

Abstract
Background
Congenital disorders of glycosylation (CDG) are a genetically heterogeneous group of disorders caused by defects in the synthesis and processing of glycoproteins. COG6‐CDG is a kind of disorder caused by conserved oligomeric golgi complex 6 (COG6) deficiency. To date, only 19 patients with COG6‐CDG have been reported.
Methods
We report a girl in a Chinese family with developmental delay, growth retardation, microcephaly, abnormal liver function, and hypohidrosis. Trio whole‐exome sequencing was performed for this patient and her parents, and the variants identified were validated by Sanger sequencing. Prenatal diagnosis was done for this family during a subsequent pregnancy. The literature review on these patients was performed by reviewing articles published in English and Chinese.
Results
Genetic sequencing identified two novel heterozygous mutations: c.428G>T (p.S143I) and c.1843C>T (p.Q615X) in the COG6 gene, inherited from her healthy parents, respectively. A total of 11 different mutations in COG6 have been reported previously, and mutations potentially affecting splicing are the most common. The main clinical features included development delay, facial dysmorphism, growth retardation, skin abnormalities (hypohidrosis), microcephaly, abnormal brain structure, liver involvement, and recurrent infections.
Conclusion
Our work broadens the mutation spectrum of COG6 gene and states the importance of whole‐exome sequencing in facilitating the definitive diagnosis of this disorder and prenatal diagnosis in a subsequent pregnancy.
Keywords: abnormal liver function, COG6, congenital disorders of glycosylation, hypohidrosis, microcephaly
We report a patient from China with growth retardation, abnormal liver function, hypohidrosis, and microcephaly. Genetic sequencing identified two novel heterozygous mutations: c.428G>T (p.S143I) and c.1843C>T (p.Q615X) in the COG6 gene, inherited from the parents. To date, only 19 patients with COG6‐CDG have been reported.

Abbreviations
- ACMG
American College of Medical Genetics and Genomics
- CDGs
congenital disorders of glycosylation
- COG6
conserved oligomeric golgi complex 6
- ECG
echocardiography
- EEG
electroencephalogram
- PCR
polymerase chain reaction
1. INTRODUCTION
Congenital disorders of glycosylation (CDGs) are a large and very heterogeneous group of metabolic disorders that comprise defects in glycosylation of proteins or lipids (Van Scherpenzeel et al., 2016). Glycosylation is the principal form of posttranslational modification of proteins and lipids, and it plays an important role in protein folding, molecular recognition, and other physiological processes (Bobby et al., 2018; Freeze et al., 2012). Because glycosylation occurs in every cell in every organism, CDGs is a multisystem disease that usually involves the nervous system, liver, heart, eyes, skeleton, and immune system (Alsubhi et al., 2017; Leroy, 2006; Li et al., 2019). The clinical phenotypes of CDG range from mild symptoms to severe multisystem dysfunction and even death (Jaeken, 2011).
Conserved oligomeric golgi (COG) complex plays an essential role in the maintenance of the structure of golgi apparatus, trafficking, and glycosylation of proteins. The COG complex is a heterooctamer containing eight different subunits (COG1–COG8) that are organized into lobe A (from COG1 to COG4) and lobe B (from COG5 to COG8) (Ungar et al., 2005). Since 2004, except for COG3, deficiency in other seven subunits have been reported to be associated with CDG (Barone et al., 2014; Haijes et al., 2018; Li et al., 2019; Lübbehusen et al., 2010; Rymen & Jaeken, 2014; Ungar et al., 2005; Wang et al., 2020).
COG6‐CDG (MIM: 614576) is a rare CDG type, since Lubbehusen and colleagues described the first COG6‐CDG in 2010 (Lübbehusen et al., 2010), to our knowledge, 19 cases have been reported (Alsubhi et al., 2017; Huybrechts et al., 2012; Li et al., 2019; Lübbehusen et al., 2010; Shaheen et al., 2013; Wu et al., 2017). In this study, we describe a Chinese girl with developmental delay, microcephaly, and abnormal liver function carrying two novel compound‐heterozygous mutations in COG6 gene (OMIM 606977) inherited from her parents, respectively. Prenatal diagnosis was performed for this family during subsequent pregnancy and revealed the fetus is carrying one mutation from her father. In addition, we summarized the clinical manifestations of patients with COG6‐CDG by reviewing articles published in English and Chinese.
