

Abstract
The efficacy of PARP-inhibitors (PARPi) is restricted by inevitable drug resistance, while its use in combination with chemotherapy and targeted agents is commonly associated with dose-limiting toxicities. Immune checkpoint blockade (ICB) has demonstrated durable responses in different solid tumors and are well-established across multiple cancers. Despite this, single agent activity is limited to a minority of patients and drug resistance remains an issue. Building on the monotherapy success of both drug classes, combining PARPi with ICB may be a safe and well tolerated strategy with the potential to improve survival outcomes. In this review, we present the preclinical, translational, and clinical data supporting the combination of DNA damage response (DDR) and ICB as well as consider important questions to be addressed with future research.
Keywords: DNA damage response, immunotherapy, PARP inhibitor, immune checkpoint inhibitor
Introduction
The field of oncology drug development has seen substantial progress in the treatment of different cancers over the past decade. Inhibitors of key DNA damage response (DDR) mechanisms, specifically against poly(ADP-ribose) polymerase (PARP), have proven to be effective and are now approved as single-agent therapies in breast, ovarian, pancreatic and castration-resistant prostate cancers (CRPC) (1). Immune check-point blockade (ICB) agents targeting cytotoxic T lymphocyte antigen 4 (CTLA-4) and programmed cell death 1/programmed cell death ligand 1 (PD-1/L1) have also emerged as effective strategies to improve survival outcomes in various solid tumors (2). However, clinical benefit for both classes of drugs is limited by innate and acquired resistance and/or drug-related toxicities. Preclinical studies have supported the mechanistic rationale for the combination of PARP inhibitors (PARPi) and immune checkpoint blockade (ICB) as a therapeutic strategy for patients with different solid tumors (3).
In this review, we discuss the preclinical, translational, and clinical studies supporting the combination of PARPi and ICB therapies as well as consider important questions to be addressed with future research.
PARP and Synthetic Lethality
The PARP family of enzymes consists of 17 proteins that play important roles in the DDR pathways for base excision repair (BER) and single strand break repair (SSB) (4–6). PARP1, the most widely studied protein of this family, is composed of a common catalytic domain, which generates poly(ADP-ribose) chains upon binding to SSB through a process known as “auto-PARylation” (4,7,8). These PARP complexes promote the downstream recruitment of SSB repair proteins and lead to the chromatin remodeling necessary in DNA damage repair (9). Without appropriate PARP1 function, the accumulation of SSB ultimately leads to replication-dependent double strand breaks (DSB) (10–12). While in normal cells these destructive DSB would be repaired by homologous recombination (HR), tumor cells with HR deficiency rely on lower fidelity repair mechanisms, and thus cannot overcome the catastrophic levels of DNA damage (1).
Under normal circumstances, the BRCA1/2 proteins restore the original DNA architecture through error-free HR at regions of DSBs (9). In the event that either of the BRCA1 or BRCA2 gene alleles undergo a germline mutation, cells must rely on the more error prone non-homologous end joining (NHEJ) repair mechanism that leads to genetic alterations, deletions and rapid cellular progression. Patients with hereditary BRCA1/2 mutations have an increased propensity for breast, ovarian, prostate and other cancers (9,13,14). Due to their genetic vulnerability, germline BRCA1/2 mutant tumors were hypothesized to be reliant on the PARP-induced SSB repair mechanism for cell survival. Indeed, the clinical application of PARPi in patients with germline BRCA1/2 mutations was developed based on the concept of “synthetic lethality”; that is, preclinical data supported the significant sensitivity of these tumor cells to the cytotoxic effects of PARP inhibition (9,15–17). Hence, BRCA mutant tumor cells have been reported to be 1000 times more sensitive to PARPi than their wild-type counterparts (16,18,19).
More recent preclinical studies have supported the importance of “PARP trapping” as the key cytotoxic event leading to cell death (10, 20–22). Without functional PARylation, the PARP1 protein is unable to dissociate from the DNA strand and the sites of DNA damage. Thus, PARP inhibition results in trapping of the PARP complex to DNA SSBs, collapse of the replication fork, and further accumulation of DNA DSB (1). Preclinical research has indicated that PARPi vary in their degree of PARP trapping function. For example, although it is potentially analogous to their clinical antitumor effects, veliparib has been shown to have reduced PARP trapping activity than the currently approved PARPi (talazoparib, niraparib, olaparib and rucaparib) (23,24).
Clinical activity of PARP inhibitors
Four randomized phase III trials —SOLO-1 (NCT01844986)I, PAOLA-1/ENGOT-OV25 (NCT02477644)II, PRIMA/ENGOT-OV26 (NCT02655016)III and VELIA/GOG-3005 (NCT02470585)IV— have demonstrated clinically significant progression-free survival (PFS) in newly diagnosed ovarian cancer patients undergoing PARPi treatment. Olaparib and rucaparib have already received FDA approval for the treatment of BRCA1/2 mutated recurrent, platinum sensitive ovarian cancer. Olaparib, niraparib and rucaparib have been approved for maintenance therapy following platinum chemotherapy in ovarian cancer (25–28). Olaparib has also led to durable antitumor responses in advanced CRPC with BRCA1/2 mutations and other HRR mutations to varying degrees, as well as, in the recurrent setting of BRCA1/2 associated breast cancer (11,29,30). Although talazoparib showed a 3-month PFS benefit in breast cancer, results from the EMBRACA study showed that the duration of response and overall survival were not significant (31).
While PARPi are, in general, a well-tolerated and effective single-agent therapy for BRCA1/2 mutated tumor types, the survival benefit has been limited to patients with hereditary breast and ovarian cancer, with platinum-sensitive ovarian cancers showing >50% response rates. Moreover, a range of different mechanisms have been observed to be responsible for both primary and acquired PARPi resistance (1). Broadening the target patient population beyond the currently approved tumor types has proven challenging as translational models and clinical data demonstrate that even BRCA1/2 mutations differ in their PARPi sensitivity between cancer types (32). Moreover, non-BRCA1/2 deficiencies in the HRR pathway may also create a BRCAness phenotype and increase tumor sensitivity to PARPi (33). Clinical data have supported the theory that patients with non-BRCA1/2 DDR mutations may potentially benefit from PARPi (28,30). In the randomized phase III PRIMA/ENGOT-OV26 (NCT02655016)III trial investigating the use of niraparib in the maintenance setting, exploratory analyses of HRD-positive subgroups showed clinical activity of niraparib regardless of germline BRCA1/2 status (28). There is, thus, an opportunity for treatment optimization by exploring other predictive biomarkers of response to PARPi and developing rational combination strategies to build on the success of PARPi monotherapy (25).
