

Abstract
Background
Microcephalic osteodysplastic primordial dwarfism type II (MOPD II) is a rare autosomal recessive disorder characterized by severe pre‐ and postnatal growth restrictions, microcephaly, skeletal dysplasia, severe teeth deformities, and typical facial features. Previous studies have shown that MOPD II is associated with mutations in the pericentrin (PCNT) gene.
Methods
We evaluated the clinical features of a 10‐year and 7‐month‐old Chinese girl with MOPD II. Subsequently, next‐generation sequencing and flow cytometry were performed to investigate genetic characteristics and the expression of PCNT protein respectively.
Results
The patient presented with short stature, microcephaly, typical craniofacial features, teeth deformity, thrombocytosis, and a delayed bone age (approximately 7 years). No abnormality in growth hormone or insulin‐like growth factor 1 was detected. Notably, the patient was found to carry a novel homozygous PCNT mutation (c.6157G>T, p.Glu2053Ter), which was inherited from her healthy heterozygous parents. Meanwhile, significant deficiency of PCNT expression was identified in the patient.
Conclusion
Our study identified a novel PCNT mutation associated with MOPD II, expanded the mutation spectrum of the PCNT gene and improved our understanding of the molecular basis of MOPD II.
Keywords: growth restriction, microcephaly, MOPD II, novel homozygous mutation, PCNT gene
We describe a rare case of MOPD II in a 10‐year and 7‐month old Chinese female. A novel homozygous mutation was identified in exon 30 of the PCNT gene (c.6157G>T, p.Glu2053Ter) by next‐generation sequencing. Meanwhile, flow cytometry revealed a deficiency in the PCNT protein expression of the patient. This is the first report of a Chinese MOPD II patient with a homozygous PCNT gene mutation.

1. INTRODUCTION
Microcephalic osteodysplastic primordial dwarfism type II (MOPD II, OMIM#210720) is a rare autosomal recessive disease mainly characterized by severe intrauterine and postnatal growth retardation, microcephaly, typical skeletal dysplasia, severe dental anomalies, and obvious facial features (Rauch, 2011). In addition, some MOPD II patients also presented with cerebrovascular disease (moyamoya disease and aneurysms), hyperopia, precocious puberty, skin pigmentation, and hematological abnormalities (thrombocytosis and leukocytosis, etc.). To date, only eight MOPD II cases have been reported in China (Chang et al., 2017; Chen et al., 2019; Li et al., 2015; Ma et al., 2021; Meng et al., 2019; Zhou et al., 2019).
MOPD II is caused by biallelic loss‐of‐function mutations of the pericentrin (PCNT) gene (Rauch et al., 2008; Willems et al., 2010). The PCNT gene, mapped to chromosome 21q22.3, spans 121.61 kb of genomic sequence and contains 47 exons (Rauch et al., 2008). The PCNT protein encoded by PCNT gene is a highly conserved coiled‐coil protein composed of 3336 amino acid residues and is ubiquitous in human tissue. It is a primary component of pericentriolar material (PCM) and plays key roles in centrosome structure and function as well as spindle assembly through anchoring the γ‐tubulin at centrosomes (Mennella et al., 2012). Centrosome, composed of the centriole and PCM, is the main microtubule organizing center and is primarily responsible for cell cycle progression (Tibelius et al., 2009; Zimmerman et al., 2004). The deficiency of PCNT protein leads to centrosome abnormalities resulting in spindle assembly abnormality and misorientation, chromosome missegregation, mitotic failure, and impaired cell cycle progression (Barbelanne & Tsang, 2014; Chen et al., 2014; Delaval & Doxsey, 2010).
In recent years, there have been only a few reports concerning the genotype‐phenotype association of MOPD II. The underlying molecular basis for most of the clinical features of MOPD II remains undefined. Herein, we described a Chinese girl suffering from MOPD II caused by a novel homozygous mutation of PCNT gene and we elucidated the clinical and genetic characteristics of this patient. To our knowledge, previously reported 8 Chinese MOPD II cases were all caused by compound heterozygous PCNT mutations and our study is the first report of a MOPD II patient with a homozygous PCNT gene mutation in China.
2. MATERIALS AND METHODS
2.1. Subjects
A 10‐year and 7‐month‐old patient and her parents were recruited from Kunming Children's Hospital. Consanguineous marriage was confirmed between the patient's parents. This study followed the principles of the 1975 Declaration of Helsinki and got official approval from the Ethics Committee of Kunming Children's Hospital. Written informed consent was obtained from the parents of the patient before collecting blood specimens and publishing the identifiable photographs of the patient in the article.
2.2. Clinical evaluation
Detailed clinical assessments were performed, including physical, imaging, and laboratory examinations. Special attention was paid to height, weight, occipitofrontal circumference, skeletal features, hair, teeth, and craniofacial features of the patient during the physical examination. Imaging examinations included bone age assessment (left hand anteroposterior x‐ray), hypophysis magnetic resonance imaging (MRI), and computed tomography (CT) of brain. The laboratory examinations included hematology and endocrine system examinations (levels of insulin‐like growth factor‐1, growth hormone, trace elements, and gonadal hormones).
2.3. Next‐generation sequencing (NGS)
We used the QIAamp DNA Mini Kit (Qiagen) to extract genomic DNA from peripheral blood. Nanodrop 2000 (Thermo Fisher Scientific), an ultramicro spectrophotometer, was applied to evaluate and quantify the extracted DNA. Qualified genomic DNA was fragmented randomly. End repair and addition of “A tails” of the DNA fragments were carried out using the Standard Library Construction Kit (MyGenostics, Inc.) and amplified by PCR. Then, 5× ligation buffer (10 μl), nuclease‐free H2O (8 μl), adapter (2 μl), and DNA ligase (5 μL) were added, and incubated at 20°C for 15 min. The genomic DNA library was amplified by PCR and purified using Ampure beads (Beckman Coulter). The ratio of magnetic beads to samples was 1:1.
