

Abstract
Despite the development of efficient anti–human immunodeficiency virus-1 (HIV-1) therapy, HIV-1 associated pathogens remain a major clinical problem. Human cytomegalovirus (CMV) is among the most common HIV-1 copathogens and one of the main causes of persistent immune activation associated with dysregulation of the immune system, cerebrovascular and cardiovascular pathologies, and premature aging. Here, we report on the development of dual-targeted drugs with activity against both HIV-1 and CMV. We synthesized seven compounds that constitute conjugates of molecules that suppress both pathogens. We showed that all seven compounds exhibit low cytotoxicity and efficiently inhibited both viruses in cell lines. Furthermore, we chose a representative compound and demonstrated that it efficiently suppressed replication of HIV-1 and CMV in human lymphoid tissue ex vivo coinfected with both viruses. Further development of such compounds may lead to the development of dual-targeted anti CMV/HIV-1 drugs.
Keywords: HIV-1, CMV, heterodimers, human tissues, viral diseases
1. INTRODUCTION
Since the start of the AIDS epidemic, coinfections with HIV-associated pathogens have become a major clinical problem [1]. In particular, human cytomegalovirus (CMV) is among the most common opportunistic infections in people living with HIV-1. Although CMV infects a wide range of the population, it is not overly pathogenic in immunocompetent people. However, in people living with HIV-1, CMV is associated with higher risk of severe non-AIDS-defining events, such as cerebrovascular and cardiovascular events [2], and non-AIDS related death. Moreover, CMV contributes significantly to immune activation, which causes dysregulation of the immune system and premature aging [3-5]. It was reported that CMV, whose seroprevalence has reached 90–100% in people living with HIV-1, has been associated with increased inflammation and inflammation-related morbidities (reviewed in [4, 6]). In coinfected individuals, HIV-1 infection leads to reactivation of CMV in blood and semen [7-10]. In turn, CMV worsens HIV-1 disease by increasing immune activation and inflammation even in ART-treated patients [3, 11-14]. Seminal shedding of CMV has been linked to higher HIV-1 viral load in semen [10, 15-17] and to increased risk of HIV-1 transmission [18-20] whether HIV-infected individuals are under treatment or not. These observations suggest that interventions aiming at reducing CMV shedding might be a meaningful HIV-1 prevention strategy in populations with high prevalence of CMV coinfection.
In this context, the use of dual-targeted antivirals against HIV-1 and CMV may give good results both for patients already infected with HIV-1 and for prevention of HIV-1 transmission. One of the ways to construct dual activity drugs is to make heterodimers of molecules that suppress corresponding pathogens. These heterodimers constitute a prodrug of parental drugs that provide certain advantages, for example a slow release of active compounds, which typically results in less toxicity than the combination of the parent drugs [21, 22].
Here, we report on the synthesis and the biological activity of heterodimers of AZT or 3TC, as typical HIV-1 reverse transcriptase inhibitors, and 1-[ω-(phenoxy)alkyl]uracil derivatives [23]. These derivatives were synthesized by our team few years ago and showed specific inhibitory properties against CMV replication in cell culture (EC50 5.5–12.0 μM). Unfortunately preclinical studies of the compounds were limited by their low solubility. So 1-[ω-(phenoxy)alkyl]uracil derivatives look perfect candidates for making prodrugs - heterodimers with NRTIs (AZT or 3TC).
2. MATERIAL AND METHODS
2.1. Chemical Synthesis
2.1.1. General
Commercial reagents Acros (Geel, Belgium), Sigma Aldrich (St. Louis, MO, USA) and Fluka (Bucharest, Romania) were used for the reactions; anhydrous solvents of high quality were used without additional purification. Silica Gel 60 0.040–0.063 mm (Merck, Darmstadt, Germany) was used for column chromatography. Silica Gel 60 F254 aluminum-backed plates (Merck, Darmstadt, Germany) were used for thin layer chromatography (TLC), and Silica Gel 60 F254 glass-backed plates (Merck, Darmstadt, Germany) for preparative layer chromatography (PLC). NMR spectra were registered on an AMX III-400 spectrometer (Bruker, Newark, Germany) with the working frequency of 400 MHz for 1H NMR (Me4Si as an internal standard for organic solvents) and 100.6 MHz for 13C NMR (with carbon-proton interaction decoupling).
High resolution mass spectra were measured on Bruker micrOTOF II instruments using electrospray ionization (ESI HRMS) using standart published conditions [24].
2.1.2. Compound Synthesis and Characterization.
2.1.2.1. Synthesis of compounds 1a-1f.
The synthesis of compounds 1a-d has been previously described (9). Compounds 1e and 1f were obtained in a similar way.
1-[10-(4-Bromophenoxy)decyl]pyrimidine-2,4(1H,3H)-dione (1e).
White solid; yield 82% (3.1 g, 7.32 mmol); mp 84-86 °C. Rf 0.50 (eluting with ethylacetate). 1H NMR (400 MHz, DMSO-d6), δ, ppm: 1.24-1.28 (10H, m, CH2 × 5), 1.36 (2H, q, J = 7.7 Hz, CH2), 1.54 (2H, q, J = 6.9 Hz, CH2), 1.67 (2H, q, J = 7.5 Hz, CH2), 3.62 (2H, t, J = 7.3 Hz, CH2), 3.91 (2H, t, J = 6.5 Hz, CH2), 5.53 (1H, dd, J = 7.8 and 2.3 Hz, H-5), 6.88 (2H, d, J = 9.0 Hz, H-3’, H-5’), 7.41 (2H, d, J = 9.1 Hz, H-2’, H-6’), 7.63 (1H, d, J = 7.8 Hz, H-6), 11.21 (1H, s, NH). 13C NMR (100.6 MHz, DMSO-d6), δ, ppm: 28.8, 29.2, 31.8, 31.9, 32.0, 32.1, 32.2, 32.3, 50.8, 71.1, 104.1, 115.1, 120.1, 135.4, 149.1, 154.3, 161.3, 167.1.
1-[12-(4-Bromophenoxy)dodecyl]pyrimidine-2,4(1H,3H)-dione (1f).
White solid; yield 80% (3.2 g, 7.09 mmol); mp 95-96.5 °C. Rf 0.54 (eluting with ethylacetate). 1H NMR (400 MHz, DMSO-d6), δ, ppm: 1.21-1.27 (14H, m, CH2 × 7), 1.37 (2H, q, J = 7.6 Hz, CH2), 1.54 (2H, q, J = 6.8 Hz, CH2), 1.67 (2H, q, J = 7.0 Hz, CH2), 3.62 (2H, t, J = 7.3 Hz, CH2), 3.92 (2H, t, J = 6.6 Hz, CH2), 5.52 (1H, dd, J = 7.9 and 2.2 Hz, H-5), 6.88 (2H, d, J = 9.0 Hz, H-3’, H-5’), 7.41 (2H, d, J = 9.0 Hz, H-2’, H-6’), 7.63 (1H, d, J = 7.8 Hz, H-6), 11.21 (1H, s, NH). 13C NMR (100.6 MHz, DMSO-d6), δ, ppm: 28.6, 29.2, 31.8, 31.9, 32.0, 32.1, 32.3, 50.8, 71.7, 104.1, 115.1, 120.1, 135.4, 149.1, 154.3, 161.3, 167.1.
2.1.2.2. General procedure for the synthesis of [3-[ω-(4-bromophenoxy)alkyl]-2,6-dioxo-3,6- dihydropyrimidin-1(2H)-yl]acetic acids 2a-f.
A mixture of 4.01 mmol of 1-[ω-(4- bromophenoxy)alkyl]uracil 1, 0.7 g (5.06 mmol) K2CO3 in 20 mL DMF was stirred at 80 °C for 1 hour, cooled to room temperature, and 0.5 ml (4.51 mmol) of ethyl bromoacetate was added. The resulting mixture was stirred at the same temperature for 20 hours. The reaction mass was evaporated in vacuo, the residue was treated with 50 mL of cold water and extracted with 1,2- dichloroethane (5 × 20 mL). The extract was evaporated under reduced pressure to give clear viscous oily liquid, which was dissolved in 15 mL of ethanol. LiOH (0.58 g, 24.22 mmol) and 10 ml of water were added to the resultant solution at room temperature under stirring. The resulting mixture was stirred at the same temperature for 24 hours. Ethanol was evaporated under reduced pressure, the residue was acidified with 6% aqueous hydrochloric acid, and the precipitate was filtered, washed on the filter with water and air dried. Yields of target acids were within 61-79%, after crystallization from a mixture ethyl acetate-hexane.
[3-[3-(4-bromophenoxy)propyl]-2,6-dioxo-3,6-dihydropyrimidin-1(2H)-yl]acetic acid (2a).
White solid; yield 61% (0.94 g, 2.45 mmol); mp 153-154 °C. Rf 0.57 (eluting with ethylacetate- iPrOH-40% aqv.NH4OH (6:9:5)). 1H NMR (400 MHz, DMSO-d6), δ, ppm: 2.04 (2H, q, J = 6.3 Hz, CH2), 3.90 (2H, t, J = 6.8 Hz, NCH2), 3.98 (2H, t, J = 6.0 Hz, OCH2), 4.42 (2H, s, CH2CO), 5.72 (1H, d, J = 7.9 Hz, H-5), 6.86 (2H, d, J = 8.9 Hz, H-3’, H-5’), 7.42 (2H, d, J = 8.9 Hz, H-2’, H-6’), 7.73 (1H, d, J = 7.9 Hz, H-6), 13.10 (1H, br.s, COOH). 13C NMR (100.6 MHz, DMSO-d6), δ, ppm: 31.1, 44.9, 49.9, 68.6, 103.3, 115.4, 120.1, 135.5, 148.2, 154.3, 161.0, 165.4, 172.5. HRMS: m/z[M + H]+ calcd for C15H15BrN2O5: 383.0237, found: 383.0237.
[3-[5-(4-bromophenoxy)pentyl]-2,6-dioxo-3,6-dihydropyrimidin-1(2H)-yl]acetic acid (2b).
