

Summary
ZUMA‐1 (NCT02348216) examined the safety and efficacy of axicabtagene ciloleucel (axi‐cel), an autologous CD19‐directed chimaeric antigen receptor (CAR)‐T cell therapy, in refractory large B‐cell lymphoma. To reduce treatment‐related toxicity, several exploratory safety management cohorts were added to ZUMA‐1. Specifically, cohort 6 investigated management of cytokine release syndrome (CRS) and neurologic events (NEs) with prophylactic corticosteroids and earlier corticosteroid and tocilizumab intervention. CRS and NE incidence and severity were primary end‐points. Following leukapheresis, patients could receive optional bridging therapy per investigator discretion. All patients received conditioning chemotherapy (days −5 through −3), 2 × 106 CAR‐T cells/kg (day 0) and once‐daily oral dexamethasone [10 mg, day 0 (before axi‐cel) through day 2]. Forty patients received axi‐cel. CRS occurred in 80% of patients (all grade ≤2). Any grade and grade 3 or higher NEs occurred in 58% and 13% of patients respectively. Sixty‐eight per cent of patients did not experience CRS or NEs within 72 h of axi‐cel. With a median follow‐up of 8·9 months, objective and complete response rates were 95% and 80% respectively. Overall, prophylactic corticosteroids and earlier corticosteroid and/or tocilizumab intervention resulted in no grade 3 or higher CRS, a low rate of grade 3 or higher NEs and high response rates in this study population.
Keywords: large B‐cell lymphoma, axi‐cel, chimaeric antigen receptor‐T cell, prophylaxis, corticosteroids, cytokine release syndrome
Introduction
The safety and efficacy of axicabtagene ciloleucel (axi‐cel), an autologous anti‐CD19 chimaeric antigen receptor (CAR)‐T cell therapy approved for treating patients with relapsed/refractory (R/R) large B‐cell lymphoma (LBCL) after two or more prior lines of systemic therapy,1, 2 was demonstrated in the pivotal phase 1/2 ZUMA‐1 study (NCT02348216).3, 4 In the ZUMA‐1 primary analysis (median follow‐up, 8·7 months), the investigator‐assessed objective response rate (ORR) was 82% and the complete response (CR) rate was 54%.4 Grade 3 or higher cytokine release syndrome (CRS) occurred in 13 patients (13%), and grade 3 or higher neurologic events (NEs) occurred in 28 patients (28%).
CRS and NEs are the adverse events (AEs) most commonly associated with anti‐CD19 CAR‐T cell therapy.5, 6 CRS is believed to result from CAR‐T cell activation upon cognate antigen recognition, causing proliferation and release of cytokines, including interferon gamma (IFN‐γ) and tumour necrosis factor α. These, in turn, activate "bystander" immune cells (e.g. myeloid lineage immune cells), which release inflammatory cytokines, including interleukin (IL) 6 — a key CRS mediator.7 CAR‐T cell levels8 and IL‐6 levels4, 8, 9 have been shown to correlate with CRS severity following CAR‐T cell infusion.
Less is known regarding aetiology of NEs.6 Associations of NEs with higher serum levels of C‐reactive protein and various cytokines have been reported.10 In ZUMA‐1, elevated levels of CAR‐T cells, IL‐2, granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) and ferritin were associated with grade 3 or higher NEs, while levels of other biomarkers (IL‐6, IL‐15, IL‐2Rα) were associated with both grade 3 or higher CRS and grade 3 or higher NEs.4
Minimising CRS and NE incidence and severity are important goals in CAR‐T therapy‐related toxicity management.11 Therefore, several exploratory safety management cohorts were added to ZUMA‐1. The cohorts were not designed for comparative purposes and no formal hypotheses were tested. Cohort 3 evaluated prophylactic use of the anticonvulsant levetiracetam starting on day 0 and the anti‐IL‐6 antibody tocilizumab on day 2.12 In cohort 3 (n = 34), relative to cohorts 1 + 2 (n = 101), prophylactic tocilizumab appeared to decrease rates of grade 3 or higher CRS (3% versus 13%), but not rates of grade 3 or higher NEs (41% versus 28%).4, 12 To investigate the effects of more general inflammatory suppression on toxicities, cohort 4 (n = 41) examined use of levetiracetam prophylaxis and earlier corticosteroid and tocilizumab intervention: corticosteroids were initiated starting at grade 1 CRS (if no improvement after 3 days) and at grade 1 NE; tocilizumab was initiated at grade 1 CRS (if no improvement after 3 days), at grade 2 or higher CRS and at grade 2 or higher NE. Rates of grade 3 or higher CRS (2%) and grade 3 or higher NEs (17%) appeared to be lower in cohort 4 versus cohorts 1 + 2. Importantly, no meaningful impact was observed on CAR‐T cell pharmacokinetics or investigator‐assessed disease response (73% ORR, 51% CR).13 To further build on these findings, the impact of adding prophylactic corticosteroids to the cohort 4 toxicity management was assessed in cohort 6.
Patients and methods
The study protocol for phase 2 cohorts 1 + 2 of the single‐arm, multicentre, registrational ZUMA‐1 study was previously described.4 Cohort 6 primarily differed from cohorts 1 + 2 in that patients received levetiracetam and corticosteroid prophylaxis and earlier corticosteroids and tocilizumab for toxicity management (Fig 1). Key similarities and differences between phase 2 cohorts 1 + 2, 4 and 6 are detailed in Table SI.
Fig 1.
