

Abstract
COVID-19 pandemic, starting from the latest 2019, and caused by SARS-CoV-2 pathogen, led to the hardest health-socio-economic disaster in the last century. Despite the tremendous scientific efforts, mainly focused on the development of vaccines, identification of potent and efficient anti-SARS-CoV-2 therapeutics still represents an unmet need. Remdesivir, an anti-Ebola drug selected from a repurposing campaign, is the only drug approved, so far, for the treatment of the infection. Nevertheless, WHO in later 2020 has recommended against its use in COVID-19. In the present paper, we describe a step-by-step in silico design of a small library of compounds as main protease (Mpro) inhibitors. All the molecules were screened by an enzymatic assay on Mpro and, then, cellular activity was evaluated using Vero cells viral infection model. The cellular screening disclosed compounds 29 and 34 as in-vitro SARS-CoV-2 replication inhibitors at non-toxic concentrations (0.32 < EC50 < 5.98 μM). To rationalize these results, additional in-vitro assays were performed, focusing on papain like protease (PLpro) and spike protein (SP) as potential targets for the synthesized molecules. This pharmacological workflow allowed the identification of compound 29, as a dual acting SARS-CoV-2 proteases inhibitor featuring micromolar inhibitory potency versus Mpro (IC50 = 1.72 μM) and submicromolar potency versus PLpro (IC50 = 0.67 μM), and of compound 34 as a selective SP inhibitor (IC50 = 3.26 μM).
Keywords: In-silico design, SARS-CoV-2 proteases dual inhibitor, Enzymatic assays, Biophysical assays, Cellular characterization
Graphical abstract

1. Introduction
SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2) is the pathogen causing Coronavirus disease 2019 (COVID-19). SARS-CoV-2 belongs to the β-coronaviridae family of which bats represent the main reservoir and many proofs suggest that human infection was a result of a zoonotic jump [1,2].
The first evidence of this pathology was recognized in Wuhan, China in the late 2019 and since this date SARS-CoV-2 has caused a worldwide outbreak as declared by WHO in March 2020. Up to now, more than 150 million of cases with about 3 million of deaths have been associated with this pathology in the whole world. The pandemic is still in progress, causing not only the hardest sanitary crisis in the last century, but also an unrecoverable socio-economic collapse. COVID-19 symptoms can range from very mild to severe and include fever, cough, tiredness and loss of taste and smell, but the infection can degenerate, spreading deeply into lungs and causing bilateral pneumonia as principal cause of death, especially in frail elderly patients [3,4].
Despite all the efforts, during the last year, addressed to build an efficient vaccine campaign, the virus spread is still ongoing and the challenge is still open. For these reasons, the main action to fight this global pandemic is based on breaking the transmission chain of SARS-CoV-2, facing not only the high infectiousness [5], but also the mutations rate, that leads to a large number of unknown, perhaps vaccine-resistant strains [6]. A proper pharmacological therapy is not currently yet available for COVID-19. Remdesivir, an RNA-dependent RNA-polymerase inhibitor developed in 2014 as anti-Ebola drug, represents the only antiviral drug approved by the FDA for the treatment of hospitalized patients with COVID-19 [7]. Nevertheless, in late 2020, WHO has issued a conditional recommendation against the use of remdesivir in hospitalized patients, as no evidences were collected concerning remdesivir improvement in survival and other outcomes in these patients [8]. Drug repurposing, in fact, is the most used approach to set up an appropriate therapy against SARS-CoV-2 due to the urgency in disposing of a molecular arsenal to contrast the epidemic. In this sense, several molecules such as favipiravir [9], aprotinin [10], prolastin [11], losmapimod [12], hydroxychloroquine [13,14] are in clinical trial, while many other drugs, developed to different purposes, are being considered as potential anti-COVID-19 treatment based on in silico studies [[15], [16], [17]]. However, drug repositioning cannot represent the only strategy and it is evident how the identification and the development of new, specific anti-SARS-CoV-2 agents are nowadays mandatory [18].
SARS-CoV-2 genome is a positive-sense RNA strand codifying for 27 non-structural and structural proteins of which four play a pivotal role in infectious cycle and, therefore, considered as principal pharmacological targets. Specifically, the spike protein (SP) is an envelope glycoprotein involved in host receptor binding, the latter identified as angiotensin-converting enzyme 2 [[15], [16], [17]]. The blockage of spike function prevents the virus entry, spread, and effectiveness [19]. RNA-dependent RNA-polymerase (RdRp) is responsible for nucleic material replication and it is the only validated target for COVID-19 pharmacological treatment so far [20,21]. Finally, exploiting host machinery, SARS-CoV-2 RNA is translated into two large polyproteins (pp1a and pp1ab) that undergo a maturation process catalyzed by two different viral proteases: the 3 chymotrypsin-like protease (3CLpro, also called main protease, Mpro) and the papain-like protease (PLpro). The first is a cysteine protease responsible for catalytic Leu-Gly cleavages at the C-terminus of pps leading to 11 non-structural, mature proteins [22,23]. The second one, PLpro, is involved in two non-structural proteins maturation by cleaving N-terminus sequences containing LeuXGlyGly↓(Ala/Leu)X motifs, where X is any type of amino acid [24,25]. The pivotal role of these enzymes together with the structural knowledge acquired about the inhibitor/protease complexes make them two of the most investigated druggable targets to contrast SARS-CoV-2.
Two classes of SARS-CoV-2 Mpro inhibitors have been developed so far: covalent and non-covalent inhibitors. The first comprises peptidomimetics designed to covalently interact with Cys145 in the catalytic site that share four features: a) moderate size; b) a group mimicking a glutamine side chain; c) a branched lipophilic group; d) a reactive electrophilic ‘warhead’, such as aldehydes, Michael acceptors, and epoxy ketones responsible for the covalent bond [25,26]. However, the possibility of non-specific biological interactions due to their huge reactivity, must be considered for the subsequent in vivo evaluation of this type of inhibitors [27,28]. Accordingly, the less reactive non-covalent inhibitors may represent safer antiviral agents. Indeed, despite the lack of covalent bond in the active site of protease, they can represent useful tools to counteract the virus as well [[29], [30], [31]].
Starting from these evidence, in the present paper we describe the design, synthesis and biological evaluation of a new series of indole-based derivatives designed as SARS-CoV-2 Mpro inhibitors. General structures of synthesized compounds are depicted in Fig. 1 . A series of peptidomimetics was synthesized starting from l-tryptophan that was functionalized with an amine moiety (R–NH2) and a natural or modified amino acid (R1-aa, Fig. 1A). Two additional small molecules were synthesized starting from methyl 1H-indole-5-carboxylate that was derivatized in positions 1 and 3 to give the corresponding aminomethyl substituted derivatives (Fig. 1B). These compounds derived from a step-by-step in silico design coupled with the evaluation of enzymatic assay results. To further investigate their pharmacological properties, the compounds were tested in a Vero cells model to evaluate their ability to reduce the plaques formations and also gaining information about their cytotoxic behavior. This workflow led to the identification of two compounds as SARS-CoV-2 proteases inhibitors. First, compound 29 emerged as powerful, non-cytotoxic, SARS-CoV-2 multitarget inhibitor, showing high affinity both for Mpro and for PLpro. Moreover, compound 34, besides its interesting cellular activity, showed a high affinity and selectivity for SP.
Fig. 1.
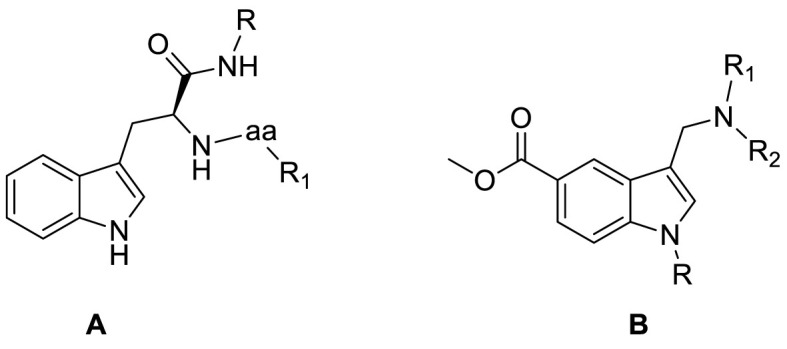
General structures of synthesized compounds.
2. Synthesis
Final compounds 7 and 22–29 were synthesized as summarized in Scheme 1 . L-Boc-Trp-OH was coupled with tert-butyl amine, 4-phenylbenzylamine or benzylamine using HOBt and HBTU as coupling agents and DIPEA as base. Amides 1–3 were thus obtained in 65–80% yields. Removal of the Boc protecting group by DCM:TFA (3:1 v:v), gave intermediates 4–6 in almost quantitative yields. The intermediates were coupled, without further purification, with different L-aminoacids (L-Boc-Pro-OH, L-Boc-Phe-OH, L-Boc-Leu-OH, L-Boc-Pra-OH or L-Boc-allylgly-OH) using the same coupling protocol described above. In this way, final compound 7 (59% of yield) and pseudo peptides intermediates 8–14 (74–82% of yields) were obtained. Removal of the Boc protection from derivatives 8–14, led to 15–21, that were further derivatized by reductive amination with 4-phenyl benzaldehyde, benzaldehyde or propionaldehyde to give final derivatives 22–28 in 55–66% yields. Alternatively, acylation of 20 with chloroacetyl chloride led to compound 29 in 62% yield.
Scheme 1.

Synthesis of final compounds 7, 22–29. Reagents and conditions: a) HOBt (1.2 eq), HBTU (1.2 eq), DIPEA (2.4 eq), amine (1.2 eq), DCM, 12h, RT b) TFA/DCM (1/3, v/v), triisopropylsilane (0.25 eq), 30–120 min, RT c) HOBt (1.0 eq), HBTU (1.0 eq), DIPEA (2.0 eq), L-Boc-aa-OH (0.83 eq), DCM, 12h, RT d) RCOH (1.2 eq), MeOH, N2 stream, RT, 12 h. Then, NaBH4 (3.0 eq), RT, 3h e) TEA (1.2 eq), chloro acetylchloride (1.2 eq), THF, RT, 20 min.
The synthetic route to obtain small molecules 34 and 35 is illustrated in Scheme 2 . Using methyl 1H-indole-5-carboxylate as starting material, N-1 alkylation was performed by reaction with isobutyl iodide or methyl iodide using sodium hydride as base. In this way, intermediates 30 and 31 were synthesized in 80 and 82% yields, respectively. Starting from these compounds, Mannich reaction was carried out [32] leading to intermediates 32 and 33 in 70% and 75% yield, respectively. Upon alkylation with benzyl bromide, final compound 34 was obtained in 78% yield, while compound 35 resulted from a coupling reaction of 33 with L-propargyl glycine as described above and was isolated in 80% yield.
Scheme 2.

Synthesis of final compounds 34 and 35. Reagents and conditions: a) NaH (1.5 eq), alkyl iodide (1.5 eq), DMF, 0 °C to RT, overnight. b) Formaldehyde (2.0 eq), TFA (2.0 eq), amine (2.0 eq), DCM, 12h, RT. c) Aldehyde (1.2 eq), MeOH, N2 stream, RT, 12 h. Then, NaBH4 (3 eq), RT, 3h d) HOBt (1.2 eq), HBTU (1.2 eq), DIPEA (2.4 eq), L-Boc-Pra-OH (1.2 eq), DCM, 12h, RT.
3. In silico design
With the aim of identifying novel antiviral agents against SARS-CoV-2, molecular docking experiments were performed against the related Mpro target (PDB code: 6M0K) [33]. Different studies focused on the discovery of new compounds able to interfere with this enzyme were recently reported, and the availability of different X-ray Mpro/inhibitor co-complex structures represented a precious source of information for orienting the design of novel compounds.
In 2020, Dai et al. reported the structure-based identification of two SARS-CoV-2 Mpro inhibitors (A1 and B1, Fig. 2 ) [33], that covalently bound Mpro due to the presence of aldehyde moieties able to react with Cys145 key residue. Also, further specific chemical functions were introduced in precise positions in order to cover the whole binding site. It is worth noting that the analysis of the active sites of Mpros revealed the high conservation among all coronavirus Mpros, and they are usually composed of four sites: S1’, S1, S2, and S4 (Fig. 2) [33,34]. The reactive Cys145 is placed in the S1’ site, S1 and S2 sites can accommodate large and cyclic systems, while S4 site was prone to interact with specific moieties, such as indole substituent identified in this study [33]. Specifically, the latter was introduced to form hydrogen bonds with key residues in the S4 site and to improve drug-like properties.
Fig. 2.

A) SARS-CoV-2 Mpro binding site; S1’, S1, S2, and S4 sites and Cys145 key residue are highlighted. B) Chemical structures of compounds A1 and B1, with specified the chemical moieties interacting with the S1’, S1, S2, and S4 Mpro sites.
Starting from these data, we performed molecular docking experiments to explore the binding mode of a series of new peptidomimetic compounds (Fig. 3 and Table S1). In details, the design and synthesis of the different derivatives were guided step-by-step by the outcomes obtained from enzymatic assays on the cognate compounds. Recursive cycles of in silico analysis, synthesis and biophysical analysis of target engagements were performed. Results are summarized in Table 1 and are compared with the IC50 of GC-376, a well-known Mpro inhibitor, used as reference compound.
Fig. 3.

A) mtrp (colored by atom type: C green, O red, N blue, polar H light grey), B) 25 (colored by atom type: C yellow, O red, N blue, polar H light grey); C) 23 (colored by atom type: C purple, O red, N blue, polar H light grey); D) 22 (colored by atom type: C pink, O red, N blue, polar H light grey); E) 7 (colored by atom type: C aqua green, O red, N blue, polar H light grey); F) 24 (colored by atom type: C light green, O red, N blue, polar H light grey); G) 27 (colored by atom type: C light brown, O red, N blue, polar H light grey); H) 26 (colored by atom type: C light violet, O red, N blue, polar H light grey); I) 28 (colored by atom type: C violet, O red, N blue, polar H light grey); J) 29 (colored by atom type: C light yellow, O red, N blue, polar H light grey); K) 34 (colored by atom type: C light blue, O red, N blue, polar H light grey); L) 35 (colored by atom type: C dark grey, O red, N blue, polar H light grey) in docking with SARS-CoV-2 Mpro (transparent molecular surface colored in grey and secondary structure colored in orange; key residues are reported as sticks and colored by atom type: C grey, O red, N blue, S yellow, polar H light grey).