White solid; yield 65% (1.07 g, 2.60 mmol); mp 144-146 °C. Rf 0.56 (eluting with ethylacetate- iPrOH-40% aqv.NH4OH (6:9:5)). 1H NMR (400 MHz, DMSO-d6), δ, ppm: 1.39 (2H, q, J = 6.6 Hz, CH2), 1.65 (2H, q, J = 7.4 Hz, CH2), 1.72 (2H, q, J = 7.3 Hz, CH2), 3.75 (2H, t, J = 7.1 Hz, NCH2), 3.93 (2H, t, J = 6.3 Hz, OCH2), 4.44 (2H, s, CH2CO), 5.74 (1H, d, J = 7.8 Hz, H-5), 6.89 (2H, d, J = 8.8 Hz, H-3’, H-5’), 7.42 (2H, d, J = 8.8 Hz, H-2’, H-6’), 7.77 (1H, d, J = 7.8 Hz, H-6), 12.90 (1H, br.s, COOH). 13C NMR (100.6 MHz, DMSO-d6), δ, ppm: 22.6, 28.37, 28.41, 41.9, 48.8, 67.8, 100.2, 112.1, 117.1, 132.4, 145.0, 151.2, 158.2, 162.3, 169.5. HRMS: m/z[M + H]+ calcd for C17H19BrN2O5: 411.0541, found: 411.0550.
[3-[6-(4-bromophenoxy)hexyl]-2,6-dioxo-3,6-dihydropyrimidin-1(2H)-yl]acetic acid (2c).
White solid; yield 79% (1.35 g, 3.17 mmol); mp 127-129 °C. Rf 0.45 (eluting with ethylacetate- iPrOH-40% aqv.NH4OH (6:9:5)). 1H NMR (400 MHz, DMSO-d6), δ, ppm: 1.30 (2H, q, J = 6.8Hz, CH2), 1.39 (2H, q, J = 6.8 Hz, CH2), 1.41 (2H, q, J = 6.6 Hz, CH2), 1.61 (2H, q, J = 7.1 Hz, CH2), 3.73 (2H, t, J = 7.1 Hz, NCH2), 3.93 (2H, t, J = 6.6 Hz, OCH2), 4.44 (2H, s, CH2CO), 5.73 (1H, d, J = 7.8 Hz, H-5), 6.89 (2H, d, J = 8.8 Hz, H-3’, H-5’), 7.42 (2H, d, J = 9.1 Hz, H-2’, H-6’), 7.76 (1H, d, J = 7.8 Hz, H-6), 12.89 (1H, br.s, COOH). 13C NMR (100.6 MHz, DMSO-d6), δ, ppm: 25.4, 25.8, 28.6, 28.7, 41.9, 48.9, 67.9, 100.2, 112.0, 117.1, 132.4, 145.0, 151.2, 158.3, 162.3, 169.5. HRMS: m/z[M + H]+ calcd for C18H21BrN2O5: 425.0707, found: 425.0707.
[3-[8-(4-bromophenoxy)octyl]-2,6-dioxo-3,6-dihydropyrimidin-1(2H)-yl]acetic acid (2d).
White solid; yield 68% (1.24 g, 2.74 mmol); mp 114-115.5 °C. Rf 0.47 (eluting with ethylacetate-iPrOH-40% aqv.NH4OH (6:9:5)). 1H NMR (400 MHz, DMSO-d6), δ, ppm: 1.25-1.31 (6H, m, 3×CH2), 1.37 (2H, q, J = 6.4 Hz, CH2), 1.58 (2H, q, J = 7.1 Hz, CH2), 1.68 (2H, q, J = 6.8 Hz, CH2), 3.71 (2H, t, J = 7.4 Hz, NCH2), 3.93 (2H, t, J = 6.6 Hz, OCH2), 4.44 (2H, s, CH2CO), 5.73 (1H, d, J = 7.8 Hz, H-5), 6.88 (2H, d, J = 8.8 Hz, H-3’, H-5’), 7.42 (2H, d, J = 9.1 Hz, H-2’, H-6’), 7.75 (1H, d, J = 7.8 Hz, H-6), 12.90 (1H, br.s, COOH). 13C NMR (100.6 MHz, DMSO-d6), δ, ppm: 25.7, 26.0, 28.6, 28.8, 28.9, 41.9, 48.9, 68.1, 100.2, 112.0, 117.0, 132.4, 145.0, 151.2, 158.3, 162.3, 169.5. HRMS: m/z[M + H]+ calcd for C20H25BrN2O5: 453.1020, found: 453.1027.
[3-[10-(4-bromophenoxy)decyl]-2,6-dioxo-3,6-dihydropyrimidin-1(2H)-yl]acetic acid (2e).
White solid; yield 61% (1.18 g, 2.45 mmol); mp 106-107.5 °C. Rf 0.49 (eluting with ethylacetate-iPrOH-40% aqv.NH4OH (6:9:5)). 1H NMR (400 MHz, DMSO-d6), δ, ppm: 1.23-1.29 (10H, m, 5×CH2), 1.37 (2H, q, J = 7.3 Hz, CH2), 1.57 (2H, q, J = 6.6 Hz, CH2), 1.67 (2H, q, J = 6.8 Hz, CH2), 3.71 (2H, t, J = 7.3 Hz, NCH2), 3.92 (2H, t, J = 6.6 Hz, OCH2), 4.44 (2H, s, CH2CO), 5.73 (1H, d, J = 7.8 Hz, H-5), 6.88 (2H, d, J = 9.0 Hz, H-3’, H-5’), 7.41 (2H, d, J = 9.0 Hz, H-2’, H-6’), 7.75 (1H, d, J = 7.8 Hz, H-6), 12.91 (1H, br.s, COOH). 13C NMR (100.6 MHz, DMSO-d6), δ, ppm: 25.8, 26.0, 28.7, 28.9 (2), 29.0, 29.1, 29.2, 41.8, 49.0, 68.1, 100.2, 112.0, 117.0, 132.4, 145.0, 151.1, 158.3, 162.3, 169.4. HRMS: m/z[M + H]+ calcd for C22H29BrN2O5: 481.1320, found: 481.1327.
[3-[12-(4-bromophenoxy)dodecyl]-2,6-dioxo-3,6-dihydropyrimidin-1(2H)-yl]acetic acid (2f).
White solid; yield 73% (1.49 g, 2.92 mmol); mp 102-103°C. Rf 0.47 (eluting with ethylacetate- iPrOH-40% aqv.NH4OH (6:9:5)) 1H NMR (400 MHz, DMSO-d6), δ, ppm: 1.22-1.28 (14H, m, 7×CH2), 1.37 (2H, q, J = 7.8 Hz, CH2), 1.57 (2H, q, J = 6.4 Hz, CH2), 1.67 (2H, q, J = 6.8 Hz, CH2), 3.70 (2H, t, J = 7.3 Hz, NCH2), 3.92 (2H, t, J = 6.6 Hz, OCH2), 4.44 (2H, s, CH2CO), 5.73 (1H, d, J = 7.8 Hz, H-5), 6.88 (2H, d, J = 9.1 Hz, H-3’, H-5’), 7.41 (2H, d, J = 9.0 Hz, H-2’, H-6’), 7.74 (1H, d, J = 7.9 Hz, H-6), 12.90 (1H, br.s, COOH). 13C NMR (100.6 MHz, DMSO-d6), δ, ppm: 25.8, 26.1, 28.7, 28.88 (2), 28.92, 29.1, 29.25 (2), 29.30, 41.8, 49.0, 68.1, 100.2, 112.0, 117.0, 132.4, 145.0, 151.1, 158.3, 162.3, 169.4. HRMS: m/z[M + H]+ calcd for C24H33BrN2O5: 411.0541, found: 411.0550.
2.1.2.3. General procedure for the synthesis of (3'-azido-3'-deoxythymidine)-2-[3-[ω-(4-bromo- phenoxy)alkyl]-2,6-dioxo-3,6-dihydropyrimidin-1(6H)-yl]acetates 3a-f.
The mixture of the corresponding acid 2a-f (0.4 mmol) and 3'-azido-2',3'-dideoxythymidine (0.4 mmol, 107 mg) was coevaporated twice with pyridine (5 mL) and then dissolved in dry pyridine (5 mL). 1,3-Dicyclohexylcarbodiimide (0.48 mmol, 99 mg) was added to the resulting solution and left for 24 hours with stirring. The solvents were evaporated and the residue was purified by PLS in a chloroform: methanol (96: 4). Yields of target products were within 57-74%, after final repurification by PLC in ethyl acetate: chloroform (3: 2) with 1% methanol added.
(3'-azido-3'-deoxytimidine)-2-(3-((4-bromophenoxy)propyl)-2,6-dioxo-2,3-dihydropyrimidin-1(6H)-yl)acetate (3a).
Light yellow oil; yield 61% (154 mg, 0.24 mmol). Rf 0.30 (eluting with CHCl3-MeOH (95:5)). 1H-NMR (400 MHz, CDCl3): 1.92 (s, 3H, CH3, Thy), 2.16-2.19 (2H, m, CH2), 2.39-2.45 (m, 2H, 2’CH2, Azt), 3.95-3.98 (m, 4H, NCH2, OCH2), 4.01-4.03 (m, 1H, 4’CH, Azt), 4.24-4.26 (m, 1H, 3’CH, Azt), 4.39-4.52 (m, 2H, 5’H-2, Azt), 4.59-4.84 (m, 2H, CH2CO), 5.73-5.75 (1H, d, J = 7.9 Hz, H-5, Ura), 6.08-6.11 ( 1H, t, J=8 Hz, 1’CH, Azt), 6.73-6.76 (2H, m, H-3’, H-5’, Ph), 7.17-7.19 (1H, d, J = 8 Hz, H-6,Ura),7.24 (s, 1H, H-6, Thy), 7.36-7.38 (2H, m, H-2’, H-6’, Ph), 8.48 (s, 1H, NH). 13C NMR (100.6 MHz, CDCl3), δ ppm: 12.63, 28.23, 37.54, 41.96, 47.61, 60.20, 63.65, 64.58, 81.90, 85.37, 101.43, 111.49, 113.60, 116.27×2, 132.56×2, 135.56, 143.57, 150.04, 151.25, 157.47, 162.36, 163.43, 167.65. HRMS: m/z[M + H]+ calcd for C25H26BrN7O8: 632.1099, found: 632.1093.