Next, the genomic DNA library was hybridized using the GenCap custom enrichment kit (MyGenostics, Inc.) at 65°C for 22 h. The biotin‐labeled probe was covalently linked with streptavidin‐modified magnetic beads to obtain the target DNA regions. The magnetic beads with the target gene were adsorbed by a magnetic rack. The adsorbed beads were washed once with WB1 Buffer (25°C, 15 min) and three times with WB3 Buffer (65°C, 10 min). The acquired DNA was purified by 80% ethanol and successively eluted with elution buffer (30 min). The purified and eluted target DNA fragments were amplified by PCR.
The Illumina NextSeq 500 sequencer (Illumina, San Diego, CA, USA) was adopted as the sequencing platform. After sequencing, Bcl2Fastq 2.18.0.12, Cutadapt 1.16, Burrows‐Wheeler aligner 0.7.10 (BWA), Samtools 1.2, Bamtools 2.4.0, Genome Analysis Toolkit software 3.7 (GATK), PolyPhen_2, SIFT, PANTHER, and Pathogenic Mutation Prediction was used for data processing and analysis. The detected mutations were analyzed using the 1000 Genomes Project Database and the SNP Database. The detection results were filtered by the characteristics of mutation location, type, frequency, and site in the databases to retain the likely pathogenic mutations. Finally, the identified mutations were compared in the Human Gene Mutation Database (HGMD; http://www.hgmd.org/) to determine whether the mutations had been reported.
2.4. Sanger sequencing
Genomic DNA from the parents of the patient were collected for Sanger sequencing to validate the mutations identified by NGS. According to the results of NGS, two primers were designed to amplify exon 30 of PCNT (NM_006031): exon 30‐forward: 5′‐GCAAGGAGATGTAAGCCTTGG‐3′; exon 30‐reverse: 5′‐TTTCTTGAATGGGAGAATCTGG‐3′. We subsequently performed PCR as follows: initial denaturation at 94°C for 5 min, followed by 32 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 45 s, and extension at 72°C for 30 s, and a final extension at 72°C for 7 min. The products were sequenced, and DNASTAR (Madison) software was applied to analyze the sequencing data.
2.5. Flow cytometry
Flow cytometry was used to examine the expression of PCNT protein in peripheral blood mononuclear cells (PBMCs) of the patient and her parents. PBMCs were collected from peripheral blood of the above subjects using lymphocyte separation medium (TBD Science, LTS1077). Then, the separated PBMCs were washed with phosphate‐buffered saline (PBS) and centrifuged (4°C, 300 g, 10 min). Cell fixation and permeabilization were performed successively using the perfix‐nc Kit (Beckman Coulter, A07803) according to the manufacturer's instructions. Next, 1:200 diluted mouse anti‐pericentrin antibody (Abcam, ab28144, UK) was added and incubated at room temperature for 30 min. After centrifugation (4°C, 360 g, 5 min), the supernatant was discarded while the precipitate was washed again and resuspended in 1ml PBS (precooling at 4°C). Finally, 1:400 diluted goat anti‐mouse IgG H&L (FITC; Abcam, ab6785) was added for staining in the dark. BD FACSCanto II flow cytometer, FACSDiva, and FlowJo v10 software were used to collect and analyze data.
3. RESULTS
3.1. Clinical characteristics
The patient was a 10‐year and 7‐month‐old girl, who was full‐term and vaginally delivered with a birth length of 30.1 cm (<−3SD) and a birth weight of 1 kg (<−3SD). The patient exhibited delayed language and motor development. Specifically, she began to lift her head at 12 months (>2 to 3 months), sit independently at 2 years (>4 to 5 months), crawl at 3 years (>5 to 12 months), stand alone at 4 years (>9 to 12 months), walk at 4.5 years (>12 to 15 months), and talk at 4 years (normal children begin to say nouns at approximately 1 year, verbs at approximately 1.5 years, and short sentences at approximately 2 years). At the age of 10 years and 7 months, the patient was admitted to Kunming Children's Hospital.
Physical examination (Figure 1) after admission showed that the patient had a short stature (81.8 cm, <−3SD), low weight (7.0 kg, <−3SD), obvious microcephaly (occipitofrontal circumference 39.1 cm, <−3SD), sparse hair (Figure 1a), small and malformed teeth with wide interdental spacing (Figure 1b), and significant facial features including prominent eyes, broad nasal bridge and root, full nose tip, as well as proportionally small ears (Figure 1a). In addition, no skin pigmentation, coxa vara or talipes equinovarus (Figure 1c) were observed. The blood pressure of the patient was 70/58 mmHg, indicating no hypertension. Bilateral breasts were in Tanner stage 2, indicating the beginning of puberty.
FIGURE 1.
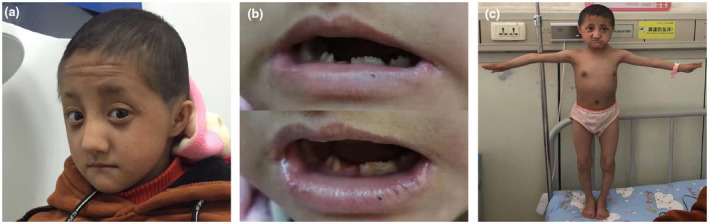
Clinical characteristics of the patient. (a) The patient showed significant craniofacial features, including microcephaly, sparse hair, prominent eyes, broad nasal bridge and root, full nose tip, as well as proportionally small ears. (b) Small and malformed teeth with wide interdental spacing were observed in the patient. (c) The patient had a short stature but no skin pigmentation, coxa vara, or talipes equinovarus
3.2. Imaging findings
Bone age assessment (left hand and wrist x‐ray) showed that ossification centers of the left carpus and ulna were apparent (the number of ossification centers of the carpus was 9), and the medial sesamoid of the thumb was not observed (Figure 2a). The bone age of the child was estimated to be approximately 7 years, which was significantly younger than the actual age of the patient, indicating a delay in ossification. Hypophysis MRI (Figure 2b) and brain CT (Figure 2c) and revealed no obvious abnormalities.
FIGURE 2.
