

Abstract
Rupintrivir targets the 3C cysteine proteases of the picornaviridae family which includes rhinoviruses and enteroviruses that cause a range of human diseases. Despite being a pan-3C protease inhibitor, rupintrivir activity is extremely weak against the homologous 3C-like protease of SARS-CoV-2. In this study the crystal structures of rupintrivir were determined bound to enterovirus 68 (EV68) 3C protease, and the 3C-like main protease (Mpro) from SARS-CoV-2. While the EV68 3C protease-rupintrivir structure was similar to previously determined complexes with other picornavirus 3C proteases, rupintrivir bound in a unique conformation to the active site of SARS-CoV-2 Mpro splitting the catalytic cysteine and histidine residues. This bifurcation of the catalytic dyad may provide a novel approach for inhibiting cysteine proteases.
Graphical Abstract
There are over 200 viral species that are known to infect humans, with three to four new human viral pathogens discovered every year 1 Viral outbreaks result in the death of hundreds to millions of people, as in the case of the coronavirus infectious disease 2019 (COVID-19) pandemic caused by SARS-CoV-2. Many of the most common human viral pathogens are from the picornaviridae family. This viral family includes rhinoviruses that cause the common cold, and enteroviruses associated with several diseases ranging from hand-foot-and-mouth disease to polio-like neuropathies. For example, the enterovirus EV68 has been linked to acute flaccid myelitis (AFM), affecting the gray matter of the spinal cord, leading to polio-like neurological symptoms such as muscle weakness. Between August 2014 and January 2020, an EV68 outbreak caused over 600 confirmed cases of AFM across 34 states 2. Unfortunately, many of the diseases caused by these viruses lack effective treatment options.
In the absence of vaccines or natural antibodies, direct acting antivirals (DAAs) are a critical therapeutic strategy to suppress viral outbreaks and treat the most vulnerable in the population. DAAs save millions of lives every year by targeting essential viral enzymes to prevent viral replication. Viral proteases, which cleave polyproteins to allow viral maturation, are key therapeutic targets. Inhibition of viral proteases with small molecule inhibitors has a proven therapeutic track record, as with HIV-1 and HCV 3–6, some of which have been shown to have potency against SARS-CoV-2 proteases 7–9. Rhinoviruses and enteroviruses have a 3C protease (3Cpro) that is necessary for viral maturation. The 3C proteases of various viral species are structurally very similar [Figure 1A]. SARS-CoV-2 has two viral proteases, the papain-like protease (PLpro) and the main protease (Mpro). Mpro is also known as the 3C-like protease because the active site domain is structurally similar to 3C proteases. SARS-CoV-2 Mpro is larger than the 3C proteases (306 AA vs ~180 AA) due to additional residues near the active site (residues 43–62) and a 100-residue domain III [Figure 1B]. The structural similarities especially in the active site suggest the potential of an effective cross-3C/3CLpro inhibitor.
Figure 1.

A) Rupintrivir in complex with viral 3C proteases of rhinovirus A, rhinovirus C, coxsackievirus, enterovirus 71, enterovirus 93, and novel enterovirus 68. The crystal structures are superimposed in cartoon representation with rupintrivir shown as green sticks (PDB: 1CQQ, 3RUO, 3SJI, 3SJO, 6KU8, 7L8H). B) Novel SARS-CoV-2 Mpro-rupintrivir crystal structure in cartoon representation with rupintrivir shown as spheres, protease residues 43–62 colored red, the domain that is structurally homologous to 3C proteases colored cyan, and domain III colored orange (PDB: 7L8I). C) Chemical structure of rupintrivir.