2. CLINICAL REPORT
The proband was admitted to the Rehabilitation Department in Wuhan Children's Hospital due to psychomotor delay at 8 months old. She was the only child of healthy nonconsanguineous parents without relevant family medical history. A small biparietal diameter in utero was detected by ultrasound examination during the last trimester of pregnancy. She was born at 39 weeks of gestation by spontaneous vaginal delivery with a birth weight of 3.25 kg, birth length of 48 cm. Head circumference (HC) and Apgar scores were not available. At the age of 1 month, her brain MRI did not reveal any obvious abnormality.
On admission, she was noticed to have growth retardation, with a height of 65 cm (3rd–10th centile), and a weight of 7 kg (3rd–10th centile), and microcephaly with an HC of 36.4 cm (3rd centile). She had obvious dysmorphic features, including a narrow forehead, hypertelorism, bulbous nose, and downslanting palpebral fissures (Figure 1a,b). She had a severe psychomotor delay, hypotonia, and was unable to control her head, turn over, and sitting steadily. Her hands were clinched and both thumbs were adducted (Figure 1c,d). She had a response to sound and her eyes could follow the light. Her skin was dry and was unable to sweat. Her liver and spleen were impalpable.
FIGURE 1.
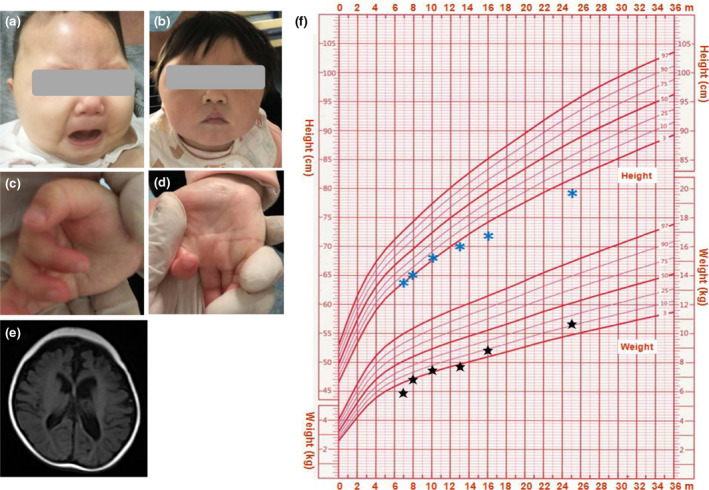
Abnormal facial appearance at the age of 8 months (a) and 20 months (b); (c,d) Adducted thumbs and camptodactyly. (e) Lesions of bilateral temporal, occipital, and parietal lobes and enlarged cerebral fissure and lateral ventricles. (f) Growth curves of this patient; the blue asterisks indicate height and the black stars indicate weight
She was extensively investigated by blood biochemistry examinations, echocardiography, electroencephalogram, brain MRI, and growth and development scale (Gesell Developmental Schedules). Blood biochemistry examinations showed abnormal liver function with elevated total billirubin (41.4 μmol/L, reference: 2–19 μmol/L), glutamic oxalacetic transaminase (231 U/L, reference: 10–40 U/L), glutamic pyruvic transaminase (72 U/L, reference: 7–45 U/L), and alkaline phosphatase (399 U/L, reference: 42–220 U/L). Renal function test showed slightly decreased blood creatinine (15.1 μmol/L, reference: 44.2–132.6 μmol/L) and urea nitrogen (1.76 mmol/L, reference: 2.9–7.1 mmol/L). The other examinations, including immune, thyroid function, coagulation, and metabolism screening test for amino acid in blood and organic acid in the urine, were normal. Echocardiography (ECG) revealed a mild atrioventricular block, and electroencephalogram (EEG) showed localized sharp waves and slow spindle waves during sleep, but no seizure. An electrocardiogram showed a shunt from left to right, indicating the likelihood of patent foramen ovale or atrial septal defect and. Brain MRI displayed softening lesion of bilateral temporal, occipital, and parietal lobes and enlarged cerebral fissure and lateral ventricles (Figure 1e). Gesell's developmental scale indicated that the patient was delayed in language, motor development, and social interaction.
She had a history of fever and abnormal liver function with a higher glutamic‐pyruvic transaminase (136 U/L) and glutamic oxalacetic transaminase (509 U/L) at the age of 7 months old, had been hospitalized in Gastrointestinal Department, and received treatment with inosine and atomolan. She also had recurrent upper respiratory tract infections, and gastrointestinal dysfunction, such as chronic diarrhea. At the age of 2 years old, her growth retardation was obvious, with a height of 79 cm (<3rd centile), a weight of 11 kg (3rd–10th centile; Figure 1f), and an HC of 41.5 cm (<3rd centile), and she was still unable to sit and walk independently.
