

Abstract
Background
Vascular Ehlers–Danlos syndrome (vEDS) is a heritable connective tissue disorder caused by defects in the type III collagen protein. It is generally considered the most severe form of Ehlers–Danlos syndrome (EDS) due to an increased risk of spontaneous artery or organ rupture. vEDS has an extremely heterogeneous presentation and muscle rupture is considered a minor diagnostic criterium.
Methods
A patient with a long history of inconclusive examinations and investigations was referred to our unit. The clinical picture was mainly characterized by muscle ruptures, whereas the cardiovascular involvement was limited to mitral regurgitation. We performed a panel analysis of genes associated with inheritable heart diseases using the TruSight Cardio kit (Illumina). A skin biopsy was then performed for functional studies to analyze the different forms of collagen molecules produced in vitro by cutaneous fibroblasts.
Results
The patient presented the novel variant c.3478A>G (p.Ile1160Val) in COL3A1 (NM_000090.3), whose pathogenicity was supported by biochemical analysis of type III collagen.
Conclusion
In this report, we describe a case of vEDS with predominant and severe musculoskeletal involvement. Our findings provide insight into genetic variants and clinical expression of vEDS, broadening the clinical scenario of the syndrome.
Keywords: COL3A1, musculoskeletal involvement, NGS, vEDS
In this report, we describe a case of vEDS with predominant and severe musculoskeletal involvement. Our findings provide insight into genetic variants and clinical expression of vEDS, enlarging the clinical scenario of the syndrome.

1. INTRODUCTION
COL3A1 (OMIM *120180) encodes for collagen pro‐α1(III) chain, also known as pro‐α1 chain of type III collagen. Pro‐α1 chains are hydroxylated, glycosylated and incorporated into trimers to form the procollagen. This molecule undergoes secretion and removal of his amino‐terminal and carboxy‐terminal ends to form tropocollagen fibers, whose association leads to the production of collagen fibers. Three α1 chains are included in a type III collagen molecule, which has a long triple‐helical domain (Kuivaniemi & Tromp, 2019). Type III collagen constitutes about 5–20% of the entire collagen content in the human body (Miller, 1988). It plays an essential role in the structural integrity of arteries, uterus, and bowel (Byers, 1999 [updated 2019]; Byers et al., 2017; Malfait et al., 2017; Malfait, 2018) and it has been also detected in skin, tendons, ligaments, and bones (Keene et al., 1991).
Heterozygous mutations in COL3A1 are associated with the vascular type of Ehlers–Danlos syndrome (vEDS). Ehlers–Danlos syndrome (EDS) is a heterogeneous group of heritable connective tissue disorders. The phenotype is variable and mainly comprises joint hypermobility, skin hyperextensibility, and tissue fragility. EDS is classified in 13 different subtypes. Among these, the vascular type is the most severe and life‐threatening due to vascular dissection, gastrointestinal perforation, or organ rupture. vEDS can also be characterized by spontaneous uterine rupture, carotid‐cavernous sinus fistula in the absence of trauma, bruising disproportionate to trauma, thin and translucent skin, spontaneous pneumothorax, acrogeria, clubbed foot, congenital hip dislocation, hypermobility of small joints, tendon and muscle rupture, keratoconus and gingival recession, and fragility (Malfait et al., 2017).
Due to the heterogeneous clinical presentation, the diagnosis of vEDS may be challenging. Therefore, it can be established through either the identification of a heterozygous causative variant in COL3A1 or the detection of abnormal synthesis and mobility of type III collagen chains on biochemical analysis of type III collagen from cultured fibroblasts (Byers, 1999 [updated 2019]).
We hereby report a novel COL3A1 variant causing vEDS with predominant and severe musculoskeletal involvement. Our findings provide insight into genetic variants and clinical expression of vEDS, broadening the clinical vEDS scenario.
2. CASE REPORT
The proband is a 35‐year‐old male with a 10‐year history of sprains and muscle ruptures. He reported six left knee sprains since the age of 17 and two ruptures of the right rectus femoris muscle at the age of 29 and 30, respectively. The patient did not mention any strenuous activity that could explain the ruptures. Given the presence of weakness and discomfort in the shoulder girdle, an ultrasonography of the left superior arm was performed, showing calcifications of the tendon of the supraspinatus muscle.
At the age of 31, the patient reported a rupture of the right biceps femoris muscle that occurred in the attempt to dismount his motorcycle. These episodes were followed by a series of spontaneous muscle ruptures of the right biceps femoris and other muscles not related to any exertion.
