholar]
- 29.Klis T., Lulinski S., Serwatowski J., Curr. Org. Chem. 2010, 14, 2549–2566. [Google Scholar]
- 30.Cid J., Carbó J. J., Fernández E., Chem. Eur. J. 2012, 18, 12794–12802. [DOI] [PubMed] [Google Scholar]
- 31.García-López D., Cid J., Marqués R., Fernández E., Carbó J. J., Chem. Eur. J. 2017, 23, 5066–5075. [DOI] [PubMed] [Google Scholar]
- 32.Cuenca A. B., Cid J., García-López D., Carbó J. J., Fernández E., Org. Biomol. Chem. 2015, 13, 9659–9664. [DOI] [PubMed] [Google Scholar]
- 33.
- 33a.Wagner M., van Eikema Hommes N. J. R., Nöth H., Schleyer P. v. R., Inorg. Chem. 1995, 34, 607–614; [Google Scholar]
- 33b.Zhao H., Dang L., Marder T. b., Lin Z., J. Am. Chem. Soc. 2008, 130, 5586–5594. [DOI] [PubMed] [Google Scholar]
- 34.
- 34a.Joannou M. V., Moyer B. S., Meek S. J., J. Am. Chem. Soc. 2015, 137, 6176–6179; [DOI] [PubMed] [Google Scholar]
- 34b.Murray S. A., Green J. C., Tailor S. B., Meek S. J., Angew. Chem. Int. Ed. 2016, 55, 9065–9069; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 9211–9215. [Google Scholar]
- 35.Joannou M. V., Moyer B. S., Goldfogel M. J., Meek S. J., Angew. Chem. Int. Ed. 2015, 54, 14141–14145; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 14347–14351. [Google Scholar]
- 36.Pelter A., Buss D., Colclough E., Singaram B., Tetrahedron 1993, 49, 7077–7103. [Google Scholar]
- 37.For some reviews reporting additional examples of nucleophilic additions of Li and Cu boryl stabilized carbanions, see:
- 37a.Miralles N., Maza R. J., Fernández E., Adv. Synth. Catal. 2018, 360, 1306–1327; [Google Scholar]
- 37b.Wu Ch., Wang J. B., Tetrahedron Lett. 2018, 59, 2128–2140; [Google Scholar]
- 37c.Nallagonda R., Padala K., Masarwa A., Org. Biomol. Chem. 2018, 16, 1050–1064. [DOI] [PubMed] [Google Scholar]
- 38.
- 38a.Zhang Ch., Wu X., Wang Ch., Zhang Ch., Qu J., Chen Y., Org. Lett. 2020, 16, 6376–6381; [DOI] [PubMed] [Google Scholar]
- 38b.Li Ch., Li M., Li J., Wu W., Jiang H., Chem. Commun. 2018, 54, 66–69. [DOI] [PubMed] [Google Scholar]
- 39.
- 39a.Aguado-Ullate S., Saureu S., Guasch L., Carbó J. J., Chem. Eur. J. 2012, 18, 995–1005; [DOI] [PubMed] [Google Scholar]
- 39b.Aguado-Ullate S., Urbano M., Villaba I., Pires E., García J. I., Bo C., Carbó J. J., Chem. Eur. J. 2012, 18, 14026–14036. [DOI] [PubMed] [Google Scholar]
- 40.MolQuO application (accessed May 2021): http://rodi.urv.es/~carbo/quadrants/index.html
- 41.Royes J., Ni S., Farré A., La–Cascia E., Carbó J. J., Cuenca A. B., Maseras F., Ferández E., ACS Catal. 2018, 8, 2833–2838. [Google Scholar]
- 42.For a review, see: Emslie D. J. H., Cowie B. E., Kolpin K. B., Dalton Trans. 2012, 41, 1101–1117. [DOI] [PubMed] [Google Scholar]
- 43.Gaussian 16, Revision A.03, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A., Jr. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, D. J. Fox, Gaussian, Inc., Wallingford CT, 2016.
- 44.R. G. Parr, W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, Oxford, 1989.
- 45.Chai J. D., Head-Gordon M., Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [DOI] [PubMed] [Google Scholar]
- 46.
- 46a.Hay P. J., Wadt W. R., J. Chem. Phys. 1985, 82, 270–283; [Google Scholar]
- 46b.Hay P. J., Wadt W. R., J. Chem. Phys. 1985, 82, 284–298; [Google Scholar]
- 46c.Hay P. J., Wadt W. R., J. Chem. Phys. 1985, 82, 299–310. [Google Scholar]
- 47.
- 47a.Höllwarth A., Böhme M., Dapprich S., Ehlers A. W., Gobbi A., Jonas V., Köler K. F., Stegmann R., Veldkamp A., Frenking G., Chem. Phys. Lett. 1993, 208, 237–240; [Google Scholar]
- 47b.Ehlers A. W., Böhme M., Dapprich S., Gobbi A., Höllwarth A., Jonas V., Köhler K. F., Stegmann R., Veldkamp A., Frenking G., Chem. Phys. Lett. 1993, 208, 111–114. [Google Scholar]
- 48.
- 48a.Gordon M. S., Chem. Phys. Lett. 1980, 76, 163–168; [Google Scholar]
- 48b.Binning R. C. J., Curtiss L. A., J. Comput. Chem. 1990, 11, 1206–1216; [Google Scholar]
- 48c.Fukui K., J. Phys. Chem. 1970, 74, 4161–4163. [Google Scholar]
- 49.Marenich A. V., Cramer C. J., Truhlar D. G. J., Phys. Chem. B 2009, 113, 6378–6396. [DOI] [PubMed] [Google Scholar]
- 50.Reed A. E., Curtiss L. A., Weinhold F., Chem. Rev. 1988, 88, 899–926. [Google Scholar]
- 51.Álvarez-Moreno M., de Graaf C., Lopez N., Maseras F., Poblet J. M., Bo C., J. Chem. Inf. Model. 2015, 55, 95–103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information