

Abstract
Laboratory mice are a crucial preclinical model system for investigating BMAd-bone and BMAd-hematopoiesis interactions. In this review, we evaluate the suitability of mice to model common human diseases related to osteopenia or hematopoietic disorders, pointing out consistencies and discrepancies among different studies, and provide insights into model selection. Species, age, sex, skeletal site, and treatment protocol should all be considered when designing future studies.
Keywords: bone marrow adipose tissue, bone marrow adipocyte, preclinical models, bone, hematopoiesis
Bone marrow adipocytes (BMAd) are a unique cell population existing within the bone marrow cavity. Although these cells were discovered over a century ago, they were largely neglected until recent decades. BMAds are no longer considered a mere space filler of the bone marrow but are now recognized as a heterogeneous cellular population that make up a distinct adipose depot and interact with surrounding bone and hematopoietic cells [1]. Due to the anatomically closed bone system and limited approaches to specifically manipulate genes or metabolism in BMAds, the regulatory mechanisms and physiological functions of BMAds remain incompletely understood. Current experimental approaches targeting BMAds also modulate adipocytes elsewhere in the body or alter gene expression in osteoblasts and marrow stromal cells. These limitations create difficulties in interpreting studies designed to investigate interactions between BMAds and bone or hematopoietic cells. In addition, the inherent differences between species may limit translation of findings from certain animal models to human physiology and pathology. Herein we summarize the most-frequently used animal models for investigating BMAd-bone and BMAd-hematopoiesis interactions, compare results from these models with human studies, and make recommendations for the most reliable animal models for preclinical studies. Discrepancies between studies often have contributions from differences in sex, age, time-course of treatment, and animal strain. These caveats are important to take into consideration when selecting a preclinical model.
Imaging methods used for in vivo and ex vivo marrow adiposity evaluation
Before we discuss the physiology of BMAds, however, a brief discussion of current methods for visualizing and quantifying bone marrow adipose tissue (BMAT) in humans and mice is warranted. BMAT can be evaluated non-invasively using magnetic resonance imaging (MRI) and computed tomography (CT)-based imaging technologies [2]. MRI-based quantitative techniques, such as water-fat imaging and 1H-MRS have advantages in assessing BMAT content and composition without ionizing radiation. Water-fat imaging is especially useful in red marrow, where BMAds are scattered with hematopoietic cells, such as in pelvis, vertebrae, ribs and proximal femurs. In addition to BMAT quantification, 1H-MRS can also assess lipid composition, such as the amount of unsaturated and saturated lipids [3]. However, MRI is less informative than CT for assessment of trabecular bone features [4]. Conventional CT integrates the density of calcified tissue with marrow components, and increased marrow fat influences the accuracy of bone mineral density (BMD) determination [5]. Dual-energy CT can be used to assess BMAT content and BMD in a single examination and provides BMAT data that closely correlate with 1H-MRS [6]. Suchacki et al. recently developed a method for visualization and quantification of human BMAT from CT data. To do so, they compared paired MRI and CT scans to identify the optimal Hounsfield Units (HU) thresholds to distinguish BMAT from red marrow and bone. With this novel method, they quantified site- and age-dependent differences in red marrow and BMAT in humans [7].
For isolated mouse bones, MRI has been used to overview the marrow fat content [8], whereas μCT is a more quantitative and accurate approach to evaluate BMAT volume in bones stained with osmium tetroxide after decalcification [9]. Moreover, Scheller et al. established nanoCT as a more precise way to quantify BMAd number and size in proximal tibia where isolated BMAds are found interspersed within regions of active hematopoiesis [10]. Of note, one of the pitfalls of osmium tetroxide staining is that it interacts with unsaturated bonds of fatty acids, which means that it endows the electron density to lipid droplets depending on lipid composition [11]. With biopsy or autopsy bone samples from patients or mice, hematoxylin and eosin (H&E) stained histological sections are a convenient, less expensive way to evaluate bone marrow hematopoietic cellularity and adiposity. Tratwal et al. developed a semi-automated digital pathology plugin named MarrowQuant, which quantifies the areas occupied by bone, BMAds, hematopoietic cells, and the interstitial/microvascular compartment. This software also provides BMAd area and size distribution for individual adipocytes [12]. Using these methods allows for quantification of BMAds in both in vivo and ex vivo studies.
Comparison of bone marrow adiposity between humans and mice
In humans, accumulation of BMAds to replace red hematopoietic marrow begins in terminal phalanges of feet and hands just prior to birth. Shortly after birth, further expansion of BMAT in long bones occurs distally to proximally and is more prevalent at distal sites. BMAT is readily detected in distal epiphyses of long bones by age seven and in the midshaft around 12–14 years old [13]. The adult distribution is first observed at about 25 years, with BMAT occupying approximately 50%–70% of the total bone marrow cavity, with at least some BMAds also present in vertebrae, sternum, ribs and pelvis [9, 13]. In lumbar vertebrae, marrow adiposity expands with age varying from 27% to 70% in individuals between 8 and 57 years of age, respectively [14]. Accumulation of BMAT with age is also influenced by sex. Female subjects younger than 55 years have approximately 6–10% lower marrow fat than age-matched males [15], whilst the sharp increase of BMAT in postmenopausal women drives about 10% higher marrow fat content than males at ages over 60 years [16]. Expansion of total BMAT volume with age is due to increased BMAd numbers and size - diameters of BMAd are ~65 mm at 82–96 years of age, compared to ~48 mm at ages of 16–29 years in Caucasians [17].
Mice are widely used in preclinical studies with advantages being their metabolic similarity to humans, shorter time of development, and ease of genetic modification. Although mice have lower bone marrow adiposity than humans, the timing and sequence in marrow BMAd development are similar. For instance, ~20% and ~60% of tibial BM cavity is filled with BMAT in 12 weeks and 56 weeks old C3H mice respectively [10]. Of note, one of the distinct differences between humans and mice is the tail. Caudal vertebrae of mice develop BMAds as early as one week after birth and are nearly fully developed at four weeks of age (Figure 1). BMAds may also be found in sacrum and lower lumbar vertebral bodies in adults, but it is rare to see BMAds in cervical and thoracic vertebrae. Only a few BMAds are visible in distal tibia one week after birth. Distal tibial BMAds are readily detectable at four weeks and continue to accumulate until the cavity is filled at around eight weeks (Figure 1) [10]. BMAT in femur and proximal tibia appears later than caudal vertebrae and distal tibia, but is detectable at four weeks or even earlier depending on sex and strain. Female mice usually develop BMAT earlier and more-extensively in proximal tibia than age-matched males [10, 18]. C57BL/6J mice have been widely used in metabolic research; however, their long bones are characterized by low trabecular and cortical bone densities, and fewer BMAds. In contrast, C3H/HeJ mice have both higher bone density and more marrow adiposity than C57BL/6J mice [10].
Figure 1. Postnatal development of bone marrow adipocytes (BMAds) in mice.