(3'-azido-3'-deoxytimidine)-2-(3-((4-bromophenoxy)pentyl)-2,6-dioxo-2,3-dihydropyrimidin-1(6H)-yl)acetate (3b).
Light yellow oil; yield 74% (195 mg, 0.30 mmol). Rf 0.31 (eluting with CHCl3-MeOH (95:5)). 1H-NMR (400 MHz, CDCl3): 1.48-1.52 (2H, m, CH2), 1.76-1.80 (m, 4H, 2CH2), 1.92 (s, 3H, CH3, Thy), 2.35-2.44 (m, 2H, 2’CH2, Azt), 3.74 −3.78 (t, J= 8 Hz, 2H, NCH2), 3.89-3.92 (m, 2H, OCH2), 4.01-4.02 (m, 1H, 4’CH, Azt), 4.23-4.25 (m, 1H, 3’CH, Azt), 4.41-4.49 (m, 2H, 5’CH2, Azt), 4.61-4.84 (m, 2H, CH2CO), 5.76-5.78 (d, 1H, J=6Hz, H-5, Ura), 6.08-6.12 (t, 1H, 1’CH, Azt), 6.73-6.75 (m, 2H, 2CH, H-3’, H-5’, Ph), 7.16-7.18 (d, 1H, H-6, J=6 Hz, Ura), 7.25 (s, 1H, H-6, Thy), 7.32-7.35 (m, 2H, 2CH2, H-2’, H-6’, Ph), 8.86 (s, 1H, NH). 13C NMR (100.6 MHz, CDCl3), δ, ppm: 12.57, 23.05, 28.68, 28.74, 37.47, 41.96, 49.85, 60.18, 63.63, 67.67, 81.82, 85.24, 101.42, 111.44, 112.88, 116.32×2, 132.30×2, 135.49, 143.05, 150.11, 151.17, 158.05, 162.34, 163.59, 167.62. HRMS: m/z[M + H]+ calcd found for C27H30BrN7O8: 660.1413, found: 660.1412.
(3'-azido-3'-deoxytimidine)-2-(3-((4-bromophenoxy)hexyl)-2,6-dioxo-2,3-dihydropyrimidin- 1(6H)-yl)acetate (3c).
Light yellow oil; yield 60% (162 mg, 0.24 mmol). Rf 0.31 (eluting with CHCl3-MeOH (95:5)). 1H-NMR (400 MHz, CDCl3): 1.37-1.41 (m, 2H, CH2), 1.47-1.52 (m, 2H, CH2) 1.70-1.78 (m, 4H, 2CH2), 1.93 (s, 3H, CH3, Thy), 2.35-2.44 (m, 2H, 2’CH2, Azt), 3.73 – 3.76 (t, J=8 Hz, 2H, NCH2), 3.89-3.39 (m, 2H, OCH2), 4.00-4.04 (m, 1H, 4’CH, Azt), 4.22-4.26 (m, 1H, 3’CH, Azt), 4.38-4.52 (m, 2H, 5’CH2, Azt), 4.61-4.85 (m, 2H, CH2CO), 5.75-5.77 (d, H, J=6 Hz, H-5, Ura), 6.08-6.12 (t, 1H, J= 8 Hz, 1’CH, Azt), 6.73-6.76 (m, 2H, H-3’,H-5’, Ph), 7.15-7.17 (d, 1H, H-6, J=8 Hz, Ura), 7.26 (s, 1H, 6CH, Thy), 7.32-7.35 (m, 2H, 2CH2, H-2’, H-6’, Ph), 8.40 (s, 1H, NH). 13C NMR (100.6 MHz, CDCl3), δ, ppm: 12.62, 25.69, 26.20, 29.02×2, 37.54, 42.01, 49.95, 60.22, 63.64, 67.94, 81.90, 85.25, 101.45, 111.50, 112.84, 116.38×2, 132.34×2, 135.48, 143.07, 150.02, 151.22, 158.19, 162.38, 163.38, 167.67. HRMS: m/z[M + H]+ calcd for C28H32BrN7O8: 674.1568, found: 674.1563.
(3'-azido-3'-deoxytimidine)-2-(3-((4-bromophenoxy)octyl)-2,6-dioxo-2,3-dihydropyrimidin- 1(6H)-yl)acetate (3d).
Light yellow oil; yield 66% (185 mg, 0.26 mmol). Rf 0.32 (eluting with CHCl3-MeOH (95:5)). 1H-NMR (400 MHz, CDCl3): 1.34-1.43 (m, 8H, 4CH2), 1.67-1.76 (m, 4H, 2CH2 ), 1.93 (s, 3H, CH3, Thy), 2.33-2.44 (m, 2H, 2’CH2, Azt), 3.73 (t, 2H, J = 7.4 Hz, NCH2), 3.88-3.91 (m, 2H, OCH2), 4.01-4.02 (m, 1H, 4’CH, Azt), 4.24-4.25 (m, 1H, 3’CH, Azt), 4.38-4.42 (m, 2H, 5’CH2, Azt), 4.49-4.85 (m, 2H, CH2CO), 5.76-5.78 (d, H, H-5, J=6 Hz, Ura), 6.10-6.12 (t, 1H, 1’CH, Azt), 6.73-6.77 (m, 2H, H-3’, H-5’, Ph), 7.16-7.17 (d, 1H, H-6, J=6 Hz, Ura), 7.27 (s, 1H, 6CH, Thy), 7.32-7.36 (m, 2H, H-2’, H-6’, Ph), 8.56 (c, 1H, NH). 13C NMR (100.6 MHz, CDCl3), δ, ppm: 12.56, 25.89, 26.35, 28.99, 29.05, 29.11, 29.13, 37.46, 41.97, 50.01, 60.20, 63.66, 68.18, 81.80, 85.14, 101.29, 111.45, 112.64, 116.37×2, 132.23×2, 135.46, 143.14, 150.23, 151.15, 158.25, 162.40, 163.76, 167.64. HRMS: m/z[M + H]+ calcd for C30H36BrN7O8: 702.1881, found: 702.1872.
(3'-azido-3'-deoxytimidine)-2-(3-((4-bromophenoxy)decyl)-2,6-dioxo-2,3-dihydropyrimidin-1(6H)-yl)acetate (3e).
Light yellow oil; yield 57% (166 mg, 0.23 mmol). Rf 0.32 (eluting with CHCl3-MeOH (95:5)). 1H-NMR (400 MHz, CDCl3): 1.25-1.41 (m, 12H, 6CH2), 1.67-1.75 (m, 4H, 2CH2), 1.92 (s, 3H, CH3, Thy), 2.35-2.44 (m, 2H, 2’CH2, Azt), 3.72 - 3.74 (m, 2H, NCH2), 3.87-3.90 (m, 2H, OCH2), 4.02 (m, 1H, 4’CH, Azt), 4.22-4.24 (m, 1H, 3’CH, Azt), 4.38-4.52 (m, 2H, 5’CH2, Azt), 4.61-4.84 (m, 2H, CH2CO), 5.76-5.78 (d, H, H-5, J=6 Hz, Ura), 6.12 (t, 1H, 1’CH, Azt), 6.73-6.75 (m, 2H, H-3’, H-5’, Ph), 7.16-7.18 (d, 1H, H-6, J=6 Hz, Ura), 7.27 (s, 1H, 6CH, Thy), 7.32-7.34 (m, 2H, H-2’, H-6’, Ph), 9.05 (s, 1H, NH). 13C NMR (100.6 MHz, CDCl3), δ, ppm: 12.73, 26.02, 26.50, 29.10, 29.19, 29.21, 29.33, 29.40, 29.45, 37.62, 42.08, 50.14, 60.28, 63.72, 68.33, 81.87, 85.18, 101.42, 111.55, 112.65, 116.42×2, 132.27×2, 135.53, 143.29, 150.26, 151.20, 158.33, 162.46, 163.75, 167.69. HRMS: m/z[M + H]+ calcd for C32H40BrN7O8: 730.2194, found: 730.2190.
(3'-azido-3'-deoxytimidine)-2-(3-((4-bromophenoxy)dodecyl)-2,6-dioxo-2,3-dihydropyrimidin-1(6H)-yl)acetate (3f).
Light yellow oil; yield 58% (176 mg, 0.23 mmol). Rf 0.33 (eluting with CHCl3-MeOH (95:5)). 1H-NMR (400 MHz, CDCl3): 1.24-1.41 (m, 16H, 8CH2), 1.72-1.75 (m, 4H, 2CH2) 1.92 (s, 3H, CH3, Thy), 2.34-2.43 (m, 2H, 2’CH2, Azt), 3.71 (t, J = 7.5 Hz, 2H, NCH2), 3.89 (t, J=6 Hz, 2H, OCH2), 4.01-4.02 (m, 1H, 4’CH, Azt), 4.23-4.25 (m, 1H, 3’CH, Azt), 4.37-4.53 (m, 2H, 5’CH2, Azt), 4.61-4.84 (m, 2H, CH2CO), 5.76 (d,1H, H-5, J=6 Hz, Ura), 6.12 (t, J=6 Hz, 1H, 1’CH, Azt), 6.73-6.76 (m, 2H, H-3’, H-5’, Ph), 7.16 (d, 1H, H-6, J=6Hz, Ura), 7.26 (s, 1H, 6CH, Thy), 7.31-7.35 (m, 2H, H-2’, H-6’, Ph), 8.97 (s, 1H, NH). 13C NMR (100.6 MHz, CDCl3), δ, ppm: 12.60, 26.04, 26.49, 29.07, 29.22×2, 29.39, 29.47, 29.54×3, 37.51, 42.01, 50.01, 60.23, 63.67, 68.35, 81.86, 85.13, 101.30, 111.52, 112.63, 116.42×2, 132.26×2, 135.44, 143.17, 150.22, 151.19, 158.34, 162.44, 163.68, 167.68. HRMS: m/z[M + H]+ calcd for C34H44BrN7O8: 758.2507, found:758.2507.
