

Abstract
Drosophilae are emerging as a valuable model to study traumatic brain injury (TBI)-induced secondary injury cascades that drive persisting neuroinflammation and neurodegenerative pathology that imposes significant risk for long-term neurological deficits. As in mammals, TBI in Drosophila triggers axonal injury, metabolic crisis, oxidative stress, and a robust innate immune response. Subsequent neurodegeneration stresses quality control systems and perpetuates an environment for neuroprotection, regeneration, and delayed cell death via highly conserved cell signaling pathways. Fly injury models continue to be developed and validated for both whole-body and head-specific injury to isolate, evaluate, and modulate these parallel pathways. In conjunction with powerful genetic tools, the ability for longitudinal evaluation, and associated neurological deficits that can be tested with established behavioral tasks, Drosophilae are an attractive model to explore secondary injury cascades and therapeutic intervention after TBI. Here, we review similarities and differences between mammalian and fly pathophysiology and highlight strategies for their use in translational neurotrauma research.
Keywords: traumatic brain injury, Drosophila, secondary injury, inflammation, innate immunity, oxidative stress, mitochondria, apoptosis, neurodegeneration
Graphical Abstract
1. Introduction
Traumatic brain injury (TBI) is currently estimated at 69 million cases per year worldwide (1), contributing to high health care expenditures, lost productivity, and diminished quality of life for the patient. TBI has become a leading cause of death and disability, with some TBI survivors experiencing problems with cognitive, sensory, and emotional processing, requiring long-term care and rehabilitation (2). Despite knowing several mechanisms involved in the development of TBI-initiated neurological and psychiatric deficits, neuroprotective post-injury treatments to lessen or prevent these deficits remains largely unrealized.
Classification of human TBI as mild, moderate, and severe is traditionally based on symptoms using the Glasgow Coma Scale (GCS) score (3). Closed head trauma can further be classified using several clinical factors involving the duration of loss of consciousness, post-traumatic amnesia, altered mental state (disoriented or confused), neurological symptoms, and neuroimaging findings. Eighty to ninety percent of TBIs are categorized as mild, primarily resulting from blunt non-penetrating head injuries due to falls, vehicle accidents, violence, contact sports, and combat activities (4). Diffuse TBI is caused by an external mechanical impact or rapid acceleration-deceleration of the head that can cause diffuse neuron, glia, and cerebrovascular injury, initiating complex and prolonged sequelae of biochemical and physiological secondary injury processes. Prolonged secondary injury cascades can promote acute and delayed neuron death, such that TBI presents more like a neurodegenerative disease than a static event (5). The progressive loss of neurons and degenerating axotomized processes initiate immune responses to clear debris; promoting chronic neuroinflammation that is purported to accelerate the onset of genetically predisposed and age-related neurodegenerative diseases, including Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), Parkinson’s disease (PD) and chronic traumatic encephalopathy (CTE) (6, 7). TBIs have been challenging to treat because the pathways, complexity, and time course of secondary injury processes directing acute and delayed disease pathogenesis are poorly understood.
TBI animal models have guided important discoveries in identifying discrete mechanisms, mapping pathological events, and testing potential disease-modifying therapies. Mammalian models allow tight control over injury severity, and they faithfully recapitulate particular aspects of clinical TBI due to the conservation of the neuro-glial-vascular unit across species. Such discoveries have led to significant advances in understanding the mechanisms leading to neurological deficits by employing a range of state-of-the-art technologies that provide exquisite detail of ongoing pathology (8). A well-established body of evidence indicates that secondary injury processes mediating chronic neuroinflammation are a primary culprit in persisting and late-onset post-TBI symptoms.
Rodent models are the primary animal model for studying TBI, where their shorter lifespan facilitates longitudinal tracking. While mammalian models are essential, the translational process, length of study duration, drug and drug delivery costs, vivarium maintenance, and requirement of highly trained personnel present financial barriers to testing both acute and longitudinal impact of secondary injury processes and long-term effectiveness of therapeutic strategies. While genetic modification is available for mice and some rats, the process requires substantial time, skill, funding, and oversight. Further, mammalian systems have redundancy in their signaling pathways that can confound outcomes due to compensation. Translating outcomes from mammalian research to clinic care remains challenging due to adaptive and maladaptive signaling cascades associated with chronic neurodegeneration and neuroinflammation. Our knowledge of secondary injury mechanisms can be expanded by complementary use of novel models to unravel the complex interactions of a wide variety of pathophysiological processes that instigate and perpetuate neurological deficits after diffuse TBI.