Table 1.
Measured activities for inhibition of SARS-CoV-2 selected protein targets. Results are reported as average ± SD.
|
Compound |
Structure | Target protein |
||
|---|---|---|---|---|
|
Mpro |
PLpro |
SP |
||
| IC50 (μM) | IC50 (μM) | KD (μM) | ||
| GC-376 | 0.57 ± 0.15 | – | – | |
| GRL-0617 | – | 1.67 ± 0.63 | – | |
| 7 | 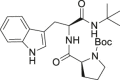 |
5.01 ± 2.31 | >25 | >25 |
| 22 | 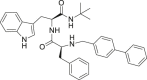 |
>25 | >25 | 19.05 ± 0.41 |
| 23 | 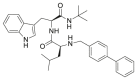 |
1.73 ± 0.89 | >25 | >25 |
| 24 | 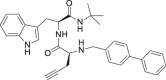 |
>25 | >25 | >25 |
| 25 | 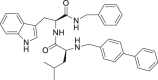 |
23.61 ± 8.72 | >25 | – |
| 26 | 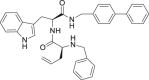 |
>25 | >25 | >25 |
| 27 | 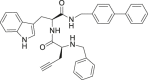 |
2.86 ± 1.42 | >25 | – |
| 28 | 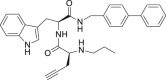 |
22.65 ± 9.82 | >25 | – |
| 29 | 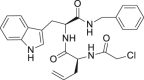 |
1.72 ± 0.75 | 0.67 ± 0.59 | > 25 |
| 34 | 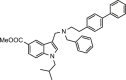 |
>25 | >25 | 3.26 ± 0.11 |
| 35 | 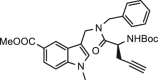 |
>25 | >25 | 11.41 ± 0.36 |
In this way, the combination of in silico and experimental testing allowed the gradual generation of a small library of compounds sharing similar chemical functions and exhibiting promising activities against SARS-CoV-2 Mpro. Specifically, we firstly evaluated whether the l-tryptophan fragment could show a similar interaction network established by the reference compounds A1 and B1, in particular regarding the placement of the indole function in the binding site. Unfortunately, molecular docking calculations highlighted that the indole moiety of capped L-Trp (mtrp) was placed in the S2 instead of S4 site (Fig. 3A). In order to force the 1H-indol-3-yl moiety in the S4 site, the L-Trp starting fragment was decorated introducing voluminous substituents at both the amino (N-) and carboxyl (C-) termini. After different attempts, satisfactory outcomes were obtained by linking the L-Leu at the L-Trp N-terminus and an N-benzyl moiety at the L-Trp C-function terminus. The introduction of a ([1,1′-biphenyl]-4-yl) substituent at the L-Leu N-terminus led to compound 25, showing a good accommodation in the Mpro binding site (Fig. 3B). This compound was indeed able to cover S1’, S1, S2, and S4 sites while also establishing a set of interactions with His41 and Cys145 (belonging to the catalytic dyad), Phe140, Glu166, and Gln189. The promising binding of compound 25 to Mpro was confirmed by enzymatic assays (Table 1), obtaining an IC50 value in the medium micromolar range of activity. Also, the careful analysis of the sampled binding poses of 25 disclosed that the terminal benzyl moiety could be replaced by a shorter and bulky substituent, finally leading to compound 23 (Fig. 3C) featuring a tert-butyl moiety. As expected, 23 showed a remarkable inhibitory activity against Mpro in the low micromolar range (1.73 μM, Table 1). The introduction of a bulkier and aromatic substituent (compound 22, featuring the L-Phe residue instead of the L-Leu) determined a totally different predicted binding mode (Fig. 3D), thus indicating the poor ability of 22 in interfering with Mpro activity, as confirmed by enzymatic assays (Table 1). On the other hand, compound 7 showed an interaction network and a total shape of the molecule similar to that previously obtained for 23 (Fig. 3E). Such computational indications were confirmed by detecting the promising ability of 7 in interfering with the enzyme activity (Table 1).
Starting from the most promising compound 23 (Fig. 3C), we then wondered whether the introduction of a reactive chemical function able to covalently bind the Cys145 could lead to an improved biological activity. With this aim, the isobutyl moiety from L-Leu residue in 23 was replaced with a propyn-2-yl function (compound 24), thus taking into account the reactivity of alkyne group with cysteine residues, as widely reported [[35], [36], [37]]. On the other hand, the analysis of the covalent complex revealed the loss of a series of key interactions with the key residues belonging to S1’, S1, S2, and S4 sites as previously detected for 23 (Fig. 3F), and confirmed by poor outcomes from enzymatic assays (Table 1). Starting from compound 24, we introduced the ([1,1′-biphenyl]-4-yl) substituent at the C-terminus while using a smaller benzyl moiety at the N-terminus (compound 27). Covalent docking calculations showed a binding mode compatible with that previously observed for 23 (Fig. 3G), confirmed by the promising data from enzymatic experiments (Table 1). On the other hand, compound 26, differing from 27 for the presence of an alkene function instead of the alkyne one, usually showing a reversible covalent behavior [38], showed a poor interference with the protein counterpart, as expected. Specifically, while covalent docking calculations revealed an interaction pattern similar to that established by 27, the poses related to the non-covalent complex highlighted the loss of most of the interactions with the protein counterpart (e.g., His41) (Fig. 3H) and, for these reasons, we speculated that the observed poorer activity could be ascribed to this latter binding mode. The replacement of the benzyl substituent in 27 with an alkyl function (compound 28) determined the loss of a series of key interactions with the protein counterpart (Fig. 3I), again leading to a reduced inhibitory activity. Instead, the introduction of a further putative covalent attachment point (α-chloroketone moiety, compound 29) led to a promising binding related to the irreversible complex (Fig. 3J), subsequently corroborated by enzymatic assay data (Table 1).
Eventually, two small molecules (compounds 34 and 35), obtained according to the synthetic route reported in the chemistry section (Table 1), were evaluated. Specifically, the indole function was modified introducing alkyl substituents on the basic nitrogen while also showing an ester function at C-5 position. As expected, molecular docking calculations highlighted that such modifications led to a poor predicted binding with the protein counterpart, especially for what concerned the interaction network in the S4 site, since the modified indole moiety pointed in a region between the S2 and S1’ site (Fig. 3K). For what concerns compound 35, the poses arising from covalent docking calculations again highlighted the contacts between the modified indole moiety with the residues close to the S1 site, thus again not in accordance with the interaction network detected for the most promising compounds (e.g., 7, 23, 27, 29) (Fig. 3L). As expected, for both the compounds, biological experiments highlighted the absence of inhibitory activity (Table 1).
4. Pharmacological characterization in SARS-CoV-2 transfected cells
To further expand the results obtained in-vitro, the synthesized compounds were challenged in a more relevant biological environment. A cellular screening using Vero cells transfected with two different clinical isolates of SARS-CoV-2 (UC-1074 and UC-1075 strains) was performed. The EC50 of the synthesized compounds was calculated along with the determination of cell morphology and cell growth. Remdesivir was used as positive control. Results obtained are summarized in Table 2 .
Table 2.
Cellular assays results.
| Compound | Antiviral activity EC50a UC-1074 #1 strain Vero cells |
Antiviral activity EC50a UC-1075 #1 strain Vero cells |
Cytotoxicity |
|
|---|---|---|---|---|
| 15 μl/10 mL | 100 μl/10 mL | Cell morphology (MCC)b | Cell growth (CC50)c | |
| Remdesivir | 0.87 μM | 0.61 μM | >40 μM | >40 μM |
| 7 | >100 μM | >100 μM | >100 μM | >100 μM |
| 22 | 2.19 μM | 2.01 μM | > 100 μM | 1.49 μM |
| 23 | >4 μM | >4 μM | 20 μM | 10.61 μM |
| 24 | >4 μM | >4 μM | 20 μM | 4.67 μM |
| 25 | >4 μM | >20 μM | ≥20 μM | 34.70 μM |
| 26 | 63.14 μM | >20 μM | ≥100 μM | 78.37 μM |
| 27 | 8.94 μM | 10.94 μM | ≥ 100 μM | 51.52 μM |
| 28 | >20 μM | >20 μM | >100 μM | 42.35 μM |
| 29 | 0.32 μM | 1.37 μM | 100 μM | 38.67 μM |
| 34 | 5.98 μM | 0.44 μM | 100 μM | 31.53 μM |
| 35 | 4 μM | 20 μM | 100 μM | 8.09 μM |
Effective concentration required to reduce virus plaque formation by 50%. Virus input was 100 CCID50.
Minimum cytotoxic concentration that causes a microscopically detectable alteration of cell morphology.
Cytotoxic concentration required to reduce cell growth by 50%.
Despite the remarkable activity showed in enzymatic assays, compounds 7 and 23 largely failed to inhibit viral plaque formation with compound 23 showing high cytotoxicity. These data could be probably justified on the basis of the predominant lipophilic character of the two compounds, determining unfavorable pharmacokinetic properties as well as precipitation and/or aggregation in the cellular medium [39,40]. On the other hand, compounds 27 and 29 confirmed the antiviral activity showed in-vitro. In particular, compound 29 potency was comparable to remdesivir and 28-fold higher than compound 27 over two different SARS-CoV-2 strains, with negligible cytotoxicity. Moreover, derivatives 22 and 34, that were unable to antagonize Mpro in-vitro, showed cellular antiviral activity. Besides results for derivative 22 were largely affected by high cytotoxicity (Table 2), compound 34 antiviral activity was worth of further investigations.
Thus, we questioned whether the synthesized compounds were also able to bind different SARS-CoV-2 target proteins, namely PLpro and SP. As reported in Table 1, compound 29 showed remarkable affinity for the PLpro with lower EC50 than the reference compound GRL-0617. This dual inhibitory effect is likely responsible for the higher cellular antiviral activity of 29, when compared to 23, highlighting the potential of dual Mpro and PLpro inhibitors as anti SARS-CoV-2 therapeutics.
In order to rationalize these data, molecular docking calculations were performed for compound 29 against SARS-CoV-2 PLpro. Specifically, two different models, related to the non-covalent and covalent binding, were generated. In the non-covalent ligand/protein complex (PDB code: 7JIW) [41], compound 29 was accommodated in the binding site of the protein establishing a large set of interactions, namely π-π stacking with Tyr268, π-cation with Arg166, H-bonds with Gln269 and Thr301 (Fig. 4 A). Furthermore, the comparison of the binding modes of the tested compound and of the compound VBY originally co-crystallized in the protein structure highlighted similar ligand shapes [41], thus confirming the promising interaction of 29 with SARS-CoV-2 PLpro. On the other hand, to shed light on the hypothetical covalent binding, the SARS-CoV-2 PLpro covalently co-complexed with a peptide inhibitor (PDB code: 6WX4) through the reactive Cys111 residue was accounted [24]. In this case, the output poses arising from covalent docking calculations highlighted a different binding mode and, in particular, the indole and benzyl functions were oriented towards the external part of the protein, while also establishing H-bonds with Gly271 and His272 (belonging to the catalytic triad of this enzyme), and π-π stacking with Trp106 (Fig. 4B, Table S2).
Fig. 4.

A) non-covalent and B) covalent molecular docking complexes between 29 (colored by atom type: C light yellow, O red, N blue, polar H light grey) and SARS-CoV-2 PLpro (transparent molecular surface colored in grey and secondary structure colored in green; key residues are reported as sticks and colored by atom type: C grey, O red, N blue, S yellow, polar H light grey).∖
Interestingly, we found that the cellular inhibitory effects of compound 34, is due to its ability to selectively inhibit SARS-CoV-2 spike protein in the low micromolar range (Table 1). The small molecule 34 could be thus considered an interesting chemotype for the development of a new class of anti-SARS-CoV-2 agents, selectively inhibiting SP.
Molecular docking calculations were performed accounting the SP receptor-binding domain structure released by Wang et al. (PDB code: 6LZG) originally co-complexed with ACE2 [42]. In silico experiments highlighted the accommodation of 34 featuring a good shape complementarity with the external surface of SP and a large network of contacts with some residues responsible of the SP-ACE2 protein-protein interaction (Fig. 5 ). Specifically, 34 made π-π stacking with Phe459 and Tyr348 and H-bond with Glu484 as well as a large set of hydrophobic and polar contacts with Tyr449, Leu455, Phe486, Gln493, and Ser494 (Fig. 5, Table S3).
Fig. 5.

Molecular docking complex between 34 (colored by atom type: C light blue, O red, N blue, polar H light grey) and SARS-CoV-2 SP (transparent molecular surface colored in grey and secondary structure colored in light yellow; key residues are reported as sticks and colored by atom type: C grey, O red, N blue, S yellow, polar H light grey).
Finally, compounds 27, 29 and 34 were also tested for their antiviral activity over Vero cells infected with CMV and VZV strains, to assess their selectivity. The compounds proved to be selective SARS-CoV-2 antiviral agents with no effects over cytomegalovirus and varicella zoster virus (data not shown).
5. Discussion
Different druggable targets of SARS-CoV-2 have been identified so far and several modulators have been described [15,16,[43], [44], [45]]. First attempts were focused on SARS-CoV-2 spike protein, RdRp (RNA dependent RNA polymerase), and Mpro targeting [46]. Despite little evidence has been collected about the clinical efficacy of these molecules, researchers largely agree about their limitations due to the rapid viral mutations, underlying resistance. In this context, the development of multitarget inhibitors, able to target different proteins belonging to the viral machinery has been explored as a suitable strategy to overcome viral resistance, improve the overall anti-viral efficacy and largely reduce the multi-drug dose burden [[47], [48], [49], [50]]. Moreover, less focus has been given, initially, to other important proteases, such as PLpro, that equally play a pivotal role in the viral replication processes [46,48]. Mpro and PLpro represent two cysteine proteases of the virus, that process the C-terminal of replicase apo-polyprotein to yield the active functional proteins necessary for viral replication and, hence, for infection spreading. Thus, simultaneous inhibition of these proteases may significantly hamper the viral machinery and represents a valuable therapeutic option to SARS-CoV-2 tackling. Some of such dual inhibitors, obtained by drug repurposing or de-novo synthesis, have been yet described. In particular, a library of ebselen derivatives has been synthesized to specifically identify dual Mpro and PLpro inhibitors [51]. These molecules showed nanomolar potency over Mpro, while were thousand-fold less potent over PLpro. Cellular screenings have not been performed for these molecules and there is a general lack of knowledge concerning their efficacy and safety in a relevant biological environment, though ebselen is largely considered non-cytotoxic. On the other hand, drug repurposing in silico protocols have highlighted some natural and synthetic products as dual Mpro and PLpro inhibitors [52,53]. Ginkgolic and anacardic acids, in particular, showed moderate potencies in enzymatic assays, especially over PLpro, cellular plaque formation IC50s in the high micromolar range, but also comparable CC50 values [52]. Ma and coworkers also showed that several compounds identified through in silico screening as potential multitarget proteases inhibitors completely lack of cellular antiviral activity, despite the high potencies showed in enzymatic assays. This is probably due to nonspecific oxidation or alkylation of the cysteine residue in the protease catalytic pocket by reactive compounds [53]. In this context compound 29 represents an advancement of the existing knowledge, being a synthetic molecule with a comparable in-vitro inhibitory potency against Mpro and PLpro and a remarkable ability in reducing viral plaques formation in Vero cells. These antiviral properties are further strengthened by reduced cytotoxicity and good selectivity over other viruses, making derivative 29 an interesting hit compound for the development of anti-SARS-CoV-2 therapeutics. In silico studies, indeed, provided important SAR clues for the rational development of a new class of specific dual proteases inhibitors. On the other side, the extended investigations carried on the synthesized compounds, also led to the identification of compound 34 as a promising selective SARS-CoV-2 spike protein inhibitor, deserving further development in consideration of the binding mode described and the specific drug/target interactions evidenced.