2.1.2.4. Synthesis of compound 4c.
((−)-L-2′,3′-dideoxy-3′-thiacytidine)-2-(3-((4-bromophenoxy)hexyl)-2,6-dioxo-2,3-dihydropyrimidin-1(6H)-yl)acetate (4c).
Light yellow oil; yield 30% (27 mg, 0.04 mmol). Rf 0.5 (eluting with CHCl3-MeOH (95:5)). 1H-NMR (400 MHz, CDCl3): 1.36-1.43 (m, 2H, CH2), 1.47-1.55 (m, 2H, CH2) 1.72-1.79 (m, 4H, 2CH2), 3.01-3.07 (m, 1H, 2’CH2, 3TC), 3.49-3.55 (m, 1H, 2’CH2, 3TC), 3.73-3.78 (t, J=8 Hz, 2H, NCH2), 3.89-3.93 (t, 2H, OCH2), 4.44-4.56 (m, 2H, 5’CH2, 3TC), 4.69-4.82 (m, 2H, CH2CO), 5.33-5.36 (m, 1H, 4’CH2, 3TC), 5.76-5.79 (d, H, J=6 Hz, H-5, 3TC), 5.93-5.96 (d, 1H, J=6 Hz, H-5,Ura), 6.30-6.34 (t, 1H, J=8 Hz, 1’CH, 3TC), 6.73-6.79 (m, 2H, H-3’,H-5’, Ph), 7.20-7.22 (d, 1H, H-6, J=8 Hz, Ura), 7.33-7.38 (m, 2H, 2CH2, H-2’, H-6’, Ph), 7.63-7.65 (d, 1H, H-6, J=8 Hz, 3TC). 13C NMR (100.6 MHz, CDCl3), δ, ppm: 25.6, 26.1, 28.9×2, 37.6, 41.8, 49.8, 65.3, 67.9, 82.1, 87.7, 95.2, 101.3, 112.7, 116.3×2, 132.2×2, 140.6, 143.1, 151.1, 155.4, 158.1,162.4, 165.7, 167.5. HRMS: m/z[M + H]+ calcd for C26H30BrN5O7S: 636.1122, found: 636.1127.
2.2. Hydrolysis of the synthesized compounds by esterase from porcine liver
Hydrolysis of the compounds was assayed in 30 μl 50 mM Tris-HCl buffer pH 8.2 containing NaCl 250 mM, CaCl2 6 mM, esterase 5.2 units/test, different concentrations of compounds 3b- 3d and 4c (2 - 10 mM) in methanol. The reactions were proceeded at 37 °C for 0-18 h. Reaction products were separated by TLC in chloroform-ethanol 32:1 for compounds 3b and 3d, or chloroform-methanol 95:5 for compounds 3c and 4c. The compounds 1b-d, 2-2d, AZT and 3TC were used as references. Rf of hydrolysis products: 3b – 0.72, 2b – 0.31, 1b – 0.72, 3c – 0.69, 2c – 0.03, 1c – 0.44, 4c – 0.50, 3TC – 0.30, 3d – 0.80, 2d – 0.38, 1d – 0.60, AZT – 0.51. The products were identified by mass-spectrometry. Retention time: 3.4 min for 2',3'-dideoxy-3'-azidothymidine 2b - 4.1 min, 2c - 4.3 min, 2d - 4.7 min, 3b - 6.3 min, 3c - 6.5 min, 3d - 7.1 min. Retention time for 4c was 7.5 min, methanol gradient elution.
2.3. Cells and reagents
Human T lymphocyte MT-4 cell line was obtained from Dr. D. Richman (NIH/AIDS reagent program, division of AIDS, NIAID, NIH). The fibroblastic WS1 cell line was obtained from ATCC (Manassas, VA). MT-4 was cultured in RPMI 1640 FBS 10% while WS1 was cultured in DMEM FBS 10% (FBS, Gemini Products, Sacramento, CA) respectively. Human lymphoid tissues were surgically removed during routine tonsillectomies at Children’s National Medical Center (Washington, DC) and obtained within 4-5h of excision according to an NIH Institutional review Board-approved protocol. Tonsils were cut in 2-3-mm tissue-blocks. Nine blocks were placed on a collagen sponge at the air-liquid interface and cultured for 12 to 15 days in 6-well plates with a change of medium every 3 days. Tissue blocks were cultured in RPMI 1640 supplemented with 15% FBS, L-Glutamine, essential amino acids, pyruvate, 50 ng/mL gentamycin and 50 ng/mL Fungizone. Typically 3 wells of 9 tissue blocks were used per experimental condition.
The prototypical X4 HIV-1 variant X4LAI.04 (stock at 50 ng of p24gag /mL) was obtained from VQA Laboratory (Chicago, IL).
CMV AD169 (TCID50 of 104,3/0,2 mL) used to infect WS1 cell lines was obtained from The State Virus Collection of the Gamaleya National Research Center for Epidemiology and Microbiology of the Ministry of Healthcare of the Russian Federation, Moscow, Russia (NRCEM, Moscow, Russia).
2.4. Antiviral activity
2.4.1. Anti HIV-1 activity in MT-4 cell cultures.
The anti HIV-1 activity of all seven newly synthesized compounds was evaluated in MT-4 cell cultures. MT-4 cells (100 μL at 107 cells/mL) were incubated with 100 μL of HIV-1Lai.04 for 90 min at 37°C. The excess of virus was washed off twice with 25 mL of PBS and resuspended. After resuspending cell pellet with 1 mL of complete medium 100 μL of infected cells were added per well followed by the addition of compound diluted in 900 uL of complete medium. After 3 days of culture, p24gag concentration was assessed.
2.4.2. Anti HIV-1 activity in Human tissue culture ex vivo.
27 tonsillar tissue blocks were pretreated with compound 3c at different concentrations overnight and then inoculated by application of 7 μL of X4LAI.04 viral stock (50 ng/mL) on top of each block. Compound 3c was re-added every 3 days when medium was changed and the tissue blocks were cultured for 12 days. HIV-1 replication was assessed by measuring p24gag concentration in tissue culture. HIV inhibition was calculated as percent of HIV replication in tissue blocks cultured in presence of drugs compared to HIV replication in tissue blocks cultured with DMSO control.
2.4.3. Anti-CMV activity in WS1 cell cultures.
We evaluated the anti-CMV activity of heterodimers 3a-3f and their parental molecules 1d, 1f, and 2a-2f using a standard plaque reduction assay. The human embryo fibroblasts WS1 cell monolayers were infected with CMV AD169 (TCID50 104.3/0,2 mL) and incubated for 1 h at 37°C under 5% CO2. Then, the infected cells were washed twice with PBS and cultured in cell culture media containing different concentrations of compounds. Cells were cultured at 37°C for 5 days. We calculated the plaque inhibition rate using the standard method as described previously [25]. The EC50 (50% effective concentration) of tested compounds was calculated by regression analysis.
2.4.4. Anti-CMV/HIV-1 activity of heterodimer 3c and 4c in coinfected human tissues ex vivo.
27 tonsillar tissue blocks were simultaneously infected with 7 μL of HIV-1 (X4LAI.04) and 5 μL of CMV (AD169) viral stocks. Tissue cultures were pre-treated 12 h prior to viral infection with compound 3c or 4c. Medium was changed every 3 days. Tissue blocks were cultured for 15 days. The anti-HIV-1 and anti CMV activities of 3c or 4c was evaluated by comparing HIV or CMV replication in tissues cultured in presence of drugs versus HIV or CMV replication in tissue-control.
2.4.5. Antiviral Activity Evaluation.
Productive HIV-1 infection was evaluated from measurements of p24gag antigen in culture supernatant by Luminex, as previously described [26]. Productive CMV replication was evaluated by measurement of CMV DNA copies present in culture supernatant using real-time polymerase chain reaction as previously described [27]. Antiviral activity was either expressed as (i) EC50s (heterodimer concentration that inhibits viral replication by 50%) or (ii) viral inhibition expressed as percent of control (untreated infected condition). Means ± SD of EC50 for each compound are presented in Table 1 or giving in the text (compound 4c). Each experiment was performed between 2 to 5 times.
Table 1.
Anti-HIV-1 activity and cytotoxicity of synthesized heterodimers and the products of their hydrolysis in the MT-4 cell line.
| Compound s |
Antiviral activitya EC50 ± SD (μM) |
Cytotoxicityb CC50 (μM) |
|---|---|---|
| 3a | 1.96 ± 0.78 | 82.1 |
| 3b | 0.61 ± 0.31 | >100 |
| 3c | 0.53 ± 0.29 | >100 |
| 3d | 0.23 ± 0.11 | >100 |
| 3e | 0.44 ± 0.29 | >100 |
| 3f | 0.21 ± 0.1 | >100 |
| AZT | 3 nM (95%CI: 1-16)1 | >1001 |
| 2c | >100 | >100 |
EC50: 50% Effective concentration or compound concentration required to inhibit HIV-1 replication by 50%. (2 < n < 5)
CC50: 50% Cytotoxic concentration or compound concentration that decreases cell viability by 50%.
See Vanpouille et al. (2014); Antiviral Res 109, 125-131
2.5. Cell Viability
2.5.1. Viability assay in MT-4 cell cultures.
Cell viability was performed using a NucleoCounter® NC-100™ automated cell counting system (ChemoMetec, Lillerød, Denmark) using nucleocassette pre-loaded with the propidium iodide (PI) fluorescent dye for dead cells. Viability of cells was calculated by comparing the number of total cells (obtained by lysing cells before measurement) and the number of dead cells (no pre-treatment). Each condition was run in duplicate. Cell viability for each concentration of each compound was expressed as percentage of cells in untreated control.
