

Abstract
Mass spectrometry (MS)-based proteomic profiling of whole proteome and protein posttranslational modifications (PTMs) is a powerful technology to measure the dynamics of proteome with high throughput and deep coverage. The reproducibility of quantification benefits not only from the fascinating developments in high performance liquid chromatography (LC) and high resolution MS with enhanced scan rates, but also from the invention of multiplexed isotopic labeling strategies, such as the tandem mass tags (TMT). In this chapter, we introduce a 16-plex TMT-LC/LC-MS/MS protocol for proteomic profiling of biological and clinical samples. The protocol includes protein extraction, enzymatic digestion, PTM peptide enrichment, TMT labeling, two-dimensional reverse-phase liquid chromatography fractionation coupled with tandem mass spectrometry (MS/MS) analysis, followed by computational data processing. In general, more than 10,000 proteins and tens of thousands of PTM sites (e.g. phosphorylation and ubiquitination) can be confidently quantified. This protocol provides a general protein measurement tool, enabling the dissection of protein dysregulation in any biological samples and human diseases.
Keywords: Mass spectrometry, Proteomics, Proteome, Posttranslational modifications, Phosphorylation, Ubiquitination, Ubiquitin, Tandem mass tag, Liquid chromatography, Database
1. Introduction
Mass spectrometry has become a versatile tool for analyzing proteins from small-scale assays to genome-wide studies, including protein identification, quantification, posttranslational modifications, protein interaction networks, and systems biology of cells and tissues (1-4). Although the top-down approach of directly analyzing intact proteins is dramatically improved (5), the bottom-up approach of analyzing proteolytic peptides is a sensitive and robust method with high proteome coverage (1,6). During last few years, implementation of enhanced two-dimensional liquid chromatography and high resolution mass spectrometry have dramatically improved the bottom-up proteomics platform, allowing proteome analysis at a scale comparable to transcriptomics analysis (7,8). Deep posttranslational modifications (PTM) analysis (e.g., phosphoproteome and ubiquitinome) of mammalian tissues has also been accomplished (9,10) (see also Chapters 8, 13 – 14, 16). Maturation of bottom-up proteomics enables a wide range of applications to profile disease tissues, such as cancer (11-14) and Alzheimer’s disease (AD) (15,16), to gain fundamental insights into molecular pathogenesis for novel therapeutic strategies.
In addition to protein identification, a number of MS-based techniques have been developed for quantitative untargeted proteomics. Label-free quantification can be achieved by data-dependent acquisition (DDA) methods (see Chapters 8 – 10, 13 – 17, 20 – 21, 26), including the comparison of spectral counts (17) or signal intensities of precursor ions (18), as well as the data-independent acquisition (DIA) method (19) (see Chapters 8, 16, 22 - 24), such as SWATH-MS-based comparison of signal intensities of product ions (20). The label-free method is straightforward, affordable, and capable of generating reproducible data with one-dimensional LC-MS/MS, especially when sample processing is highly automated (21), however, the method variation increases with two-dimensional LC fractionation of unlabeled peptides. To overcome the limitation, stable isotope labeling methods have been introduced to differentially label proteins or digested peptides (2), followed by pooling prior to LC fractionation to reduce experimental variation. The chemical labeling of proteins/peptides can occur in vivo by Stable Isotope Labeling with Amino acids in Cell culture (SILAC) (2) (see Chapters 8, 13, 18), or in vitro by tagging proteolytic peptides (see Chapters 8 – 10, 12, 14 – 15, 17, 25 – 26, 28), using TMT (22), Isobaric Tags for Relative and Absolute Quantitation (iTRAQ) (23), and DiLeu labeling methods (24). SILAC metabolic labeling process, however, can also introduce experimental variation and cannot be readily applied to clinical specimens. Instead, the TMT isobaric labeling method has emerged as a popular quantitative proteomics strategy (25) and is under continuous active development.
Like the existing TMT10/11-plex reagents, the newly released 16-plex TMT (TMTpro) reagents consist of a mass reporter, a mass balance group and a reactive group (26) (Figure 1). The digested peptides from different samples are differentially labeled with the TMT reagents, mixed and then processed as a single sample for LC/LC-MS/MS analysis. The same peptides in different samples that carry different isobaric tags are chemically identical and non-distinguishable during LC separation and display the same peaks in the precursor ion scans by mass spectrometry. Importantly, during the fragmentation of precursor ions (e.g. MS2 or MS3), the reporter ions of these isobaric tags are generated to show different mass and their intensities are used for quantification. This minimizes missing values in the same batch of up to 16 samples, and allows precise quantification due to reduced experimental variation (27). A well-known caveat of the TMT method is that the noise levels due to co-eluted interfering ions often lead to ratio compression, underestimating the protein difference, particularly in complex protein samples (28). The distorted ratios can be rescued by multiple strategies including pre-MS extensive fractionation (29), MS settings with small MS2 isolation window (29) or gas-phase isolation (30), or post-MS corrections by subtracting interference (29,31). In addition, complement reporter ion clusters can be accurately quantified during MS analysis (32), while multistage MS3-based technique can almost eliminate the effect of ratio compression (28).
Fig. 1.
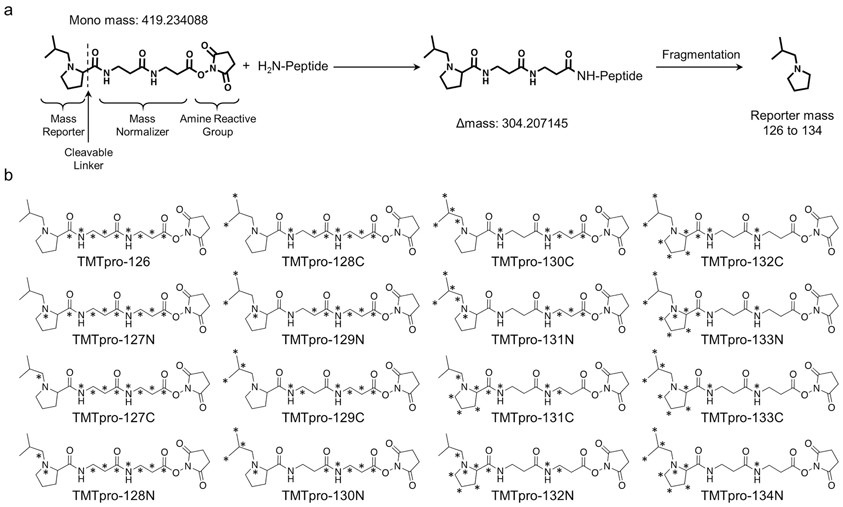
Structure of the 16-plex TMT reagent.