6. Experimental section
6.1. General
All reagents and solvents used were purchased from Sigma-Aldrich (Milan, Italy). Reactions were performed under magnetic stirring in round-bottomed flasks unless otherwise noted. Moisture sensitive reactions were conducted in oven-dried glassware under nitrogen stream, using distilled solvents. Purifications were conducted on the Biotage Isolera One flash purification system, using prepacked KP-sil columns, (Biotage, Uppsala, Sweden). TLC analysis were performed on precoated glass silica gel plates 60 (F254, 0.25 mm, VWR International). 1D-NMR spectra were recorded with Bruker Avance (400 MHz) spectrometer, at room temperature. Chemical shifts were reported in δ values (ppm) relative to internal Me4Si for 1H and 13C NMR. J values were reported in hertz (Hz). The following abbreviations are used to describe peaks: s (singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quartet), and m (multiplet). HR-MS experiments were performed using an LTQ-Orbitrap-XL-ETD mass spectrometer (Thermo Scientific, Bremen, Germany), using electrospray ionization. Elemental analysis was carried on using a PerkinElmer 2400 Series II CHNS/O analyzer. Results obtained were within ±0.4% of theoretical values.
6.1.1. General procedure A: coupling reactions
1 mmol of various N-L-Boc amino acids was dissolved in dichloromethane and HOBt (1.2 eq), HBTU (1.2 eq), DIPEA (2.4 eq) and the corresponding amine (1.2 eq) were added and stirred at room temperature for 12 h. Then, the solvent was evaporated in vacuum, and the residue was dissolved in dichloromethane and washed with water (3 × 200 mL), a saturated solution of NaHCO3 (3 × 200 mL), and a solution of citric acid (10% w:w, 3 × 200 mL). The organic phase was extracted, dried over Na2SO4, filtered, and concentrated under vacuum. The crude products were purified by flash chromatography using mixtures of n-hexane/ethyl acetate as mobile phase [54].
6.1.2. General procedure B: Boc removal
The N-Boc protected intermediate (0.2 mmol) was dissolved in a mixture of TFA/DCM (1/3, v/v), and added with triisopropylsilane (0.25 equiv). Reaction was stirred at room temperature for 2h. Then, a solution of NaOH (2 N) was added until pH 7. The mixture was diluted with water and dichloromethane, and the organic phase was extracted, dried over Na2SO4, filtered, and concentrated under vacuum. The intermediates obtained were not further purified.
6.1.3. General procedure C: reductive amination
The proper intermediate (1 mmol) was dissolved in MeOH dry and 1.2 equivalents of the proper aldehyde were added. The mixture was allowed to react for 12h under nitrogen stream, at room temperature. Then, 3 equivalents of NaBH4 were added portionwise and the mixture was stirred for further 3h. The reaction was quenched by 10% aqueous solution of citric acid, the solvent was evaporated in vacuum, and the residue was dissolved in dichloromethane and washed with water (3 × 200 mL). Organic layer was separated, dried over anhydrous Na2SO4, filtered, and evaporated in vacuo. The crude products were purified by column chromatography using mixtures of ethyl acetate/n-hexane as eluent.
6.1.4. General procedure D: N-alkylation
Methyl indole-5-carboxylate (1.0 mmol) was dissolved in anhydrous DMF under magnetic stirring at 0 °C. To this solution, 1.5 equivalents of NaH and 1.5 equivalents of iodomethane or 1-iodo-2-methylpropane in DMF were added dropwise and the reaction was warmed to room temperature and maintained under stirring overnight. The reaction was quenched by 10% aqueous solution of citric acid and washed with brine. Organic layer was separated, dried over anhydrous Na2SO4, filtered, and evaporated in vacuo. Crude products were purified by column chromatography.
6.1.5. General procedure E: Mannich reactions
A solution of formaldehyde (2.0 eq), trifluoroacetic acid (2.0 eq) and amine (2.0 eq) in dichloromethane was stirred at room temperature for 30 min. Then, a solution of the proper 1,5-disubstituted indole (1 mmol) in dichloromethane was added and the mixture was stirred for 12h. The reaction was quenched by 10% aqueous solution of sodium bicarbonate and washed with brine, dried over anhydrous Na2SO4 and filtered. Organic phase was evaporated in vacuum and 3-aminomethyl indole derivatives were obtained after purification by flash chromatography.
6.1.5.1. tert-butyl (1-(tert-butylamino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)carbamate (1)
Synthesized according to the general procedure A, using Boc-L-Trp-OH and tert-butylamine as starting materials, FC in n-hexane/ethyl acetate 3/1, Rf = 0.35. Yellowish oil (65% yield). 1H NMR (400 MHz, CDCl3): δ: 1.14 (s, 9H, CH 3); 1.46 (s, 9H, CH 3); 3.07–3.12 (m, 1H, CH 2a); 3.30–3.35 (m, 1H, CH 2b); 4.36 (bs, 1H, CH); 5.37 (bs, 1H, NH); 7.06 (s, 1H aryl); 7.15 (t, 1H, aryl, J = 7.2 Hz); 7.22 (t, 1H, aryl, J = 7.4 Hz); 7.39 (d, 1H, aryl, J = 8.2 Hz); 7.72 (d, 1H, aryl, J = 6.8 Hz); 8.43 (bs, 1H, NH). HR-MS m/z calcd for C20H30N3O3 [(M + H)]+: 360.2282; found 360.2277.
6.1.5.2. (S)-tert-butyl (1-(([1,1′-biphenyl]-4-ylmethyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)carbamate (2)
Intermediate 2 was obtained according to the general procedure A, starting from Boc-L-Trp-OH and 4-phenyl benzylamine, FC in n-hexane/ethyl acetate 3/1, Rf = 0.4. Yellowish oil (82% yield). 1H NMR (400 MHz, CDCl3): δ: 1.44 (s, 9H, CH 3); 3.22 (dd, 1H, CH 2a , J’ = 7.7, J” = 14.4 Hz); 3.38 (dd, 1H, CH 2b, J’ = 5.3, J” = 14.4 Hz); 4.30–4.36 (m, 2H, CH 2); 4.49 (bs, 1H, CH); 5.20 (bs, 1H, NH); 6.05 (bs, 1H, NH); 7.02 (s, 1H aryl); 7.17 (d, 1H, aryl, J = 7.6 Hz); 7.19 (t, 1H, aryl, J = 7.8 Hz); 7.24 (t, 1H, aryl, J = 7.8 Hz); 7.35–7.40 (m, 2H, aryl); 7.44–7.48 (m, 5H, aryl); 7.56 (d, 2H, aryl, J = 8.5 Hz); 7.71 (d, 1H, aryl, J = 7.8 Hz); 8.07 (bs, 1H, NH). HR-MS m/z calcd for C29H32N3O3 [(M + H)]+: 470.2438; found 470.2445.
6.1.5.3. (S)-tert-butyl (1-(benzylamino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)carbamate (3)
Synthesized according to the general procedure A, starting from Boc-L-Trp-OH and benzylamine.
FC in n-hexane/ethyl acetate 3/1, Rf = 0.4. Yellowish oil (80% yield). 1H NMR (400 MHz, CD3OD): δ: 1.40 (s, 9H, CH 3); 3.10 (dd, 1H, CH 2a, J’ = 6.6, J” = 13.8 Hz); 3.25 (dd, 1H, CH 2b, J’ = 6.9, J” = 14.4 Hz); 4.19–4.23 (m, 1H, CH); 4.34–4.41 (m, 2H, CH 2); 7.02–7.06 (m,5H, aryl); 7.12 (t, 1H, aryl, J = 7.1 Hz); 7.20–7.25 (m, 2H, aryl); 7.37 (d, 1H, aryl, J = 8.1 Hz); 7.62 (d, 1H, aryl, J = 7.8 Hz). HR-MS m/z calcd for C23H28N3O3 [(M + H)]+: 394.2125; found 394.2114.
6.1.5.4. tert-butyl (1-(tert-butylamino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)carbamate (4)
Intermediate 4 was synthesized according to the general procedure B, starting from 1. White powder (90% yield). 1H NMR (400 MHz, CDCl3): δ: 1.22 (s, 9H, CH 3); 3.10 (dd, 1H, CH 2a J’ = 7.8, J” = 14.5 Hz); 3.30 (dd, 1H, CH 2b J’ = 5.9, J” = 14.5 Hz); 3.86 (t, 1H, CH, J = 6.7 Hz); 5.09 (bs, 2H, NH 2); 6.78 (s, 1H aryl); 7.10–7.17 (m, 2H, aryl); 7.36 (d, 1H, aryl, J = 8.1 Hz); 7.64 (d, 1H, aryl, J = 7.8 Hz); 8.92 (bs, 1H, NH). HR-MS m/z calcd for C15H22N3O [(M + H)]+: 260.1757; found 260.1750.
6.1.5.5. (S)–N-([1,1′-biphenyl]-4-ylmethyl)-2-amino-3-(1H-indol-3-yl)propanamide (5)
Intermediate 5 was synthesized according to the general procedure B, starting from 2. White powder (91% yield). 1H NMR (400 MHz, CD3OD): δ: 3.27–3.35 (m, 1H, CH 2a); 3.42 (dd, 1H, CH 2b, J’ = 7.6, J” = 14.4 Hz); 4.16 (t, 1H, CH, J = 7.4 Hz); 4.31 (d, 1H, CH 2a , J = 14.8 Hz); 4.40 (d, 1H, CH 2b, J = 14.8 Hz); 7.07–7.19 (m, 6H aryl); 7.41–7.45 (m, 3H, aryl); 7.50 (d, 2H, aryl, J = 8.2 Hz); 7.58 (d, 2H, aryl, J = 7.2 Hz); 7.66 (d, 1H, aryl, J = 7.9 Hz). HR-MS m/z calcd for C24H24N3O [(M + H)]+: 370.1914; found 370.1907.
6.1.5.6. (S)-2-Amino-N-benzyl-3-(1H-indol-3-yl)propanamide (6)
Intermediate 6 was synthesized according to the general procedure B, starting from 3. White powder (95% yield). 1H NMR (400 MHz, CD3OD): δ: 3.05 (dd, 1H, CH 2a, J’ = 5.8, J” = 10.4 Hz); 3.20 (dd, 1H, CH 2b, J’ = 6.0, J” = 10.4 Hz); 3.69 (t, 1H, CH, J = 5.2 Hz); 4.25 (d, 1H, CH 2a, J = 12.6 Hz); 4.35 (d, 1H, CH 2b, J = 12.6 Hz); 7.02–7.07 (m, 5H, aryl); 7.12 (t, 1H, aryl, J = 7.2 Hz); 7.20–7.26 (m, 2H, aryl); 7.39 (d, 1H, aryl, J = 8.2 Hz); 7.64 (d, 1H, aryl, J = 8.0 Hz). HR-MS m/z calcd for C18H20N3O [(M + H)]+: 294.1601; found 294.1610.
6.1.5.7. tert-butyl 2-((1-(tert-butylamino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)carbamoyl)pyrrolidine-1-carboxylate (7)
Final product 7 was synthesized starting from 1 and L-Boc-Pro-OH, following the procedure A. FC dichloromethane/methanol 4.8/0.2, Rf = 0.40. Yellow oil (59% yield). (A) 1H NMR (400 MHz, CD3OD): δ: 1.27 (s, 9H, CH 3); 1.29 (s, 9H, CH 3); 1.64–1.89 (m, 2H, CH 2); 2.11–2.20 (m, 2H, CH 2); 3.03–3.39 (m, 4H, CH 2); 4.08–4.10 (m, 1H, CH); 4.54–4.57 (m, 1H, CH); 7.05–7.17 (m, 3H, aryl); 7.33–7.40 (m, 1H, aryl); 7.60–7.69 (m, 1H, aryl). (B) 1H NMR (400 MHz, CD3OD): δ: 1.21 (s, 9H, CH 3); 1.34 (s, 9H, CH 3); 1.64–1.89 (m, 4H, CH 2); 3.03–3.39 (m, 4H, CH 2); 4.14–4.16 (m, 1H, CH); 4.63–4.65 (m, 1H, CH); 7.05–7.17 (m, 3H, aryl); 7.33–7.40 (m, 1H, aryl); 7.60–7.69 (m, 1H, aryl).13C NMR (100 MHz, CD3OD) δ 22.3, 23.0, 23.9, 26.2, 27.1, 27.4, 28.0, 29.5, 29.7, 30.9, 54.4, 60.4, 60.7, 80.1, 108.4, 109.4, 110.8, 111.2, 117.7, 118.1, 118.4, 118.8, 121.1, 121.3, 123.3, 123.5, 124.7, 127.6, 136.6, 154.6, 155.2, 171.3, 173.2, 173.8. HR-MS m/z calcd for C25H37N4O4 [(M + H)]+: 457.2809; found 457.2801.
