

Abstract
Angiogenesis factors are widely known to promote tumor growth by increasing tumor angiogenesis in the tumor microenvironment, however, little is known whether their intracellular function is involved in tumorigenesis. Here we show that AGGF1 acts as a tumor suppressor by regulating p53 when acting inside tumor cells. AGGF1 antagonizes MDM2 function to inhibit p53 ubiquitination, increases the acetylation, phosphorylation, stability and expression levels of p53, activates transcription of p53 target genes, and regulates cell proliferation, cell cycle, and apoptosis. AGGF1 also interacts with p53 through the FHA domain. Somatic AGGF1 variants in the FHA domain in human tumors, including p.Q467H, p.Y469 N, and p.N483T, inhibit AGGF1 activity on tumor suppression. These results identify a key role for AGGF1 in an AGGF1-MDM2-p53 signaling axis with important functions in tumor suppression, and uncover a novel trans-tumor-suppression mechanism dependent on p53. This study has potential implications in diagnosis and therapies of cancer.
Keywords: FHA domain, p53 dependent, Ubiquitination, Translational modification, Somatic AGGF1 variants
1. Introduction
The AGGF1 gene, previously referred to as VG5Q, encodes an angiogenic factor with both a FHA domain and a G-patch domain [33]. Homozygous Aggf1 knockout (KO) mice died before embryonic day 8.5, and one third of heterozygous KO mice also died embryonically, indicating that Aggf1 is an essential gene for life [42]. AGGF1 has been shown to stimulate angiogenesis in vitro and in vivo, to regulate differentiation of veins and development of intersegmental vessels in zebrafish by activating AKT signaling, and to specify the differentiation of hemangioblasts (multipotent precursor cells that can differentiate into both hematopoietic and endothelial cells) [4,21,23,24,42]. Recently, we showed that systemic injection of recombinant AGGF1 protein restored cardiac structure and function in a mouse model for coronary artery disease (CAD) and myocardial infarction (MI) by inducing autophagy via JNK signaling and in a transverse aortic constriction (TAC) mouse model for cardiac hypertrophy and heart failure by regulating a novel noncanonical ERK-CHOP ER stress signaling pathway, and blocked restenosis and neointima formation after vascular injury by inhibiting vascular smooth muscle cell phenotypic switching and proliferation [23, 37,39].
TP53 (encoding p53) is the most commonly mutated tumor suppressor gene in human cancer. p53 acts as a transcription factor, and activates numerous target genes involved in regulation of many cellular progresses, including cell differentiation, cell cycle, apoptosis, DNA repair, cell senescence [32]. As a crucial tumor suppressor, p53 is regulated by posttranslational modifications such as phosphorylation, acetylation and ubiquitination [30]. The expression level, stability and transcription activation of p53 are modulated by the posttranslational modifications [1,29]. The ubiquitination of p53 is mediated by MDM2, an E3 ubiquitin ligase that plays an important role in regulation of p53 nuclear export and degradation [41]. Phosphorylation and acetylation of p53 can also affect its expression level and stability. TP53 is mutated in more than 50% of human cancer [2]. Similar to TP53, somatic AGGF1 variants were identified in many human cancer tissue samples in public databases. Therefore, we hypothesized that AGGF1 may act like TP53 to play a crucial role in regulating tumor cell functions.
In addition to the extracellular and paracrine functions, we previously showed that AGGF1 was highly expressed in nuclei [33], which was later confirmed by another group [28]. Nuclear expression of AGGF1 led to the proposal that it may have an additional function(s) inside cells [18,28]. Here, we describe an important intracellular function of AGGF1 in regulation of p53 stability and expression and provide a mechanistic link to this function in human tumors.
2. Materials and methods
2.1. Cell culture and transfection
HCT116 human colorectal cancer cells, NCI–H1299 human non-small lung cancer cells, and B16–F0 murine melanoma cells were cultured in DMEM media (Invitrogen, Carlsbad, CA) supplemented with 10% FBS (Hyclone, Logan, UT). Cells were maintained at 37 °C with 10% CO2. Lipofectamine 2000 (Invitrogen) was used to transfect cells according to the manufacturer’s instructions.
2.2. Plasmids and siRNAs
The pET-28VG5Q-wt plasmid with the full-length human AGGF1 cDNA cloned into pET-28b was described previously [33]. The AGGF1 cDNA was PCR-amplified and cloned into pEGFP-C1, resulting in an expression plasmid for EGFP-tagged AGGF1, pEGFP-C1-AGGF1. The AGGF1 cDNA was PCR-amplified and cloned into pcDNA3.1-FLAG, resulting in an expression plasmid for FLAG-tagged AGGF1, pFLAG-AGGF1. Serial N-terminal and C-terminal deletions of AGGF1 were created by PCR with different combinations of primers and pEGFP-C1-AGGF1 plasmid DNA as the template, and cloned into pCMV-C-HA (Applied Biosystems, Foster City, CA, USA), resulting in a series of pCMV-C-HA-AGGF1 expression plasmids for HA-tagged AGGF1 mutants with different deletions. The primers used to create pCMV-C-HA-AGGF1 deletion plasmids were shown in Table S2. The FHA domain (amino acids 435–508) was amplified by PCR and cloned into pEGFP-C1-AGGF1, resulting in an expression plasmid for EGFP-tagged FHA domain of AGGF1, pEGFP-C1-AGGF1-FHA. The expression plasmid for p53, pCDNA3.1-P53 and pEGFP-P53, an expression plasmid for Myc-tagged Ubiquitin (pCMV-Myc-Ub), and a luciferase reporter plasmid which contained the luciferase reporter gene driven by a basic promoter element joined to 14 repeats of the p53 binding site, p53-Luc, were kindly provided by Prof. Mugen Liu at Huazhong University of Science and Technology. The TP53 cDNA was amplified by PCR from pEGFP-P53 and cloned into pCMV-C-HA, resulting in an expression plasmid for HA-tagged p53, pCMV-HA-p53. The pGPH1/GFP/shNcontrol and pGPH1/GFP/shAGGF1 were from Genepharm.
AGGF1 missense mutations were introduced into pFLAG-AGGF1 and pEGFP-C1-AGGF1 using the PCR-based mutagenesis method as described [3,6,17,31,43]. siRNAs for knockdown of AGGF1 (siAGGF1) and control siRNA (siNC) were from Guangzhou RioboBio.
2.3. Quantitative real-time RT-PCR analysis
Real-time RT-PCR analysis was carried out as described [16,23,27, 37,39]. In brief, total RNA was extracted from cells using the Trizol Reagent kit (Invitrogen, Carlsbad, California) according to the manufacturer’s instructions. Two micrograms of total RNA samples were reverse-transcribed using a reverse transcription kit (Promega). Quantitative PCR was performed using the ABI 7900HT system. PCR reactions were carried out in 10 μl reactions using SYBR Green PCR master mix (Roche) and 0.2 mM specific primers.
2.4. Western blot analysis
Western blot analysis was carried out as described previously [13,22, 34,40]. The monoclonal antibody (mAb)against p53 (DO-1) was from Santa Cruz Biotechnology. Polyclonal antibodies (pAb) against p21, Bax, GAPDH, and α-tubulin were from Cell Signaling Technology. A series of Phospho-p53 pAbs were also from Cell Signaling Technology. Three Acetyl-p53 pAbs were from Epitomics, and the anti-EGFP mAb was from Abcam. The anti-FLAG antibody, mouse IgG, and rabbit IgG were from Santa Cruz Biotechnology. The anti-HA and anti-Myc mAb were from MBL. The goat anti-rabbit HRP conjugated secondary antibody and goat anti-mouse HRP conjugated secondary antibody were from Millipore.
2.5. Immunostaining
HCT116 cells were co-transfected with pEGFP-C1-AGGF1 and pCMV-HA-p53, fixed with 4% paraformaldehyde, permeabilized with 0.1% TritonX-100 in PBS, and blocked with 0.5% BSA in PBS. The cells were incubated with the primary anti-HA antibody (1:100, Proteintech) at 4 °C overnight, and then with the TRITC labeled goat anti-rabbit secondary antibody (Life Technologies). Nuclei were counterstained with DAPI (Invitrogen). Co-localization of AGGF and p53 were then analyzed under a FV1000 confocal microscope as described [7,21,43].
2.6. Co-Immunoprecipitation (Co-IP)
Co-IP was carried out as described previously [10,17,35]. HCT116 cells were co-transfected with pEGFP-P53 and pFLAG-AGGF1, pEGFP-P53 and wild type (WT) or different mutant pCMV-C-HA-AGGF1 expression plasmids, or pEGFP-P53 and pEGFP-C1-AGGF1-FHA. The transfected cells were lysed in lysis buffer (1% NP-40, 50 mM Tris-Hcl [pH 8.0], 150 mM NaCl, 10 mM NaF, 1 mM Na3VO4, phenylmethane-sulfonylfluoride, and Roche EDTA-free protease inhibitors). Cellular lysates were sonicated and centrifuged to remove insoluble materials, and then incubated with protein-A agarose beads (Santa Cruz) at 4 °C for 1 h. The cleared lysates (1 ml) were divided into identical aliquots of 500 μl and incubated with 1 μg of the antibody for immunoprecipitation (anti-EGFP, anti-HA, anti-FLAG or p53 and mouse or rabbit IgG as negative control) at 4 °C overnight. Samples were incubated with protein G sepharose (GE Healthcare) for 2–4 h, and washed. The bound proteins were eluted with 2X SDS-PAGE buffer and separated by SDS-PAGE, transferred to nitrocellulose membranes (Millipore) and probed with Western blotting antibodies (anti--FLAG, anti-GFP or anti-p53).
2.7. Chromatin immunoprecipitation analysis (ChIP)
ChIP analysis was performed as described [10,25,36,44]. Briefly, HCT116 cells in 10 mm wells were transfected with expression plasmids or siRNAs for 48 h. Chromatin immunoprecipitation was carried out using the Millipore EZ-CHIP™ kit (Catalog#17–371). Chromatins were sonicated into short pieces, and immunoprecipitated with an anti-p53 antibody (Santa Cruze DO-1). The precipitated chromatin (DNA) was used for quantitative real-time PCR (qPCR) to detect the p53-binding sites. The primers used for qPCR were shown in methods.
2.8. Cell proliferation assays
Cell proliferation was monitored using the EdU incorporation assay with the Cell-Light™ EdU Apollo 567 in vitro imaging kit following the manufacturer’s instructions (Ribobio) [26,36–38]. HCT116 cells and H1299 cells in 24-well plates were transfected with expression plasmids or siRNAs for 48 h, and incubated with 50 μM EdU for 2 h, fixed with 4% paraformaldehyde for 30 min at 37 °C, and washed twice with PBS containing 0.5% Triton X-100. Then, 100 μl of Apollo 567 stain reaction buffer was added. The cells were incubated for 30 min, stained with 100 μl of Hoechst for 30 min at room temperature, and imaged under a confocal microscope. The rate of cell proliferation was computed with the percentage of EdU-positive red cells over Hoechst stained cells (blue) × 100%.
2.9. Cell cycle assays
The cell cycle analysis was carried out using the Cell Cycle kit (BD) as described [26,36–38]. In brief, HCT116 and H1299 cells were transfected with expression plasmids or siRNAs for 48 h, washed with PBS, trypsinized, and fixed in 70% ice-cold ethanol. The cells were then incubated with RNase (100 mg/ml) and propidium iodide (4 mg/ml) in PBS. Cell cycle phases were detected with a flow cytometer (FC500; Beckman Coulter) and analyzed by CXP software.