(a) Unlabeled structure of the 16-plex TMT reagent, the mass of the reagent, mass shift after labeling, and the mass of the reporter ion are shown.
(b) Heavy isotope labeled structures of the TMT16 reagents.
In this chapter, we introduce a detailed 16-plex TMT (TMTpro) method for profiling the whole proteome and the ubiquitinome, as an example of PTM analysis. Similar to other PTM profiling, ubiquitinome profiling often involves peptide-level enrichment by using an antibody recognizing the ubiquitin remnant motif di-GG tag on lysine residues of substrates after tryptic digestion (K-ε-GG motif) (10,33,34), enabling measurements of over 10,000 ubiquitination sites in mammalian cells and tissues (35,36). The TMT-LC/LC-MS/MS method includes (in order) protein extraction from cells and tissues, protein digestion, peptide desalting, PTM enrichment, 16-plex TMT labeling, basic pH reverse-phase (RP) LC fractionation, and acidic pH RPLC-MS/MS (Figure 2). Additionally, the computational data processing is discussed in detail for large-scale analysis (Figure 3).
Fig. 2.
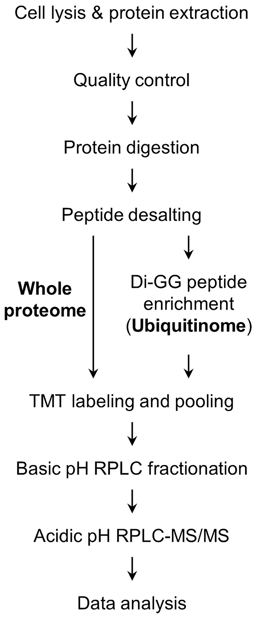
Experimental design of TMT-LC/LC-MS/MS for profiling the whole proteome and ubiquitinome.
Fig. 3.
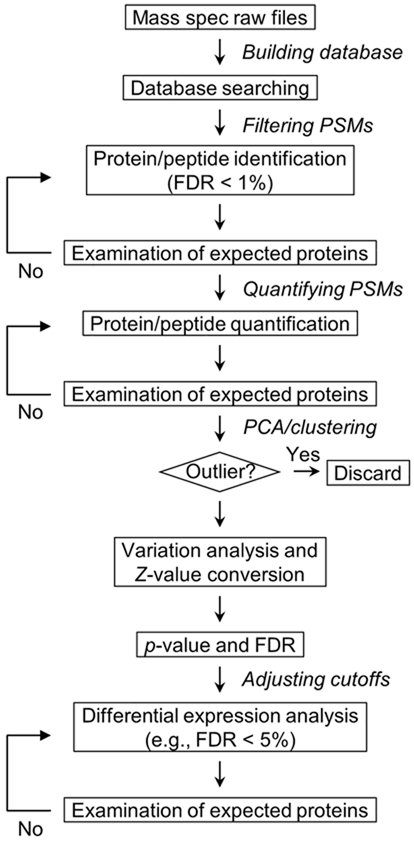
Data processing strategies for large-scale proteomic analysis.
2. Materials
2.1. Protein Extraction from Cells and Tissues
Human postmortem brain tissues of normal control or Alzheimer’s disease: frontal cortex with a well-characterized pathology record, stored at −80 °C. The samples are used as an example experiment in this method. Other tissues or cells may be processed similarly.
Lysis buffer: 8 M urea (see Note 1), 50 mM [4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid] (HEPES), pH 8.5, 0.5% sodium deoxycholate, 1 × PhosSTOP phosphatase inhibitor cocktail (Roche), and 1 mM dithiothreitol (DTT). For ubiquitinome analysis, DTT is replaced by 50 μM (final) PR-619 ((2,6-diamino-5-thiocyanatopyridin-3-yl) thiocyanate) and 100mM (final) iodoacetamide (IAA) (see Note 2).
0.5 mm diameter glass beads.
Bullet Blender (Next Advance).
Protein quantification standard: 20 mg/mL bovine serum albumin (BSA).
BCA Protein Assay Kit (Thermo Fisher Scientific).
NuPAGE Novex 4–12% Bis-Tris Gel (Life Technologies).
GelCode Blue Stain (Thermo Fisher Scientific).
2.2. Protein In-Solution Digestion
Digestion enzymes: Lys-C (FUJIFILM Wako) and trypsin (Promega).
Dilution buffer: 50 mM HEPES, pH 8.5.
Peptide disulfide bond reduction: 1 M DTT in 50 mM HEPES.
Peptide cysteine alkylation: 1 M IAA in 50 mM HEPES (see Note 3).
Acetonitrile (ACN, HPLC grade).
2.3. Peptide Desalting
Desalting columns: 500 mg Sep-Pak C18 desalting cartridge (Waters, ~1 mL of bed volume), 200 mg Sep-Pak C18 desalting cartridge (Waters, ~400 μL of bed volume), Ultra MicroSpin C18 columns (The Nest Group, 25 μL of bed volume), C18 StageTips (Thermo Fisher Scientific) (see Note 4).
Vacuum manifold.
Trifluroacetic acid (TFA).
Methanol (HPLC grade).
Equilibration and wash buffer: 0.1% TFA.
Elution buffer: 60% ACN, 0.1% TFA.
SpeedVac concentrator.
2.4. Enrichment of di-GG Peptides and di-GG Peptides Depletion Test
PTMScan® Ubiquitin Remnant Motif (K-ε-GG) antibody (Cell Signaling Technology).