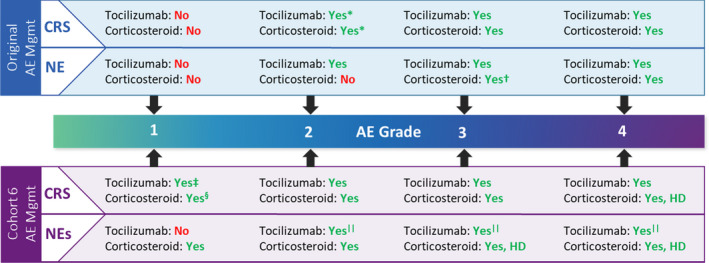
Toxicity management in ZUMA‐1. *Only in case of comorbidities or older age. †Only if no improvement with tocilizumab, use standard dose. ‡If no improvement after 24 h. §If no improvement after three days. ||Only for grade 2 or higher NEs with concurrent CRS. AE, adverse event; CRS, cytokine release syndrome; HD, high dose; Mgmt, management; NE, neurologic event. [Colour figure can be viewed at wileyonlinelibrary.com]
Patients
In cohort 6, eligible patients had histologically confirmed R/R LBCL after two or more lines of systemic therapy, were refractory to first‐line therapy [defined as best response of progressive disease (PD) or stable disease (SD) to four or more cycles of first‐line therapy with SD duration at most six months], or had PD or relapsed ≤12 months after autologous stem cell transplant (SCT). Additional inclusion/exclusion criteria are available in Data S1. All patients provided written informed consent. The study was approved by the institutional review board at each site and was conducted in accordance with the Good Clinical Practice guidelines of the International Conference on Harmonization.
Treatment
Bridging therapy was allowed per investigator discretion (e.g. patients with bulky or rapidly progressing disease at baseline) after leukapheresis (Data S1). Patients received a conditioning regimen of cyclophosphamide (500 mg/m2/d) and fludarabine (30 mg/m2/d) on days − 5 to − 3 and 1 dose of axi‐cel (target dose, 2 × 106 CAR‐T cells/kg; minimum dose, 1 × 106 CAR‐T cells/kg; maximum flat dose for patients >100 kg, 2 × 108 CAR‐T cells) on day 0. Patients received once‐daily oral dexamethasone 10 mg on days 0 (before axi‐cel), 1 and 2. Starting on day 0, patients received levetiracetam (750 mg orally or intravenously twice daily). If no grade 2 or higher NE occurred, the levetiracetam dose was tapered and discontinued as clinically indicated per investigator discretion. If previously discontinued, levetiracetam was reinitiated at the onset of subsequent grade 2 or higher NEs.
Corticosteroid therapy was used to manage all grade ≥1 NEs, all grade 2 or higher CRS and grade 1 CRS if there was no improvement after three days (Fig 1; Table SII). Tocilizumab was used to manage grade 2 or higher NEs with concurrent CRS, all grade 2 or higher CRS, and grade 1 CRS if there was no improvement after 24 h.
End‐points and analyses
No formal hypothesis was tested; all end‐points were analyzed descriptively. The primary end‐points were the incidence and severity of CRS and NEs. CRS was defined and graded per modified Lee 2014 criteria.11 NEs were identified by specific search strategy per Topp et al 14 and graded for severity per Common Terminology Criteria for Adverse Events version 4.03.15 Secondary end‐points included investigator‐assessed ORR (CR plus partial response) per revised International Working Group Response Criteria for Malignant Lymphoma,16 duration of response (DOR), progression‐free survival (PFS), overall survival (OS), incidence of AEs and levels of anti‐CD19 CAR‐T cells and cytokines in blood (Data S1). Confidence intervals (CIs) for ORRs were generated by the Clopper‐Pearson method, and CIs and landmark estimates of DOR, PFS and OS were generated by Kaplan–Meier (KM) survival method. For patients who underwent SCT before documented progression, DOR and PFS were censored at SCT date. Exploratory end‐points included biomarker assessments (Data S1).
The modified intent‐to‐treat (mITT) population, comprising all enrolled patients treated with axi‐cel at a dose of ≥1 × 106 CAR‐T cells/kg, was used for response‐based end‐points. All patients treated with any dose of axi‐cel were included in the safety analysis set. Tumour burden was measured by sum of product diameters of target lesions.16 For patients who received bridging therapy (excluding corticosteroids alone), baseline tumour burden was measured after bridging therapy, but before conditioning therapy.
Propensity score matching (PSM) analysis
Exploratory retrospective PSM analysis17, 18 was performed to descriptively compare results for cohort 6 and cohorts 1 + 2 (primary analysis)4 after balancing for the following key baseline disease characteristics: tumour burden, International Prognostic Index score, number of prior lines of chemotherapy, disease stage and lactate dehydrogenase level (Data S1).
Results
Patient disposition and baseline and product characteristics
Beginning on 1 May 2019, 62 patients were screened; 42 were enrolled and leukapheresed (Figure S1). Of 42 enrolled patients, 40 (95%) received conditioning chemotherapy and axi‐cel. The manufacturing success rate was 98% (n = 41/42). Two patients did not receive axi‐cel due to product not available and sponsor decision (n = 1 each). The 40 axi‐cel‐treated patients comprised both the mITT and safety analysis populations. Twenty‐one patients (53%) received bridging therapy before axi‐cel, most commonly corticosteroids (23%), bendamustine/rituximab plus corticosteroids (10%) and bendamustine/rituximab alone (8%; Table SIII). Median tumour burden was not significantly reduced with bridging therapy. One patient did not receive corticosteroid prophylaxis per protocol on days 1 and 2 because of site error. The data cut‐off date was 16 June 2020; the median follow‐up was 8·9 months (range, 6·0–12·1 months).
The median patient age was 64·5 years (range, 37–85 years); 65% of patients had stage III or IV disease, 38% had three or more prior therapies and 43% had PD as best response to most recent chemotherapy (Table I). Pre‐infusion product characteristics (total number of T cells, per cent transduction, IFN‐γ production and T‐cell phenotypes) are shown in Table SIV.
Table I.
Patient and disease characteristics at baseline.
| Characteristic | Cohort 6 (n = 40) |
|---|---|
| Age | |
| Median (range), years | 64·5 (37–85) |
| ≥65 years, n (%) | 20 (50) |
| Male sex, n (%) | 23 (58) |
| ECOG performance status score of 1, n (%) | 22 (55) |
| Disease stage, n (%) | |
| I or II | 14 (35) |
| III or IV | 26 (65) |
| IPI score, n (%) | |
| 0–2 | 22 (55) |
| 3–4 | 18 (45) |
| Number of prior lines of chemotherapy, n % | |
| 1 | 2 (5) |
| 2 | 23 (58) |
| 3 | 12 (30) |
| 4 | 2 (5) |
| ≥5 | 1 (3) |
| Prior autologous SCT, n (%) | 10 (25) |
| PD as best response to most recent chemotherapy, n (%)* | 17 (43) |
| Median (range) tumour burden by SPD,† mm2 | 1 184 (116–17 057) |
| Median (range) LDH, U/l | 236 (155–2 042) |
| Median (range) ferritin, ng/ml | 364 (13–1 748) |
| Refractory subgroup, n (%) | |
| Primary refractory | 2 (5) |
| Refractory ≥ second‐line therapy | 23 (58) |
| Relapsed ≥ second‐line therapy | 7 (18) |
| Relapsed post‐ASCT | 8 (20) |
ASCT, autologous stem cell transplant; ECOG, Eastern Cooperative Oncology Group; IPI, International Prognostic Index; LDH, lactate dehydrogenase; PD, progressive disease; SCT, stem cell transplant; SPD, sum of the products of diameters.