In an attempt to cure the common cold, Pfizer developed rupintrivir [Figure 1C], an irreversible covalent inhibitor designed to target the 3C cysteine protease of human rhinovirus B 10. Rupintrivir inhibits several rhinovirus serotypes and patient isolates with an EC50 between 3 and 183 nM11. In 2003, a phase 2 randomized, double-blind, placebo-controlled study showed that rupintrivir caused significant prophylaxis and daily symptom reduction12 and had good safety and pharmacokinetics in healthy volunteers13. Rupintrivir was later shown to potently inhibit the 3C proteases of several enterovirus strains with a low nanomolar EC50 11 14–15 with a similar binding mode [Figure 1A]. Despite their similarities however, rupintrivir only weakly inhibits the Mpro of SARS-CoV-1 and 2 with an IC50 of >100 μM and 68 ± 7 μM, respectively 16–17. However, the binding mode of rupintrivir underlying this potency loss was not clear as the structure of SARS-CoV-2 Mpro in complex with rupintrivir had not been determined.
In this study, we present the crystal structures of both EV68 3Cpro and SARS-CoV-2 Mpro with rupintrivir bound at the active site. EV68 3Cpro binds rupintrivir in the previously observed canonical binding mode, however in the SARS-CoV-2 Mpro complex rupintrivir adopts a unique binding mode. The SARS-CoV-2 Mpro-rupintrivir complex structure reveals how both the inhibitor and the protease active site undergo conformational changes to accommodate rupintrivir by bifurcating the catalytic dyad.
To elucidate the structure and binding mode of rupintrivir in inhibiting coronaviral Mpro, the crystal structure of rupintrivir in complex with SARS-CoV-2 Mpro was determined. We also determined the crystal structure of rupintrivir bound to enteroviral EV68 3Cpro for comparison. The inhibitor binding modes and catalytic dyad side chain conformations were validated by composite omit maps with and without simulated annealing [Figure S1]. Additional details regarding diffraction data and inhibitor modeling are provided in the materials and methods section and Table S2.
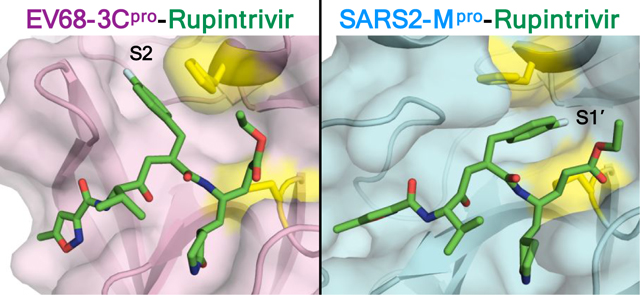
The EV68 3Cpro-rupintrivir complex structure was determined to 1.95 Å resolution (Rfactor/Rfree = 15.8/18.6%) in the space group R3 with two molecules in the asymmetric unit (AU). As expected, rupintrivir’s binding conformation in EV68 3Cpro active site is identical to all other previously published 3C proteases, with the P2 fluorophenylalanine located in the S2 subsite [Figure 2A]. Unlike some of the other 3Cpro-rupintrivir complexes18, our structure has the catalytic histidine (His40) in only one conformation. The P2 fluorophenylalanine fits in the hydrophobic channel [Figure 2B] and the fluorine participates in a halogen bonding interaction with Thr129 while pi-stacking with the catalytic His40 [Figure 2C]. Other hydrogen bonds between the inhibitor and protease include several interactions to the backbone atoms of Gly164 and Asn165 which likely interact with the cleavage sites [Figure 2D].
Figure 2.

A,E) Rupintrivir and catalytic residues in the crystal structures of enterovirus 68 3C protease (EV68 3Cpro) and SARS-CoV-2 Mpro (SARS2-Mpro), shown within the electron density. The 2Fo-Fc direct maps are depicted as grey mesh contoured at 1.0 σ while the Fo-Fc difference maps have positive density depicted as green mesh contoured at 3.0 σ and negative density as red mesh contoured at −3.0 σ. B,F) Binding mode of rupintrivir at the active site of proteases in the crystal structures. Rupintrivir shown as sticks and protease active site shown as cartoon with transparent surface. C,G) Residues involved in hydrogen bonding with rupintrivir are shown as sticks. D,H) Inter-molecular hydrogen bonds between rupintrivir and protease in EV68 3Cpro and SARS-CoV-2 Mpro crystal structures. PDB: 7L8H (left) and 7L8I (right).