3. METHODS
3.1. Trio whole‐exome sequencing and Sanger sequencing verification of the COG6 genetic mutation
After obtaining written informed consent, genomic DNAs of this patient and her parents were extracted from whole blood samples using the QIAamp Blood DNA mini kit (Qiagen) according to the manufacturer's instructions. Whole‐exome sequencing and subsequent data analysis were conducted with the help of the third‐party medical testing laboratory (Chigene Lab). Candidate variants in the COG6 gene were verified by Sanger sequencing using self‐designed primers. The primers for amplification of the COG6 gene (NG_028352.1) were synthesized by Sangon Biotechnology Co. Ltd. The primer sequences for exon 4 were forward 5′‐GAG TTC CAT AGA GTG ATC TC‐3′ and reverse 5′‐CAT CAT TTC TGA ACT CCA CAG C‐3′, and the primer sequences for exon 19 were forward 5′‐GAT TAA CTG TGT AGC CAT ATA GTG‐3′ and reverse 5′‐GGA TTC ATC ACG GCT GCA TAC‐3′. The target fragments were amplified using polymerase chain reaction (PCR) and the products were sequenced using an ABI 3500DX sequencer (Applied Biosystems). Prenatal diagnosis for this family was performed by amniocentesis in the subsequent pregnancy.
4. RESULTS
4.1. Whole‐exome sequencing and prenatal diagnosis
Whole‐exome sequencing detection and subsequent data analysis found two heterozygous mutations: c.428G>T (p.S143I) in exon 4 and c.1843C>T (p.Q615X) in exon 19 of COG6 (NM_020751.3) in the patient, inheriting from his mother and his father (Figure 2a,b), respectively, which was confirmed by Sanger sequencing. Two mutations are not listed in any database (i.e., gnomAD, ClinVar, and 1000 genomes). Bioinformatic analysis showed these two residues are conserved among different species (Figure 2c). According to the standards and guidelines recommended by the American College of Medical Genetics and Genomics (ACMG) (Richards et al., 2015), these two mutations were classified as likely pathogenic and pathogenic, respectively.
FIGURE 2.
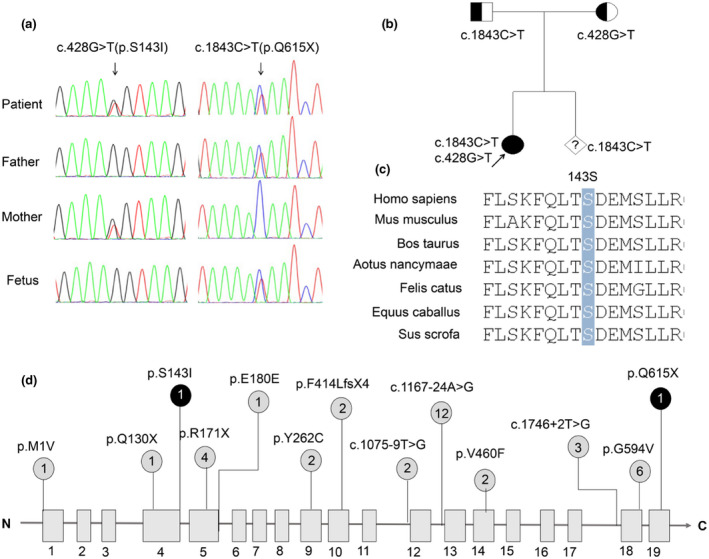
(a) Sanger sequencing of COG6 mutations in this family, (b) genealogical tree of this family, (c) conservation analysis of COG6 protein among different species. The 143rd amino acid is indicated by a blue bar and conserved throughout all indicated species. (d) Distribution of variants within the COG6 gene. Variants described previously are in gray, and variants in our study are in black. The numbers in circle indicate that the total alleles of each variant reported in COG‐CDG patients
After 1 year, the mother was pregnant again, and ultrasonographic screening during the subsequent pregnancy did not reveal any abnormal findings. The gene testing using amniotic fluid cells at 16 weeks of pregnancy showed that the fetus only carried the c.1843C>T mutation (Figure 2a). and continued the pregnancy. Following up, the child was born and no abnormality is found at present.