2.5.2. Viability assay in WS1 cell cultures.
Compound cytotoxicity was also tested by MTT method. WS1 cells were cultured in 96 well plates containing different concentrations of compounds in the cell culture medium at 37 °C in 5% CO2 atmosphere. Three days later, the cell culture medium was replaced with 50 μL of fresh medium containing 1 mg/mL of MTT solution (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide). Cells were incubated at 37 °C for 4 h, and 100 μL of acid-isopropanol (HCl 0.1N in isopropanol) was added to each well. After ensuring that all crystals were dissolved, the absorbance in the plates were measured using an automatic plate reader (TECAN, Switzerland) at 570 nm test wavelength and a 690 nm reference wavelength. The 50% cytotoxic concentration (CC50) was defined as the compound concentration that caused a 50% reduction in the number of viable cells.
2.5.3. Viability assay in human lymphoid tissue.
To assay viability of cells in tissues ex vivo, tissue blocks were digested with collagenase IV at day 12. Cells from 3c-(10 μM) treated tissue blocks and untreated-control tissues were stained with anti-CD3QD655, anti-CD4QD605, anti-CD8QD800, anti-CD45RA-FITC, anti-CCR7-PECy7, anti-CXCR4-PE, and anti-CCR5-APC antibodies (stain 1) or stained with anti–CD3-QD655, anti–CD4-QD605, anti–CD8-QD800, anti-CD25-APC, anti-CD38-PE, anti-CD95-PE-Cy7, and anti-HLA-DR-BV570 antibodies (stain 2) (B&D Biosciences or Caltag laboratories, CA). Data were processed on a NovoCyte cytometer (AceaBiosciences, Inc). The depletion of each CD3+ lymphocytic cell subsets was assessed by using Trucount beads (Becton-Dickinson, NJ).
3. RESULTS
3.1. Chemical synthesis
The reaction of 1-[ω-(4-bromophenoxy)alkyl]uracils 1a-f with ethyl bromoacetate in the presence of K2CO3 in DMF, followed by alkaline hydrolysis at room temperature, gave the corresponding [2,6-dioxo-3-[ω-(4-bromophenoxy)alkyl]-3,6-dihydropyrimidine-1(2H)-yl] acetic acids 2a-f in one-pot with 60–79% yields (Scheme 1). The target conjugates 3a-f were prepared by condensation of 2',3'-dideoxy-3'-azidothymidine and acids 2a-f in the presence of dicyclohexylcarbodiimide (DCC) in pyridine. The yields of 5'-[2,6-dioxo-3-(ω-phenoxyalkyl)-3,6-dihydropyrimidine-1(2H)-yl]acetates of 2',3'-dideoxy-3'-azidothymidine (3a-f) were 52-74% after isolation and purification by column chromatography on silica gel.
Scheme 1.
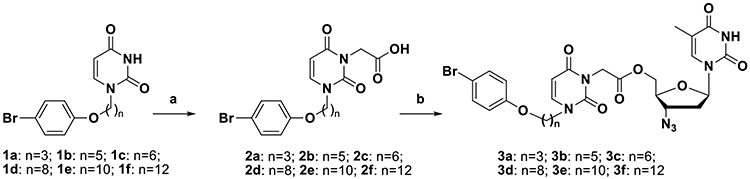
Synthesis of conjugates 3a-f. Reagents and conditions: (a) 1. BrCH2COOEt, K2CO3, DMF; 2. LiOH, H2O/EtOH; (b) AZT, DCC, Py.
((−)-L-2′,3′-dideoxy-3′-thiacytidine)-2-(3-((4-bromophenoxy)hexyl)-2,6-dioxo-2,3-dihydropyrimidin-1(6H)-yl)acetate (4c) was synthesized in a similar manner starting from 3TC and 2c.
3.2. Hydrolysis of the synthesized compounds by esterase from porcine liver
We assumed that the compounds 3a-f could be hydrolyzed enzymatically, for instance by esterases, and generate biologically active components. Towards this goal, we investigated the hydrolysis of compounds by porcine esterase. We showed that reaction with esterase generated two main products identified by TLC and high-resolution mass spectrometry as 2',3'-dideoxy-3'-azidothymidine and the corresponding 1-[ω-(4- bromophenoxy)alkyl]uracil acetic acids 2b-2d, as well as 5-10% of unidentified products. It should be noted that T1/2 for 3b and 3c was about 7-8 hrs, but T1/2 for with longer linker was 10-12 hrs although hydrolysable bonds were the same. T1/2 for 4c was about 24 hrs. Thymidine diacetate was used as a positive control; its hydrolysis was totally completed after 5-6 hours under the same conditions.
3.3. Heterodimers suppressed HIV-1 replication in MT-4 cell cultures
HIV inhibition of all six AZT-based newly-developed anti-CMV/HIV-1 heterodimers was evaluated in MT-4 cells inoculatyed with HIV-1LAI.04. In this system, HIV-1 replication, evaluated as p24gag accumulation in culture medium over 3 days, reached on average (195.8 ± 26.5) x 103 ng/mL p24gag (n=3). All six tested compounds efficiently and dose-dependently suppressed HIV-1 replication with EC50 ranging from 0.21 μM to 1.96 μM (Table 1; Fig. S1).
3.4. Heterodimers suppressed CMV replication in human embryo fibroblast cultures
The anti-CMV activity of the heterodimers 3a-f as well as their hydrolysis products 2a-f were tested for inhibition of CMV replication in human embryo fibroblast (WS1) cultures infected with CMV (AD169) (Table 2). EC50 of heterodimers 3a-3f ranged from 3.2 to 12.2 μM and showed similar activity to that of control compound ganciclovir (EC50 2 μM), 1d and 1f (EC50 13.1 and 3.1 μM). Compounds 2c and 2b were less active (EC50 78.1 and 104 μM, respectively); compounds 2a, 2e, and 2f were inactive. The only active compound among acids 2a-2f was 2d, with EC50 11.9 μM.
Table 2.
Anti-CMV activity and cytotoxicity of synthesized heterodimers and the products of their hydrolysis in the human fibroblast WS1 cell line infected with CMV AD169.
| Compounds | Antiviral activitya EC50 ± SD (μM) |
Cytotoxicityb CC50 (μM) |
Controls | Antiviral activitya EC50 ± SD (μM) |
Cytotoxicityb CC50 (μM) |
|---|---|---|---|---|---|
| 3a | 3.2 ± 0.8 | 347 | 2a | NA | 965 |
| 3b | 4.4 ±2.1 | 173 | 2b | 103.6±12.2 | 749.4 |
| 3c | 7.9 ± 3.0 | 393 | 2c | 78.1±18.3 | 724 |
| 3d | 12.2 ± 1.7 | 600 | 2d | 11.9±2.1 | 90.5 |
| 3e | 3.2 ± 0.5 | 182 | 2e | NA | 164 |
| 3f | 5.1 ± 1.2 | 253 | 2f | NA | 22 |
| GCVc | 2.0 ± 0.5 | 500 |
EC50: 50% Effective concentration or compound concentration that inhibits CMV replication by 50% (n=3)
CC50: 50% Cytotoxic concentration or compound concentration required to decrease cell viability by 50%.
The common anti-CMV drug ganciclovir (GCV) was used as control of the experimental system.
3.5. AZT-based heterodimers exhibit low cytotoxicity
In principle, viral inhibition could be the result of cell toxicity. To exclude the possibility that the HIV-1 inhibition reported here was due to cell death, we cultured MT-4 cells in the presence of increasing concentrations of each heterodimer and assessed cell viability after 3 days of culture. Five out of six compounds did not show any significant cytotoxicity, even at 100 μM (Table 1; Fig. S2). Compound 3a showed some cell toxicity, killing around 50% of cells at 100 μM (500 times its EC50). Compounds 3c and 3f, however, were two of the best, with very low cytotoxicity, even at 100 μM (respectively 9.0 ± 3.9% and 10.3 ± 4.2% at 100 μM) (Table 1; Fig. S2).
3.6. Heterodimer 3c suppressed the replication of HIV-1 in human tonsillar tissue ex vivo
All our newly developed anti CMV/anti-HIV-1 compounds efficiently inhibited HIV-1 in cell cultures (MT-4) Fig. S1. We chose one of them, compound 3c, to investigate its anti-HIV-1 activity in tonsillar tissue ex-vivo. This system of culture was shown to retain tissue cytoarchitecture and to support HIV-1 replication without exogenous activation [29]. Tonsillar tissue blocks were pre-treated with 3c overnight and inoculated with HIV-1. In control untreated HIV-1-infected cultures, the cumulative HIV-1 replication ([p24gag]) levels in tissues from different donors varied from 2.5 to 97.9 ng/mL, as typically observed in human tissues ex vivo, where cell activation and coreceptor expression are highly variable from donor to donor. Compound 3c at concentrations ranging from 1 nM to 10 μM inhibited HIV-1 replication in a dose-dependent manner by 0 ± 0.5%, 17.9 ± 8.2%, 31.4 ± 7.7%, 76.5 ± 3.3%, and 96.2 ± 1.0%, respectively (n=9 (except for 1 nM (n=2)), p>0.05; p>0.05; p<0.05; p<0.05; p<0.05) (Fig. 2). The EC50 of AZT alone in human tissue ex vivo was previously reported (3.1 nM ((95% CI (0.63–15.7)) [30].
Figure 2. Inhibition by heterodimer 3c of HIV-1 replication in human tonsillar tissues.
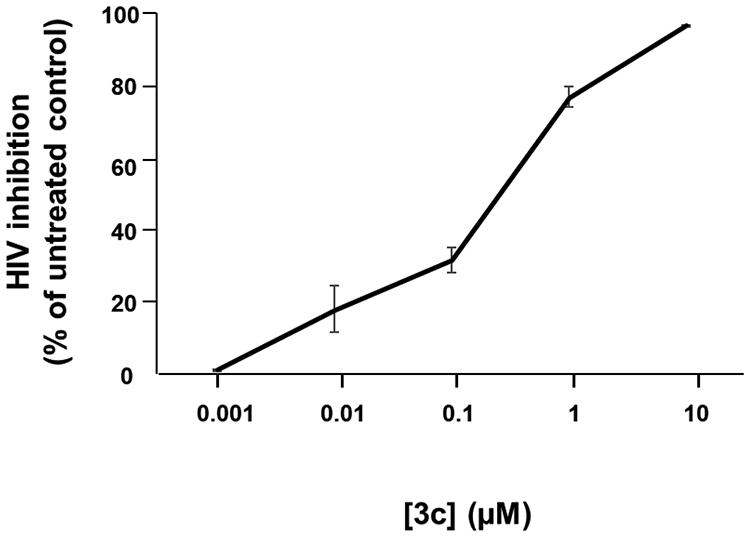
27 tonsillar blocks from each of 6 donors were infected with X4LAI.04 and incubated with 3c at five concentrations ranging from 1 nM to 10 μM for 12 days or used as untreated controls. We evaluated HIV-1 replication from p24gag core antigen present in tissue culture medium using a Luminex bead-based assay. The inhibition of HIV-1 was expressed as percentage of donor-matched untreated control for each concentration of 3c. Presented are means ± SEM of HIV-1 inhibition.