Immuno-affinity purification (IAP) buffer: 50 mM [3-(N-morpholino)propanesulfonic acid] (MOPS), pH 7.2, 10 mM sodium phosphate and 50 mM NaCl.
Wash buffer: Phosphate-buffered saline (PBS).
Elution buffer: 0.15% TFA.
C18 StageTips (Thermo Fisher Scientific).
1% formic acid.
40% ACN.
5% formic acid.
2.5. TMT16 Labeling of Peptides and Desalting
Peptide labeling buffer: 50 mM HEPES, pH 8.5.
16-plex TMT Isobaric Mass Tagging Kit (Thermo Fisher Scientific).
Anhydrous ACN.
Quenching solution: 5% hydroxylamine.
Desalting columns: 500 mg Sep-Pak C18 desalting cartridge (Waters, ~1 mL of bed volume), 200 mg Sep-Pak C18 desalting cartridge (Waters, ~400 μL of bed volume), C18 StageTips (Thermo Fisher Scientific).
Methanol (HPLC grade).
Equilibration and wash buffer: 5% ACN, 0.1% TFA (see Note 5).
Elution buffer: 60% ACN, 0.1% TFA.
2.6. Offline Basic pH Reverse-Phase Liquid Chromatography Fractionation
Waters XBridge C18 column, 3.5 μm particle size, 4.6 mm × 25 cm or 2.1 mm × 15 cm (see Note 6).
Buffer A: 10 mM ammonium formate, adjust pH to 8.0 by 28% ammonium hydroxide.
Buffer B: 10 mM ammonium formate, 90% ACN, adjust pH to 8.0 by 28% ammonium hydroxide.
Wash solution: 25% isopropanol, 25% methanol, 25% ACN and 25% water.
High-performance liquid chromatography (HPLC) system (e.g. Agilent 1220 Infinity LC System).
Fraction collector (e.g. Gilson FC 203B).
2.7. Acidic pH RPLC-MS/MS Analysis
New Objective empty column: 75 μm I.D. × 50 cm, 15 μm tip orifice.
1.9 μm particle size C18 resin (Dr. Maisch GmbH Germany).
Butterfly portfolio column heater.
HPLC system (e.g. Waters ACQUITY UPLC or Dionex Ultimate 3000 RSLCnano System).
Tandem MS instrument (e.g. Thermo Q Exactive HF or Fusion).
Sample loading solution: 5% formic acid.
Sample vials with 0.2 mL bottom-spring polypropylene inserts.
Buffer A: 5% dimethyl sulfoxide and 0.2% formic acid.
Buffer B: 65% ACN, 5% dimethyl sulfoxide and 0.2% formic acid.
2.8. MS Data Analysis
Database downloaded from the UniProt website (https://www.uniprot.org/downloads).
Proteomics software suite for data processing: JUMP software suite (29,37,38) or Proteome Discover (Thermo Scientific) (see also Chapter 28).
A computer cluster for data processing.
3. Methods
3.1. Protein Extraction from Cells and Tissues
3.1.1. Whole Proteome
Cut the frozen brain tissue (~10 mg) from a defined brain region (e.g. prefrontal cortex) on a metal plate on dry ice (see Note 7). Weigh the tissue and transfer immediately to a pre-cooled Eppendorf tube on dry ice followed by the addition of glass beads (~20% of the volume of lysate).
Add freshly prepared lysis buffer (10 μL per mg tissue) into the tube. Generally, 1 mg tissue yields 50-100 μg total proteins. The final protein concentration is 5-10 μg/μL. For whole proteome analysis, usually 100 μg protein per sample is sufficient.
Seal the tube caps with parafilm and put the tubes in a Bullet Blender in 4 °C cold room. Lyse the tissues under speed 8 with the cycle time of 30 sec on and 5 sec off until the sample is homogenized (~5 cycles).
Make aliquots for the lysates (see Note 8). Small aliquots (e.g. 10 μL) can be used for the analysis of protein concentration and the evaluation of protein quality (i.e. Western blot validation of positive control proteins) (see Note 9). Large aliquots (e.g. 50 μL) are for whole proteome analysis. Freeze the aliquots immediately on dry ice and store at −80 °C until further use.
Measure the protein concentrations by the BCA assay (protocol provided by the manufacturer). Considering that the BCA assay may be affected by non-protein reducing components in the tissue lysate, we often confirm the protein concentration by a short SDS gel staining method (39), in which a serial dilution of standard protein is used for the establishment of a standard curve (e.g., BSA titrations of 0.1, 0.3, 1, 3 and 10 μg).
3.1.2. Ubiquitinome
Due to the nature of low abundance / stoichiometry of ubiquitination on proteins and high activity of deubiquitinating enzymes (DUBs) in cells, lysis protocol is modified to limit DUB activities and maximize recovery of ubiquitinated proteins/peptides (40,41).
Lyse ~40 mg brain tissue per sample with DUB inhibitors (e.g. PR-619 and IAA) as described in section 3.1.1. Dissolve lyophilized Lys-C in lysis buffer (1 μg/μL) and keep at 4 °C for use in next step.
After making small aliquots (e.g., 10 μL) for protein quality control analysis, add 10 μg Lys-C to the remaining tissue lysates (at least 2 mg protein per sample) immediately and incubate at room temperature. The rapid digestion eliminates the impact of the DUB activities.
Determine the protein amount in the cell lysates as described in section 3.1.1 using the small aliquots. Then add additional Lys-C as well as 10% ACN to the lysates at a final protein/Lys-C ratio of 100:1 (w/w) and incubate for 3 h at room temperature. Store the digested lysates at −80 °C.
3.2. Protein In-Solution Digestion
For whole proteome samples, add Lys-C into each lysate at a protein/Lys-C ratio of 100:1 (w/w). Then, add ACN to a final concentration of 10%. Incubate at room temperature for 3 h (42). For ubiquitinome samples, directly go to the next step.
Dilute the lysates with 50 mM HEPES (pH 8.5) to lower the urea concentration to 2M. Add trypsin into the diluted lysates at a protein/trypsin ratio of 50:1 (w/w) and incubate at room temperature overnight.