5. DISCUSSION
Glycosylation is an essential biological process in cells for various protein or lipid modifications and ~2% of human genes encode glycosylation‐related proteins (Apweiler et al., 1999). CDGs affect many organs and it is difficult to make a definitive diagnosis because of lack of uniformity in clinical manifestations. In this study, we reported a Chinese patient with growth retardation, facial dysmorphism, microcephaly, developmental delay, abnormal liver function, hypohidrosis, recurrent infection, and gastrointestinal dysfunction. Two variants in COG6 gene were found in this patient, and bioinformatic analysis suggested that these two variants are likely pathogenic and pathogenic, respectively. One is nonsense mutation, c.1843C>T (p.Q615X), which gives rise to premature termination of translation and truncated protein, whereas missense mutation, c.428G>T (p.S143I), may result in misfolding and degradation of the mutated protein. Another COG6 missense mutation, c.1646G>T(p.G594V), was proved to lead to decreased protein due to instability of the COG6 mRNA (Lübbehusen et al., 2010). The missense mutation, c.428G>T (p.S143I), in our patient, caused polar amino acids to be replaced by hydrophobic amino acids, nonpolar amino acids, which could affect the structure or stability of protein, consequently, disrupt the function of COG6 protein.
As shown in Figure 2d, to date, a total of 13 different variants have been reported in 20 COG6‐CDG patients from Saudi (9), Morocco (4), Turkey (3), Bulgaria (1), and China (3), respectively. Of these variants, 3 are splicing variants, 3 are nonsense mutations, and 5 are missense mutations, 1 is an insertion of one base which results in a frameshift and immediate stop codon, and 1 is synonymous mutation (Alsubhi et al., 2017; Huybrechts et al., 2012; Li et al., 2019; Lübbehusen et al., 2010; Wu et al., 2017). The splicing variant c.1167‐24A>G, which results in a frameshift and insertion of the premature stop codon (NM_020751.2: p.G390Ffs*6), was the most common and found to be homozygous in six patients with COG‐CDG (Alsubhi et al., 2017; Shaheen et al., 2013), which qualifies c.1167‐24A>G as a hotspot variant in the COG6 gene. Interestingly, of all 20 patients with COG‐CDG, 13 are homozygous carriers for COG6 mutations (Alsubhi et al., 2017; Huybrechts et al., 2012; Shaheen et al., 2013; Ungar et al., 2005), and 10 out of 13 are from Middle East, thus, intraethnic marriage may be an important factor for the relative prevalence of this disorder in these regions. It should be noted that one patient was the sister of the patient (Lübbehusen et al., 2010). She did not have a genetic test and died at the age of 5 weeks. According to her clinical manifestations, this patient was suspected to carry the same mutation (c.1646G>T) as her elder sister.
We summarized the clinical manifestations of patients reported by reviewing the literatures and found that microcephaly is a consistent clinical hallmark of COG6‐CDG, which could be found at birth or in later development. It is proposed that microcephaly or other cortical dysplasia is likely due to reduced brain growth or damaged migration of neurons and formation of synapses during brain development (Kodera et al., 2015; Rymen et al., 2015). As shown in Figure 3, the most frequent features of COG6‐CGD patients are developmental disability (18/18) followed by facial dysmorphisms (17/18) and growth retardation (15/17). Skin abnormalities are also very common, including hyperkeratosis/hyperthermia, dry skin, orange peel skin, red rash, and hypohidrotic ectodermal dysplasia. Hypohidrosis may be caused by abnormal function of sweat glands as a normal structure and the density of eccrine sweat glands was observed in skin biopsy (Rymen & Jaeken, 2014). Recurrent infections and liver involvement are severe complications, which could be a critical cause of death (Rymen et al., 2015). Heart defects, including atrial septum defect, ventricular septal defect, and patent ductus arteriosus, are another feature and observed in more than half of COG6‐CDG patients (9/14). In addition, COG6‐CDG patients have some other symptoms, such as hypotonia, skeletal and gastrointestinal abnormalities, epilepsy, and vision and hearing problems. Compared to these patients with COG6‐CDG, our patient had the typical clinical manifestations, such as developmental delay, facial dysmorphism, growth retardation, hypotonia, hypohidrosis, microcephaly, abnormal brain imaging, liver dysfunction, recurrent infection, and gastrointestinal dysfunction. However, there is no obvious heart problem, vision defect, hearing loss, and epilepsy. Combined the genetic variants identified in this patient, the diagnosis of COG6‐CDG was made (Table 1).
FIGURE 3.
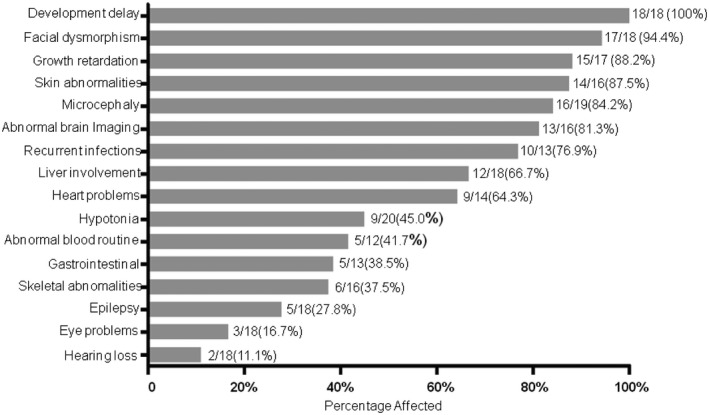
Phenotype summary from the 20 COG6‐CDG cases
TABLE 1.
