

Abstract
Background
We report the molecular analysis of the DMD gene in a group of Peruvian patients with Duchenne/Becker dystrophinopathy. This is the first study to thoroughly characterize mutations in this population.
Methods
We used the combination of multiplex ligation‐dependent probe amplification (MLPA) and sequencing analysis of the DMD gene. We recruited Peruvian patients in 2 years from reference national hospitals. We performed DNA tests in 152 patients, checking first exon deletion/duplication by MLPA, and subsequently, if negative, samples were sequenced to detect point mutations.
Results
The average age for diagnosis was 9.8 years, suggesting a delay for timely diagnosis and care. We found causal DMD mutations in 125 patients: 72 (57.6%) exon deletions/duplications (41.6% deletions, 16.0% duplications), and 53 (42.4%) point mutations (27.2% nonsense, 9.6% small indels, and 5.6% splice site).
Conclusion
Due to our genetic background, we expected a higher number of novel and recurrent causal mutations in our sample. Results showed 16% of novel mutations, similar to other well‐studied populations.
Keywords: Becker muscular dystrophy, Duchenne muscular dystrophy, molecular diagnosis, multiple ligation probe amplification, targeted Next Generation Sequencing
This study characterizes DMD/DMB mutations in Peru. This population has been poorly studied at the genetic level. We want to obtain a landscape of mutations affecting patients and give them treatment options with therapies currently under use in other countries. We found that deletions of the DMD gene, are predominant (41.6%) and point mutations with more than 40% of cases, explain the other cases. Duplications (16%) come in third place in this study. Regarding point mutations, the nonsense type (27%) is the most frequent change found.
1. INTRODUCTION
Mutations in the DMD gene can cause different phenotypes, called dystrophinopathies, which range from severe forms, such as Duchenne muscular dystrophy (DMD, OMIM # 310200), to milder forms, such as Becker muscular dystrophy (BMD, OMIM 300376) and X‐linked dilated cardiomyopathy 3B (CMD3B, OMIM # 302045). DMD is a severe, progressive disease affecting fibers of the skeletal and cardiac muscles. Its estimated incidence rate ranges from 1/3500 to 1/6000 globally among newborn boys (Emery, 1991; Miller et al., 2006). DMD has an early onset, and affected individuals typically have short lifespans of 20–30 years (Darras et al., 2000). Proximal muscle weakness becomes apparent around 3–5 years of age, with tiptoe walking and a positive Gowers’ sign. Various features, such as pseudohypertrophic calves, contractures, scoliosis, and the need for mechanical support for ambulation and breathing, are most commonly observed in affected individuals, and cardiomyopathy is the most common cause of death (Hotta, 2015). BMD has a later onset, with milder skeletal muscle weakness and loss of ambulation after the age of 15 or late adulthood (Dent et al., 2005; Okubo et al., 2016). Mutations in the DMD gene in BMD usually lead to an abnormal version of dystrophin, which retains some function; DMD mutations typically prevent any functional dystrophin from being produced (Darras et al., 2000).
The DMD gene, located on Xp21.1, is the largest in the human genome, with about 2.4 Mb and 79 exons, and codes for the dystrophin protein (Koenig et al., 1987). Dystrophin acts as a molecular shock absorber for muscle fiber contraction and physically anchors the cellular skeletal actin fibers and sarcolemma membranes in muscle fibers. It is also necessary for the structural and functional activities of other proteins, such as α‐actin, β‐actin, and dystroglycan, which form the dystrophin‐associated protein complex (Hotta, 2015; Toksoy et al., 2019).
Dystrophinopathies are caused by variants leading to frameshifts or premature stop codons, resulting in a lack of physical or functional dystrophin (Monaco et al., 1988; Zatz et al., 2016). Most reports in various populations show that, among DMD mutations, whole‐exon deletions/duplications are more prevalent (~60%). Smaller variations (i.e., one or a few nucleotides) found within or around exons, account for about 20% of cases according to some authors (Bennett et al., 2001) or ~20%–35% of pathogenic variants (Aartsma‐Rus et al., 2016; Toksoy et al.,l., 2019). DNA analysis is necessary to identify the causative mutations, establish individual prognoses, program coherent therapy according to disease progression, provide genetic counseling, and guide family planning (Hoffman & Giron, 2001). Ad hoc‐targeted therapies are also underway for specific mutations, such as exon skipping and reading through stop codons (Fairclough et al., 2013; Van Vliet et al., 2008).
The current study aimed to identify genetic mutations among individuals with suspected dystrophinopathy in the main clinical centers of Lima, Peru. It has been shown that the ancestry of the Peruvian population is ~70% of Native South Americans; in Lima, this background fluctuates from 70% to 80% (Sandoval et al., 2013). We analyzed DNA samples from 152 individuals who were clinically diagnosed with dystrophinopathy. Due to the admixed and predominantly Native South American ancestry of the population, as well as the scant information available, we wanted to perform a first look at the type of mutations found in our sample.
2. MATERIALS AND METHODS
2.1. Subjects and recruitment strategy
We recruited 152 unrelated male patients with the main inclusion criteria of the referral for suspicion of DMD/BMD from services of Pediatric Neurology or Genetics from national reference health centers, belonging either to the Social Security System (EsSalud): Hospital Nacional Edgardo Rebagliati Martins, Hospital Nacional Guillermo Almenara Irigoyen or to the Ministerio de Salud System: Instituto Nacional de Salud del Niño Breña and Instituto Nacional de Ciencias Neurológicas, from August 2015 to June 2018. The clinicians comply to assess at least three inclusion criteria: infants with delay in ambulation, children‐teenagers with loss of ambulation, pseudohypertrophic calves, Gowers’ sign, electromyographic pattern concordant with DMD/BMD, CPK higher than 3000 U/L, and family history (Table S1). The study was performed from August 2015 to June 2018.
2.2. Blood sampling and DNA extraction
A total of 3 ml of peripheral blood was collected in EDTA tubes. DNA was extracted using the salting‐out methodology with minor modifications (Miller et al., 1988).
2.3. MLPA DMD assays
Assays were performed as described in the MLPA DNA Protocol version MDP‐v.003 (MRC‐Holland). An ABI‐3500 Genetic Analyzer™ was used to detect and quantify products. Following the reactions, Coffalyzer. Net software (Coffa & van den Berg, 2011) was used to establish deletion/duplication of the exon of the DMD gene (Table S2). Cases with single‐exon deletion were checked with sequencing as suggested by the manufacturer.