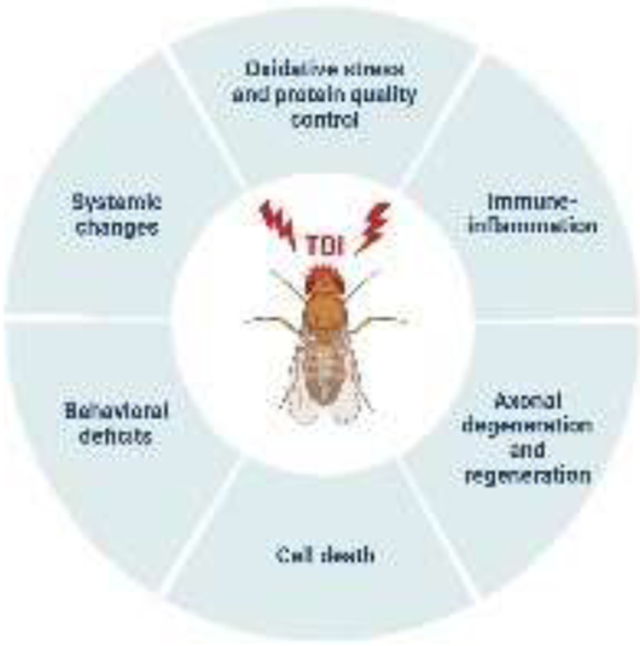
Drosophila offers the unique advantage of a model system with an array of genetic and biological approaches that can be utilized to rapidly uncover longitudinal pathophysiological processes associated with a heterogeneous TBI that underlie neurological diseases. Herein we discuss the secondary pathomechanisms that play a role in the long-term effects of TBI. We review what is currently known about central nervous system (CNS) primary injuries that drive secondary reactions due to alterations in signaling cascades that operate after TBI. We introduce current methods used to induce TBI in Drosophila and summarize new developments that highlight the utility of Drosophila as a model for TBI as cellular and molecular players in neuroinflammation and immune response, axonal degeneration and regeneration, cell death mechanisms, and peripheral effects. Finally, we present future perspectives on how studies in fly models will reveal crucial functions of genes that regulate underlying secondary injury cascades to infer translatability of these invertebrate studies to rodent and human pathophysiology. Unless otherwise stated, we focus on diffuse TBI.
2. TBI pathophysiology
2.1. Primary injury and secondary injury cascades
Primary injury during TBI is caused by biomechanical forces such as direct contact, rotational acceleration/deceleration, compression, torsion, or blast wave. In mammals, diffuse TBI is multifocal and imparts shear, tensile and compressive strains. It commonly produces damage to long-distance white matter connections via diffuse axonal injury (DAI), which includes damage to the neurofilament subunits and the axolemma adjacent to the nodes of Ranvier. Regions of gray-white matter interface and associated capillaries are vulnerable to shear forces resulting in petechial hemorrhage. Other forms of primary cellular injury include dendritic and synaptic damage and disruption. Movement of the brain within the skull can cause mechanical depolarization of neurons and release high concentrations of excitatory neurotransmitters that exacerbate cellular and tissue damage and contribute to blood-brain barrier permeability (8). TBI also causes dysregulation of cerebral blood flow and metabolism, which typically decreases after brain injury contributing to a mild ischemic effect (9). Together, these primary injuries instigate a number of secondary injury cascades implicated in the chronic morbidity associated with TBI (see Figure 1).
Figure 1.