At the age of 30, the patient started complaining about generalized muscle pain and progressive hyposthenia in his proximal lower limbs. The x‐ray of the hip bones was normal. The neurological examination and the electrophysiological studies of the limbs were negative. A spinal MRI revealed several disc herniations for which the patient had undergone surgery, without pain relief. Since the muscular pain was invalidating, an opioid therapy was started leading to partial remission.
Due to the lack of an underlying diagnosis, even after a series of clinical evaluations, the patient came to our attention.
On physical examination, the proband was slender and considerably taller than his parents (185 cm vs. 160 cm of the mother and 161 cm of the father). He reported a growing spurt at the age of 16, with a gain of 22 cm in 1 year (from 160 to 182 cm of height). Other findings were pectus excavatum, arm span of 187 cm (arm span/height ratio: 1.01), flat feet, and cutaneous striae on the hips and the thighs.
On the clinical suspicion of Marfan syndrome, the Marfan Systemic Score (Dietz, 2001 [updated 2017]) was assessed and a score of 4/23 was obtained, making this diagnosis unlikely. Moreover, the ocular examination was normal and the ultrasound of the abdomen did not reveal any vascular abnormality. Serum IGF1 levels, assayed to rule out a pituitary adenoma as the cause of the patient's growing spurt, was normal.
Further examinations were performed to investigate the cardiovascular system. An ECG revealed right branch block and left branch hemiblock with a right axis deviation (−120°). An echocardiography showed mild mitral regurgitation and delayed diastolic relaxation. These findings were confirmed during the following 3 years of cardiologic follow‐up, without significant variations.
Vascular echo‐Doppler showed normal structure and diameters of the aorta and the supra‐aortic trunks, as well as of the upper and lower limbs’ arteries.
After coming to our attention, the patient started presenting paroxysmal hypertensive episodes. Further workup, aimed at ruling out pheochromocytoma, revealed normal plasma and urinary metanephrines and no pathologic accumulation of fluorodeoxyglucose (FDG) at the PET scan examination. Repeated Holter monitoring blood pressure showed that daytime values of heart rate and blood pressure (bp) were set at the upper normal levels and there was a significant variability in daytime blood pressure rates with a physiological reduction at night (this pattern is defined as “dipper status”). One year later the variation between daytime and nighttime bp levels further increased (i.e., “extreme dipper”). A therapy with lisinopril failed to guarantee a complete pressure control, so the patient was referred to cardiological follow‐up. A switch to carvedilol caused the onset of faintings (min bp: 40 mmHg), so lisinopril was reintroduced. The opioid therapy for chronic pain is still ongoing.
Although the family history was totally negative for collagen‐related diseases, the recurrence of sprains and the evidence of cutaneous striae on clinical examination were suggestive of a collagenopathy with cardiac involvement.
Therefore, we considered the possibility of a cardiac‐valvular form of EDS, so we performed a panel analysis of genes associated with inheritable heart diseases. Genomic DNA was extracted from peripheral blood samples using standard procedure. Briefly, the exonic and flanking splice junction regions of genes of interest were captured using the TruSight Cardio kit (Illumina) and sequenced on a MiSeq Illumina system with 150bp paired‐end reads. Reads were aligned to human genome build GRCh37/UCSC hg19 and analyzed for sequence variants using a custom‐developed analysis tool (Sana et al., 2016), selecting collagenopathy genes to create a virtual panel. Mean coverage was 285x and >30x for each target region.
On note, FBN1 gene was included in the panel and was normal, furtherly disproving the initial suspect of Marfan syndrome. The analysis showed the presence of the heterozygous variant c.3478A>G (p.Ile1160Val) in COL3A1 (NM_000090.3).
This variant has never been reported neither in literature nor in the major genetic databases (gnomAD, ExAC). Following ACMG2015 criteria, the mutation was classified as a variant of uncertain significance. It is predicted as neutral by some bioinformatics tools (Provean, Polyphen 2, Mutation Assessor and Mutpred) and damaging by others (SIFT, Mutation Taster and CADD).
In HGMD database, 708 COL3A1 mutations, mostly missense, are listed. Several missense mutations associated with vEDS have been described in the 10‐amino acid regions upstream and downstream isoleucine 1160 (Figure 1), suggesting the presence of a potential hot‐spot region at this level. This residue of isoleucine is highly conserved, particularly in mammals (Figure 2).
FIGURE 1.