For patients who had not relapsed post‐ASCT.
Restaged after bridging, except for two patients without post‐bridging assessment in which case the screening assessments were used for baseline.
Safety
AEs occurred in all treated patients. The most frequent any‐grade AEs were pyrexia (85%), hypotension (55%) and neutropenia (50%; Table II). Grade 3 or higher AEs were reported in all treated patients — the most frequent were neutropenia (45%), neutrophil count decreased (33%), anaemia (20%) and white blood cell count decreased (20%). Three patients (8%) had fatal treatment‐emergent AEs: one respiratory failure due to ongoing respiratory infection (day 91; related to axi‐cel), one urosepsis (day 107; unrelated to axi‐cel) and one unknown AE, identified per public records that occurred after the patient withdrew consent to undergo autologous SCT. Prolonged grade 3 or higher cytopenias present on or after day 30 post‐axi‐cel infusion were reported in 18 patients (45%): neutropenia (30%; 0% febrile), thrombocytopenia (20%) and anaemia (10%; Table SV). Overall, 50% of patients experienced infections, with median time to onset of 42·5 days (range, 3–219 days) post‐axi‐cel infusion; 20% of patients had grade 3 or higher infections.
Table II.
Treatment‐emergent adverse events of any grade occurring in ≥15% of patients and grade 3 or higher adverse events occurring in >10% of patients.
| n (%) | Any grade | Worst grade 3 | Worst grade 4 |
|---|---|---|---|
| Any | 40 (100) | 10 (25) | 26 (65) |
| Pyrexia | 34 (85) | 5 (13) | 0 |
| Hypotension | 22 (55) | 5 (13) | 0 |
| Neutropenia | 20 (50) | 3 (8) | 15 (38) |
| Fatigue | 18 (45) | 1 (3) | 0 |
| Confusional state | 15 (38) | 1 (3) | 0 |
| Constipation | 15 (38) | 0 | 0 |
| Nausea | 14 (35) | 1 (3) | 0 |
| Anaemia | 13 (33) | 8 (20) | 0 |
| Headache | 13 (33) | 0 | 0 |
| Neutrophil count decreased | 13 (33) | 4 (10) | 9 (23) |
| Diarrhoea | 11 (28) | 1 (3) | 0 |
| Hypokalaemia | 11 (28) | 2 (5) | 0 |
| Hypophosphataemia | 11 (28) | 6 (15) | 0 |
| Thrombocytopenia | 10 (25) | 5 (13) | 2 (5) |
| Tremor | 9 (23) | 1 (3) | 0 |
| Chills | 8 (20) | 0 | 0 |
| Decreased appetite | 8 (20) | 0 | 0 |
| Hypoxia | 8 (20) | 2 (5) | 1 (3) |
| Vomiting | 8 (20) | 1 (3) | 0 |
| White blood cell count decreased | 8 (20) | 2 (5) | 6 (15) |
| Arthralgia | 7 (18) | 0 | 0 |
| Dyspnoea | 7 (18) | 1 (3) | 0 |
| Leukopenia | 7 (18) | 1 (3) | 5 (13) |
| Aphasia | 6 (15) | 1 (3) | 0 |
| Cough | 6 (15) | 0 | 0 |
| Dizziness | 6 (15) | 0 | 0 |
| Hypogammaglobulinaemia | 6 (15) | 0 | 0 |
| Hyponatraemia | 6 (15) | 2 (5) | 0 |
| Insomnia | 6 (15) | 0 | 0 |
| Muscular weakness | 6 (15) | 1 (3) | 0 |
| Platelet count decreased | 6 (15) | 0 | 4 (10) |
| Somnolence | 6 (15) | 0 | 0 |
| Tachycardia | 6 (15) | 1 (3) | 0 |
CRS occurred in 32 patients (80%); all cases were grade 1 or 2 (Table III). The most common CRS symptoms among patients with CRS were pyrexia (97%), hypotension (53%) and hypoxia (19%). Median time to CRS onset was 5·0 days (range, 1–15 days); median duration was 4·0 days (range, 1–11 days). All CRS‐related events had resolved by the data cut‐off.
Table III.
Incidence, severity, onset, and duration of CRS and NEs.
| TEAE | Cohort 6 (n = 40) |
|---|---|
| CRS | |
| Any, n (%) | 32 (80) |
| Worst grade 1, n (%) | 14 (35) |
| Worst grade 2, n (%) | 18 (45) |
| Worst grade 3, n (%) | 0 |
| Worst grade 4, n (%) | 0 |
| Worst grade 5, n (%) | 0 |
| Median (range) time to onset of any‐grade CRS, days | 5·0 (1–15) |
| Median (range) duration, days | 4·0 (1–11) |
| Neurologic events | |
| Any, n (%) | 23 (58) |
| Worst grade 1, n (%) | 10 (25) |
| Worst grade 2, n (%) | 8 (20) |
| Worst grade 3, n (%) | 3 (8) |
| Worst grade 4, n (%) | 2 (5) |
| Worst grade 5, n (%) | 0 |
| Median (range) time to onset of any‐grade NE, days | 6·0 (2–162) |
| Median (range) duration, days | 18·5 (1–103) |
CRS, cytokine release syndrome; NE, neurologic event; TEAE, treatment‐emergent adverse event.