2.10. Apoptosis assays
Apoptosis assays were performed as described [26,36–38]. In brief, HCT116 and H1299 cells were transfected with expression plasmids or siRNAs for 48 h, and resuspended in binding buffer. Cells were stained with phycoerythrin (PE)-conjugated annexin V and 7-aminoactinomycin D (7-AAD) (Keygentec) and analyzed by a Coulter FC500 instrument (Beckman Coulter). The flow cytometry data were analyzed using CXP software (Beckman Coulter). We calculated the percentage of apoptotic cells by the percentage of cells in the upper-right quadrant (annexin V-positive, PI-positive) plus cells in the lower right quadrant (annexin V-positive, PI-negative) over the total number of cells.
2.11. Transcription activation assays for p53
The transcription activation activity of p53 was determined using luciferase assays with a p53-luc reporter from Stratagene. The p53-luc reporter contains a firefly luciferase reporter gene driven by a basic promoter element and a TATA box, which are joined to a tandem of 14 repeats of a p53 binding element (TGCCTGGACTTGCCTGG), and was kindly provided by Prof. Zhiyong Ma at Chinese Academy of Agricultural Science. The pRL-TK plasmid (internal control) containing Renilla luciferase (Promega) was co-transfected with p53-luc into cells and luciferase assays were performed as described [8–10,25,31,44].
2.12. Soft agar colony formation assays
Cells were transfected with AGGF1 expression plasmids or siRNAs for 24 h in six-well plates and harvested as single-cell suspension in a concentration of 1 × 106 cells/ml). We poured 1 ml of 0.6% agarose gel pre well prepared by 2X full media and 1.2% agarose solution into six-well culture plates for form the bottom layer of agar. After the agar was solidified, we loaded 0.3% agarose gel containing 1 × 104 cells/well to the plates as the top layer, incubated the plates for 3 weeks at 37 °C in a humidified incubator. Plates were washed once with 0.85 M NaCl, stained with crystal violet (Sigma) dissolved in ethanol for 15 min, and washed to remove excess crystal violet. The number of colonies containing >50 cells was scored. Each experiment was performed twice in triplicates.
2.13. Tumor cell invasion assays
HCT116 cells were transfected with WT or mutant AGGF1 expression plasmids vs. empty vector control pCMV-3 × FLAG for 48 h, and starved without FBS for 12 h. Corning-Costar 3494 Transwells were pre-coated with 50 mg/l of Matrigel (BD) diluted at 1:8 with DMEM, and inserted into the chamber of 12-well plates filled with 500 μl of DMEM with FBS. Transwells were washed with DMEM without FBS, and then cells in DMEM without FBS were loaded into Transwells, and allowed to migrate for 12–48 h in a 37 °C incubator with 5% CO2. The bottom filters of Transwells were fixed with 4% formaldehyde for 10 min, and the cells located in the lower filters were stained with 0.1% crystal violet for 20 min, washed with PBS, and photographed under a TI-80 microscope (Nikon).
2.14. In vivo tumor growth assays in mice
The mouse studies were approved by the Ethics Committee of Huazhong University of Science and Technology. HCT116 cells were transfected with wild type or mutant AGGF1 expression plasmids, and AGGF1 siRNA or control siRNA. A total of 5 × 106 cells in 0.2 ml of PBS were injected subcutaneously into the subcutaneous pocket of each BALB/C-nunu athymic mouse (6-week old, 18.0 ± 2.0 g), and tumor growth was monitored. For B16–F0 tumor growth analysis, cells were transfected with wild type or mutant AGGF1 expression plasmids. A total of 5 × 106 cells in 0.2 ml of PBS were injected subcutaneously into the subcutaneous pocket of each C57BL/6 mouse (6-week old, 18.0 ± 2.0 g), and tumor growth was monitored. For tumor growth measurements, tumor diameters were measured with vernier calipers every 2 days, and tumor volumes were calculated using formula V (mm3) = (width2 × length)⁄2. The mice were sacrificed at the end of each study, and tumors were removed, weighed, photographed and fixed for further characterization.
2.15. Statistical analysis
All experiments were repeated independently at least three times starting with the preparation of plasmid DNA from different bacterial colonies. The data were presented as mean ± SEM. Statistical analysis was performed using the Student’s t-test. For comparisons of more than two groups, one-way ANOVA was employed. A P value of <0.05 was considered to be statistically significant.
3. Results
3.1. Overexpression of AGGF1 suppresses tumor growth and knockdown of AGGF1 expression promotes tumor growth
Our analysis of public human tumor mutation databases identified 180 somatic mutations in AGGF1 across cancers in cBioportal (http://www.cbioportal.org/) [11] and 191 somatic AGGF1 mutations in the COSMIC database (https://cancer.sanger.ac.uk/cosmic/gene/analysis?ln=AGGF1) (Fig. S1 and Table S1). For the 10528 TCGA pan-cancer samples in cBioPortal, 1.1% harbor an AGGF1 somatic mutation (not including copy number alterations) (Fig. S2). Identified AGGF1 mutations are missense mutations with few hotspots (Fig. S2), frequent homozygous deletions and amplifications (Fig. S2). The frequency of AGGF1 mutations was more than 1% of total tissue samples examined in five different types of cancers, including malignant melanoma (2.97%), endometrial cancer (2.66%), skin cancer (2.16%), cancer in large intestine (1.74%), and biliary tract cancer (1.02%) (Table S1). These data support the hypothesis that AGGF1 is involved tumorigenesis.
To examine the role of AGGF1 in cancer development and determine whether it functioned as an oncogene or a tumor suppressor, we carried out an in vitro soft agar colony formation assay using HCT116 human colon cancer cells as AGGF1 somatic mutations were identified frequently in colon cancer (1.74% of samples, Table S1). Overexpression of AGGF1 dramatically inhibited colony formation of HCT116 cells, whereas silencing of AGGF1 expression promoted colony formation, suggesting a role in tumor suppression (Fig. 1A–B). In vivo in human tumor xenografts in BALB/C-nunu mice, HCT116 cells with overexpression of AGGF1 showed significantly reduced tumor growth, whereas HCT116 cells with knockdown of AGGF1 expression showed significantly increased tumor growth (Fig. 1C–E).
Fig. 1. Overexpression of AGGF1 suppresses tumor growth and knockdown of AGGF1 promotes tumor growth.
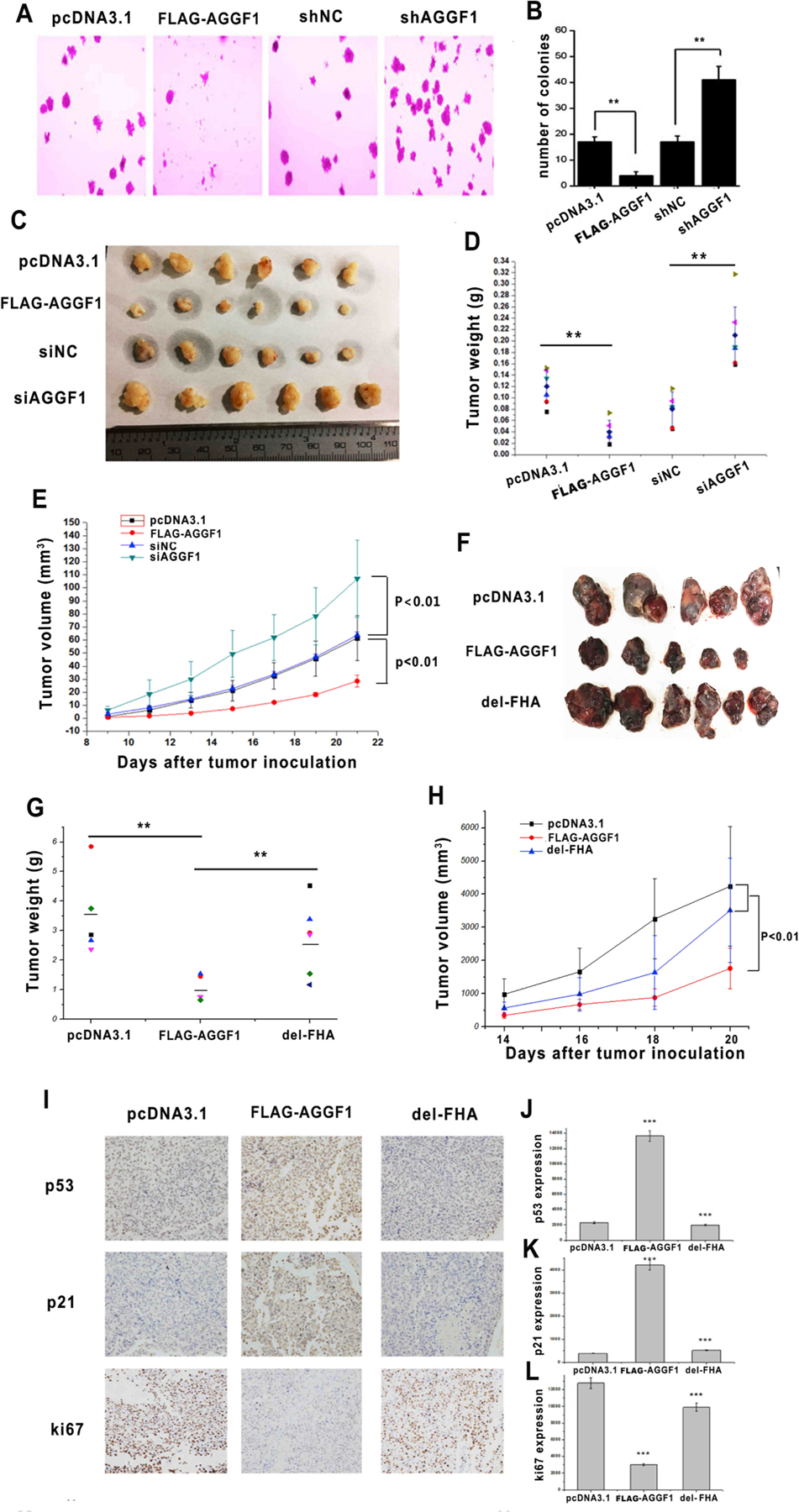
(A) Representative images of soft agar colony-formation assays with HCT116 cells transfected with an expression plasmid for FLAG-tagged AGGF1 vs. empty vector control (pcDNA3.1), AGGF1 shRNA or negative control shRNA. **P < 0.01, n = 3/group; NS, not significant.
(B) Quantification and statistical analysis of data from (A). (C-E) Transfected HCT116 tumor cells were implanted in nude mice (xenograft tumor model), and tumor growth was monitored (E). At day 22, the mice were sacrificed, tumors were excised, photographed (C), weighed and compared (D). **P < 0.01, n = 6/group. (F–H) Tumor growth analysis in C57BL/6J mice with B16–F0 cells transfected with an expression plasmid for FLAG-tagged wild type AGGF1, mutant AGGF1 without the FHA domain (del-FHA) or empty vector control pcDNA3.1 (CTRL). **P < 0.01, n = 5–6/group. (I) Representative images from immunostaining of tumor sections with anti-p53, anti-p21 or anti-Ki67 antibody. (J-L) Quantified data of images as in (I) are shown for p53 (J), p21 (K), and Ki67 (L). ***P < 0.001, n = 6/group.