The SARS-CoV-2 Mpro-rupintrivir complex structure was determined in two different space groups. The structure in the P21 space group had 2.10 Å resolution (Rfactor/Rfree = 20.6/26.3%) with two monomers in the AU while the structure in the P21212 space group was determined to 2.45 Å resolution (Rfactor/Rfree = 22.4/28.1%) with a single molecule in the AU. Contrary to all previous rupintrivir structures with 3C proteases, in complex with SARS-CoV-2 Mpro the P2 fluorophenylalanine moiety is intercalated between the catalytic residues (His41 and Cys145 in Mpro) pointing toward the S1’ subsite [Figure 2E–F] instead of binding in the S2 subsite. This was true for both monomers in the structure solved in the P21 space group, as well as in the structure solved in the P21212 space group, both of which were validated by omit maps [Figure S1]. Thus this unique conformation is present in all three complex structures determined. In this binding mode, rather than occupying the S2 subsite, the P2 fluorophenylalanine splits the catalytic active site, stacking against His41 [Figure 2F–G].
The major alteration in the binding mode of rupintrivir in the active site of Mpro is the conformation of the fluorophenylalanine which normally occupies the S2 subsite when bound to 3C proteases. While the majority of the SARS-CoV-2 Mpro substrates have leucine as the P2 residue, the S2 subsite also accommodates phenylalanine and valine19. However, with the fluorophenylalanine, which introduces polarity, the interactions become unfavorable in the hydrophobic S2 subsite. In SARS-CoV-2 Mpro there is a lack of space to create polar interactions while optimally maintaining the other backbone hydrogen bonds [Figure S2], despite some polar atoms such as the side chain hydroxyl of Tyr54 in the back of the S2 pocket. Instead, the P2 fluorophenylalanine is oriented toward the S1’ pocket, bifurcating the two catalytic residues. In this unique binding mode, the P2 ring makes pi-stacking interactions with the catalytic histidine, but the fluorine atom does not have any polar interactions in the S1’ pocket [Figure 2G]. This alternative orientation of the P2 group and lack of specific fluorine interactions likely contribute to rupintrivir’s reduced potency against SARS-CoV-2 Mpro.
In addition to those of the P2 moiety, other interactions of rupintrivir are also compromised when bound to SARS-CoV-2 Mpro compared with the EV68 3Cpro-rupintrivir complex. When bound to EV68 3Cpro rupintrivir forms a network of five hydrogen bonds through the P3-P4 moieties with the protease active site, while with SARS-CoV-2 Mpro only two are formed. In particular, the hydrogen bond formed in EV68 3Cpro complex with the backbone nitrogen of Gly128 is missing in Mpro as the two enzymes are distinct around that region of the protease fold [Figure 2C–D vs 2G-H]. These losses in specific hydrogen bonding may also contribute to the dramatic loss in rupintrivir potency.