Changes in TBI pathophysiological biomarkers over time post-injury. Graphs are not representative of actual fold changes (modified from Bramlett and Dietrich, 2015 and Simon et al., 2017) (31, 32). Created with BioRender.com
Increased extracellular levels of glutamate cause uncontrolled and excessive stimulation of glutamate receptors, leading to hyperexcitability and spreading depolarization, increasing the global energy demand. At the same time, decreased cerebral blood flow can cause a deficit of oxygen, resulting in an energy deficiency and metabolic crisis, where glycolysis is reduced, and anaerobic glycolysis reciprocally increases for days post-injury (10). Massive depolarization increases extracellular potassium and intracellular calcium and sodium, causing increased levels of Ca2+ in mitochondria that trigger the release of cytochrome C and apoptosis-inducing factors (AIF) into the extracellular space and initiates cell death signaling (11). As mitochondrial respiration increases to meet the energy demand, it produces excess reactive oxygen species (ROS) beyond the ability of antioxidants (e.g., glutathione peroxidase, superoxide dismutase) to neutralize (12). Calcium also activates numerous cellular pathways that stimulate the activation of protein kinase signaling, calpains, caspases, and enzymes, including nitric oxide synthase (NOS), which produces free radicals. Free radicals and ROS damage nucleic acids, inhibit the electron-transport chain, and cause oxidative damage to phospholipids, proteins, and some enzymes (11). Damaged biomolecules and organelles increase and offset protein homeostasis impacting the ubiquitin-proteasome and autophagy-lysosome pathways, where reduced proteasome activity and increased autophagic flux are impaired after experimental TBI (13-16). Despite the survival of axotomized and damaged neurons, secondary molecular cascades, immune responses, and altered afferent/efferent signaling can further compromise cellular stability and tip the balance towards cell death for months and years post-injury (17). Delayed cell death prolongs neurodegeneration and the activation of microglia and astrocytes associated with the neuroinflammatory response (18).
Neuroinflammation, which involves local and peripherally-derived immune responses, is an important aspect of secondary injury after TBI (19). Moreover, growing evidence indicates a strong link between neurological deficits and abnormalities in immune mechanisms that most likely involve both innate and adaptive immunity. The innate immune response is responsible for the rapid initial response to the primary injury and facilitates activation of the adaptive immune response (20). Cells involved in adaptive immunity include T- and B-lymphocytes, which have been implicated in both neuroprotection and neurodegeneration depending on the context in which they are activated (21). Yet, the mechanisms underpinning innate immunity following TBI are confounded by the complexity of the cell-mediated adaptive immune component, in part because they overlap. Therefore, it is of great therapeutic relevance to determine the effects of TBI on mediators of innate immunity in pathways that are highly conserved across species. Because the cellular effectors of innate immunity can determine the inflammatory phenotype, their involvement in secondary pathological processes has implications on the chronic responses after injury.
In mammals, there is still much to learn about the innate immune response to TBI. TBI causes DAI (hallmark pathology) that is characterized by impaired axonal transport leading to progressive axonal swelling, disconnection, and Wallerian degeneration of the distal axon (22, 23). Within minutes to hours post-TBI, these stressed, injured, and dying cells can release damage-associated molecular patterns (DAMPS; aka alarmins, IL-1α, S100 proteins, IL-33) and pathogen-associated molecular patterns (PAMPS). DAMPS and PAMPS are ligands for pattern recognition receptors (PRRs), including Toll-like receptor (TLR), NOD-like receptor (NLR), AIM2-like receptor (ALR), RIG-I-like receptor (RLR), and C-type lectin receptor (CLR) families, which are also important for activation of microglia and astrocytes, stimulating infiltration of monocytes, and recruitment of microglia (24). DAMPS and PAMPS also stimulate pathways that lead to the activation of nuclear factor-κB (NF-κB), AP-1, and IL-1β to mediate inflammation, inflammasomes, and apoptosis in response to brain injury (25). In addition to phagocytosis of dead tissue, infiltrating monocytes and activated microglia release tumor necrosis factor-α (TNF-α). Levels of the pro-inflammatory cytokine, TNFα, increase after TBI and are associated with other neurodegenerative disorders with prolonged activation of microglia and astrocytes (26). Along with activating similar pathways as PAMPS and DAMPS, TNF can stimulate the c-Jun N-terminal Kinase (JNK) pathway with the adaptor protein TRAF6, which also positively regulates signaling cascades for NF-κB and AP-1 and promotes apoptosis and autophagy after TBI (27-30).
The Janus kinase/signal transducer and activator of transcription proteins (JAK/STAT) pathway is important for both pro-inflammatory and anti-inflammatory signaling, where it has been implicated in the promotion of neurite outgrowth, cellular differentiation, proliferation, cell survival, and inhibitory neurotransmission (33, 34). Following experimental TBI, the JAK/STAT pathway activated within 3h. Pharmacological manipulation that increased activation of the JAK/STAT pathway supported benefits, including reduced apoptosis and less tissue loss for acute cellular events (34, 35). However, inhibition demonstrated improvement in epilepsy and a prolonged improvement in vestibular function in other brain injury models (36). A better understanding of this pathway and the longitudinal impact on activation and inhibition after TBI may be valuable to guiding circuit reorganization and repair.