Moreover, as malignant melanoma is also frequently associated with AGGF1 somatic mutations (2.97% of samples, Table S1), we characterized the role of AGGF1 in B16–F0 murine melanoma cells. In a C57BL/6 Xenograft tumor model, B16–F0 cells with overexpression of AGGF1 showed significantly reduced tumor growth, however, B16–F0 cells with overexpression of mutant AGGF1 with the critical FHA domain deleted failed to inhibit tumor growth (Fig. 1F–H). Altogether, these data suggest that AGGF1 is a tumor suppressor.
3.2. AGGF1 expression is positively correlated with the expression levels of p53 and p21, but negatively with cell proliferation in tumors
To identify the molecular mechanism by which AGGF1 suppresses tumor growth, we examined the expression levels of p53 and p21. Immunostaining of tumor sections from B16–F0 xenografts showed significantly increased expression levels of p53 and p21 in tumors with overexpression of AGGF1, however, the effect was abolished in tumors with overexpression of mutant AGGF1 without the FHA domain (Fig. 1I–L). The expression levels of p53 and p21 were negatively correlated with the immunostaining signal for cell proliferation marker Ki67 (Fig. 1I and L).
3.3. AGGF1 increases p53 expression by enhancing its stability
Western blot analysis showed that in HCT116 cells, knockdown of AGGF1 expression by two different siRNAs reduced the expression level of p53 (Fig. 2A), whereas overexpression of AGGF1 increased the expression level of p53 (Fig. 2B). Both Western blot and real-time RT-PCR analyses showed that overexpression of AGGF1, but not mutant AGGF1 without the FHA domain, significantly reduced the expression levels of p53-downstream genes p21 and Bax, whereas knockdown of AGGF1 expression significantly reduced the expression level of p21 and Bax (Fig. 2C–D). Interestingly, real-time RT-PCR analysis showed that either knockdown or overexpression of AGGF1 did not affect the expression level of p53 mRNA (Fig. 2E). These data suggest that the regulation of p53 expression by AGGF1 is post-transcriptional.
Fig. 2. AGGF1 increases the expression level of p53 by promoting its stability.
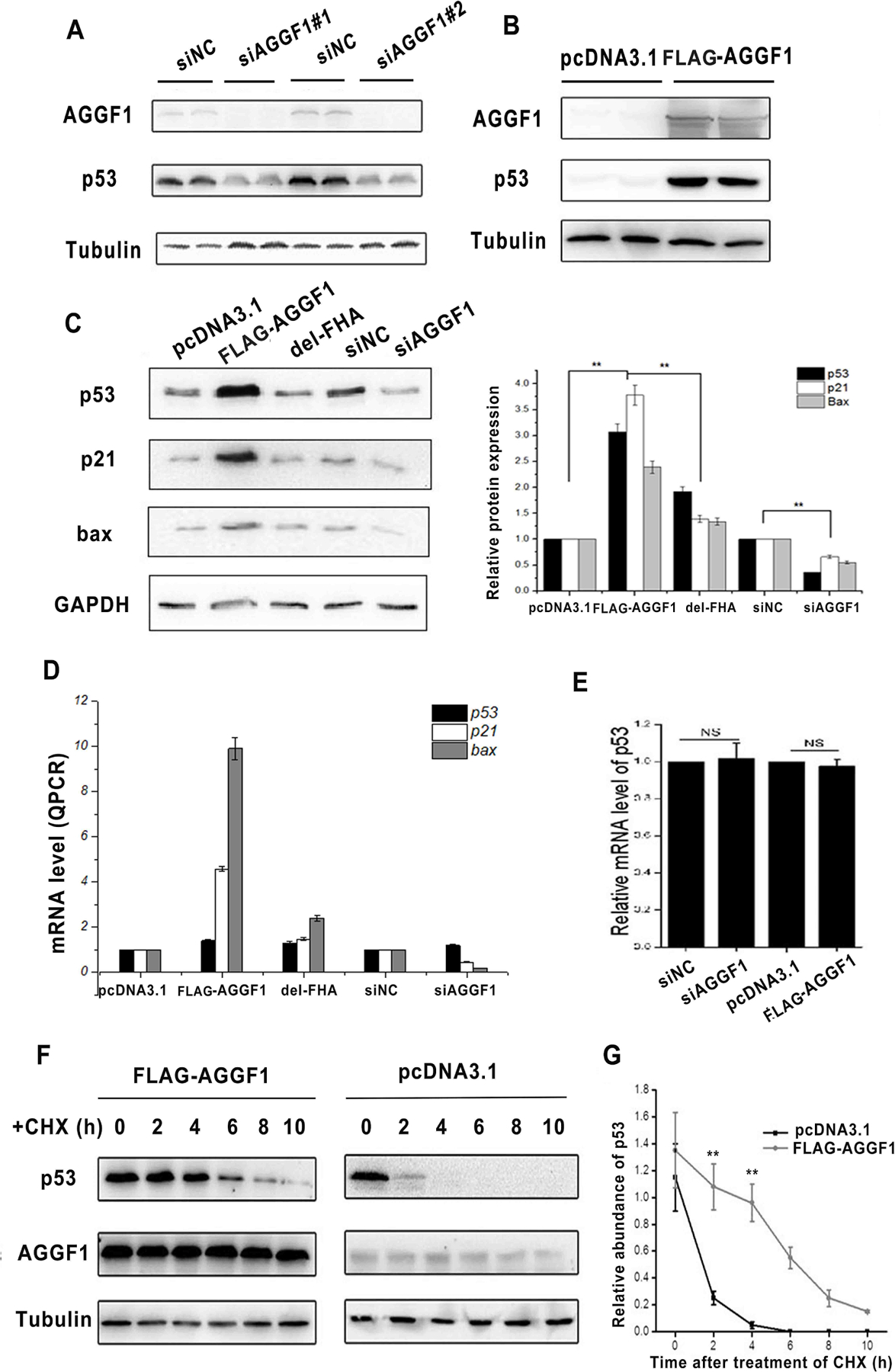
(A) Western blot analysis for AGGF1 and p53 with HCT116 cells transfected with two AGGF1 specific siRNAs (siAGGF1) and negative control scramble siRNAs (sicontrol). Mock, cells without any treatment; tubulin, loading control.
(B) Western blot analysis for AGGF1 and p53 with HCT116 cells transfected with pcDNA3.1-FLAG-AGGF1 or pcDNA3.1.
(C) Western blot analysis for p53, p21 and Bax with HCT116 cells with overexpression of wild type AGGF1, mutant AGGF1 without FHA domain (AGGF1-delFHA), and knockdown of AGGF1 using AGGF1 siRNA (siAGGF1) or control siRNA (sinc). Quantified data re shown at the bottom.
(D) Real time RT-PCR analysis of mRNA expression levels of TP53 coding for p53, CDKN1A coding for p21, and BAX coding for Bax for transfected HCT116 cells with different plasmids or siRNAs.
(E) Real time RT-PCR analysis of TP53 mRNA expression levels for transfected HCT116 cells.
(F) p53 stability assays. HCT116 cells were transfected with a FLAG-AGGF1 expression plasmid or pcDNA3.1 control, treated with CHX for 0–10 h, and used for Western blot analysis for p53, AGGF1, and tubulin.
(G) Quantified data from (F). *P < 0.01, **P < 0.01, n = 3/group.
We hypothesized that AGGF1 increased p53 expression by enhancing the stability of p53. HCT116 cells were transfected with an AGGF1 expression plasmid, empty vector control pcDNA3.1 and blank control (endogenous system), treated with CHX, an inhibitor of protein biosynthesis, and analyzed for the decay of p53 at different time points (Fig. 2F and Fig. S5). Overexpression of AGGF1 dramatically increased the stability of p53 protein (Fig. 2G).
3.4. Molecular mechanism by which AGGF1 increases stability of p53
To identify the molecular mechanism by which AGGF1 increased the stability of p53, we studied the effect of MG132, a proteasome inhibitor. The effect of AGGF1 knockdown on downregulation of p53 was reversed by MG132 (Fig. 3A), suggesting that AGGF1 increases p53 stability through the ubiquitin–proteasome degradation pathway. Ubiquitination assays showed that overexpression of AGGF1 markedly reduced the ubiquitination level of p53 (Fig. 3B), and increased p53 acetylation at K382, K305, and K373 (Fig. 3C), and increased P53 phosphorylation at S15, S20, S392, and S46 (Fig. 3D).
Fig. 3. AGGF1 increases p53 expression by inhibiting its ubiquitination via antagonizing MDM2 function.
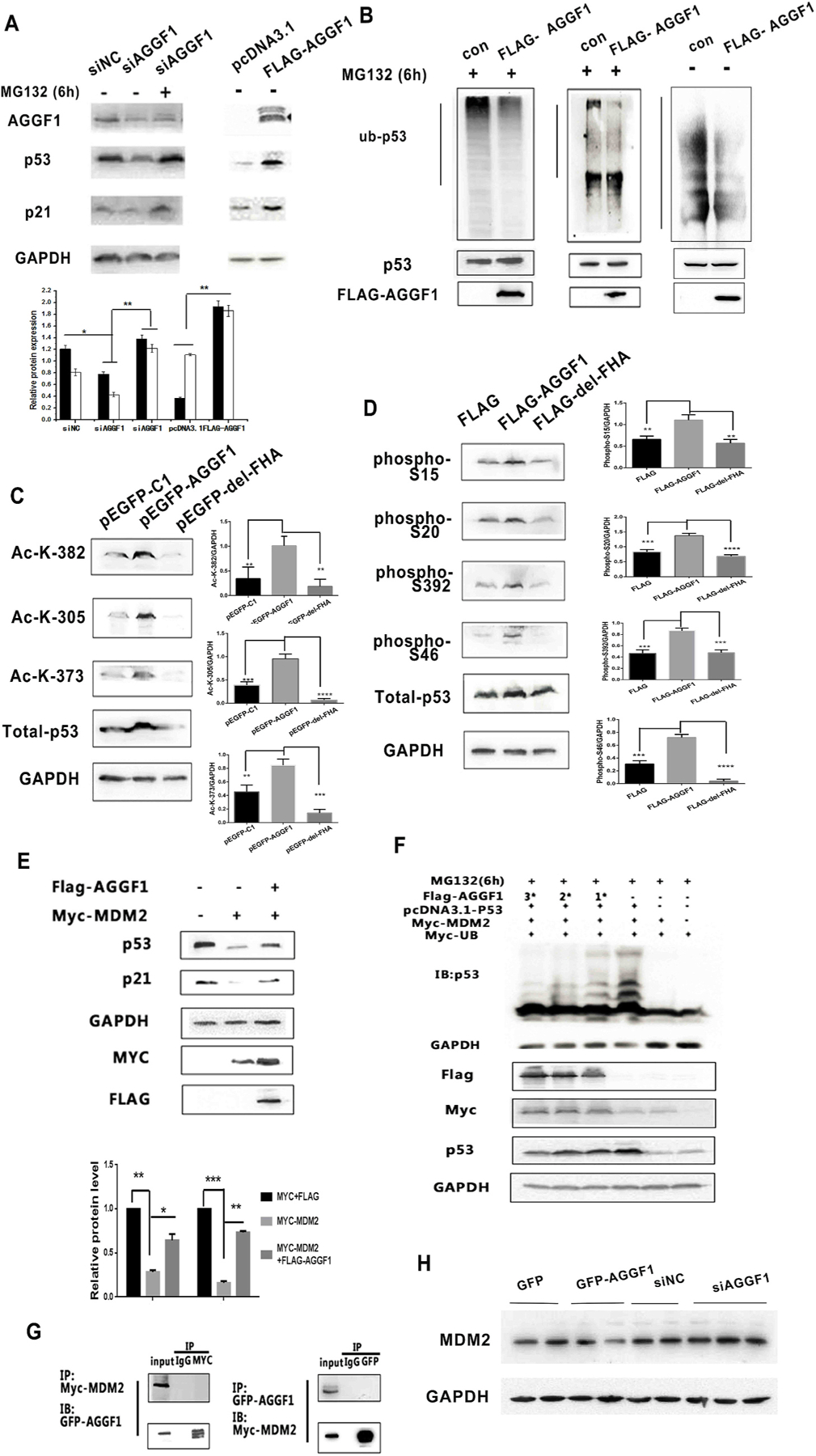
(A) MG132 treatment rescued the decreased p53 and p21 expression levels induced by AGGF1 siRNA (siAGGF1) as compared to siNC control siRNA. GAPDH was used as loading control.