Twenty‐three patients (58%) experienced NEs, including three patients (8%) with worst‐grade 3 NEs and two patients (5%) with worst‐grade 4 NEs. The most frequent any‐grade NE symptoms among patients with NEs were confusional state (38%), tremor (23%) and aphasia and somnolence (each 15%). Among grade 3 or higher NEs, the most common symptoms among patients were seizure (8%; two of three patients were receiving levetiracetam prophylaxis at the time) and mental status change (5%). Median time to NE onset was 6·0 days (range, 2–162 days); median duration was 18·5 days (range, 1–103 days). At data cut‐off, NEs had resolved in 20 of 23 patients (Table SVI). Notably, 68% of patients (n = 27/40) did not experience either NEs or CRS within 72 h (days 0–3) of axi‐cel infusion.
Excluding prophylaxis, corticosteroids were used to treat CRS, NEs and other events in 17 patients (43%), 16 patients (40%) and 1 (3%) patient respectively. The median cumulative cortisone‐equivalent corticosteroid dose required to treat CRS or NEs was 1 878 mg (n = 24), with median time to onset of 5·0 days (range, 3–9 days) post‐infusion. The median cumulative cortisone‐equivalent corticosteroid dose was 1 252 mg including prophylaxis (n = 40) and 2 504 mg excluding prophylaxis (n = 25; Table SVII). As 15 patients received corticosteroids only as prophylaxis and no additional corticosteroids for AE management, the median cumulative dose shifted to a smaller value when prophylactic doses were included versus excluded. Tocilizumab (≥1 dose) was used to treat CRS in 23 patients (58%) and NE with concurrent grade 2 CRS in one patient (3%).
Efficacy
The investigator‐assessed ORR was 95% (95% CI, 83–99%), and the CR rate was 80% (95% CI, 64–91%; Fig 2A). ORR and CR rates were 100% and 73%, respectively, in the 15 patients who received corticosteroid prophylaxis only and were 92% and 84%, respectively, in the 25 patients who received corticosteroids for prophylaxis and toxicity management.
Fig 2.
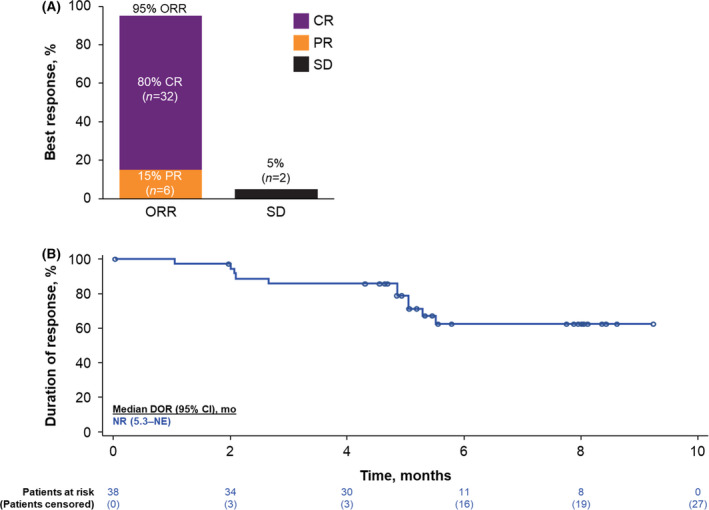
(A) Objective response rates (ORRs) and (B) duration of response (DOR) in patients achieving an objective response in ZUMA‐1 cohort 6. For patients who underwent stem cell transplantation before documented progression, DOR was censored at the date of stem cell transplantation. CR, complete response; NE, not estimable; NR, not reported; PR, partial response; SD, stable disease. [Colour figure can be viewed at wileyonlinelibrary.com]
Median DOR [KM DOR median follow‐up, 5·8 months (95% CI, 5·1–8·0 months)] was not reached; the KM estimate of the six‐month DOR rate was 62·4% (95% CI, 41·6–77·6%; Fig 2B). As of the data cut‐off date, 62·5% of treated patients remained in ongoing response. Median PFS and OS were not reached (Figure S2). KM estimates of the six‐month PFS and OS rates were 72·2% (95% CI, 54·1–84·1%) and 87·3% (95% CI, 72·1–94·5%) respectively. One patient proceeded to SCT post‐axi‐cel.
Biomarker analyses
Median peak CAR‐T cell expansion was observed within 2 weeks post‐axi‐cel infusion (64·4 cells/µl blood; Fig 3A). Median levels of inflammatory serum biomarkers previously shown to be associated with severe NEs and/or CRS4 — such as IFN‐γ, IL‐2, GM‐CSF, and ferritin — peaked within 8 days post‐axi‐cel infusion (Fig 3B).
Fig 3.
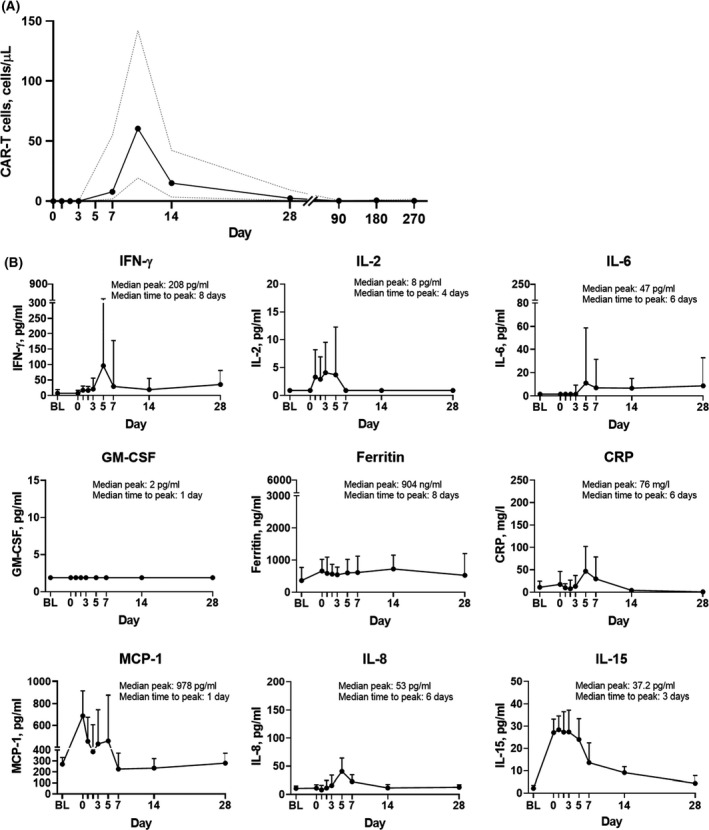
Levels of (A) blood chimaeric antigen receptor (CAR)‐T cells and (B) key soluble serum biomarkers over time. Dashed lines (A) and bars (B) represent the interquartile range. BL, baseline; CRP, C‐reactive protein; GM‐CSF, granulocyte‐macrophage colony‐stimulating factor; IFN, interferon; IL, interleukin; MCP‐1, monocyte chemoattractant protein 1.