2.2. Secondary injury mechanisms increase the risk for neurological deficits
The high degree of complexity of multiple processes involving molecular and circuit-level components after TBI has long-term implications for the development of neurological deficits. ROS are responsible for processes that lead to an accumulation of protein aggregates involved in secondary pathogenic processes that form hallmark deposits after TBI that are major risk factors for developing chronic neurodegenerative diseases like CTE, frontotemporal dementia, and AD (37-39) . Dopamine neurons are vulnerable to oxidative stress, with implications for loss of dopamine neurons associated with PD (40). Multiple secondary injury mechanisms serve as mediators of apoptosis, resulting in delayed apoptosis. Delayed onset of neuron degeneration increases the temporal window for neurodegenerative processes and prolongs the activation of glia and other associated neuroinflammatory processes, corroborating clinical pathology (41, 42). Activated astrocytes and microglia also mediate adaptive and maladaptive regenerative and compensatory processes post-injury (43-45). Neuroplastic regenerative responses manifest as dendritic and synaptic sprouting (46), indicating axonal and dendritic growth and connection in surviving axotomized neurons (47). This novel connectivity does not reconstitute the same level of pre-injury circuit integrity, contributing to the appearance of novel or increased severity of symptoms reported pre-TBI (8). Damage and reorganized circuits can prevent or decrease the effectiveness of feedback regulation of hypothalamic-pituitary axes, preventing the return to homeostasis and giving rise to affective and other neuroendocrine-related symptoms (44, 48-52). Increased circulating inflammatory mediators can also influence peripheral organs, like the gut, contributing to leaky gut syndrome that can instigate peripheral inflammatory responses and microbiome dysbiosis, which forms precursors for neurotransmitters and hormones (53, 54). These distinct processes occur in parallel, where biomolecules from secondary injury cascades often overlap or have multiple targets. The interacting secondary injury factors give rise to an array of acute and chronic behavioral deficits that include impairments of cognitive, affective, sensory, motor functions, and sleep-wake rhythms. Clinically, this creates challenges for identifying TBI-induced symptoms, diagnosing appropriate treatment, and distinguishing TBI-induced symptoms from other non-TBI related neurological deficits, where a better understanding of the relevant signaling cascades may be relevant to preventative treatments or better symptom management for improved long-term outcomes and treatment.
3. Modeling TBI in Drosophila
As a human TBI model, the fly offers a simplistic platform to recapitulate head injuries at controlled levels of force and acceleration to study injury mechanisms that are not yet well understood. The advantages of the fruit fly as a human disease model have been well recognized in modern biology, as approximately 75% of human genes have a fly ortholog. Drosophila has emerged as an excellent model system for several neurological disorders with a broad measure of validity in the context of genetics (construct validity) and simple and complex phenotypic behaviors (face validity) to test potential candidates for therapeutic efficacy (predictive validity). A range of recent technological advancements allow genetic and molecular manipulations that isolate pathophysiological pathways, thus providing a more comprehensive understanding of complex disorders (55). Thousands of genetically manipulated Drosophila stocks are commercially available, facilitating observation of cell-specific gene function/activity and organelle markers (https://bdsc.indiana.edu/). Flies are inexpensive to maintain and have identifiable, behaviorally conserved brain circuitry. A relatively short lifespan facilitates longitudinal aging studies, making them particularly attractive models of neurodegenerative disease. Given these useful features and to address specific scientific questions, flies are uniquely positioned to fill the knowledge gaps left out in mammalian TBI models. To this end, multiple TBI models have been developed for the assessment of TBI in Drosophila.