For whole proteome samples, reduce the digested peptides with 1 mM DTT for 2 h and alkylate cysteine residues with 10 mM IAA for 30 min in the dark. Quench the alkylation by 30 mM DTT and incubating at room temperature for another 30 min. For ubiquitinome samples, directly quench the alkylation by 30 mM DTT.
Terminate the digestion by adding TFA to 0.5% (v/v). Check the pH of the mixture to ensure the pH is lower than 3.
3.3. Peptide Desalting
To limit the loss of peptides during the desalting process, we choose C18 desalting cartridges or spin columns with binding capacity matched with the amount of peptides to be desalted (see Note 4).
3.3.1. Whole Proteome
Centrifuge the tryptic peptides at 21,000 × g for 10 min to remove any insoluble materials.
Wash Ultra MicroSpin C18 desalting columns with 5 × bed volumes of elution buffer (60% ACN, 0.1% TFA) three times by spinning at 500 × g for 30 sec.
Equilibrate the columns with 5 × bed volumes of equilibration and wash buffer (0.1% TFA) three times by spinning at 500 × g for 30 sec.
Transfer the supernatant of samples to the C18 desalting columns and spin at 100 × g for 3 min.
Wash the columns three times with 5 × bed volumes of equilibration and wash buffer (0.1% TFA) by spinning at 500 × g for 30 sec.
Elute peptides with 3 × bed volumes of elution buffer (60% ACN, 0.1% TFA) by spinning at 100 × g for 3 min.
Dry the eluted peptides in SpeedVac and store the peptides at −80 °C for further TMT labeling. (see Note 11).
3.3.2. Ubiquitinome
Centrifuge the tryptic peptides at 21,000 × g for 10 min to remove insoluble materials.
Condition 500 mg Sep-Pak C18 desalting cartridges on vacuum manifold by washing the cartridges with 5 × bed volumes of methanol twice, 5 × bed volumes of elution buffer (60% ACN, 0.1% TFA) twice and 5 × bed volumes of equilibration and wash buffer (0.1% TFA) twice.
Transfer the supernatant of samples to the C18 cartridges. Adjust the vacuum pressure of manifold to let samples flow through the cartridges at the slowest speed (dropwise). Keep the flow through until the desalting is done successfully. Wash the cartridge at least three times with 5 × bed volumes of equilibration and wash buffer (0.1% TFA).
Elute peptides with 3 × bed volumes of elution buffer (60% ACN, 0.1% TFA).
Dry the eluted peptides in SpeedVac and store the peptides at −80 °C for di-GG peptides enrichment. (see Note 11).
3.4. Enrichment of di-GG Peptides
This section is only for ubiquitinome analysis. Skip this section and move to section 3.5 for whole proteome analysis (see Note 10).
Dissolve each of the sixteen desalted peptide samples in 1 mL of ice-cold IAP buffer by vortexing and adjust pH to 7.2 if necessary (see Note 11).
Centrifuge at 21,000 × g for 10 min to remove insoluble materials. Aliquot 1 μL as input for testing the efficiency of di-GG peptide enrichment (see Note 12).
Tap the tube of ubiquitin remnant motif (K-ε-GG) antibody to mix the beads well and transfer 40 μL of slurry into an Eppendorf tube (see Note 13).
Wash the slurry three times with 1 mL cold IAP buffer and settle the beads down by spinning at 2,000 × g for 1 min and remove the supernatant.
Transfer peptide solution into the first tube that contains antibody slurry and put the mixture on a 360-degree rotator and rotate for 2 h at 4 °C.
Centrifuge at 2,000 × g for 2 min to remove supernatant and keep the supernatant at −80 °C as flow-through for the depletion test (see Note 12).
Spin the remaining beads again at 2,000 × g for 2 min to remove residual solution.
Wash the beads twice with 1 mL cold IAP buffer, thrice with 1 mL cold PBS by inverting the tubes six times and then remove supernatant after centrifuging at 2,000 × g for 2 min.
Add 50 μL of elution buffer into beads and gently tap the bottom of the tube to mix the solution with beads. Remain at room temperature for 5 min (see Note 14).
Centrifuge at 2,000 × g to settle the beads and transfer the eluent to another tube.
Repeat steps 9-10 and combine both eluents.
Desalt input and flow-through by C18 StageTips (see section 3.3.1) and the eluents are analyzed by LC/MS-MS for di-GG peptide depletion test (see Note 12).
Based on the depletion test result, if there is still K48 chain and K63 chain in the flow-through, a second round of enrichment may be necessary. In general, we find that ~240 μg of antibody is enough to deplete K48 chain peptide in 4 mg of total peptides from cell lysate. However, it is necessary to perform an individual pilot depletion test for each sample.
3.5. TMTpro (TMT16) Labeling of Peptides and Desalting
3.5.1. Whole proteome
Resuspend the desalted peptides with 50 mM HEPES, pH 8.5, examine the pH by pH paper and take ~1 μg unlabeled peptides for TMT labeling efficiency test.
Dissolve the TMT16 reagents in anhydrous acetonitrile (10 μg/μL) immediately before use and make aliquots if necessary.
For the labeling of the whole proteome sample, add TMT16 reagents to each sample at a reagent/substrate ratio of 1.5:1 (w/w) and incubate at room temperature for 30 min. Take ~1 μg labeled peptides for TMT labeling efficiency test and keep the remaining peptides at −80 °C without quenching the reaction.
Perform the labeling efficiency test. Desalt all the aliquots of labeled and unlabeled samples with C18 StageTips (see section 3.3.1) followed by analyzing the labeled and unlabeled samples using LC-MS/MS (see Note 15). If the TMT labeling is complete, the unlabeled peptide peaks should not be seen in the labeled samples.
If the labeling is completed, quench the reaction by adding the quenching solution (5% hydroxylamine) and incubate for 15 min at room temperature. If not, additional TMT reagents can be used to further label the peptide samples.