Immuno-oncology and immune checkpoint blockade
Immuno-oncology (IO) therapy targets the complex adaptive changes in the tumor microenvironment upon antigen recognition (34). An interesting IO target is the Cytotoxic T lymphocyte-associated protein 4 (CTLA-4), a co-inhibitory molecule found on the cell surface of regulatory T cells that interacts with antigen presenting cells (APC) (35). In conjunction with the downregulation of co-stimulatory CD80 and CD86 cells, CTLA-4 suppresses T cell function and mitigates the amplitude of the cytotoxic T cell response (34, 35). Murine models have confirmed that the inhibitory effect of CTLA-4 on T cell activation results in tumor growth and fatal organ dysfunction (37). Ipilimumab was the first therapeutic anti-CTLA-4 antibody approved by the FDA for clinical use in metastatic melanoma (2,38). By inhibiting cell tolerance of tumor infiltrating lymphocytes (TILs) through the CTLA-4 blockade, patients using ipilimumab had improved survival outcomes compared to those undergoing standard treatment (36). However, given the complicated factors that regulate the transport and expression of the molecule, the mechanism of action has yet to be well-defined (39).
In a similar manner, targeting the PD-1/L1 immune checkpoint has led to patient benefit and FDA approval of multiple PD-1/L1 inhibitors. PD-1/L1 are expressed by primed T cells in the setting of cytokine release and tissue inflammation. Upon PD-1/L1 binding, T cell tolerance and subsequent cellular apoptosis occurs (39). The downregulation of T cells with PD-1/L1 blockade, with a significant reduction of the lympho-proliferative reaction compared to mice with CTLA-4 blockade, has been confirmed in murine models (40). Clinical trials have supported the safety and efficacy of blocking PD-1/L1 in order to restore TIL function and immune effect on tumor cells in vivo (39). Anti-PD-1 antibodies such as nivolumab and pembrolizumab have demonstrated improved patient outcomes in metastatic melanoma, lung cancer, renal cell carcinoma, head and neck cancers, bladder cancer and gastric cancers (41). While PD-L1 inhibitors such as durvalumab, atezolizumab and avelumab are FDA approved for non-small cell lung cancer (NSCLC) and metastatic urothelial carcinoma (42), the PD-1 inhibitor cemiplimab is approved in metastatic cutaneous squamous cell carcinoma (43). Thus far, gene sequencing of different solid tumors seems to indicate that a high mutational tumor burden may be a predictive marker of response to PD-1/L1 inhibitors (44).
Combining PARP inhibition and immunotherapy, a rational approach
Despite the approval of different PD-1/L1 agents for the treatment of multiple cancer types, only a minority of patients will benefit from a monotherapy approach (38,42). This is exemplified in gynecologic cancers, where the use of PD-1/L1 agents has demonstrated a modest 10–15% response rates in ovarian, cervical and endometrial cancers (45,46). Recently, we have observed a shift towards developing rational combinatorial strategies to enhance the efficacy of ICBs, including combinations with PARPi (47).
As depicted in Figure 1, DNA damage is recognized by the immune system through damage-associated molecular patterns (DAMP), such as the cytosolic DNA - cyclic GMP-AMP synthase complex (cGAS). Recruitment and activation of the STING (stimulator of interferon genes) pathway promotes a significant antitumor immune response when high levels of DNA damage and cell death are prevalent (48,49). As previously discussed, the inhibition of PARP1 in the setting of HR deficiency (e.g. BRCA1/2 mutations) leads to the accumulation of DNA DSB and replication fork collapse; the subsequent accumulation of cytosolic DNA and overwhelming tumor mutational burden (TMB) activates the cGAS-STING pathway, enhances T cell infiltration and induces type 1 IFN signaling, thereby resetting or priming the tumor microenvironment (48,50,51).
Figure 1.
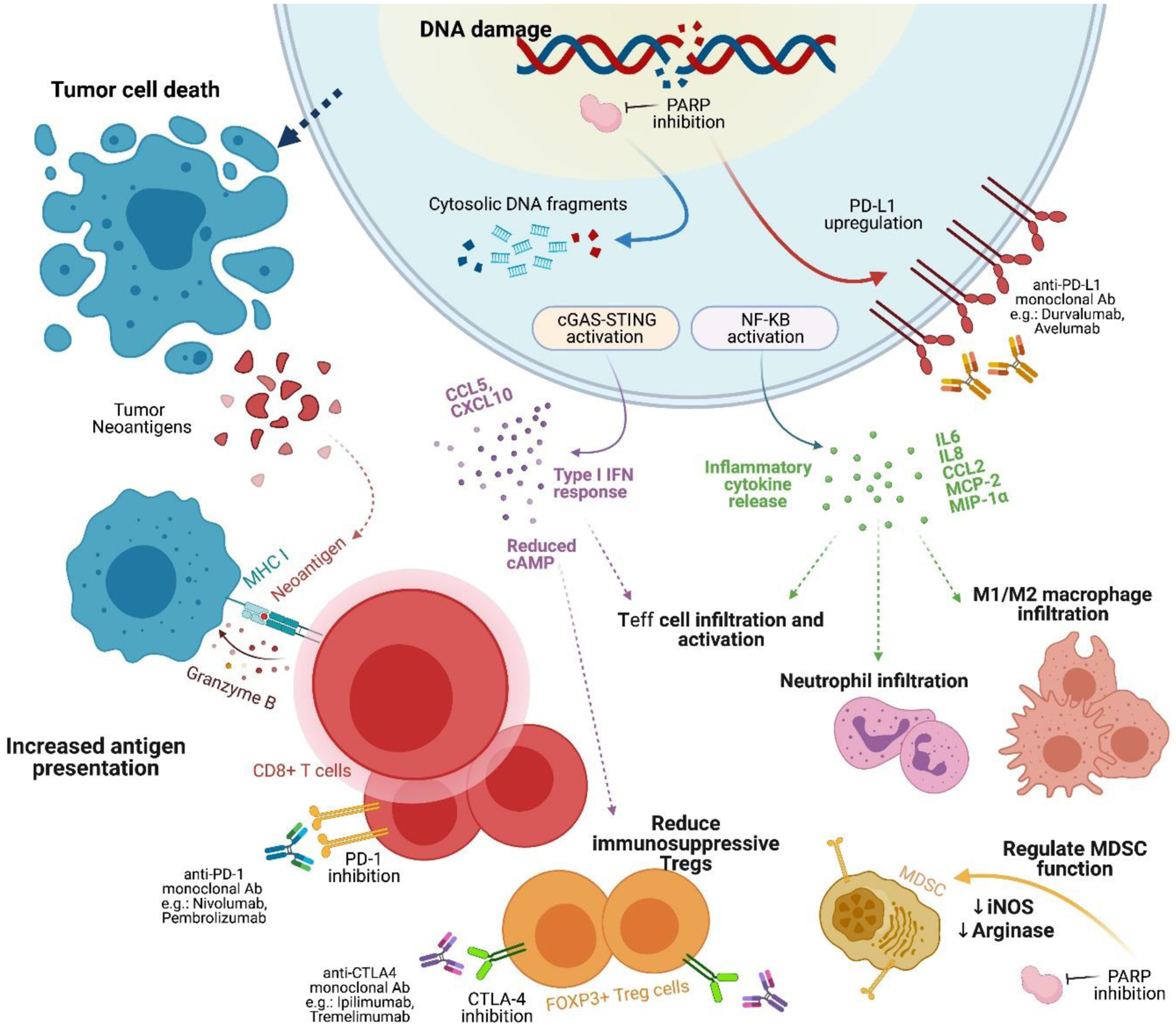
Mechanisms of DNA damage, repair and immune priming underlying PARPi and ICB combination therapy.