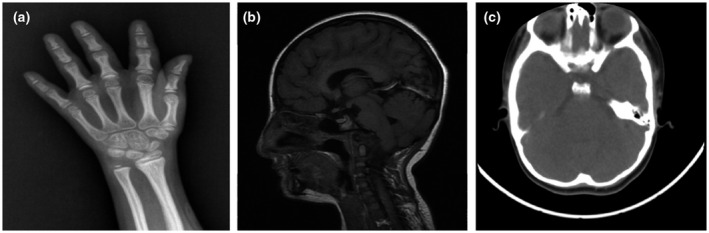
Imaging findings of the patient. (a) Bone age of the patient was examined through left hand and wrist X‐ray. Ossification centers of the left carpus and ulna were apparent (the number of ossification centers of the carpus was nine), and the medial sesamoid of the thumb was not observed. (b) Hypophysis MRI showed no obvious abnormalities. (c) Brain CT revealed no definite pathological signs. CT, computed tomography; MRI, magnetic resonance imaging
3.3. Laboratory findings
3.3.1. Hematological parameters
The patient was found to have thrombocytosis without leukocytosis or anemia. Specifically, the platelet count was significantly increased (705 × 109/L; reference range, 100–300 × 109/L), while the leukocyte count (8.55 × 109/L; reference range, 4–10 × 109/L) and the level of hemoglobin (118 g/L; reference range, 110–160g/L) were normal. In addition, the lymphocyte count (3.63 × 109/L; reference range, 1–3 × 109/L), monocyte count (0.89 × 109/L; reference range, 0.2–0.7 × 109/L), lymphocyte percentage (42.50%; reference range, 20–40%), monocyte percentage (10.40%; reference range, 3–8%), and plateletcrit (0.65; reference range, 0.108–0.282) of the patient were increased to varying degrees, while the neutrophil percentage (43.50%; reference range, 50–70%), mean corpuscular hemoglobin concentration (303.00 g/L; reference range, 320–360 g/L), mean platelet volume (9.2 fl; reference range, 9.4–12.5 fl), and platelet distribution width (10.00 fl; reference range, 15.5–18.1 fl) were decreased. Besides, the fasting blood glucose value (4.7 mmol/L) of the patient was in the normal range (3.9–5.8 mmol/L). No obvious abnormality was found in the rest of the hematological parameters.
3.3.2. Endocrine assessments
Insulin‐like growth factor‐1 (IGF‐1) testing showed no abnormality (375 ng/mL; reference range, 111–551 ng/ml). Growth hormone (GH) stimulation testing showed that the peak value at 90 min was 26.22 ng/mL (>10 ng/ml), indicating normal GH secretion. Moreover, the levels of trace elements (lead, zinc, iron, and copper) in the patient were normal. Sex hormone levels were also evaluated. In detail, the levels of luteinizing hormone (4.23 IU/L; reference range, 0.1–0.4 IU/L), follicle‐stimulating hormone (14.77 IU/L; reference range, 0.4–6.6 IU/L), and estradiol (113.3 pmol/L; reference range, 22–99 pmol/L) were increased, while the level of total testosterone (0.09 nmol/L, reference range 0.22–2.9 nmol/L) was decreased. Pituitary prolactin (474.70 mIU/L; reference range, 62–504 mIU/L) and progesterone (0.74 nmol/L; reference range, 0.7–4.3 nmol/L) were within normal ranges. Fluctuations in the levels of these gonadal hormones were normal due to the onset of puberty in the patient.
3.4. A novel pathogenic mutation associated with MOPD II was identified in the PCNT gene of the patient
A novel homozygous nonsense mutation was identified in the PCNT gene of the patient (Ⅱ‐1) through gene sequencing. Specifically, there was a single‐nucleotide substitution from “G” to “T” at position of c.6157 in exon 30 of the PCNT gene (c.6157G>T). This mutation altered the glutamate to a premature termination codon at the site of 2053, leading to PCNT protein truncation (p.Glu2053Ter, “p.” for “protein sequence”) (Figure 3a). To date, there is no report of this mutation in the HGMD professional database. Meanwhile, this is the first homozygous mutation of the PCNT gene reported in China and this mutation was likely to be the cause of MOPD II. The result of NGS was validated by Sanger sequencing. The sequencing results of her family members (Figure 3a) showed that the mutation was inherited from her parents as both her father (Ⅰ‐1) and mother (Ⅰ‐2) had a heterozygous c.6157G>T mutation in PCNT gene. Because MOPD II is an autosomal recessive disease, we speculated that the patient suffered from MOPD II and the homozygous PCNT c.6157G>T mutation led to the onset of the disease, while her parents are healthy carriers of this causative mutation (Figure 3b).
FIGURE 3.