Rupintrivir is a potent antiviral protease inhibitor that successfully targets a whole family of picornaviridae 3Cpro enzymes, promising that a pan-viral DAA is not out of reach. In this study we determined the crystal structure of the EV68 3Cpro-rupintrivir complex and demonstrated that rupintrivir binds in the canonical mode for piconaviral 3Cpro complexes. The SARS-CoV-2 Mpro is a 3C-like protease with a homologous domain harboring a similar active site, but rupintrivir is not a potent Mpro inhibitor17. The molecular basis for the lack of potency was revealed by determining the structure of SARS-CoV-2 Mpro-rupintrivir complex here, which shows a loss of hydrogen bonding network throughout the active site as well as a unique binding mode involving repositioning of the P2 fluorophenylalanine moiety. While this manuscript was under review, a crystal structure of the of SARS-CoV-2 Mpro-rupintrivir complex (PDB-ID: 7P35, 20 ) was released where the P2 moiety was accommodated at the S2 site with considerable rearrangement of both the protease and the inhibitor with overall weakened interactions [Figure S2], suggesting a dynamic equilibrium of P2 orientations. Orientation of the P2 group toward the S1’ subsite in our structure presents opportunities to optimize interactions in this region and potentially grow the inhibitor toward the primed side of the active site. When bound to Mpro rupintrivir’s P2 group splits the catalytic active site of this cysteine protease, physically separating the catalytic dyad. This bifurcation orients the catalytic histidine side chain in a conformer distal from the cysteine. Previous structures have observed multiple conformers for the catalytic histidine in some of which water molecules were observed separating the catalytic residues [Figure S4]. Additionally, recent computational work confirmed the mobility of the catalytic dyad21. This conformational plasticity is consistent with rupintrivir’s binding mode observed in our structure, which can be further exploited in inhibitor design. To our knowledge splitting a catalytic active site by an inhibitor has not previously been observed and may represent a new strategy for disrupting cysteine proteases in the design of pan-viral protease inhibitors.
Materials and Methods
EV68–3Cpro Expression and Purification
The gene encoding the EV68–3Cpro was subcloned in the IPTG-inducible pET28 expression vector. 1 μL of DNA was added to E. coli BL21-DE3 cells and incubated on ice for 20 min. The cells were heat shocked at 42°C for 30 sec, 200 μL of SOC media were added and the cells were grown at 37°C for 1 hr on a shaker. The entire 200 μL were then plated on an LB/Kan plate and grown overnight at 37°C. The following morning, one colony was inoculated in 10 mL of LB in an Erlenmeyer flask and kanamycin was added to a final concentration of 35 μg/mL. The cells were grown at 37°C for 10 hr on a shaker. The entirety of the 10 mL was then added to 285 mL of LB, kanamycin was added to a final concentration of 35 μg/mL and grown at 37°C overnight. 6 L of cells were grown at 37°C in the presence of kanamycin until an OD600 of 0.45 and then the temperature was dropped to 18°C until the cells reached an OD600 of 0.6 where the cells were induced with 250 mL of 1 mM IPTG. Cells were grown overnight at 18°C. Cells grown in 6 L of TB were thawed at RT, resuspended in EV Ni1 buffer [50 mM HEPES pH 7.5, 300 mM NaCl, 10 mM Imidazole, and 2 mM DTT], and lysed with a cell disruptor. Cell debris was removed by centrifugation at 25000 rcf at 4°C for 1 hr. A 1 mL HisTrap crude FF column was equilibrated in a 5x column volume of EV Ni1 buffer. The soluble lysate was loaded onto the HisTrap column and the flow-through was collected. The column was then washed with 100 mL of EV Ni1 buffer and the wash was collected. The protein was eluted with 30mL of EV Ni2 buffer [50mM HEPES pH 7.5, 300 mM NaCl, 250 mM Imidazole, and 2 mM DTT] and the elution was collected in the presence of 50 units of thrombin. The collected elution was dialyzed overnight against 2 L of EV SEC buffer [25 mM HEPES pH 7.5, 300 mM NaCl, and 2 mM DTT] at 4°C. Post dialysis, any precipitate was removed by centrifugation and the protein was concentrated to 1.25mg/mL. For crystallography, an additional purification step was performed with a Pharmacia Superdex 75 16/60 FPLC column equilibrated in EV SEC buffer. EV68–3Cpro fractions purified from the size exclusion column were concentrated to 0.92 mg/mL - 6.2 mg/mL using an Amicon Ultra-15 10kDa device (Millipore) for crystallization.