Take half of each TMT labeled sample and pool all the 16 samples to make a mixture “mix1”. For whole proteome samples, take ~1 μg pooled peptides for LC-MS/MS analysis after desalting. Calculate and compare the average intensity of each TMT16 reporter ion. If the discrepancies of intensity among samples are larger than 5%, adjust the intensity by adding individual remaining TMT labeled samples into “mix1” according to the calculated average intensity to make a “mix2”. Repeat the adjustment until all samples are mixed equally.
Desalt the pooled TMT16 labeled peptides (see section 3.3.2). Note that the wash buffer (5% ACN, 0.1% TFA) for TMT16 labeled peptides contains a higher level of ACN than the regular wash buffer (0.1% TFA) for unlabeled peptides, because the 5% ACN is required to remove the hydrophobic derivatives of quenched TMT16 reagents.
3.5.2. Ubiquitinome
As global ubiquitinome varies dramatically under different stress conditions such as heat shock, proteasome inhibition, etc., the normalization of sample loading bias cannot be performed the same as for the whole proteome analysis. Here we only adjust the mixing amount for the replicates in the 16-plex samples to reduce experimental variations that occur in sample preparation steps for the ubiquitinome samples.
Add 800 μg TMT16 reagent to the di-GG peptides that are enriched from 4 mg peptides and incubate at room temperature for 30 min. Take 1% unlabeled and labeled di-GG peptides for TMT labeling efficiency test. In the meantime, keep the remaining peptides at −80 °C.
For TMT16 mixing ratio adjustment, check the discrepancies of intensity only for the replicate samples and adjust mixing volume as described in section 3.5.1.
3.6. Offline Basic pH RPLC Fractionation
Prepare a Waters XBridge C18 column (3.5 μm particle size, 4.6 mm × 25 cm).
Dissolve the mixture of TMT16 labeled peptides with basic pH RPLC buffer A (10 mM ammonium formate, pH 8.0).
Centrifuge the peptides at 21,000 × g for 10 min to remove precipitates.
Wash the LC sample loop with methanol, water and buffer A sequentially using 3 × loop volume.
Inject 100 μL of wash solution (25% isopropanol, 25% methanol, 25% ACN and 25% water) to clean the column and then equilibrate the column with 95% buffer A for 1 h with a flow rate of 0.4 mL/min.
The whole proteome samples are fractionated using the following gradient: 5% buffer B for 10 min, 5-15% buffer B for 2 min, 15–45% buffer B for 148 min and 45–95% buffer B for 5 min, at the flow rate of 0.4 mL/min. Collect the fractions every 1 min by a fraction collector and concatenate to 40 fractions in 4 cycles.
All the fractions are dried in SpeedVac and stored at −80 °C for further LC-MS/MS analysis.
For ubiquitinome analysis, switch to a 2.1 mm × 15 cm C18 column and flow rate to 0.2 mL/min. The di-GG peptides are fractionated in a 40 min gradient with 15–45% buffer B. Collect the fractions every 1 min by a fraction collector and concatenate to 10 fractions in 4 cycles.
3.7. Acidic pH RPLC-MS/MS Analysis
Pack the LC-MS/MS columns with 1.9 μm C18 resin. The fractions of whole proteome are analyzed on 75 μm x 15 cm C18 columns and the fractions of ubiquitinome samples are analyzed on 75 μm x 50 cm C18 columns. Heat the column at 65 °C with a butterfly portfolio heater to reduce the backpressure.
Inject 100 ng of rat brain or BSA tryptic peptides to evaluate the performance of LC/MS system.
Dissolve the basic pH RPLC fractions in the sample loading solution (5% formic acid). Centrifuge the samples at 21,000 × g for 5 min and transfer 5 μL supernatant to inserts.
Whole proteome analysis: load ~1 μg for each fraction. Peptides are eluted in a 60 min gradient of 18–45% buffer B at 0.25 μL/min flow rate. The gradient range may be slightly adjusted according to specific samples and different LC-MS/MS settings. The mass spectrometer settings include MS1 scans (60,000 resolution, scan range 450–1600 m/z, 1 × 106 AGC and 50 ms maximal ion time) and 20 data-dependent MS2 scans (60,000 resolution, scan range starting from 120 m/z, 1 × 105 AGC, ~110 ms maximal ion time, 32% normalized collision energy (NCE), 1.0 m/z isolation window, 0.2 m/z isolation offset, exclude isotopes on, and 10 sec dynamic exclusion) (see Note 16).
For ubiquitinome analysis: reconstitute the dried di-GG peptides in 2 μL of 5% formic acid and load all 2 μL onto the column. Elute peptides by 18–45% buffer B in 150 min at ~0.125 μL/min flow rate. Operate mass spectrometer in data-dependent mode with a survey scan in Orbitrap (60,000 resolution, scan range 450–1600 m/z, 1 × 106 AGC target, ~50 ms maximal ion time) and 20 data-dependent MS2 scans (60,000 resolution, scan range starting from 120 m/z, 1 × 105 AGC target, ~110 ms maximal ion time, 32% NCE, 1.0 m/z isolation window, 0.2 m/z isolation offset, exclude isotopes on, and 10 sec dynamic exclusion).
3.8. MS Data Analysis
3.8.1. Database Search
The MS raw files are converted to mzXML files and searched against the human protein database using our in-house developed JUMP pipeline (29,37,38). The database is generated by combining Swiss-Prot, TrEMBL, and UCSC databases, removing redundancy (83,955 entries for human proteins) and adding customized protein sequences which are not contained in the reference databases, including but not limited to protease cleaved proteins, proteins with single nucleotide polymorphisms and all possible contaminants. Search parameters include 10 ppm mass tolerance for precursor ions and 15 ppm for fragment ions, full trypticity with two maximal missed cleavages, and three maximal modification sites. TMT16 modification on Lys residues and N termini (+304.20715 Da) and carbamidomethylation of Cys residues (+57.02146 Da) are used as static modifications, while Met oxidation (+15.99492 Da) is set as dynamic modification. For ubiquitinome analysis, the maximal missed cleavage sites are set to three and di-GG modification on Lys residues (+114.04293 Da) is set as an additional dynamic modification.