3.1. Head and body injuries
The high impact trauma (HIT) device inflicts an impact acceleration-deceleration injury to flies by deflecting and releasing a plastic vial rapidly onto a polyurethane foam pad, modeling head and/or body injuries that occur in motor vehicle accidents and sports injuries (56). The HIT model reproduces closed head injury characteristic of blunt non-penetrating head trauma. The angle of spring deflection can be changed to alter impact velocity, and repetitive strikes can be administered to increase injury severity. HIT induces temporary incapacitation, locomotor dysfunction, mitochondrial protein oxidation, and brain neuropil vacuole formation (56-58). Heads of young and older flies exposed to HIT injury have increased Relish (Relish is the fly NF-κB ortholog) and antimicrobial peptide (AMP) expression within twenty-four hours of injury, and AMP expression levels correlate with mortality (56, 59-62). Interestingly, changes in larval length, early eclosion, and behavioral deficits have been observed in offspring of HIT-injured flies (63). The HIT model induces high variability in injury severity and lacks control of rebound injury. A caveat for this model is that variability in reproducibility across laboratories can be introduced by components of the HIT device, including the type of spring used and the thickness and consistency of the foam padding, thus requiring optimization of each HIT device. A major advantage of the HIT model is its relatively low price, ease of setup and operation, and high throughput.
The bead ruptor platform model incorporates a homogenizer platform to induce repetitive impact to a large number of flies in screwcap tubes (64). The model is well suited to study the functional consequences of repetitive brain injury in the absence of anesthesia. Video acquisition allows calculation of fly velocity and frame-by-frame analysis detection of head, thorax, and abdominal contact with the tube. The degree of injury can be controlled by adjusting the rotation speed and injury exposure time. Bead ruptor platform-induced TBI damages neural processes, induces sleep disturbances, and increases acute and sustained AMP expression (64). As with the HIT model, the bead ruptor platform causes a combination of head and body injury. This model also allows observation of the structural and functional consequences of repetitive injuries. While both head-body injury models generate variable injury severity within a single experiment, the force of injury can be more tightly controlled than with the less expensive HIT device.
3.2. Head-specific injury models
For the Strike Device injury model, direct head impact is delivered when pressurized CO2 is applied to the head of an anesthetized fly that is immobilized by a fly holder attached to a syringe barrel (65). The burst of gas creates a very rapid, localized acceleration-deceleration and replicates diffuse injury commonly observed in humans. This technique reduces damage to other parts of the body and allows for delivery of measurable impacts restricted to the head, while controlling for severity, time between impact, and the number of impacts. Injured flies have locomotor deficits and reduced life span (65). Because the direct impact to the head is more controlled and reproducible in this model, it allows more precise mapping of the CNS response to injury. This model is relatively expensive, requires more technical equipment, and has relatively low throughput.
Another closed head injury device that reduces body injury and models human diffuse TBI consists of a pull-type solenoid powered block to inflict a precisely controlled strike to the head of individually restrained flies (66). Flies are collected using an aspirator and transferred into a restrainer (pipette tip) to expose the fly’s head. The head is subjected to impact by a controlled current which drives the brass trapezoid-shaped block into the head. Injury-induced phenotypes include increased mortality and motor deficits. This closed head injury model provides the opportunity to avoid anesthesia, but the procedure is labor-intensive, low throughput, time-consuming, and requires specialized equipment.
The controlled compression head-specific injury model mimics compression loads with a blunt impact as used in rodent TBI models. Here, variable voltage to a power amplifier causes a piezoelectric actuator to bend rapidly, creating a linear displacement and compression of an immobilized fly head against a metal plate (67, 68). The compression impact can be adjusted for mild, moderate, and severe injury levels to induce ataxia, neuronal degeneration, and reduced lifespan. Severe injury induces cognitive and memory acquisition deficits as well as transient blood-brain barrier permeability and decreased macrophage-mediated debris clearance in the brain. Additionally, the controlled compression injury can induce transient increases in lysosome number, proteasome activity, and antioxidant mediator glutathione S transferase, all of which indicate oxidative stress (68).
4. Insights gained from Drosophila TBI models
4.1. Oxidative stress and protein quality control system
Mitochondrial respiration is the primary source of ROS in cells. Oxidative stress occurs when antioxidant systems fail to control levels of ROS, which react with and damage phospholipids, nucleic acids, and proteins. The accumulation of free radicals and oxidative modification of fatty acids generates mitochondrial lipid peroxides that can trigger deadly changes in membrane permeability and a feedforward production of ROS by the electron transport chain, which itself is susceptible to oxidation (reviewed in (69)). Oxidative stress is implicated in the propagation of neurodegenerative diseases via secondary injury processes following TBI (reviewed in (12)). Cyclooxygenase disruption has been reported in 24 hours post-TBI fly brain, leading to oxidative stress and decreased ATP production (70). Increased mitochondrial aging and a transient increase in antioxidant demand, both symptoms of oxidative stress, have also been reported in flies exposed to TBI (68, 71). Similar events have been identified in neurodegenerative diseases and disease models (40, 72-76).
