

Abstract
Introduction
Although over 1000 hemoglobin (Hb) variants were identified so far, Hb Port Phillip compound with α‐thalassemia deletion had no reported before.
Methods
Two patients and the associated families from Guangdong province in China were recruited. Hematological parameters were determined by blood routine examination and hemoglobin electrophoresis. Genotyping was performed by Gap‐PCR and Sanger sequencing.
Results
One patient was diagnosed as Hb Port Phillip, while her daughter was compounded with ‐α4.2 deletion, with normal Hb level (150 g/L), mean corpuscular volume (MCV) 108.4 fl and mean corpuscular hemoglobin (MCH) (30.5 pg). Another patient was diagnosed as compound Hb Port Phillip and ‐‐SEA deletion. This proband presented with more severe α‐thalassemia trait than the patient compounded with ‐α4.2 deletion, with hemoglobin 80 g/L, MCV 61.7 fl, and MCH 18.7 pg.
Conclusion
Here we first time identified two patients compound with Hb Port Phillip and ‐α4.2 and ‐‐SEA deletions, respectively, which had never been reported. Our study widens the genotypes of hemoglobinopathy and provides reference for genetic counselling and prenatal diagnosis in this population.
Keywords: blood, hemoglobin variants, prenatal diagnosis, thalassemia
Hemoglobin variants are a group of hereditary hemoglobin diseases and the phenotype ranges from asymptomatic to severe clinical manifestations. Hb Port Phillip was reported with decreased hemoglobin stability and presented as hypochromic polyglobulia microcytic red blood cells. In this study, we first time identified two probands compound with Hb Port Phillip and ‐α4.2 and ‐‐SEA deletions, respectively, and provided reference for genetic counselling and prenatal diagnosis in Chinese population.

Haemoglobinopathy, with a high rate (about 7% of the world's population are carriers for important hemoglobin disorders) around the world (Weatherall, 2008), is a kind of single gene deficient disease that caused by deletions or mutations on α‐ (HBA, (MIM 141800–141850)) or β‐ globin gene (HBB, (MIM 141900)) (Xiong et al., 2010; Xu et al., 2004). Thalassemia and abnormal hemoglobin are included. To date, more than 1300 hemoglobin variants have been reported (Globin Gene Server, http://globin.bx.psu.edu) (Giardine et al., 2011; Hardison et al., 2002). Caused by deficiencies on globin gene, hemoglobin variants are a group of hereditary hemoglobin diseases that could change the primary structure of globin and further resulting in the molecular structure of hemoglobin (Thom et al., (2013)). A rare Hb variant, Hb Port Phillip (α91 (FG3) Leu → Pro, HBA2: c.275T>C (NM_000517.6(HBA2_V001): c.275T>C)(GRCh37)), caused by a missense mutation (CTT>CCT) at the codon 91 of the HBA2 (MIM 141850), has been reported with decreased hemoglobin stability, which was resulted from the loss of haem contact and general perturbation that results from the substitution of a proline for the leucine (Brennan et al., 1977). Steven O. Brennan et al reported that the patient with Hb Port Phillip presented with hypochromic polyglobulia microcytic red blood cells (RBCs) (Brennan et al., 1977). To date, due to the conventional methods usually detect the common genotypes of thalassemia, Hb Port Phillip compounded with α‐thalassemia deletions has not been reported. In this study, we identified two probands with Hb Port Phillip that compounded with ‐α4.2 or ‐‐SEA deletions from two Chinese families.