3.8.2. Peptide-Spectrum Match (PSM) Filtering
The resulting PSMs are first filtered by precursor ion mass accuracy, and then grouped by peptide length, tryptic ends, modifications, miscleavage sites, and precursor ion charge state. The data are further filtered with the JUMP-based matching scores (Jscore and ΔJn) to reduce false discovery rate (FDR) below 1% for either proteins (whole proteome analysis) or peptides (ubiquitinome analysis), based on the target-decoy strategy (43,44). The shared peptides from numerous homologous proteins are matched to the protein with the top PSM number according to the rule of parsimony. If some positive control proteins are missing at the filtering steps, the FDR may be reduced to 5% with manual examination to see if these proteins/peptides can be salvaged.
3.8.3. TMT-based Quantification
Proteins or peptides quantification is performed in the following steps based on our previous method (29): (i) Extract TMT reporter ion intensities of each PSM. (ii) Correct the raw intensities based on isotopic distribution of each labeling reagent (e.g. TMT16-126 generates 92.6%, 7.2%, and 0.2% of 126, 127C, and 128C m/z ions, respectively). (iii) Discard PSMs of very low intensities (e.g. minimum intensity of 1,000 and median intensity of 5,000). (iv) Minimize sample loading bias by normalization with the trimmed median intensity of all PSMs. (v) Calculate the mean-centered intensities across samples (e.g. relative intensities between each sample and the mean). (vi) Summarize protein relative intensities by averaging related PSMs. (vii) Finally derive protein absolute intensities by multiplying the relative intensities by the grand-mean of three most highly abundant PSMs. In addition, y1 ion-based correction is also used for TMT quantification analysis (29). The intensities of positive control proteins should be checked and matched to Western blot (see also Chapter 3) results to see if there is any error of sample mislabeling.
In general, the analysis of quantification is carried out with data without missing values. However, PSMs with missing values in specific labeling channels may be examined under certain conditions (e.g. knockout of positive control proteins/genes in the samples). Our JUMP software generates a separate table containing all PSMs with the missing values for manual evaluation. We then evaluate the quality of samples by statistical methods such as PCA or clustering analysis to determine if there are any outliers in the samples or mislabeling of the samples. After removing all the outliers, protein fold change and p values of different comparisons are calculated based on the protein signal intensities. The log2(fold change) values are further transformed to z scores to reduce the impact of ratio compression (29), which is based on the calculated experimental standard deviation between replicated samples. Finally, the differential expression analysis is performed by applying different combinations of cutoff values (e.g., FDR (Benjamini-Hochberg corrected p) < 0.05, and z > 2). The cutoffs are often adjusted to include known positive proteins that are dysregulated in the samples.
Acknowledgements
This work was partially supported by the National Institutes of Health (R01GM114260, R01AG047928, R01AG053987, and RF1AG064909) and ALSAC (American Lebanese Syrian Associated Charities). The MS analysis was performed in the Center of Proteomics and Metabolomics at St. Jude Children’s Research Hospital, partially supported by NIH Cancer Center Support Grant (P30CA021765).
Footnotes
The solution form of urea is not stable during longtime storage and can decompose into ammonium cyanate. Ammonium cyanate reacts with Lys residues to induce protein carbamylation. Heating accelerates this process. Thus, lysis buffer should be prepared in fresh form or stored at −80 °C if necessary. All digestion steps are carried out at room temperature instead of 37 °C.
Some of the DUBs are still active in 8 M urea and can remove ubiquitin modifications within short period of time. Add DUBs inhibitors (PR-619 and IAA) in the lysis buffer just before tissue lysis if ubiquitinome analysis is performed. Also, adding Lys-C to cell lysate immediately after protein extraction can minimize the activities of DUBs. Skip these steps if only whole proteome analysis is performed.
IAA modification on lysine residues may introduce ubiquitination artifact mimics (45). This reaction does not occur at room temperature (46), and the pseudo GG peptides are not recognized by the K-ε-GG motif antibody (47). Thus, IAA is used in the protocol. Or use other IAA alternatives such as iodoacetic acid or 2-chloroacetamide instead in ubiquitinome analysis (48).
Selecting appropriate cartridges or columns can improve the recovery of peptides during desalting and fractionation. In general, the binding capacity of C18 resin is about 10 μg peptides per 1 μL of cartridges bed volume. The loading capacity of a C18 LC column is around 3 μg of peptides per 1 μL of column bed volume (42).
Byproducts in TMT reaction (side reaction products with H2O and quenching reaction product with hydroxylamine) also bind to C18 resins and affect subsequent LC/LC-MS/MS analysis. These byproducts can be removed by extensively washing the desalting C18 cartridges with wash buffer (5% ACN and 0.1% TFA, 10 × bed volumes).
Use different offline basic pH LC columns for the whole proteome and the ubiquitinome analysis based on the loading amount of peptides (42).
To reduce the variations caused by heterogeneity of brains tissues, the collected tissue samples can be pre-pulverized and mixed thoroughly before tissue lysis. Meanwhile, it is essential to include at least two replicates for each biological condition in the TMT design for statistical analysis.
Do not centrifuge the lysate at high speed to remove the insoluble proteins. Digestion of these insoluble proteins with trypsin can increase the number of identified proteins (49).
Quality control (QC) steps (i.e. Western blot of positive and negative control proteins) before TMT labeling is necessary to avoid wasting of reagents and time on low-quality samples. QC results can also be used to identify potential sample mislabeling during sample processing.
The enrichment of ubiquitinated peptides should be performed before TMT labeling because the di-GG peptides are not recognized by Ubiquitin remnant motif K-ε-GG antibodies after TMT labeling. For other PTM analysis, such as phosphorylation and methylation, the enrichment is often performed after TMT labeling to eliminate experimental variation during the enrichment.
Desalted peptides should be dried completely to remove trace amount of TFA. Residual TFA may reduce pH lower than 7 when the peptides are dissolved in the IAP buffer, which may affect the interaction between di-GG peptides and antibody. It is important to confirm the pH value before the enrichment.