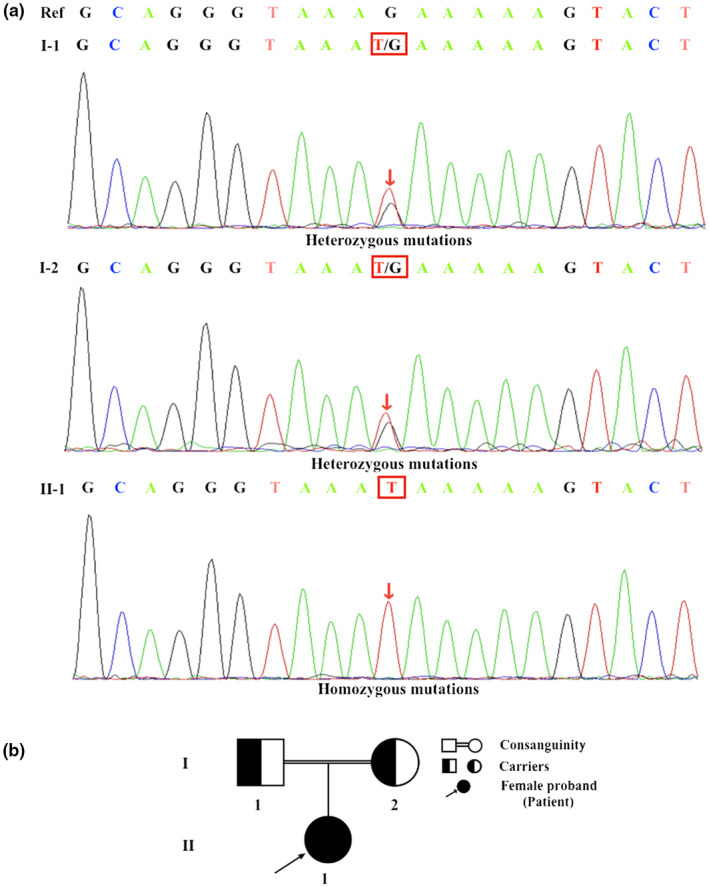
Mutational analysis of all subjects. (a) The patient (II‐1) was found to have a novel homozygous PCNT mutation (c.6157G>T, p.Glu2053Ter), which was inherited from her healthy heterozygous parents (father, I‐1; mother, I‐2). Nucleotide 6157 in the coding region was changed from guanine (G) to thymine (T) (c.6157G>T), which altered the glutamate to a premature termination codon at the site of 2053 (p.Glu2053Ter). Mutation sites are indicated by red arrows and squares. (b) Pedigree of the family. The father (I‐1) and mother (I‐2) were consanguineous. They were confirmed to be healthy carriers of this mutation without any symptoms, while their daughter (II‐1) carrying the homozygous PCNT mutation showed typical symptoms of MOPD II
3.5. The expression of PCNT protein was significantly decreased in PBMCs from the patient
In order to further verify the pathogenicity of the causative mutation identified by genetic testing, the expression levels of PCNT protein in PBMCs isolated from all subjects were measured by flow cytometry. The flow cytometry results of the patient revealed that the peak for PCNT‐positive cells nearly overlapped with the blank control peak (PCNT‐negative cells), indicating that there was almost no PCNT expression (0.82%) in PBMCs of the patient (Figure 4). However, the results of her parents showed that the peaks for PCNT‐positive cells partially shifted to right compared with the blank control peak and the expressions of PCNT protein in her father and mother were 46.3% and 42.0%, respectively.
FIGURE 4.
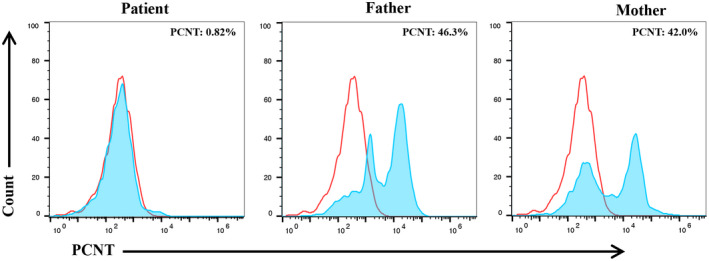
The expressions of PCNT protein in PBMCs isolated from the patient and her parents. In the patient, the peak for PCNT‐positive cells nearly overlapped with the blank control peak. The percentage of PCNT‐positive cells was 0.82%. In her parents, the peaks for PCNT‐positive cells partially shifted to right compared with the blank control peak and the percentages of PCNT‐positive cells were 46.3% (in the father) and 42.0% (in the mother), respectively. The peaks of the blank control and the subjects are marked in red and blue, respectively. PBMCs, peripheral blood mononuclear cells
4. DISCUSSION
MOPD II is a rare single‐gene genetic disease characterized by severe intrauterine and postnatal growth restriction accompanied by microcephaly (Klingseisen & Jackson, 2011). Patients with MOPD II will not grow taller than 110 cm even as adults. Both intensified nutrition and regulation with GH cannot alter the final growth, and GH therapy can even aggravate insulin resistance (Bober et al., 2012). PCNT protein plays an essential role in centrosome function, mitotic spindle assembly and the regulation of mitosis (Kim & Rhee, 2014), and the mutations of PCNT gene have been verified to have a close relationship with MOPD II. Here, we reported a rare case of MOPD II with a novel homozygous PCNT mutation (c.6157G>T, p.Glu2053Ter), which was inherited from the healthy carrier parents of the patient.
In our study, the patient exhibited significant short stature accompanied by microcephaly but no lack of GH or IGF‐1. MRI revealed no hypophysis dysplasia. Therefore, we could exclude that the short stature of this patient was caused by hypophysis dwarfism or GH resistance syndrome (Lindsey & Mohan, 2016). The patient showed dental deformity and distinctive facial features, including prominent eyes, broad nasal bridge and root, full nose tip, and proportionally small ears. The clinical manifestations of the patient are consistent with previous publications about MOPD II (Hall et al., 2004; Rauch et al., 2008). Moreover, thrombocytosis and delayed ossification, two common abnormalities of MOPD II, were also found in the patient. Subsequently, a novel homozygous nonsense mutation (c.6157G>T, p. Glu2053Ter) on exon 30 of the PCNT gene in this patient was identified through genetic sequencing. This mutation led to an amino acid change from glutamate to a premature termination codon, causing translational termination of PCNT protein. Finally, the altered expression of PCNT protein was validated by flow cytometry. The result showed that expression of PCNT protein was significantly decreased in the patient (0.82%) compared to her parents (46.3% and 42.0%) and confirmed the effect of this mutation on the expression of PCNT protein. Based on the clinical and genetic characteristics, the patient was finally diagnosed as MOPD II.