Clinical characteristics of patients with COG6‐CDG
| Patients | Present study | P1 (Rymen, et al) | P2 (Rymen, et al) | P3 (Rymen, et al) | P4 (Rymen, et al) | P5 (Rymen, et al) | P6 (Rymen, et al) | P7 (Rymen, et al) | P8 (Lübbehusen et al) | P9 (Huybrechts, et al) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sex | F | F | M | F | M | M | F | F | F | F | |
| Age of onset | 8 months | 1 month | 1 year | 6 months | 21 years | 3 months | At birth | 1 month | NA | 3 months | |
| Age of being reported | 20 months | – | – | – | 21 years | – | – | 12 years | – | 6 years | |
| Age of death | – | 1 month | 12 months | 15 months | – | 14 months | 5 weeks | – | 5 weeks | – | |
| Ethnicity | Chinese | Bulgarian | Turkish | Turkish | Moroccans | Moroccans | Moroccans | Turkish | Turkish | Moroccans | |
| Facial dysmorphism | + | + | + | + | + | – | NA | + | NA | + | |
| Developmental delay | + | + | + | + | + | + | NA | + | NA | Mild | |
| Growth retardation | + | + | + | + | – | – | NA | + | NA | + | |
| Microcephaly | + | + | + | + | + | + | + | + | NA | + | |
| Hypotonia | – | + | + | – | – | – | + | – | – | – | |
| Brain MRI | Lesions of bilateral temporal, occipital, and parietal lobes | Agenesis of corpus callosum | Asymmetric lateral ventricle and Cerebrospinal fluid increase | Atrophy of brain and cerebellum | NA | NA | – | Atrophy of cerebral cortex | NA | – | |
| Congenital heart disease | ASD/PFO | ASD, PDA | NA | ASD, PDA | NA | NA | ASD | VSD | NA | NA | |
| Hepatobiliary abnormalities | Cholestasis, abnormal liver function | HMG, Cholestasis | HMG | HMG | HMG | HMG, Cholestasis, Hepatic failure | HMG, Cholestasis | HMG, Cholestasis, Cirrhosis | Cholestasis | Cirrhosis | |
| Gastrointestinal | – | NA | Chronic diarrhea | Chronic diarrhea | – | NA | Intestinal ischemia | Chronic diarrhea | NA | Enteritis | |
| Recurrent infection | + | – | + | + | + | + | – | + | – | + | |
| Skin abnormalities | – | NA | NA | Hyperkeratosis | Hyperkeratosis | Dry skin | NA | Orange peel skin | NA | NA | |
| Blood routine examination | – | Thrombocytopenia | Anemia, thrombocytopenia | Thrombocytopenia, leukocytosis | Thrombocytopenia, pancytopenia | NA | NA | Mild pancytopenia | NA | NA | |
| skeleton | NA | Joint contracture, adduction of thumb | – | Postaxial polydactyly | – | – | NA | Scoliosis | NA | Postaxial polydactyly | |
| Seizure | Abnormal EEG | + | – | – | – | – | – | – | + | NA | |
| Hearing/ophthalmological abnormality | – | Optic atrophy | Hearing loss | NA | Hearing loss | NA | – | – | – | – | |
| Others | – | Hyperechoic kidney, hypotonia | NA | Immunodeficiency | Monocytosis | NA | Thymus hyperplasia, adrenal hypoplasia | Unilateral renal hypoplasia | intracranial hemorrhage | Combined immunodeficiency | |
| Allele 1 | c.1843C>T (p.Q615X) | c.511C>T (p.R171X) | c.1746+2T>G | c.1238_1239insA (p.F414Lfs*4) | c.1646G>T (p.G594V) | c.1646G>T (p.G594V) | NA* | c.511C>T (p.R171X) | c.1646G>T (p.G594V) | c.1646G>T (p.G594V) | |
| Allele 2 | c.428G>T (p.S143I) | c.511C>T (p.R171X) | c.1746+2T>G | c.1238_1239insA (p.F414Lfs*4) | c.785A>G (p.T262C) | c.785A>G (p.T262C) | NA | c.1746+2T>G | c.1646G>T (p.G594V) | c.1646G>T (p.G594V) | |