2.4. Sequencing assays
Eighty DMD‐affected individuals with negative MLPA results were sent for targeted NGS, using either the Ion Ampliseq™ Inherited Diseases Panel (Life Technologies™) performed by Macrogen Inc., or a custom DMD gene panel.
Variants detected by targeted NGS sequencing were validated by Sanger sequencing.
2.5. Assessment of novel variations
Variations were searched in databases, including the NCBI (NCBI.nlm.nih.gov), the Leiden Open Variation Database (LOVD3), the Exome Variant Server (evs.gs.washington.edu/EVS/), and the UMD‐TREAT‐NMD DMD mutations database (Universal Mutation Database—Translational Research in Europe for the Assessment and Treatment of Neuromuscular Disorders). The corroborated novel variants were deposited at the NCBI site and analyzed for pathogenicity using in silico web tools such as PredictSNP2, MutationTaster Variant Effect Predictor (VEP), and SIFT indel LoF Score Human Splice Finder (Bendl et al., 2014; Desmet et al., 2009; Hu & Ng, 2013; McLaren et al., 2016; Pagel et al., 2017; Schwarz et al., 2014). The 2015 American College of Medical Genetics and Genomics criteria were also used for variant classification and correct nomenclature (Richards et al., 2015).
3. RESULTS
We performed molecular tests for DMD mutations in 152 individuals with suspicion of dystrophinopathy. We found mutations in 125 individuals (82.2%), whereas 27 (17.7%) showed no DMD mutation with the tests performed.
3.1. Subjects and clinical status
A total of 152 unrelated male individuals who were clinically diagnosed with dystrophinopathy were enrolled in the study (Table 1). The study was performed between August 2015 and June 2018. Among the participants with available data (135 cases), the average age at the last examination was 9.8 years old, and CPK values (108 cases) were 10,162 U/L on average. Neuroconduction studies and EMG performed in 67 patients showed a myopathic pattern in 62 cases (92%). Regarding ambulation capacity (114 cases), 76 (66.6%) of patients were ambulatory, and 38 (33.3%) were unable to walk. Family history was registered for 94 individuals, and 32 (34%) of them declared having at least one affected relative, whereas 62 (66%) reported no other affected relative. We relied on the expert clinical opinion of each neuromuscular physician that contributed samples.
TABLE 1.
Percentage of mutations found for the DMD gene by MLPA and targeted NGS for 152 analyzed individuals
| No of cases | Percentage | |
|---|---|---|
| Total number of analyzed patients | 152 | |
| Through the technique MLPA | ||
| Exon deletions | 52 | 34.2 |
| Deletions, one exon | 11 | 7.2 |
| Deletions, multiexon | 41 | 27 |
| Exon duplications | 20 | 13.2 |
| Duplications, one exon | 8 | 5.3 |
| Duplications, multiexon | 12 | 7.9 |
| Through NGS | ||
| Point mutation | 53 | 34.8 |
| Nonsense | 34 | 22.4 |
| Frameshift | 12 | 7.8 |
| Splice site | 7 | 4.6 |
| None | 27 | 17.8 |
3.2. Analysis of DMD exon copy by MLPA
Of the 152 patients with DMD/BMD phenotypes, we identified 72 exon deletion/duplication mutations by MLPA and corroborated these by resequencing single‐exon deletions. Forty‐one multiexon deletions and 15 single‐exon deletions were initially observed. Four single‐exon deletions turned out to be false due to the presence of small mutations at the annealing site of the respective MLPA primers: two nonsense (patients DMD‐62 and DMD‐125) and two frameshifts (DMD‐110 [deletion] and DMD‐184 [novel insertion]). Twelve multiple exon duplications and eight single‐exon duplications were also identified. Four of these multiexon deletions, two multiexon duplications, and one mutation frameshift are novel mutations (Table 1 and Figure 1).
FIGURE 1.
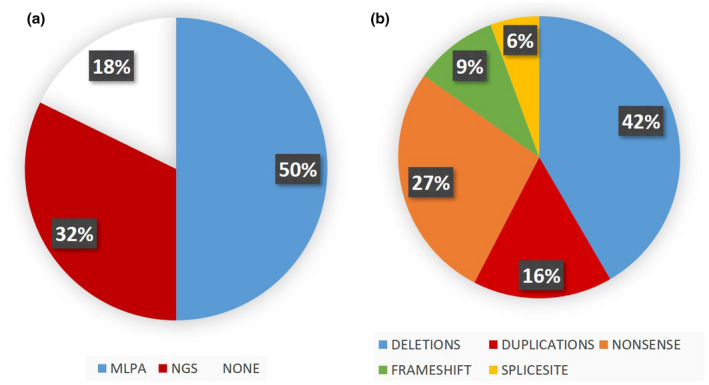
Graphical representation of the 152 dystrophinopathy studied patients, A. Shows the percentage diagnosed using MLPA and NGS, and the DMD negative patients (n = 27). B. Shows percentages of different types of mutations found in DMD using both techniques on 125 patients.
Recurrent MLPA mutations in unrelated affected individuals were distributed as follows: three deletions from exons 45 to 50, three deletions from exons 48 to 50, three deletions from exons 48 to 52, three deletions in exon 52, and five duplications in exon 2 (Figure 1). In four affected individuals, proximal deletions involved the Dp427c promoter through exon 2 (DMD‐132 and DMD‐202), exon 29 (DMD‐80), and exon 44 (DMD‐133), respectively. One case showed a terminal deletion from exon 58 to exon 79 (DMD‐77). In another (DMD‐154), there was a double duplication from exons 12 to 43 (in frame) and exons 49 to 52 (out of frame) (Figure 2). Sixty‐one (84.7%) affected individuals had exon deletions or duplications, which resulted in out‐of‐frame products. Six (8.3%) MLPA‐positive individuals had in‐frame exon deletions or duplications on the DMD gene, and the remaining five (7%) corresponded to exon deletions involving the promoter (four) and one terminal deletion. The results showed “hotspot” variations, involving exons 46–52 in the central region of the DMD gene (38 patients) and another small peak from exons 18–29 (10 patients). For duplications, no striking differences appeared, but small peaks on exons 2 and 52 were observed (Figure 1).
FIGURE 2.