6.1.5.8. tert-butyl (1-((1-(tert-butylamino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)amino)-1-oxo-3-phenylpropan-2-yl)carbamate (8)
Obtained from 4 and L-Boc-Phe-OH following the general procedure A. FC in n-hexane/ethyl acetate 3/1, Rf = 0.44. Yellowish oil (78% yield). 1H NMR (400 MHz, DMSO): δ: 1.18 (s, 9H, CH 3); 1.29 (s, 9H, CH 3); 2.67–2.73 (m, 1H, CH 2a); 2.89 (dd, 1H, CH 2b J’ = 4.2, J” = 13.8 Hz); 2.95–3.07 (m, 2H, CH 2); 4.08–4.13 (m, 1H, CH); 4.48–4.53 (m, 1H, CH); 6.92–6.99 (m, 2H aryl); 7.06 (t, 1H, aryl, J = 7.9 Hz); 7.14–7.26 (m, 3H, aryl); 7.31 (d, 1H, aryl, J = 8.0 Hz); 7.43 (s, 1H, aryl); 7.58 (d, 1H, aryl, J = 8.0 Hz); 7.80 (d, 1H, aryl, J = 7.8 Hz); 10.81 (bs, 1H, NH). HR-MS m/z calcd for C29H39N4O4 [(M + H)]+: 507.2966; found 507.2974.
6.1.5.9. tert-butyl (1-((1-(tert-butylamino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (9)
Obtained from 4 and L-Boc-Leu-OH following the general procedure A. FC in n-hexane/ethyl acetate 2/1, Rf = 0.35. Yellowish oil (74% yield). 1H NMR (400 MHz, CDCl3): δ: 0.92–0.95 (m, 6H, CH 3); 1.17 (s, 9H, CH 3); 1.35 (s, 9H, CH 3); 1.64–1.67 (m, 1H, CH); 1.79–1.87 (m, 2H, CH 2); 3.08 (dd, 1H, CH 2a J’ = 8.0, J” = 14.4 Hz); 3.39–3.44 (m, 1H, CH 2b); 4.03–4.07 (m, 1H, CH); 4.63 (q, 1H, CH, J = 7.9 Hz); 4.71 (d, 1H, NH, J = 6.9 Hz); 5.62 (bs, 1H, NH); 7.09 (s, 1H, aryl); 7.17 (t, 1H, aryl, J = 7.6 Hz); 7.23 (d, 1H, aryl, J = 7.4 Hz); 7.39 (d, 1H, aryl, J = 8.0 Hz); 7.73 (d, 1H, aryl, J = 7.6 Hz); 8.22 (bs, 1H, NH). HR-MS m/z calcd for C26H41N4O4 [(M + H)]+: 473.3122; found 473.3109.
6.1.5.10. tert-butyl ((S)-1-(((S)-1-(tert-butylamino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)amino)-1-oxopent-4-yn-2-yl)carbamate (10)
Compound 10 was obtained using general procedure A, starting from intermediate 4 which was reacted with N-Boc-l-propargylglycine. FC in n-hexane/ethyl acetate 2/1, Rf = 0.36. Yellowish oil (80% yield). 1H NMR (400 MHz, CDCl3): δ: 1.18 (s, 9H, CH 3); 1.36 (s, 9H, CH 3); 2.08 (s, 1H, CH); 2.56–2.62 (m, 1H, CH 2a); 2.81–2.85 (m, 1H, CH 2b); 3.08 (dd, 1H, CH 2a J’ = 7.8, J” = 14.4 Hz); 3.46–3.51 (m, 1H, CH 2b); 4.22–4.24 (m, 1H, CH); 4.63–4.69 (m, 1H, CH); 5.24 (d, 1H, NH, J = 4.8 Hz); 5.62 (bs, 1H, NH); 7.09 (s, 1H, aryl); 7.16 (t, 1H, aryl, J = 7.9 Hz); 7.23 (t, 1H, aryl, J = 7.8 Hz); 7.39 (d, 1H, aryl, J = 8.0 Hz); 7.75 (d, 1H, aryl, J = 7.7 Hz); 8.43 (bs, 1H, NH). HR-MS m/z calcd for C25H35N4O4 [(M + H)]+: 455.2653; found 455.2660.
6.1.5.11. tert-butyl ((S)-1-(((S)-1-(([1,1′-biphenyl]-4-ylmethyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)amino)-1-oxopent-4-en-2-yl)carbamate (11)
Obtained from 5 and Boc-L-allylgly-OH following the general procedure A. FC in hexane/ethyl acetate 3/1, Rf = 0.45. White powder (75% yield). 1H NMR (400 MHz, CD3OD): δ: 1.36 (s, 9H, CH 3); 2.21–2.26 (m, 1H, CH 2a); 2.28–2.36 (m, 1H, CH 2b); 3.02 (dd, 1H, CH 2a, J’ = 7.4, J” = 14.4 Hz); 3.14 (dd, 1H, CH 2b, J’ = 6.5, J” = 14.4 Hz); 3.96–4.02 (m, 1H, CH); 4.21–4.32 (m, 2H, CH 2); 4.62 (q, 1H, CH, J = 7.2 Hz); 4.96–5.04 (m, 2H, CH 2); 5.61–5.73 (m, 1H, CH); 6.87 (d, 1H, aryl, J = 8.1 Hz); 6.99 (t, 1H, aryl, J = 7.5 Hz); 7.07 (t, 1H, aryl, J = 7.8 Hz); 7.13–7.15 (m, 1H aryl); 7.34–7.38 (m, 2H, aryl); 7.46 (t, 2H, aryl, J = 7.5 Hz); 7.52 (d, 2H, aryl, J = 8.2 Hz); 7.59–7.64 (m, 2H, aryl); 7.93 (d, 1H, aryl, J = 8.0 Hz); 8.43 (t, 1H, aryl, J = 5.6 Hz); 10.9 (s, 1H, NH). HR-MS m/z calcd for C34H39N4O4 [(M + H)]+: 567.2966; found 567.2958.
6.1.5.12. tert-butyl ((S)-1-(((S)-1-(([1,1′-biphenyl]-4-ylmethyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)amino)-1-oxopent-4-yn-2-yl)carbamate (12)
Compound 12 was synthesized in 82% yield starting from intermediate 5 and Boc-L-Pra-OH following the general procedure A. FC in n-hexane/ethyl acetate 3/1, Rf = 0.45. White powder. 1H NMR (400 MHz, CD3OD): δ: 1.32 (s, 9H, CH 3); 2.35 (s, 1H, CH); 2.54–2.65 (m, 2H, CH 2); 3.27–3.37 (m, 2H, CH 2); 4.15 (t, 1H, CH, J = 6.2 Hz); 4.25–4.37 (m, 2H, CH 2); 4.73 (t, 1H, CH, J = 6.0 Hz); 7.04–7.15 (m, 4H, aryl); 7.33 (t, 1H, aryl, J = 7.4 Hz); 7.37–7.47 (m, 6H, aryl); 7.58 (d, 2H, aryl, J = 9.2 Hz); 7.65 (d, 1H, aryl, J = 7.6 Hz). HR-MS m/z calcd for C34H37N4O4 [(M + H)]+: 565.2809; found 565.2815.
6.1.5.13. tert-butyl ((S)-1-(((S)-1-(benzylamino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)amino)-1-oxopent-4-en-2-yl)carbamate (13)
Obtained from coupling reaction (procedure A) between 6 and Boc-L-allylgly-OH. FC in hexane/ethyl acetate 2/1, Rf = 0.40. White powder (78% yield). 1H NMR (400 MHz, CD3OD): δ: 1.27 (s, 9H, CH 3); 2.25–2.34 (m, 1H, CH 2a); 2.44–2.48 (m, 1H, CH 2b); 3.28 (d, 2H, CH, J = 4.6 Hz); 4.02 (t, 1H, CH, J = 5.2 Hz); 4.21–4.26 (m, 1H, CH 2a); 4.32 (dd, 1H, CH 2b, J’ = 3.4, J” = 11.9 Hz); 4.70 (t, 1H, CH, J = 4.5 Hz); 5.04–5.11 (m, 2H, CH 2); 5.67–5.75 (m, 1H, CH); 7.03–7.10 (m, 5H, aryl); 7.14 (t, 1H, aryl, J = 5.6 Hz); 7.20–7.25 (m, 2H, aryl); 7.38 (d, 1H, aryl, J = 6.5 Hz); 7.63 (d, 1H, aryl, J = 6.2 Hz). HR-MS m/z calcd for C28H35N4O4 [(M + H)]+: 491.2653; found 491.2661.
6.1.5.14. tert-butyl ((S)-1-(((S)-1-(benzylamino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (14)
The intermediate 14 was obtained using L-Boc-Leu-OH and 6 as starting material, following general procedure A. FC in hexane/ethyl acetate 2/1, Rf = 0.45. White powder (77% yield). 1H NMR (400 MHz, DMSO): δ: 0.79 (d, 3H, CH 3, J = 6.4 Hz); 0.83 (d, 3H, CH 3, J = 6.5 Hz); 1.24–1.35 (m, 12H, CH, CH 2, CH 3); 3.01–3.15 (m, 2H, CH 2); 3.90–3.96 (m, 1H, CH); 4.22 (d, 2H, CH 2, J = 5.6 Hz); 4.59 (q, 1H, CH, J = 6.9 Hz); 6.97 (t, 1H, aryl, J = 6.9 Hz); 7.04–7.11 (m, 3H, aryl); 7.18–7.25 (m, 3H, aryl); 7.33 (d, 1H, aryl, J = 8.0 Hz); 7.57 (d, 1H, aryl, J = 7.7 Hz); 7.81 (d, 1H, aryl, J = 8.0 Hz); 8.40 (t, 1H, NH, J = 5.1 Hz); 10.82 (s, 1H, NH). HR-MS m/z calcd for C29H38N4O4 [(M + H)]+: 506.2888; found 506.2899.
6.1.5.15. 2-Amino-N-(1-(tert-butylamino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-3-phenylpropanamide (15)
Intermediate 15 was synthesized according to the general procedure B, starting from 8. White powder (95% yield). FC in hexane/ethyl acetate 7/3, Rf: 0.47. Yellow oil (55% yield). 1H NMR (400 MHz, CDCl3): δ: 1.17 (s, 9H, CH 3); 2.71 (dd, 1H, CH 2a J’ = 8.3, J” = 13.6 Hz); 3.06 (dd, 1H, CH 2b J’ = 8.0, J” = 14.5 Hz); 3.13 (dd, 1H, CH 2a J’ = 4.9, J” = 13.8 Hz); 3.23 (dd, 1H, CH 2b, J’ = 6.5, J” = 14.6 Hz); 3.73 (t, 1H, CH, J = 5.3 Hz); 4.62 (q, 1H, CH, J = 7.6 Hz); 5.63 (bs, 1H, NH); 7.05 (s, 1H, aryl); 7.11–7.30 (m, 6H, aryl); 7.35 (d, 1H, aryl, J = 8.0 Hz); 7.68 (d, 1H, aryl, J = 8.0 Hz); 7.88 (d, 1H, aryl, J = 7.1 Hz); 8.29 (bs, 1H, NH). HR-MS m/z calcd for C24H31N4O2 [(M + H)]+: 407.2442; found 407.2451.
6.1.5.16. 2-Amino-N-(1-(tert-butylamino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-4-methylpentanamide (16)
Intermediate 16 was synthesized according to the general procedure B, starting from 9. White powder (94% yield). 1H NMR (400 MHz, CD3OD): δ: 0.91 (d, 3H, CH 3, J = 6.5 Hz); 0.94 (d, 3H, CH 3, J = 6.5 Hz); 1.39–1.48 (m, 1H, CH 2a); 1.54–1.61 (m, 1H, CH 2b); 1.65–1.71 (m, 1H, CH); 3.24 (dd, 1H, CH 2a J’ = 7.8, J” = 14.7 Hz); 3.34 (dd, 1H, CH 2b, J’ = 5.5, J” = 14.7 Hz); 3.56 (dd, 1H, CH, J’ = 6.0, J” = 8.4 Hz); 7.03 (t, 1H, aryl, J = 7.9 Hz); 7.11 (t, 1H, aryl, J = 8.1 Hz); 7.13 (s, 1H, aryl); 7.37 (d, 1H, aryl, J = 8.1 Hz); 7.55 (d, 1H, aryl, J = 7.9 Hz). HR-MS m/z calcd for C21H33N4O2 [(M + H)]+: 373.2598; found 373.2604.
6.1.5.17. tert-butyl ((S)-1-(((S)-1-(tert-butylamino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)amino)-1-oxopent-4-yn-2-yl)carbamate (17)
Intermediate 17 was synthesized according to the general procedure B, starting from 10. White powder (92% yield). 1H NMR (400 MHz, CDCl3): δ: 1.15 (s, 9H, CH 3); 1.96 (t, 1H, CH, J = 2.6 Hz); 2.47–2.54 (m, 1H, CH 2a); 2.62–2.68 (m, 1H, CH 2b); 3.12 (dd, 1H, CH 2a, J’ = 8.3, J” = 14.4 Hz); 3.30 (dd, 1H, CH 2b J’ = 6.7, J” = 15.0 Hz); 3.49 (dd, 1H, CH, J’ = 4.6, J” = 7.1 Hz); 4.63 (q, 1H, CH, J = 8.1 Hz); 5.57 (bs, 1H, NH); 7.07 (s, 1H, aryl); 7.14 (t, 1H, aryl, J = 7.2 Hz); 7.21 (t, 1H, aryl, J = 7.0 Hz); 7.37 (d, 1H, aryl, J = 8.0 Hz); 7.74 (d, 1H, aryl, J = 7.8 Hz); 8.49 (bs, 1H, NH). HR-MS m/z calcd for C20H27N4O2 [(M + H)]+: 355.2129; found 355.2136.
6.1.5.18. (S)–N-((S)-1-(([1,1′-biphenyl]-4-ylmethyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-2-aminopent-4-enamide (18)
Intermediate 18 was synthesized according to the general procedure B, starting from 11. White powder (89% yield). 1H NMR (400 MHz, CD3OD): δ: 1.95–2.03 (m, 1H, CH 2a); 2.33–2.38 (m, 1H, CH 2b); 3.18–3.21 (m, 1H, CH); 3.26–3.35 (m, 2H, CH 2); 4.28–4.40 (m, 2H, CH 2); 4.93–5.04 (m, 3H, CH 2 and CH); 5.45–5.55 (m, 1H, CH); 6.94 (s, 1H, aryl); 7.06–7.11 (m, 3H, aryl); 7.19 (t, 1H, aryl, J = 7.7 Hz); 7.34–7.39 (m, 3H, aryl); 7.43–7.47 (m, 4H aryl); 7.56 (d, 1H, aryl, J = 7.2 Hz); 7.68 (d, 1H, aryl, J = 8.0 Hz); 8.14 (d, 1H, NH, J = 8.2 Hz); 8.95 (s, 1H, NH). HR-MS m/z calcd for C29H31N4O2 [(M + H)]+: 467.2442; found 467.2436.