It is worth noting that MOPD II is characterized by high clinical heterogeneity. Apart from the clinical manifestations presented by our patient, MOPD II can be accompanied by other abnormalities. We collected characteristic clinical manifestations of eight MOPD II cases previously reported in other studies and compared them to our case (Table 1). Specifically, some skeletal changes, such as talipes equinovarus and hip pathologies (including coxa vara, hip dislocation/subluxation, and hip dysplasia), were respectively observed in Patient 6 and 7 but not in our case (Patient 1). The most detailed research of MOPD II with hip pathologies was completed by Karatas et al. (2014). Their results showed that approximately 67% of patients with MOPD II suffered from hip pathologies. MOPD II can also be associated with precocious puberty (in Patient 2 and 3), insulin resistance (in Patients 3 and 5) and several hematological abnormalities other than thrombocytosis (in our patient), including leukocytosis and anemia (in Patient 4). In the study of Hall et al. (2004), 13 of 22 male patients developed secondary sex characteristics between the ages of 11 and 13, and 6 of 21 girls had their first menstruation at approximately 9 years of age. In our case, the 10‐year and 7‐month‐old girl had normal breast development and gonadal hormone levels without precocious puberty. As indicated by Huang‐Doran et al. (2011), precocious puberty can be caused by insulin resistance, which is a very common symptom in MOPD II. According to their report, 86% of patients with MOPD II exhibited insulin resistance, which can lead to type II diabetes and acanthosis nigricans. Despite the absence of clear molecular mechanism of PCNT deficiency causing severe insulin resistance, the partial failure of adipocyte differentiation was considered to have the major contribution to this (Huang‐Doran et al., 2011). The clinical signs associated with insulin resistance such as abnormal fasting blood glucose levels and hypertension as well as acanthosis nigricans were not found in our patient, but fasting blood glucose, insulin levels, and blood pressure will continue to be monitored regularly in the daily clinical management of our patient. Regarding hematological abnormalities, the research of Unal et al. (2014) revealed that 87.5% of MOPD II patients exhibited thrombocytosis, 75% had leukocytosis, 62.5% had both thrombocytosis and leukocytosis, and 25% had anemia. Their study suggested a significant effect of PCNT protein on the proliferation and differentiation of hematopoietic cells. In addition, both Patients 8 and 9 had cerebrovascular disease. PCNT protein plays a crucial role in cerebrovascular development and the deficiency of PCNT protein is recognized as a major cause of cerebrovascular disease in MOPD II patients. According to previous reports, 25–50% of patients with MOPD II had cerebrovascular anomalies, including moyamoya disease and aneurysm (Bober et al., 2010; Brancati et al., 2005; Dieks et al., 2014; Teo et al., 2016). Without timely and effective intervention, the above cerebrovascular anomalies may lead to ischemic stroke, hemiplegia, and even death with a mortality rate of 23% (Huang et al., 2017; Kılıç et al., 2012; Perry et al., 2013). Fortunately, no pathological signs of cerebrovascular disease were found in our patient. As many researchers/clinicians believe that it is necessary to perform magnetic resonance angiogram (MRA) or CT angiography every year for patients diagnosed with MOPD II (Bober et al., 2010; Teo et al., 2016), we will continue monitoring the vessels to ensure vascular health of our patient. In addition to the above‐mentioned symptoms, some MOPD II patients may present with varying degrees of psychomotor retardation. Majewski and Goecke. (1998) reported three MOPD II cases with remarkable psychomotor retardation, which was also observed in Patients 1, 6, and 8, but not in Patients 2, 5, and 7. (The psychomotor development of Patients 3, 4, and 9 was not described in the corresponding literatures.) Mutations in the PCNT gene and consequent PCNT protein defects may cause disruption of neuronal growth and circuitry as well as neurodevelopmental deficiency, thus resulting in psychomotor retardation. In summary, the clinical manifestations of MOPD II are highly heterogeneous, indicating the complex physiological functions of the PCNT protein. Furthermore, the mutations identified in the PCNT gene of these eight patients are different (genetic information of Patient 7 was unknown). This result reflected high genetic heterogeneity of PCNT gene in MOPD II and suggested the necessity of molecular genetic analysis in precise diagnosis of MOPD II.
TABLE 1.
Clinical and genetic characteristics of the patients with MOPD II
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | Patient 8 | Patient 9 | |
|---|---|---|---|---|---|---|---|---|---|
| Gender | Female | Female | Female | Male | Male | Male | Female | Male | Female |
| Age | 10 years and 7 months | 6 years and 11 months | 10 years and 5 months | 6 months | 13 years | 1 year and 6 months | 11 months | 3 years and 2 months | 17 years |
| Clinical data | |||||||||
| Short stature | + | + | + | + | + | + | + | + | + |
| Length | 81.8 cm | 82.2 cm | 84.8 cm | NK | 123.0 cm | 55.0 cm | 56.0 cm | 60.4 cm | 98.0 cm |
| Weight | 7.0 kg | 7.7 kg | 8.6 kg | NK | 30.8 kg | 4.1 kg | 6.0 kg | 5.0 kg | 8.3 kg |
| Microcephaly | + | + | + | + | + | + | + | + | + |
| Occipitofrontal circumference | 39.1 cm | NK | 43.0 cm | NK | 47.6 cm | 34.5 cm | 41.0 cm | 40.2 cm | 35.0 cm |
| Psychomotor retardation | + | − | NK | NK | − | + | − | + | NK |
| Typical facial features | + | + | + | + | + | + | + | + | + |
| Dental anomalies | + | + | + | − | + | − | + | + | + |
| Talipes equinovarus | − | − | NK | − | − | + | + | − | − |
| Hip pathology | − | − | NK | − | − | − | + | − | − |
| Cerebrovascular abnormalities | − | − | − | − | − | − | NK | + | + |
| Precocious puberty | − | + | + | NK | − | − | NK | NK | − |
| Insulin resistance | − | − | + | NK | + | − | NK | NK | − |
| Impaired glucose tolerance | − | + | + | NK | + | − | NK | NK | − |
| Hypertension | − | − | − | NK | + | − | NK | + | − |
| Skin pigmentation | − | − | + | − | + | − | + | − | + |
| Thrombocytosis | + | − | − | + | − | − | NK | − | − |
| Leukocytosis | − | − | − | + | − | − | NK | − | − |
| Anemia | − | − | − | + | − | − | NK | − | − |
| Genetic characteristics | |||||||||
| PCNTa mutations | c.6157G>T (exon 30) | c.1828dupT; c.1207+1G>A | c.6711delG (exon 30) | c.2037 A>T (exon 13) | c.3103C>T; c.502C>T | c.7960G>T; c.9419T>A | NK | c.1527_1528insA (exon 10) | c.9842A>C (exon 45); c.6619Del‐C (exon 30); c.9366‐9381Del‐16 (exon 41) |
| Mutation type | Homozygous | Compound heterozygous | Homozygous | Homozygous | Compound heterozygous | Compound heterozygous | NK | Homozygous | Compound heterozygous |
| Reference | This study | Ma et al. (2021) | Huang‐Doran et al. (2011) | Unal et al. (2014) | Chang et al. (2017) | Zhou et al. (2019) | Kraft et al. (2000) | Piane et al. (2009) | Li et al. (2015) |
Abbreviations: −, absent; +, present; NK, not known.