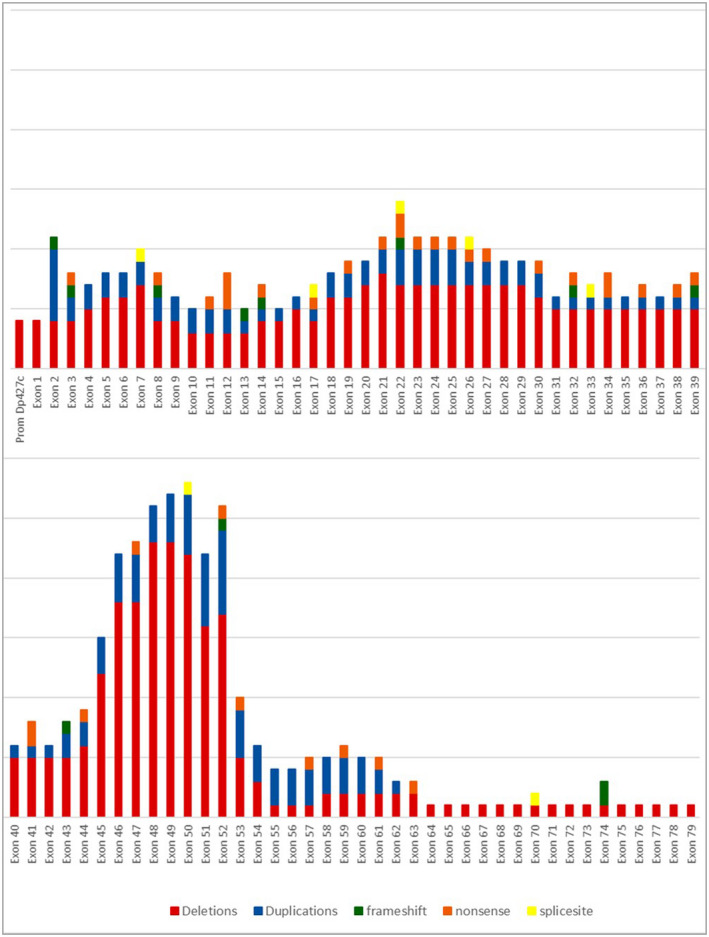
Representation of DMD positive individuals with variants found in different exons of the DMD gene in our sample.
3.3. Targeted NGS DMD gene sequence analysis
Targeted NGS Ampliseq™ Inherited Diseases Panel (16 patients) or a custom DMD gene panel (60 patients) analysis was performed in the 76 MLPA‐negative cases. In 49 affected individuals, small DMD‐pathogenic mutations were found, including 32 stop codons, 10 deletions/insertions of one to four nucleotides, and seven splice variations, and all corroborated by Sanger sequencing. Twenty‐seven individuals exhibited no changes in DMD when checked using the chosen Ampliseq™ or the DMD gene panel. Thirteen new variants were also detected through targeted NGS analysis. These are: DMD‐10, DMD‐34, DMD‐36, DMD‐43, DMD‐44, DMD‐60, DMD‐123, DMD‐126, DMD‐127, DMD‐141, DMD‐166; DMD‐170, and DMD‐184 (Table 2).
TABLE 2.
Clinical and molecular characterization of novel DMD mutations
| Clinical characteristics | Results | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No | USMP code | Sex | Age | CPK (U/L) | Electromyography | Ambulation | Other family | S Molecular tests | Type of mutation | Exon affected | HGNC name variant | Protein | Phenotype | |
| 1 | DMD‐10 | M | 9.0 | N.A. | N.A. | NO | YES | NGS | Frameshift deletion | Exon 43 | c.6253 delT | p.Try2085Glyfs*28 | DMD | |
| 2 | DMD‐34 | M | 7.0 | 9525 | N.A. | YES | NO | NGS | Nonsense | Exon 34 | c.4840G>T | p.Gly1614* | DMD | |
| 3 | DMD‐36 | M | 8.0 | 4312 | N.A. | YES | NO | NGS | Nonsense | Exon 22 | c.2945T>A | p.Leu982* | DMD | |
| 4 | DMD‐43 | M | 6.0 | 34733 | N.A. | YES | YES | NGS | Frameshift deletion | Exon 52 | c.7618_7619 delAA | p.Lys2540Aspfs*7 | DMD | |
| 5 | DMD‐44 | M | 6.0 | 8396 | Positive | NO | YES | NGS | Nonsense | Exon 26 | c.3490A>T | p.Gln1163* | DMD | |
| 6 | DMD‐60 | M | 8.0 | Elevated | N.A. | N.A. | YES | NGS | Frameshift deletion | Exon 74 | c.10409delT | p.Leu3470Cysfs*1 | DMD | |
| 7 | DMD‐64 | M | 20.0 | 5166 | Positive | NO | NO | MLPA | Deletion | Exons 14–48 | c.1603‐?_7098+?del | p.? | BMD | |
| 8 | DMD‐77 | M | 14.0 | 29560 | Positive | YES | NO | MLPA | Deletion | Exons 58–79 | c.8548‐?_*2691del | p.? | DMD | |
| 9 | DMD‐80 | M | 12.0 | 1470 | Positive | NO | YES | MLPA | Deletion | Dp427c‐Exon 29 | c.−128297_4071+?del | p.? | DMD | |
| 10 | DMD‐123 | M | 11.0 | N.A. | N.A. | NO | N.A. | NGS | Nonsense | Exon 19 | c.2348 C>G | p.Ser783* | DMD | |
| 11 | DMD‐125 | M | 4.0 | 27528 | N.A. | YES | NO | MLPA | Nonsense | Exon 41 | c.5812 G>T | p.Glu1938* | DMD | |
| 12 | DMD‐126 | M | 8.8 | 18080 | N.A. | YES | N.A. | NGS | Frameshift deletion | Exon 8 | c.803delT | p.Leu268Tryfs*14 | DMD | |
| 13 | DMD‐127 | M | 6.0 | 15113 | N.A. | YES | N.A. | NGS | Frameshift deletion | Exon 14 | c.1672delC | p.Leu558Phefs*13 | DMD | |
| 14 | DMD‐140 | M | 8.0 | Elevated | Positive | YES | N.A. | MLPA | Duplication | Exons 43–52 | c.6118‐?_7660+?dup | p.? | DMD | |
| 15 | DMD‐141 | M | 11.0 | 9038 | N.A. | NO | NO | NGS | Nonsense | Exon 53 | c.7768G>T | p.Glu2590* | DMD | |
| 16 | DMD‐154 | M | 7.0 | 11899 | Positive | YES | NO | MLPA | Duplication | Exons 12–43 and 49–52 | c.(1332‐?_6290+?dup; 7099‐?_7660+?dup) | p.? | DMD | |
| 17 | DMD‐166 | M | 6.0 | 11,000 | Positive | YES | NO | NGS | Splicesite | Intron 33 | c.4675‐1G>A | p.? | DMD | |
| 18 | DMD‐170 | M | 7.0 | 22377 | N.A. | YES | NO | NGS | Frameshift deletion | Exon 2 | c.44delA | p.Asp15Valfs*10 | DMD | |
| 19 | DMD‐184 | M | 11.1 | N.A. | N.A. | NO | N.A. | MLPA | Frameshift insertion | Exon 3 | c.131insT | p.Leu44Leufs*44 | DMD | |
| 20 | DMD‐199 | M | 5.0 | 18467 | N.A. | N.A. | NO | MLPA | Deletion | Exons 45–62 | c.6439‐?_9224+?del | p.? | DMD | |
3.4. Presence of small mutations
In our 152 cases, we have 52 exon deletions (34.2%), 20 exon duplications (13.2%), 53 point mutations (34.8%), and 27 samples (17.8%) without DMD mutations (Table 1). Considering the 125 patients with proven DMD mutations (by MLPA, Sanger, and targeted NGS tests), the distribution is as follows: 52 (41.6%) exon deletions, 20 (16%) exon duplications, 34 (27.2%) nonsense variations, 12 (9.6%) small indels, and 7 (5.6%) splice site variations. No missense mutations of relevance to the phenotype were found in our analysis.