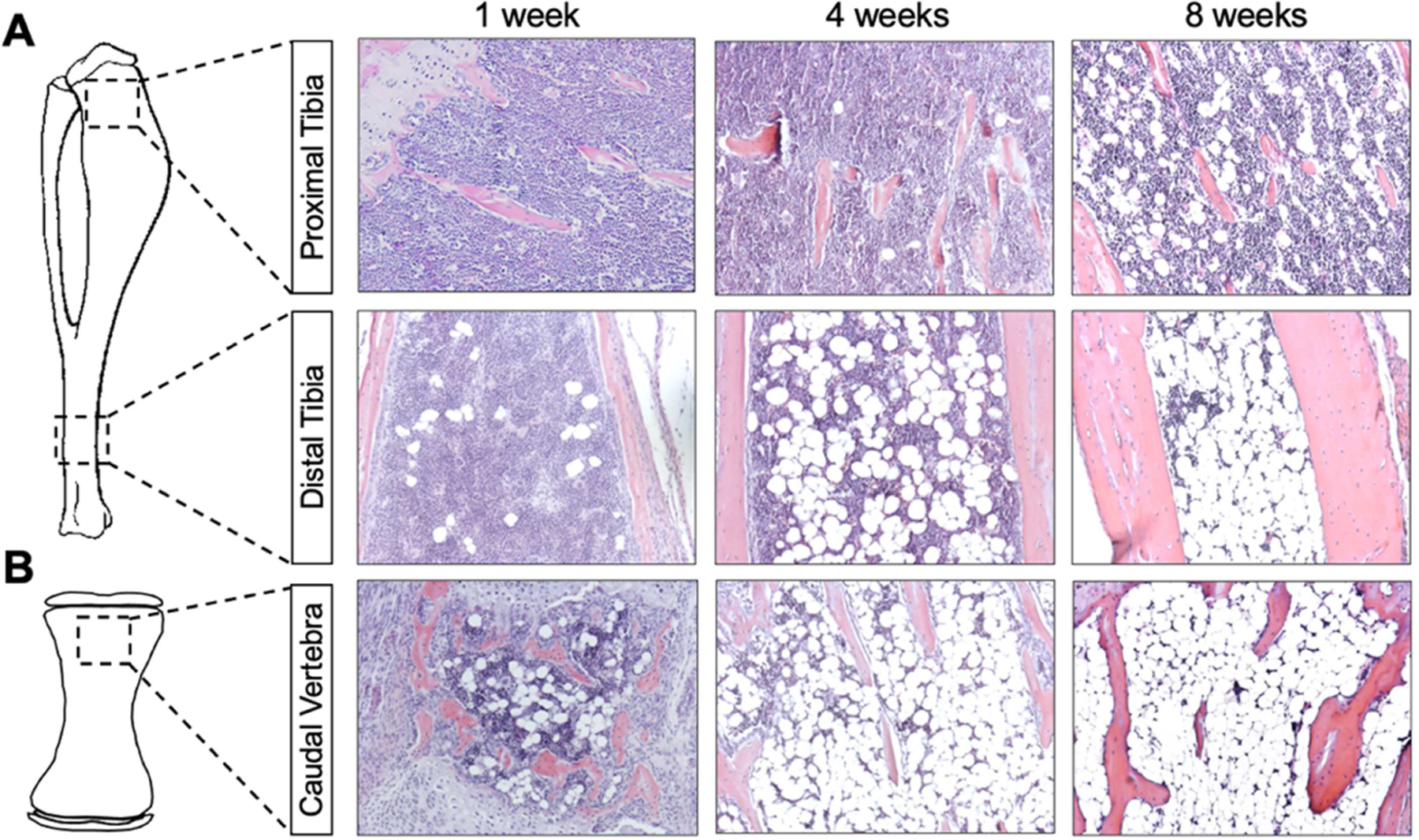
Mouse long bones (A) and 3rd caudal vertebrae (B) were collected at 1, 4 and 8 weeks after birth. Bones were decalcified with 14% EDTA for 3 weeks and used for paraffin sectioning. Slides were stained with hematoxylin (nuclei) and eosin (cytoplasm). Photomicrographs were taken under 200x magnification.
As first characterized by Tavassoli [19] and extended by our group, BMAds can be broadly classified into two groups. BMAds that are interspersed in hematopoietic cells are labile or regulated (rBMAds), and can be lost in response to phenylhydrazine-induced hemolysis, fasting, lactation, bariatric surgery, granulocyte colony-stimulating factor (G-CSF), and chronic central leptin administration. This population is readily expanded in response to aging, obesity, caloric restriction, ovariectomy, irradiation and thiazolidinedione treatment. In contrast, stable or constitutive BMAds (cBMAds), which appear early in life, are densely packed and have an appearance indistinguishable from white adipose tissue (WAT). As suggested by their nomenclature, cBMAds are largely not influenced by the variables above, although recent work has shown loss of cBMAds with bariatric surgery and G-CSF administration [10, 19]. Because rBMAds exist proximally to commonly analyzed bone regions and can be altered by myriad physological and phathological conditions, most preclinical models have focused on the changes in rBMAds, which are found in proximal tibia, distal femur and axial skeleton.
Animal models for investigation of BMAT functions in bone homeostasis
The physical location of BMAds adjacent to osteoblasts/osteoclasts has led many to speculate that BMAT influences development of bone diseases. The tenet of BMAd-bone interaction is supported by many clinical studies that marrow fat content is inversely related to trabecular BMD, microstructure variables and ex vivo failure load [20]. Although this relationship has been observed in a wide variety of animal models, there remains a lack of direct evidence for a causal relationship.
Age is a complex factor for interactions between BMAds and bone.
Distinct from the continual increase of BMAT in humans throughout life, BMD and bone mineral content (BMC) peak around 30 years of age [16, 21] and then decline with age. Therefore, BMAT expansion occurs concurrently with bone mass accrual in children and young individuals [22] but expansion is associated with bone loss after 30 years of age. It is important to note, however, that cross-sectional studies demonstrate an inverse relationship between BMD and BMAT at all ages [23–26]. In rodents, rBMAT increases until at least 56 weeks of age [10]. How bone variables are influenced by age is dependent on site and type of bone. For example, cortical bone generally reaches maximum mineral density and thickness at 24 weeks of age, whereas age of maximal trabecular bone number and volume varies between six and 24 weeks, depending on strain and location [27, 28]. Trabecular bone is thus not an ideal choice for age-associated bone studies. Of note, most BMAd-bone interaction studies were performed when mice were younger than 24 weeks, a time in which both BMAT and bone are still developing.
Overall, the development of BMAT throughout life and loss of bone mass with aging are largely consistent between humans and rodents. Although BMAT amount adversely associates with bone mass at all stages of life, whether age also plays an independent role is an open question. To address this, Almeida et al. deleted PPARγ in BMAds to decrease baseline BMAT, and block the increase in BMAT that normally occurs with aging. However, even in the absence of BMAT, these mice still had age-related trabecular and cortical bone loss [29]. A caveat to this study is that the mouse model used also deleted PPARγ in some stromal cells and osteoblasts. Further mechanistic research on how age drives BMAT expansion is required to understand the relationship to bone loss. Age is an important variable to consider in experimental design or data interpretation.
High fat diet (HFD)-induced obesity poorly replicate clinical observations in bone.
Human obesity increases adiposity, not only of subcutaneous and visceral depots, but also of bone marrow. Although the myriad side-effects of obesity on cardiovascular diseases, glucose and lipid disorders have been extensively studied, effects on bone are still debated. Quality of bone should ideally be determined by geometry, cortical thickness and porosity, trabecular bone morphology, and intrinsic properties of bony tissue; however, quality in clinical practice is usually estimated indirectly by BMD. Overweight/obese patients generally have higher BMD than lean individuals [30] due to heavier mechanical loading and lower bone turnover [31]. However, fracture risk differs across skeletal sites, with significant interaction between BMI and BMD.