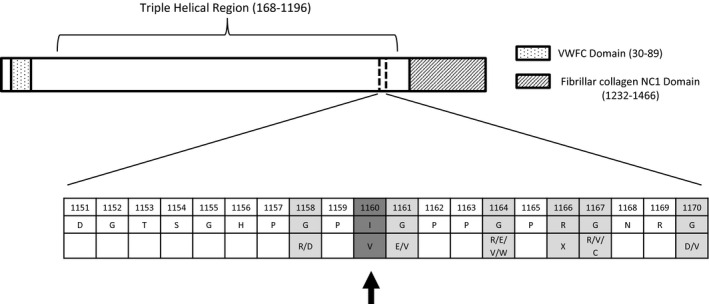
Schematic representation of collagen α1(III) chain: a 10‐amino acid regions upstream and downstream p.Ile1160Val is focused in detail to show all the nearby mutations
FIGURE 2.

Isoleucine 1160 is highly conserved, particularly in mammals
Further workup was aimed at investigating the pathogenicity of the variant.
The segregation analysis, performed only in the mother (father died at 59 years of lung cancer), was negative, partially supports the de novo origin of the variant.
Afterward, a skin sample of the patient was achieved to perform a functional study on the fibroblasts. The fibroblasts cultures were grown and maintained in Dulbecco modified Eagle's medium (DMEM) with 10% fetal calf serum. Type I, III, and V collagens were purified from medium and cell layer as described by Valli et al. (1991) and then separated by gel electrophoresis. Gels were processed for fluorography using standard procedures and submitted to quantitation by using the program Image J released from National Institute of Health, USA. The reference range of type III/I collagen ratio has been determined by measuring this ratio in samples collected from healthy subjects over a period of 3 years. This analysis showed delayed electrophoretic mobility of type III collagen (Figure 3). A decreased amount of α1(III) chain, as confirmed by the densitometric scan of the fluorogram, was also detected.
FIGURE 3.
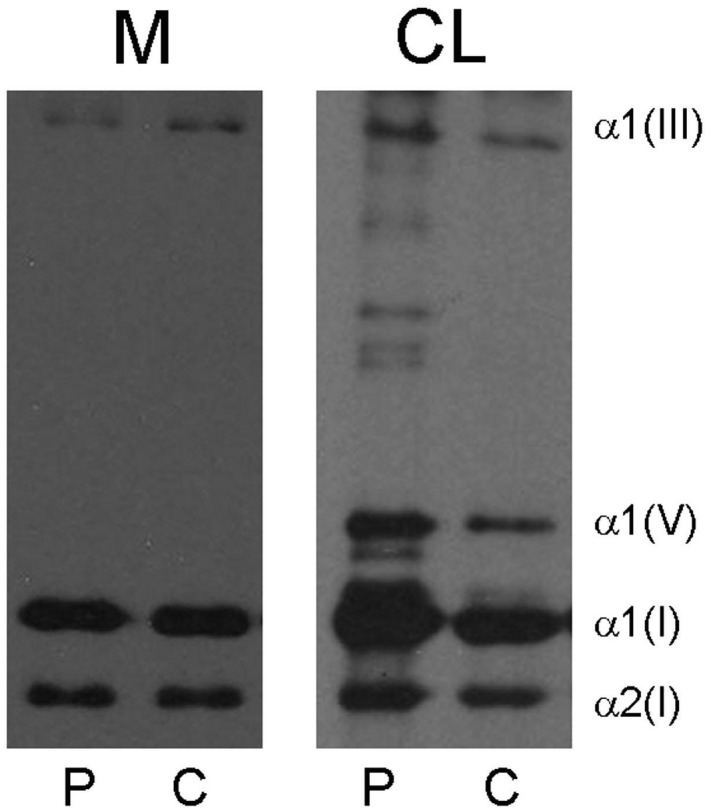
Six percent sodium dodecyl sulfate‐polyacrylamide gel electrophoresis of pepsin‐treated collagen secreted by fibroblasts (M) and retained in cell layer (CL) of a patient affected by vEDS (P) and an age‐matched control (C). In the first and in the third lanes a delay in the electrophoretic mobility of α1(III) chain is observed. Furthermore, a decreased amount of α1(III) chain can be detected
In the hypothesis that the electrophoretic abnormalities were due to post‐transcriptional modifications of the mRNA we analyzed the mutation through “Human Splicing Finder” (Figure 4), which predicted the creation of a cryptic donor splice site (CCCGTTGGA).
FIGURE 4.