RefSeq reference transcript: NM_006031 (PCNT).
In addition, due to the consanguineous marriage in this family, there was also a possibility of other genetic defects. Two heterozygous mutations of the STIL gene (NM_001048166), including paternal c.1226G>C in exon 11 and maternal c.1136C>T in exon 11, were found in our patient (Table 2). Previous studies have shown that mutations in the STIL gene are closely related to microcephaly primary hereditary type 7 (MCPH7, OMIM#181590), a neural developmental disorder characterized by microcephaly and significant mental retardation but no short stature (Kakar et al., 2015). Reduced cerebral structures (including the cortex and white matter), corpus callosum hypoplasia, simplified gyral pattern, and periventricular neuronal heterotopias are common neuroimaging abnormalities in MCPH patients (Naveed et al., 2018). However, our patient had short stature and no brain parenchymal abnormalities were found through brain CT. Therefore, some clinical features and imaging findings of our patient did not fully conform to the characteristics of MCPH7. This compound heterozygous mutation in STIL gene (c.1226G>C/c.1136C>T) has uncertain significance to the phenotype, and more in‐depth research is still needed in the future.
TABLE 2.
Possibly harmful gene mutations
| Gene | Mutations | Chromosomal location | Exons | Mutation types | Frequencya |
|---|---|---|---|---|---|
| STIL (NM_001048166) | c.1226G>C | chr1‐47748039 | Exon11 | Heterozygous | 0.0001 |
| c.1136C>T | chr1‐47748129 | Exon11 | Heterozygous | 0.0029 |
Frequencies of mutations in healthy populations.
5. CONCLUSION
In conclusion, our patient was diagnosed with MOPD II caused by a novel homozygous mutation of the PCNT gene. Researching and reporting this novel pathogenic mutation will expand the mutation spectrum of PCNT gene and facilitate the genetic counseling and prenatal diagnosis of MOPD II. Future studies should be focused on exploring the clear genotype–phenotype correlation and the detailed molecular mechanisms associated with MOPD II.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest regarding the publication of this manuscript.
AUTHORS’ CONTRIBUTIONS
Li, Haifeng Liu, and Na Tao participated in study design, analysis and interpretation of the results, manuscript preparation, data collection and processing, and statistical analysis. Yan Wang was responsible for revising the manuscript. Yang, Xiaoli He, Yu Zhang, and Yuantao Zhou participated in data collection and processing; Xiaoning Liu, Xingxing Feng, Meiyuan Sun, Fang Xu, and Yanfang Su participated in sample collection and processing.
ETHICS STATEMENT
This study was approved by the Ethics Committee of Kunming Children's Hospital. Meanwhile, the patient's informed consent has been submitted to the Submission System.
ACKNOWLEDGMENTS
We sincerely thank all the family members for their participation and support in this study.
Liu, H., Tao, N., Wang, Y., Yang, Y., He, X., Zhang, Y., Zhou, Y., Liu, X., Feng, X., Sun, M., Xu, F., Su, Y., & Li, L. (2021). A novel homozygous mutation of the PCNT gene in a Chinese patient with microcephalic osteodysplastic primordial dwarfism type II. Molecular Genetics & Genomic Medicine, 9, e1761. 10.1002/mgg3.1761
Haifeng Liu, Na Tao and Yan Wang contributed equally to this work.
Funding information
The National Natural Science Foundation of China, Grant/Award Number: 81560262, 81960294, 82060192 and 82060291; The Basic Applied Study Planning Projects of Yunnan Province, Grant/Award Number: 2018FB130; The Joint Special Fund for Basic Research from Yunnan Provincial Science and Technology and Kunming Medical University, Grant/Award Number: 2017FE467‐149; The High Level Health and Family Planning Technical Personnel Training Projects of Yunnan Province, Grant/Award Number: H‐201626; Kunming Research Center for Exosome Immunotherapy of Malignant Tumors in Children, Grant/Award Number: 2018‐SW(R)‐5; Kunming Health Science and Technology Talent Project‐100 Projects, Grant/Award Number: 2016‐SW(P)‐15; Kunming Science and Technology Planning Projects, Grant/Award Number: 2019‐1‐S‐25318000001078; Yunnan Key Laboratory of Children's Major Disease Research and Yunnan Medical Center for Pediatric Diseases.
DATA AVAILABILITY STATEMENT
Not applicable. The conclusions of the manuscript are based on relevant data, which are available in this manuscript.