Is it possible for mice to model the complexity between obesity and bone biology observed in humans? Induction of obesity with HFD is commonly used for investigation of metabolic syndrome, and also for effects on BMAT expansion and bone metabolism. Although BMAT expansion varies from 12 to 16 weeks, it has been consistently observed in different strains. However, effects of obesity on bone is complex with considerations including age of HFD feeding, types of diet, mouse strain and sex. For example, HFD feeding for 12 weeks induces BMAT expansion and loss of trabecular bone with milder effects on cortical bones in C57BL/6J and FVB mice initially of ~8 weeks of age [32–34]. In contrast, if started at ~11 weeks of age, HFD feeding increases bone mass of C57BL/6J, LG/J and SM/J mice [35, 36]. To complicate matters further, feeding C57BL/6J with HFD for 12 weeks from 3 weeks of age causes no changes to trabecular and cortical bone [37]. Overall, these discrepancies between different studies suggest that investigators should be cautious about using obesity as a preclinical model to study effects of BMAT accumulation on bone metabolism. Further standardization of HFD feeding models with respect to age, diet, and strain is warranted, and may improve our ability to model complex interactions between marrow adiposity and bone.
Genetic models of obesity have also been used to study relationships between BMAd and osteopenia. For example, leptin-deficient ob/ob mice have a marked increase in BMAd number. Although femora are shorter with less bone mass, trabecular bone of vertebra is increased [38]. Leptin administration restores femoral length, BMD and BMC in ob/ob mice, suggesting that leptin is critical for bone development [39]. Because leptin receptor is expressed and functional in bone marrow stromal cells, BMAds, osteoblasts and chondrocytes, the reasons underlying osteopenia in ob/ob mice are unclear. Therefore, care should be taken when considering genetic models to investigate interactions between BMAds and bone cells.
Caloric restriction of young male mice mimics the consequences on bone of anorexia nervosa.
In the 1970s, it was first noted that anorexia nervosa patients had BM hypoplasia with expansion and a change in character of BMAT [40]. This observation was built on in the 1980s with multiple groups finding that these patients also have osteoporosis/osteopenia [41, 42]. Moreover, increased marrow adiposity and bone loss are associated with elevated cortisol and other hormonal changes [43]. Clinical work in anorexia nervosa patients noted an inverse relationship between marrow fat content and BMD [44]. Anorexia nervosa is a disease in which restriction of calories is a symptom of a psychological disorder. Interestingly, caloric restriction (CR) is thought to have health benefits including reduced adiposity, improved metabolism and increased lifespan; however, a downside includes effects on bone. In non-obese younger adults randomly assigned to 25% CR or ad libitum for two years, CR significantly decreased BMD at lumbar spine, total hip, femoral neck and other regional hip sites at 12 and 24 months [45]. The CR group also had increases in cortisol and adiponectin, but lower levels of circulating leptin and physical activity [45]. Obese humans over 65 who voluntarily underwent weight loss generally had reduced BMD in total hip and femoral neck, but with no change or higher BMD in L1-L4 [46].
Effect of CR on bone and marrow niche of humans is effectively modeled by CR in young male rodents [8, 18]. Expansion of BMAT with CR has been consistently observed across age, sex and durations from 6 to 19 weeks, reduction of bone mass is not uniformly observed. CR of male mice consistently causes loss of bone when they are actively growing through three weeks to about three months of age [8, 47]; however, there are few effects on bone in the 13–17 month range, and CR actually increases bone mass of male mice at two years of age [48]. In contrast, female mice have much milder bone changes despite the significant changes in BMAT after CR. Although CR causes bone loss in female mice in very young actively-growing mice, effects on bone gradually diminish with age [49, 50]. It is possible that estrogen contributes to differences between sexes. In both sexes, changes in body composition and hormones are shared between mouse CR and human anorexia nervosa, including lower adipose mass and leptin, and higher cortisol levels. Despite the fact that the incidence of anorexia nervosa is much higher in women, use of CR in female mice may not be a useful model for studying bone loss. However, it may be that mechanistic insights can be gained through use of younger male mice where bone loss with CR is observed.
Estrogen deficiency may be an ideal model to study BMAd-bone interaction.
In women with postmenopausal osteoporosis, bone loss occurs due to increased bone turnover, and is accompanied by an increase of BMAT. Bone marrow fat content is negatively associated with BMD at lumbar spine and hip [51]. Estrogen treatment increases bone formation markers, and inhibits bone resorption markers [52]. In mice, ovariectomy is classically used to study effects of estrogen deficiency. Consistent with menopause, ovariectomy causes BMAT expansion and bone loss in mice. An increase in BMAT is observed as early as four weeks after ovariectomy, although bone loss varies depending on age, bone site and strain [53–55]. Ovariectomy of C57BL/6J mice at eight weeks causes substantial cancellous bone loss, whereas at 12 weeks it results in only moderate bone loss and at 16 weeks bone loss is very mild [56]. Moreover, C57BL/6J mice have greater bone loss than BALB/c and outbred (ICR and Kunming) strains [56]. Trabecular bone loss is generally detected between 4–12 weeks of surgery in proximal tibial metaphysis and lumbar spine [54, 57]. Consistent with human studies, there is an inverse relationship between marrow fat expansion and bone loss [53, 58]. Ovariectomy at young age is thus a promising murine model to study mechanisms of BMAd-bone interaction during estrogen deficiency. However, Iwaniec et al. found that mice without BMAT still lost bone following ovariectomy [57], which suggests that under these conditions, gain of BMAT is independent of bone mass, or that estrogen plays independent roles in bone homeostasis. Further work is required to pinpoint mechanistic relationships between BMAT expansion and bone loss.
Irradiation causes transient BMAT expansion and long-term bone loss.
Increased fracture risk is commonly observed in cancer patients who have undergone radiotherapy, particularly with irradiation of bone. Hypocellularity is frequently present in irradiated bones and is often associated with a disproportionate accumulation of collagen and BMAT at ~6 months, which then progressively worsens [59, 60]. The timing and degree of reduced bone mass is variable after radiotherapy, but 12 months is often sufficient to observe a reduction of BMD and increase of fracture risk [61, 62]. Although mechanisms of radiotherapy-induced bone loss have not been unraveled completely, impairment of osteoblasts and activation of osteoclasts have been proposed. Effects of antiresorptive agents (i.e. bisphosphonates and RANKL inhibitors) or osteoanabolic agents (i.e. parathyroid hormone and sclerostin antibody) have been well studied in rodents, but have not been translated to humans to date.
Mice serve as a suitable preclinical model for irradiation-induced effects on bone and the marrow niche. Consistent with BMAT expansion in humans, irradiation of mice causes a rapid and transient ~10-fold increase in rBMAT, peaking at around two weeks after irradiation but still well above baseline at eight weeks [63–65]. Irradiation also causes dose-dependent loss of bone, with 1 Gy causing a 28% reduction and 2 Gy causing a ~40% decline in tibial cancellous bone volume fraction within 10 days [66], which persists for at least 110 days [67]. Rapid reduction of trabecular bone mass is due to resorption, with elevated osteoclast numbers observed at 1 to 2 weeks post-radiation therapy, followed by long-term osteoclast depletion [68]. In vivo studies indicate radiotherapy-induced bone loss is ameliorated in mice treated with bisphosphonates [69, 70]. In addition to elevated bone resorption, decreased active osteoblasts and bone formation following radiation have also been reported [71], which is strengthened by the observation that PTH1–34 alleviates radiotherapy-induced bone loss by preventing apoptosis of osteoblasts and osteocytes, and attenuating radiation-induced bone marrow adiposity [72]. Although radiation therapy in humans and rodents causes rapid BMAT expansion and bone loss, whether increased BMAT causes depletion of bone remains an open question. This reflects our limited knowledge of mechanisms for accumulation of BMAT and the function of BMAT within the marrow niche.