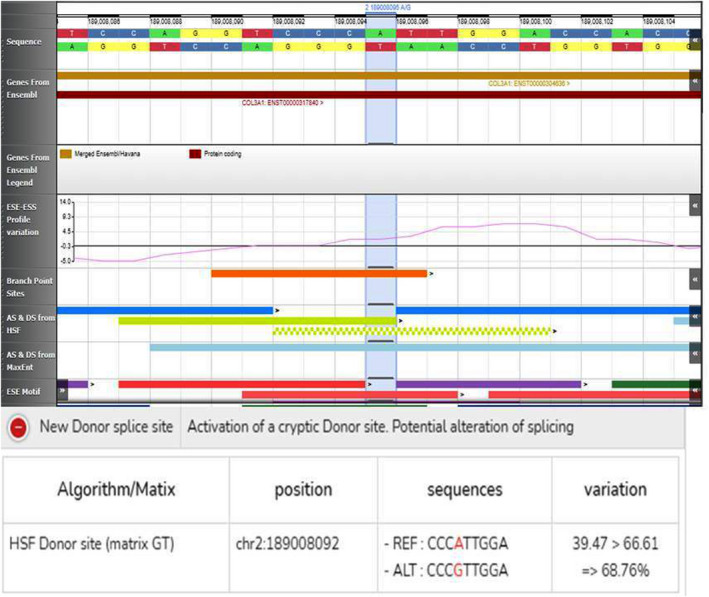
Computational analysis of the c.3478A>G variant in COL3A1 (NM_000090.3) through “Human Splicing Finder”: the A>G transition potentially leads to the activation of a cryptic donor splice site
The employment of this cryptic site may result in the synthesis of shortened chains with consequent alteration at a post‐translational level, especially in the trimer folding process. Although the prediction made by the software was weak, alternative splicing have been considered as an additional cause of the electrophoretic delay and further mRNA analysis on fibroblasts of the patient has been performed.
Further mRNA analysis on fibroblasts of the patient shows no modifications on mRNA length through electrophoresis and Sanger analysis revealing no splicing defects.
Nevertheless, adding the finding of the protein study as a parameter in the computational analysis, the variant can be classified as likely pathogenic according to ACMG2015 criteria.
A muscular MRI of neck, upper, and lower limbs with axial and coronal T1 and T2‐STIR weighted sequences (Figure 5) was performed to seek the consequences of type III collagen abnormality on the muscle tissue level. This investigation did not show a selective pattern of muscle involvement, but only diffuse slight signs of fat substitution, mainly in gluteus and soleus muscles, bilaterally. No signs of fat substitution in the other muscle districts examined were reported, neither muscle edema could be detected.
FIGURE 5.
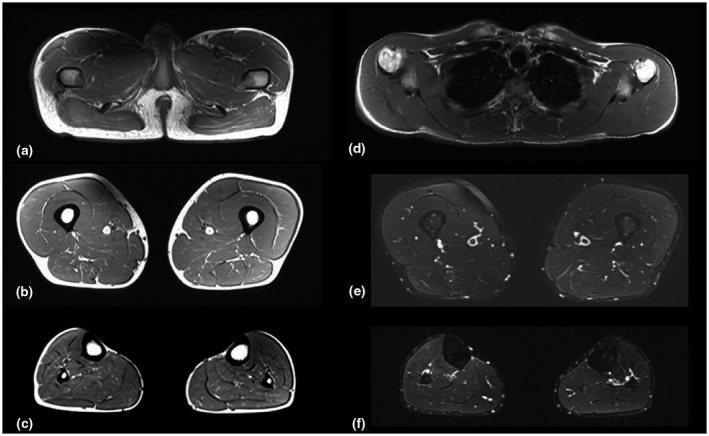
(a–f). Axial T1‐SE (a,b,c,d) and T2‐STIR sequences (e and f) of the pelvic girdle (a), shoulder girdle (d), thigh (b and e), leg (c and f), bilaterally. Muscle trophism and signal are normal at the level of the thigh (b) and shoulder girdle (d), whereas slight hyperintensity can be detected at the level of glutei maximi (Mercuri score 1) (a) and of the muscles of the posterior compartment of the legs (c). No muscle edema could be detected in the inferior limbs (e and f)
3. DISCUSSION
Ehlers–Danlos syndrome (EDS) is a heterogeneous group of heritable connective tissue disorders with 13 clinically distinct subtypes (Malfait et al., 2017). Vascular EDS (vEDS) is caused by heterozygous pathogenic variants in COL3A1 gene (Pepin et al., 2000). COL3A1 encodes for the α1 chain of type III collagen, which assembles into a homotrimer to form type III collagen molecules. Since it is a primary structural component of blood vessels and hollow organs, reduced or abnormal type III collagen leads to an increased risk of spontaneous artery or organ ruptures. For this reason, vEDS is generally considered the most severe form of EDS and it is associated with a decreased life expectancy (Oderich et al., 2005).