6.1.5.19. (S)–N-((S)-1-(([1,1′-biphenyl]-4-ylmethyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-2-aminopent-4-ynamide (19)
Intermediate 19 was synthesized according to the general procedure B, starting from 12. White powder (95% yield). 1H NMR (400 MHz, CD3OD): δ: 2.49 (s, 1H, CH); 2.66 (dd, 1H, CH 2a, J’ = 7.2, J” = 14.8 Hz); 2.75 (dd, 1H, CH 2b, J’ = 5.1, J” = 19.7 Hz); 3.21 (dd, 1H, CH 2a, J’ = 6.9, J” = 14.4 Hz); 3.29–3.36 (m, 1H, CH 2b); 3.83 (t, 1H, CH, J = 5.4 Hz); 4.25 (d, 1H, CH 2a, J = 15.0 Hz); 4.37 (d, 1H, CH 2b, J = 15.0 Hz); 4.78 (t, 1H, CH, J = 7.3 Hz); 7.03–7.15 (m, 4H aryl); 7.35 (t, 1H, aryl, J = 7.6 Hz); 7.37–7.49 (m, 6H, aryl); 7.58 (d, 2H, aryl, J = 9.2 Hz); 7.66 (d, 1H, aryl, J = 7.6 Hz). HR-MS m/z calcd for C29H29N4O2 [(M + H)]+: 465.2285; found 465.2292.
6.1.5.20. (S)-2-Amino-N-((S)-1-(benzylamino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)pent-4-enamide (20)
Intermediate 20 was synthesized according to the general procedure B, starting from 13. White powder (93% yield). 1H NMR (400 MHz, CD3OD): δ: 2.55–2.61 (m, 1H, CH 2a); 2.65–2.69 (m, 1H, CH 2b); 3.19 (dd, 1H, CH 2a, J’ = 5.5, J” = 11.3 Hz); 3.31 (dd, 1H, CH 2b, J’ = 6.3, J” = 10.9 Hz); 3.95 (t, 1H, CH, J = 5.5 Hz); 4.25 (dd, 1H, CH 2a, J’ = 3.7, J” = 11.8 Hz); 4.31 (dd, 1H, CH 2b, J’ = 4.4, J” = 12.0 Hz); 4.77 (t, 1H, CH, J = 6.0 Hz); 5.21–5.27 (m, 2H, CH 2); 5.72–5.80 (m, 1H, CH); 7.02–7.07 (m, 3H, aryl); 7.14 (t, 1H, aryl, J = 5.7 Hz); 7.21–7.26 (m, 4H, aryl); 7.39 (d, 1H, aryl, J = 6.4 Hz); 7.66 (d, 1H, aryl, J = 6.3 Hz). HR-MS m/z calcd for C23H27N4O2 [(M + H)]+: 391.2129; found 391.2135.
6.1.5.21. (S)-2-Amino-N-((S)-1-(benzylamino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-4-methylpentanamide (21)
Intermediate 21 was synthesized according to the general procedure B, starting from 14. White powder (95% yield). 1H NMR (400 MHz, CD3OD): δ: 0.87 (d, 3H, CH 3, J = 6.6 Hz); 0.90 (d, 3H, CH 3, J = 6.5 Hz); 1.20–1.28 (m, 1H, CH 2a); 1.38–1.46 (m, 1H, CH 2b); 1.56–1.67 (m, 1H, CH); 3.27–3.37 (m, 3H, CH and CH 2); 4.25 (d, 1H, CH 2a, J = 15.0 Hz); 4.32 (d, 1H, CH 2b, J = 15.0 Hz); 4.72 (t, 1H, CH, J = 7.2 Hz); 7.01–7.13 (m, 6H, aryl); 7.20–7.26 (m, 2H, aryl); 7.36 (d, 1H, aryl, J = 8.1 Hz); 7.64 (d, 1H, aryl, J = 7.8 Hz). HR-MS m/z calcd for C24H31N4O2 [(M + H)]+: 407.2442; found 407.2435.
6.1.5.22. (S)-2-(([1,1′-biphenyl]-4-ylmethyl)amino)-N-((S)-1-(tert-butylamino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-3-phenylpropanamide (22)
Final product 22 was synthesized in 64% yield starting from 15 and 4-phenylbenzaldehyde, following the general procedure C. FC in hexane/ethyl acetate 1/1, Rf = 0.35. Yellowish powder. 1H NMR (400 MHz, CDCl3): δ: 1.18 (s, 9H, CH 3); 2.61 (dd, 1H, CH 2a, J’ = 9.3, J” = 13.8 Hz); 3.11–3.17 (m, 2H, CH 2b and CH 2a); 3.34–3.44 (m, 3H, CH, CH 2b and CH 2a); 3.53, (d, 1H, CH 2a, J = 13.4 Hz); 3.70 (d, 1H, CH 2b, J = 13.6 Hz); 4.69 (dd, 1H, CH, J’ = 8.1, J” = 14.3 Hz); 5.59 (bs, 1H, NH); 7.06 (d, 3H, aryl, J = 8.1 Hz); 7.12–7.30 (m, 6H, aryl); 7.39 (t, 2H, aryl, J = 8.1 Hz); 7.44–7.48 (m, 4H, aryl); 7.58 (d, 2H, aryl, J = 8.4 Hz); 7.76 (d, 1H, aryl, J = 7.8 Hz); 8.07 (d, 1H, aryl, J = 5.8 Hz); 8.39 (bs, 1H, NH). 13C NMR (100 MHz, CDCl3) δ 28.2,28.5, 39.0, 51.1, 52.1, 54.0, 63.0, 111.21, 111.28, 119.2, 119.8, 122.3, 123.2, 127.0, 127.3, 127.5, 128.5, 128.8, 1292, 136.3, 137.2, 138.1, 140.1, 140.9. HR-MS m/z calcd for C37H40N4O2 [(M + H)]+: 573.3230; found 573.3238.
6.1.5.23. 2-(([1,1′-biphenyl]-4-ylmethyl)amino)-N-(1-(tert-butylamino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-4-methylpentanamide (23)
Obtained from 16 and 4-phenylbenzaldehyde following the general procedure C. FC in hexane/ethyl acetate 1/1, Rf = 0.35. Yellowish powder (56% yield). 1H NMR (400 MHz, CDCl3): δ: 0.74 (d, 3H, CH 3, J = 6.4 Hz); 0.81 (d, 3H, CH 3, J = 6.4 Hz); 1.16–1.24 (m, 1H, CH); 1.40–1.45 (m, 1H, CH 2a); 1.47–1.56 (m, 1H, CH 2b); 3.07 (dd, 1H, CH 2a, J’ = 8.0, J” = 14.4 Hz); 3.17–3.20 (m, 1H, CH); 3.24 (dd, 1H, CH 2b, J’ = 6.5, J” = 14.5 Hz); 3.54 (d, 1H, CH 2a, J = 12.9 Hz); 4.59 (q, 1H, CH); 5.54 (bs, 1H, NH); 7.03 (s, 1H, aryl); 7.06 (t, 1H, aryl, J = 7.7 Hz); 7.13 (t, 1H, aryl, J = 7.2 Hz); 7.18–7.21 (m, 2H, aryl); 7.27 (d, 1H, aryl, J = 8.3 Hz); 7.35 (t, 2H, aryl, J = 7.5 Hz); 7.43–7.49 (m, 4H, aryl); 7.69 (d, 1H, aryl, J = 7.7 Hz); 7.85 (d, 1H, aryl, J = 7.9 Hz); 8.06 (s, 1H, NH). 13C NMR (100 MHz, CDCl3) δ 25.0, 28.2, 28.5, 29.7, 42.4, 51.2, 52.1, 54.0, 60.8, 111.2, 111.4, 119.2, 119.8, 122.3, 123.1, 127.1, 127.3, 128.8, 129.0, 136.3, 140.4, 140.8, 170.2. HR-MS m/z calcd for C34H43N4O2 [(M + H)]+: 539.3381; found 539.3390.
6.1.5.24. (S)-2-(([1,1′-biphenyl]-4-ylmethyl)amino)-N-((S)-1-(tert-butylamino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)pent-4-ynamide (24)
Synthesized according to the general procedure C, starting from intermediate 17 and 4-phenylbenzaldehyde. FC in hexane/ethyl acetate 1/1, Rf = 0.30. White powder (57% yield). 1H NMR (400 MHz, CDCl3): δ: 1.24 (s, 9H, CH 3); 2.32 (s, 1H, CH); 2.43–2.58 (m, 2H, CH 2); 3.17–3.26 (m, 2H, CH 2); 3.32–3.34 (m, 1H, CH); 3.55 (dd, 2H, CH 2, J’ = 13.2, J” = 15.2 Hz); 4.69 (t, 1H, CH, J = 7.3 Hz); 7.03 (t, 1H, aryl, J = 7.9 Hz); 7.11 (t, 1H, aryl, J = 7.9 Hz); 7.14 (s, 1H, aryl); 7.23 (d, 2H, aryl, J = 8.2 Hz); 7.33 (d, 2H, aryl, J = 8.9 Hz); 7.42 (t, 2H, aryl, J = 7.4 Hz); 7.49 (d, 2H, aryl, J = 8.2 Hz); 7.58 (d, 2H, aryl, J = 7.2 Hz); 7.68 (d, 1H, aryl, J = 7.6 Hz). 13C NMR (100 MHz, CDCl3) δ 21.7, 27.4, 28.0, 50.8, 54.2, 58.1, 59.6, 71.1, 79.2, 109.5, 110.9, 118.3, 121.1, 123.3, 126.5, 126.8, 127.5, 128.6, 136.7, 138.2, 139.9, 140.8, 171.3, 173.2. HR-MS m/z calcd for C33H37N4O2 [(M + H)]+: 521.2911; found 521.2918.
6.1.5.25. (S)-2-(([1,1′-biphenyl]-4-ylmethyl)amino)-N-((S)-1-(benzylamino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-4-methylpentanamide (25)
Synthesized according to the general procedure C, starting from intermediate 21 and 4-phenylbenzaldehyde. FC in hexane/ethyl acetate 1/1, Rf = 0.48. White powder (60% yield). 1H NMR (400 MHz, CD3OD): δ: 0.78 (d, 3H, CH 3, J = 6.6 Hz); 0.88 (d, 3H, CH 3, J = 6.6 Hz); 1.28–1.40 (m, 2H, CH 2); 1.56–1.63 (m, 1H, CH); 3.13 (t, 1H, CH, J = 6.8 Hz); 3.21 (dd, 1H, CH 2a, J’ = 7.9, J” = 14.5 Hz); 3.28 (dd, 1H, CH 2b, J’ = 6.6, J” = 14.6 Hz); 3.48 (dd, 2H, CH 2, J’ = 13.1, J” = 25.1 Hz); 4.26 (d, 1H, CH 2a, J = 15.0 Hz); 4.36 (d, 1H, CH 2b, J = 15.0 Hz); 4.82 (t, 1H, CH, J = 7.8 Hz); 7.03–7.14 (m, 6H, aryl); 7.17–7.27 (m, 5H, aryl); 7.31–7.36 (m, 1H, aryl); 7.41–7.47 (m, 4H, aryl); 7.57 (d, 2H, aryl, J = 8.5 Hz); 7.67 (d, 1H, aryl, J = 7.8 Hz). 13C NMR (100 MHz, CD3OD) δ 21.3, 21.9, 24.5, 27.9, 42.5, 42.7, 51.2, 53.7, 60.0, 109.4, 111.0, 118.2, 118.5, 121.1, 123.3, 126.5, 126.7, 126.84, 127.04, 128.1, 128.4, 128.7, 136.7, 138.1, 138.4, 139.8, 140.8, 172.4, 175.9. HR-MS m/z calcd for C37H40N4O2 [(M + H)]+: 573.3224; found 573.3230.
6.1.5.26. (S)–N-((S)-1-(([1,1′-biphenyl]-4-ylmethyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-2-(benzylamino)pent-4-enamide (26)
Final product 26 was synthesized as a yellow powder in 66% yield starting from 18 and benzaldehyde and following the general procedure C. FC in hexane/ethyl acetate 1/1, Rf = 0.45. 1H NMR (400 MHz, CD3OD): δ: 2.19–2.31 (m, 2H, CH 2); 3.07 (dd, 1H, CH 2a, J’ = 8.3, J” = 14.4 Hz); 3.18–3.23 (m, 4H, CH 2b, CH 2 and CH); 4.25 (q, 2H, CH 2, J = 15.0 Hz); 4.73–4.76 (m, 1H, CH); 4.88–4.94 (m, 2H, CH 2); 5.47–5.57 (m, 1H, CH); 6.93–7.02 (m, 7H aryl); 7.07 (d, 2H, aryl, J = 8.2 Hz); 7.14 (d, 2H, aryl, J = 2.8 Hz); 7.21 (t, 1H, aryl, J = 6.8 Hz); 7.30 (t, 2H, aryl, J = 7.4 Hz); 7.38 (d, 2H, aryl, J = 8.2 Hz); 7.45 (d, 2H, aryl, J = 7.2 Hz); 7.57 (d, 1H, aryl, J = 7.6 Hz). 13C NMR (100 MHz, CD3OD) δ 28.1, 36.4, 42.4, 50.9, 54.0, 60.2, 109.4, 111.1, 118.09, 118.20, 118.6, 121.2, 123.4, 126.49, 126.66, 126.91, 127.6, 128.23, 128.43, 128.7, 132.5, 136.7, 137.2, 140.0, 140.7, 172.1. HR-MS m/z calcd for C36H37N4O2 [(M + H)]+: 557.2911; found 557.2918.