3.7. Heterodimer 3c was not cytotoxic in human lymphoid tissues ex vivo
As an adequate model to assess the efficiency of newly-synthesized drugs or microbicides [30-32], the system of human tissue explants is also a trademark system to evaluate potential cytotoxicity of various drugs for different cell subsets. Since HIV-1 mainly replicates in lymphocytes, here we used human tonsillar tissues ex vivo to assess the effect of 3c on cell a variety of lymphocytic cell subsets. At the end of the culture (day 12), cells were isolated from control tissue blocks not treated or from tissue blocks treated with 3c at 10 μM, a concentration that completely abrogated HIV-1 replication in this system. Cells were stained with different cell markers, and we evaluated the effect of 3c on cell viability by comparing the numbers of cells in tonsillar tissue blocks treated with 3c and with those in untreated tonsillar tissue control. We evaluated the depletion of T cells (CD3+), CD3+CD4+ cells, CD3+CD8+ cells, and various subsets of CD4+ T cells: CD4+ expressing HIV-1 coreceptor CXCR4 or CCR5, CD45+CCR7+ (Tnaïve), CD45RA-CCR7+, CD45RA-CCR7−, CD45RA+CCR7−, CD4+ expressing different markers of activation (CD25+, CD38+, CD95+, HLA-DR+). Figure 3 shows that 3c, even at a concentration of 10 μM, didn’t significantly deplete any cells (n=7, p<0.05).
Figure 3. Cell toxicity of heterodimer-inoculated ex vivo tissue cultures.
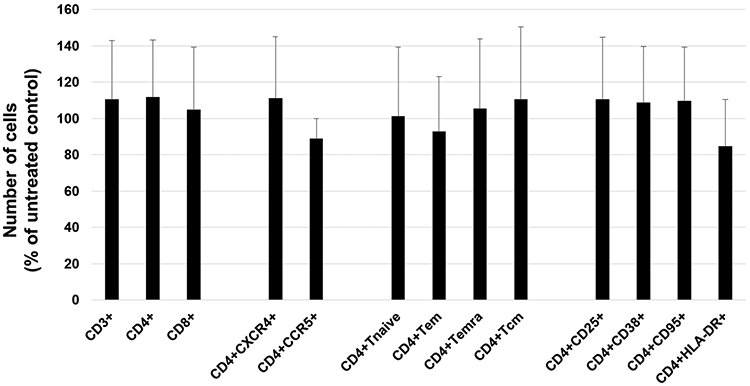
At day 12 of culture, 27 human lymphoid tissue blocks treated or not with 3c were stained with anti-CD3QD655, anti-CD4QD605, anti-CD8QD800, anti-CD45RA-FITC, anti-CCR7-PECy7, anti-CXCR4-PE, and anti-CCR5-APC antibodies (stain 1) or stained with anti-CD3-QD655, anti-CD4-QD605, anti-CD8-QD800, anti-CD25-APC, anti-CD38-PE, anti-CD95-PE-Cy7, and anti-HLA-DR-BV570 antibodies (stain 2). Means ± SEM (error bars) of various cell subsets compared to matched untreated tissue performed with tissues from seven different donors are presented. Cell depletion for each cell subset was normalized by tissue-block weights.
3.8. Heterodimer 3c suppressed CMV and HIV-1 replication in coinfected lymphoid tissues ex vivo
CMV reactivation is a major problem in immunocompromised people, including people living with HIV-1. To mimic more closely the in vivo situation where HIV-1 and CMV both replicate in coinfected individuals, we coinfected human tonsillar tissues ex vivo with both viruses and investigated the effect of compound 3c, as a representative of our newly synthesized heterodimers. Both HIV-1 and CMV replication in coinfected tissues were typical of replication of these viruses in singly infected human tissues. The cumulative HIV-1 replication levels in coinfected tonsillar tissues varied from 10.3 to 83.3 ng/mL p24gag, while the cumulative CMV replication levels varied from 3,226 to 32,667 CMV DNA copies/μL (n=6). In HIV-1/CMV-coinfected human lymphoid tissue, 3c retained its inhibitory activity against both viruses: 10 μM 3c inhibited the cumulative production of p24gag by 97.2 ± 2.1% (n=6; p<0.05) (Fig. 4A) and the number of CMV DNA copies/uL by 86.9 ± 3.3% (n=6; p<0.05) (Fig. 4B). In coinfected tissues, AZT and 3TC controls inhibited HIV-1 replication respectively by 97.0 ± 1.6% and 99.7 ± 0.2% (n=3; p<0.05). Control compounds 1c and 2c inhibited CMV respectively by 92.8 ± 2.1% and 68.6 ± 11.3% (n=3; p<0.05). AZT and 3TC have no effect on CMV replication [28]. Compound 1c has been reported to be inactive against HIV-1 [23], while 2c was tested and did not inhibit HIV-1 either (Table 1).
Figure 4. Suppression of HIV-1 and CMV replication in coinfected tonsillar tissues.
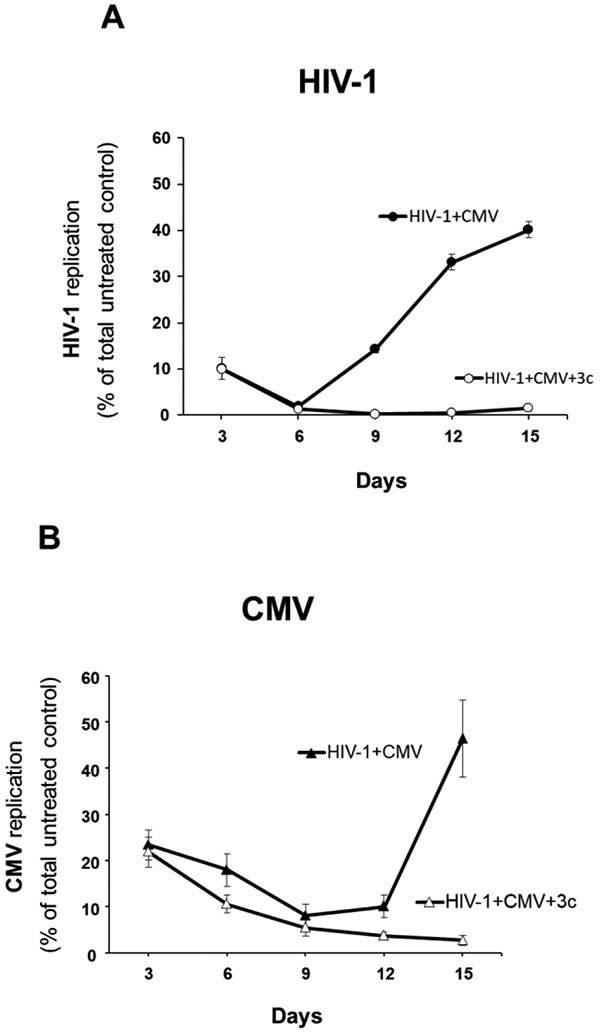
(A) Blocks of human tissue were co-inoculated with HIV-1 (X4LAI.04) and CMV (AD169) and cultured for 15 days at 37°C. Tissues were treated with 3c at 10 μM or left untreated (control). We evaluated HIV-1 replication by measuring p24gag in culture medium, using a Luminex bead-based assay. (B) The replication of CMV was measured with real-time PCR for viral DNA accumulated in culture medium. For each day, presented are means ± SEM of viral replication as percent of cumulative virus produced in untreated control condition in 6 tonsillar tissues from 6 different donors.
3.9. Heterodimer 4c suppressed CMV and HIV-1 replication
As a proof-of-concept, next we synthesized heterodimer 4c, the equivalent of 3c using 3TC as the anti-HIV-1 molecule instead of AZT (Fig. 5A). EC50 of 4c in MT-4 cell cultures was 2.0 μM (95% CI (1.4–2.8) (Fig. 5B). CC50 of 4c was greater than 100 μM. The EC50 and CC50 of the 3TC control were 17 nM (n=3) and >100 μM, respectively [30]. In human tissue ex vivo infected with HIV-1, compound 4c at concentrations ranging from 1 nM to 10 μM inhibited HIV-1 replication in a dose-dependent manner by 0.6 ± 0.2%, 14.6 ± 6.5%, 45.7 ± 3.7%, 88.6 ± 2.9%, and 99.6 ± 0.1%, respectively (n=4; p>0.05; p>0.05; p<0.05; p<0.05; p<0.05) (Fig. 5C). The EC50 of 3TC alone in human tissue ex vivo was previously reported (16.6 nM (95% CI (2.7–101.6)) [30].
Figure 5. Inhibition of HIV-1 and CMV replication by heterodimer 4c.