Abbreviations: Ab, antibody; cGAS, cyclic GMP/AMP synthase; CTLA-4; cytotoxic T lymphocyte-associated antigen 4; ICB, immune checkpoint blockade; IFN, interferon; iNOS, inducible NO synthase; MDSC, myeloid-derived stem cell; NF-κB, nuclear factor-κB; PARPi, poly(ADP ribose) polymerase inhibitor; PD-L1, programmed cell death ligand 1; STING, stimulator of interferon genes; Teff cell, T effector cell; Treg, T regulatory cell.
Recent preclinical studies have suggested potential synergy between PARPi and ICBs. For instance, talazoparib has demonstrated efficacy in activating the STING pathway, and thus promoting T cell infiltration and activity in murine models (49). In a murine model of BRCA1-deficient TNBC, antitumor efficacy was due to the olaparib-induced specific recruitment of CD8+ T cells through the cGAS-STING pathway (51). PARPi have also been shown to enhance immunosuppression by upregulating PD-L1 expression in breast cancer cell lines through the inactivation of GSK3β (52). A high TMB, neoantigen release, and upregulation of PD-L1 expression driven by PARP inhibition suggest a microenvironment of increased immunogenicity; such microenvironment presents a potential opportune role for ICB and improved prognosis (3,24,53).
In a similar manner, BRCA1/2 and certain HR deficient tumors have been associated with a higher tumor-specific neoantigen load, as well as, higher TIL and PD-1/L-1 expression than HR-proficient tumors (54). It has also been suggested that BRCA1/2 deficient tumors promote activation of the cGAS-STING pathway (55). CTLA-4 has also been shown to suppress the antitumor immune response downstream from PARP inhibition. Although the use of CTLA-4 antibodies in murine models of ovarian cancer have shown relative resistance, preclinical studies in BRCA1-deficient mice with ovarian cancer have achieved improved survival with the combination of veliparib and anti-CTLA-4. This synergistic response has been associated with increased T-cell infiltration and IFN-y production in the peritoneal microenvironment. Both the elevated levels of IFN-y in human BRCA1 cells treated with PARPi and the expression of CTLA-4 in ascites of patients with ovarian cancer support the rationale for combining these types of agents (56). Currently, two phase 1 trials combining PARPi and anti-CTLA-4 therapy are underway: while one combines tremelimumab and olaparib in gBRCAm recurrent ovarian cancers (NCT02571725)V, the other combines radiation with durvalumab +/− tremelimumab or olaparib in SCLC (NCT03923270)VI (53, 54).
In line with the potential for catastrophic DNA damage to promote immune priming, preclinical studies in breast, ovarian and colorectal murine models have showed promising antitumor activity and treatment efficacy directly associated with high TMB when combining anti-PD-1/L1 with olaparib, talazoparib and rucaparib, respectively (58).
Thus far, two phase II trials have obtained early results from the combination of PARPi and ICB agents. In one hand, the MEDIOLA phase II basket trial studied the combination of olaparib and durvalumab for advanced solid cancers, including triple negative breast (TNBC), ovarian, cervical and uterine cancers (NCT02734004)VII. For patients with germline BRCA1/2 mutations and platinum-sensitive disease, ORR and disease control rate at 12 weeks were 63% and 81%, respectively. The ORR of patients pre-treated with one or two prior lines of chemotherapy was 68% (59). On the other hand, the open-label, phase II TOPACIO trial (NCT02657889)VIII assessed the effects of niraparib in combination with pembrolizumab (200 mg oral daily of niraparib and 200 mg IV on day 1 of each 21-day cycle of pembrolizumab) in women with advanced or metastatic TNBC or recurrent ovarian carcinoma, irrespective of BRCA1/2 status. As anticipated, patients with BRCA1/2 mutated cancers had better ORR (45%) and disease control rates (73%) compared to the entire study cohort (25% and 68%, respectively). However, the TOPACIO trial showed clinical benefit beyond what is expected with monotherapy in patients holding the BRCA wild type gene and in patients that are HR-proficient (ORR 24% and 27%, respectively) (60). Early data from this trial suggested that the combination of PARP inhibitor and anti-PD-1/L1 may be beneficial in patients regardless of mutational status or even in the case of platinum sensitivity (61). In both of these trials, the most common grade 3 toxicities included anemia, fatigue and thrombocytopenia, as well as, immune-related toxicities at rates comparable to single-agent PARPi and PD-1/L1 inhibitor therapies (48,60). Further studies combining PARPi and immunotherapeutic agents are shown in Table 1.
Table 1.