EV68–3Cpro Rupintrivir Co-crystallization
Discovery of the condition producing cocrystals of EV68–3Cpro and rupintrivir without seeding was achieved using the PACT Premier Screen (Molecular Dimensions), in Well F4, consisting of 20 % (w/v) PEG 3350, 0.1M Bis-Tris propane pH 6.5, and 0.2M Potassium thiocyanate, with a protease concentration of 6.6 mg/mL and 10-fold molar excess of Rupintrivir. Crystals from well F4 of the PACT Premier screen were used for microseeding. Cocrystals were grown at RT by hanging drop vapor diffusion method in 24-well VDX handing-drop trays (Hampton Research) with protease concentrations of 0.92, 2.1, 3.76, and 6.2 mg/mL and 10-fold molar excess of rupintrivir. Crystallization drops were set with the reservoir solution consisting of 10–30% PEG 3350, 0.2 M Potassium thiocyanate and 0.1M Bis-Tris propane pH 6.5 set with 2 μL of well solution and 1 μL protein microseeded with a cat whisker. The condition that produced the best crystals for data collection was 15% (w/v) PEG 3350 with a protease concentration of 3.76 mg/mL and 10-fold molar excess of rupintrivir. Data was collected at 100 K using cryogenic conditions containing the precipitant solution supplemented with 25% glycerol.
SARS-CoV-2-Mpro Expression and Purification
SARS-CoV-2 Mpro was expressed and purified as previously described22. Briefly, the plasmid was transformed into E. coli strain HI-Control™ BL21(DE3) (Lucigen). The transformed cells were precultured at 37 °C in LB medium with ampicillin (100 μg/mL) overnight, and the cell culture was inoculated into TB medium containing 50 mM sodium phosphate (pH 7.0) and ampicillin (100 Hg/mL). When 0D600 value reached ~2.0, 0.5 mM IPTG was added to induce SARS2-Mpro expression and the cell culture was further incubated overnight at 20°C. Cells were harvested by centrifugation at 5000 rpm for 20 min, resuspended in lysis buffer (50 mM Tris-HCl (pH 8.0), 400 mM NaCl, 1 mM TCEP) and lysed by a cell disruptor. The lysate was clarified by ultracentrifugation at 18000 rpm for 50 min. The supernatant was loaded onto a HisTrap FF column (GE Healthcare) equilibrated with lysis buffer, washed with lysis buffer and followed by elution using elution buffer (50 mM Tris-HCl pH 8.0, 400 mM NaCl, 500 mM imidazole, 1 mM TCEP) with a linear gradient of imidazole ranging from 0 mM to 500 mM. The fractions of Mpro-His tag were mixed with GST-PreScission protease-His-tag at a molar ratio of 5:1 to remove the C-terminal His tag. The PreScission-treated Mpro was applied to the nickel column to remove the GST-PreScission protease-His-tag and protein with uncleaved His-tag. The His-tag cleaved Mpro in the flow-through was further purified by size-exclusion chromatography (HiLoad™ 16/60 Superdex 75, GE Healthcare) and stored in 20 mM HEPES pH 7.5, 150 mM NaCl, 1 mM TCEP.
SARS-CoV-2 Mpro Crystallization
SARS-CoV-2 Mpro cocrystallized in multiple conditions. One condition producing large plate-like crystals was discovered using the MCSG-1 crystal screen (Anatrace), Well B2, containing 25% (w/v) PEG 3350 and 0.1 M bis-tris-methane pH 5.5 and 0.2 M sodium chloride. The SARS2-Mpro-Rupintrivir cocrystals were grown at room temperature by hanging drop vapor diffusion method in a 24-well VDX hanging-drop tray (Hampton Research). The P21 crystals grew at 22% (w/v) PEG 3350 with a protease concentration of 6.0 mg/mL with 6-fold molar excess of Rupintrivir (10% DMSO). The P21212 crystals grew at 19% (w/v) PEG 3350 with a protease concentration of 12.0 mg/mL with 3-fold molar excess of Rupintrivir (10% DMSO). Both conditions were mixed with the precipitant solution at a 1:1 ratio (1 μL:1 μL) and micro-seeded (1:1 – 1:4 dilution) with a cat whisker. Crystals appeared overnight and grew to diffraction quality within a week. The P21212 crystals were unexpected and had a needle morphology. As data was collected at 100 K, cryogenic conditions consisted of the precipitant solution supplemented with 25% glycerol.