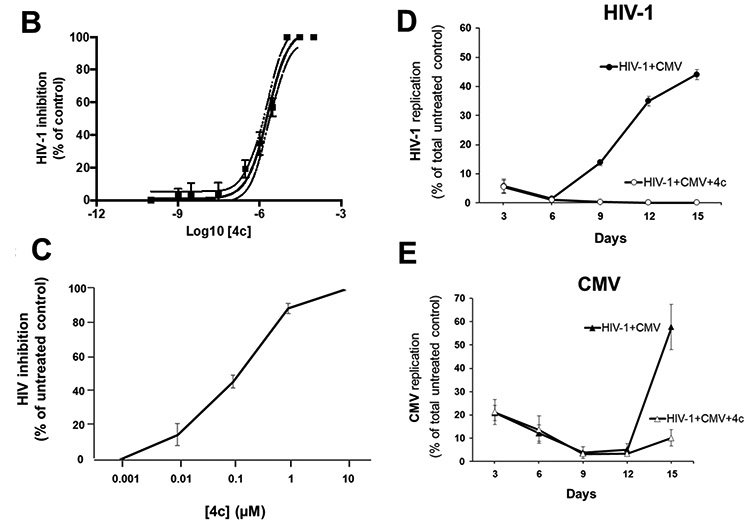
(A) Structure of the compounds used in the synthesis of the 3TC-based heterodimer 4c or resulting from its hydrolysis. (B) Anti HIV-1 activity of heterodimer 4c in MT-4 cell cultures. Graph was obtained by fitting the data points to a sigmoidal dose-response curve using Prism software. EC50 of 4c in MT-4 cell cultures was 2.0 μM (95% CI (1.4–2.8). (C) Inhibition of HIV replication in human tonsillar tissues. Human tonsillar tissues ex vivo were inoculated with X4LAI.04 and incubated with 4c at five concentrations ranging from 1 nM to 10 μM for 12 days. HIV-1 inhibition was expressed as percentage of donor-matched untreated control for each concentration of 4c. Means ± SEM of HIV-1 inhibition in tonsillar tissues blocks from 3 donors, relative to results for matched untreated tissues are presented. (D & E) Suppression of HIV-1 and CMV replication in coinfected tonsillar tissues. As for heterodimer 3c, tonsils were coinfected with HIV-1 (X4LAI.04) and CMV (AD169) and treated with 4c 10 μM or left untreated (control). (D) The replication of HIV-1 was assessed by measuring p24gag using a Luminex bead-based assay. (E) Replication of CMV was evaluated by real-time PCR by measuring viral DNA in culture medium. For each day, means ± SEM of viral replication as percent of total virus produced in untreated control condition in tonsillar tissues from 3 different donors are presented.
In HIV-1/CMV-coinfected human lymphoid tissue, 4c retained its inhibitory activity against both viruses: 10 μM 4c inhibited the cumulative production of p24gag by 99.4 ± 0.4% (n=3; p<0.05) (Fig. 5D) and the number of CMV DNA copies/μL by 78.7 ± 1.7% (n=3; p<0.05) (Fig. 5E). In these coinfected tissues, 3TC control inhibited HIV-1 replication by 99.7 ± 0.2% (n=3; p<0.05).
4. DISCUSSION
Here, we present newly-designed anti CMV/anti-HIV-1 heterodimers which are prodrugs of corresponding antiviral agents. A prodrug strategy is a well-known and effective way to overcome shortcomings of active compounds such as poor solubility and low bioavailability. Also, it can improve the pharmacokinetic profile and make drug delivery to the target more efficient [30, 33, 34].
AZT, an HIV-1 nucleoside reverse transcriptase inhibitor, was chosen as anti-HIV-1 agent because of our previous experience in synthesizing its effective prodrugs [30, 34]. AZT had been approved by the FDA and has been in clinical use for many years but its pharmacokinetic profile with jumps in blood levels results in toxicity and in emergence of HIV-1 resistant strains. On the other hand, nonnucleoside inhibitors of CMV are hydrophobic, have poor water solubility and some of them were cytotoxic. Therefore, we suggested that synthesizing mutual prodrugs may solve the problems of both antiviral agents. We decided to connect two active parts of prodrugs with acetic acid as a short linker. Six nonnucleoside inhibitors of CMV with linkers of different lengths, connecting uracil and p-bromophenoxy fragment, were used, and thus six corresponding target prodrugs were synthesized with good yields in two step-synthesis. The cytotoxicity and the antiviral activity of the compounds 1a-1f, which were chosen as anti-CMV molecules in the design of our heterodimers, were all described in our previous publication [23].
We tested the efficiency of these six compounds in inhibiting HIV-1 replication in cell cultures of MT-4 cells, a lymphocytic cell line known to support a high level of HIV-1 replication, and in cultures of WS1 cells, a fibroblastic cell line known to support CMV replication. All six compounds were active with similar inhibitory capabilities against both HIV-1 and CMV and were generally not cytotoxic. To further characterize the biological activity of the heterodimers, we chose one of them, 3c, for analysis in human tonsillar tissues ex vivo.
Specifically, we tested the antiviral activity and tissue cytotoxicity in a system of human lymphoid tissue ex vivo. This system of culture is an adequate model for preclinical drug testing, as it does not require cell activation or stimulation. Moreover, this system retains the original cytoarchitecture of the tissue and the expression of key cell-surface molecules that are important for viral infections [30]. Although the range of viral production can be large in human tissue ex vivo, as observed in patients in vivo, the kinetics of replication are very similar from tissue-donor to tissue-donor [29].
Ex vivo human lymphoid tissues have previously been used for microbicide pretesting [29, 32]. By specifically investigating the effect of the representative compound 3c on different T-cell subpopulations, we confirmed the low cytotoxicity of our chimeric compound in human lymphoid tissue.
The dual inhibitory activity of our compounds is an important feature, as the goal of developing such drugs is to interrupt the vicious circle of mutual facilitation between HIV-1 and CMV. Indeed, although both viruses have evolved complex strategies that manipulate our immune system to their own advantage, CMV infection alone is often asymptomatic. However, when present together, CMV infections can be particularly devastating, leading to severe organ disease and death. We found that in experimental control condition (no compound 3c), the replication of both viruses was typical of viral replication observed in human tissue ex vivo [29]. In tissues treated with 3c, both viruses were fully suppressed, thus confirming the hydrolysis of 3c in two active compounds inside cells.
Similar results were also obtained with compound 4c, for which AZT was replaced by 3TC, thus validating our strategy.
5. CONCLUSION
We designed, synthesized, and tested biological activities of new compounds active against HIV-1 and CMV, which is one of the major HIV-1 copathogens. The heterodimers described here are the first dual-targeted compounds supressing both HIV-1 and CMV. Further development and testing of the biological activity of such compounds and optimization of their pharmacokinetic profiles may lead to the development of dual-targeted anti CMV/HIV-1 antivirals.
Supplementary Material
Figure 1. Structure of the compounds used for the synthesis of heterodimer 3c or resulting from its hydrolysis.

The anti-CMV compound 1a-1f were combined with the anti HIV-1 compound AZT to synthesize six AZT-based heterodimers 3a-3f. Upon hydrolysis, heterodimers 3a-3f generate AZT and the anti CMV compounds 2a-2f. The anti HIV-1 activity of all heterodimers and AZT are presented in Table 1. The anti CMV compounds 1a-1f had no anti HIV-1 activity, as previously reported [23]. The anti CMV compound 2c (chosen as a representative of the group of compounds 2a-2f) also did not have any anti HIV-1 activity (see Table 1). The anti CMV activities of the compounds 1a-1f are in the range 4–20 μM [23]. The anti-CMV activity of the heterodimers 3a-3f and compounds 2a-2f are presented in Table 2. AZT does not have any anti CMV activity [28].
We synthetized seven dual-targeted anti-CMV/HIV heterodimers.
AZT or 3TC were used as anti HIV molecules while 1-[ω-(phenoxy)alkyl]uracil derivatives were used as anti CMV molecules.
These compounds inhibited CMV and HIV respectively in WS1 and MT-4 cell cultures.
Both AZT and 3TC-based heterodimers inhibited CMV and HIV replication in coinfected human lymphoid tissues.
ACKNOWLEDGEMENT
We thank Dr. Meghan Delaney and her entire staff at the department of Pathology of Children’s National Medical Center for their generous assistance in obtaining human tonsillar tissues.
The work of CV, RP, VM, and LM was supported by the NIH office of AIDS research (Intramural-to-Russian (I-to-R)) Program and the NICHD Intramural Program.
The work of AK, AM, MN, EM, MP, MK, and SK was supported by the Russian Foundation for Basic Research Projects № 17-54-30016 NIH and 18-29-08010.