| Patients | P10 (Shaheen, et al) | P11 (Li, et al) | P12 (Alsubhi, et al) | P13 (Alsubhi, et al) | P14 (Alsubhi, et al) | P15 (Alsubhi, et al) | P16 (Alsubhi, et al) | P17 (Alsubhi, et al) | P18 (Alsubhi, et al) | P19 (Wu, et al) |
|---|---|---|---|---|---|---|---|---|---|---|
| Sex | M | M | M | M | F | M | F | F | M | F |
| Age of onset | 1 week | 1 month | NA | NA | NA | NA | NA | NA | NA | 2 months |
| Age of being reported | 13 years | 28 months | – | 13 years | 2 years | 8 years | 4 years | 3 years | 7 years | – |
| Age of death | – | – | 3 months | – | – | – | – | – | – | Dead (unclear detail) |
| Ethnicity | Saudi | Chinese | Saudi | Saudi | Saudi | Saudi | Saudi | Saudi | Saudi | Chinese |
| Facial dysmorphism | + | + | + | + | + | + | + | + | + | + |
| Developmental delay | + | + | + | + | + | + | + | + | + | NA |
| Growth retardation | NA | + | + | + | + | + | + | + | + | + |
| Microcephaly | – | Mild | + | + | – | + | + | + | + | – |
| Hypotonia | – | – | + | + | + | – | + | + | + | – |
| Brain MRI | NA | abnormal signaling of the long T1 and long T2 in the right frontal region | Agenesis of corpus callosum | Hypo−myelination | PVL, reduced WM, thin splenium of corpus callosum | – | Delayed myelination, thin corpus callosum | Thin corpus callosum | Brain atrophy, PVL, bright T2 along corticospinal tract | Bilateral ventricles are slightly full |
| Congenital heart disease | NA | VSD/PFO | Dysplastic aortic valve, PDA | – | VSD/PFO | – | – | – | – | VSD/PFO |
| Hepatobiliary abnormalities | NA | – | – | – | – | – | – | – | NA | Abnormal liver function |
| Gastrointestinal | NA | – | NA | NA | NA | NA | NA | NA | NA | Gas accumulation |
| Recurrent infection | + | + | NA | NA | NA | NA | NA | NA | NA | + |
| Skin abnormalities | Hyperkeratosis | red rash | Lipodystrophy | HH | HH | HH | – | Lipodystrophy | HH | Hypohidrotic ectodermal dysplasia |
| Blood routine examination | NA | NA | NA | NA | NA | NA | NA | NA | NA | Anemia |
| Skeleton | NA | – | + | – | – | + | – | – | – | – |
| Seizure | NA | + | – | + | – | – | – | – | – | – |
| Hearing and eye problems | – | – | Strabismus | – | – | + | – | – | Bilateral ptosis | – |
| Others | NA | Elevation of myocardial enzymes | Cerebellar hypoplasia | Cerebellar hypoplasia | NA | NA | NA | Autism | Left kidney crystal | |
| Allele 1 | c.1167‐24A>G (p.G390FfsX6) | c.1A>G (p.M1V) | c.1378G>T (p.V460F) | c.1167‐24A>G (p.G390FfsX6) | c.1167‐24A>G (p.G390FfsX6) | c.1167‐24A>G (p.G390FfsX6) | c.1167‐24A>G (p.G390FfsX6) | c.1075‐9T>G | c.1167‐24A>G (p.G390FfsX6) | c.511C>T (p.R171X) |
| Allele 2 | c.1167‐24A>G (p.G390FfsX6) | c.388C>T (p.Q130X) | c.1378G>T (p.V460F) | c.1167‐24A>G (p.G390FfsX6) | c.1167‐24A>G (p.G390FfsX6) | c.1167‐24A>G (p.G390FfsX6) | c.1167‐24A>G (p.G390FfsX6) | c.1075‐9T>G | c.1167‐24A>G (p.G390FfsX6) | c.540G>A (p.E180E) |
HH, Hypohydrosis; HMG, hepatomegaly; NA, not available.
P6 was the sister of P8, dead at the age of 5 weeks, and not done genetic test. GenBank reference sequence and version number: NG_028352.1.