Data Collection and Structure Determination
EV68 3Cpro-rupintrivir cocrystals and SARS-CoV-2 Mpro-rupintrivir cocrystals in the P21212 space group were flash frozen in liquid nitrogen and diffraction data was collected at the National Synchrotron Light Source II at the Brookhaven National Laboratory, Beamline 17-ID-1 (AMX). Synchrotron diffraction intensities were indexed, integrated, and scaled using XDS 23. The SARS2-Mpro-Rupintrivir cocrystals in the P21 space group were flash frozen under a cryostream when mounting the crystal at our in-house Rigaku_Saturn944 X-ray system and these diffraction intensities were indexed, integrated, and scaled using HKL3000 24. The structures were solved using molecular replacement with PHASER 25 using either an Mpro monomer (PDB: 7L0D)22 or an EV68–3Cpro monomer (PDB: 3ZV8)26. Model building and refinement were performed using Coot27 and Phenix28. During refinement, all crystals utilized optimized stereochemical weights. The EV68–3Cpro structure required a merohedral twin law (h,-h-k,-l) during refinement. The Rupintrivir ligand was designed in Maestro and the output sdf file was used in the Phenix program eLBOW29 to generate the cif file containing atomic positions and constraints necessary for ligand refinement. Iterative rounds of crystallographic refinement were carried out until convergence was achieved. To limit bias throughout the refinement process, five percent of the data was reserved for the free R-value calculation30. Modeling of Rupintrivir and catalytic histidines were further validated with omit maps with and without simulated annealing. MolProbity31 was applied to evaluate the final structures before deposition in the PDB32–33. Structure analysis, superposition and figure generation was done using PyMOL. X-ray data collection and crystallographic refinement statistics are presented in the Supporting Information [Table S2].
Supplementary Material
Acknowledgement
This research was funded by National Institute of General Medical Sciences, grant numbers R01 GM135919 and R35 GM118112; National Institute of Allergy and Infectious Diseases grant number R21 AI149716. This research used beamline 17-ID-1 (AMX) of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. The Center for BioMolecular Structure (CBMS) is primarily supported by the National Institutes of Health, National Institute of General Medical Sciences (NIGMS) through a Center Core P30 Grant (P30GM133893), and by the DOE Office of Biological and Environmental Research (KP1605010).
Footnotes
Supporting Information
Figures depicting the electron density map around the inhibitor (Figure S1), comparison of 3Cpro and Mpro active sites (Figure S2), catalytic histidine in 3Cpro crystal structures (Figure S3); Table S1: X-ray data collection and crystallographic refinement statistics.
Accession Codes
The structures described here have been deposited as PDB entries 7L8H, 7L8I and 7L8J.