6.1.5.27. (S)–N-((S)-1-(([1,1′-biphenyl]-4-ylmethyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-2-(benzylamino)pent-4-ynamide (27)
Final product 27 was synthesized in 68% yield starting from 19 and benzaldehyde, following the general procedure C. FC in hexane/ethyl acetate 1/1, Rf = 0.40. White powder (55% yield). 1H NMR (400 MHz, CD3OD): δ: 2.54 (s, 1H, CH); 2.79 (dd, 2H, CH 2, J’ = 2.7, J” = 6.5 Hz); 3.17 (dd, 1H, CH 2a, J’ = 9.4, J” = 14.5 Hz); 3.38 (dd, 1H, CH 2b, J’ = 6.2, J” = 14.8 Hz); 3.43 (d, 1H, CH 2a, J = 13.0 Hz); 3.63 (d, 1H, CH 2b, J = 13.0 Hz); 3.83 (t, 1H, CH, J = 6.5 Hz); 4.42 (q, 2H, CH 2, J = 15.0 Hz); 5.01 (dd, 1H, CH, J’ = 6.0, J” = 9.4 Hz); 7.06–7.09 (m, 2H aryl); 7.13 (d, 2H, aryl, J = 7.0 Hz); 7.19 (s, 1H, aryl); 7.23–7.26 (m, 3H, aryl); 7.38–7.46 (m, 6H, aryl); 7.55 (d, 2H, aryl, J = 8.2 Hz); 7.60 (d, 2H, aryl, J = 7.2 Hz); 7.73–7.76 (m, 1H, aryl). 13C NMR (100 MHz, CD3OD) δ 20.3, 28.3, 42.4, 49.8, 54.4, 57.7, 73.8, 109.5, 111.2, 118.2, 118.6, 121.2, 123.5, 126.5, 126.7, 127.0, 127.4, 127.7, 128.4, 128.8, 129.4, 129.8, 130.3, 136.7, 137.2, 140.1, 140.7, 165.7, 171.6. HR-MS m/z calcd for C36H34N4O2 [(M + H)]+: 555.2755; found 555.2763.
6.1.5.28. (S)–N-((S)-1-(([1,1′-biphenyl]-4-ylmethyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-2-(propylamino)pent-4-ynamide (28)
Final product 28 was synthesized in 64% starting from 19 and propionaldehyde following the general procedure C. FC in hexane/ethyl acetate 1/2, Rf = 0.35. White powder. 1H NMR (400 MHz, CD3OD): δ: 0.95 (t, 3H, CH 3, J = 7.4 Hz); 1.36–1.42 (m, 2H, CH 2); 1.54–1.58 (m, 2H, CH 2); 2.60 (s, 1H, CH); 2.80 (dd, 1H, CH2a, J’ = 7.4, J” = 17.5 Hz); 2.88 (dd, 1H, CH2b , J’ = 8.1, J” = 17.5 Hz); 3.20 (dd, 1H, CH 2a , J’ = 7.0, J” = 14.3 Hz); 3.31–3.37 (m, 1H, CH 2b); 4.05 (dd, 1H, CH, J’ = 5.4, J” = 7.4 Hz); 4.25 (d, 1H, CH 2a, J = 15.1 Hz); 4.36–4.40 (m, 1H, CH 2b); 4.78–4.80 (m, 1H, CH); 7.03–7.15 (m, 4H aryl); 7.34 (t, 1H, aryl, J = 7.4 Hz); 7.38–7.48 (m, 6H, aryl); 7.58 (d, 2H, aryl, J = 8.5 Hz); 7.67 (d, 1H, aryl, J = 7.9 Hz). 13C NMR (100 MHz, CD3OD) δ 12.8, 21.1, 27.9, 34.4, 42.4, 51.4, 54.8, 73.7, 104.5, 109.1, 111.0, 118.0, 118.5, 121.2, 123.4, 126.5, 126.9, 127.3, 127.5, 128.4, 136.7, 139.9, 140.7, 167.0, 171.8. HR-MS m/z calcd for C32H35N4O2 [(M + H)]+: 507.2755; found 507.2747.
6.1.5.29. (S)–N-((S)-1-(benzylamino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)-2-(2-chloroacetamido)pent-4-enamide (29)
Intermediate 20 (1 mmol) was dissolved in THF and added with TEA (1.2 eq), and chloro acetylchloride (1.2 eq). The mixture was allowed to react for 20 min. Afterward, the reaction mixture was diluted with dichloromethane (20 mL), and the resulting solution was washed successively with water (2 × 25 mL), dried over Na2SO4 and evaporated to dryness. The crude products were purified by flash chromatography using n-hexane/ethyl acetate 1/1, Rf = 0.38. White powder (62% yield). 1H NMR (400 MHz, DMSO): δ: 2.24–2.31 (m, 1H, CH 2a); 2.37–2.44 (m, 1H, CH 2b); 2.99 (dd, 1H, CH 2a, J’ = 8.1, J” = 14.5 Hz); 3.15 (dd, 1H, CH 2b, J’ = 4.6, J” = 14.5 Hz); 4.09 (dd, 2H, CH 2, J’ = 13.1, J” = 15.8 Hz); 4.24–4.26 (m, 2H, CH 2); 4.38–4.45 (m, 1H, CH); 4.56–4.61 (m, 1H, CH); 5.00 (dd, 2H, CH 2, J’ = 17.2, J” = 25.5 Hz); 5.61–5.71 (m, 1H, CH); 6.98 (t, 1H, aryl, J = 7.8 Hz); 7.06 (t, 1H, aryl, J = 7.0 Hz); 7.10–7.13 (m, 2H, aryl); 7.19–7.28 (m, 3H, aryl); 7.34 (d, 1H, aryl, J = 8.1 Hz); 8.25 (d, 1H, aryl, J = 8.0 Hz); 8.40 (t, 1H, aryl, J = 5.9 Hz); 10.83 (s, 1H, NH). 13C NMR (100 MHz, DMSO) δ 28.3, 36.9, 40.5, 42.5, 43.0, 52.7, 54.1, 110.3, 111.7, 118.1, 118.9, 121.3, 124.1, 127.1, 127.4, 127.8, 128.6, 134.3, 136.5, 139.6, 166.1, 170.7, 171.6. HR-MS m/z calcd for C25H27ClN4O3 [(M + H)]+: 467.1844; found 467.1852.
6.1.5.30. Methyl 1-isobutyl-1H-indole-5-carboxylate (30)
Intermediate 30 was obtained according to the general procedure D, starting from methyl indole-5-carboxylate and 1-iodo-2-methylpropane, as previously described [31]. FC in n-hexane/ethyl acetate 3/1, Rf = 0.4. Yellowish oil (80% yield). 1H NMR (400 MHz, CDCl3): δ: 0.94 (d, 6H, CH 3, J = 6.7 Hz); 2.17–2.24 (m, 1H, CH); 3.93 (d, 2H, CH 2, J = 7.3 Hz); 3.96 (s, 3H, CH 3); 6.61 (d, 1H, aryl, J = 3.2 Hz); 7.14 (d, 1H, aryl, J = 3.1 Hz); 7.35 (d, 1H, aryl, J = 8.7 Hz); 7.94 (d, 1H, aryl, J = 10.1 Hz); 8.44 (s, 1H, aryl). HR-MS m/z calcd for C14H18NO2 [(M + H)]+: 232.1332; found 232.1340.
6.1.5.31. Methyl 1-methyl-1H-indole-5-carboxylate (31)
Intermediate 31 was obtained according to the general procedure D, starting from methyl indole-5-carboxylate and iodomethane, as previously described [30]. FC in n-hexane/ethyl acetate 3/1, Rf = 0.4. Yellowish oil (82% yield). 1H NMR (400 MHz, CDCl3): δ: 3.85 (s, 3H, CH 3); 3.92 (s, 3H, CH 3); 6.57 (d, 1H, aryl, J = 3.4 Hz); 7.26 (d, 1H, aryl, J = 3.4 Hz); 7.43 (d, 1H, aryl, J = 9.2 Hz); 7.87 (d, 1H, aryl, J = 9.6 Hz); 8.32 (s, 1H, aryl). HR-MS m/z calcd for C11H12NO2 [(M + H)]+: 190.0863; found 190.0871.
6.1.5.32. Methyl 3-((([1,1′-biphenyl]-4-ylmethyl)amino)methyl)-1-isobutyl-1H-indole-5-carboxylate (32)
The compound 32 was obtained using general procedure E, starting from intermediate 30 which was reacted with 2-([1,1′-biphenyl]-4-yl)ethan-1-amine. FC in dichloromethane/methanol 4.8/0.2. Rf = 0.42. Yellowish oil (70% yield). 1H NMR (400 MHz, CDCl3): δ: 3.83 (s, 3H, CH 3); 3.86 (s, 2H, CH 2); 3.93 (s, 3H, CH 3); 3.99 (s, 2H, CH 2); 7.28 (s, 1H, aryl); 7.32–7.38 (m, 5H, aryl); 7.43 (d, 1H, aryl, J = 8.8 Hz); 7.88 (d, 1H, aryl, J = 9.2 Hz); 8.35 (s, 1H, aryl). HR-MS m/z calcd for C28H31N2O2 [(M + H)]+: 427.2380; found 427.2389.
6.1.5.33. Methyl 3-(((2-([1,1′-biphenyl]-4-yl)ethyl)amino)methyl)-1-isobutyl-1H-indole-5-carboxylate (33)
The compound 33 was obtained using general procedure E, starting from intermediate 31 which was reacted with benzylamine. FC in dichloromethane/methanol 4.8/0.2. Rf = 0.45. Yellowish oil (75% yield). 1H NMR (400 MHz, CDCl3): δ: 0.94 (d, 6H, CH 3, J = 6.6 Hz); 2.14–2.25 (m, 1H, CH); 2.94 (t, 2H, CH 2, J = 7.3 Hz); 3.06 (t, 2H, CH 2, J = 6.9 Hz); 3.89 (d, 2H, CH 2, J = 7.3 Hz); 3.94 (s, 3H, CH 3); 4.09 (s, 2H, CH 2); 7.10 (s, 1H, aryl); 7.31–7.33 (m, 4H, aryl); 7.36 (d, 1H, aryl, J = 7.4 Hz); 7.45 (t, 2H, aryl, J = 7.8 Hz); 7.55 (d, 2H, aryl, J = 8.2 Hz); 7.60 (d, 2H, aryl, J = 8.9 Hz); 7.94 (d, 1H, aryl, J = 10.1 Hz); 8.43 (s, 1H, NH). HR-MS m/z calcd for C29H33N2O2 [(M + H)]+: 441.2537; found 441.2549.
6.1.5.34. Methyl 3-((([1,1′-biphenyl]-4-ylmethyl)(benzyl)amino)methyl)-1-isobutyl-1H-indole-5-carboxylate (34)
Final compound 34 was obtained following general procedure C, using as starting materials intermediate 32 and benzaldehyde. FC in dichloromethane/methanol 4.8/0.2. Rf = 0.27. Yellowish oil (78% yield). 1H NMR (400 MHz, CDCl3): δ: 0.94 (d, 6H, CH 3, J = 6.7 Hz); 2.15–2.25 (m, 1H, CH); 2.86–2.89 (m, 2H, CH 2); 2.94–2.98 (m, 2H, CH 2); 3.80 (s, 2H, CH 2); 3.89 (d, 2H, CH 2 , J = 7.4 Hz); 3.94 (s, 2H, CH 2); 3.99 (s, 3H, CH 3); 7.02 (s, 1H, aryl); 7.23 (d, 2H, aryl, J = 8.1 Hz); 7.28–7.41 (m, 5H, aryl); 7.45–7.53 (m, 6H, aryl); 7.64 (d, 2H, aryl, J = 7.1 Hz); 7.98 (d, 1H, aryl, J = 8.7 Hz); 8.54 (s, 1H, aryl). 13C NMR (100 MHz, CDCl3) δ 203, 29.7, 33.2, 49.2, 51.8, 54.1, 55.2, 58.6, 65.9, 109.2, 114.0, 120.8, 123.0, 126.9, 127.8, 128.3, 128.4, 128.9, 129.4, 138.7, 139.3, 140.0, 141.1, 168.3. HR-MS m/z calcd for C35H38N2O2 [(M + H)]+: 531.3001; found 531.2994.
6.1.5.35. (S)-Methyl 3-((N-benzyl-2-((tert-butoxycarbonyl)amino)pent-4-ynamido)methyl)-1-methyl-1H-indole-5-carboxylate (35)
Final compound 35 was obtained following general procedure A, using as starting materials intermediate 33 and Boc-L-Pra-OH. FC in dichloromethane/methanol 4.8/0.2. Rf = 0.3. White powder (80% yield). 1H NMR (400 MHz, CD3OD): δ: 1.34 (s, 9H, CH 3); 2.59–2.68 (m, 3H, CH 2 and CH); 3.79 (s, 3H, CH 3); 3.92 (s, 3H, CH 3); 4.51–4.63 (m, 4H CH 2); 5.06 (t, 1H, CH, J = 6.5 Hz); 7.19–7.44 (m, 5H, aryl); 7.88 (t, 2H, aryl, J = 7.1 Hz); 8.21 (s, 1H, aryl); 8.29 (s, 1H, aryl). 13C NMR (100 MHz, CD3OD) δ 22.0, 27.2, 31.61, 31.74, 39.2, 42.1, 49.2, 49.9, 51.0, 79.3, 111.1, 120.8, 121.1, 122.1, 122.6, 126.8, 127.2, 128.1, 128.4, 130.0, 130.8, 136.4, 136.9, 139.8, 155.8, 168.5, 171.4. HR-MS m/z calcd for C29H33N3O5 [(M + H)]+: 504.2493; found 504.2501.
6.1.6. Enzymatic assays
6.1.6.1. Mpro enzymatic assay
The assay was performed in a volume of 25 μL in black 384-well OptiPlate. A fluorescent FRET substrate (DABCYL-KTSAVLQSGFRKME-EDANS) harboring the cleavage site of SARS-COV-2 Mpro and aqueous buffer solution (40 mM Tris-HCl, pH 8.0, 110 mM NaCl, 2.2 mM KCl, 20% glycerol, 3 mM DTT, 8 mM maltose) were used for the inhibition assay (BPS Bioscience 3CL Protease, MBP-tagged Assay). Mpro recombinant protease, at a final concentration of 150 ng per reaction, was preincubated for 30 min at room temperature with the compounds at different concentrations. Finally, the reaction was initiated by adding 5 μl of the FRET substrate to each well (final concentration, 50 μM). Buffer with the same amount of DMSO (1%) was used as control and Mpro inhibitor GC376 is also included as a positive control. The plate was covered with a TopSeal™-A PLUS sealing film to prevent contamination and evaporation of the samples and incubated for 4 h at room temperature in subdued light.
The fluorescence signals (excitation/emission, 360 nm/460 nm) of released EDANS were read using a PerkinElmer EnSight multimode plate reader. The experiments were performed in triplicate. The IC50 values were calculated using GraphPad Prism 8.0 software by nonlinear regression of dose-response inhibition.