Two patients from two unrelated families were recruited in this study. Informed consent was obtained from all the patients in this study and the study was conducted in accordance with the Declaration of Helsinki. Peripheral blood (PB) and umbilical cord blood (CB) samples were collected, and hematological parameters were determined by using a Sysmex XN5000 automated hematology analyzer (Sysmex Corporation), which has high precision and reproducibility. Hb quantification was performed by automated capillary electrophoresis system (CE) (Sebia Capillarys 2), which has high sensitivity. Genomic DNA from PB and amniotic fluid were extracted by using Magpure tissue & Blood DNA KF Kit (MD5111, Magen) and NucleoSpin Blood kit (740951.250, MACHEREY‐NAGEL), respectively. We used Gap‐PCR to detect the common α‐thalassemia mutations with kit from Yaneng Biosciences in China. Sanger sequencing was performed to detect the mutation in HBA1 (MIM 141800) and HBA2 genes (PCR primers were as follow: HBA1: forward primer 5′‐TGGAGGGTGGAGACGTCCTG‐3′; reverse primer 5′‐TCCATCCCCTCCTCCCGCCCCTGCCTTTTC‐3′. HBA2: forward primer 5′‐TGGAGGGTGGAGACGTCCTG‐3′; reverse primer 5′‐CCATTGTTGGCACATTCCGG‐3′).
Patient 1 was a 27‐year‐old pregnant woman from Huizhou, Guangdong province, China, and genetic analysis showed that she was heterozygous for the Hb Port Phillip (α91 (FG3) Leu → Pro, HBA2: c.275T>C). This patient did not have any history of blood transfusion. She came to our hospital for prenatal diagnosis. Hematology examinations were performed on the family including the proband, her parents, husband and brother (Figure 1a). Sanger sequencing was performed to support the diagnosis of the proband. There was a heterozygous substitution (HBA2: c.275T>C) at codon 91 of the HBA2 gene that was previously reported as Hb Port Phillip was detected in Patient 1 by Sanger sequencing, with low hemoglobin level (103 g/L) and normal MCV (86.0 fL) and MCH (28.4 pg). Hb quantification analysis revealed that there was an abnormal Hb (Hb X, 8.4%) in the Z8 region (Figure 1b). The members of her family were recruited to analysis (hematological data were showed in Table 1). We found that her mother also had an abnormal Hb (7.3%) in the same region (I1, Figure 1b), while her father was normal, which indicated that the mutation was inherited from her mother (Figure 1a). Her husband (II 3) was detected as Hb H disease with the ‐α4.2 and ‐‐SEA deletions, resulting in α‐thalassemia intermedia. Her fetus was diagnosed by using amniotic fluid. Homozygous of Hb Port