3.5. In silico predictive testing of novel mutations
Different types of functional predictive software were used to test the recorded 14 novel variants’ pathogenicity. (Table 3). For the seven small deletions/insertions (one or two nucleotides), all predictors were in concordance with a pathogenic effect due to nonsense‐mediated mRNA decay (NMD), inferred by MutationTaster and SIFT indel, with a confidence score of 0.858 and a loss of function (LoF) prediction of 0.342. Similarly, for the six new nonsense variants, all predictors were in concordance with the pathogenic effect supported by NMD (confidence scores of 1–0.858) and the LoF score (0.342). PredictSNP2 suggested a deleterious effect for all of them with an expected accuracy of 58%–81% (Table 3).
TABLE 3.
In silico analysis of new DMD nonsense and frameshift deletion variants found in affected individuals using different web tools for pathogenicity prediction
| No | USMP code | Mutation type | Exon affected | Nucleotide change | ACMG | In silico pathogenic supporting | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Classification | Criteria | Mutation taster | SIFT indel | PredictSNP2 | LoFT | Human splice finder | |||||
| 1 | DMD‐10 | Frameshift deletion | EXON 43 | c.6253delT | Likely pathogenic | PVS1, PM2 | Disease causing | Damaging | N.A. | Possibly damaging | N.A. |
| 2 | DMD‐34 | Nonsense | EXON 34 | c.4840G>T | Pathogenic | PVS1, PM2, PP3 | Disease causing | N.A. | Deleterious | Possibly damaging | N.A. |
| 3 | DMD‐36 | Nonsense | EXON 22 | c.2945T>A | Pathogenic | PVS1, PM2, PP3 | Disease causing | N.A. | N.A. | Possibly damaging | N.A. |
| 4 | DMD‐43 | Frameshift deletion | EXON 52 | c.7618_7619delAA | Pathogenic | PVS1, PM2, PP3 | Disease causing | Damaging | N.A. | Possibly damaging | N.A. |
| 5 | DMD‐44 | Nonsense | EXON 26 | c.3490A>T | Pathogenic | PVS1, PM2, PP3 | Disease causing | N.A. | Deleterious | Possibly damaging | N.A. |
| 6 | DMD‐60 | Frameshift deletion | EXON 74 | c.10409delT | Pathogenic | PVS1, PM2, PP3 | Disease causing | Damaging | N.A. | Possibly damaging | N.A. |
| 7 | DMD‐123 | Nonsense | Exon 19 | c.2348 C>G | Pathogenic | PVS1, PM2, PP3 | Disease causing | N.A. | Deleterious | Possibly damaging | N.A. |
| 8 | DMD‐125 | Nonsense | Exon 41 | c.5812 G>T | Pathogenic | PVS1, PM2, PP3 | Disease causing | N.A. | Neutral | Possibly damaging | N.A. |
| 9 | DMD‐126 | Frameshift deletion | EXON 8 | c.803delT | Likely pathogenic | PVS1, PM2 | Disease causing | Damaging | N.A. | Possibly damaging | N.A. |
| 10 | DMD‐127 | Frameshift deletion | EXON 14 | c.1672delC | Likely pathogenic | PVS1, PM2 | Disease causing | Damaging | N.A. | Possibly damaging | N.A. |
| 11 | DMD‐141 | Nonsense | EXON 53 | c.7768G>T | Pathogenic | PVS1, PM2, PP3 | Disease causing | N.A. | Deleterious | Possibly damaging | N.A. |
| 12 | DMD‐166 | Splice Site | INTRON 33 | c.4675‐1G>A | Pathogenic | PVS1, PM2, PP3 | Disease causing | N.A. | N.A. | N.A. | Splicing alteration |
| 13 | DMD‐170 | Frameshift deletion | EXON 2 | c.44delA | Pathogenic | PVS1, PM2, PP3 | Disease causing | Damaging | N.A. | Possibly damaging | N.A. |
| 14 | DMD‐184 | Frameshift Insertion | EXON 3 | c.131insT | Pathogenic | PVS1, PM1, PM2 | Disease causing | Damaging | N.A. | Possibly damaging | N.A. |
4. DISCUSSION
In this study, we analyzed the mutational profile of the DMD gene in 152 Peruvian patients suspected of dystrophinopathy. All samples went through MLPA, and targeted NGS if no deletions/duplications were found, determining causal mutations for 125 patients. Our center was the first to introduce the MLPA methodology for DMD/BMD patients in Peru, and between mid‐2015 and mid‐2018, we performed the molecular analyses for patients sent by our clinicians. The only previously reported analysis of Peruvian patients with clinical DMD diagnoses appeared in two studies using multiplex polymerase chain reaction (PCR) to detect exon deletions for DMD (Abarca Barriga, 2016; Rojas et al., 2012). As expected, no information was obtained about the extent of the deletions or the exon duplications and small mutations (Beggs & Kunkel, 1990; Chamberlain et al., 1988). The patients studied in the present study are part of a majority around the globe that is underrepresented by the “European bias” and is missing diversity data in human genetics studies (Sirugo et al., 2019). To the present authors’ knowledge, this is the first molecular report to use MLPA and targeted NGS of DMD variants to study dystrophinopathies on a Peruvian population.