Anti-diabetic thiazolidinediones (TZDs) stimulate BMAT accumulation and bone loss in humans and mice.
Patients with type 2 diabetes mellitus (T2DM) have an increased risk of fragility fractures despite normal or higher BMD. Thiazolidinediones (TZDs) are anti-diabetic drugs that increase insulin sensitivity by activating PPARγ [73]. However, TZDs cause side effects including weight gain, bone loss and increased fracture risks [74]. Severity of TZD-induced osteoporosis is higher in women, the aged, diabetics and with longer treatments [74]. Of note, significant bone loss induced by TZD appears as early as 14 weeks in postmenopausal women [75]. TZD therapy has been associated with a reduction in bone formation marker in female and males, and a significant increase in the levels of the resorption marker in women only [76], which may partially explain gender-differences. Clinical trials have generally paid little attention to BMAT changes with TZDs; however, one study found that pioglitazone increases bone marrow fat in patients with T2DM after 6 months treatment [77], whereas another study observed the opposite - that spine fat decreases with rosiglitazone treatment for 14 weeks [78]. In addition to the specific TZD evaluated, this discrepancy may because the former study recruited diabetic adults from both sexes, whereas the latter only included healthy postmenopausal women. Further clinical trials are needed to investigate whether TZDs cause substantial changes in human BM adiposity.
In rodents, treatment with rosiglitazone causes bone loss and greater marrow adiposity within four weeks [79, 80]. The mechanisms of TZD-induce bone loss relate to bone remodeling - TZDs decrease osteoblast function and suppress new bone formation, whilst increasing bone resorption. Aging influences the mechanisms by which TZDs cause bone loss. Young growing mice show only a mild decrease in bone formation rate. However, adult mice have significant bone loss with rosiglitazone treatment, mainly due to attenuated bone formation, whereas in aged mice, bone loss was associated with increased osteoclastogenesis [81]. In vitro studies demonstrated that activation of PPARg by TZDs promotes commitment of progenitor cells to the adipocytic lineage while inhibiting osteoblastogenesis [82], which may explain how these drugs decrease bone formation and increase fragility.
The effects of TZDs on BMD and marrow adiposity also vary by mouse strain, which is thus an important consideration for experimental design and interpretation. C57BL/6J male mice treated with rosiglitazone showed both bone loss and increased marrow adiposity in both adult and old mice but not in young growing animals [81]. Rosiglitazone also caused loss of bone and increased marrow adiposity in five month old Swiss-Webster mice [79]. Although C3H/HeJ mice showed a significant reduction of BMD in response to rosiglitazone, less striking changes in BMAT were observed than in C57BL/6J mice. In DBA/2J and A/J mice, skeletal effects are milder compared with C57BL/6J and C3H/HeJ mice [83]. Taken together, most animal studies indicate that TZDs expand BMAT and deplete bone. Future studies using BMAd-specific mouse model to knockout PPARg will give us more insights about whether expansion of BMAT is required for bone loss.
Effects of lipodystrophy on bone and marrow adiposity depend on underlying genetic cause and often differ between humans and mice.
Congenital generalized lipodystrophy (CGL) is a rare condition characterized by an almost total lack of WAT. Instead of storing fat in adipocytes, these patients store fat elsewhere in the body, including liver and muscle, which causes serious metabolic dysfunction. Four types of CGL have been described, which are distinguished by their genetic causes. Mutations in the AGPAT2, BSCL2, CAV1, and CAVIN1 genes cause CGL types 1 through 4, respectively. In addition to peripheral adipose loss, CGL1 and CGL2 patients have obvious loss of BMAT, whereas, CGL3 and CGL4 patients have well-preserved bone marrow adiposity. CGL1 and CGL2 patients are more likely to form bone cysts after puberty, which can cause individuals to be prone to spontaneous fractures. CGL4 is associated with muscle weakness, delayed development and joint abnormalities. Although CGL patients generally have bone deterioration, increased BMC and BMD have also been found [84, 85].
Mice deficient for AGPAT2 and BSCL2 have lipodystrophy phenotypes similar to CGL1 and CGL2, with significant loss of WAT and BMAT [86, 87]; however, no bone data is available on these models. Similar to CGL3 patients, knockout mice have a loss of WAT yet retain normal BMAT content and unchanged tibial trabecular and mid-diaphysis bone mass at 16–17 weeks of age [10]. Of note, CAV1 knockout mice at five to eight weeks of age have increased bone formation rate and stiffness, causing bone to mature more rapidly [88]. CAVIN1/PTRF knockout mice lost rBMAT in proximal tibia, but cBMAT of distal tibia and caudal vertebrae was preserved. Corresponding to loss of rBMAT, an increase of trabecular bone was observed in proximal tibia [10]. Other transgenic mouse models of congenital or acquired lipodystrophy have been created, including A-ZIP/F-1 (AZIP) [89], adiponectin-driven PPARγ, kindlin-2 or LMNA knockout [90, 91] (Corsa et al.-under revision), and DTA expressing model [92]. These models consistently show loss of WAT and BMAT, and increased bone mass and formation or even osteopetrosis, which supports the common observation that BMAT negatively correlates with bone mass. Due to inconsistency in evaluation of bone parameters in lipodystrophy patients and animal studies, further studies are required to establish suitability of specific mouse models for preclinical studies. Moreover, the global metabolic disorders of lipodystrophy patients and mice make mechanistic interpretations of effects on bone challenging.
Animal models used for studying BMAd-hematopoiesis interactions
In adults, ~70% of bone marrow is filled with BMAT, which means that hematopoietic cellularity in 30% of bone marrow volume is responsible for generation of red blood cells and the majority of lymphocytes. Clinical work in premenopausal women demonstrate an inverse relationship between BMAT and circulating red and white blood cells [93]. Further, estrogen treatment of anorexia nervosa resulted in a reduction of BMAT and an increase in white and red blood cells [93]. BMAds interact extensively with maturing cells of the myeloid/granulocyte lineage and are closely associated with erythroblast islands [94]. A single BMAd interacts with more than 100 hematopoietic cells through direct cell-cell contact and indirect signals [94]. Our own studies demonstrate that myeloid cell expansion induced by G-CSF administration depletes BMAT dramatically [95], suggesting that BMAds may serve as energy sources during hematopoietic cell proliferation. However, work in a variety of preclinical models suggests that debates about whether BMAT plays beneficial or detrimental roles in hematopoiesis are far from over.
Aging increases BMAT and alters hematopoietic cell fate determination in humans and rodents.