We thank Dr. Paul Solyev for the assistance with HRMS registration and interpretation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
REFERENCES
- [1].Lisco A, Vanpouille C, Margolis L, War and peace between microbes: HIV-1 interactions with coinfecting viruses, Cell Host Microbe, 6 (2009) 403–408. 10.1016/j.chom.2009.10.010. [DOI] [PubMed] [Google Scholar]
- [2].Lichtner M, Cicconi P, Vita S, Cozzi-Lepri A, Galli M, Lo Caputo S, Saracino A, De Luca A, Moioli M, Maggiolo F, Marchetti G, Vullo V, d'Arminio Monforte A, Study IF, Cytomegalovirus coinfection is associated with an increased risk of severe non-AIDS-defining events in a large cohort of HIV-infected patients, J Infect Dis, 211 (2015) 178–186. 10.1093/infdis/jiu417. [DOI] [PubMed] [Google Scholar]
- [3].Barrett L, Stapleton SN, Fudge NJ, Grant MD, Immune resilience in HIV-infected individuals seronegative for cytomegalovirus, AIDS, 28 (2014) 2045–2049. 10.1097/QAD.0000000000000405. [DOI] [PubMed] [Google Scholar]
- [4].Freeman ML, Lederman MM, Gianella S, Partners in Crime: The Role of CMV in Immune Dysregulation and Clinical Outcome During HIV Infection, Curr HIV/AIDS Rep, 13 (2016) 10–19. 10.1007/s11904-016-0297-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tavenier J, Margolick JB, Leng SX, T-cell immunity against cytomegalovirus in HIV infection and aging: relationships with inflammation, immune activation, and frailty, Med Microbiol Immunol, 208 (2019) 289–294. 10.1007/s00430-019-00591-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gianella S, Letendre S, Cytomegalovirus and HIV: A Dangerous Pas de Deux, J Infect Dis, 214Suppl 2 (2016) S67–74. 10.1093/infdis/jiw217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Speck CE, Coombs RW, Koutsky LA, Zeh J, Ross SO, Hooton TM, Collier AC, Corey L, Cent A, Dragavon J, Lee W, Johnson EJ, Sampoleo RR, Krieger JN, Risk factors for HIV-1 shedding in semen, Am J Epidemiol, 150 (1999) 622–631. 10.1093/oxfordjournals.aje.a010061. [DOI] [PubMed] [Google Scholar]
- [8].Lisco A, Munawwar A, Introini A, Vanpouille C, Saba E, Feng X, Grivel JC, Singh S, Margolis L, Semen of HIV-1-infected individuals: local shedding of herpesviruses and reprogrammed cytokine network, J Infect Dis, 205 (2012) 97–105. 10.1093/infdis/jir700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gianella S, Morris SR, Anderson C, Spina CA, Vargas MV, Young JA, Richman DD, Little SJ, Smith DM, Herpes viruses and HIV-1 drug resistance mutations influence the virologic and immunologic milieu of the male genital tract, AIDS, 27 (2013) 39–47. 10.1097/QAD.0b013e3283573305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gianella S, Massanella M, Richman DD, Little SJ, Spina CA, Vargas MV, Lada SM, Daar ES, Dube MP, Haubrich RH, Morris SR, Smith DM, California T Collaborative Treatment Group, Cytomegalovirus replication in semen is associated with higher levels of proviral HIV DNA and CD4+ T cell activation during antiretroviral treatment, J Virol, 88 (2014) 7818–7827. 10.1128/JVI.00831-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sylwester AW, Mitchell BL, Edgar JB, Taormina C, Pelte C, Ruchti F, Sleath PR, Grabstein KH, Hosken NA, Kern F, Nelson JA, Picker LJ, Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects, J Exp Med, 202 (2005) 673–685. 10.1084/jem.20050882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wittkop L, Bitard J, Lazaro E, Neau D, Bonnet F, Mercie P, Dupon M, Hessamfar M, Ventura M, Malvy D, Dabis F, Pellegrin JL, Moreau JF, Thiebaut R, Pellegrin I, S.e.A. Groupe d'Epidemiologie Clinique du, Effect of cytomegalovirus-induced immune response, self antigen-induced immune response, and microbial translocation on chronic immune activation in successfully treated HIV type 1-infected patients: the ANRS CO3 Aquitaine Cohort, J Infect Dis, 207 (2013) 622–627. 10.1093/infdis/jis732. [DOI] [PubMed] [Google Scholar]
- [13].Freeman ML, Mudd JC, Shive CL, Younes SA, Panigrahi S, Sieg SF, Lee SA, Hunt PW, Calabrese LH, Gianella S, Rodriguez B, Lederman MM, CD8 T-Cell Expansion and Inflammation Linked to CMV Coinfection in ART-treated HIV Infection, Clin Infect Dis, 62 (2016) 392–396. 10.1093/cid/civ840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Patel EU, Gianella S, Newell K, Tobian AA, Kirkpatrick AR, Nalugoda F, Grabowski MK, Gray RH, Serwadda D, Quinn TC, Redd AD, Reynolds SJ, Elevated cytomegalovirus IgG antibody levels are associated with HIV-1 disease progression and immune activation, AIDS, 31 (2017) 807–813. 10.1097/QAD.0000000000001412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sheth PM, Danesh A, Sheung A, Rebbapragada A, Shahabi K, Kovacs C, Halpenny R, Tilley D, Mazzulli T, MacDonald K, Kelvin D, Kaul R, Disproportionately high semen shedding of HIV is associated with compartmentalized cytomegalovirus reactivation, J Infect Dis, 193 (2006) 45–48. 10.1086/498576. [DOI] [PubMed] [Google Scholar]
- [16].Gianella S, Anderson CM, Vargas MV, Richman DD, Little SJ, Morris SR, Smith DM, Cytomegalovirus DNA in semen and blood is associated with higher levels of proviral HIV DNA, J Infect Dis, 207 (2013) 898–902. 10.1093/infdis/jis777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gianella S, Anderson CM, Var SR, Oliveira MF, Lada SM, Vargas MV, Massanella M, Little SJ, Richman DD, Strain MC, Perez-Santiago J, Smith DM, Replication of Human Herpesviruses Is Associated with Higher HIV DNA Levels during Antiretroviral Therapy Started at Early Phases of HIV Infection, J Virol, 90 (2016) 3944–3952. 10.1128/JVI.02638-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Griffiths PD, CMV as a cofactor enhancing progression of AIDS, J Clin Virol, 35 (2006) 489–492. 10.1016/j.jcv.2005.10.016. [DOI] [PubMed] [Google Scholar]
- [19].Gianella S, Morris SR, Vargas MV, Young JA, Callahan B, Richman DD, Little SJ, Smith DM, Role of seminal shedding of herpesviruses in HIV Type 1 Transmission, J Infect Dis, 207 (2013) 257–261. 10.1093/infdis/jis683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gianella S, Scheffler K, Mehta SR, Little SJ, Freitas L, Morris SR, Smith DM, Seminal Shedding of CMV and HIV Transmission among Men Who Have Sex with Men, Int J Environ Res Public Health, 12 (2015) 7585–7592. 10.3390/ijerph120707585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Poorvashree J, Suneela D, Novel drug delivery of dual acting prodrugs of hydroxychloroquine with aryl acetic acid NSAIDs: Design, kinetics and pharmacological study, Drug Deliv Transl Res, 7 (2017) 709–730. 10.1007/s13346-017-0420-5. [DOI] [PubMed] [Google Scholar]
- [22].Cheng AV, Wuest WM, Signed, Sealed, Delivered: Conjugate and Prodrug Strategies as Targeted Delivery Vectors for Antibiotics, ACS Infect Dis, 5 (2019) 816–828. 10.1021/acsinfecdis.9b00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Novikov MS, Babkov DA, Paramonova MP, Khandazhinskaya AL, Ozerov AA, Chizhov AO, Andrei G, Snoeck R, Balzarini J, Seley-Radtke KL, Synthesis and anti-HCMV activity of 1-[omega-(phenoxy)alkyl]uracil derivatives and analogues thereof, Bioorg Med Chem, 21 (2013) 4151–4157. 10.1016/j.bmc.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Khandazhinskaya AL, Alexandrova LA, Matyugina ES, Solyev PN, Efremenkova OV, Buckheit KW, Wilkinson M, Buckheit RW, Chernousova LN, Smirnova TG, Andreevskaya SN, Leonova OG, Popenko VI, Kochetkov SN, Seley-Radtke KL, Novel 5-Norcarbocyclic Pyrimidine Derivatives as Antibacterial Agents, Molecules, 23 (2018) ARTN 3069 10.3390/molecules23123069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Buzdin AA, Artcibasova AV, Fedorova NF, Suntsova MV, Garazha AV, Sorokin MI, Allina D, Shalatonin M, Borisov NM, Zhavoronkov AA, Kovalchuk I, Kovalchuk O, Kushch AA, Early stage of cytomegalovirus infection suppresses host microRNA expression regulation in human fibroblasts, Cell Cycle, 15 (2016) 3378–3389. 10.1080/15384101.2016.1241928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Biancotto A, Brichacek B, Chen SS, Fitzgerald W, Lisco A, Vanpouille C, Margolis L, Grivel JC, A highly sensitive and dynamic immunofluorescent cytometric bead assay for the detection of HIV-1 p24, J Virol Methods, 157 (2009) 98–101. 10.1016/j.jviromet.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lisco A, Vanpouille C, Tchesnokov EP, Grivel JC, Biancotto A, Brichacek B, Elliott J, Fromentin E, Shattock R, Anton P, Gorelick R, Balzarini J, McGuigan C, Derudas M, Gotte M, Schinazi RF, Margolis L, Acyclovir is activated into a HIV-1 reverse transcriptase inhibitor in herpesvirus-infected human tissues, Cell Host Microbe, 4 (2008) 260–270. 10.1016/j.chom.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Freitas VR, Fraser-Smith EB, Chiu S, Michelson S, Schatzman RC, Efficacy of ganciclovir in combination with zidovudine against cytomegalovirus in vitro and in vivo, Antiviral Res, 21 (1993) 301–315. 10.1016/0166-3542(93)90009-8. [DOI] [PubMed] [Google Scholar]
- [29].Grivel JC, Margolis L, Use of human tissue explants to study human infectious agents, Nat Protoc, 4 (2009) 256–269. 10.1038/nprot.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Vanpouille C, Khandazhinskaya A, Karpenko I, Zicari S, Barreto-de-Souza V, Frolova S, Margolis L, Kochetkov S, A new antiviral: chimeric 3TC-AZT phosphonate efficiently inhibits HIV-1 in human tissues ex vivo, Antiviral Res, 109 (2014) 125–131. 10.1016/j.antiviral.2014.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Vanpouille C, Lisco A, Derudas M, Saba E, Grivel JC, Brichacek B, Scrimieri F, Schinazi R, Schols D, McGuigan C, Balzarini J, Margolis L, A new class of dual-targeted antivirals: monophosphorylated acyclovir prodrug derivatives suppress both human immunodeficiency virus type 1 and herpes simplex virus type 2, J Infect Dis, 201 (2010) 635–643. 10.1086/650343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Andrei G, Lisco A, Vanpouille C, Introini A, Balestra E, van den Oord J, Cihlar T, Perno CF, Snoeck R, Margolis L, Balzarini J, Topical tenofovir, a microbicide effective against HIV, inhibits herpes simplex virus-2 replication, Cell Host Microbe, 10 (2011) 379–389. 10.1016/j.chom.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Calogeropoulou T, Detsi A, Lekkas E, Koufaki M, Strategies in the design of prodrugs of anti-HIV agents, Curr Top Med Chem, 3 (2003) 1467–1495. [DOI] [PubMed] [Google Scholar]
- [34].Khandazhinskaya A, Matyugina E, Shirokova E, Anti-HIV therapy with AZT prodrugs: AZT phosphonate derivatives, current state and prospects, Expert Opin Drug Metab Toxicol, 6 (2010) 701–714. 10.1517/17425251003713501. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.