(B) AGGF1 deubiquintinates p53. H1299 cells with overexpression of p53 were transfected with a FLAG-AGGF1 expression plasmid or empty vector control (Con), treated with MG132 or without MG132 (served as a control), and lysed for immunoprecipitation with an anti-p53 antibody. The precipitate was used for Western blot analysis with an anti-Ubiquitin or anti-p53 antibody. Bottom: Total cellular lysates were used for Western blot analysis to show overexpression of FLAG-AGGF1 and p53.
(C-D) H1299 cells were co-transfected with pcDNA3.1-p53 together with expression plasmid for GFP-AGGF1 or mutant GFP-AGGF1 without the FHA domain (GFP- del-FHA) for 48 h and used for Western blot analysis for acetylated p53 at different amino acid residues (C) and phosphorylated p53 at different amino acid residues (D). Total p53 and GAPDH were used as controls.
(E) Western blot analysis showing that MYC-MDM2 reduces expression of p53 and p21, however, the effect was reversed by overexpression of FLAG-AGGF1 in HCT116 cells.
(F) Western blot analysis showing that MYC-MDM2 increases p53 ubiquitination, however, the effect was reversed by FLAG-AGGF1 in a dose-dependent manner in HCT116 cells.
(G) Co-IP for MYC-MDM2 and GFP-AGGF1 in HCT116 cells
(H) Western blot analysis showing that the effects of FLAG-AGGF1 and AGGF1 siFNA on the expression level of MDM2 in HCT116 cells. GAPDH was used as loading control.
Ubiquitination of p53 is regulated by the E3-ubiquitin ligase MDM2. Overexpression of MDM2 downregulated the expression levels of p53 and its downstream target p21 (Fig. 3E), however, the effect was reversed by AGGF1 (Fig. 3E). These data suggest that AGGF1 regulates p53 expression by antagonizing the effect of MDM2. The conclusion is further supported by the data from p53 ubiquitination assays using Western blot analysis (Fig. 3F). HCT116 cells were transfected with expression plasmid for MYC-UB, MYC-MDM2, p53, and varying amounts of FLAG-AGGF1 (1x, 2x and 3x). As shown in Fig. 3F, overexpression of MYC-MDM2 induced dramatic ubiquitination of p53, which was diminished by overexpression of AGGF1 in a dose-dependent manner. Altogether, these data suggest that AGGF1 antagonizes the function of MDM2, thereby inhibiting ubiquitination of p53, and increasing the stability and expression level of p53.
To determine how AGGF1 antagonizes the effect of MDM2, we first examined whether AGGF1 interacts with MDM2. Co-IP studies with cells with co-expression of MYC-tagged MDM2 and EGFP-tagged AGGF1 revealed that anti-GFP failed to precipitate MYC-MDM2, and anti-MYC failed to precipitate GFP-AGGF1 (Fig. 3G). The data suggest that AGGF1 does not interact with MDM2. Second, we determined whether AGGF1 regulates the expression level of MDM2. Western blot analysis showed that overexpression of AGGF1 decreased MDM2 expression, and knockdown of AGGF1 expression by siRNA increased the expression level of MDM2 in HCT116 cells (Fig. 3H). The data suggest that AGGF1 antagonizes the effect of MDM2 and affects the ubiquitination, stability and expression level of p53 by reducing the expression level of MDM2.
3.5. AGGF1 interacts with p53
To further characterize how AGGF1 affects the ubiquitination, stability and expression level of p53, we determined whether there is interaction between AGGF1 and p53. HCT116 cells were co-transfected with FLAG-tagged AGGF1 and HA-tagged p53 expression plasmids, and immunostaining showed that AGGF co-localized with p53 in the nucleus (Fig. 4A). Co-immunoprecipitation (Co-IP) showed that EGFP-p53 successfully precipitated FLAG-AGGF1, and reciprocal Co-IP found that FLAG-AGGF1 successfully precipitated EGFP-p53 (Fig. 4B). Co-IP with serial N-terminal deletions of AGGF1 mapped the interaction domain for p53 between amino acid residues 435 and 509 (Fig. 4C–D), and similar analysis with serial C-terminal deletions mapped the p53 binding domain between amino acid residues 434 and 508 (Fig. 4C–D). The region between amino acids 434 and 509 is the FHA domain of AGGF1. Co-IP showed that the AGGF1 FHA domain interacted with p53 (Fig. 4E). These data suggest that p53 interacts with the FHA domain of AGGF1. Therefore, AGGF1 may act as a chaperone for p53 to stabilize p53.
Fig. 4. AGGF1 interacts with p53.
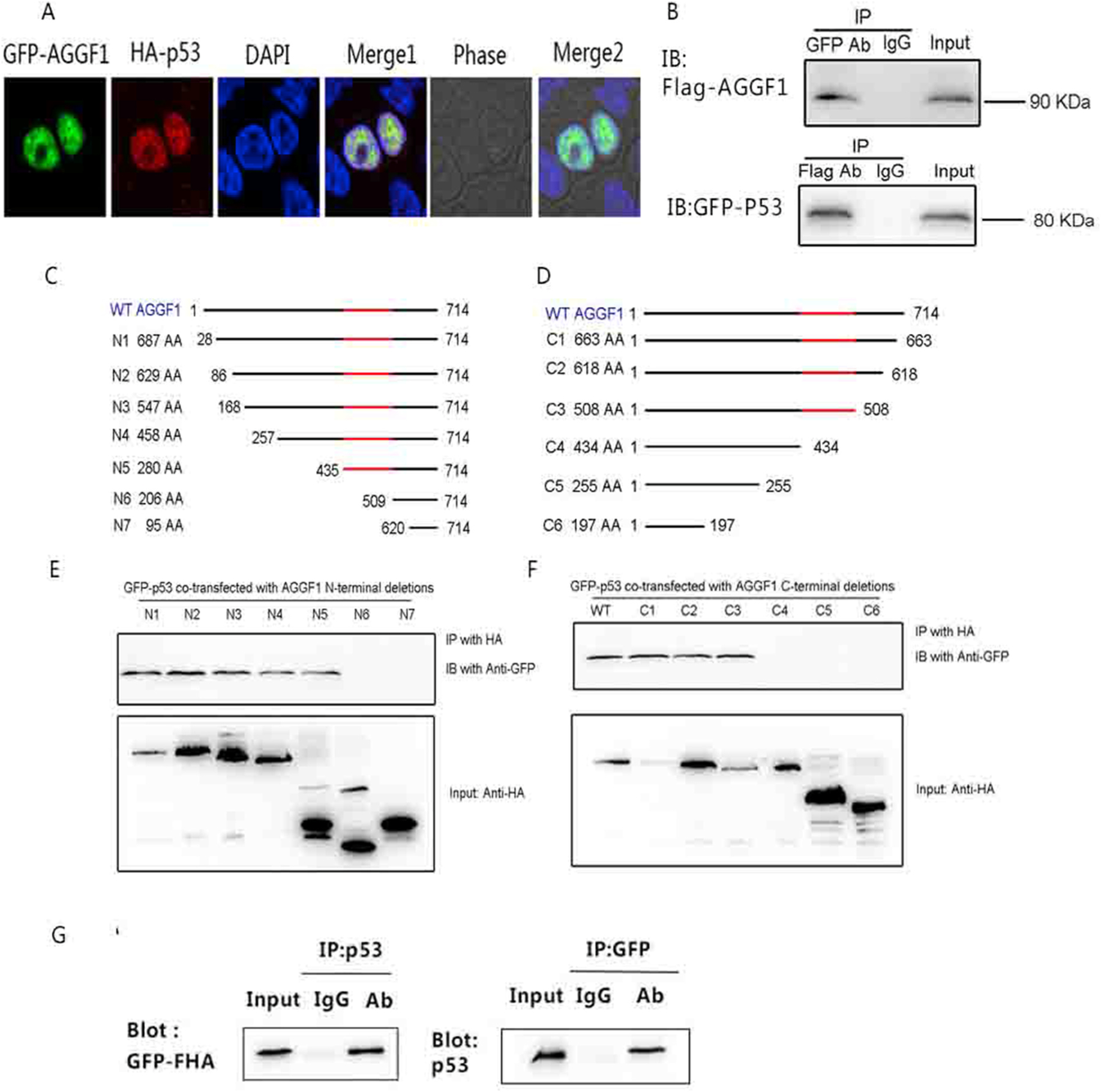
(A) Immunostaining images showing co-localization of AGGF1 and p53 in nuclei in HEK116 cells transfected with with pEGFP-C1-AGGF1 (green) and pCMV-N-HA-p53 (red). The nuclei were stained with DAPI.
(B) Co– IP showing interaction between AGGF1 and p53 in HCT116 cells co-transfected with pcDNA3.1-FLAG-AGGF1 and PEGFP-N1-P53.
(C) Co-IP analysis between EGFP-tagged p53 and seven N-terminal serial AGGF1 deletions (HA-tagged).
(D) Co-IP analysis between EGFP-tagged p53 and six C-terminal serial AGGF1 deletions (HA-tagged). (I) Co-IP analysis with HCT116 cells co-transfected with pEGFP-FHA for overexpression of the AGGF1 FHA domain only and pcDNA3.1-P53.. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.6. In vivo functional characterization AGGF1 somatic mutations from human cancer patients
We next sought to characterize the functional effects of human cancer-associated AGGF1 somatic mutations from the COSMIC database. We focused on six missense mutations identified in the FHA domain of AGGF1, including p.R447Q, p.G437E, p.Q467H, p.Y469 N, p.N483T, and p.V497I (Table S2 and Fig. 5A). HCT116 cells were transfected with WT or different mutant AGGF1 expression plasmids individually (empty vector as negative control), implanted in BALB/C-nunu mice, and analyzed for tumor growth. Compared with negative control, overexpression of AGGF1 dramatically inhibited tumor growth, however, the effect of AGGF1 was significantly attenuated by mutations p.Q467H, p. Y469 N and p.N483T, but not by mutations p.R447Q and p.V497I (Fig. 5A–C). Consistent with the data from tumor growth, mutations p. Q467H, p.Y469 N, and p.N483T, but not mutations p.R447Q and p. V497I, reduced the expression level of p53 and p21 (Fig. 5D). Mutation p.G437E significantly reduced the expression level of p53 and p21 compared with WT AGGF1, however, its effect on tumor growth was increased in three mice but not in three other mice (Fig. 5A–D). We also analyzed the effect of AGGF1 mutations on tumor invasion. Overexpression of AGGF1 in HCT116 cells inhibited tumor invasion, however the effect was abolished by the deletion of the FHA domain (Fig. 5E). The effect of AGGF1 on tumor invasion was also attenuated by mutations p.G437E, p.Q467H, p.Y469 N, and p.N483T, but not by mutations p. R447Q and p.V497I. Together, these data suggest that some somatic mutations in the FHA domain of AGGF1, including mutations p.Q467H, p.Y469 N, p.N483T, and probably p.G437E, are functional pathogenic variants that promote tumor growth. In aggregate, the data support the hypothesis that AGGF1 is a tumor suppressor. It is also important note that as expected, not all AGGF1 genomic variants in the FHA domain from cancer patients are disease-causing mutations.