Out of 125 patients with DMD mutations, we found that 41.6% of participants exhibited exon deletions, 16% duplications, and 42.4% small mutations. Percentages differ from results reported for other populations, which show a higher number of exon deletions/duplications and a smaller mutation prevalence of <25%. For example, reports of the LOVD3, and the UMD‐TREAT‐NMD DMD variation database, each with several thousands of mutations, report 66%–69% exon deletions and 22%–20% small mutations, respectively (Aartsma‐Rus et al., 2006; Bladen et al., 2015). Exon deletions have also been found in 60%–70% of several hundred samples collected in China and Japan (Chen et al., 2014; Takeshima et al., 2010). While we were presenting this report, Neri et al. (2020) published a study on 1902 DMD Italian patients, which found a higher incidence of small mutations (32%) and observed regional variation in the distribution of DMD mutations within the country, claiming distinctive ethnic backgrounds. On the other hand, Flanigan et al. (2009) reported 55% of deletion/duplications and 46% of point mutations in a group of more than 1000 patients, suggesting an enrichment “for point mutations during those years when sequence analysis of the DMD gene was not widely available.” We do not have an explanation for the lower prevalence of large exon deletions and a higher prevalence of small mutations in this research and this could be due to one of the two explanations, either a regional variation, similar to Neri et al. (2020), or a bias by selecting samples that had previous MLPA deletion/duplication or Multiplex‐PCR analysis, similar to Flanigan et al. (2009). Until now, some hospitals in our country still use the Multiplex PCR approach due to the high cost of NGS target and MLPA technologies. This is why we may have this selection bias that apparently enriches the point mutations. Our methodology (MLPA followed by target NGS) gives a better approach to an accurate molecular diagnosis.
Considering the “Reading Frame Rule” hypothesis (Monaco et al., 1988), we identified 47 events among the internal exon deletions (excluding deletions of a promoter or terminal regions), 42 of which were out of frame and five that were in‐frame. According to the hypothesis, we should have 42 patients with DMD and five with BMD phenotypes. In the clinical examinations of the five in‐frame exon deletion patients, three of them complied with the criteria for BMD phenotype, patient DMD‐64, 20 y.o. (novel mutation, deletion exons 14–48), patient DMD‐222 (deletion exons 45–47), 38 y.o., and patient DMD 242 (deletion exons 45–49), 30 y.o., the last two with preserved ambulation. In one case, we found a 25‐year old patient (DMD‐230) with an in‐frame duplication event of exons 2–7 with BMD phenotype, who lost ambulation at age 18.
Two other in‐frame patients were clinically diagnosed with DMD. Patient DMD‐40, 9 y.o., had a relatively large deletion (exons 5–44) and other cases of DMD in the maternal family. The mutation was previously described as causal of DMD by Flanigan et al. (2009). Another patient DMD‐192, 8 y.o, with a deletion of exon 16, also had a family history of DMD. This mutation was described as causal of DMD by Covone et al. (1991). Concerning the 42 out‐of‐frame exon deletions, we did not find any patients with a clinical assignment of BMD by our collaborators. Other authors have reported exceptions to the Reading Frame Rule (Flanigan et al., 2009).
We report 20 novel causal mutations on the DMD gene with 4 large exon deletions, two exon duplications, and 14 small mutations. Because of the scarcity of studies on populations with an Amerindian genetic background, we expected to find a higher proportion of novel mutations in our sample (16%). Other populations with more exhaustive studies have similar percentages. For example, studies of 576 Spanish families (Juan‐Mateu et al., 2015), 177 families in Brazil (De Almeida et al., 2017), and 132 families in China (Wang et al., 2019) found 14.7%, 10.73%, and 18%, respectively, had novel mutations. Our novel mutations appear to reflect the background of new mutations in all global populations. Ancestry studies are necessary to assess the origin (Native, European, African, or Asian) of these mutations. As described before, small mutations are distributed along with all exons of the gene, with no “hotspots” in a specific region (Flaigan et al, 2009).
Definitive assessment tests for DMD/BMD search for either the lack of dystrophin or the presence of mutations in the DMD gene (Bushby et al., 2010) is achieved by DNA testing, because of its relative simplicity and versatility in addition to its usefulness for family counseling, prenatal diagnosis, and the promising therapies based on DMD mutations. The average age for molecular DMD assessment among the sampled patients was 9.8 years compared with the average of 5 years in the USA and the UK (Bushby et al., 2010). One limitation of this study is the “rare disease” nature of dystrophinopathies in Peru, where the scarcity of specialists who can identify the disease affects the time of diagnosis. This delay (4.8 years after the global consensus) affects opportune clinical interventions and physical therapy (Bushby et al., 2010; Janssen et al., 2005; López‐Hernández et al., 2015). In addition, the lack of routine DNA testing for the DMD gene in Peru reduces the number of reported cases.
Another limitation is the heterogeneity of the protocols or guidelines in Peruvian healthcare institutions as well as complementary laboratory tests (i.e., CPK or EMG), which preclude collecting consistent and homogeneous information for all patients. Patients are also typically lost during the follow‐up period with their physicians, switching to different systems and institutions in the national healthcare structure. Not all individuals have access to molecular diagnosis due to the relatively high cost of tests, which are performed in commercial laboratories outside Peru. The molecular tests presented here are the first local MLPA and sequencing analyses of dystrophinopathy among Peruvians and were performed at no charge to the families. Therefore, this initiative has benefited dozens of Peruvian families, providing them with the correct diagnoses at the molecular level. In terms of genetic counseling, we are offering it to some families that request it, as part of another study (Bazalar et al., 2020). In addition, we must consider that other neuromuscular diseases may be present in the sample within the 27 “non‐DMD mutation group.” For example, in 16 patients, where the “Ampliseq Inherited Disease Panel” (400 diseases) was used, we found mutations on EMD, ATXN1, and ATXN2. Also, there is a small percentage of cases where intronic mutations may cause the disease. These mutations are not detected by the methods we used (Flanigan et al., 2009).