References
- 1.Woolhouse M; Scott F; Hudson Z; Howey R; Chase-Topping M, Human viruses: discovery and emergence. Philos Trans R Soc LondB Biol Sci 2012, 367 (1604), 2864–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park SW; Pons-Salort M; Messacar K; Cook C; Meyers L; Farrar J; Grenfell BT, Epidemiological dynamics of enterovirus D68 in the United States and implications for acute flaccid myelitis. Sci TranslMed 2021, 13 (584). [DOI] [PubMed] [Google Scholar]
- 3.Lamarre D; Anderson PC; Bailey M; Beaulieu P; Bolger G; Bonneau P; Bos M; Cameron DR; Cartier M; Cordingley MG; Faucher AM; Goudreau N; Kawai SH; Kukolj G; Lagace L; LaPlante SR; Narjes H; Poupart MA; Rancourt J; Sentjens RE; St George R; Simoneau B; Steinmann G; Thibeault D; Tsantrizos YS; Weldon SM; Yong CL; Llinas-Brunet M, An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature 2003, 426 (6963), 186–189. [DOI] [PubMed] [Google Scholar]
- 4.Surleraux DL; Tahri A; Verschueren WG; Pille GM; de Kock HA; Jonckers TH; Peeters A; De Meyer S; Azijn H; Pauwels R; de Bethune MP; King NM; Prabu-Jeyabalan M; Schiffer CA; Wigerinck PB, Discovery and selection of TMC114, a next generation HIV-1 protease inhibitor. J Med Chem 2005, 48 (6), 1813–22. [DOI] [PubMed] [Google Scholar]
- 5.El Bouzidi K; White E; Mbisa JL; Sabin CA; Phillips AN; Mackie N; Pozniak AL; Tostevin A; Pillay D; Dunn DT, HIV-1 drug resistance mutations emerging on darunavir therapy in Pi-naive and-experienced patients in the UK. Journal of Antimicrobial Chemotherapy 2016, 71 (12), 3487–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Meyer S; Azijn H; Surleraux D; Jochmans D; Tahri A; Pauwels R; Wigerinck P; de Béthune M-P, TMC114, a novel human immunodeficiency virus type 1 protease inhibitor active against protease inhibitor-resistant viruses, including a broad range of clinical isolates. Antimicrobial agents and chemotherapy 2005, 49 (6), 2314–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mengist HM; Dilnessa T; Jin T, Structural Basis of Potential Inhibitors Targeting SARS-CoV-2 Main Protease. Front Chem 2021, 9, 622898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghahremanpour MM; Tirado-Rives J; Deshmukh M; Ippolito JA; Zhang CH; Cabeza de Vaca I; Liosi ME; Anderson KS; Jorgensen WL, Identification of 14 Known Drugs as Inhibitors of the Main Protease of SARS-CoV-2. ACS Med Chem Lett 2020, 11 (12), 2526–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bafna K; White K; Harish B; Rosales R; Ramelot TA; Acton TB; Moreno E; Kehrer T; Miorin L; Royer CA; Garcia-Sastre A; Krug RM; Montelione GT, Hepatitis C virus drugs that inhibit SARS-CoV-2 papain-like protease synergize with remdesivir to suppress viral replication in cell culture. Cell Rep 2021, 35 (7), 109133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matthews DA; Dragovich PS; Webber SE; Fuhrman SA; Patick AK; Zalman LS; Hendrickson TF; Love RA; Prins TJ; Marakovits JT; Zhou R; Tikhe J; Ford CE; Meador JW; Ferre RA; Brown EL; Binford SL; Brothers MA; DeLisle DM; Worland ST, Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc Natl Acad Sci U S A 1999, 96 (20), 11000–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Binford SL; Maldonado F; Brothers MA; Weady PT; Zalman LS; Meador JW 3rd; Matthews DA; Patick AK, Conservation of amino acids in human rhinovirus 3C protease correlates with broad-spectrum antiviral activity of rupintrivir, a novel human rhinovirus 3C protease inhibitor. Antimicrob Agents Chemother 2005, 49 (2), 619–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayden FG; Turner RB; Gwaltney JM; Chi-Burris K; Gersten M; Hsyu P; Patick AK; Smith GJ 3rd; Zalman LS, Phase II, randomized, double-blind, placebo-controlled studies of ruprintrivir nasal spray 2-percent suspension for prevention and treatment of experimentally induced rhinovirus colds in healthy volunteers. Antimicrob Agents Chemother 2003, 47 (12), 3907–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsyu PH; Pithavala YK; Gersten M; Penning CA; Kerr BM, Pharmacokinetics and safety of an antirhinoviral agent, ruprintrivir, in healthy volunteers. Antimicrob Agents Chemother 2002, 46 (2), 392–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang XN; Song