Bone marrow cellularity declines with aging of humans and mice, although hematopoietic stem cell (HSC) numbers increase [96]. Generally, HSCs in young humans and animals are quiescent and serve as a reservoir for long-term storage; however, aged HSCs have a higher rate of cell division and proliferation [97]. HSCs differentiate into myeloid progenitor cells and lymphoid progenitor cells. In humans, hematopoietic changes with aging include declines in lymphopoiesis [98], and increases in frequency of myelodysplastic syndromes, myeloproliferative disorders, and myeloid malignancies [99]. HSCs from the elderly exhibit more myeloid-biased differentiation potential, and do not engraft or generate lymphoid progeny as efficiently in xenotransplanted-immunodeficient mice [97]. In aged mice, BMAT accumulation is accompanied by a decrease in B-lineage progenitor cells [100], and HSCs are more likely to differentiate into myeloid progenitors [101]. Because effects of age on BMAT and hematopoiesis are largely shared between humans and mice, the interactions between these cellular populations can be studied in mice as a preclinical model.
BMAT may influence hematopoietic regeneration following irradiation.
As discussed above, irradiation causes a rapid accumulation of BMAT, which is accompanied by a decrease of hematopoietic cellularity within three days, followed by a gradual and near total recovery of hematopoiesis by 28 days in mice [102]. Irradiation of A-ZIP/F1 mice, which are devoid of all adipocytes including BMAds, demonstrated that in long bones, BMAds are important for HSC maintenance and hematopoietic regeneration by secreting stem cell factor [65]. In contrast, BMAds negatively regulate HSC frequency and hematopoietic regeneration in adipocyte-enriched caudal vertebrae [65]. These observations suggest that interactions between BMAd and hematopoietic cellularity may differ between sites. Most irradiation studies are followed by bone marrow transplantation. Using this approach, Naveiras et al. found that marrow engraftment is accelerated after irradiation in fatless A-ZIP/F1 mice and PPARγ inhibitor-treated mice [89]. Consistent with this approach, co-transplantation of HSCs with bone marrow-derived preadipocytes greatly impaired hematopoiesis [103]. Again, further investigation is required to establish mechanisms and functions of BMAT expansion after irradiation. It is likely that BMAds play roles in hematopoietic regeneration by secreting adipokines/cytokines and/or changing the microenvironment of bone marrow.
Hematological malignancy models may be valuable to explore the relationship between BMAds and malignant cell proliferation/survival.
Human epidemiological data show that obesity, which correlates positively with higher bone marrow adiposity, is associated with an increased risk of developing leukemia and relapse [104, 105]. These data suggest that adipose tissue plays a direct or indirect role in development of acute myeloid leukemia (AML), acute lymphocytic leukemia (ALL), chronic myeloid leukemia (CML), chronic lymphocytic leukemia (CLL) and multiple myeloma (MM). For AML, it has been demonstrated that BMAds within and immediately surrounding the tumor microenvironment support the survival and proliferation of malignant cells during the initial stage of leukemia. The extensive proliferation of leukemic blasts in the bone marrow reduces size of adipocytes by activating lipolysis, with transfer of fatty acids from adipocytes to AML blasts [106]. Importantly, presence of small BMAd predict poor prognoses of AML patients [107]. Obese adults diagnosed with ALL have a 50% higher likelihood of relapse than lean adults. ALL cell injection into obese mice does not affect the time to develop leukemia, but the chemotherapy efficacy is impaired in diet-induced obese mice [108]. MM is typically a disease of the elderly with a median age of diagnosis of 65 years. As discussed, BMAT gradually accumulates in bone marrow cavities as part of the aging process. It is likely that functional interactions between BMAds and MM cells exist. In vitro studies demonstrated that adipocytes support the proliferation and migration of MM cells whereas they inhibit MM cell apoptosis [109]. Although the preponderance of studies support the idea that BMAT provides a suitable microenvironment for tumor growth and protects tumor cells from treatments, activation of PPARγ rescued healthy hematopoietic output and repressed leukemic growth in both patients [110] and mice [111]. These studies cannot exclude the possibility that PPARγ agonists may directly affect leukemic cells per se, rather than working through BMAds. Overall, it appears that BMAds play an active role in generation of a metabolic niche suitable for maintenance of malignant blasts in BM. Preclinical models using malignant cell injection may be suitable to explore targets for preventing or repressing leukemic cell proliferation and survival.
Summary
Preclinical models of aging, obesity, caloric restriction, estrogen deficiency, irradiation, TZD treatment, and lipodystrophy generally share the inverse relationship between BMAT and bone found in humans and mice. Of these, aging, estrogen deficiency, irradiation, and TZD treatment appear to be particularly good preclinical models, whereas caution should be exercised for effects of obesity and lipodystrophy. The relationship between BMAT and hematopoiesis with aging, irradiation and malignancy can also be modeled adequately in preclinical models, although much less work has been performed in this area. Discrepancies between preclinical models and humans can be minimized in some cases by careful choice of species, sex, age, strain, site-specificity and treatment protocol. Understanding mechanistic relationships between BMAT and either bone or hematopoietic cells remains in its infancy, and requires more sophisticated animal models to investigate functional interactions between BMAd and cells of the marrow niche.
Figure 2. Suitability of preclinical models for BMAd-bone and BMAd-hematopoiesis studies.

Bone marrow adipocytes (BMAds) may serve as energy source or secrete adipokines/cytokines to influence bone resorption, bone formation, hematopoietic stem cell (HSC) maintenance and hematopoiesis. Many preclinical models have been used to investigate the relationship between BMAds and bone/hematopoiesis. However, each one has their own pros and cons. We reviewed the literature and ranked these models based on their consistencies with human studies and repeatability between different studies. + indicates less-recommended and ++++ indicates well-recommended.
Practice Points:
Many clinical studies have demonstrated inverse relationships between BMAT and bone/hematopoiesis homeostasis; however, direct evidence for causal relationships require further experimentation.
Animal models have been widely used for preclinical studies, but thoughtful consideration of species, sex, age, strain and site-specificity may increase relevance to humans.
Research agenda:
Basic research into relationships between bone marrow adiposity and bone loss or hematopoietic disorders is critical for understanding pathologies of bone-related diseases.
To avoid the systematic effects of treatments on bones and hematopoiesis, BMAd-specific animal model is necessary for future studies.