Intracellular protein turnover reduces oxidative stress by eliminating damaged proteins and depolarized mitochondria, promoting cell function and survival. After TBI, oxidative stress disproportionately increases levels of oxidized dysfunctional proteins (misfolded) that can evade degradation systems and promote toxic protein aggregate formation (77). Disruptions in the ubiquitin-proteasome system, chaperone-mediated autophagy, and macroautophagy have been heavily implicated in neurodegenerative processes, including TBI (14, 78). Interestingly, mild TBI triggers a sustained increase in autophagosome marker autophagy gene 8a (atg8a), the Drosophila ortholog of mammalian GABA type A receptor-associated protein (GABARAP) and autophagosome substrate receptor ref(2)p (fly ortholog of p62) (64). Moderate and severe TBI can transiently increase the presence of LysoTracker®-positive lysosomes and degradation of CL1-GFP, which is recognized by E3 ubiquitin ligases (68), while mild TBI has no effect on LysoTracker® staining. Cytoplasmic aggregates called “stress granules” containing the fly homolog of ALS-associated transactive response DNA-binding protein-43 (TDP-43) appear in the fly brain within 24 hours after repetitive TBI, and their presence increases with the number of hits (79). Taken together, these reports suggest that autophagy demand could increase with the severity of brain injury. Rapamycin administration or overexpression of atg8a reduces stress granule formation; thus, autophagy machinery may be overwhelmed after TBI or inhibited by it (79). Additional studies featuring the wide variety of available fly models of neurodegenerative diseases have the potential to strengthen our understanding of the mechanisms by which TBI predisposes patients to a variety of neurodegenerative diseases (80). While Drosophila TBI can trigger autophagy, severe brain injury may produce an insurmountable burden on autophagy machinery, warranting therapeutic strategies promoting autophagy, particularly in early disease stages.
4.2. Immune response and inflammation
Inflammation is a highly conserved cellular response to stressful stimuli such as infection and tissue damage that plays a critical role in the clearance of the pathogen or tissue debris, followed by the re-establishment of tissue homeostasis. The acute inflammatory response is initiated by so-called ‘danger signals,’ including PAMPs and DAMPs released by injured neurons. These molecules interact with PRRs on a variety of cells to generate early inflammatory mediators, that if appropriately controlled, play an important role in the eradication of the pathogen and/or clearance of cellular debris. If not appropriately controlled, however, the inflammatory response can become chronic, which may be associated with exacerbation of tissue injury rather than promoting tissue repair. Several clinical and experimental studies have shown that TBI induces a sustained neuroinflammatory response and overactivation of microglia that continues years after the insult - leading to chronic neurodegeneration, progression of dementia, and traumatic encephalopathy (32, 81, 82).
The fact that Drosophila is devoid of adaptive immune reactions offers a simplistic yet well-characterized model of highly conserved innate immunity relative to the human. Pioneering studies identified several multifaceted innate defense responses involving systemic and cellular reactions as players in the first line of defense in Drosophila, with most components of innate immunity evolutionarily conserved across species (83-85). In Table 1, the homologs for molecules of these pathways are defined for Drosophilae and mammals. In Drosophila, the two main systemic innate immune response pathways include the immune deficiency (Imd) and the Toll pathway. In the Imd pathway, binding of the cell surface receptor, Peptidoglycan recognition protein LC (PGRP-LC) activates Imd, which is a death domain-containing protein sharing homology with the mammalian receptor-interacting protein (RIP) (86). In mammals, RIP functions in a complex with the TNF-α receptor; thus, the Imd pathway in Drosophila is considered the significant equivalent of the TNF-α receptor signaling in mammals, while the Toll signaling pathway is similar to the mammalian TLR pathway involving myeloid differentiation primary response protein 88 (MyD88) (87, 88). These Imd and Toll signaling pathways function in parallel and induce the nuclear translocation of NF-κB homologs which activate the transcription of overlapping but different effector proteins, including AMPs, which are ubiquitously expressed in all multicellular organisms from insects to humans (89-93). In Drosophila, chronic expression of NF-κB-induced effector proteins in the absence of infection or overt trauma has been associated with the development or progression of neurodegenerative phenotypes (94-97). In mammals, NF-κB signaling is an important regulator of TBI-induced changes in the phenotypic plasticity of microglia and macrophages that results in the release of proinflammatory factors and free radical formation (81, 98-100).