Phillip compounded with heterozygous of ‐α4.2 were found in her fetus (Figure 1c). This was the first time to identified Hb Port Phillip compounded with α‐thalassemia deletion in Chinese population. Umbilical cord blood was used to analyzed after her daughter was bored. However, we observed that the Hb level (150 g/L), MCV (108.4 fL) and MCH (30.5 pg) were normal in her daughter (III1, Table 1). The hematological parameters of her daughter were different from the data we observed in patients with Hb Port Phillip (Table 1), which showed hypochromic polyglobulia microcytic RBCs. We reasoned that this may because the data was obtained from a new bored infant, whose red blood cells were usually bigger than normal adult people.
FIGURE 1.
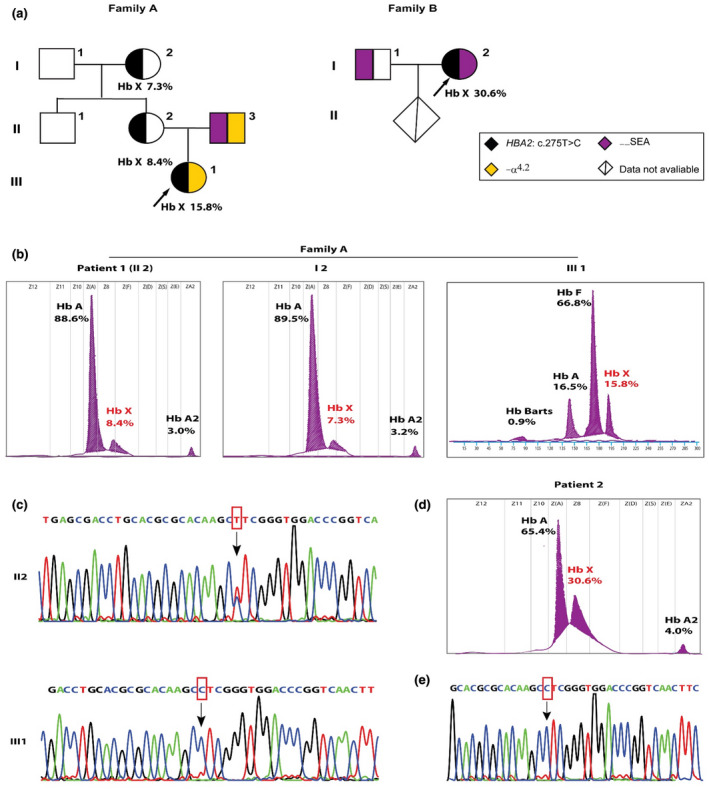
Phenotypic and genotypic analysis in two patients and their families. (a) Pedigrees of the two families. The arrows indicate the probands. The HBA2: c.275C>T mutation in the first proband was inherited from the patient 1. (b) Hemoglobin components measured by CE in family A. HbX indicated Hb Port Phillip. Data in III1 was measured by using cord blood. (c) Sanger sequencing analysis of patient 1 (II2) and her daughter (III1), who was compounded with Hb Port Phillip homozygous and ‐α4.2 deletion. The arrow indicated the mutation HBA2: c.275C>T. (d) Hemoglobin analysis of patient 2 who was compounded with Hb Port Phillip and ‐‐SEA deletion. (e) Sanger sequencing analysis of patient 2
TABLE 1.
The phenotypic and genotypic data of two patients and their families
| ID | Hb (g/L) | MCV (fl) | MCH (pg) | HbA (%) | HbA2 (%) | HbX | HBA genotype | HBB genotype |
|---|---|---|---|---|---|---|---|---|
| Family A | ||||||||
| I‐1 | 167 | 89.7 | 31.3 | 97.2 | 2.8 | — | αα/αα | N/N |
| I‐2 | 120 | 85.0 | 27.3 | 89.5 | 3.2 | 7.3 | ααPort Phillip/αα | N/N |
| II‐1 | 167 | 88.0 | 29.9 | 97.1 | 2.9 | — | αα/αα | N/N |
| II‐2 | 103 | 83.6 | 26.8 | 88.6 | 3.0 | 8.4 | ααPort Phillip/αα | N/N |
| II‐3 | ‐α4.2/αα | N/N | ||||||
| III‐1a | 150 | 108.4 | 30.5 | 16.5 | 0.9(Hb Bart's) | 15.8 | ‐α4.2/ααPort Phillip | N/N |
| Family B | ||||||||
| I‐1 | 126 | 86.0 | 27.6 | 97.4 | 2.6 | — | ‐α4.2/αα | N/N |
| I‐2a | 80 | 61.7 | 18.7 | 65.4 | 4.0 | 30.6 | ‐‐SEA/ααPort Phillip | N/N |
| Hb Phnom Penhb | 99.0 | 60.7 | 19.8 | — | 1.6 | 98.2 | ‐‐SEA/ααPhnom Penh | N/N |
| Hb Hekinan (n = 3)b | 126.0±4.0 | 65.6±3.4 | 21.3±4.4 | — | 2.2±0.2 | 97.2±0.7 | ‐‐SEA/ααHekinan | N/N |
Probands; N, normal HBB allele.
Data were derived from Supan Fucharoen 8, 9 and showed as mean ± SD.