Acknowledgements
This work was supported by grants from the NIH to OAM (DK092759; DK121759, DK125513, AG069795), and from the American Diabetes Association (1-18-PDF-087) to ZL.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Li Z, et al. , Development, regulation, metabolism and function of bone marrow adipose tissues. Bone, 2018. 110: p. 134–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singhal V and Bredella MA, Marrow adipose tissue imaging in humans. Bone, 2019. 118: p. 69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ren J, et al. , Composition of adipose tissue and marrow fat in humans by 1H NMR at 7 Tesla. J Lipid Res, 2008. 49(9): p. 2055–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guerri S, et al. , Quantitative imaging techniques for the assessment of osteoporosis and sarcopenia. Quant Imaging Med Surg, 2018. 8(1): p. 60–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goodsitt MM, Johnson RH, and Chesnut CH 3rd, A new set of calibration standards for estimating the fat and mineral content of vertebrae via dual energy QCT. Bone Miner, 1991. 13(3): p. 217–33. [DOI] [PubMed] [Google Scholar]
- 6.Bredella MA, et al. , Marrow Adipose Tissue Quantification of the Lumbar Spine by Using Dual-Energy CT and Single-Voxel (1)H MR Spectroscopy: A Feasibility Study. Radiology, 2015. 277(1): p. 230–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *7.Suchacki KJ, et al. , Bone marrow adipose tissue is a unique adipose subtype with distinct roles in glucose homeostasis. Nat Commun, 2020. 11(1): p. 3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *8.Devlin MJ, et al. , Caloric restriction leads to high marrow adiposity and low bone mass in growing mice. J Bone Miner Res, 2010. 25(9): p. 2078–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *9.Scheller EL, et al. , Use of osmium tetroxide staining with microcomputerized tomography to visualize and quantify bone marrow adipose tissue in vivo. Methods Enzymol, 2014. 537: p. 123–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *10.Scheller EL, et al. , Region-specific variation in the properties of skeletal adipocytes reveals regulated and constitutive marrow adipose tissues. Nat Commun, 2015. 6: p. 7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng J, et al. , Quantitative electron microscopy shows uniform incorporation of triglycerides into existing lipid droplets. Histochem Cell Biol, 2009. 132(3): p. 281–91. [DOI] [PubMed] [Google Scholar]
- 12.Tratwal J, et al. , MarrowQuant Across Aging and Aplasia: A Digital Pathology Workflow for Quantification of Bone Marrow Compartments in Histological Sections. Front Endocrinol (Lausanne), 2020. 11: p. 480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *13.Kricun ME, Red-yellow marrow conversion: its effect on the location of some solitary bone lesions. Skeletal Radiol, 1985. 14(1): p. 10–9. [DOI] [PubMed] [Google Scholar]
- 14.Liney GP, et al. , Age, gender, and skeletal variation in bone marrow composition: a preliminary study at 3.0 Tesla. J Magn Reson Imaging, 2007. 26(3): p. 787–93. [DOI] [PubMed] [Google Scholar]
- 15.Kugel H, et al. , Age- and sex-specific differences in the 1H-spectrum of vertebral bone marrow. J Magn Reson Imaging, 2001. 13(2): p. 263–8. [DOI] [PubMed] [Google Scholar]
- 16.Griffith JF, et al. , Bone marrow fat content in the elderly: a reversal of sex difference seen in younger subjects. J Magn Reson Imaging, 2012. 36(1): p. 225–30. [DOI] [PubMed] [Google Scholar]
- 17.Allen JE, et al. , Fat cells in red bone marrow of human rib: their size and spatial distribution with respect to the radon-derived dose to the haemopoietic tissue. Int J Radiat Biol, 1995. 68(6): p. 669–78. [DOI] [PubMed] [Google Scholar]
- 18.Cawthorn WP, et al. , Expansion of Bone Marrow Adipose Tissue During Caloric Restriction Is Associated With Increased Circulating Glucocorticoids and Not With Hypoleptinemia. Endocrinology, 2016. 157(2): p. 508–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tavassoli M, Marrow adipose cells. Histochemical identification of labile and stable components. Arch Pathol Lab Med, 1976. 100(1): p. 16–8. [PubMed] [Google Scholar]
- 20.Karampinos DC, et al. , Association of MRS-Based Vertebral Bone Marrow Fat Fraction with Bone Strength in a Human In Vitro Model. J Osteoporos, 2015. 2015: p. 152349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu J, et al. , Peak Bone Mass and Patterns of Change in Total Bone Mineral Density and Bone Mineral Contents From Childhood Into Young Adulthood. J Clin Densitom, 2016. 19(2): p. 180–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moore SG and Dawson KL, Red and yellow marrow in the femur: age-related changes in appearance at MR imaging. Radiology, 1990. 175(1): p. 219–23. [DOI] [PubMed] [Google Scholar]
- 23.Shen W, et al. , Comparison of the relationship between bone marrow adipose tissue and volumetric bone mineral density in children and adults. J Clin Densitom, 2014. 17(1): p. 163–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Di Iorgi N, et al. , Reciprocal relation between marrow adiposity and the amount of bone in the axial and appendicular skeleton of young adults. J Clin Endocrinol Metab, 2008. 93(6): p. 2281–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shen W, et al. , MRI-measured bone marrow adipose tissue is inversely related to DXA-measured bone mineral in Caucasian women. Osteoporos Int, 2007. 18(5): p. 641–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen W, et al. , Relationship between MRI-measured bone marrow adipose tissue and hip and spine bone mineral density in African-American and Caucasian participants: the CARDIA study. J Clin Endocrinol Metab, 2012. 97(4): p. 1337–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *27.Papageorgiou M, et al. , Age- and Strain-Related Differences in Bone Microstructure and Body Composition During Development in Inbred Male Mouse Strains. Calcif Tissue Int, 2020. 106(4): p. 431–443. [DOI] [PubMed] [Google Scholar]
- 28.Halloran BP, et al. , Changes in bone structure and mass with advancing age in the male C57BL/6J mouse. J Bone Miner Res, 2002. 17(6): p. 1044–50. [DOI] [PubMed] [Google Scholar]
- 29.Almeida M, et al. , Increased marrow adipogenesis does not contribute to age-dependent appendicular bone loss in female mice. Aging Cell, 2020. 19(11): p. e13247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Evans AL, et al. , Bone density, microstructure and strength in obese and normal weight men and women in younger and older adulthood. J Bone Miner Res, 2015. 30(5): p. 920–8. [DOI] [PubMed] [Google Scholar]
- 31.Viljakainen H, et al. , Suppressed bone turnover in obesity: a link to energy metabolism? A case-control study. J Clin Endocrinol Metab, 2014. 99(6): p. 2155–63. [DOI] [PubMed] [Google Scholar]
- 32.Scheller EL, et al. , Changes in Skeletal Integrity and Marrow Adiposity during High-Fat Diet and after Weight Loss. Front Endocrinol (Lausanne), 2016. 7: p. 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tencerova M, et al. , High-Fat Diet-Induced Obesity Promotes Expansion of Bone Marrow Adipose Tissue and Impairs Skeletal Stem Cell Functions in Mice. J Bone Miner Res, 2018. 33(6): p. 1154–1165. [DOI] [PubMed] [Google Scholar]
- 34.Devlin MJ, et al. , Differential effects of high fat diet and diet-induced obesity on skeletal acquisition in female C57BL/6J vs. FVB/NJ Mice. Bone Rep, 2018. 8: p. 204–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lecka-Czernik B, et al. , High bone mass in adult mice with diet-induced obesity results from a combination of initial increase in bone mass followed by attenuation in bone formation; implications for high bone mass and decreased bone quality in obesity. Mol Cell Endocrinol, 2015. 410: p. 35–41. [DOI] [PubMed] [Google Scholar]