Current trials with PARP inhibitor and immunotherapy combination
| Combination | Trial | Population | |
|---|---|---|---|
| olaparib + durvalumab | NCT03594396 | Phase I/II | Resectable stage II/III TNBC |
| MEDIPAC (NCT03772561) | Phase I | Advanced or metastatic solid tumors | |
| NCT03842228 | Phase I | DDR-mutated unresectable, advanced or metastatic solid tumors | |
| NCT03775486 | Phase II | Metastatic NSCLC | |
| NCT03579784 | Phase II | Unresectable or recurrent gastric carcinoma | |
| NCT03810105 | Phase II | Recurrent prostate cancer | |
| DOLAF (NCT04053322) | Phase II | Locally advanced or metastatic ER+ HER2- breast cancer | |
| DAPPER (NCT03851614) | Phase II | Locally advanced or metastatic MMR proficient CRC, pancreatic adenocarcinoma or leiomyosarcoma | |
| DOMEC (NCT03951415) | Phase II | Recurrent, refractory or metastatic endometrial cancer or carcinosarcoma of the endometrium | |
| DORA (NCT03167619) | Phase II | Locally advanced or metastatic platinum-treated TNBC | |
| MEDIOLA (NCT02734004) | Phase II | gBRCAm platinum-sensitive ovarian cancer; gBRCAm HER2- breast cancer; platinum-resistant relapsed gastric cancer | |
| NCT03801369 | Phase II | Metastatic TNBC | |
| BAYOU (NCT03459846) | Phase II | Advanced or metastatic platinum-ineligible urothelial carcinoma | |
| NCT02484404 | Phase II | Advanced, recurrent or metastatic ovarian, TNBC, lung, prostate, CRC or solid tumors | |
| NEODURVARIB (NCT03534492) | Phase II | Resectable urothelial cancer | |
| DUO-O (NCT03737643) | Phase III | Newly diagnosed advanced ovarian, fallopian tube or primary peritoneal carcinoma or carcinosarcoma | |
| DUO-E (NCT04269200) | Phase III | Newly diagnosed advanced or recurrent endometrial carcinoma | |
| olaparib + pembrolizumab | KEYNOTE-365 (NCT02861573) | Umbrella study | Previously treated mCRPC |
| olaparib + tremelimumab | NCT02571725 | Phase I | BRCA1/2 mutated recurrent ovarian cancer |
| olaparib + tremelimumab + durvalumab | NCT02953457 | Phase I/II | DDR mutated recurrent ovarian, fallopian tube or primary peritoneal cancer |
| niraparib + pembrolizumab | TOPACIO-KEYNOTE 162 (NCT02657889) | Phase II | Advanced or metastatic TNBC or platinum-resistant ovarian cancer |
| niraparib + PD-1 inhibitor | NCT03308942 | Phase II | Locally advanced or metastatic NSCLC |
| niraparib + TSR-042 niraparib + TSR-042 + platinum-based therapy | NCT03307785 NCT03602859 | Phase I/II Phase III | Solid tumors Nonmucinous epithelial ovarian cancer |
| niraparib + atezolizumab | ANITA (NCT03598270) | Phase III | Recurrent ovarian, fallopian tube, or primary peritoneal carcinoma |
| rucaparib + nivolumab | NCT03572478 | Phase I/II | mCRPC or recurrent endometrial cancer |
| ATHENA (NCT03522246) | Phase III | Front line ovarian cancer | |
| CheckMate 9KD (NCT03338790) | Phase III | mCRPC | |
| rucaparib + atezolizumab | ARIANES (NCT04276376) | Phase II | DDR-deficient or platinum sensitive solid tumors |
| rucaparib + atezolizumab | EndoBARR (NCT03694262) | Phase II | Recurrent progressive endometrial carcinoma |
| talazoparib + avelumab | Javelin PARP Medley (NCT03330405) | Phase Ib/II | Previously treated advanced solid tumors |
| Javelin BRCA/ATM (NCT03565991) | BRCA/ATM-mutant solid tumors | ||
| Javelin BRCA/ATM (NCT03637491) | Ras-mutant solid tumors | ||
| Javelin Ovarian PARP 100 (NCT03642132) | Phase III | Front line ovarian cancer | |
| BGB-A317 + BGB-290 | NCT02660034 | Basket study | Ovarian cancer, TNBC, mCRPC, bladder cancer, SCLC, HER2 (−) gastric cancer, pancreatic cancer, other solid tumors |
| veliparib + atezolizumab | NCT02849496 | HER2 (−), BRCA-mutant TNBC | |
| pamiparib + tislelizumab | NCT02660034 | Phase I | Previously treated advanced solid tumors |
Abbreviations: MMR (mismatch repair), CRC (colorectal cancer), mCRPC (metastatic castration-resistant prostate cancer), TNBC (triple-negative breast cancer), SCLC (small cell lung cancer)
Key considerations when combining PARPi and immune checkpoint inhibitors
Patient Selection
To maximize the number of patients who may benefit from combined PARP and immune checkpoint inhibition, we must optimize patient selection through the use of response and resistance predictive biomarkers (9). While tumors with other known mechanisms of HR deficiency or “BRCAness” may also benefit from exploiting the synthetic lethality relationship with PARP inhibitors beyond BRCA1/2 mutations, empirically targeting other HRR alterations has yet to show a comparable level of clinical benefit (15). What seems to be even more challenging is the definition of robust response predictive biomarker for the combination of PARPi and PD-1/L1 inhibitors since it will probably require a combination of genomics and the functionality of different biomarkers.
The significance of tumor type, platinum status and molecular subtype has been assessed in trials evaluating PARPi/IO combination therapies in ovarian, breast and prostate cancers. While the TOPACIO and MEDIOLA trials suggest potential treatment efficacy in HR-proficient patients in advanced breast and ovarian cancers, similar results have yet to be seen in CRPC (62). While an early trial suggested that patients with CRPC with HRR defects showed greater benefit with the olaparib/durvalumab combination than patients with intact or unknown DDR status (PFS 16.1 months v 4.8 months), these early clinical data have not been recapitulated in larger clinical studies (63). The reasons for this are not currently evident. Early studies suggested that patients with fewer peripheral myeloid-derived suppressor cells and with alterations in DDR genes were more likely to respond (63). Early changes in circulating tumor cell counts and in both innate and adaptive immune characteristics were also associated with response. Similar translational studies from a larger group of patients to investigate the underlying mechanisms of response and resistance to this PARPi/IO combination in CRPC will be important, for example to determine if there is an inherent immune exclusion that is specific to CRPC that is not apparent in breast and ovarian cancers.
While the majority of trials have assessed or are evaluating the PARPi/IO combination in platinum-sensitive, high grade serous ovarian cancer, the TOPACIO study is one of only a few trials that have included platinum-resistant patients. Yet, it remains to be defined how the response to the combination of PARPi and ICB would be in clear cell and endometrioid ovarian cell types that are refractory to platinum-based chemotherapy
Tumor type
Several strategies have been utilized to select patients for the combination of ICB with PARPi or other inhibitors of DDR pathways. Thus far, early phase clinical trials have been initiated in multiple tumor types, including both BRCA1/2-lineage (e.g. breast, ovarian and prostate cancers) and non-BRCA1/2-lineage tumor types. To date, combinations of PARPi with ICB have yet to demonstrate meaningful antitumor efficacy in early phase trials recruiting non-BRCA lineage tumors, such as small cell lung cancer and gastric cancer, where generally low response rates have been reported across studies (64). Yet, amongst these trials, reports of durable responders who do not harbor non-BRCA1/2 tumors and/or underlying molecular markers of HRD have been noted, suggesting that other unknown factors are sensitizing these tumors to the combination (64,65). In a phase II trial of SCLC treated with durvalumab and olaparib, despite the low objective response rate (ORR) of only 10.5% in the overall trial cohort, clinical benefit with durable responses lasting > 8 months was observed in 2 patients who had previously received ICB (64).
DNA damage repair pathway genes, HRD status
Across reported trials investigating combinations of PARPi + ICB, the presence of BRCA1/2 mutation, HR deficiency, or other alterations in DDR related genes appear to be associated with treatment response. The phase II MEDIOLA trial showed high ORR (63%) and DCR (80%) for gBRCA1/2-mutant breast and ovarian cancer treated with durvalumab plus olaparib (59). In metastatic CRPC, durvalumab plus olaparib showed prostate specific antigen (PSA) responses in 71% (12/17) of patients of prostate cancer group (63). 30% of responders harbored germline alterations in DDR related genes (1 patient with deleterious mutation in NBN and 3 patients with frameshift indels in BRCA2), while 17% of responders were homozygous for somatic BRCA2 alterations. Of these responding patients, three patients -all of whom harbored somatic or germline mutations in HRR related genes- had durable responses exceeding 12 months (63). Despite these promising early clinical data, larger studies did not appear to recapitulate this high PSA response rate (64). In a phase I/II study of the same drug combination in SCLC, the sole patient who achieved a RECIST complete response had a deleterious somatic BRCA1 mutation (NCT02484404)IX (66). The ultimate challenge will be to determine whether these responses are due to the antitumor activity of the PARPi or the respective contribution of the PD-1/L1 inhibitor. Establishing the contribution of components remains a key challenge for the PARPi and ICB combination.