ZG; Jiang T; Shi BS; Hu YW; Yuan ZH, Rupintrivir is a promising candidate for treating severe cases of Enterovirus-71 infection. World J Gastroenterol 2010, 16 (2), 201–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun L; Meijer A; Froeyen M; Zhang L; Thibaut HJ; Baggen J; George S; Vernachio J; van Kuppeveld FJ; Leyssen P; Hilgenfeld R; Neyts J; Delang L, Antiviral Activity of Broad-Spectrum and Enterovirus-Specific Inhibitors against Clinical Isolates of Enterovirus D68. Antimicrob Agents Chemother 2015, 59 (12), 7782–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shie JJ; Fang JM; Kuo TH; Kuo CJ; Liang PH; Huang HJ; Wu YT; Jan JT; Cheng YS; Wong CH, Inhibition of the severe acute respiratory syndrome 3CL protease by peptidomimetic alpha,beta-unsaturated esters. BioorgMed Chem 2005, 13 (17), 5240–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vatansever EC; Yang K; Kratch KC; Drelich A; Cho CC; Mellott DM; Xu S; Tseng CK; Liu WR, Targeting the SARS-CoV-2 Main Protease to Repurpose Drugs for COVID-19. bioRxiv 2020. [Google Scholar]
- 18.Wang J; Fan T; Yao X; Wu Z; Guo L; Lei X; Wang J; Wang M; Jin Q; Cui S, Crystal structures of enterovirus 71 3C protease complexed with rupintrivir reveal the roles of catalytically important residues. J Virol 2011, 85 (19), 10021–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ullrich S; Nitsche C, The SARS-CoV-2 main protease as drug target. Bioorg Med Chem Lett 2020, 30 (17), 127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fabrega-Ferrer M; Perez-Saavedra J; Herrera-Morande A; Coll M, 7P35 Structure of the SARS-CoV-2 3CL protease in complex with rupintrivir. July 21, 2021. ed.; RCSB PDB, 2021. [Google Scholar]
- 21.Verma N; Henderson JA; Shen J, Proton-Coupled Conformational Activation of SARS Coronavirus Main Proteases and Opportunity for Designing Small-Molecule Broad-Spectrum Targeted Covalent Inhibitors. J Am Chem Soc 2020, 142 (52), 21883–21890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lockbaum GJ; Reyes AC; Lee JM; Tilvawala R; Nalivaika EA; Ali A; Kurt Yilmaz N; Thompson PR; Schiffer CA, Crystal Structure of SARS-CoV-2 Main Protease in Complex with the Non-Covalent Inhibitor ML188. Viruses 2021, 13 (2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kabsch W, Xds. Acta Crystallogr D Biol Crystallogr 2010, 66 (Pt 2), 125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Otwinowski Z; Minor W, [20] Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 25.McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ, Phaser crystallographic software. JAppl Crystallogr 2007, 40 (Pt 4), 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tan J; George S; Kusov Y; Perbandt M; Anemuller S; Mesters JR; Norder H; Coutard B; Lacroix C; Leyssen P; Neyts J; Hilgenfeld R, 3C protease of enterovirus 68: structure-based design of Michael acceptor inhibitors and their broad-spectrum antiviral effects against picornaviruses. J Virol 2013, 87 (8), 4339–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Emsley P; Cowtan K, Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 2004, 60 (Pt 12 Pt 1), 2126–32. [DOI] [PubMed] [Google Scholar]
- 28.Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH, PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 2010, 66 (Pt 2), 213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moriarty NW; Grosse-Kunstleve RW; Adams PD, electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr D Biol Crystallogr 2009, 65 (Pt 10), 1074–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brunger AT, Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 1992, 355 (6359), 472–5. [DOI] [PubMed] [Google Scholar]
- 31.Davis IW; Leaver-Fay A; Chen VB; Block JN; Kapral GJ; Wang X; Murray LW; Arendall WB 3rd; Snoeyink J; Richardson JS; Richardson DC, MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 2007, 35 (Web Server issue), W375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE, The Protein Data Bank. Nucleic Acids Res 2000, 28 (1), 235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berman H; Henrick K; Nakamura H, Announcing the worldwide Protein Data Bank. Nat Struct Biol 2003, 10 (12), 980. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.