REFERENCES
- Barbelanne, M., & Tsang, W. Y. (2014). Molecular and cellular basis of autosomal recessive primary microcephaly. BioMed Research International, 2014, 547986. 10.1155/2014/547986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bober, M. B., Khan, N., Kaplan, J., Lewis, K., Feinstein, J. A., Scott, C. I.Jr., & Steinberg, G. K. (2010). Majewski osteodysplastic primordial dwarfism type II (MOPD II): expanding the vascular phenotype. American Journal of Medical Genetics. Part A, 152A(4), 960–965. 10.1002/ajmg.a.33252 [DOI] [PubMed] [Google Scholar]
- Bober, M. B., Niiler, T., Duker, A. L., Murray, J. E., Ketterer, T., Harley, M. E., Alvi, S., Flora, C., Rustad, C., Bongers, E. M., Bicknell, L. S., Wise, C., & Jackson, A. P. (2012). Growth in individuals with Majewski osteodysplastic primordial dwarfism type II caused by pericentrin mutations. American Journal of Medical Genetics. Part A, 158A(11), 2719–2725. 10.1002/ajmg.a.35447 [DOI] [PubMed] [Google Scholar]
- Brancati, F., Castori, M., Mingarelli, R., & Dallapiccola, B. (2005). Majewski osteodysplastic primordial dwarfism type II (MOPD II) complicated by stroke: clinical report and review of cerebral vascular anomalies. American Journal of Medical Genetics. Part A, 139(3), 212–215. 10.1002/ajmg.a.31009 [DOI] [PubMed] [Google Scholar]
- Chang, G., Li, J., Wang, J., Wang, X., Ding, Y., Cheng, Q., Li, X., & Shen, Y. (2017). Novel compound heterozygous mutations of the PCNT gene in one Chinese boy with microcephalic osteodysplastic primordial dwarfism typeⅡ: Case report and literature review. Chinese Journal of Endocrinology and Metabolism, 33, 47–51. 10.3760/cma.j.issn.1000-6699.2017.01.008 [DOI] [Google Scholar]
- Chen, C. T., Hehnly, H., Yu, Q., Farkas, D., Zheng, G., Redick, S. D., Hung, H. F., Samtani, R., Jurczyk, A., Akbarian, S., Wise, C., Jackson, A., Bober, M., Guo, Y., Lo, C., & Doxsey, S. (2014). A unique set of centrosome proteins requires pericentrin for spindle‐pole localization and spindle orientation. Current Biology: CB, 24(19), 2327–2334. 10.1016/j.cub.2014.08.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, W. J., Huang, F. C., & Shih, M. H. (2019). Ocular characteristics in a variant microcephalic primordial dwarfism type II. BMC Pediatrics, 19(1), 329. 10.1186/s12887-019-1685-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaval, B., & Doxsey, S. J. (2010). Pericentrin in cellular function and disease. The Journal of Cell Biology, 188(2), 181–190. 10.1083/jcb.200908114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieks, J. K., Baumer, A., Wilichowski, E., Rauch, A., & Sigler, M. (2014). Microcephalic osteodysplastic primordial dwarfism type II (MOPD II) with multiple vascular complications misdiagnosed as Dubowitz syndrome. European Journal of Pediatrics, 173(9), 1253–1256. 10.1007/s00431-014-2368-5 [DOI] [PubMed] [Google Scholar]
- Hall, J. G., Flora, C., Scott, C. I.Jr, Pauli, R. M., & Tanaka, K. I. (2004). Majewski osteodysplastic primordial dwarfism type II (MOPD II): natural history and clinical findings. American Journal of Medical Genetics. Part A, 130A(1), 55–72. 10.1002/ajmg.a.30203 [DOI] [PubMed] [Google Scholar]
- Huang, S., Guo, Z. N., Shi, M., Yang, Y., & Rao, M. (2017). Etiology and pathogenesis of Moyamoya Disease: An update on disease prevalence. International Journal of Stroke, 12(3), 246–253. 10.1177/1747493017694393 [DOI] [PubMed] [Google Scholar]
- Huang‐Doran, I., Bicknell, L. S., Finucane, F. M., Rocha, N., Porter, K. M., Tung, Y. C., Szekeres, F., Krook, A., Nolan, J. J., O'Driscoll, M., Bober, M., O'Rahilly, S., Jackson, A. P., & Semple, R. K.; Majewski Osteodysplastic Primordial Dwarfism Study Group . (2011). Genetic defects in human pericentrin are associated with severe insulin resistance and diabetes. Diabetes, 60(3), 925–935. 10.2337/db10-1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakar, N., Ahmad, J., Morris‐Rosendahl, D. J., Altmüller, J., Friedrich, K., Barbi, G., Nürnberg, P., Kubisch, C., Dobyns, W. B., & Borck, G. (2015). STIL mutation causes autosomal recessive microcephalic lobar holoprosencephaly. Human Genetics, 134(1), 45–51. 10.1007/s00439-014-1487-4 [DOI] [PubMed] [Google Scholar]
- Karatas, A. F., Bober, M. B., Rogers, K., Duker, A. L., Ditro, C. P., & Mackenzie, W. G. (2014). Hip pathology in Majewski osteodysplastic primordial dwarfism type II. Journal of Pediatric Orthopedics, 34(6), 585–590. 10.1097/BPO.0000000000000183 [DOI] [PubMed] [Google Scholar]
- Kılıç, E., Utine, E., Unal, S., Haliloğlu, G., Oğuz, K. K., Cetin, M., Boduroğlu, K., & Alanay, Y. (2012). Medical management of moyamoya disease and recurrent stroke in an infant with Majewski osteodysplastic primordial dwarfism type II (MOPD II). European Journal of Pediatrics, 171(10), 1567–1571. 10.1007/s00431-012-1732-6 [DOI] [PubMed] [Google Scholar]
- Kim, S., & Rhee, K. (2014). Importance of the CEP215‐pericentrin interaction for centrosome maturation during mitosis. PLoS One, 9(1), e87016. 10.1371/journal.pone.0087016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingseisen, A., & Jackson, A. P. (2011). Mechanisms and pathways of growth failure in primordial dwarfism. Genes & Development, 25(19), 2011–2024. 10.1101/gad.169037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft, C. N., Diedrich, O., Wagner, U., & Schmitt, O. (2000). Differential diagnostic considerations in microcephalic dwarfism. Zeitschrift Fur Orthopadie Und Ihre Grenzgebiete, 138(2), 126–130. 10.1055/s-2000-10126 [DOI] [PubMed] [Google Scholar]