Interestingly, the response rates in the TOPACIO trial were similar regardless of BRCA mutation status or HRD assessed using the Myriad Mychoice® HRD test (67). Other markers of HRD such as BROCA targeted sequencing and RAD51 immunohistochemistry have also failed to discriminate between responders and non-responders. ORR in the BRCA-wildtype/HR-proficient group was 19%, similar to the ORR seen in the BRCA-mutated group (18%) and the HR-deficient group (14%). Surprisingly, out of 8 patients with durable responses greater than 6 months, 5 patients were BRCA-wildtype (67). The investigators were subsequently able to demonstrate that positivity for a specific mutational signature associated with HRD, termed signature 3 (Sig3), was predictive for clinical benefit and improved PFS after treatment with niraparib and pembrolizumab (median PFS of 5.0 months v 2.2 months for Sig3-positive and negative patients, respectively; HR 0.37, 95% CI 0.17–0.80, p=0.0005) (68). Sig 3 is characterized by a high number of larger deletions (up to 50 base pairs) with overlapping microhomology at breakpoint junctions, reflecting dependence on NHEJ or microhomology-mediated end joining mechanisms of DNA repair, leading to retained sensitivity to PARPi from non-restored or incompletely restored HR in platinum-resistant ovarian cancers (67). Single-cell imaging analysis on the TOPACIO trial further showed that Sig3-positive tumors displayed higher PD-L1 expression on tumor cells and macrophages, suggesting increased immunogenicity in these tumors (69).
Platinum sensitivity
Platinum sensitivity, irrespective of underlying BRCA1/2 mutation status, has been well recognized as a biomarker for PARPi response in ovarian cancer, leading to the regulatory approval of olaparib, niraparib and rucaparib monotherapy as maintenance therapy in platinum-sensitive recurrent ovarian cancer (28,70,71). Similarly, in BRCA1/2-mutated pancreatic cancer, prior platinum-sensitivity was noted amongst all documented rucaparib-responders on an open-label phase II trial (NCT02042378)X (72).
Amongst reported trials, limited clinical efficacy has been demonstrated for PARPi + ICB combinations in platinum-resistant ovarian and gastric cancer, with these cohorts demonstrating ORR of 14% (NCT02484404) and 10% (NCT02734004) for the durvalumab plus olaparib combination (73,74). Conversely, in updated results from the phase II MEDIOLA trial of 32 platinum-sensitive ovarian cancer patients, a 12-week disease control rate of 81% and ORR of 63% were reported (75). However, as all patients recruited harbored germline BRCA1/2 mutations, the precise role of platinum-sensitivity in predicting response to PARPi and immune checkpoint inhibition strategies in the context of BRCA1/2-wildtype status remains unclear, and few reported trials have recruited platinum-sensitive BRCA1/2-wildtype ovarian cancer patients to such combinations at present.
Notably, on the TOPACIO trial, differences in immune microenvironmental gene expression patterns were observed between chemotherapy-naïve and platinum-pre-treated tumor samples. Platinum-exposed tumors displayed higher scores for immune-related pathways as well as higher immune cell-type scores on RNA sequencing using a Nanostring platform (67). Single cell imaging also confirmed that platinum-treated tumors had higher proportions of immune cells comprising primarily of macrophages, CD4+ and CD8+ T cells, compared to the chemotherapy-naïve ones. However, these differences in the immune phenotype between platinum-treated and chemotherapy-naïve samples were not associated with differential responses to niraparib plus pembrolizumab therapy (67).
Immune profiling
Several trials have reported correlative immune analysis of on-treatment tumor and blood sampling. For instance, despite the fact that Sig3 positivity was identified as a surrogate for enhanced immunogenicity and HRD, as well as, a potential predictive biomarker of response, Sig3 positivity failed to capture all responders for the niraparib plus pembrolizumab combination in the TOPACIO trial. Immunogenomic profiling and multiplexed single-cell imaging performed on tumor samples retrieved during the course of the same trial made possible the identification of a positive immune score as a marker of interferon-primed exhausted effector CD8+ T-cells in the tumor microenvironment. Termed “combined score”, the presence of the immune score and/or Sig3 positivity captured all objective response (p=0.01), and it was correlated with PFS (HR 0.32 (95%CI 0.15–0.70, p = 0.002). In contrast, the absence of both markers was associated with non-response to niraparib plus pembrolizumab. On single-cell spatial imaging, prominent interactions between PD-L1 positive macrophages/tumor cells with exhausted CD8+ T cells were noted in treatment responders and in Sig3-positive tumors, further highlighting the role of exhausted CD8+ T cells in determining antitumor responses to the niraparib plus pembrolizumab combination (67).
Another phase II trial in recurrent ovarian cancer that studied the combination of durvalumab and olaparib showed significantly upregulation of IFNγ, CXCL9 and CXCL10, as well as, an increase in systemic IFNγ and TNFα production (76). Although increased systemic IFNγ correlated with improved PFS (HR 0.37, p=0.02) and clinical benefit, high local IFNγ levels did not correlate with clinical benefit (76). Furthermore, a positive baseline IFN-γ signature or baseline 18-gene immune signature did not correlate with treatment response (77). Although treatment with durvalumab plus olaparib in SCLC increased concentrations of IFNγ, IL-6 and IL-10 indicating immune activation, higher cytokine levels at baseline -and their change during treatment- did not correlate significantly with clinical benefit (66). In recurrent ovarian cancer, two other markers, higher TILs at baseline and immunoreactive (C2 subtype) by transcriptomic analysis, were noted to be significantly associated with clinical benefit to durvalumab plus olaparib (76). The correlation of high TILs with antitumor response to PARPi + ICB combinations has similarly been reported in SCLC, whereby regressing tumors had pre-existing dense T cell infiltration and further increased after exposure to durvalumab plus olaparib (66). Conversely, non-responders displayed an immune desert phenotype (78). Other immunological biomarkers of treatment response were described in the phase II trial of olaparib and durvalumab in CRPC (NCT02484404)IX. Patients with lower baseline levels of myeloid-derived suppressor cells (MDSCs) and increased markers of T cell activation (namely increased Ki67+ PDL1+ cells within CD4+ and CD8+ T cell populations and increased HLA-DR expression) were shown to have prolonged PFS (p=0.04) (65). Within the first 15 days of treatment, early increases in the expression of CD83 on CD141+ dendritic cells, which reflects dendritic cell maturity and support of Th1 polarization, were also associated with longer PFS (79).