PSM analysis
The incidence of grade 3 or higher CRS and grade 3 or higher NEs observed in cohort 6 (0% and 13% respectively) was numerically lower compared with cohorts 1 + 2 (13% and 28% respectively).4 However, cohort 6 was not designed to provide statistical comparison with cohorts 1 + 2, and the patients in these cohorts were not balanced with respect to key baseline characteristics. To verify the primary analysis findings, PSM was performed using known predictive and prognostic markers for clinical outcomes in LBCL and with CAR‐T cell therapy (cohorts 1 + 2 and other studies).4, 9, 19, 20, 21 Standardised mean difference18, 22 values demonstrated that covariates were appropriately balanced after PSM (Table SVIII). Baseline disease characteristics were comparable between 32 matched patients each in cohort 6 and cohorts 1 + 2 (Table SIX). Eight patients in cohort 6 were excluded due to non‐availability of matched patients in cohorts 1 + 2 using a caliper criterion during PSM. Efficacy and safety outcomes, CAR‐T cell levels and soluble serum biomarkers are summarised in Table SX. Differences in incidence of grade 3 or higher CRS and time to onset of CRS observed between patients in cohorts 1 + 2 and cohort 6 before PSM were maintained after PSM. The incidence, severity and onset of NEs were generally similar between cohort 6 and cohorts 1 + 2 following PSM. Clinical efficacy (ongoing response rates) in cohort 6 remained comparable to that observed in cohorts 1 + 2 before and after PSM and was corroborated by lower levels of soluble inflammatory biomarkers and comparable peak CAR‐T cell levels versus those in cohorts 1 + 2 before and after PSM. The median cumulative cortisone‐equivalent corticosteroid dose required to manage CRS or NEs was lower in cohort 6 (1878 mg) than in matched cohorts 1 + 2 (7418 mg).
Discussion
Optimal CRS and NE management in patients treated with CAR‐T cell therapy remains a subject of ongoing research,5, 6, 8, 23, 24 and the effects of corticosteroid use on outcomes remain unclear in retrospective analyses.25, 26 Relative to the initial management scheme used in ZUMA‐1 cohorts 1 + 2,3, 4 ZUMA‐1 cohort 4 explored earlier corticosteroid and/or tocilizumab intervention.13 The present cohort 6 study investigated the effects of prophylactic corticosteroid use and early corticosteroid and/or tocilizumab intervention on the incidence of CRS and NEs in patients with R/R LBCL.
In the absence of randomised data, the cohort 6 results suggest that prophylactic corticosteroids and early corticosteroid and/or tocilizumab intervention may benefit axi‐cel‐treated patients. Foremost, incidence rates of grade 3 or higher CRS were 13%, 2% and 0% in cohorts 1 + 2, 4 and 6 respectively.4, 13 The fact that no cases of grade 3 or higher CRS occurred among patients treated with prophylactic corticosteroids is encouraging, as is the progressively shorter median CRS duration (8, 7 and 4 days, in cohorts 1 + 2, 4 and 6 respectively) and delayed time to CRS onset (median 2, 2 and 5 days, in cohorts 1 + 2, 4 and 6 respectively).4, 13 Moreover, the incidence of grade 3 or higher NEs in cohort 6 (13%) was comparable to that of cohort 4 (17%), and both rates were numerically lower than in cohorts 1 + 2 (28%). However, the median duration of NEs in cohort 6 was numerically longer than that previously reported in cohorts 4 and 1 + 2 (18·5, 8 and 13 days respectively). Of note, n =27/40 patients (68%) in cohort 6 did not experience either CRS or NEs within 72 h (days 0–3) of axi‐cel infusion; these results pave the way for optimisation of corticosteroid utilisation, which may increase the proportion of patients that can be managed in an outpatient setting early post‐axi‐cel infusion.
Corticosteroid prophylaxis and early corticosteroid and/or tocilizumab intervention did not appear to compromise axi‐cel efficacy. At a median study follow‐up of 8·9 months, the ORR (95%) and CR rates (80%) were higher than corresponding rates in the primary analyses of cohorts 1 + 2 (82% and 54% respectively4) or 4 (73% and 51% respectively13). With longer follow‐up for cohorts 1 + 2 (median 27·1 months), the ORR was 83%, the CR rate was 58%, the median PFS was 5·9 months, and the median OS had not yet been reached.3 In a subsequent follow‐up (median 51·1 months) analysis, median OS was 25·8 months and the KM estimate of the four‐year OS rate was 44%.27 Additional follow‐up of cohort 6 patients will confirm if prophylactic corticosteroids improve long‐term safety without compromising durability of responses.
Notably, prolonged use of high‐dose corticosteroids for NE treatment is hypothesised to affect CAR‐T cell expansion and therefore clinical outcomes of treated patients.28 Although glucocorticoids induce lymphocyte apoptosis,29 and immunosuppressive agents may diminish CAR‐T cell expansion,30 no negative impact of corticosteroids on CAR‐T cell pharmacokinetics was noted in this study. However, the lower levels of soluble biomarkers, including inflammatory cytokines, observed in cohort 6 versus cohorts 1 + 2 suggest that systemic corticosteroids may exert an immunomodulatory effect that reduces immune cell‐related inflammation more than CAR‐T cell expansion.31 Notably, the median cumulative cortisone‐equivalent corticosteroid dose required to treat CRS or NEs was numerically lower in cohort 6 (1878 mg) versus cohorts 1 + 2 (7418 mg). Overall, prophylactic corticosteroids and earlier corticosteroid and/or tocilizumab intervention resulted in a lower cumulative corticosteroid dose (overall and to treat CRS or NEs) without negatively affecting CAR‐T cell pharmacokinetics or efficacy (ongoing response rates).