Fig. 5. Human tumor somatic mutations in the AGGF1 FHA domain attenuate the tumor suppressing effect of AGGF1.
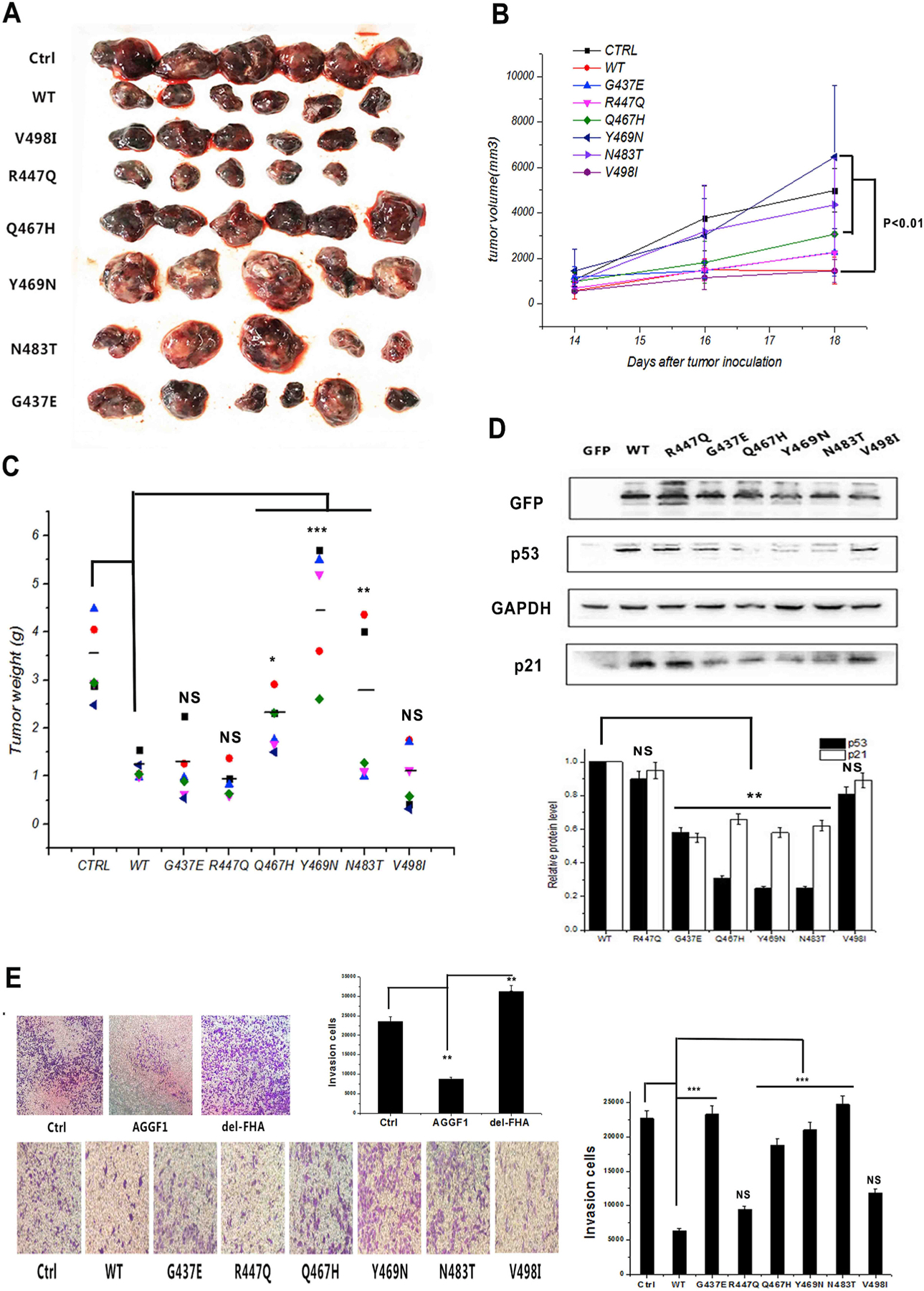
(A-C) Tumor growth analysis in C57BL/6J mice with B16–F0 cells with overexpression of wild type AGGF1 (WT vs. control Ctrl) and six AGGF1 somatic mutations in the FHA domain. (A) Images of tumors excised from mice at the end of the study (day 18). (B) Tumor growth as measured by the tumor volume at different time points. (C) Tumor weight at the end of the study. B16–F0 cells transfected with an expression plasmid for FLAG-tagged wild type AGGF1, mutant AGGF1 without the FHA domain (del-FHA) or empty vector control pcDNA3.1 (CTRL). **P < 0.01, n = 5–6/group.
(D) Western blot analysis showing the effects of six AGGF1 mutations in the FHA domain on the expression levels of p53 and p21. GAPDH was sued as loading control. Quantifications were normalized to GAPDH and show as relative density (bottom).
(E) Representative images for tumor cell invasion assays using HCT116 cells transfected with an expression plasmid for wild type AGGF1, mutant AGGF1 with the FHA domain deleted or empty vector control (Ctrl). Quantification of invasion cells was shown (right).
3.7. Tumor suppressor effect of AGGF1 is dependent on expression of p53
We next questioned whether the tumor suppressor effect of AGGF1 is dependent on p53. We assayed the transcription activation activity of p53 using a p53-Luc reporter with the luciferase reporter gene driven by a basic promoter element joined to 14 repeats of the p53 binding site. Co-transfection of the p53-Luc reporter and an expression plasmid for FLAG-AGGF1 in HCT116 cells with a p53 expression plasmid significantly enhanced the transcriptional activity of p53, whereas a p53 inhibitor pifithrin-α reversed the effect induced by AGGF1 (Fig. 6A). Knockdown of AGGF1 expression in HCT116 cells by an AGGF1 specific siRNA significantly inhibited p53 transcription activation (Fig. 6A). Real-time RT-PCR analysis showed that knockdown of AGGF1 expression downregulated the expression levels of p53 downstream genes, including genes encoding p21, Puma, Noxa, Bax, Xpc, p53R2, c-Fos, Gadd45 and DRAM1, in HCT116 cells, whereas overexpression of AGGF1 increased the expression levels of these genes (Fig. 6B). ChIP analysis showed that overexpression of AGGF1 increased the binding of p53 to promoters of genes coding for p21, Puma, Bax, and Noxa in HCT116 cells, whereas knockdown of AGGF1 significantly reduced the binding of p53 to its target promoters (Figs. S4A–4D). Interestingly, the effect of AGGF1 on transcription activation of p53 was completely abolished in a p53-null cell line H1299, which lacks endogenous p53 (Fig. 6A). Similarly, the effects of AGGF1 on cell proliferation (Fig. 6C–F), cell cycle progression (Fig. 6G and H), and apoptosis (Fig. 6I–K) were observed only in HCT116 cells with p53, but not in H1299 cells without p53. These data suggest that the effects of AGGF1 on transcription activation of p53 downstream genes, tumor cell proliferation, cell cycle regulation, and apoptosis are dependent on the presence of p53.
Fig. 6. AGGF1 regulates tumor cell growth depending on p53. (A) HCT116 cells with a high level of p53 expression were co-transfected with a TP53 promoter luciferase reporter (p53-luc), pRL-TK, a FLAG-AGGF1 expression plasmid vs. pcDNA3.1 vector control or AGGF1 siRNA (siAGGF1) vs. negative control siRNA (siNC) for 48 h, and lysed to prepare cellular extracts for luciferase assays to measure the transcription activation activity of p53 (left). PFT-α, an inhibitor of p53. Similar assays using H1299 cells without expression of p53 (right).
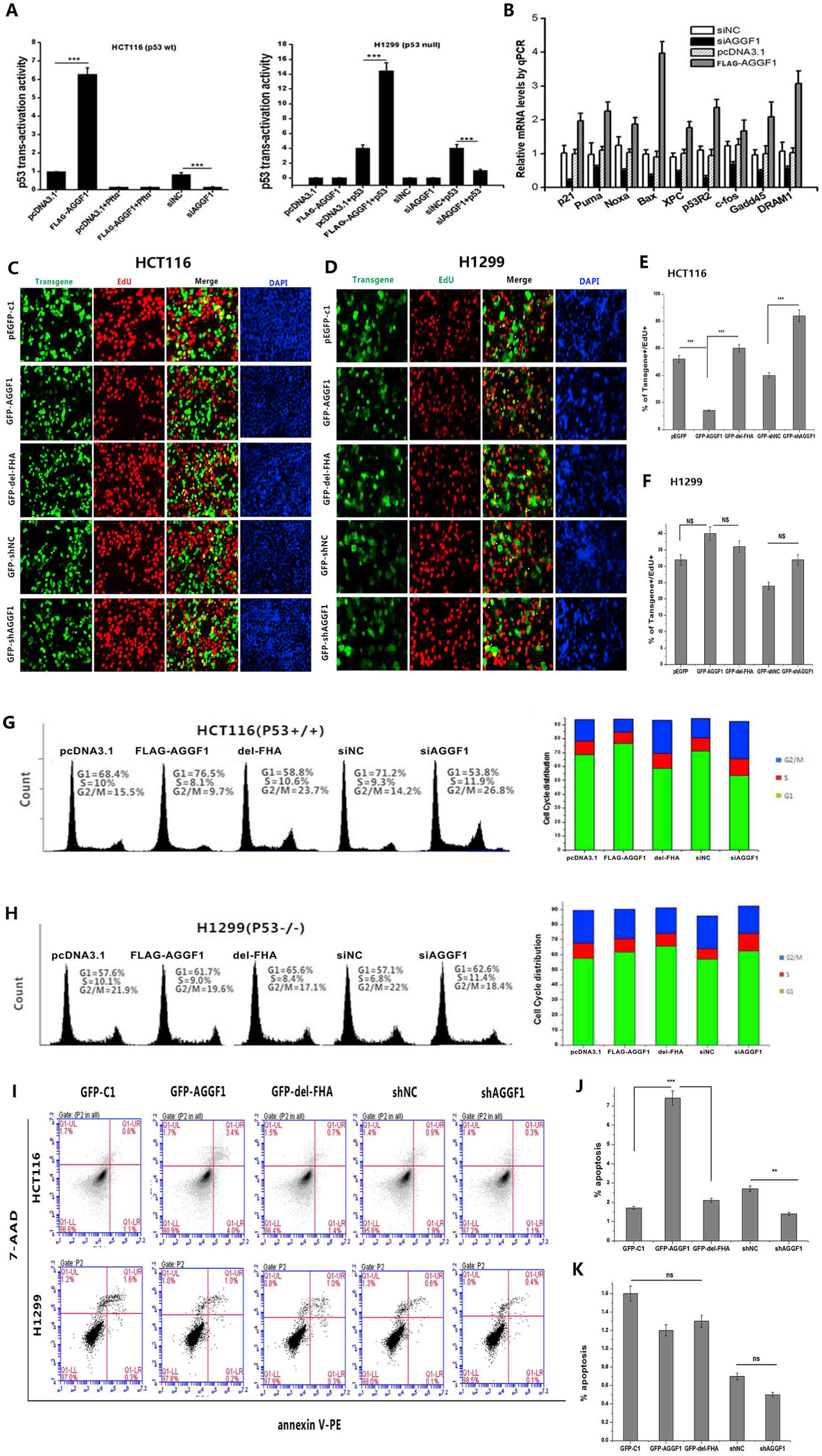
(B) Real-time RT-PCR analysis for TP53 downstream genes in HCT116 cells with overexpression of FLAG-AGGF1 vs. pcDNA3.1 control or knockdown of AGGF1 using siAGGF1 vs. siNC.