Table 1.
Vertebrate/mammalian homologs of Drosophila immune signaling molecules. Homologs listed were obtained from FlyBase, unless otherwise noted in the main text of the manuscript. Table subheadings indicate the relevant section of the manuscript in which they are discussed.
| HOMOLOGS | |
|---|---|
| Drosophila melanogaster | Vertebrate/mammalian |
|
4.2 Immune Response and Inflammation
Imd signaling | |
| PGRP-LC (Peptidoglycan recognition protein LC) | PGLYRP1 (peptidoglycan recognition protein 1) |
| Fadd (Fas-associated death domain-containing protein) | FADD |
| Diap2 (Death-associated inhibitor of apoptosis 2) | BIRC2 (Baculoviral IAP repeat-containing 2) |
| Tak1 (Transforming growth factor-b activated kinase 1) | MAP3K7 (mitogen-activated protein kinase kinase kinase 7) |
| IKKb | IKBKB (inhibitor of nuclear factor-kappa B kinase subunit beta) |
| key (kenny) | IKBKG (inhibitor of nuclear factor-kappa B kinase regulatory subunit gamma) |
| Dredd (Death related ced-3/Nedd2-like caspase) | CASP10/CASP8 (Caspase-10/Caspase-8) |
| casp (caspar) | FAF1 (human Fas-associated factor 1) |
| Rel (Relish) | NF-kB1 (p105)/NF-kB 2 (p100) |
| Toll signaling | |
| T1 (Toll) | TLRs (Toll-like receptors) |
| MyD88 | MYD88 (MYD88 innate immune signal transduction adaptor) |
| pll (pelle) | IRAK4 (IL-1 Receptor-associated kinase 4) |
| cact (cactus) | NFKBIA (NF-kB inhibitor alpha) |
| Dif (Dorsal-related immunity factor) | RELA (RELA proto-oncogene, NF-kB subunit/p65); RELB |
| dl (dorsal) | REL (REL proto-oncogene, NF-kB subunit); RELA |
| JNK signaling | |
| hep (hemipterous) | MAP2K7 (mitogen-activated protein kinase kinase 7) |
| Mkk4 (MAP kinase kinase 4) | MAP2K4 (mitogen-activated protein kinase kinase 4) |
| bsk (basket) | MAPK10/MAPK8 (mitogen-activated protein kinase 10/8) |
| Jra (Jun-related antigen) | JUN (Jun proto-oncogene, AP-1 transcription factor subunit) |
| kay (kayak) | FOS (Fos proto-oncogene, AP-1 transcription factor subunit) |
| JAK/STAT signaling | |
| dome (domeless) | PTPRQ (Protein tyrosine phosphatase receptor type Q) |
| hop (hopscotch) | JAK1/JAK2 (Janus kinase 1/2) |
| Stat92E (Signal-transducer and activator of transcription protein at 92E) | STAT (Signal transducer and activator of transcription) |
| Socs36E (Suppressor of cytokine signaling at 36E) | SOCS (Suppressor of cytokine signaling ) |
| Tep1/Tep2 (Thioester-containing protein 1, 2) | CD109 (a2-macroglobulin/C3 family) |
| HDAC1 (Histone deacetylase 1) | HDAC1 (Histone deacetylase 1) |
| Dsp1 (Dorsal switch protein 1) | HMGB3 (high mobility group box 3) |
| 4.3 Axonal Degeneration and Regeneration | |
| drpr (draper) | MEGF10 (multiple EGF like domains 10) |
| Shark (SH2 ankyrin repeat kinase) | ZAP70 (zeta chain of T cell receptor-associated protein kinase 70) |
| Src42a (Src oncogene at 42A) | FRK (Fyn related Src family tyrosine kinase) |
| ced-6 | GULP1 (GULP PTB domain-containing engulfment adaptor 1) |
| egr (eiger) | TNFSF (tumor necrosis factor superfamily) |
| wgn (wengen) | NGFR (nerve growth factor receptor); TNFRSF (TNF receptor superfamily) |
| Traf6 (TNF receptor-associated factor 6) | TRAF6 (TNF receptor-associated factor 6) |
| 4.4 Cell Death | |
| AIF (Apoptosis-inducing factor) | AIFM1 (Apoptosis-inducing factor 1, mitochondrial) |
| Buffy | BOK (Bcl-2-related ovarian killer protein) |
| Cyt-c-d (Cytochrome c distal) | CYSCS (cytochrome c) |
| Cyt-c-p (Cytochrome c proximal) | CYSCS (cytochrome c) |
| Dark (Death-associated APAF1-related killer) | APAF-1 (Apoptotic protease-activating factor 1) |
| DrICE (Death related ICE-like caspase) | CASP3/6/7 (Caspase 3/6/7) |
| Dronc (Death regulator Nedd2-like caspase) | CASP2/9 (Caspase 2/9) |