It is critical to evaluate the efficiency of antibody-based enrichment of di-GG peptides. K48 and K63 chains consist of a substantial portion of total ubiquitinome. Comparison of K48 and K63 chain intensity between input and flow-through by MS can help estimate the enrichment efficiency of ubiquitinome.
Among the two commercially available K-GG monoclonal antibodies, the antibody from Lucerna was generated against GG-modified histone, the one from Cell Signaling Technology was produced against the sequence CXXXXXXKGGXXXXXX (X = any amino acid except Cys and Trp). Both antibodies gave similar and partially overlapping results in a comparative study, but the antibody from Lucerna led to ~30% less coverage of the ubiquitinome. Thus, we describe the use of the antibody from Cell Signaling Technology in this chapter. The antibody can distinguish K-ε-GG peptides from M1-GG peptides (linear peptide modified on the N-terminal amine group) (47). In addition to the antibody-based enrichment, ubiquitinome may be analyzed through ubiquitinated protein purification using a range of affinity reagents (50,51), but the purification is typically complicated by co-purified unmodified proteins (52).
The K-GG antibody is non-covalently attached to protein A beads. Thus, the antibody is co-eluted with di-GG peptides and may affect the LC-MS/MS analysis. During TMT labeling process, the antibody is labeled with TMT reagent and become highly hydrophobic. The TMT labeled antibody is removed by desalting, as it binds to C18 resin and is not eluted. The alternative way to limit the antibody’s impact on LC-MS/MS is to crosslink the antibody to beads with dimethyl pimelimidate (DMP) prior to enrichment.
When performing the labeling efficiency test, TMT labeled samples should be analyzed before unlabeled samples to reduce the impact of LC carryover on the calculation of labeling efficiency.
The MS parameters are optimized for TMT16 labeled peptides based on our previous settings for TMT11 labeled peptides (8). Compared to TMT11, TMT 16 has an increased mass, and therefore MS1 scan range starts from a higher m/z (450 instead of 410). The MS2 ion time (110 ms) matches the time required to obtain MS scans at the 60,000 resolution on a Q Exactive HF MS (53). The NCE is optimized to balance peptide fragment ion intensity and TMT reporter ion intensity. A lower collision energy (32%) is used in TMT16 experiments and 35% in TMT11 studies (26). In addition, we use a relatively short dynamic exclusion time (10 s) because for extensively fractionated samples (40 fractions), the MS time is sufficient to scan all detected precursor ions. During the LC gradient, the peak width is around 20 s, so 10 sec exclusion allows at least two MS2 scans for each precursor ion: one at the beginning of the peak and the other at the center point of the peak to improve the MS2 signal.
References
- 1.Zhang Y, Fonslow BR, Shan B et al. (2013) Protein analysis by shotgun/bottom-up proteomics. Chem Rev 113(4):2343–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aebersold R, Mann M (2016) Mass-spectrometric exploration of proteome structure and function. Nature 537(7620):347–355. [DOI] [PubMed] [Google Scholar]
- 3.Huttlin EL, Bruckner RJ, Paulo JA et al. (2017) Architecture of the human interactome defines protein communities and disease networks. Nature 545(7655):505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu J, Peng J, Chi H (2019) Systems immunology: Integrating multi-omics data to infer regulatory networks and hidden drivers of immunity. Curr Opin Sys Biol 15:19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toby TK, Fornelli L, Kelleher NL (2016) Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu Rev Anal Chem (Palo Alto Calif) 9(1):499–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peng J, Gygi SP (2001) Proteomics: the move to mixtures. J Mass Spectrom 36(10):1083–1091. [DOI] [PubMed] [Google Scholar]
- 7.Wang H, Yang Y, Li Y et al. (2015) Systematic optimization of long gradient chromatography mass spectrometry for deep analysis of brain proteome. J Proteome Res 14(2):829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bai B, Tan H, Pagala VR et al. (2017) Deep Profiling of Proteome and Phosphoproteome by Isobaric Labeling, Extensive Liquid Chromatography, and Mass Spectrometry. Methods Enzymol 585:377–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huttlin EL, Jedrychowski MP, Elias JE et al. (2010) A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143(7):1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim W, Bennett EJ, Huttlin EL et al. (2011) Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell 44(2):325–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mertins P, Mani DR, Ruggles KV et al. (2016) Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534(7605):55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vasaikar S, Huang C, Wang X et al. (2019) Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 177(4):1035–1049 e1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stewart E, McEvoy J, Wang H et al. (2018) Identification of Therapeutic Targets in Rhabdomyosarcoma through Integrated Genomic, Epigenomic, and Proteomic Analyses. Cancer Cell 34(3):411–426 e419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang H, Diaz AK, Shaw TI et al. (2019) Deep multiomics profiling of brain tumors identifies signaling networks downstream of cancer driver genes. Nat Commun 10(1):3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bai B, Hales CM, Chen PC et al. (2013) U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer's disease. Proc Natl Acad Sci U S A 110(41):16562–16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bai B, Wang X, Li Y et al. (2020) Deep Multilayer Brain Proteomics Identifies Molecular Networks in Alzheimer’s Disease Progression. Neuron [Epub ahead of print]:online 8 January 2020. [Google Scholar]
- 17.Liu H, Sadygov RG, Yates JR 3rd (2004) A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem 76(14):4193–4201. [DOI] [PubMed] [Google Scholar]
- 18.Cox J, Hein MY, Luber CA et al. (2014) Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13(9):2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Venable JD, Dong MQ, Wohlschlegel J et al. (2004) Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat Methods 1(1):39–45. [DOI] [PubMed] [Google Scholar]