(C) Representative images of cell proliferation assays using EdU cell proliferation assays, which measure the incorporation of EdU into newly synthesized DNA in HCT116 cells transfected with plasmids for overexpression of GFP-AGGF1 or mutant GFP-AGGF1 without the FHA domain vs. empty pEGFP-c1 vector control, shRNA for AGGF1 (GFP-shAGGF1) vs. empty vector control (GFP-shNC).
(E) Experimental data as in (C) were quantified and plotted.
|(D) Similar cell proliferation assays but in H1299 cells without p53 expression.
(F) Experimental data as in (D) were quantified and plotted. ***, P < 0.001, n = 3/group; NS, not significant.
(G) Representative flow cytometry images of cell cycle assays using HCT116 cells transfected with plasmids for overexpression of GFP-AGGF1 or mutant GFP-AGGF1 without the FHA domain vs. empty pEGFP-c1 vector control, shRNA for AGGF1 (GFP-shAGGF1) vs. empty vector control (GFP-shNC). Experimental data were quantified and plotted.
(H) Similar cell cycle assays but in H1299 cells without p53 expression. Experimental data were quantified and plotted. ***, P < 0.001, n = 3/group; NS, not significant.
(I) Representative images of apoptosis assays using HCT116 cells transfected with plasmids for overexpression of GFP-AGGF1 or mutant GFP-AGGF1 without the FHA domain vs. empty pEGFP-c1 vector control, shRNA for AGGF1 (GFP-shAGGF1) vs. empty vector control (GFP-shNC) (top). Similar cell cycle assays but in H1299 cells without p53 expression (bottom).
(J) Experimental data as in (I, top) were quantified and plotted.
(K) Experimental data as in (I, bottom) were quantified and plotted. ***, P < 0.001, n = 3/group; NS, not significant.
4. Discussion
In aggregate, the data in this study provide important evidence to show that AGGF1 is a candidate tumor suppressor dependent on p53. AGGF1-related tumor suppression activities such as inhibition of tumor cell proliferation, cell cycle regulation and activation of apoptosis are all dependent on p53 (Fig. 6). Analysis of AGGF1 and TP53 mutations in 10,500 TCGA PanCancer samples showed that there was a significant co-occurrence between these mutations (Fig. S3). Tumors may be caused by a two-hit model involving one mutation in AGGF1 and one mutation in the TP53 tumor suppressor gene. AGGF1 mutations reduce the expression level of p53 as shown in Fig. 5, which may be sufficient to cause tumor development when combined with a single mutation in p53. Future studies are needed to test this interesting hypothesis.
TP53 is mutated in >50% of all human cancers [12,20]. However, many more tumor suppressors continue to be identified, which provide unprecedented insights into the molecular mechanisms of tumor growth and suppression. For example, in 2018, Hindupur et al. showed that the protein histidine phosphatase LHPP was a new tumor suppressor [14]. Also in 2018, Cho et al. showed that recurrent mutations in the promoter of LncRNA gene PVT1 were found in human cancers, and that the PVT promoter regulates MYC transcription to exert a tumor suppressor function [5]. In 2017, Keckesova et al. showed that mitochondrial protein LACTB is a new tumor suppressor [19]. For human cancers negative for p53 mutations, pathogenic variants in other tumor suppressors such as AGGF1, LHPP, PVT, LACTB, and other unidentified genes may play important roles in tumor formation. For AGGF1, more systematic studies are needed to determine the exact frequencies of AGGF1 mutations in various tumors, and identify interesting and/or unique correlations of these mutations with regard to tumor grade, metastasis/stage, drug-resistance, and other features as compared to mutations in TP53 and other tumor suppressor genes.
In this study, we focused on two major tumor cell types, HCT116 and B16–F0, as malignant melanoma and colorectal cancer showed more frequent AGGF1 somatic mutations (2.97% and 1.74% of tested tumor samples, respectively; Table S1). However, other tumor cells may also be susceptible to the tumor suppressing functions of AGGF1 as identified in HCT116 and B16–F0 cells. This will require further studies. Alternatively, other tumor cell types may respond to AGGF1 differently. For example, we found that most of MCF-7 cells with overexpression of AGGF1 died (data not shown). Furthermore, AGGF1 was shown to regulate cell proliferation and migration and other important cellular processes by regulating the phosphorylation of catalytic p110α subunit and p85α regulatory subunit of PI3K, AKT, GSK3β, p70S6K, ERK1/2, and JNK [4,15,23,37,39,42]. Some of these signaling pathways may also be involved in tumor suppressing activities of AGGF1 in tumor cells. Moreover, there are many mutations that are located outside of the AGGF1-p53 interaction domain, i.e. the FHA domain. Some of these mutations may promote tumor growth by p53-independent mechanisms.
In summary, here we describe a new tumor suppressor protein function of AGGF1 and we propose a novel signaling mechanism of tumor suppression. We show, using in vivo, in vitro, and human cancer mutations, that AGGF1 is a potent inhibitor of proliferation of multiple types of tumor cells. Importantly, we show that three somatic mutations in the FHA domain of AGGF1 identified in human cancers (p.Q467H, p. Y469 N, and p.N483T) are functional pathogenic variants that attenuate the tumor suppressing activity of AGGF1. An interesting aspect of AGGF1-mediated tumor suppression is its cell type-specific activity in tumor cells with the presence of p53, but not in cells without p53 expression. An unexpected finding of this study is that an angiogenic factor can function as a tumor suppressor, which may act as a braking system to minimize the effects of angiogenic factors in the stroma to stimulate tumor growth. As angiogenic factor AGGF1 is currently being developed into therapies to treat human diseases such as CAD and MI, cardiac hypertrophy and heart failure, ischemia-reperfusion, peripheral vascular disease, and restenosis after vascular injury [23,24,37,39,42], the finding that AGGF1 acts as a tumor suppressor when overexpressed in tumor cells may minimize some concerns regarding stimulation of tumor growth by angiogenic factors, which may be advantageous for AGGF1 during evolution.
Overall, our study identifies a novel tumor suppressor for AGGF1 which works through a p53 signaling axis to suppress tumor formation. Furthermore, this work provides important insights into how the pro-growth pathways of angiogenic factors on tumor growth may be balanced by control of other pathways that suppress tumor formation and growth.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Key resources table
| REAGENT | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| mouse IgG | Santa Cruz Biotechnology | sc-2025 |
| rabbit IgG | Santa Cruz Biotechnology | sc-2027 |
| HA-tag | MBL | 561 |
| Myc-tag | MBL | M047–3 |
| FLAG-tag | Proteintech | 66008–2-Ig |
| GFP-tag | MBL | 598 |
| p53 (DO-1) | Santa Cruz Biotechnology | sc-126 |