PD-L1 expression and TMB have been identified as putative predictive biomarkers of response for ICB therapy selection in NSCLC, however, these have not been consistently shown to have predictive potential in the context of PARPi and ICB combination strategies. Although Sig3 positive tumors in the TOPACIO trial displayed higher PD-L1 expression on tumor cells and macrophages, neither PD-L1 expression itself, nor TMB correlated with treatment benefit (67). Similarly, in SCLC and prostate cancer, no clear correlation between PD-L1 expression and treatment response has been demonstrated (65,66).
Concluding Remarks
Despite FDA approval for use of PARP inhibitors in HR deficient CRPC and BRCA1/2 mutated ovarian, breast and pancreatic tumors, the efficacy of single agent use and combination therapy has been limited by drug resistance and toxicity. Similarly, durable responses with ICB single therapy have been limited to a minority of patients and tumor types. Robust preclinical data have suggested activation of the cGAS-STING pathway by PARP inhibition, shifting the tumor immune microenvironment from one of low-level, chronic inflammation to high TMB and PD-L1 activation —thereby creating an opportune environment to exploit ICB. This mechanistic rationale for the synergistic combination of PARPi and ICB has been supported by clinical trials showing improved survival outcomes in breast, ovarian, prostate and SCLC. The addition of PD-1/L-1 blockade to PARP inhibitor therapy has proven to be a well-tolerated combination amongst the available clinical data. Moreover, there is evidence to support utilizing this combination treatment in patients with wildtype BRCA1/2.
While BRCA1/2 and other HRR mutations encompassing “BRCAness” have been used as predictive biomarkers of response for PARPi, the utility of PARP inhibition and combination therapy has shown efficacy beyond those with BRCA1/2 deficiencies. In order to optimize the potential of combination anticancer therapy, the focus should move towards more sophisticated strategies for patient selection. The TOPACIO and MEDIOLA trials compare outcomes by tumor type, platinum sensitivity and molecular subtypes; however, the results of these subgroup analyses have not been consistent across tumor subtypes. Similarly, high TMB and PD-1 are not perfect predictors of response for IO therapy. Attention to elevated immune profiling and biomarkers of both treatment response and resistance is needed to guide clinical trials towards outcomes of relevant depth and duration beyond HRD status. Several important questions remain, as noted in the Outstanding Questions section below.
PARP resistance continues to be a clinically significant phenomenon that we are still working on deciphering. Looking at additional agents—chemotherapy, radiation, targeted therapy— to enhance antitumor efficacy by rationally adding combination treatments is pertinent. As with any combination treatment, efficacy would need to be balanced against safety and tolerability.
Outstanding Questions.
How can we compare the predictive potential of response predictive biomarkers, positive immune scores or single-call spatial imaging for PARPi/ICB?
What other robust response predictive biomarkers can be used to optimize patient selection and maximize the number of patients who may benefit from for PARPi/IO combinations?
What other strategies for patient selection can be used to consistently compare patient outcomes across tumor subtypes?
How does the clinical benefit between patients with HRR defects and patients with intact or unknown DDR status compare in larger clinical studies that use different types of combination therapies?
How does platinum-resistant patients respond to the combination of PARPi and ICB in the context of BRCA1/2-wildtype status?
Highlights.
Poly(ADP-ribose) polymerase inhibitors (PARPi) are approved by the US Food and Drug Administration as monotherapy for ovarian, breast and pancreatic tumors harboring BRCA1/2 mutations, as well as castration-resistant prostate cancers with homologous recombination repair (HRR) mutations.
Immune checkpoint blockade (ICB) -through the use of programmed cell death 1/programmed cell death ligand 1 (PD-1/L1) inhibitors- has demonstrated durable responses in different solid tumors. Several PD-1/L1 inhibitors are FDA approved for approved for non-small cell lung cancer and metastatic urothelial carcinoma and metastatic cutaneous squamous cell carcinoma.
Preclinical data support a synergistic relationship between PARPi and ICB; PARPi-mediated DNA damage modulates the tumor immune microenvironment though different molecular and cellular mechanisms, such as increasing genomic instability, immune pathway activation, and PD-L1 expression on cancer cells, all of which may increase responsiveness to ICB.
Early data from a phase I trial suggested that the combination of PARP inhibitor and anti-PD-1/L1 may be beneficial in patients regardless of mutational status or even in the case of platinum sensitivity. Thus far, it has been shown that combining PARPi with ICB may be a safe and well tolerated strategy, with the potential to improve survival outcomes in a broad population of patients with BRCA1/2 mutations.
Acknowledgments
Melissa Pham is supported by the National Institute of Health (T32 CA101642/CA/NCI NIH HHS/United States). Natalie Y.L. Ngoi is supported by the National Medical Research Council, Singapore (MOH-FLWSHP19may-0006). David S.P. Tan is supported by the National Medical Research Council, Singapore (CSAINV16may008), and has received charitable research funding from the Pangestu Family Foundation Gynaecological Cancer Research Fund. Guang Peng is supported by MD Anderson Cancer Center Support grant (NCI CA016672), the US Department of Defense Research Program (OC140431), and the National Institute of Health (R01 CA181663). Timothy A. Yap is supported by MD Anderson Cancer Center Support grant (NIH/NCI P30 CA016672), the US Department of Defense Ovarian Cancer Research Program (OC200482) and the V Foundation Clinical Scholar Program (VC2020-001).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Resources
References
- 1.Brown JS, O’Carrigan B, Jackson SP, Yap TA. Targeting DNA repair in cancer: Beyond PARP inhibitors. Cancer Discov. 2017;7(1):20–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pardoll D. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, et al. PARPi triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Res. 2019;79(2):311–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cerrato A, Morra F, Celetti A. Use of poly ADP-ribose polymerase [PARP] inhibitors in cancer cells bearing DDR defects: The rationale for their inclusion in the clinic. J Exp Clin Cancer Res [Internet]. 2016;35(1):1–13. Available from: 10.1186/s13046-016-0456-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Satoh M, Lindahl T. Role of poly(ADP-ribose) formation in DNA repair. Nature. 1992;356(6367):356–8. [DOI] [PubMed] [Google Scholar]