Although tendon/muscle ruptures are included in the minor diagnostic criteria of vEDS (Malfait et al., 2017), predominant or exclusive musculoskeletal involvement is not a common clinical presentation of the disease. Therefore, in absence of family history, the diagnosis of vEDS can be challenging in a patient who only presents muscle ruptures, even if they are multiple and not associated with any major trauma.
When faced with this phenotype, Marfan syndrome and collagenopathy should be both included in the differential diagnosis, since they share similar clinical characteristics.
Indeed, after the exclusion of Marfan syndrome (more frequently associated with musculoskeletal involvement) through the assessment of Marfan Systemic Score and/or FBN1 analysis, clinicians should consider collagenopathy in its different, and perhaps atypical, forms.
A musculoskeletal involvement of different degrees characterizes all the EDS subtypes with a less severe impairment in the vEDS than in the other subtypes (Malfait et al., 2017).
vEDS was the EDS subtype which could fit in the clinical features of the patient herein described. Indeed, he presented mitral regurgitation (if progressive, it is considered a major criterium for this EDS subtype) together with other minor criteria such as pectus excavatum, pes planus, and joint dislocations. Being the most severe form of EDS, vEDS is generally ruled out in the diagnostic pathway. However, given the extremely variable phenotype, vEDS should be considered even if only one minor criterium (muscle ruptures) is present.
Therefore, we suggest that, even when suspecting a specific subtype of EDS, clinicians should consider the analysis of a next‐generation sequencing gene panel including all forms of collagenopathy.
With our report, we describe a novel variant of COL3A1 in a non‐glycine codon, whose pathogenic role has been supported by collagen analysis.
In literature, most mutations described involve glycine residues which are essential for triple helix of type III collagen structure. In the last few years mutations in codons different from glycine have been described associated with various EDS phenotypes (Ghali et al., 2019). Several mutations in different domains of the protein have been described mostly associated with vascular and classical EDS subtypes. Familial and sporadic cases have been reported with inter‐familial variability. The prevalence of vEDS phenotype associated to COL3A1 and described in the literature could possibly be due to an enrollment bias; indeed, all the COL3A1 analyses have been performed on well‐defined EDS subtypes patients (see Table 1 for further details).
TABLE 1.
COL3A1 no‐glycine substitutions and relative phenotype as described in literature
| Mutation | References | Sex | Age | Phenotype | Familiarity | Relatives |
|---|---|---|---|---|---|---|
| E241K | Angwin et al., (2020) | F | 30 | vEDS | sporadic | |
| E241K | Ghali et al., (2019) | F | 33 | cEDS | sporadic | |
| E241K | Ghali et al., (2019) | M | 54 | cEDS | sporadic | |
| R271Q | Frank et al., (2015) | F | 45 | vEDS | sporadic | |
| R271Q | Frank et al., (2015) | F | 47 | vEDS | sporadic | |
| N389Y | Frank et al., (2015) | F | 44 | vEDS | familial | Family 1 |
| N389Y | Frank et al., (2015) | F | 49 | vEDS | familial | Family 2 |
| N389Y | Frank et al., (2015) | F | 70 | vEDS | familial | Family 3 |
| E682K | Ghali et al., (2019) | M | 50 | vEDS/cEDS | familial | Family 4 |
| E682K | Ghali et al., (2019) | M | 73 | EDS | familial | Family 4 |
| E682K | Ghali et al., (2019) | F | 65 | EDS | familial | Family 4 |
| E682K | Ghali et al., (2019) | M | 74 | EDS | familial | Family 4 |
| E682K | Ghali et al., (2019) | M | 3 | EDS | familial | Family 4 |
| E682K | Ghali et al., (2019) | F | 35 | cEDS | familial | Family 4 |
| E682K | Ghali et al., (2019) | M | 44 | cEDS | familial | Family 4 |
| E682K | Ghali et al., (2019) | F | 8 | EDS | familial | Family 4 |
| E682K | Ghali et al., (2019) | F | 5 | EDS | familial | Family 4 |
| E682K | Ghali et al., (2019) | M | 37 | vEDS | sporadic | |
| E682K | Ghali et al., (2019) | F | 22 | cEDS | familial | Family 5 |
| E682K | Ghali et al., (2019) | F | 14 | EDS | familial | Family 5 |