| p21 | Epitomics | 3733–1 |
| Bax (D2E11) | Cell Signaling Technology | 5023S |
| Gapdh | ABMART | M20006 |
| p53 phospho pAbs (S46) | Cell Signaling Technology | 2521P |
| p53 phospho pAbs (S15) | Cell Signaling Technology | 9284P |
| p53 phospho pAbs (S20) | Cell Signaling Technology | 9287P |
| p53 phospho pAbs (S392) | Cell Signaling Technology | 9281P |
| p53 acetyl pAbs (K382) | Epitomics | 2485-S |
| p53 acetyl pAbs (K373) | Epitomics | 2204-S |
| p53 acetyl pAbs (K305) | Epitomics | 3308–1 |
| goat anti-rabbit HRP conjugated secondary antibody | Thermo | ZB-2301 |
| goat anti-mouse HRP conjugated secondary antibody | Thermo | ZB-2305 |
| Critical Commercial Assays | ||
| EZ-CHIP™ | Millipore | 17–371 |
| BD Pharmingen™ Cell Cycle Kit | BD biosciences | 558662 |
| BD Pharmingen™ FITC Annexin V | BD biosciences | 556420 |
| Cell-Light™ EdU Apollo®567 In Vitro Imaging Kit | Ribobio | C10310–1 |
| SYBR Green PCR master mix | Roche | 4913914001 |
| experimental models: cell lines | ||
| HCT116 | China Center for Type CultureCollection | N/A |
| NCI–H1299 | China Center for Type CultureCollection | N/A |
| B16–F0 | China Center for Type CultureCollection | N/A |
|
experimental models: mouse
model
| ||
| BALB/C-nunu athymic mouse
Experimental animal center of Wuhan university C57BL/6 mouse
Experimental animal center of Wuhan university | ||
|
Primers for cloning
plasmids
| ||
| pCMV-C-HA- AGGF1 |
F:GCGCGCAGATCTATGGCCTCGGAGGCGCCGTCC R:GCGCGCGGATCCCTCTAAAGTCCCTTTTACCCAAGG |
|
| pCMV-C-HA- AGGF1-N1 |
F:GCGCGCAGATCTGAGCCTGAGCTGGCCCAG R:GCGCGCGGATCCCTCTAAAGTCCCTTTTACCCAAGG |
|
| pCMV-C-HA- AGGF1-N2 |
F:GCGCGCAGATCTAATAAAAAGTCTGATGTAG R:GCGCGCGGATCCCTCTAAAGTCCCTTTTACCCAAGG |
|
| pCMV-C-HA- AGGF1-N3 |
F:GCGCGCAGATCTGCAGAAGCGGCTGTATCACAGA R:GCGCGCGGATCCCTCTAAAGTCCCTTTTACCCAAGG |
|
| pCMV-C-HA- AGGF1-N4 |
F:GCGCGCAGATCTGATTTGCAACCTTATCCGACTTC R:GCGCGCGGATCCCTCTAAAGTCCCTTTTACCCAAGG |
|
| pCMV-C-HA AGGF1-N5 |
F:GCGCGCAGATCTATTGGAAGAGAAAAGGATATGG R:GCGCGCGGATCCCTCTAAAGTCCCTTTTACCCAAGG |
|
| pCMV-C-HA- AGGF1-N6 |
F:GCGCGCAGATCTGAAACTGTCTTATCCTTTCACATTC R:GCGCGCGGATCCCTCTAAAGTCCCTTTTACCCAAGG |
|
| pCMV-C-HA- AGGF1-N7 |
F:GCGCGCAGATCTAGCAACAAAGGTCGGAAGATGT R:GCGCGCGGATCCCTCTAAAGTCCCTTTTACCCAAGG |
|
| pCMV-C-HA AGGF1-C1 |
F:GCGCGCAGATCTATGGCCTCGGAGGCGCCGTCC R:GCGCGCGGATCCGCCTGTCCCCAAGCCTGCATG |
|
| pCMV-C-HA AGGF1-C2 |
F:A
GCGCGCAGATCTTGGCCTCGGAGGCGCCGTCC R:GCGCGCGGATCCATCAGTAATTTCAGAATGAAC |
|
| pCMV-C-HA AGGF1-C3 |
F:GCGCGCAGATCTATGGCCTCGGAGGCGCCGTCC R:GCGCGCGGATCCTCCAATTTTGACTTCATCTCCA |
|
| pCMV-C-HA AGGF1-C4 |
F:GCGCGCAGATCTATGGCCTCGGAGGCGCCGTCC R:GCGCGCGGATCCTGTAGCAGGGTTTACAGCAGTAA |
|
| pCMV-C-HA AGGF1-C5 |
F:GCGCGCAGATCTATGGCCTCGGAGGCGCCGTCC R:GCGCGCGGATCCTACTCGAGAATGAAACTGATAAC |
|
| pCMV-C-HA AGGF1-C6 |
F:GCGCGCAGATCTATGGCCTCGGAGGCGCCGTCC R:GCGCGCGGATCCTGCAGCTCTCAAACTTTCAG |
|
|
Primers for
qPCR
| ||
| p21 (NM_078467) | F:CGATGGAACTTCGACTTTGTCA R:GCACAAGGGTACAAGACAGTG |
|
| p53 (NM_001126118) | F:GAGGTTGGCTCTGACTGTACC R:TCCGTCCCAGTAGATTACCAC |
|
| Bax (NM_138763) | F:CCCGAGAGGTCTTTTTCCGAG R:CCAGCCCATGATGGTTCTGAT |
|
| Puma (NM_001127240) | F:GACCTCAACGCACAGTACGAG R:AGGAGTCCCATGATGAGATTGT |
|
| Noxa (NM_021127) | F:ACCAAGCCGGATTTGCGATT R:ACTTGCACTTGTTCCTCGTGG |
|
| Xpc (NM_004628) | F:CTTCGGAGGGCGATGAAAC R:TTGAGAGGTAGTAGGTGTCCAC |
|
| c-Fos (NM_005252) | F:CACTCCAAGCGGAGACAGAC R:AGGTCATCAGGGATCTTGCAG |
|
| Gadd45 (NM_001199741) | F:GAGAGCAGAAGACCGAAAGGA R:CAGTGATCGTGCGCTGACT |
|
| DRAM1 (NM_018370) | F:CGTCAGCCGCCTTCATTATCT R:TCCAAGCACTAAAGACACCAAG |
|
| MDM2 (NM_002392) | F:GGCAGGGGAGAGTGATACAGA R:GAAGCCAATTCTCACGAAGGG |
|
| APAF1 (NM_001160) | F:GTCACCATACATGGAATGGCA R:CTGATCCAACCGTGTGCAAA |
|
| RRM2B (NM_001172478) | F:ATTGGGCCTTGCGATGGATA R:GAGTCCTGGCATAAGACCTCT |
|
| AGGF1 (NM_018046) | F:TGGTCCAACACTAAGTAAGGAGG R:CCCTACGTTTTCCAGCTCTATCT |
|
| GAPDH (NM_001256799) | F:GGAGCGAGATCCCTCCAAAAT R:GGCTGTTGTCATACTTCTCATGG |
|
| ACTB (NM_001256799) | F:CATGTACGTTGCTATCCAGGC R:CTCCTTAATGTCACGCACGAT |
|
|
Primers for point
mutation
| ||
| p.E402* | F:ATTAAGACAAAATTTGGCCCCCATGTA R:AAATTTTGTCTTAATCTTCATCCTCACT |
|
| p.R566* | F:ATGAGTAAAATATGGTTTACAGAATACAG R:CCATATTTTACTCATATTTTCTTTAATTC |
|
| p.E616* | F:TTAAATTACTGATAGCAACAAAGGTCGG R:TGCTATCAGTAATTTAAGAATGAACAGAT |
|
| p.G437E | F:TGTAAACCCTGCTACAATTGAAAGAGAA R:ATATCCTTTTCTCTTTCAATTGTAGCA |
|
| p.R447Q | F:AGGATATGGAACATACTCTCCAAATCCCTGA R:GACACCAACTTCAGGGATTTGGAGAGTATG |
|
| p.Q467H | F:ATTTTGACCATGACTTACACAGTTATGT R:CAAGGACATAACTGTGTAAGTCATGGT |
|
| p.Y469 N | F:GTAATGTCCTTGTGGATCAAGGCAGTCA R:GATCCACAAGGACATTACTTTGTAAGTC |
|
| p.N483T | F:CTGGAAAACAGATTCTTCAGCCGAAA R:TGAAGAATCTGTTTTCCAGTAACAATTG |
|
| p.V498I | F:CTTACATACTTGAGCATGGAGATGAAG R:CATGCTCAAGTATGTAAGGGTCACAT |
|
|
Primers for CHIP-qPCR
analysis
| ||
| P21 PROMOTER (p53 binding site and non-specific site) |
P21–P53–F:CGATGGAACTTCGACTTTGTCA P21–P53-R:GCACAAGGGTACAAGACAGTG P21-NS-F:GAGTCCTGTTTGCTTCTGGGCA P21-NS-R:CTGCATTGGGGCTGCCTATGTA |
|
| BAX PROMOTER (p53 binding site and non-specific site) |
BAX-P53-F:AGCGTTCCCCTAGCCTCTTT BAX-P53-R:GCTGGGCCTGTATCCTACATTCT BAX–NS–F:GCGGGCGCCTATAGTCTCAGCT BAX–NS–R:ACGAGAAAGCCACCCCCATGA |
|
| PUMA PROMOTER (p53 binding site and non-specific site) |
PUMA-P53-F:TGTCCATGGTGTGGATTTGCG PUMA-P53-R:AGACACCGGGACAGTCGGACA PUMA–NS–F:GGGAGATTTCACGTGAGATATAG PUMA–NS–R:ACTCACCTTCCAGTGCCTAGTGTG |
|
| NOXA PROMOTER (p53 binding site and non-specific site) |
NOXA-P53-F:CAGCGTTTGCAGATGGTCAA NOXA-P53-R:CCCCGAAATTACTTCCTTACAAAA NOXA–NS–F:GCACTAGCCATTACACCCCGTCTC NOXA–NS–R:CCACCCTCCGCCCATACCT |
|
| GADD45 PROMOTER (p53 binding site and non-specific site) |
GADD45-P53-F:AGCGGAAGAGATCCCTGTGA GADD45-P53-R:CGGGAGGCAGGCAGATG GADD45-NS-F:GGCCCACATCTAAGACCAGCT GADD45-NS-R:CCTCATGTACTTTGGCAATAA |
|
| GAPDH PROMOTER | F:TACTAGCGGTTTTACGGGCG R:TCGAACAGGAGGAGCAGAGAGCGA |
|
Acknowledgements
We thank Mugen Liu and Zhiyong Ma for some of the plasmids and other members of Center for Human Genome Research for discussions and help.
Funding
This work was supported by the China National Natural Science Foundation Key grants (81630002 and 31430047) (C.X.) and Young Scholar grant (81902860) (W.S.), and Hubei Province Natural Science Program Innovative Team grant (2017CFA014) (C.X.)" should read "This work was supported by the China National Natural Science Foundation Key grants (81630002 and 31430047) (C.X.), Young Scholar grant (81902860) (W.S.), Hubei Province Natural Science Program Innovative Team grant (2017CFA014) (C.X.), and in part by an NIH/NCI Cancer Center Support Grant (P30CA008748) (B.W.).
Abbreviations
- KTS
Klippel-Trenannary syndrome
- RT-qPCR
Reverse transcription quantitative real-time PCR
- ChIp
Chromatin immunoprecipitation
- Co-IP
Co-Immunoprecipitation
- KO
mice knockout mice
- CAD
coronary artery disease
- MI
myocardial infarction
- TAC
transverse aortic constriction
Footnotes
Declaration of competing interest
The authors declare no competing interests.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.canlet.2020.10.014.