Although it is clear that all novel exon deletions/duplications or point mutations described here had phenotypic impacts, in silico data with different predictors were used to assess their pathogenicity (Table 3). In our case, it can be difficult to correlate the causative mutation with the phenotype of DMD or BMD because most patients did not have proper follow‐up clinical visits after initial diagnosis. Natural history studies with longitudinal data for each case harboring a novel variant will be important in determining genotype–phenotype correlations.
Regarding therapies for specific mutations—such as drugs that read through premature stop codons, antisense oligonucleotide‐exon skipping, and stem cells—there are, as yet, no local trials but increased information related to molecular diagnoses will help health authorities better understand the issue. Creating a database with these variations will become a priority for physicians and health authorities to depict the spectrum of variations found in Peru, and certainly, it will have an impact in other Andean countries due to our common history. In our sample, it would ideally help 34 patients who could benefit from the readthrough stop codon therapy and nine patients with the exon skipping of exon 51 strategy.
In conclusion, this is the first report of an analysis performed among Peruvian patients with clinical diagnoses of dystrophinopathy from different health institutions in Lima. In 125 patients, we obtained definitive diagnoses by DMD mutations: 41.6% exon deletions, 16% exon duplications, and 42.4% of point mutations. With this study, we hope to raise awareness of the importance of genetic diagnoses at an earlier age and achieve the possibility of treatment regarding the therapies that help patients achieve a better quality of life.
CONFLICT OF INTEREST
The authors declare that no conflict of interest could be perceived as prejudicial to the impartiality of the reported research.
ETHICAL COMPLIANCE
The study protocol was executed in accordance with the recommendations and under supervision of the “Comité Nacional de Ética en Investigación de la Facultad de Medicina Humana de la Universidad de San Martín de Porres” [International Committee of Research Ethics of the Faculty of Human Medicine of the University] (IRB IORB00003251 OHRP/FDA Universidad de San Martín de Porres). All subjects and one parent per subject provided written informed consent in accordance with the Declaration of Helsinki.
AUTHORS’ CONTRIBUTIONS
M.G. was the primary researcher responsible for recruiting affected individuals and explaining results to families; she also wrote the manuscript. F.H. performed MLPA and subsequent analyses and helped write the manuscript. A.E. implemented the MLPA technique and analyses and helped write the discussion section. D.O. helped with the bioinformatics process and mutation search from files. D.R. implemented tests for DMD and in silico new variation analyses and helped write the discussion section. M.T., B.G., A.P., C.C., and H.A. provided sample and clinical data collected from participants. M.C., V.M., and M.D. provided sample and clinical data collected from participants and helped write the discussion section. The M.C. and V.M also provided DNA aliquots collected from participants at the neurological tertiary centre. L.C. and J.L., provided sample and clinical data and helped with clinical diagnosis. R.F. acted as a research advisor, helped write the discussion section, and reviewed the manuscript. R.S. and V.B. provided help with MLPA and Sanger sequencing to check mutations. All authors have read and approved the manuscript.
Supporting information
Table S1
Table S2
ACKNOWLEDGMENTS
The authors thank the participants and their relatives, the ADM‐Peru, the physicians, including R. Yábar, J. Toro, A. Tori, R. Caparó, S. Samalvides, G. Chávez, and M. Chávez, and others who helped with sample collection and clinical exams. They also thank PTC Therapeutics and authorities at the Facultad de Medicina, Universidad de San Martín de Porres, who helped fund this study.
Guevara‐Fujita, M. L., Huaman‐Dianderas, F., Obispo, D., Sánchez, R., Barrenechea, V., Rojas‐Málaga, D., Estrada‐Cuzcano, A., Trubnykova, M., Cornejo‐Olivas, M., Marca, V., Gallardo, B., Dueñas‐Roque, M., Protzel, A., Castañeda, C., Abarca, H., Celis, L., La Serna‐Infantes, J., & Fujita, R. (2021). MLPA followed by target‐NGS to detect mutations in the dystrophin gene of Peruvian patients suspected of DMD/DMB. Molecular Genetics & Genomic Medicine, 9, e1759. 10.1002/mgg3.1759
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are openly available in zenodo at https://doi.org/10.5281/zenodo.4741000, reference number 4741001.
REFERENCES
- Aartsma‐Rus, A., Ginjaar, I. B., & Bushby, K. (2016). The importance of genetic diagnosis for Duchenne muscular dystrophy. Journal of Medical Genetics, 53(3), 145–151. 10.1136/jmedgenet-2015-103387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aartsma‐Rus, A., Van Deutekom, J. C. T., Fokkema, I. F., Van Ommen, G.‐J.‐B., & Den Dunnen, J. T. (2006). Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading‐frame rule. Muscle & Nerve, 34(2), 135–144. 10.1002/mus.20586 [DOI] [PubMed] [Google Scholar]
- Abarca Barriga, H. H. (2016). Análisis retrospectivo de las características clínicas y moleculares de 40 pacientes con distrofia muscular de Duchenne y de Becker en el Hospital Nacional Guillermo Almenara Irigoyen, 1997 – 2007. M.Sc. thesis. Lima, Perú: Universidad Nacional Mayor de San Marcos, Facultad de Ciencias Biológicas, Unidad de Posgrado, 66 h.