As reported by Rymen and her colleagues, patients with splice site mutation had mild clinical phenotype especially in patients with c.1167‐24A>G mutation (Rymen et al., 2015). All six patients with homozygous c.1167‐24A>G mutation had only mild to moderate developmental delay and mental retardation and no abnormality in liver function, gastrointestinal system, and skeletal system (Alsubhi et al., 2017; Li et al., 2019; Shaheen et al., 2013). Only one of the six patients had heart problems, skeletal abnormalities, or recurrent infection, but all six patients presented skin abnormalities. However, both patients with homozygous loss‐of‐function mutations p.Arg171* or p.Phe414Leufs*4 die at the age of 1 month and 15 months due to respiratory or liver failure, respectively (Rymen et al., 2015). So, all findings suggest that patients’ harboring nonsense mutations may present more severe clinical manifestations. These results need to be further confirmed by studying more patients.
6. CONCLUSION
We identified two compound heterozygous variants in COG6 gene in a patient with developmental delay, facial dysmorphism, microcephaly, recurrent infection, and hypohidrosis. A prenatal diagnosis was performed by amniocentesis for this family in the subsequent pregnancy. Our work broadens the mutation spectrum of COG6 gene and also emphasizes the importance of whole‐exome sequencing in definitive diagnosis and prenatal diagnosis in the subsequent pregnancy.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The study was done after obtaining the ethics approval from the institutional review board of Wuhan Children's Hospital, Tongji Medical College, Huazhong University of Science and Technology (approval number 2019011), and written informed consent from the patient's parents.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
Study concepts: XH, PZ. Study design: PZ, PH, JC. Literature research: LZ. Clinical information collection: HP, JC. Data acquisition: PZ, SL. Data analysis/interpretation: LT, YH. Manuscript preparation: PZ, PH. Manuscript editing/revision/review: XH, JC. All authors have read and approved the manuscript.
ACKNOWLEDGMENTS
We thank the patient and her parents for their participation in this study.
Zhao, P., Zhang, L., Tan, L., Luo, S., Huang, Y., Peng, H., Cao, J., & He, X. (2021). Genetic analysis and prenatal diagnosis in a Chinese with growth retardation, abnormal liver function, and microcephaly. Molecular Genetics & Genomic Medicine, 9, e1751. 10.1002/mgg3.1751
Hanming Peng, Jiangxia Cao, and Xuelian He are equally contributed to this work.
Funding information
This work is supported by the grants of Wuhan Municipal Health Commission (no. WX19C19), Youth Program of National Natural Science Foundation of China (no. 81700302), and Natural Science Foundation of Hubei Province (2017CFB322).
DATA AVAILABILITY STATEMENT
The data and materials are submitted and are available on reasonable request.
REFERENCES
- Alsubhi, S., Alhashem, A., Faqeih, E., Alfadhel, M., Alfaifi, A., Altuwaijri, W., Alsahli, S., Aldhalaan, H., Alkuraya, F. S., Hundallah, K., Mahmoud, A., Alasmari, A., Mutairi, F. A., Abduraouf, H., AlRasheed, L., Alshahwan, S., & Tabarki, B. (2017). Congenital disorders of glycosylation: The Saudi experience. American Journal of Medical Genetics Part A, 173(10), 2614–2621. 10.1002/ajmg.a.38358 [DOI] [PubMed] [Google Scholar]
- Apweiler, R., Hermjakob, H., & Sharon, N. (1999). On the frequency of protein glycosylation, as deduced from analysis of the SWISS‐PROT database. Biochimica Et Biophysica Acta, 1473(1), 4–8. 10.1016/s0304-4165(99)00165-8 [DOI] [PubMed] [Google Scholar]
- Barone, R., Fiumara, A., & Jaeken, J. (2014). Congenital disorders of glycosylation with emphasis on cerebellar involvement. Seminars in Neurology, 34(3), 357–366. 10.1055/s-0034-1387197 [DOI] [PubMed] [Google Scholar]
- Bobby, G., Ng, B. G., & Freeze, H. H. (2018). Perspectives on glycosylation and its congenital disorders. Trends in Genetics, 34(6), 466–476. 10.1016/j.tig.2018.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeze, H. H., Eklund, E. A., Ng, B. G., & Patterson, M. C. (2012). Neurology of inherited glycosylation disorders. The Lancet. Neurology, 11(5), 453–466. 10.1016/S1474-4422(12)70040-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haijes, H. A., Jaeken, J., Foulquier, F., & van Hasselt, P. M. (2018). Hypothesis: lobe A (COG1‐4)‐CDG causes a more severe phenotype than lobe B (COG5‐8)‐CDG. Journal of Medical Genetics, 55(2), 137–142. 10.1136/jmedgenet-2017-104586 [DOI] [PubMed] [Google Scholar]