- 6.Howard S, Yanez D, Stark J. DNA damage response factors from diverse pathways, including DNA crosslink repair, mediate alternative end joining. PLoS Genet. 2015;11(1):e1004943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huarte E, Cubillos-Ruiz JR, Nesbeth YC, Scarlett UK, Martinez DG, Buckanovich RJ, et al. Depletion of dendritic cells delays ovarian cancer progression by boosting antitumor immunity. Cancer Res [Internet]. 2008/09/05. 2008;68(18):7684–91. Available from: https://www.ncbi.nlm.nih.gov/pubmed/18768667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barkauskaite E, Jankevicius G, Ahel I. Structures and Mechanisms of Enzymes Employed in the Synthesis and Degradation of PARP-Dependent Protein ADP-Ribosylation. Mol Cell [Internet]. 2015;58(6):935–46. Available from: 10.1016/j.molcel.2015.05.007 [DOI] [PubMed] [Google Scholar]
- 9.Lord CJ, Ashworth A. PARP Inhibitors: The First Synthetic Lethal Targeted Therapy. Science (80- ) [Internet]. 2017;355(6330):1152–8. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6175050/pdf/emss-79613.pdf [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murai J, Huang SYN, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72(21):5588–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rouleau M, Patel A, Hendzel M, Kaufmann S, Poirier G. PARP inhibition: PARP1 and beyond. Nat Rev Cacner. 2010;10:293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chaudhuri A, Nussenzweig A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling Arnab. Nat Rev Mol Cell Biol. 2017;18(10):610–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;7(2):263–72. [DOI] [PubMed] [Google Scholar]
- 14.Moynahan ME, Cui TY, Jasin M. Homology-directed DNA repair, mitomycin-C resistance, and chromosome stability is restored with correction of a Brca1 mutation. Cancer Res. 2001;61(12):4842–50. [PubMed] [Google Scholar]
- 15.Brown JS, O’Carrigan B, Jackson SP, Yap TA. Targeting DNA repair in cancer: Beyond PARP inhibitors. Cancer Discov. 2017;7(1):20–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fong P, Boss D, Yap T, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of Poly(ADP-Ribose) Polymerase in Tumors from BRCA Mutation Carriers. N Engl J Med. 2009;361(2):123–34. [DOI] [PubMed] [Google Scholar]
- 17.Pearl L, Schierz A, Ward S, Al-Lazikani B, Pearl F. Therapeutic opportunities within the DNA damage response. Nat Rev Cancer. 2015;15(3):166–80. [DOI] [PubMed] [Google Scholar]
- 18.Farmer H, McCabe N, Lord C, Tutt A, Johnson D, Richardson T, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–21. [DOI] [PubMed] [Google Scholar]
- 19.Bryant H, Schultz N, Thomas H, Parker K, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–7. [DOI] [PubMed] [Google Scholar]
- 20.Shen Y, Rehman FL, Feng Y, Boshuizen J, Bajrami I. BMN 673 , a novel and highly potent PARP1 / 2 inhibitor for the treatment of human cancers with DNA repair deficiency. 2019;19(18):5003–15. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6485449/pdf/emss-54129.pdf [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murai J, Huang SN, Renaud A, Zhang Y, Ji J, Morris J, et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. 2015;13(2):433–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pommier Y, O’Connor M, de Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med. 2016;8(362):362ps17. [DOI] [PubMed] [Google Scholar]
- 23.Murai J. Targeting DNA repair adn replication stress in the treatment of ovarian cancer. Int J Clin Oncol. 2017;22(4):619–28. [DOI] [PubMed] [Google Scholar]
- 24.Patel M, Nowsheen S, Maraboyina S, Xia F. The role of poly(ADP-ribose) polymerase inhibitors in the treatment of cancer and methods to overcome resistance: a review. Cell Biosci [Internet]. 2020;10(1):1–12. Available from: 10.1186/s13578-020-00390-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mirza M, Coleman R, Gonzalez-Martin A, Moore K, Colombo N, Ray-Coquard I, et al. The forefront of ovarian cancer therapy: update on PARP inhibitors. Ann Oncol. 2020;S0923–7534(20):39891–4. [DOI] [PubMed] [Google Scholar]
- 26.Coleman RL, Fleming GF, Brady MF, Swisher EM, Steffensen KD, Friedlander M, et al. Veliparib with first-line chemotherapy and as maintenance therapy in ovarian cancer. N Engl J Med. 2019;381(25):2403–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moore K, Colombo N, Scambia G, Kim BG, Oaknin A, Friedlander M, et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2018;379(26):2495–505. [DOI] [PubMed] [Google Scholar]
- 28.Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154–64. [DOI] [PubMed] [Google Scholar]
- 29.Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377(6):523–33. [DOI] [PubMed] [Google Scholar]
- 30.Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373(18):1697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prasanna T, Wu F, Khanna KK, Yip D, Malik L, Dahlstrom JE, et al. Optimizing poly (ADP-ribose) polymerase inhibition through combined epigenetic and immunotherapy. Cancer Sci. 2018;109(11):3383–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jonsson P, Bandlamudi C, Cheng ML, Srinivasan P, Chavan SS, Friedman ND, et al. Tumour lineage shapes BRCA-mediated phenotypes. Nature. 2019;571(7766):576–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pilié PG, Gay CM, Byers LA, O’Connor MJ, Yap TA. PARP inhibitors: Extending benefit beyond BRCA-mutant cancers. Clin Cancer Res. 2019;25(13):3759–71. [DOI] [PubMed] [Google Scholar]
- 34.Hoos A. Development of immuno-oncology drugs - from CTLA4 to PD1 to the next generations. Nat Rev Drug Discov. 2016;15:235–47. [DOI] [PubMed] [Google Scholar]
- 35.Perkins D, Wang Z, Donovan C, He H, Mark D, Guan G, et al. Regulation of CTLA-4 expression during T cell activation. J Immunol. 1996;156(11):4154–9. [PubMed] [Google Scholar]
- 36.Thaxton JE, Li Z. To affinity and beyond: Harnessing the T cell receptor for cancer immunotherapy. Hum Vaccines Immunother. 2014;10(11):3313–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tivol E, Borriello F, Schweitzer A, Lynch W, Bluestone J, Sharpe A. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–7. [DOI] [PubMed] [Google Scholar]
- 38.Granier C, Karaki S, Roussel H, Badoual C, Tran T, Anson M, et al. Cancer immunotherapy: Rational and recent breakthroughs. Rev Med Interne. 2016;37(10):694–700. [DOI] [PubMed] [Google Scholar]
- 39.Velcheti V, Schalper K. Basic overview of current immunotherapy approaches in cancer. In: American Society of Clinical Oncology Educational Book. 2016. p. 298–308. [DOI] [PubMed] [Google Scholar]
- 40.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–51. [DOI] [PubMed] [Google Scholar]
- 41.Vaddepally RK, Kharel P, Pandey R, Garje R, Chandra AB. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers (Basel). 2020;12(3):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Z, Song W, Rubinstein M, Liu D. Recent updates in cancer immunotherapy: A comprehensive review and perspective of the 2018 China Cancer Immunotherapy Workshop in Beijing. J Hematol Oncol. 2018;11(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Migden MR, Rischin D, Schmults CD, Guminski A, Hauschild A, Lewis KD, et al. PD-1 blockade with cemiplimab in advanced cutaneous squamous-cell carcinoma. N Engl J Med. 2018;379(4):341–51. [DOI] [PubMed] [Google Scholar]
- 44.Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16(11):2598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pakish JB, Jazaeri AA. Immunotherapy in Gynecologic Cancers: Are We There Yet? Curr Treat Options Oncol. 2017;18(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Levinson K, Dorigo O, Rubin K, Moore K. Immunotherapy in Gynecologic Cancers: What We Know Now and Where We Are Headed. Am Soc Clin Oncol Educ B. 2019;(39):e126–40. [DOI] [PubMed] [Google Scholar]