- Beggs, A. H., & Kunkel, L. M. (1990). Improved diagnosis of Duchenne/Becker muscular dystrophy. Journal of Clinical Investigation, 85(3), 613–619. 10.1172/JCI114482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendl, J., Stourac, J., Salanda, O., Pavelka, A., Wieben, E. D., Zendulka, J., Brezovsky, J., & Damborsky, J. (2014). PredictSNP: Robust and accurate consensus classifier for prediction of disease‐related mutations. PLoS Computational Biology., 10(1), e1003440. 10.1371/journal.pcbi.1003440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, R. R., Darras, B. T., & Kunkel, L. M. (2001). Detection of mutations in the dystrophin gene via automated DHPLC screening and direct sequencing. BMC Genetics, 2, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bladen, C. L., Salgado, D., Monges, S., Foncuberta, M. E., Kekou, K., Kosma, K., Dawkins, H., Lamont, L., Roy, A. J., Chamova, T. & Guergueltcheva, V. (2015). The TREAT‐NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Human Mutation, 36(4), 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushby, K., Finkel, R., Birnkrant, D. J., Case, L. E., Clemens, P. R., Cripe, L., Kaul, A., Kinnett, K., McDonald, C., Pandya, S., Poysky, J., Shapiro, F., Tomezsko, J., Constantin, C., & DMD Care Considerations Working Group (2010). Diagnosis and management of Duchenne Muscular Dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. The Lancet Neurology, 9(1), 77–93. [DOI] [PubMed] [Google Scholar]
- Chamberlain, J. S., Gibbs, R. A., Rainer, J. E., Nguyen, P. N., & Thomas, C. (1988). Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucleic Acids Research, 16(23), 11141–11156. 10.1093/nar/16.23.11141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C., Ma, H., Zhang, F., Chen, L., Xing, X., Wang, S., Zhang, X., & Luo, Y. (2014). Screening of Duchenne muscular dystrophy (DMD) mutations and investigating its mutational mechanism in Chinese individuals affected. PLoS One. 9(9):e108038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffa, J., & van den Berg, J. (2011). Analysis of MLPA data using novel software Coffalyser.NET by MRC – Holland. modern approaches to quality control, Dr. Ahmed Badr Eldin (Ed.), ISBN: 978‐953‐307‐971–4, In Tech.
- Covone, A. E., Lerone, M., & Romeo, G. (1991). Phenotype‐phenotype correlation and germline mosaicism in DMD/BMD patients with deletions of the dystrophin gene. Human Genetics, 87, 353–360. [DOI] [PubMed] [Google Scholar]
- Darras, B. T., Urion, D. K., & Ghosh, P. S. (2000). [Updated 2018 Apr 26]. Dystrophinopathies. In Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Bean L. J. H., Mirzaa G., & Amemiya A. (Eds.), GeneReviews® [Internet] (pp. 1993–2019). University of Washington. https://www.ncbi.nlm.nih.gov/books/NBK1119/ [PubMed] [Google Scholar]
- De Almeida, P. A. D., Machado‐Costa, M. C., Manzoli, G. N., Ferreira, L. S., Rodrigues, M. C. S., Bueno, L. S. M., Saute, J. A., Pinto Vairo, F., Matte, U. S., Siebert, M., & Cossio, S. L. (2017). Genetic profile of Brazilian individuals affected with dystrophinopathies. Clinical Genetics, 92(2), 199–203. [DOI] [PubMed] [Google Scholar]
- Dent, K. M., Dunn, D. M., von Niederhausern, A. C., Aoyagi, A. T., Kerr, L., Bromberg, M. B., Hart, K. J., Tuohy, T., White, S., den Dunnen, J. T., Weiss, R. B., & Flanigan, K. M. (2005). Improved molecular diagnosis of dystrophinopathies in an unselected clinical cohort. American Journal of Medical Genetics, 134A, 295–298. 10.1002/ajmg.a.30617 [DOI] [PubMed] [Google Scholar]
- Desmet, F. O., Hamroun, D., Lalande, M., Collod‐Béroud, G., Claustres, M., & Béroud, C. (2009). Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Research, 37, e67. 10.1093/nar/gkp215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery, A. E. H. (1991). Population frequencies of inherited neuromuscular diseases—A world survey. Neuromuscular Disorders, 1(1), 19–29. 10.1016/0960-8966(91)90039-U [DOI] [PubMed] [Google Scholar]
- Fairclough, R. J., Wood, M. J., & Davies, K. E. (2013). Therapy for Duchenne muscular dystrophy: renewed optimism from genetic approaches. Nature Reviews Genetics, 14(6), 373–378. 10.1038/nrg3460 [DOI] [PubMed] [Google Scholar]
- Flanigan, K. M., Dunn, D. M., von Niederhausern, A., Soltanzadeh, P., Gappmaier, E., Howard, M. T., Sampson, J. B., Mendell, J. R., Wall, C., King, W. M., Pestronk, A., Florence, J. M., Connolly, A. M., Mathews, K. D., Stephan, C. M., Laubenthal, K. S., Wong, B. L., Morehart, P. J., Meyer, A., … Weiss, R. B. (2009). Mutational spectrum of DMD mutations in dystrophinopathy patients: Application of modern diagnostic techniques to a large cohort. Human Mutation, 30, (12), 1657–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazalar‐Montoya, J., Illanes‐Manrique, M., Inca‐Martinez, M., Marca, V., Huaman‐Dianderas, F., Guevara‐Fujita, M. L., Fujita, R., & Cornejo‐Olivas, M. (2020). Asesoramiento genético a una portadora asintomática de DMD: Primer caso reportado en el Sistema deSalud Pública del Perú [Genetic counseling to a DMD asymptomatic carrier: First case report in the Peruvian public healthcare system]. Revista De Neuro‐Psiquiatria, 83(4), 278–283. [Google Scholar]
- Hoffman, E. P., & Giron, J. (2001). Muscular dystrophy: methods and protocols. Human Press. [Google Scholar]
- Hotta, A. (2015). Genome editing gene therapy for Duchenne muscular dystrophy. Journal of Neuromuscular Diseases, 2(4), 343–355. 10.3233/JND-150116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, J., & Ng, P. C. (2013). SIFT Indel: Predictions for the functional effects of amino acid insertions/deletions in proteins. PLoS One, 8(10), e77940. 10.1371/journal.pone.0077940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen, B., Hartmann, C., Scholz, V., Jauch, A., & Zschocke, J. (2005). MLPA analysis for the detection of deletions, duplications and complex rearrangements in the dystrophin gene: Potential and pitfalls. Neurogenetics, 6(1), 29–35. [DOI] [PubMed] [Google Scholar]