- 20.Ludwig C, Gillet L, Rosenberger G et al. (2018) Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial. Mol Syst Biol 14(8):e8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bache N, Geyer PE, Bekker-Jensen DB et al. (2018) A Novel LC System Embeds Analytes in Pre-formed Gradients for Rapid, Ultra-robust Proteomics. Mol Cell Proteomics 17(11):2284–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thompson A, Schafer J, Kuhn K et al. (2003) Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem 75(8):1895–1904. [DOI] [PubMed] [Google Scholar]
- 23.Ross PL, Huang YN, Marchese JN et al. (2004) Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics 3(12):1154–1169. [DOI] [PubMed] [Google Scholar]
- 24.Frost DC, Greer T, Li L (2015) High-resolution enabled 12-plex DiLeu isobaric tags for quantitative proteomics. Anal Chem 87(3):1646–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rauniyar N, Yates JR 3rd (2014) Isobaric labeling-based relative quantification in shotgun proteomics. J Proteome Res 13(12):5293–5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thompson A, Wolmer N, Koncarevic S et al. (2019) TMTpro: Design, Synthesis, and Initial Evaluation of a Proline-Based Isobaric 16-Plex Tandem Mass Tag Reagent Set. Anal Chem 91(24):15941–15950. [DOI] [PubMed] [Google Scholar]
- 27.Hogrebe A, von Stechow L, Bekker-Jensen DB et al. (2018) Benchmarking common quantification strategies for large-scale phosphoproteomics. Nat Commun 9(1):1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ting L, Rad R, Gygi SP et al. (2011) MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat Methods 8(11):937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niu M, Cho JH, Kodali K et al. (2017) Extensive Peptide Fractionation and y1 Ion-Based Interference Detection Method for Enabling Accurate Quantification by Isobaric Labeling and Mass Spectrometry. Anal Chem 89(5):2956–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wenger CD, Lee MV, Hebert AS et al. (2011) Gas-phase purification enables accurate, multiplexed proteome quantification with isobaric tagging. Nat Methods 8(11):933–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Savitski MM, Mathieson T, Zinn N et al. (2013) Measuring and managing ratio compression for accurate iTRAQ/TMT quantification. J Proteome Res 12(8):3586–3598. [DOI] [PubMed] [Google Scholar]
- 32.Wuhr M, Haas W, McAlister GC et al. (2012) Accurate multiplexed proteomics at the MS2 level using the complement reporter ion cluster. Anal Chem 84(21):9214–9221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peng J, Schwartz D, Elias JE et al. (2003) A proteomics approach to understanding protein ubiquitination. Nat Biotechnol 21(8):921–926. [DOI] [PubMed] [Google Scholar]
- 34.Udeshi ND, Mani DR, Eisenhaure T et al. (2012) Methods for Quantification of in vivo Changes in Protein Ubiquitination following Proteasome and Deubiquitinase Inhibition. Mol Cell Proteomics 11 (5):148–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Udeshi ND, Svinkina T, Mertins P et al. (2013) Refined preparation and use of anti-diglycine remnant (K-epsilon-GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol Cell Proteomics 12(3):825–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rose CM, Isasa M, Ordureau A et al. (2016) Highly Multiplexed Quantitative Mass Spectrometry Analysis of Ubiquitylomes. Cell Syst 3(4):395–403 e394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang X, Li Y, Wu Z et al. (2014) JUMP: a tag-based database search tool for peptide identification with high sensitivity and accuracy. Mol Cell Proteomics 13(12):3663–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Y, Wang X, Cho JH et al. (2016) JUMPg: An Integrative Proteogenomics Pipeline Identifying Unannotated Proteins in Human Brain and Cancer Cells. J Proteome Res 15(7):2309–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu P, Duong DM, Peng JM (2009) Systematical Optimization of Reverse-Phase Chromatography for Shotgun Proteomics. J Proteome Res 8(8):3944–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peng J, Cheng D (2005) Proteomic analysis of ubiquitin conjugates in yeast. Methods Enzymol 399:367–381. [DOI] [PubMed] [Google Scholar]
- 41.Na CH, Jones DR, Yang Y et al. (2012) Synaptic protein ubiquitination in rat brain revealed by antibody-based ubiquitome analysis. J Proteome Res 11(9):4722–4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pagala VR, High AA, Wang X et al. (2015) Quantitative protein analysis by mass spectrometry. Methods Mol Biol 1278:281–305. [DOI] [PubMed] [Google Scholar]
- 43.Peng J, Elias JE, Thoreen CC et al. (2003) Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J Proteome Res 2(1):43–50. [DOI] [PubMed] [Google Scholar]
- 44.Elias JE, Gygi SP (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods 4(3):207–214. [DOI] [PubMed] [Google Scholar]
- 45.Nielsen ML, Vermeulen M, Bonaldi T et al. (2008) Iodoacetamide-induced artifact mimics ubiquitination in mass spectrometry. Nat Methods 5(6):459–460. [DOI] [PubMed] [Google Scholar]
- 46.Xu P, Duong DM, Seyfried NT et al. (2009) Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 137(1):133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bustos D, Bakalarski CE, Yang Y et al. (2012) Characterizing ubiquitination sites by peptide based immunoaffinity enrichment. Mol Cell Proteomics 11(12):1529–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen PC, Na CH, Peng J (2012) Quantitative proteomics to decipher ubiquitin signaling. Amino Acids 43(3):1049–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pirmoradian M, Budamgunta H, Chingin K et al. (2013) Rapid and deep human proteome analysis by single-dimension shotgun proteomics. Mol Cell Proteomics 12(11):3330–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu P, Peng J (2006) Dissecting the ubiquitin pathway by mass spectrometry. Biochim Biophys Acta 1764(12):1940–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao Y, Li Y, Zhang C et al. (2016) Enhanced Purification of Ubiquitinated Proteins by Engineered Tandem Hybrid Ubiquitin-binding Domains (ThUBDs). Mol Cell Proteomics 15(4):1381–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seyfried NT, Xu P, Duong DM et al. (2008) Systematic approach for validating the ubiquitinated proteome. Anal Chem 80(11):4161–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kelstrup CD, Jersie-Christensen RR, Batth TS et al. (2014) Rapid and Deep Proteomes by Faster Sequencing on a Benchtop Quadrupole Ultra-High-Field Orbitrap Mass Spectrometer. Journal of Proteome Research 13(12):6187–6195. [DOI] [PubMed] [Google Scholar]