- Li, F. F., Wang, X. D., Zhu, M. W., Lou, Z. H., Zhang, Q., Zhu, C. Y., Feng, H. L., Lin, Z. G., & Liu, S. L. (2015). Identification of two novel critical mutations in PCNT gene resulting in microcephalic osteodysplastic primordial dwarfism type II associated with multiple intracranial aneurysms. Metabolic Brain Disease, 30(6), 1387–1394. 10.1007/s11011-015-9712-y [DOI] [PubMed] [Google Scholar]
- Lindsey, R. C., & Mohan, S. (2016). Skeletal effects of growth hormone and insulin‐like growth factor‐I therapy. Molecular and Cellular Endocrinology, 432, 44–55. 10.1016/j.mce.2015.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, Y., Xu, Z., Zhao, J., & Shen, H. (2021). Novel compound heterozygous mutations of PCNT gene in MOPD type II with central precocious puberty. Gynecological Endocrinology, 37(2), 190–192. 10.1080/09513590.2020.1827382 [DOI] [PubMed] [Google Scholar]
- Majewski, F., & Goecke, T. O. (1998). Microcephalic osteodysplastic primordial dwarfism type II: report of three cases and review. American Journal of Medical Genetics, 80(1), 25–31. [DOI] [PubMed] [Google Scholar]
- Meng, L., Tu, C., Lu, G., Lin, G., & Tan, Y. (2019). Novel biallelic PCNT deletion causing microcephalic osteodysplastic primordial dwarfism type II with congenital heart defect. Science China. Life Sciences, 62(1), 144–147. 10.1007/s11427-018-9329-3 [DOI] [PubMed] [Google Scholar]
- Mennella, V., Keszthelyi, B., McDonald, K. L., Chhun, B., Kan, F., Rogers, G. C., Huang, B., & Agard, D. A. (2012). Subdiffraction‐resolution fluorescence microscopy reveals a domain of the centrosome critical for pericentriolar material organization. Nature Cell Biology, 14(11), 1159–1168. 10.1038/ncb2597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naveed, M., Kazmi, S. K., Amin, M., Asif, Z., Islam, U., Shahid, K., & Tehreem, S. (2018). Comprehensive review on the molecular genetics of autosomal recessive primary microcephaly (MCPH). Genetics Research, 100, e7. 10.1017/S0016672318000046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry, L. D., Robertson, F., & Ganesan, V. (2013). Screening for cerebrovascular disease in microcephalic osteodysplastic primordial dwarfism type II (MOPD II): an evidence‐based proposal. Pediatric Neurology, 48(4), 294–298. 10.1016/j.pediatrneurol.2012.12.010 [DOI] [PubMed] [Google Scholar]
- Piane, M., Della Monica, M., Piatelli, G., Lulli, P., Lonardo, F., Chessa, L., & Scarano, G. (2009). Majewski osteodysplastic primordial dwarfism type II (MOPD II) syndrome previously diagnosed as Seckel syndrome: report of a novel mutation of the PCNT gene. American Journal of Medical Genetics. Part A, 149A(11), 2452–2456. 10.1002/ajmg.a.33035 [DOI] [PubMed] [Google Scholar]
- Rauch, A. (2011). The shortest of the short: pericentrin mutations and beyond. Best Practice & Research Clinical Endocrinology & Metabolism, 25(1), 125–130. 10.1016/j.beem.2010.10.015 [DOI] [PubMed] [Google Scholar]
- Rauch, A., Thiel, C. T., Schindler, D., Wick, U., Crow, Y. J., Ekici, A. B., van Essen, A. J., Goecke, T. O., Al‐Gazali, L., Chrzanowska, K. H., Zweier, C., Brunner, H. G., Becker, K., Curry, C. J., Dallapiccola, B., Devriendt, K., Dorfler, A., Kinning, E., Megarbane, A., … Reis, A. (2008). Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science (New York, N.Y.), 319(5864), 816–819. 10.1126/science.1151174 [DOI] [PubMed] [Google Scholar]
- Teo, M., Johnson, J. N., Bell‐Stephens, T. E., Marks, M. P., Do, H. M., Dodd, R. L., Bober, M. B., & Steinberg, G. K. (2016). Surgical outcomes of Majewski osteodysplastic primordial dwarfism Type II with intracranial vascular anomalies. Journal of Neurosurgery. Pediatrics, 25(6), 717–723. 10.3171/2016.6.PEDS16243 [DOI] [PubMed] [Google Scholar]
- Tibelius, A., Marhold, J., Zentgraf, H., Heilig, C. E., Neitzel, H., Ducommun, B., Rauch, A., Ho, A. D., Bartek, J., & Krämer, A. (2009). Microcephalin and pericentrin regulate mitotic entry via centrosome‐associated Chk1. The Journal of Cell Biology, 185(7), 1149–1157. 10.1083/jcb.200810159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unal, S., Alanay, Y., Cetin, M., Boduroglu, K., Utine, E., Cormier‐Daire, V., Huber, C., Ozsurekci, Y., Kilic, E., Simsek Kiper, O. P., & Gumruk, F. (2014). Striking hematological abnormalities in patients with microcephalic osteodysplastic primordial dwarfism type II (MOPD II): A potential role of pericentrin in hematopoiesis. Pediatric Blood & Cancer, 61(2), 302–305. 10.1002/pbc.24783 [DOI] [PubMed] [Google Scholar]
- Willems, M., Geneviève, D., Borck, G., Baumann, C., Baujat, G., Bieth, E., Edery, P., Farra, C., Gerard, M., Héron, D., Leheup, B., Le Merrer, M., Lyonnet, S., Martin‐Coignard, D., Mathieu, M., Thauvin‐Robinet, C., Verloes, A., Colleaux, L., Munnich, A., & Cormier‐Daire, V. (2010). Molecular analysis of pericentrin gene (PCNT) in a series of 24 Seckel/microcephalic osteodysplastic primordial dwarfism type II (MOPD II) families. Journal of Medical Genetics, 47(12), 797–802. 10.1136/jmg.2009.067298 [DOI] [PubMed] [Google Scholar]
- Zhou, Y., Xu, W., & Zhang, G. (2019). Microcephalic osteodysplastic primordial dwarfism type II in a child. Chinese Journal of Pediatrics, 57(4), 295–297. 10.3760/cma.j.issn.0578-1310.2019.04.014 [DOI] [PubMed] [Google Scholar]
- Zimmerman, W. C., Sillibourne, J., Rosa, J., & Doxsey, S. J. (2004). Mitosis‐specific anchoring of gamma tubulin complexes by pericentrin controls spindle organization and mitotic entry. Molecular Biology of the Cell, 15(8), 3642–3657. 10.1091/mbc.e03-11-0796 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable. The conclusions of the manuscript are based on relevant data, which are available in this manuscript.