- 36.Silva MJ, et al. , Effects of High-Fat Diet and Body Mass on Bone Morphology and Mechanical Properties in 1100 Advanced Intercross Mice. J Bone Miner Res, 2019. 34(4): p. 711–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doucette CR, et al. , A High Fat Diet Increases Bone Marrow Adipose Tissue (MAT) But Does Not Alter Trabecular or Cortical Bone Mass in C57BL/6J Mice. J Cell Physiol, 2015. 230(9): p. 2032–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hamrick MW, et al. , Leptin deficiency produces contrasting phenotypes in bones of the limb and spine. Bone, 2004. 34(3): p. 376–83. [DOI] [PubMed] [Google Scholar]
- 39.Steppan CM, et al. , Leptin is a potent stimulator of bone growth in ob/ob mice. Regul Pept, 2000. 92(1–3): p. 73–8. [DOI] [PubMed] [Google Scholar]
- 40.Cornbleet PJ, Moir RC, and Wolf PL, A histochemical study of bone marrow hypoplasia in anorexia nervosa. Virchows Arch A Pathol Anat Histol, 1977. 374(3): p. 239–47. [DOI] [PubMed] [Google Scholar]
- 41.Rigotti NA, et al. , Osteoporosis in women with anorexia nervosa. N Engl J Med, 1984. 311(25): p. 1601–6. [DOI] [PubMed] [Google Scholar]
- 42.Treasure J, Fogelman I, and Russell GF, Osteopaenia of the lumbar spine and femoral neck in anorexia nervosa. Scott Med J, 1986. 31(3): p. 206–7. [PubMed] [Google Scholar]
- 43.Biller BM, et al. , Mechanisms of osteoporosis in adult and adolescent women with anorexia nervosa. J Clin Endocrinol Metab, 1989. 68(3): p. 548–54. [DOI] [PubMed] [Google Scholar]
- 44.Bredella MA, et al. , Increased bone marrow fat in anorexia nervosa. J Clin Endocrinol Metab, 2009. 94(6): p. 2129–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Villareal DT, et al. , Effect of Two-Year Caloric Restriction on Bone Metabolism and Bone Mineral Density in Non-Obese Younger Adults: A Randomized Clinical Trial. J Bone Miner Res, 2016. 31(1): p. 40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang BC and Villareal DT, Weight Loss-Induced Reduction of Bone Mineral Density in Older Adults with Obesity. J Nutr Gerontol Geriatr, 2019. 38(1): p. 100–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hamrick MW, et al. , Caloric restriction decreases cortical bone mass but spares trabecular bone in the mouse skeleton: implications for the regulation of bone mass by body weight. J Bone Miner Res, 2008. 23(6): p. 870–8. [DOI] [PubMed] [Google Scholar]
- 48.Tatsumi S, et al. , Life-long caloric restriction reveals biphasic and dimorphic effects on bone metabolism in rodents. Endocrinology, 2008. 149(2): p. 634–41. [DOI] [PubMed] [Google Scholar]
- 49.Devlin MJ, et al. , Daily leptin blunts marrow fat but does not impact bone mass in calorie-restricted mice. J Endocrinol, 2016. 229(3): p. 295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McGrath C, et al. , Exercise Degrades Bone in Caloric Restriction, Despite Suppression of Marrow Adipose Tissue (MAT). J Bone Miner Res, 2020. 35(1): p. 106–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Milisic L, Vegar-Zubovic S, and Valjevac A, Bone marrow adiposity is inversely associated with bone mineral density in postmenopausal females. Med Glas (Zenica), 2020. 17(1): p. 15–21. [DOI] [PubMed] [Google Scholar]
- 52.Limonard EJ, et al. , Short-Term Effect of Estrogen on Human Bone Marrow Fat. J Bone Miner Res, 2015. 30(11): p. 2058–66. [DOI] [PubMed] [Google Scholar]
- 53.Martin RB and Zissimos SL, Relationships between marrow fat and bone turnover in ovariectomized and intact rats. Bone, 1991. 12(2): p. 123–31. [DOI] [PubMed] [Google Scholar]
- 54.Lei Z, Xiaoying Z, and Xingguo L, Ovariectomy-associated changes in bone mineral density and bone marrow haematopoiesis in rats. Int J Exp Pathol, 2009. 90(5): p. 512–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bouxsein ML, et al. , Ovariectomy-induced bone loss varies among inbred strains of mice. J Bone Miner Res, 2005. 20(7): p. 1085–92. [DOI] [PubMed] [Google Scholar]
- 56.Zhou S, et al. , Age-dependent variations of cancellous bone in response to ovariectomy in C57BL/6J mice. Exp Ther Med, 2018. 15(4): p. 3623–3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Iwaniec UT and Turner RT, Failure to generate bone marrow adipocytes does not protect mice from ovariectomy-induced osteopenia. Bone, 2013. 53(1): p. 145–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kurabayashi T, et al. , Effects of a beta 3 adrenergic receptor agonist on bone and bone marrow adipocytes in the tibia and lumbar spine of the ovariectomized rat. Calcif Tissue Int, 2001. 68(4): p. 248–54. [DOI] [PubMed] [Google Scholar]
- 59.Marx RE and Johnson RP, Studies in the radiobiology of osteoradionecrosis and their clinical significance. Oral Surg Oral Med Oral Pathol, 1987. 64(4): p. 379–90. [DOI] [PubMed] [Google Scholar]
- 60.Curi MM, et al. , Histopathologic and Histomorphometric Analysis of Irradiation Injury in Bone and the Surrounding Soft Tissues of the Jaws. J Oral Maxillofac Surg, 2016. 74(1): p. 190–9. [DOI] [PubMed] [Google Scholar]
- 61.Yaprak G, et al. , Osteoporosis development and vertebral fractures after abdominal irradiation in patients with gastric cancer. BMC Cancer, 2018. 18(1): p. 972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kwon JW, et al. , Pelvic bone complications after radiation therapy of uterine cervical cancer: evaluation with MRI. AJR Am J Roentgenol, 2008. 191(4): p. 987–94. [DOI] [PubMed] [Google Scholar]
- 63.Wright LE, et al. , Single-Limb Irradiation Induces Local and Systemic Bone Loss in a Murine Model. J Bone Miner Res, 2015. 30(7): p. 1268–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Green DE, et al. , Altered composition of bone as triggered by irradiation facilitates the rapid erosion of the matrix by both cellular and physicochemical processes. PLoS One, 2013. 8(5): p. e64952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *65.Zhou BO, et al. , Bone marrow adipocytes promote the regeneration of stem cells and haematopoiesis by secreting SCF. Nat Cell Biol, 2017. 19(8): p. 891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kondo H, et al. , Total-body irradiation of postpubertal mice with (137)Cs acutely compromises the microarchitecture of cancellous bone and increases osteoclasts. Radiat Res, 2009. 171(3): p. 283–9. [DOI] [PubMed] [Google Scholar]
- 67.Hamilton SA, et al. , A murine model for bone loss from therapeutic and space-relevant sources of radiation. J Appl Physiol (1985), 2006. 101(3): p. 789–93. [DOI] [PubMed] [Google Scholar]
- 68.Oest ME, et al. , Longitudinal Effects of Single Hindlimb Radiation Therapy on Bone Strength and Morphology at Local and Contralateral Sites. J Bone Miner Res, 2018. 33(1): p. 99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Willey JS, et al. , Risedronate prevents early radiation-induced osteoporosis in mice at multiple skeletal locations. Bone, 2010. 46(1): p. 101–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Keenawinna L, et al. , Zoledronic acid prevents loss of trabecular bone after focal irradiation in mice. Radiat Res, 2013. 180(1): p. 89–99. [DOI] [PubMed] [Google Scholar]
- 71.Szymczyk KH, Shapiro IM, and Adams CS, Ionizing radiation sensitizes bone cells to apoptosis. Bone, 2004. 34(1): p. 148–56. [DOI] [PubMed] [Google Scholar]