| E682K | Ghali et al., (2019) | M | 50 | cEDS | familial | Family 5 |
| P806L | Weerakkody et al., (2016) | F | 38 | vEDS / hEDS overlap | sporadic | |
| R1082Q | Frank et al., (2015) | M | 60 | vEDS | sporadic | |
| E1171K | Frank et al., (2015), Henneton et al., (2019), Ghali et al., (2019) | M | 22 | vEDS | sporadic | |
| E1171K | Angwin et al., (2020) | M | 25 | vEDS | sporadic | |
| E1171K | Angwin et al., (2020) | F | 49 | vEDS | sporadic | |
| E1171K | Ghali et al., (2019) | F | 53 | cEDS | familial | Family 6 |
| E1171K | Ghali et al., (2019) | M | 27 | cEDS | familial | Family 6 |
| A1203T | Frank et al., (2015) | F | 54 | vEDS | sporadic | |
| P1258S | Frank et al., (2015) | F | 51 | vEDS | familial | Family 7 |
| P1258S | Frank et al., (2015) | M | 21 | asymptomatic relative | familial | Family 7 |
| A1259T | Frank et al., (2015) | F | 43 | vEDS | familial | Family 8 |
| P1270T | Frank et al., (2015) | F | 17 | vEDS | familial | Family 9 |
| P1270T | Frank et al., (2015) | F | 45 | asymptomatic relative | familial | Family 9 |
| K1273R | Frank et al., (2015) | F | 48 | vEDS | familial | Family 10 |
| K1273R | Frank et al., (2015) | F | 55 | vEDS | familial | Family 10 |
| K1273R | Frank et al., (2015) | F | 45 | vEDS | sporadic | |
| D1288V | Kerwin et al., (2008) | — | — | vEDS | — | |
| K1313R | Stembridge et al., (2015) | F | 30 | benign hypermobility | familial | Family 11 |
| K1313R | Frank et al., (2015) | F | 38 | vEDS | sporadic | |
| R1432L | Stembridge et al., (2015) | M | 39 | vEDS | familial | Family 12 |
| R1432L | Stembridge et al., (2015) | F | hEDS | familial | Family 12 | |
| P1440L | Weerakkody et al., (2016) | F | 26 | vEDS | sporadic | |
| P1440L | Stembridge et al., (2015) | F | 27 | vEDS | sporadic | |
| P1440L | Morissette et al., (2014) | F | 41 | vEDS | sporadic |
Abbreviations: cEDS, classical EDS; EDS, not specified EDS subtype; hEDS, hypermobile EDS; vEDS, vascular EDS.
Our findings broaden the phenotypic spectrum of vEDS, reporting a patient with predominant musculoskeletal and very mild cardiac involvement. The reaching of a definite diagnosis is very important for patients, particularly those with atypical clinical presentations, to avoid inappropriate therapies and to establish a correct follow‐up. Moreover, a correct diagnosis has significant relevance in genetic counseling giving a correct recurrence and procreative risk to the patient and family members.
CONFLICT OF INTEREST
The authors have no actual or potential conflicts of interest. The authors state that no approvals by ethics committee are required for the issue of this report.
AUTHOR CONTRIBUTIONS
Federica Ruscitti: conceptualization, design analysis, planning, conduct, data analysis, manuscript preparation, review of literature. Lucia Trevisan: conceptualization, design analysis, planning, conduct, data analysis, manuscript preparation, review of literature. Giulia Rosti: conduct, manuscript preparation, review of literature. Fabio Gotta: conduct, manuscript preparation. Annalia Cianflone: conduct, manuscript preparation. Alessandro Geroldi: data analysis, manuscript preparation, review of literature. Paola Origone: data analysis, manuscript preparation, review of literature. Anna Pichiecchio: data analysis, review preparation. Simona Viglio: data analysis, review preparation. Maria Iascone: conceptualization, data analysis, design analysis, planning. Paola Mandich: conceptualization, design analysis, planning, conduct, manuscript preparation.
ETHICAL COMPLIANCE
Informed consent of the patient and family members for the molecular test has been stated from a medical geneticist.
ACKNOWLEDGMENTS
The authors thank the patient and his family. This work was developed within the framework of the DINOGMI Department of Excellence of MIUR 2018‐2022 (legge 232 del 2016).
Ruscitti, F., Trevisan, L., Rosti, G., Gotta, F., Cianflone, A., Geroldi, A., Origone, P., Pichiecchio, A., Viglio, S., Iascone, M., & Mandich, P. (2021). A novel mutation in COL3A1 associates to vascular Ehlers–Danlos syndrome with predominant musculoskeletal involvement. Molecular Genetics & Genomic Medicine, 9, e1753. 10.1002/mgg3.1753
Federica Ruscitti and Lucia Trevisan should be considered joint first authors.