References
- [1].Akihiro Ito C-HL, Zhao Xuan, Saito Shin’ichi 1, Hamilton Maria H., Appella Ettore 1, Yao Tso-Pang 2, P300_CBP Mediated P53 Acetylation Is Commonly Induced by P53-Activating Agents and Inhibited by MDM2, European Molecular Biology Organization, 2001. [DOI] [PMC free article] [PubMed]
- [2].Cagatay T, Ozturk M, P53 mutation as a source of aberrant beta-catenin accumulation in cancer cells, Oncogene 21 (2002) 7971–7980. [DOI] [PubMed] [Google Scholar]
- [3].Chakrabarti S, Wu X, Yang Z, Wu L, Yong SL, Zhang C, Hu K, Wang QK, Chen Q, MOG1 rescues defective trafficking of Na(v)1.5 mutations in Brugada syndrome and sick sinus syndrome, Circ. Arrhythm. Electrophysiol 6 (2013) 392–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chen D, Li L, Tu X, Yin Z, Wang Q, Functional characterization of Klippel-Trenaunay syndrome gene AGGF1 identifies a novel angiogenic signaling pathway for specification of vein differentiation and angiogenesis during embryogenesis, Hum. Mol. Genet 22 (2013) 963–976. [DOI] [PubMed] [Google Scholar]
- [5].Cho SW, Xu J, Sun R, Mumbach MR, Carter AC, Chen YG, Yost KE, Kim J, He J, Nevins SA, Chin SF, Caldas C, Liu SJ, Horlbeck MA, Lim DA, Weissman JS, Curtis C, Chang HY, Promoter of lncRNA gene PVT1 is a tumor-suppressor DNA boundary element, Cell 173 (2018) 1398–1412 e1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Du W, Bautista JF, Yang H, ez-Sampedro A, You SA, Wang L, Kotagal P, Luders HO, Shi J, Cui J, Richerson GB, Wang QK, Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder, Nat. Genet 37 (2005) 733–738. [DOI] [PubMed] [Google Scholar]
- [7].Fan C, Chen Q, Wang QK, Functional role of transcriptional factor TBX5 in pre-mRNA splicing and holt-oram syndrome via association with SC35, J. Biol. Chem 284 (2009) 25653–25663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fan C, Duhagon MA, Oberti C, Chen S, Hiroi Y, Komuro I, Duhagon PI, Canessa R, Wang Q, Novel TBX5 mutations and molecular mechanism for Holt-Oram syndrome, J. Med. Genet 40 (2003) e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Fan C, Liu M, Wang Q, Functional analysis of TBX5 missense mutations associated with Holt-Oram syndrome, J. Biol. Chem 278 (2003) 8780–8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Fan C, Ouyang P, Timur AA, He P, You SA, Hu Y, Ke T, Driscoll DJ, Chen Q, Wang QK, Novel roles of GATA1 in regulation of angiogenic factor AGGF1 and endothelial cell function, J. Biol. Chem 284 (2009) 23331–23343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N, Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal, Sci. Signal 6 (2013) pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Giglia-Mari G, Sarasin A, TP53 mutations in human skin cancers, Hum. Mutat 21 (2003) 217–228. [DOI] [PubMed] [Google Scholar]
- [13].Han M, Zhao M, Cheng C, Huang Y, Han S, Li W, Tu X, Luo X, Yu X, Liu Y, Chen Q, Ren X, Wang QK, Ke T, Lamin A mutation impairs interaction with nucleoporin NUP155 and disrupts nucleocytoplasmic transport in atrial fibrillation, Hum. Mutat 40 (2019) 310–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hindupur SK, Colombi M, Fuhs SR, Matter MS, Guri Y, Adam K, Cornu M, Piscuoglio S, Ng CKY, Betz C, Liko D, Quagliata L, Moes S, Jenoe P, Terracciano LM, Heim MH, Hunter T, Hall MN, The protein histidine phosphatase LHPP is a tumour suppressor, Nature 555 (2018) 678–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hu FY, Wu C, Li Y, Xu K, Wang WJ, Cao H, Tian XL, AGGF1 is a novel anti-inflammatory factor associated with TNF-alpha-induced endothelial activation, Cell. Signal 25 (2013) 1645–1653. [DOI] [PubMed] [Google Scholar]
- [16].Huang Y, Wang C, Yao Y, Zuo X, Chen S, Xu C, Zhang H, Lu Q, Chang L, Wang F, Wang P, Zhang R, Hu Z, Song Q, Yang X, Li C, Li S, Zhao Y, Yang Q, Yin D, Wang X, Si W, Li X, Xiong X, Wang D, Huang Y, Luo C, Li J, Wang J, Chen J, Wang L, Wang L, Han M, Ye J, Chen F, Liu J, Liu Y, Wu G, Yang B, Cheng X, Liao Y, Wu Y, Ke T, Chen Q, Tu X, Elston R, Rao S, Yang Y, Xia Y, Wang QK, Molecular basis of gene-gene interaction: cyclic cross-regulation of gene expression and post-GWAS gene-gene interaction involved in atrial fibrillation, PLoS Genet 11 (2015), e1005393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Huang Y, Wang Z, Liu Y, Xiong H, Zhao Y, Wu L, Yuan C, Wang L, Hou Y, Yu G, Huang Z, Xu C, Chen Q, Wang QK, alphaB-crystallin interacts with Nav1.5 and regulates ubiquitination and internalization of cell surface Nav1.5, J. Biol. Chem 291 (2016) 11030–11041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kashiwada T, Fukuhara S, Terai K, Tanaka T, Wakayama Y, Ando K, Nakajima H, Fukui H, Yuge S, Saito Y, Gemma A, Mochizuki N, beta-Catenin-dependent transcription is central to Bmp-mediated formation of venous vessels, Development (Camb.) 142 (2015) 497–509. [DOI] [PubMed] [Google Scholar]
- [19].Keckesova Z, Donaher JL, De Cock J, Freinkman E, Lingrell S, Bachovchin DA, Bierie B, Tischler V, Noske A, Okondo MC, Reinhardt F, Thiru P, Golub TR, Vance JE, Weinberg RA, LACTB is a tumour suppressor that modulates lipid metabolism and cell state, Nature 543 (2017) 681–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Leroy B, Anderson M, Soussi T, TP53 mutations in human cancer: database reassessment and prospects for the next decade, Hum. Mutat 35 (2014) 672–688. [DOI] [PubMed] [Google Scholar]
- [21].Li L, Chen D, Li J, Wang X, Wang N, Xu C, Wang QK, Aggf1 acts at the top of the genetic regulatory hierarchy in specification of hemangioblasts in zebrafish, Blood 123 (2014) 501–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Li S, Xi Q, Zhang X, Yu D, Li L, Jiang Z, Chen Q, Wang QK, Traboulsi EI, Identification of a mutation in CNNM4 by whole exome sequencing in an Amish family and functional link between CNNM4 and IQCB1, Mol. Genet. Genom 293 (2018) 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lu Q, Yao Y, Hu Z, Hu C, Song Q, Ye J, Xu C, Wang AZ, Chen Q, Wang QK, Angiogenic factor AGGF1 activates autophagy with an essential role in therapeutic angiogenesis for heart disease, PLoS, Biol 14 (2016), e1002529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lu Q, Yao Y, Yao Y, Liu S, Huang Y, Lu S, Bai Y, Zhou B, Xu Y, Li L, Wang N, Wang L, Zhang J, Cheng X, Qin G, Ma W, Xu C, Tu X, Wang Q, Angiogenic factor AGGF1 promotes therapeutic angiogenesis in a mouse limb ischemia model, PloS One 7 (2012), e46998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Luo C, Pook E, Tang B, Zhang W, Li S, Leineweber K, Cheung SH, Chen Q, Bechem M, Hu JS, Laux V, Wang QK, Androgen inhibits key atherosclerotic processes by directly activating ADTRP transcription, Biochim. Biophys. Acta (BBA) - Mol. Basis Dis 1863 (2017) 2319–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Luo C, Wang F, Qin S, Chen Q, Wang Q, Coronary artery disease susceptibility gene ADTRP regulates cell cycle progression, proliferation and apoptosis by global gene expression regulation, Physiol. Genom 48 (2016) 554–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Luo CY, Wang F, Ren X, Ke T, Xu CQ, Tang B, Qin SB, Yao YF, Chen QY, Wang QK, Identification of a molecular signaling gene-gene regulatory network between GWAS susceptibility genes ADTRP and MIA3/TANGO1 for coronary artery disease, Bba-Mol Basis Dis 1863 (2017) 1640–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Major MB, Roberts BS, Berndt JD, Marine S, Anastas J, Chung N, Ferrer M, Yi X, Stoick-Cooper CL, von Haller PD, Kategaya L, Chien A, Angers S, MacCoss M, Cleary MA, Arthur WT, Moon RT, New regulators of Wnt/beta-catenin signaling revealed by integrative molecular screening, Sci. Signal 1 (2008) ra12. [DOI] [PubMed] [Google Scholar]
- [29].Kubbutat MH, Vousden KH, Ashcroft M, Regulation of p53 function and stability by phosphorylation, Mol. Cell Biol (1999). [DOI] [PMC free article] [PubMed]
- [30].Meek DW, Anderson CW, Posttranslational modification of p53: cooperative integrators of function, Cold Spring Harbor perspectives in biology 1 (2009), a000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ouyang P, Saarel E, Bai Y, Luo C, Lv Q, Xu Y, Wang F, Fan C, Younoszai A, Chen Q, Tu X, Wang QK, A de novo mutation in NKX2.5 associated with atrial septal defects, ventricular noncompaction, syncope and sudden death, Clin. Chim. Acta 412 (2011) 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Riley T, Sontag E, Chen P, Levine A, Transcriptional control of human p53-regulated genes, Nat. Rev. Mol. Cell Biol 9 (2008) 402–412. [DOI] [PubMed] [Google Scholar]
- [33].Tian XL, Kadaba R, You SA, Liu M, Timur AA, Yang L, Chen Q, Szafranski P, Rao S, Wu L, Housman DE, DiCorleto PE, Driscoll DJ, Borrow J, Wang Q, Identification of an angiogenic factor that when mutated causes susceptibility to Klippel-Trenaunay syndrome, Nature 427 (2004) 640–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wang Z, Yu G, Liu Y, Liu S, Aridor M, Huang Y, Hu Y, Wang L, Li S, Xiong H, Tang B, Li X, Cheng C, Chakrabarti S, Wang F, Wu Q, Karnik SS, Xu C, Chen Q, Wang QK, Small GTPases SAR1A and SAR1B regulate the trafficking of the cardiac sodium channel Nav1.5, Biochim. Biophys. Acta (BBA) - Mol. Basis Dis 1864 (2018) 3672–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wu L, Yong SL, Fan C, Ni Y, Yoo S, Zhang T, Zhang X, Obejero-Paz CA, Rho HJ, Ke T, Szafranski P, Jones SW, Chen Q, Wang QK, Identification of a new co-factor, MOG1, required for the full function of cardiac sodium channel Nav 1.5, J. Biol. Chem 283 (2008) 6968–6978. [DOI] [PubMed] [Google Scholar]
- [36].Xu Y, Zhou M, Wang J, Zhao Y, Li S, Zhou B, Su Z, Xu C, Xia Y, Qian H, Tu X, Xiao W, Chen X, Chen Q, Wang QK, Role of microRNA-27a in down-regulation of angiogenic factor AGGF1 under hypoxia associated with high-grade bladder urothelial carcinoma, Biochim. Biophys. Acta 1842 (2014) 712–725. [DOI] [PubMed] [Google Scholar]
- [37].Yao Y, Hu Z, Ye J, Hu C, Song Q, Da X, Yu Y, Li H, Xu C, Chen Q, Wang QK, Targeting AGGF1 (angiogenic factor with G patch and FHA domains 1) for blocking neointimal formation after vascular injury, J. Am. Heart Assoc 6 (2017), e005889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yao Y, Li H, Da X, He Z, Tang B, Li Y, Hu C, Xu C, Chen Q, Wang QK, SUMOylation of Vps34 by SUMO1 promotes phenotypic switching of vascular smooth muscle cells by activating autophagy in pulmonary arterial hypertension, Pulm Pharmacol Ther, 2019. [DOI] [PMC free article] [PubMed]
- [39].Yao Y, Lu Q, Hu Z, Yu Y, Chen Q, Wang QK, A non-canonical pathway regulates ER stress signaling and blocks ER stress-induced apoptosis and heart failure, Nat. Commun 8 (2017) 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yu G, Liu Y, Qin J, Wang Z, Hu Y, Wang F, Li Y, Chakrabarti S, Chen Q, Wang QK, Mechanistic insights into the interaction of the MOG1 protein with the cardiac sodium channel Nav1.5 clarify the molecular basis of Brugada syndrome, J. Biol. Chem 293 (2018) 18207–18217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yuan J, Luo K, Zhang L, Cheville JC, Lou Z, USP10 regulates p53 localization and stability by deubiquitinating p53, Cell 140 (2010) 384–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhang T, Yao Y, Wang J, Li Y, He P, Pasupuleti V, Hu Z, Jia X, Song Q, Tian X, Hu C, Chen Q, Wang QK, Haploinsufficiency of Klippel-Trenaunay syndrome gene Aggf1 inhibits developmental and pathological angiogenesis by inactivating PI3K and AKT and disrupts vascular integrity by activating VE-cadherin, Hum. Mol. Genet 25 (2016) 5094–5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhang X, Chen S, Yoo S, Chakrabarti S, Zhang T, Ke T, Oberti C, Yong SL, Fang F, Li L, de la FR, Wang L, Chen Q, Wang QK, Mutation in nuclear pore component NUP155 leads to atrial fibrillation and early sudden cardiac death, Cell 135 (2008) 1017–1027. [DOI] [PubMed] [Google Scholar]
- [44].Zhou B, Si W, Su Z, Deng W, Tu X, Wang Q, Transcriptional activation of the Prox 1 gene by HIF-1 alpha and HIF-2 alpha in response to hypoxia, FEBS Lett 587 (2013) 724–731. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.