- 72.Chandra A, et al. , PTH1–34 alleviates radiotherapy-induced local bone loss by improving osteoblast and osteocyte survival. Bone, 2014. 67: p. 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yki-Jarvinen H, Thiazolidinediones. N Engl J Med, 2004. 351(11): p. 1106–18. [DOI] [PubMed] [Google Scholar]
- 74.Gilbert MP and Pratley RE, The impact of diabetes and diabetes medications on bone health. Endocr Rev, 2015. 36(2): p. 194–213. [DOI] [PubMed] [Google Scholar]
- 75.Grey A, et al. , The peroxisome proliferator-activated receptor-gamma agonist rosiglitazone decreases bone formation and bone mineral density in healthy postmenopausal women: a randomized, controlled trial. J Clin Endocrinol Metab, 2007. 92(4): p. 1305–10. [DOI] [PubMed] [Google Scholar]
- 76.Zinman B, et al. , Effect of rosiglitazone, metformin, and glyburide on bone biomarkers in patients with type 2 diabetes. J Clin Endocrinol Metab, 2010. 95(1): p. 134–42. [DOI] [PubMed] [Google Scholar]
- 77.Grey A, et al. , Pioglitazone increases bone marrow fat in type 2 diabetes: results from a randomized controlled trial. Eur J Endocrinol, 2012. 166(6): p. 1087–91. [DOI] [PubMed] [Google Scholar]
- 78.Harslof T, et al. , Rosiglitazone decreases bone mass and bone marrow fat. J Clin Endocrinol Metab, 2011. 96(5): p. 1541–8. [DOI] [PubMed] [Google Scholar]
- 79.Ali AA, et al. , Rosiglitazone causes bone loss in mice by suppressing osteoblast differentiation and bone formation. Endocrinology, 2005. 146(3): p. 1226–35. [DOI] [PubMed] [Google Scholar]
- 80.Sottile V, Seuwen K, and Kneissel M, Enhanced marrow adipogenesis and bone resorption in estrogen-deprived rats treated with the PPARgamma agonist BRL49653 (rosiglitazone). Calcif Tissue Int, 2004. 75(4): p. 329–37. [DOI] [PubMed] [Google Scholar]
- 81.Lazarenko OP, et al. , Rosiglitazone induces decreases in bone mass and strength that are reminiscent of aged bone. Endocrinology, 2007. 148(6): p. 2669–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lecka-Czernik B, et al. , Divergent effects of selective peroxisome proliferator-activated receptor-gamma 2 ligands on adipocyte versus osteoblast differentiation. Endocrinology, 2002. 143(6): p. 2376–84. [DOI] [PubMed] [Google Scholar]
- *83.Ackert-Bicknell CL, et al. , Strain-specific effects of rosiglitazone on bone mass, body composition, and serum insulin-like growth factor-I. Endocrinology, 2009. 150(3): p. 1330–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Christensen JD, et al. , Bone mineral content in patients with congenital generalized lipodystrophy is unaffected by metreleptin replacement therapy. J Clin Endocrinol Metab, 2014. 99(8): p. E1493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lima JG, et al. , Bone Density in Patients With Berardinelli-Seip Congenital Lipodystrophy Is Higher in Trabecular Sites and in Type 2 Patients. J Clin Densitom, 2018. 21(1): p. 61–67. [DOI] [PubMed] [Google Scholar]
- 86.Vogel P, et al. , Pathology of congenital generalized lipodystrophy in Agpat2−/− mice. Vet Pathol, 2011. 48(3): p. 642–54. [DOI] [PubMed] [Google Scholar]
- 87.McIlroy GD, et al. , Adipose specific disruption of seipin causes early-onset generalised lipodystrophy and altered fuel utilisation without severe metabolic disease. Mol Metab, 2018. 10: p. 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rubin J, et al. , Caveolin-1 knockout mice have increased bone size and stiffness. J Bone Miner Res, 2007. 22(9): p. 1408–18. [DOI] [PubMed] [Google Scholar]
- *89.Naveiras O, et al. , Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature, 2009. 460(7252): p. 259–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang F, et al. , Lipoatrophy and severe metabolic disturbance in mice with fat-specific deletion of PPARgamma. Proc Natl Acad Sci U S A, 2013. 110(46): p. 18656–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gao H, et al. , Lipoatrophy and metabolic disturbance in mice with adipose-specific deletion of kindlin-2. JCI Insight, 2019. 4(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zou W, et al. , Congenital lipodystrophy induces severe osteosclerosis. PLoS Genet, 2019. 15(6): p. e1008244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Polineni S, et al. , Red and White Blood Cell Counts Are Associated With Bone Marrow Adipose Tissue, Bone Mineral Density, and Bone Microarchitecture in Premenopausal Women. J Bone Miner Res, 2020. 35(6): p. 1031–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Robles H, et al. , Characterization of the bone marrow adipocyte niche with three-dimensional electron microscopy. Bone, 2019. 118: p. 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li Z, et al. , G-CSF partially mediates effects of sleeve gastrectomy on the bone marrow niche. J Clin Invest, 2019. 129(6): p. 2404–2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sudo K, et al. , Age-associated characteristics of murine hematopoietic stem cells. J Exp Med, 2000. 192(9): p. 1273–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pang WW, et al. , Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci U S A, 2011. 108(50): p. 20012–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Linton PJ and Dorshkind K, Age-related changes in lymphocyte development and function. Nat Immunol, 2004. 5(2): p. 133–9. [DOI] [PubMed] [Google Scholar]
- 99.Lichtman MA and Rowe JM, The relationship of patient age to the pathobiology of the clonal myeloid diseases. Semin Oncol, 2004. 31(2): p. 185–97. [DOI] [PubMed] [Google Scholar]
- 100.Miller JP and Allman D, The decline in B lymphopoiesis in aged mice reflects loss of very early B-lineage precursors. J Immunol, 2003. 171(5): p. 2326–30. [DOI] [PubMed] [Google Scholar]
- 101.Rossi DJ, et al. , Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci U S A, 2005. 102(26): p. 9194–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yamazaki K and Allen TD, Ultrastructural and morphometric alterations in bone marrow stromal tissue after 7 Gy irradiation. Blood Cells, 1991. 17(3): p. 527–49. [PubMed] [Google Scholar]
- *103.Ambrosi TH, et al. , Adipocyte Accumulation in the Bone Marrow during Obesity and Aging Impairs Stem Cell-Based Hematopoietic and Bone Regeneration. Cell Stem Cell, 2017. 20(6): p. 771–784 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Larsson SC and Wolk A, Overweight and obesity and incidence of leukemia: a meta-analysis of cohort studies. Int J Cancer, 2008. 122(6): p. 1418–21. [DOI] [PubMed] [Google Scholar]
- 105.Castillo JJ, et al. , Obesity but not overweight increases the incidence and mortality of leukemia in adults: a meta-analysis of prospective cohort studies. Leuk Res, 2012. 36(7): p. 868–75. [DOI] [PubMed] [Google Scholar]
- 106.Shafat MS, et al. , Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood, 2017. 129(10): p. 1320–1332. [DOI] [PubMed] [Google Scholar]
- 107.Lu W, et al. , Small bone marrow adipocytes predict poor prognosis in acute myeloid leukemia. Haematologica, 2018. 103(1): p. e21–e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Behan JW, et al. , Adipocytes impair leukemia treatment in mice. Cancer Res, 2009. 69(19): p. 7867–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Caers J, et al. , Neighboring adipocytes participate in the bone marrow microenvironment of multiple myeloma cells. Leukemia, 2007. 21(7): p. 1580–4. [DOI] [PubMed] [Google Scholar]
- 110.Prost S, et al. , Erosion of the chronic myeloid leukaemia stem cell pool by PPARgamma agonists. Nature, 2015. 525(7569): p. 380–3. [DOI] [PubMed] [Google Scholar]
- 111.Boyd AL, et al. , Acute myeloid leukaemia disrupts endogenous myeloerythropoiesis by compromising the adipocyte bone marrow niche. Nat Cell Biol, 2017. 19(11): p. 1336–1347. [DOI] [PubMed] [Google Scholar]