DATA AVAILABILITY STATEMENT
The datasets generated during the current study are available from the corresponding author on reasonable request.
REFERENCES
- Angwin, C., Ghali, N., Baker, D., Brady, A. F., Pope, F. M., Vandersteen, A., Wagner, B., Ferguson, D. J. P., & van Dijk, F. S. (2020). Electron microscopy in the diagnosis of Ehlers‐Danlos syndromes: Correlation with clinical and genetic investigations. British Journal of Dermatology., 182, 698–707. 10.1111/bjd.18165 [DOI] [PubMed] [Google Scholar]
- Byers, P. H. (1999). Vascular Ehlers‐Danlos Syndrome. In Adam M. P., Ardinger H. H., Pagon R. A. et al (Eds.), GeneReviews® [Internet]. Seattle, WA: University of Washington; 1993–2021. https://www.ncbi.nlm.nih.gov/books/NBK1494/ [PubMed] [Google Scholar]
- Byers, P. H., Belmont, J., Black, J., De Backer, J., Frank, M., Jeunemaitre, X., Johnson, D., Pepin, M., Robert, L., Sanders, L., & Wheeldon, N. (2017). Diagnosis, natural history, and management in vascular Ehlers‐Danlos syndrome. American Journal of Medical Genetics. Part C, Seminars in Medical Genetics, 175(1), 40–47. 10.1002/ajmg.c.31553 [DOI] [PubMed] [Google Scholar]
- Dietz, H. (2001). Marfan Syndrome. In: Adam M. P., Ardinger H. H., Pagon R. A., et al (Eds.), GeneReviews® [Internet]. University of Washington; 1993–2021. https://www.ncbi.nlm.nih.gov/books/NBK1335/ [Google Scholar]
- Frank, M., Albuisson, J., Ranque, B., Golmard, L., Mazzella, J.‐M., Bal‐Theoleyre, L., Fauret, A.‐L., Mirault, T., Denarié, N., Mousseaux, E., Boutouyrie, P., Fiessinger, J.‐N., Emmerich, J., Messas, E., & Jeunemaitre, X. (2015). The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers‐Danlos syndrome. European Journal of Human Genetics, 23, 1657–1664. 10.1038/ejhg.2015.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghali, N., Baker, D., Brady, A. F., Burrows, N., Cervi, E., Cilliers, D., Frank, M., Germain, D. P., Hulmes, D. J. S., Jacquemont, M.‐L., Kannu, P., Lefroy, H., Legrand, A., Pope, F. M., Robertson, L., Vandersteen, A., von Klemperer, K., Warburton, R., Whiteford, M., & van Dijk, F. S. (2019). Atypical COL3A1 variants (glutamic acid to lysine) cause vascular Ehlers‐Danlos syndrome with a consistent phenotype of tissue fragility and skin hyperextensibility. Genetics in Medicine, 21(2081–2091), 10.1038/s41436-019-0470-9 [DOI] [PubMed] [Google Scholar]
- Henneton, P., Juliette Albuisson, J., Adham, S., Legrand, A., Mazzella, J. M., Jeunemaitre, X., & Frank, M. (2019). Accuracy of clinical diagnostic criteria for patients with vascular Ehlers‐Danlos syndrome in a tertiary referral centre. Circulation: Genomic and Precision Medicine, 12, e001996. 10.1161/CIRCGEN.117.001996 [DOI] [PubMed] [Google Scholar]
- Keene, D. R., Sakai, L. Y., & Burgeson, R. E. (1991). Human bone contains type III collagen, type VI collagen, and fibrillin: type III collagen is present on specific fibers that may mediate attachment of tendons, ligaments, and periosteum to calcified bone cortex. Journal of Histochemistry and Cytochemistry, 39(1), 59–69. 10.1177/39.1.1983874 [DOI] [PubMed] [Google Scholar]
- Kerwin, W., Pepin, M., Mitsumori, L., Yarnykh, V., Schwarze, U., & Byers, P. (2008). MRI of great vessel morphology and function in Ehlers‐Danlos syndrome type IV. International Journal of Cardiovascular Imaging, 24, 519–528. 10.1007/s10554-007-9283-z [DOI] [PubMed] [Google Scholar]
- Kuivaniemi, H., & Tromp, G. (2019). Type III collagen (COL3A1): Gene and protein structure, tissue distribution, and associated diseases. Gene, 707, 151–171. 10.1016/j.gene.2019.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malfait, F. (2018). Vascular aspects of the Ehlers‐Danlos Syndromes. Matrix Biology, 71–72, 380–395. 10.1016/j.matbio.2018.04.013 [DOI] [PubMed] [Google Scholar]