Because the safety management cohorts were not designed or powered for statistical comparisons with each other or with the pivotal cohorts, differences in baseline characteristics and cohort sizes should be considered when making comparisons. To overcome these limitations and reduce bias in the absence of a randomised trial, PSM17, 18 was applied to cohorts 1 + 2 and cohort 6. This statistical method adjusts for potential imbalances in baseline disease characteristics between cohorts, thereby providing a more balanced and robust comparison.18, 32 The aforementioned differences in CRS toxicity outcomes observed between patients in cohort 6 and cohorts 1 + 2 before and after PSM, together with generally similar NE toxicity after PSM, suggest a benefit of early AE intervention and corticosteroid prophylaxis. Furthermore, response rates in cohort 6 were comparable to those observed in cohorts 1 + 2 before and after PSM and were corroborated by CAR‐T cell pharmacokinetics.
In summary, the use of prophylactic corticosteroids and earlier corticosteroid and/or tocilizumab intervention for toxicity management resulted in no cases of grade 3 or higher CRS, delayed CRS onset, and generally similar NE toxicity without adversely affecting CAR‐T cell pharmacokinetics or efficacy outcomes for axi‐cel‐treated patients.
Data sharing statement
Gilead is committed to sharing clinical trial data with external medical experts and scientific researchers in the interest of advancing public health. As such, Gilead shares anonymised individual patient data (IPD) upon request or as required by law and/or regulation. Qualified external researchers may request IPD for studies of Gilead compounds approved in the United States and the European Union with a marketing authorisation date on or after 1 January 2014 and are publicly listed on clinicaltrials.gov or the European Union‐Clinical Trials Register (EU CTR). For studies of newly approved compounds or indication, the IPD will be available for request six months after US Food and Drug Administration (FDA) and European Medicines Agency (EMA) approval. Such requests are at Gilead’s discretion and are dependent on the nature of the request, the merit of the research proposed, availability of the data, and the intended use of the data. If Gilead agrees to the release of clinical data for research purposes, the requestor will be required to sign a data sharing agreement, to ensure protection of patient confidentiality before the release of any data.
Funding information
The study was supported by Kite, a Gilead Company.
Author contributions
OOO, JJK and TvM designed the study; all authors provided study material or patients, collected or assembled data, participated in drafting and revising the manuscript, approved the final version of the manuscript, and are accountable for all aspects of this work.
Conflict of interest
OOO: consultancy or advisory role for Kite, a Gilead Company, Pfizer, Spectrum Pharmaceuticals, Legend, and Bayer. KB: honoraria from Kite, a Gilead Company, Takeda, Roche, and Celgene; consultancy or advisory role for Kite, a Gilead Company, Takeda, Roche, and Sandoz; travel support from Roche. JM: honoraria from Kyowa and Seattle Genetics; consultancy or advisory role for Pharmacyclics, Bayer, Kite, a Gilead Company, Pfizer, Janssen, Juno/Celgene, Bristol‐Myers Squibb, Kyowa, Alexion, Fosun Kite, Innovent, Seattle Genetics, and BeiGene; speakers’ bureau participation for Kite, a Gilead Company, Kyowa, Bayer, Pharmacyclics/Janssen, Seattle Genetics, Acrotech/Aurobindo, BeiGene, Verastem, AstraZeneca, Juno/Celgene/Bristol Myers Squibb, Genentech/Roche, and AbbVie; research funding from Bayer, Kite, a Gilead Company, Juno/Celgene/Bristol‐Myers Squibb, Merck, Portola, Incyte, Genentech, Pharmacyclics, Seattle Genetics, Janssen, and Millennium. SdG: honoraria from Gilead Sciences, AbbVie, and Janssen; consultancy or advisory role for Gilead Sciences, AbbVie, and Janssen. JMV: honoraria from Kite, a Gilead Company, AbbVie, Epizyme, Janssen, AstraZeneca, Verastem, Miltenyi, Loxo, Allogene, Wugen, Celgene, Roche, Genentech, and Karyopharm; consultancy or advisory role for AbbVie, Janssen, Kite, a Gilead Company, AstraZeneca, Verastem, Miltenyi, Loxo, Allogene, Wugen, Roche, Genentech, Karyopharm, and Morphosis; research funding from AstraZeneca, Bristol‐Myers Squibb, Incyte, Kite, a Gilead Company, Novartis, Seattle Genetics, Loxo, and Epizyme; travel support from Roche Pakistan. NLB: consultancy or advisory role for ADC Therapeutics, Roche/Genentech, and Seattle Genetics; research funding from ADC Therapeutics, Autolus, Bristol‐Myers Squibb, Celgene, Forty Seven, Genentech, Janssen, Kite, a Gilead Company, Merck, Millennium, Pharmacyclics, and Seattle Genetics. YL: consultancy or advisory role for Kite, a Gilead Company, Janssen, Novartis, Celgene, Bluebird Bio, Juno, Legend, Sorrento, Gamida Cell, and Vineti; research funding from Kite, a Gilead Company, Janssen, Celgene, Bluebird Bio, Merck, and Takeda. AD: consultancy or advisory role for Janssen and Kite, a Gilead Company. PAM: employment with Colorado Blood Cancer Institute Medical Group; consultancy or advisory role for, speakers' bureau participation for, and research funding from Kite, a Gilead Company. AHG: employment with Regional Cancer Care Associates/OMI; leadership role at and stock or other ownership in Cancer Outcome Tracking Analysis (COTA); consultancy or advisory role for AstraZeneca, Celgene, Gilead Sciences, Kite, a Gilead Company, and Xcenda; research funding from Acerta, AstraZeneca, Celgene, Constellation, Genentech‐Hoffmann La Roche, Infinity, and Infinity Verastem. MJK: honoraria from, consultancy or advisory role for, and travel support from Kite, a Gilead Company, Novartis, and Miltenyi; research funding from Roche, Takeda, and Celgene. CAJ: honoraria from Kite, a Gilead Company, Celgene, Novartis, Pfizer, Precision Biosciences, Nkarta, Lonza, and AbbVie; consultancy or advisory role for and travel support from Kite, a