6.1.6.2. PLpro enzymatic assay
The assay was performed in a volume of 50 μL in black 96-well OptiPlate. A fluorometric peptide Z-Arg-Leu-Arg-Gly-Gly-AMC (Z-RLRGG-AMC) was used as substrate in PLpro enzymatic assay (BPS Bioscience Papain-like Protease Assay: Deubiquitinase Activity). Upon cleavage by PLpro, the fluorescence of the AMC moiety dramatically raises. For steady state measurement, the enzyme was incubated for 30 min at 37 °C (final concentration, 25 ng per reaction) with the compounds at different concentrations in assay buffer (40 mM Tris-HCl, pH 8, 110 mM NaCl, 2.2 mM KCl, 0.04% Tween-20, 3 mM DTT, 20% glycerol and 115 mM Imidazole). Then, the reaction was initiated by adding 10 μl of the substrate to each well (final concentration, 250 nM). Buffer with the same amount of DMSO (1%) was used as control and PLpro inhibitor GRL0617 is also included as a positive control. The plate was covered with a TopSeal™-A PLUS sealing film to prevent contamination and evaporation of the samples and incubated in the dark for 50 min at 37 °C. The fluorescence signals (excitation/emission, 360 nm/460 nm) were read using a PerkinElmer EnSight multimode plate reader. The experiments were performed in triplicate. The IC50 values were calculated using GraphPad Prism 8.0 software by nonlinear regression of dose-response inhibition.
6.1.7. SPR binding assay
SARS-Cov-2 Spike protein (SP) was acquired by Genscript Biotech, NE (cat. no. Z03501-1). Series S Sensor Chip CM5 8 (cat. no. BR100530), His Capture Kit (cat. no. 28995056), Amine Coupling Kit (cat. no. BR100050), HBS-P (cat. no. BR100368) were purchased from Cytiva.
The affinity of synthetic compounds for SARS-Cov-2 Spike protein (SP) was determined by SPR using a Biacore T200 (Cytiva) optical biosensor equipped with research-grade CM5 (Carboxy Methyl Dextran) sensor chip. Prior to the immobilization of the SP protein, a pH scouting was performed as follows. Solutions of 1.25 μM of the ligand in 10 mM sodium acetate with pH values ranging from 4.47 to 6 were prepared and injected onto the surface. The SP protein (1.25 μM in 10 mM sodium acetate, pH 4.59) was immobilized by using standard amine-coupling protocol to obtain densities of 11500 RU. HBS-P buffer (0.01 M HEPES pH 7.4, 0.15 M NaCl, 0.005% (v/v) Surfactant P20) diluted 10 × with Milli-Q water and supplemented with 5% DMSO was used as a running buffer. Stock solutions of compounds in 100% DMSO were prepared (10 mM). Running buffer was injected at a flow rate of 10 μL/min over the chip to clean and equilibrate the immobilizes sensor surface, then a solvent correction was performed as indicated in Laboratory Guideline 29-0057-18 AA, GE Healthcare Life Sciences. A series of increasing concentrations of compounds (0.75–100 μM) diluted in the ligand buffer were injected at 25 °C with a flow rate of 20 μL/min for 90s (association phase), and then the buffer alone was injected for 600 s (dissociation phase). A regeneration step was not necessary. The first channel was used as a reference surface. The experiments were performed in triplicate. The equilibrium dissociation constants (KD) and kinetic dissociation (kd) and association (ka) constants were calculated from the sensorgrams by global fitting of a 1:1 binding model using evaluation software (v3.1) provided with the Biacore T200 instrument (Cytiva).
6.1.8. Cellular assay
SARS-CoV-2 antiviral assay Vero cells (ATCC-CCL81) were grown in Dulbecco's Modified Eagle's Medium (DMEM, ThermoFisher, Belgium) supplemented with 10% fetal calf serum (FCS), 2 mM l-glutamine, 0.1 mM non-essential amino acids, 1 mM sodium pyruvate, and 10 mM HEPES at 37 °C in a 5% CO2 humidified atmosphere. The SARS-CoV-2 strains UC-1074 and UC-1075 were isolated from the nasopharyngeal swabs of two COVID-19 patients that had a Ct of 19 and 22, respectively, for the detection of the SARS-CoV-2 E protein by real-time reverse transcription PCR (RT-qPCR). The infectious virus titer of the clinical isolates was determined in Vero cells and expressed as 50% cell culture infectious dose (CCID50) per mL. The titers of the viral stocks were 1.58E+06 (UC-1074) and 1.08E+04 (UC-1075) TCID50/mL. For the antiviral assays, Vero cells were seeded in 96-well plates at a density of 1 × 104 cells per well in DMEM 10% FCS medium. After 24 h growth, the medium was removed, and cells were treated with different compound concentrations in DMEM 2% FCS and mocked-infected or SARS-CoV-2-infected with 100 CCID50/well (final volume 200 μl/450 well). After 5 days of incubation at 37 °C, viral CPE was recorded microscopically and the 50% effective concentration (EC50) was calculated for each compound and remdesivir (reference anti-SARS-CoV-2 compound). In parallel, the cytotoxic effects of the compounds were assessed by evaluating the MCC (minimum cytotoxic concentration that causes a microscopically detectable alteration of cell morphology). The effects of the compounds on cell growth were determined by counting the number of cells with a Coulter counter in mock-infected cultures and expressed as the cytostatic concentration required to reduce cell growth by 50% (CC50). All SARS-CoV-2-related work was conducted in the high-containment BSL3 facilities of the KU Leuven Rega Institute (3CAPS) under licenses AMV 30112018 SBB 219 2018 0892 and AMV 23102017 SBB 219 2017 0589 according to institutional guidelines.
6.1.9. Computational details
3D structures of SARS-CoV-2 Mpro in complex with A1 antagonist (FJC) (PDB code: 6M0K) [33] and of SARS-CoV-2 PLpro in complex with non-covalent inhibitor VBY (PDB code: 7JIW) [40] and with covalent peptidic inhibitor VIR251 (PDB code: 6WX4) [24], and of SARS-CoV-2 spike protein (PDB code: 6LZG) [41] were prepared using the Schrödinger Protein Preparation Wizard workflow [55]. Specifically, water molecules and the co-complexed compounds/counterparts (ACE2 in the case of spike protein) were deleted, cap termini were included, all hydrogen atoms were added, and bond orders were assigned. Eventually, the prepared.pdb files were converted to the final.mae files. The grids accounted for the subsequent molecular docking calculations were generated analyzing the positions of the related co-crystallized compounds.
The focused library of investigated compounds (See Results and Discussion) was prepared using LigPrep software (Schrodinger Suite) [56]. Specifically, all the possible tautomers and protonation states (pH = 7.4 ± 1.0) were generated for each compound, and the obtained structures were minimized using the OPLS 2005 force field.
Molecular docking experiments were performed using Glide software (Schrödinger Suite), using the Extra Precision [XP] mode [57]. In details, 20,000 poses were kept in the starting phase of docking 1200 poses for energy minimization were selected. The scoring window for keeping the initial poses was set to 400.0 and a scaling factor of 0.8 related to van der Waals radii with a partial charge cutoff of 0.15, basing on a 0.5 kcal/mol rejection cutoff for the obtained minimized poses, was considered. In the output file, 10 poses for each compound were saved.
Covalent docking experiments were performed using Glide software (Schrödinger Suite) [57]. Cys145 was set as the reactive protein residue for SARS-CoV-2 Mpro and Cys111 for SARS-CoV-2 PLpro, whereas the specific reaction type was selected in the related panel according to the specific ligand chemical features. When needed, the specific.cdock “custom chemistry” file was generated. In the output file, 10 poses for each compound were saved.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors are grateful to M Brecht Dirix for excellent technical assistance in the biological assays.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2021.113863.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Dhama K., Patel S.K., Sharun K., Pathak M., Tiwari R., Yatoo M.I., Malik Y.S., Sah R., Rabaan A.A., Panwar P.K., Singh K.P., Michalak I., Chaicumpa W., Martinez-Pulgarin D.F., Bonilla-Aldana D.K., Rodriguez-Morales A.J. SARS-CoV-2 jumping the species barrier: zoonotic lessons from SARS, MERS and recent advances to combat this pandemic virus. Trav. Med. Infect. Dis. 2020;37:101830. doi: 10.1016/j.tmaid.2020.101830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tiwari R., Dhama K., Sharun K., Iqbal Yatoo M., Malik Y.S., Singh R., Michalak I., Sah R., Bonilla-Aldana D.K., Rodriguez-Morales A.J. COVID-19: animals, veterinary and zoonotic links. Vet. Q. 2020;40:169–182. doi: 10.1080/01652176.2020.1766725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Larsen J.R., Martin M.R., Martin J.D., Kuhn P., Hicks J.B. Modeling the onset of symptoms of COVID-19. Front Public Health. 2020;8:473. doi: 10.3389/fpubh.2020.00473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Triggle C.R., Bansal D., Ding H., Islam M.M., Farag E., Hadi H.A., Sultan A.A. A comprehensive review of viral characteristics, transmission, pathophysiology, immune response, and management of SARS-CoV-2 and COVID-19 as a basis for controlling the pandemic. Front. Immunol. 2021;12:631139. doi: 10.3389/fimmu.2021.631139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Falahi S., Kenarkoohi A. Transmission routes for SARS-CoV-2 infection: review of evidence. New Microbes New Infect. 2020;38:100778. doi: 10.1016/j.nmni.2020.100778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.team E.e. Updated rapid risk assessment from ECDC on the risk related to the spread of new SARS-CoV-2 variants of concern in the EU/EEA – first update. Euro Surveill. 2021;26:2101211. doi: 10.2807/1560-7917.ES.2021.26.3.2101211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eastman R.T., Roth J.S., Brimacombe K.R., Simeonov A., Shen M., Patnaik S., Hall M.D. Remdesivir: a review of its discovery and development leading to emergency use authorization for treatment of COVID-19. ACS Cent. Sci. 2020;6:672–683. doi: 10.1021/acscentsci.0c00489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y., Zhang D., Du G., Du R., Zhao J., Jin Y., Fu S., Gao L., Cheng Z., Lu Q., Hu Y., Luo G., Wang K., Lu Y., Li H., Wang S., Ruan S., Yang C., Mei C., Wang Y., Ding D., Wu F., Tang X., Ye X., Ye Y., Liu B., Yang J., Yin W., Wang A., Fan G., Zhou F., Liu Z., Gu X., Xu J., Shang L., Zhang Y., Cao L., Guo T., Wan Y., Qin H., Jiang Y., Jaki T., Hayden F.G., Horby P.W., Cao B., Wang C. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2020;395:1569–1578. doi: 10.1016/S0140-6736(20)31022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dabbous H.M., Abd-Elsalam S., El-Sayed M.H., Sherief A.F., Ebeid F.F.S., El Ghafar M.S.A., Soliman S., Elbahnasawy M., Badawi R., Tageldin M.A. Efficacy of favipiravir in COVID-19 treatment: a multi-center randomized study. Arch. Virol. 2021;166:949–954. doi: 10.1007/s00705-021-04956-9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10.Bojkova D., Bechtel M., McLaughlin K.M., McGreig J.E., Klann K., Bellinghausen C., Rohde G., Jonigk D., Braubach P., Ciesek S., Munch C., Wass M.N., Michaelis M., Cinatl J., Jr. Aprotinin inhibits SARS-CoV-2 replication. Cells. 2020;9 doi: 10.3390/cells9112377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McElvaney O.J., O'Connor E., McEvoy N.L., Fraughan D.D., Clarke J., McElvaney O.F., Gunaratnam C., O'Rourke J., Curley G.F., McElvaney N.G. Alpha-1 antitrypsin for cystic fibrosis complicated by severe cytokinemic COVID-19. J. Cyst. Fibros. 2021;20:31–35. doi: 10.1016/j.jcf.2020.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grimes J.M., Grimes K.V. p38 MAPK inhibition: a promising therapeutic approach for COVID-19. J. Mol. Cell. Cardiol. 2020;144:63–65. doi: 10.1016/j.yjmcc.2020.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Covid R., Treatments C. Use of hydroxychloroquine in hospitalised COVID-19 patients is associated with reduced mortality: findings from the observational multicentre Italian CORIST study. Eur. J. Intern. Med. 2020;82:38–47. doi: 10.1016/j.ejim.2020.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meo S.A., Klonoff D.C., Akram J. Efficacy of chloroquine and hydroxychloroquine in the treatment of COVID-19. Eur. Rev. Med. Pharmacol. Sci. 2020;24:4539–4547. doi: 10.26355/eurrev_202004_21038. [DOI] [PubMed] [Google Scholar]
- 15.Ou X., Liu Y., Lei X., Li P., Mi D., Ren L., Guo L., Guo R., Chen T., Hu J., Xiang Z., Mu Z., Chen X., Chen J., Hu K., Jin Q., Wang J., Qian Z. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020;11:1620. doi: 10.1038/s41467-020-15562-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xie Y., Karki C.B., Du D., Li H., Wang J., Sobitan A., Teng S., Tang Q., Li L. Spike proteins of SARS-CoV and SARS-CoV-2 utilize different mechanisms to bind with human ACE2. Front Mol Biosci. 2020;7:591873. doi: 10.3389/fmolb.2020.591873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Di Micco S., Musella S., Scala M.C., Sala M., Campiglia P., Bifulco G., Fasano A. In silico analysis revealed potential anti-SARS-CoV-2 main protease activity by the zonulin inhibitor larazotide acetate. Front Chem. 2021;8:628609. doi: 10.3389/fchem.2020.628609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sultana J., Crisafulli S., Gabbay F., Lynn E., Shakir S., Trifirò G. Challenges for drug repurposing in the COVID-19 pandemic era. Front. Pharmacol. 2020;11:1657. doi: 10.3389/fphar.2020.588654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang Y., Yang C., Xu X.F., Xu W., Liu S.W. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020;41:1141–1149. doi: 10.1038/s41401-020-0485-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.FDA's Approval of Veklury (Remdesivir) for the Treatment of COVID-19—The Science of Safety and Effectiveness. https://www.fda.gov/drugs/drug-safety-and-availability/fdas-approval-veklury-remdesivir-treatment-covid-19-science-safety-and-effectiveness lastly accessed 07/19/2021.