- Malfait, F., Francomano, C., Byers, P., Belmont, J., Berglund, B., Black, J., Bloom, L., Bowen, J. M., Brady, A. F., Burrows, N. P., Castori, M., Cohen, H., Colombi, M., Demirdas, S., De Backer, J., De Paepe, A., Fournel‐Gigleux, S., Frank, M., Ghali, N., … Tinkle, B. (2017). The 2017 international classification of the Ehlers‐Danlos syndromes. American Journal of Medical Genetics. Part C, Seminars in Medical Genetics, 175(1), 8–26. 10.1002/ajmg.c.31552 [DOI] [PubMed] [Google Scholar]
- Miller, E. J. (1988). Collagen types: structure, distribution, and functions. In Nimni M. E. (Ed.), Collagen: Volume 1: Biochemistry (pp. 139–156). CRC Press. [Google Scholar]
- Morissette, R., Schoenhoff, F., Xu, Z., Shilane, D. A., Griswold, B. F., Chen, W., Yang, J., Zhu, J., Fert‐Bober, J., Sloper, L., Lehman, J., Commins, N., Van Eyk, J. E., & McDonnell, N. B. (2014). Transforming growth factor‐β and inflammation in vascular (type IV) Ehlers‐Danlos syndrome. Circulation: Cardiovascular Genetics, 7, 80–88. 10.1161/CIRCGENETICS.113.000280 [DOI] [PubMed] [Google Scholar]
- Oderich, G. S., Panneton, J. M., Bower, T. C., Lindor, N. M., Cherry, K. J., Noel, A. A., Kalra, M., Sullivan, T., & Gloviczki, P. (2005). The spectrum management and clinical outcome of Ehlers‐Danlos syndrome type IV: A 30‐year experience. Journal of Vascular Surgery, 42, 98–106. 10.1016/j.jvs.2005.03.053 [DOI] [PubMed] [Google Scholar]
- Pepin, M., Schwarze, U., Superti‐Furga, A., & Byers, P. H. (2000). Clinical and genetic features of Ehlers‐Danlos syndrome type IV, the vascular type. New England Journal of Medicine, 342, 673–680. 10.1056/NEJM200003093421001 [DOI] [PubMed] [Google Scholar]
- Sana, M. E., Quilliam, L. A., Spitaleri, A., Pezzoli, L., Marchetti, D., Lodrini, C., Candiago, E., Lincesso, A. R., Ferrazzi, P., & Iascone, M. (2016). A novel HRAS mutation independently contributes to left ventricular hypertrophy in a family with a known MYH7 mutation. PLoS One, 11(12), e0168501. 10.1371/journal.pone.016850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stembridge, N. S., Vandersteen, A. M., Ghali, N., Sawle, P., Nesbitt, M., Pollitt, R. C., Ferguson, D. J. P., Holden, S., Elmslie, F., Henderson, A., Hulmes, D. J. S., & Pope, F. M. (2015). Clinical, structural, biochemical and X‐ray crystallographic correlates of pathogenicity for variants in the C‐propeptide region of the COL3A1 gene. American Journal of Medical Genetics, 167A, 1763–1772. 10.1002/ajmg.a.37081 [DOI] [PubMed] [Google Scholar]
- Valli, M., Mottes, M., Tenni, R., Sangalli, A., Gomez Lira, M., Rossi, A., Antoniazzi, F., Cetta, G., & Pignatti, P. F. (1991). A de novo G to T transversion in a pro‐alpha 1 (I) collagen gene for a moderate case of osteogenesis imperfecta. Substitution of cysteine for glycine 178 in the triple helical domain. Journal of Biological Chemistry, 266(3), 1872–1878. [PubMed] [Google Scholar]
- Weerakkody, R. A., Vandrovcova, J., Kanonidou, C., Mueller, M., Gampawar, P., Ibrahim, Y., Norsworthy, P., Biggs, J., Abdullah, A., Ross, D., Black, H. A., Ferguson, D., Cheshire, N. J., Kazkaz, H., Grahame, R., Ghali, N., Vandersteen, A., Pope, F. M., & Aitman, T. J. (2016). Targeted next‐generation sequencing makes new molecular diagnoses and expands genotype–phenotype relationship in Ehlers‐Danlos syndrome. Genetics in Medicine, 18, 1119–1127. 10.1038/gim.2016.14 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated during the current study are available from the corresponding author on reasonable request.