- Huybrechts, S., De Laet, C., Bontems, P., Rooze, S., Souayah, H., Sznajer, Y., & Goyens, P. (2012). Deficiency of subunit 6 of the conserved oligomeric golgi complex (COG6‐CDG): Second patient, different phenotype. JIMD Reports, 4, 103–108. 10.1007/8904_2011_79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeken, J. (2011). Congenital disorders of glycosylation (CDG): It’s (nearly) all in it! Journal of Inherited Metabolic Disease, 34, 853–858. 10.1007/s10545-011-9299-3 [DOI] [PubMed] [Google Scholar]
- Kodera, H., Ando, N., Yuasa, I., Wada, Y., Tsurusaki, Y., Nakashima, M., Miyake, N., Saitoh, S., Matsumoto, N., & Saitsu, H. (2015). Mutations in COG2 encoding a subunit of the conserved oligomeric golgi complex cause a congenital disorder of glycosylation. Clinical Genetics, 87(5), 455–460. 10.1111/cge.12417 [DOI] [PubMed] [Google Scholar]
- Leroy, J. G. (2006). Congenital disorders of N‐glycosylation including diseases associated with O‐ as well as N‐glycosylation defects. Pediatric Research, 60(6), 643–656. 10.1203/01.pdr.0000246802.57692.ea [DOI] [PubMed] [Google Scholar]
- Li, G., Xu, Y., Hu, X., Li, N., Yao, R., Yu, T., Wang, X., Guo, W., & Wang, J. (2019). Compound heterozygous variants of the COG6 gene in a Chinese patient with deficiency of subunit 6 of the conserved oligomeric Golgi complex (COG6‐CDG). European Journal of Medical Genetics, 62(1), 44–46. 10.1016/j.ejmg.2018.04.017 [DOI] [PubMed] [Google Scholar]
- Lübbehusen, J., Thiel, C., Rind, N., Ungar, D., Prinsen, B. H., de Koning, T. J., van Hasselt, P. M., & Körner, C. (2010). Fatal outcome due to deficiency of subunit 6 of the conserved oligomeric Golgi complex leading to a new type of congenital disorders of glycosylation. Human Molecular Genetics, 19(18), 3623–3633. 10.1093/hmg/ddq278 [DOI] [PubMed] [Google Scholar]
- Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier‐Foster, J., Grody, W. W., Hegde, M., Lyon, E., Spector, E., Voelkerding, K., & Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rymen, D., & Jaeken, J. (2014). Skin manifestations in CDG. Journal of Inherited Metabolic Disease, 37(5), 699–708. 10.1007/s10545-014-9678-7 [DOI] [PubMed] [Google Scholar]
- Rymen, D., Winter, J., Van Hasselt, P. M., Jaeken, J., Kasapkara, C., Gokçay, G., Haijes, H., Goyens, P., Tokatli, A., Thiel, C., Bartsch, O., Hecht, J., Krawitz, P., Prinsen, H. C. M. T., Mildenberger, E., Matthijs, G., & Kornak, U. (2015). Key features and clinical variability of COG6‐CDG. Molecular Genetics and Metabolism, 116(3), 163–170. 10.1016/j.ymgme.2015.07.003 [DOI] [PubMed] [Google Scholar]
- Shaheen, R., Ansari, S., Alshammari, M. J., Alkhalidi, H., Alrukban, H., Eyaid, W., & Alkuraya, F. (2013). A novel syndrome of hypohidrosis and intellectual disability is linked to COG6 deficiency. Journal of Medical Genetics, 50(7), 431–436. 10.1136/jmedgenet-2013-101527 [DOI] [PubMed] [Google Scholar]
- Ungar, D., Oka, T., Vasile, E., Krieger, M., & Hughson, F. M. (2005). Subunit architecture of the conserved oligomeric Golgi complex. The Journal of Biological Chemistry, 280(38), 32729–32735. 10.1074/jbc.M504590200 [DOI] [PubMed] [Google Scholar]
- Van Scherpenzeel, M., Willems, E., & Lefeber, D. J. (2016). Clinical diagnostics and therapy monitoring in the congenital disorders of glycosylation. Glycoconjugate Journal, 33(3), 345–358. 10.1007/s10719-015-9639-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X., Han, L., Wang, X. Y., Wang, J. H., Li, X. M., Jin, C. H., & Wang, L. (2020). Identification of Two Novel Mutations in COG5 Causing Congenital Disorder of Glycosylation. Frontiers in Genetics, 11, 168. 10.3389/fgene.2020.00168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, B. B., Li, W. X., Yang, L., Wang, H. J., & Zhou, W. H. (2017). A case of neonatal congenital disorder of glycosylation caused by COG6 gene mutation and literature review. Chinese Journal of Evidence‐Based Pediatrics, 12(1), 49–53. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data and materials are submitted and are available on reasonable request.