Patient 2 was a 19‐year‐old pregnant woman from Shenzhen, Guangdong, China. She came to Shenzhen Longgang maternal and child health care hospital for thalassemia screening. Her husband was diagnosed as heterozygous for ‐‐SEA deletion. She was diagnosed as heterozygous of ‐‐SEA deletion by using Gap‐PCR. Interestingly, the hemoglobin quantification analysis showed an abnormal Hb (HbX, 30.6%) nearby HbA (Figure 1D), at the same region as patient 1. Therefore, she came to our hospital for further analysis. By using Sanger sequencing, we found a heterozygous substitution (HBA2: c.275T>C) at codon 91 of the HBA2 gene in this patient (Figure 1E). Taking together, she was diagnosed as compounded heterozygous of ‐‐SEA deletion and Hb Port Phillip. Interestingly, compared with the heterozygous of Hb Port Phillip or the patients mentioned above, phenotypic parameters in this patient were more severe, with Hb 80 g/L, MCV 61.7 fl and MCH 18.7 pg. However, she had no history of blood transfusion. We also compared the hematological parameters with Hb Port Phillip reported by Jian Li (Chin J Evid Based Pediatr, August 2014) and Hb Phnom Penh [α117(GH5)‐Ile‐α118(H1)(α1), NP_000549.1(HBA1_V001):p.Phe118_Thr119insI] (Singha et al., 2013) and Hb Hekinan [α27(B8)Glu → Asp(α2), NM_000517.6(HBA2_V001):c.84G>C] (Fucharoen et al., 2003) and found Hb Port Phillip compounded with ‐‐SEA deletion caused more clinical manifestation severity (Table 1).
Hb variants usually result in change in globin structure, thus affecting the stability of globin and making it easier to degrade. The phenotype of Hb variants ranges from asymptomatic to severe clinical manifestations (Indrak et al., 2018). Hb Port Phillip was caused by a substitution of C>T at codon 91 of the HBA2 gene. Previous study reported this Hb variant was resulted from the loss of haem contact and general perturbation that results from the substitution of a proline for the leucine (Brennan et al., 1977).
In this study, we identified 2 patients compound with Hb Port Phillip and ‐α4.2 and ‐‐SEA deletions, respectively, for the first time. The patient 1 with Hb Port Phillip presented with hypochromic polyglobulia microcytic red blood cells (RBCs), which was consistent with the previous reported by Brennan et al., (1977). Interestingly, III3, the daughter of patient 1 in the first family, compounded with Hb Port Phillip and ‐α4.2 deletion, had no anemia manifestation, with normal Hb (150 g/L), MCV (108.4 fL) and MCH (30.5 pg). We reasoned that this may be due to the data obtained from a new born infant, whose red blood cells were usually bigger than normal adult people. Long‐term observation and further data should be collected to evaluate the clinical manifestation of this compounded genotype. What enrolled our interest was that patient 2 had a more severe clinical manifestation when compounded with ‐‐SEA deletion. Her hemoglobin was only 80 g/L, while the MCV and MCH were 61.7 fl and 18.7 pg, respectively. Compared with the data in patients with Hb Port Phillip reported by Jian Li and other Hb variants, we found more severity in clinical manifestation in Hb Port Phillip compounded with ‐‐SEA deletion. Unfortunately, the information of patient 2’s fetus was unavailable so that we did not know about the genotype or further detail. Given that High Performance Liquid Chromatograph (HPLC) could not identify this Hb variant with a substitution of Proline for Leucine, Hb Port Phillip may be overlooked during the laboratory process. Thus, it is vital to use CE to analyze the hemoglobin during prenatal diagnosis.
In summary, we identified two patients compounded for Hb Port Phillip and ‐α4.2 or ‐‐SEA deletions in Chinese population for the first time. Giving the variants of clinical features and phenotypes of Hb Port Phillip compounded with α‐thalassemia deletions, more insights should be required during the process of prenatal.
CONFLICT OF INTEREST
The authors declare no competing financial interests.
ACKNOWLEDGEMENT
We thank the patients for their willingness to participate in this study. Financial supports from the Science and Technology Program of Guangzhou, China (grant no. 202002030390) is gratefully acknowledged.
Li Du and Xiuqin Bao contributed equally to this study.
Funding information
This study is funded by the Science and Technology Program of Guangzhou, China (grant no. 202002030390).
Contributor Information
Jianhong Chen, Email: cjhong2004@hotmail.com.
Aihua Yin, Email: yinaiwa@126.com.
DATA AVAILABILITY STATEMENT
All the data in this study were available in the figures and tables in the manuscript. The oxygen affinity status of Hb Port Phillip would be good data to have, however, we were unable to get it due to COVID.