Gilead Company, Celgene, Novartis, Bristol‐Myers Squibb, Precision Biosciences, Nkarta, and Lonza; speakers' bureau participation for Axis and Clinical Care Options; research funding from Pfizer. UF: honoraria from Kite, a Gilead Company. MCM: consultancy or advisory role for Gilead Sciences, Janssen Cilag, and Servier; research funding and travel support from Celgene. CT: honoraria from Gilead Sciences, Novartis, Celgene, Roche, and Janssen; consultancy or advisory role for Celgene, Roche, Novartis, and Janssen; research funding from Roche; travel support from Novartis, Roche, Gilead Sciences, and Janssen. JMT: stock or other ownership in Genmab, Corvus, Marker Therapeutics, and Bluebird Bio; consultancy or advisory role for Kite, a Gilead Company, Celgene, Immune Design, and Celldex Therapeutics; research funding from Bristol‐Myers Squibb, Kite, a Gilead Company, Spectrum Pharmaceuticals, and Merck; travel support from Bristol‐Myers Squibb and Kite, a Gilead Company. PS: honoraria from and consultancy or advisory role for MorphoSys, Karyopharm, and Crispr; researching funding from Kite, a Gilead Company, Incyte, Amgen, Gamida Cell, Macrogenics, Cellectar, and Bristol‐Myers Squibb. IA: no relevant financial relationships to disclose. DT: consultancy or advisory role for Partner, Takeda, EUSA, Kite, a Gilead Company, Kyowa Kirin, and Magenta; speakers' bureau participation for Takeda and Kite, a Gilead Company; research funding from Bristol‐Myers Squibb, Kite, a Gilead Company, Genentech, Incyte, and Fate. JJK, JM, YZ, and LG: employment with Kite, a Gilead Company; stock or other ownership in Gilead Sciences. ZB: employment with Kite, a Gilead Company; stock or other ownership in OmniacPharmConsult Ltd. JMR: former employment with Kite, a Gilead Company. LJ: employment with Kite, a Gilead Company, and Trillium Therapeutics; stock or other ownership in and patents, royalties, or other intellectual property from Trillium Therapeutics. TvM: honoraria from Celgene; consultancy or advisory role for Kite, a Gilead Company.
Supporting information
Data S1. Supplementary appendix.
Table SI. Summary of key similarities and differences between ZUMA‐1 phase 2 cohorts 1 + 2, 4, and 6.
Table SII. Guidelines for tocilizumab and corticosteroid management of CRS and NE. Does not include prophylactic corticosteroid use, which was per protocol for all patients.
Table SIII. Summary of bridging regimens.
Table SIV. Summary of product characteristics.
Table SV. Summary of cytopenias present on or after day 30 after axi‐cel infusion.
Table SVI. Unresolved neurologic events at data cut‐off.
Table SVII. Summary of corticosteroid use, excluding prophylaxis.
Table SVIII. Standardised mean difference between patients in cohorts 1 + 2 and cohort 6 before and after propensity score matching.
Table SIX. Comparison of baseline and product characteristics between patients in cohorts 1 + 2 and cohort 6 before and after propensity score matching.
Table SX. Comparison of efficacy and safety outcomes and CAR‐T cell and soluble serum biomarker levels between patients in cohorts 1 + 2 and cohort 6 before and after propensity score matching.
Fig S1. Patient disposition.
Fig S2. Investigator‐assessed (A) progression‐free survival and (B) overall survival in the modified intent‐to‐treat patient population (n = 40).
Acknowledgements
The authors thank all investigators, coordinators, study site personnel and, especially, the patients and their families for participating in this study; Lianqing Zheng, PhD, and Rong Chu, PhD — both formerly of Kite, a Gilead Company — for their statistical contributions; and Anne Kerber, MD, and Remus Vezan, MD, PhD — of and formerly of, respectively, Kite, a Gilead Company — for their contributions to the study design. Editorial and writing assistance was provided by Robert Rydzewski, CMPP, Skye Geherin, PhD, and Ashley Skorusa, PhD, of Nexus Global Group Science, a Vaniam Group Company, with funding provided by Kite, a Gilead Company.
References
- 1.YESCARTA® (axicabtagene ciloleucel) [summary of product characteristics]. Amsterdam, the Netherlands: Kite Pharma EU B.V.; 2018. [Google Scholar]
- 2.YESCARTA® (axicabtagene ciloleucel) Prescribing information. Kite Pharma, Inc; 2021. [Google Scholar]
- 3.Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long‐term safety and activity of axicabtagene ciloleucel in refractory large B‐cell lymphoma (ZUMA‐1): a single‐arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019;20(1):31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T‐cell therapy in refractory large B‐cell lymphoma. N Engl J Med. 2017;377(26):2531–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garcia Borrega J, Gödel P, Rüger MA, Onur ÖA, Shimabukuro‐Vornhagen A, Kochanek M, et al. In the eye of the storm: immune‐mediated toxicities associated with CAR‐T cell therapy. Hemasphere. 2019;3(2):e191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neelapu SS. Managing the toxicities of CAR T‐cell therapy. Hematol Oncol. 2019;37(Suppl 1):48–52. [DOI] [PubMed] [Google Scholar]
- 7.Shimabukuro‐Vornhagen A, Gödel P, Subklewe M, Stemmler HJ, Schlößer HA, Schlaak M, et al. Cytokine release syndrome. J Immunother Cancer. 2018;6(1):56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Santomasso B, Bachier C, Westin J, Rezvani K, Shpall EJ. The other side of CAR T‐cell therapy: cytokine release syndrome, neurologic toxicity, and financial burden. Am Soc Clin Oncol Educ Book. 2019;39:433–44. [DOI] [PubMed] [Google Scholar]
- 9.Locke FL, Rossi JM, Neelapu SS, Jacobson CA, Miklos DB, Ghobadi A, et al. Tumor burden, inflammation, and product attributes determine outcomes of axicabtagene ciloleucel in large B‐cell lymphoma. Blood Adv. 2020;4(19):4898–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gust J, Taraseviciute A, Turtle CJ. Neurotoxicity associated with CD19‐targeted CAR‐T cell therapies. CNS Drugs. 2018;32(12):1091–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Locke FL, Neelapu SS, Bartlett NL, Lekakis LJ, Jacobson CA, Braunschweig I, et al. Preliminary results of prophylactic tocilizumab after axicabtagene ciloleucel (axi‐cel; KTE‐C19) treatment for patients with refractory, aggressive non‐Hodgkin lymphoma (NHL). Blood. 2017;130(Suppl 1):1547. [Google Scholar]