- Juan‐Mateu, J., Gonzalez‐Quereda, L., Rodriguez, M. J., Baena, M., Verdura, E., Nascimento, A., Ortez, C., Baiget, M., & Gallano, P. (2015). DMD mutations in 576 dystrophinopathy families: A step forward in genotype‐phenotype correlations. PLoS ONE, 10(8), e0135189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig, M., Hoffman, E. P., Bertelson, C. J., Monaco, A. P., Feener, C., & Kunkel, L. M. (1987). Complete cloning of the duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell, 50(3), 509–517. 10.1016/0092-8674(87)90504-6 [DOI] [PubMed] [Google Scholar]
- López‐Hernández, L. B., Gómez‐Díaz, B., Luna‐Angulo, A. B., Anaya‐Segura, M., Bunyan, D. J., Zúñiga‐Guzman, C., Escobar‐Cedillo, R., Roque‐Ramírez, B., Ruano‐Calderón, L., Rangel‐Villalobos, H., López‐Hernández, J., Estrada‐Mena, F., García, S., & Coral‐Vázquez, R. (2015). Comparison of mutation profiles in the duchenne muscular dystrophy gene among populations: implications for potential molecular therapies. International Journal of Molecular Sciences, 16(12), 5334–5346. 10.3390/ijms16035334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren, W., Gil, L., Hunt, S. E., Riat, H. S., Ritchie, G. R., Thormann, A., Flicek, P., & Cunningham, F. (2016). The ensemble variant effect predictor. Genome Biology, 17(1):122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, L. A., Romitti, P. A., Cunniff, C., Druschel, C., Mathews, K. D., Meaney, F. J., , Matthews, D., Kantamneni, J., Feng, Z.‐F., Zemblidge, N., Miller, T. M., Andrews, J., Fox, D., Ciafaloni, E., Pandya, S., Montgomery, A., & Kenneson, A. (2006). The muscular Dystrophy Surveillance Tracking and Research Network (MD STARnet): Surveillance methodology. Birth Defects Research Part A: Clinical and Molecular Teratology, 76(11), 793–797. 10.1002/bdra.20279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, S. A., Dykes, D. D., & Polesky, H. F. (1988). A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Research, 16(3), 1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco, A. P., Bertelson, C. J., Liechti‐Gallati, S., Moser, H., & Kunkel, L. M. (1988). An explanation for the phenotypic differences between individuals affected bearing partial deletions of the DMD locus. Genomics, 2(1):90–95. [DOI] [PubMed] [Google Scholar]
- Neri, M., Rossi, R., Trabanelli, C., Mauro, A., Selvatici, R., Falzarano, M. S., Spedicato, N., Margutti, A., Rimessi, P., Fortunato, F., Fabris, M., Gualandi, F., Comi, G., Tedeschi, S., Seia, M., Fiorillo, C., Traverso, M., Bruno, C., Giardina, E., … Ferlini, A. (2020). The genetic landscape of dystrophin mutations in Italy: A nationwide study. Frontiers in Genetics, 11, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okubo, M., Minami, N., Goto, K., Goto, Y., Noguchi, S., Mitsuhashi, S., & Nishino, I. (2016). Genetic diagnosis of Duchenne/Becker muscular dystrophy using next‐generation sequencing: validation analysis of DMD mutations. Journal of Human Genetics, 61(6), 483. 10.1038/jhg.2016.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagel, K. A., Pejaver, V., Lin, G. N., Nam, H. J., Mort, M., Cooper, D. N., Sebat, J., Iakoucheva, L. M., Mooney, S. D., & Radivojac, P. (2017). When loss‐of‐function is loss of function: assessing mutational signatures and impact of loss‐of‐function genetic variants. Bioinformatics, 33(14), i389–i398. 10.1093/bioinformatics/btx272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier‐Foster, J., Grody, W. W., Hegde, M., Lyon, E., Spector, E., Voelkerding, K., & Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–423. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas, D., Narvaja, M. E., Rivas, L., Guevara‐Fujita, M. L., Castañeda, C., & Fujita, R. (2012). Implementación de la Prueba del Multiplex PCR para el Gen DMD en Pacientes con sospecha de Distrofia Muscular de Duchenne/Becker y la identificación de una deleción de los exones 48‐51. Horizonte Médico, 12:8–15. [Google Scholar]
- Sandoval, J. R., Salazar‐Granara, A., Acosta, O., Castillo‐Herrera, W., Fujita, R., Pena, S. D., & Santos, F. R. (2013). Tracing the genomic ancestry of Peruvians reveals a major legacy of pre‐Columbian ancestors. Journal of Human Genetics, 58(9), 627–634. 10.1038/jhg.2013.73 [DOI] [PubMed] [Google Scholar]
- Schwarz, J. M., Cooper, D. N., Schuelke, M., & Seelow, D. (2014). MutationTaster2: Mutation prediction for the deep‐sequencing age. Nature Methods, 11, 361–362. 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- Sirugo, G., Williams, S. M., & Tishkoff, S. A. (2019). The missing diversity in human genetic studies. Cell, 177(1), 26–31. 10.1016/j.cell.2019.02.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshima, Y., Yagi, M., Okizuka, Y. O., Awano, H., Zhang, Z., Yamauchi, Y., Nishio, H., & Matsuo, M. (2010). Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center. Journal of Human Genetics, 55, 379–388. 10.1038/jhg.2010.49 [DOI] [PubMed] [Google Scholar]
- Toksoy, G., Durmus, H., Aghayev, A., Bagirova, G., Sevinc Rustemoglu, B., Basaran, S., Avci, S., Karaman, B., Parman, Y., Altunoglu, U., Yapici, Z., Tekturk, P., Deymeer, F., Topaloglu, H., Kayserili, H., Oflazer‐Serdaroglu, P., & Uyguner, Z. O. (2019). 2019 Mutation spectrum of 260 dystrophinopathy patients from Turkey and important highlights for genetic counseling. Neuromuscular Disorders, 29(8), 601–613. 10.1016/j.nmd.2019.03.012 [DOI] [PubMed] [Google Scholar]
- Van Vliet, L., de Winter, C. L., van Deutekom, J. C., van Ommen, G.‐J.‐B., & Aartsma‐Rus, A. (2008). Assessment of the feasibility of exon 45–55 multiexon skipping for duchenne muscular dystrophy. BMC Medical Genetics, 9(1), 45–55. 10.1186/1471-2350-9-105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L., Xu, M., Li, H., He, R., Lin, J., Zhang, C., & Zhu, Y. (2019). Genotypes and phenotypes of DMD small mutations in chinese patients with dystrophinopathies. Frontiers in Genetics, 10, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zatz, M., Passos‐Bueno, M. R., & Vainzof, M. (2016). Neuromuscular disorders: genes, genetic counseling and therapeutic trials. Genetics and Molecular Biology, 39(3), 339–348. 10.1590/1678-4685-GMB-2016-0019 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Table S2
Data Availability Statement
The data that support the findings of this study are openly available in zenodo at https://doi.org/10.5281/zenodo.4741000, reference number 4741001.