REFERENCES
- Brennan, S. O., Tauro, G. P., & Melrose, W. (1977). Haemoglobin Port Phillip alpha91 (FG3) Leu replaced by Pro, a new unstable haemoglobin. FEBS Letters, 81, 115–117. [DOI] [PubMed] [Google Scholar]
- Fucharoen, S., Changtrakun, Y., Ratanasiri, T., Fucharoen, G., & Sanchaisuriya, K. (2003). Complex interaction of Hb Hekinan [alpha27(B8) Glu‐Asp] and Hb E [beta26(B8) Glu‐Lys] with a deletional alpha‐thalassemia 1 in a Thai family. European Journal of Haematology, 70, 304–309. [DOI] [PubMed] [Google Scholar]
- Giardine, B., Borg, J., Higgs, D. R., Peterson, K. R., Philipsen, S., Maglott, D., Singleton, B. K., Anstee, D. J., Basak, A. N., Clark, B., Costa, F. C., Faustino, P., Fedosyuk, H., Felice, A. E., Francina, A., Galanello, R., Gallivan, M. V. E., Georgitsi, M., & Gibbons, R. J., …, Patrinos, G. P. (2011). Systematic documentation and analysis of human genetic variation in hemoglobinopathies using the microattribution approach. Nature Genetics, 43, 295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardison, R. C., Chui, D. H., Giardine, B., Riemer, C., Patrinos, G. P., Anagnou, N., Miller, W., & Wajcman, H. (2002). HbVar: A relational database of human hemoglobin variants and thalassemia mutations at the globin gene server. Human Mutation, 19, 225–233. [DOI] [PubMed] [Google Scholar]
- Indrak, K., Divoka, M., Pospisilova, D., Cermak, J., Belickova, M., Horvathova, M., & Divoky, V. (2018). Hemoglobinopathies. Vnitr Lek, 64, 476–487. [PubMed] [Google Scholar]
- Singha, K., Srivorakun, H., Fucharoen, G., Changtrakul, Y., Komwilaisak, P., Jetsrisuparb, A., Puangplruk, R., & Fucharoen, S. (2013). Association of Hb Thailand [alpha56(E5)Lys–>Thr] and Hb Phnom Penh [alpha117(GH5)‐Ile‐alpha118(H1)] with alpha(0)‐thalassemia: molecular and hematological features and differential diagnosis. Hemoglobin, 37, 37–47. [DOI] [PubMed] [Google Scholar]
- Thom, C. S., Dickson, C. F., Gell, D. A., & Weiss, M. J. (2013). Hemoglobin variants: biochemical properties and clinical correlates. Cold Spring Harbor Perspectives in Medicine, 3, a011858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherall, D. J. (2008). Hemoglobinopathies worldwide: present and future. Current Molecular Medicine, 8, 592–599. [DOI] [PubMed] [Google Scholar]
- Xiong, F., Sun, M., Zhang, X., Cai, R., Zhou, Y., Lou, J., Zeng, L., Sun, Q., Xiao, Q., Shang, X., Wei, X., Zhang, T., Chen, P., & Xu, X. (2010). Molecular epidemiological survey of haemoglobinopathies in the Guangxi Zhuang Autonomous Region of southern China. Clinical Genetics, 78, 139–148. [DOI] [PubMed] [Google Scholar]
- Xu, X. M., Zhou, Y. Q., Luo, G. X., Liao, C., Zhou, M., Chen, P. Y., Lu, J. P., Jia, S. Q., Xiao, G. F., Shen, X., Li, J., Chen, H. P., Xia, Y. Y., Wen, Y. X., Mo, Q. H., Li, W. D., Li, Y. Y., Zhuo, L. W., Wang, Z. Q., … Zhong, M. (2004). The prevalence and spectrum of alpha and beta thalassaemia in Guangdong Province: implications for the future health burden and population screening. Journal of Clinical Pathology, 57, 517–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All the data in this study were available in the figures and tables in the manuscript. The oxygen affinity status of Hb Port Phillip would be good data to have, however, we were unable to get it due to COVID.