- 47.Lee L, Matulonis U. Immunotherapy and radiation combinatorial trials in gynecologic cancer: A potential synergy? Gynecol Oncol. 2019;154(1):236–45. [DOI] [PubMed] [Google Scholar]
- 48.Stewart RA, Pilie PG, Yap TA. Development of PARP and immune-checkpoint inhibitor combinations. Cancer Res. 2018;78(24):6717–25. [DOI] [PubMed] [Google Scholar]
- 49.Chen Q, Sun L, Chen Z. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol. 2016;17:1142–9. [DOI] [PubMed] [Google Scholar]
- 50.Chabanon RM, Muirhead G, Krastev DB, Adam J, Morel D, Garrido M, et al. PARP inhibition enhances tumor cell–intrinsic immunity in ERCC1-deficient non– small cell lung cancer. J Clin Invest. 2019;129(3):1211–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pantelidou C, Sonzogni O, Taveira MDO, Mehta AK, Kothari A, Wang D, et al. Parp inhibitor efficacy depends on CD8+ T-cell recruitment via intratumoral sting pathway activation in brca-deficient models of triple-negative breast cancer. Cancer Discov. 2019;9(6):722–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jiao S, Xia W, Yamaguchi H, Wei Y, Chen M-K, Hsu J-M, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppresion. Clin Cancer Res. 2017;23(14):3711–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gubin MM, Artyomov MN, Mardis ER, Schreiber RD. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest. 2015;125(9):3413–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Strickland KC, Howitt BE, Shukla SA, Rodig S, Ritterhouse LL, Liu JF, et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget. 2016;7(12):13587–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barber GN. STING: Infection, inflammation and cancer. Nat Rev Immunol. 2015;15(12):760–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Higuchi T, Flies D, Marjon N, Mantia-Smaldone G, Ronner L, Gimotty P, et al. CTLA-4 Blockade Synergizes Therapeutically with PARP Inhibition in BRCA1-Deficient Ovarian Cancer. Cancer Immunol Res. 2015;3(11):1257–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Knelson EH, Patel SA, Sands JM. Parp inhibitors in small‐cell lung cancer: Rational combinations to improve responses. Cancers (Basel). 2021;13(4):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mosely SIS, Prime JE, Sainson RCA, Koopmann JO, Wang DYQ, Greenawalt DM, et al. Rational selection of syngeneic preclinical tumor models for immunotherapeutic drug discovery. Cancer Immunol Res. 2017;5(1):29–41. [DOI] [PubMed] [Google Scholar]
- 59.Drew Y, de Jonge M, Hong S, Park Y, Wolfer A, Brown J et al. An open-label, phase II basket study of olaparib and durvalumab (MEDIOLA): results in germline BRCA-mutated (gBRCAm) platinum-sensitive relapsed (PSR) ovarian cancer (OC). Gynecol Oncol. 2018;149:246–7. [Google Scholar]
- 60.Konstantinopoulos PA, Waggoner S, Vidal GA, Mita M, Moroney JW, Holloway R, et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination with Pembrolizumab in Patients with Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019;5(8):1141–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boussios S, Karihtala P, Moschetta M, Karathanasi A, Sadauskaite A, Rassy E, et al. Combined strategies with poly (ADP-ribose) polymerase (PARP) inhibitors for the treatment of ovarian cancer: A literature review. Diagnostics. 2019;9(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kaufman B, Shapira-Frommer R, Schmutzler R, Audeh M, Friedlander M, Balmana J, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33:244–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Karzai F, Madan R, Owens H, Couvillon A, Hankin A, Williams M et al. A phase 2 study ofolaparib and durvalumab in metastatic castrate-resistant prostate cancer (mCRPC) in an unselected population. J Clin Oncol. 2018;36:163. [Google Scholar]
- 64.Peyraud F, Italiano A. Combined parp inhibition and immune checkpoint therapy in solid tumors. Cancers (Basel). 2020;12(6):1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Karzai F, VanderWeele D, Madan R, et al. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J Immunother Cancer. 2018;6:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thomas A, Vilimas R, Trindade C, et al. Duvalumab in Combination with Olaparib in Patients with Relapsed SCLC: Results from a Phase II Study. J Thorac Oncol. 2019;14:1447–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Farkkila A, Gulhan D, Casado J, et al. Immunogenomic profiling determines responses to combined PARP and PD-1 inhibition in ovarian cancer. Nat Commun. 2020;11:1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Polak P, Kim J, Braunstein L, et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat Genet. 2017;49:1476–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Elbaek C, Petrosius V, Sorensen C. WEE1 kinase limits CDK activities to safeguard DNA replication and mitotic entry. Mutat Res. 2020;819–820:111694. [DOI] [PubMed] [Google Scholar]
- 70.Coleman R, Oza A, Lorusso D, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390:1949–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012;366:1382–92. [DOI] [PubMed] [Google Scholar]
- 72.Shroff R, Hendifar A, McWilliams R, et al. Rucaparib Monotherapy in Patients with Pancreatic Cancer and a Known Deleterious BRCA Mutation. JCO Precis Oncol. 2018;1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zimmer A, Nichols E, Cimino-Mathews A, Peer C, Cao L, Lee M, et al. Phase I study of the PD-L1 inhibitor, durvalumab (MEDI4736; D) in combination with a PARP inhibitor, olaparib (O) or a VEGFR inhibitor, cediranib (C) in women’s cancers (NCT02484404). J Immunother Cancer. 2019;7(1):197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bang Y, Kaufman B, Geva R, Stemmer S, Hong S, Lee J, et al. An open-label, phase II basket study of olaparib and durvalumab (MEDIOLA): Results in patients with relapsed gastric cancer. J Clin Oncol. 2019;37(4). [Google Scholar]
- 75.Drew Y, Kaufman B, Banerjee S, Lortholary A, Hong SH, Park YH, et al. Phase II study of olaparib + durvalumab (MEDIOLA): Updated results in germline BRCA-mutated platinum-sensitive relapsed (PSR) ovarian cancer (OC). Ann Oncol [Internet]. 2019;30(October):v485–6. Available from: 10.1093/annonc/mdz253.016 [DOI] [Google Scholar]
- 76.Lampert E, Zimmer A, Padget M, Cimino-Mathews A, Jayakumar N, Liu Y, et al. Combination of PARP Inhibitor Olaparib, and PD-L1 Inhibitor Durvalumab, in Recurrent Ovarian Cancer: a Proof-of-Concept Phase II Study. Clin Cancer Res. 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ayers M, Lunceford J, Nebozhyn M, et al. IFN-gama-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127:2930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Domchek S, Postel-Vinay S, Im S, et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): an open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020;21:1155–64. [DOI] [PubMed] [Google Scholar]
- 79.Munster P, Mita M, Mahipal A, et al. First-In-Human Phase I Study of A Dual mTOR Kinase and DNA-PK Inhibitor (CC-115) In Advanced Malignancy. Cancer Manag Res. 2019;11:10463–76. [DOI] [PMC free article] [PubMed] [Google Scholar]