- 21.Kokic G., Hillen H.S., Tegunov D., Dienemann C., Seitz F., Schmitzova J., Farnung L., Siewert A., Hobartner C., Cramer P. Mechanism of SARS-CoV-2 polymerase stalling by remdesivir. Nat. Commun. 2021;12:279. doi: 10.1038/s41467-020-20542-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gioia M., Ciaccio C., Calligari P., De Simone G., Sbardella D., Tundo G., Fasciglione G.F., Di Masi A., Di Pierro D., Bocedi A., Ascenzi P., Coletta M. Role of proteolytic enzymes in the COVID-19 infection and promising therapeutic approaches. Biochem. Pharmacol. 2020;182:114225. doi: 10.1016/j.bcp.2020.114225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Q., Kang C. Progress in developing inhibitors of SARS-CoV-2 3C-like protease. Microorganisms. 2020;8:1250. doi: 10.3390/microorganisms8081250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rut W., Lv Z., Zmudzinski M., Patchett S., Nayak D., Snipas S.J., El Oualid F., Huang T.T., Bekes M., Drag M., Olsen S.K. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: a framework for anti-COVID-19 drug design. Sci Adv. 2020;6 doi: 10.1126/sciadv.abd4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zaidman D., Gehrtz P., Filep M., Fearon D., Prilusky J., Duberstein S., Cohen G., Owen D., Resnick E., Strain-Damerell C., Lukacik P., Barr H., Walsh M.A., von Delft F., London N. bioRxiv; 2020. An Automatic Pipeline for the Design of Irreversible Derivatives Identifies a Potent SARS-CoV-2 Mpro Inhibitor. 2020.2009.2021.299776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arafet K., Serrano-Aparicio N., Lodola A., Mulholland A.J., González F.V., Świderek K., Moliner V. Mechanism of inhibition of SARS-CoV-2 Mpro by N3 peptidyl Michael acceptor explained by QM/MM simulations and design of new derivatives with tunable chemical reactivity. Chem. Sci. 2021;12:1433–1444. doi: 10.1039/d0sc06195f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paul A.S., Islam R., Parves M.R., Mamun A.A., Shahriar I., Hossain M.I., Hossain M.N., Ali M.A., Halim M.A. Cysteine focused covalent inhibitors against the main protease of SARS-CoV-2. J. Biomol. Struct. Dyn. 2020:1–20. doi: 10.1080/07391102.2020.1831610. [DOI] [PubMed] [Google Scholar]
- 28.Hoffman R.L., Kania R.S., Brothers M.A., Davies J.F., Ferre R.A., Gajiwala K.S., He M., Hogan R.J., Kozminski K., Li L.Y., Lockner J.W., Lou J., Marra M.T., Mitchell L.J., Jr., Murray B.W., Nieman J.A., Noell S., Planken S.P., Rowe T., Ryan K., Smith G.J., 3rd, Solowiej J.E., Steppan C.M., Taggart B. Discovery of ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19. J. Med. Chem. 2020;63:12725–12747. doi: 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lockbaum G.J., Reyes A.C., Lee J.M., Tilvawala R., Nalivaika E.A., Ali A., Kurt Yilmaz N., Thompson P.R., Schiffer C.A. Crystal structure of SARS-CoV-2 main protease in complex with the non-covalent inhibitor ML188. Viruses. 2021;13:174. doi: 10.3390/v13020174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang C.H., Stone E.A., Deshmukh M., Ippolito J.A., Ghahremanpour M.M., Tirado-Rives J., Spasov K.A., Zhang S., Takeo Y., Kudalkar S.N., Liang Z., Isaacs F., Lindenbach B., Miller S.J., Anderson K.S., Jorgensen W.L. Potent noncovalent inhibitors of the main protease of SARS-CoV-2 from molecular sculpting of the drug perampanel guided by free energy perturbation calculations. ACS Cent. Sci. 2021;7:467–475. doi: 10.1021/acscentsci.1c00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kitamura N., Sacco M.D., Ma C., Hu Y., Townsend J.A., Meng X., Zhang F., Zhang X., Ba M., Szeto T., Kukuljac A., Marty M.T., Schultz D., Cherry S., Xiang Y., Chen Y., Wang J. Expedited approach toward the rational design of noncovalent SARS-CoV-2 main protease inhibitors. J. Med. Chem. 2021 doi: 10.1021/acs.jmedchem.1c00509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ostacolo C., Di Sarno V., Lauro G., Pepe G., Musella S., Ciaglia T., Vestuto V., Autore G., Bifulco G., Marzocco S., Campiglia P., Gomez-Monterrey I.M., Bertamino A. Identification of an indol-based multi-target kinase inhibitor through phenotype screening and target fishing using inverse virtual screening approach. Eur. J. Med. Chem. 2019;167:61–75. doi: 10.1016/j.ejmech.2019.01.066. [DOI] [PubMed] [Google Scholar]
- 33.Dai W., Zhang B., Jiang X.M., Su H., Li J., Zhao Y., Xie X., Jin Z., Peng J., Liu F., Li C., Li Y., Bai F., Wang H., Cheng X., Cen X., Hu S., Yang X., Wang J., Liu X., Xiao G., Jiang H., Rao Z., Zhang L.K., Xu Y., Yang H., Liu H. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang H., Xie W., Xue X., Yang K., Ma J., Liang W., Zhao Q., Zhou Z., Pei D., Ziebuhr J., Hilgenfeld R., Yuen K.Y., Wong L., Gao G., Chen S., Chen Z., Ma D., Bartlam M., Rao Z. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005;3:e324. doi: 10.1371/journal.pbio.0030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim H., Hwang Y.S., Kim M., Park S.B. Recent advances in the development of covalent inhibitors. Rsc Med Chem. 2021;12:1037–1045. doi: 10.1039/d1md00068c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mons E., Jansen I.D.C., Loboda J., van Doodewaerd B.R., Hermans J., Verdoes M., van Boeckel C.A.A., van Veelen P.A., Turk B., Turk D., Ovaa H. The alkyne moiety as a latent electrophile in irreversible covalent small molecule inhibitors of cathepsin K. J. Am. Chem. Soc. 2019;141:3507–3514. doi: 10.1021/jacs.8b11027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sommer S., Weikart N.D., Linne U., Mootz H.D. Covalent inhibition of SUMO and ubiquitin-specific cysteine proteases by an in situ thiol-alkyne addition. Bioorg. Med. Chem. 2013;21:2511–2517. doi: 10.1016/j.bmc.2013.02.039. [DOI] [PubMed] [Google Scholar]
- 38.Krishnan S., Miller R.M., Tian B., Mullins R.D., Jacobson M.P., Taunton J. Design of reversible, cysteine-targeted Michael acceptors guided by kinetic and computational analysis. J. Am. Chem. Soc. 2014;136:12624–12630. doi: 10.1021/ja505194w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Locatelli C., Rosso R., Santos-Silva M.C., de Souza C.A., Licínio M.A., Leal P., Bazzo M.L., Yunes R.A., Creczynski–Pasa T.B. Ester derivatives of gallic acid with potential toxicity toward L1210 leukemia cells. Bioorg. Med. Chem. 2008;16:3791–3799. doi: 10.1016/j.bmc.2008.01.049. [DOI] [PubMed] [Google Scholar]
- 40.Liao T.T., Wang L., Jia R.W., Fu X.H., Chua H. Lipophilic organic pollutants induce changes in phospholipid and membrane protein composition leading to Vero cell morphological change. J Environ Sci Heal B. 2014;49:760–768. doi: 10.1080/03601234.2014.929868. [DOI] [PubMed] [Google Scholar]
- 41.Osipiuk J., Azizi S.A., Dvorkin S., Endres M., Jedrzejczak R., Jones K.A., Kang S., Kathayat R.S., Kim Y., Lisnyak V.G., Maki S.L., Nicolaescu V., Taylor C.A., Tesar C., Zhang Y.A., Zhou Z., Randall G., Michalska K., Snyder S.A., Dickinson B.C., Joachimiak A. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 2021;12:743. doi: 10.1038/s41467-021-21060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Q., Zhang Y., Wu L., Niu S., Song C., Zhang Z., Lu G., Qiao C., Hu Y., Yuen K.-Y., Wang Q., Zhou H., Yan J., Qi J. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell. 2020;181:894–904. doi: 10.1016/j.cell.2020.03.045. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O'Meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L., Tummino T.A., Huttenhain R., Kaake R.M., Richards A.L., Tutuncuoglu B., Foussard H., Batra J., Haas K., Modak M., Kim M., Haas P., Polacco B.J., Braberg H., Fabius J.M., Eckhardt M., Soucheray M., Bennett M.J., Cakir M., McGregor M.J., Li Q., Meyer B., Roesch F., Vallet T., Mac Kain A., Miorin L., Moreno E., Naing Z.Z.C., Zhou Y., Peng S., Shi Y., Zhang Z., Shen W., Kirby I.T., Melnyk J.E., Chorba J.S., Lou K., Dai S.A., Barrio-Hernandez I., Memon D., Hernandez-Armenta C., Lyu J., Mathy C.J.P., Perica T., Pilla K.B., Ganesan S.J., Saltzberg D.J., Rakesh R., Liu X., Rosenthal S.B., Calviello L., Venkataramanan S., Liboy-Lugo J., Lin Y., Huang X.P., Liu Y., Wankowicz S.A., Bohn M., Safari M., Ugur F.S., Koh C., Savar N.S., Tran Q.D., Shengjuler D., Fletcher S.J., O'Neal M.C., Cai Y., Chang J.C.J., Broadhurst D.J., Klippsten S., Sharp P.P., Wenzell N.A., Kuzuoglu-Ozturk D., Wang H.Y., Trenker R., Young J.M., Cavero D.A., Hiatt J., Roth T.L., Rathore U., Subramanian A., Noack J., Hubert M., Stroud R.M., Frankel A.D., Rosenberg O.S., Verba K.A., Agard D.A., Ott M., Emerman M., Jura N., von Zastrow M., Verdin E., Ashworth A., Schwartz O., d'Enfert C., Mukherjee S., Jacobson M., Malik H.S., Fujimori D.G., Ideker T., Craik C.S., Floor S.N., Fraser J.S., Gross J.D., Sali A., Roth B.L., Ruggero D., Taunton J., Kortemme T., Beltrao P., Vignuzzi M., Garcia-Sastre A., Shokat K.M., Shoichet B.K., Krogan N.J. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sadegh S., Matschinske J., Blumenthal D.B., Galindez G., Kacprowski T., List M., Nasirigerdeh R., Oubounyt M., Pichlmair A., Rose T.D., Salgado-Albarran M., Spath J., Stukalov A., Wenke N.K., Yuan K., Pauling J.K., Baumbach J. Exploring the SARS-CoV-2 virus-host-drug interactome for drug repurposing. Nat. Commun. 2020;11:3518. doi: 10.1038/s41467-020-17189-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou Y., Hou Y., Shen J., Huang Y., Martin W., Cheng F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020;6:14. doi: 10.1038/s41421-020-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sternberg A., McKee D.L., Naujokat C. Novel drugs targeting the SARS-CoV-2/COVID-19 machinery. Curr. Top. Med. Chem. 2020;20:1423–1433. doi: 10.2174/1568026620999200517043137. [DOI] [PubMed] [Google Scholar]
- 47.Mitra K., Ghanta P., Acharya S., Chakrapani G., Ramaiah B., Doble M. Dual inhibitors of SARS-CoV-2 proteases: pharmacophore and molecular dynamics based drug repositioning and phytochemical leads. J. Biomol. Struct. Dyn. 2020:1–14. doi: 10.1080/07391102.2020.1796802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rajpoot S., Alagumuthu M., Baig M.S. Dual targeting of 3CLpro and PLpro of SARS-CoV-2: a novel structure-based design approach to treat COVID-19. Curr Res Struct Biol. 2021;3:9–18. doi: 10.1016/j.crstbi.2020.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baby K., Maity S., Mehta C.H., Suresh A., Nayak U.Y., Nayak Y. SARS-CoV-2 entry inhibitors by dual targeting TMPRSS2 and ACE2: an in silico drug repurposing study. Eur. J. Pharmacol. 2021;896:173922. doi: 10.1016/j.ejphar.2021.173922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Naik B., Gupta N., Ojha R., Singh S., Prajapati V.K., Prusty D. High throughput virtual screening reveals SARS-CoV-2 multi-target binding natural compounds to lead instant therapy for COVID-19 treatment. Int. J. Biol. Macromol. 2020;160:1–17. doi: 10.1016/j.ijbiomac.2020.05.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zmudzinski M., Rut W., Olech K., Granda J., Giurg M., Burda-Grabowska M., Zhang L., Sun X., Lv Z., Nayak D., Kesik-Brodacka M., Olsen S.K., Hilgenfeld R., Drag M. bioRxiv; 2020. Ebselen Derivatives Are Very Potent Dual Inhibitors of SARS-CoV-2 Proteases - PLpro and Mpro in in Vitro Studies. 2020.2008.2030.273979. [Google Scholar]
- 52.Chen Z., Cui Q., Cooper L., Zhang P., Lee H., Chen Z., Wang Y., Liu X., Rong L., Du R. Ginkgolic acid and anacardic acid are specific covalent inhibitors of SARS-CoV-2 cysteine proteases. Cell Biosci. 2021;11:45. doi: 10.1186/s13578-021-00564-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ma C., Hu Y., Townsend J.A., Lagarias P.I., Marty M.T., Kolocouris A., Wang J. Ebselen, disulfiram, carmofur, PX-12, tideglusib, and shikonin are nonspecific promiscuous SARS-CoV-2 main protease inhibitors. ACS Pharmacol Transl Sci. 2020;3:1265–1277. doi: 10.1021/acsptsci.0c00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Temussi P.A., Grimaldi M., Stillitano I., Amodio G., Santoro A., Buonocore M., Moltedo O., Remondelli P., D'Ursi A.M. Structural basis of antiviral activity of peptides from MPER of FIV gp36. PloS One. 2018;13 doi: 10.1371/journal.pone.0204042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schrödinger Release 2021-2: Protein Preparation Wizard. Epik, Schrödinger, LLC; New York, NY: 2021. Impact, Schrödinger, LLC, New York, NY; Prime, Schrödinger, LLC, New York, NY, 2021. [Google Scholar]
- 56.Schrödinger Release 2021-2. LigPrep, Schrödinger, LLC; New York, NY: 2021. [Google Scholar]
- 57.Schrödinger Release 2021-2: Glide. Schrödinger, LLC; New York, NY: 2021. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.