- 13.Topp M, Van Meerten T, Houot R, Minnema MC, Milpied N, Lugtenburg PJ, et al. Earlier steroid use with axicabtagene ciloleucel (Axi‐Cel) in patients with relapsed/refractory large B cell lymphoma. Blood. 2019;134(Suppl 1):243. [Google Scholar]
- 14.Topp MS, Gökbuget N, Stein AS, Zugmaier G, O'Brien S, Bargou RC, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B‐precursor acute lymphoblastic leukaemia: a multicentre, single‐arm, phase 2 study. Lancet Oncol. 2015;16(1):57–66. [DOI] [PubMed] [Google Scholar]
- 15.U.S. Department of Health and Human Services . Common Terminology Criteria for Adverse Events (CTCAE) Version 4.03. 2010. National Institutes of Health National Cancer Institute. [Google Scholar]
- 16.Cheson BD, Pfistner B, Juweid ME, Gascoyne RD, Specht L, Horning SJ, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25(5):579–86. [DOI] [PubMed] [Google Scholar]
- 17.Rosenbaum PR, Rubin DB. The central role of the propensity score in observational studies for causal effects. Biometrika. 1983;70(1):41–55. [Google Scholar]
- 18.Austin PC. An introduction to propensity score methods for reducing the effects of confounding in observational studies. Multivariate Behav Res. 2011;46(3):399–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buecklein V, Blumenberg V, Ackermann J, Schmidt C, Rejeski K, Mueller N, et al. Single‐center experience with axicabtagene‐ciloleucel (axi‐cel) and tisagenlecleucel (tisa‐cel) for relapsed/refractory diffuse large B‐cell lymphoma: comparable response rates and manageable toxicity. Blood. 2020;136(Suppl 1):34–5. [Google Scholar]
- 20.Vercellino L, Di Blasi R, Kanoun S, Tessoulin B, Rossi C, D'Aveni‐Piney M, et al. Predictive factors of early progression after CAR T‐cell therapy in relapsed/refractory diffuse large B‐cell lymphoma. Blood Adv. 2020;4(22):5607–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.International Non‐Hodgkin’s Lymphoma Prognostic Factors Project . A predictive model for aggressive non‐Hodgkin's lymphoma. N Engl J Med. 1993;329(14):987–94. [DOI] [PubMed] [Google Scholar]
- 22.Austin PC. A critical appraisal of propensity‐score matching in the medical literature between 1996 and 2003. Stat Med. 2008;27(12):2037–49. [DOI] [PubMed] [Google Scholar]
- 23.Freyer CW, Porter DL. Cytokine release syndrome and neurotoxicity following CAR T‐cell therapy for hematologic malignancies. J Allergy Clin Immunol. 2020;146(5):940–8. [DOI] [PubMed] [Google Scholar]
- 24.Riegler LL, Jones GP, Lee DW. Current approaches in the grading and management of cytokine release syndrome after chimeric antigen receptor T‐cell therapy. Ther Clin Risk Manag. 2019;15:323–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nastoupil LJ, Jain MD, Feng L, Spiegel JY, Ghobadi A, Lin YI, et al. Standard‐of‐care axicabtagene ciloleucel for relapsed or refractory large B‐cell lymphoma: results from the US Lymphoma CAR T Consortium. J Clin Oncol. 2020;38(27):3119–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strati P, Ahmed S, Furqan F, Fayad LE, Lee HJ, Iyer SP, et al. Prognostic impact of corticosteroids on efficacy of chimeric antigen receptor T‐cell therapy in large B‐cell lymphoma. Blood. 2021. 10.1182/blood.2020008865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jacobson CA, Locke FL, Ghobadi A, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long‐term survival and gradual recovery of B cells in patients with refractory large B cell lymphoma treated with axicabtagene ciloleucel (axi‐cel). Blood. 2020;136(Suppl 1):40–2. [Google Scholar]
- 28.Strati P, Nastoupil LJ, Westin J, Fayad LE, Ahmed S, Fowler NH, et al. Clinical and radiologic correlates of neurotoxicity after axicabtagene ciloleucel in large B‐cell lymphoma. Blood Adv. 2020;4(16):3943–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmidt S, Rainer J, Ploner C, Presul E, Riml S, Kofler R. Glucocorticoid‐induced apoptosis and glucocorticoid resistance: molecular mechanisms and clinical relevance. Cell Death Differ. 2004;11(Suppl 1):S45–55. [DOI] [PubMed] [Google Scholar]
- 30.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu S, Deng B, Yin Z, Pan J, Lin Y, Ling Z, et al. Corticosteroids do not influence the efficacy and kinetics of CAR‐T cells for B‐cell acute lymphoblastic leukemia. Blood Cancer J. 2020;10(2):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Z, Kim HJ, Lonjon G, Zhu Y. Balance diagnostics after propensity score matching. Ann Transl Med. 2019;7(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplementary appendix.
Table SI. Summary of key similarities and differences between ZUMA‐1 phase 2 cohorts 1 + 2, 4, and 6.
Table SII. Guidelines for tocilizumab and corticosteroid management of CRS and NE. Does not include prophylactic corticosteroid use, which was per protocol for all patients.
Table SIII. Summary of bridging regimens.
Table SIV. Summary of product characteristics.
Table SV. Summary of cytopenias present on or after day 30 after axi‐cel infusion.
Table SVI. Unresolved neurologic events at data cut‐off.
Table SVII. Summary of corticosteroid use, excluding prophylaxis.
Table SVIII. Standardised mean difference between patients in cohorts 1 + 2 and cohort 6 before and after propensity score matching.
Table SIX. Comparison of baseline and product characteristics between patients in cohorts 1 + 2 and cohort 6 before and after propensity score matching.
Table SX. Comparison of efficacy and safety outcomes and CAR‐T cell and soluble serum biomarker levels between patients in cohorts 1 + 2 and cohort 6 before and after propensity score matching.
Fig S1. Patient disposition.
Fig S2. Investigator‐assessed (A) progression‐free survival and (B) overall survival in the modified intent‐to‐treat patient population (n = 40).