| Diap1 (Death-associated inhibitor of apoptosis 1) | XIAP (X-linked inhibitor of apoptosis) |
| Dcp-1 (Death caspase-1) | CASP3/6/7 (Caspase 3/6/7) |
| Debcl (Death executioner Bcl-2) | BOK (Bcl-2-related ovarian killer protein) |
| HTRA2 (HTRA2-related serine protease) | HTRA2 |
| Tor (Target of rapamycin) | MTOR (Mammalian target of rapamycin) |
Numerous studies in Drosophila suggest that TBI activates both the Imd and the Toll pathways. The study of innate immune pathway activation in flies after TBI has begun to elucidate the potential role of AMP gene expression on various behavioral factors and pathological injury outcomes. Although there is some discrepancy across studies, there is general agreement that AMP genes related to both Toll (e.g., Drosomycin, Metchnikowin) and Imd (e.g., Diptericin, Attacin) pathway activation are upregulated in the heads of TBI flies as early as one to two hours post-injury (56, 60, 64, 101). The majority of studies suggest that expression is maintained for at least 24 hours post-injury and potentially as long as 72 hours post-injury before returning to baseline (56, 66, 102). Furthermore, these injury-induced effects appear to be independent of injury model as similar observations have been reported with the HIT device (56, 60, 101), the Omni bead homogenizer (64), a head-only model (66), and a penetrating injury model (102). Few studies have evaluated long-term changes in AMP gene expression following TBI in Drosophila, and those that have yielded conflicting results with one study reporting that expression of all AMP genes returned to baseline at 7 days post-injury (66) and others reporting a secondary increase in AMP genes at 7 days post-injury (64). Overexpression of Metchnikowin increases TBI-induced mortality and the absence of Relish, the NF-κB homolog in the Imd pathway, reduces TBI-induced mortality (62, 66), which implicates injury-induced activation of NF-κB signaling in secondary injury processes with deleterious consequences.
Two additional innate immune pathways present in Drosophila include the JNK and the JAK/STAT pathways (Figure 2). Much less is known regarding the role of these pathways in Drosophila TBI (dTBI), although they are both activated in response to axonal injury in the Drosophila CNS. In Drosophila, the highly conserved, canonical JNK pathway is activated when Eiger, the Drosophila homolog of mammalian TNF, binds to its cognate receptor, Wengen. With the help of the adaptor protein, TNF receptor-associated factor 6 (Traf6), Drosophila transforming growth factor activated kinase 1 (dTAK1; homologous to the mammalian MAP kinase kinase kinase 7) (103) is activated. Drosophila TAK1 is also activated downstream of Imd as a separate branch of the Imd pathway. Activation of dTAK1 results in the phosphorylation off hemipterous (homolog of mammalian JNKK, or MKK7), which then phosphorylates basket (JNK homolog), that translocates to the nucleus to activate transcription of AP-1-related genes. The dAP-1 is a heterodimer formed by Jra (the homolog of mammalian c-Jun) and kayak (homolog of mammalian c-fos). Jra expression increases following dTBI (101), indicating that the JNK pathway is indeed activated by dTBI. Several factors influence the effects of this pathway on the outcome from TBI. If the innate immune response is adequately controlled, low levels of ROS will result in transient activation of JNK signaling, which is important for wound healing and may serve a neuroprotective function. An increase in glutathione-S-transferase (Gst) genes has been reported following diffuse TBI (68), suggesting that an antioxidant response is initiated following CNS injury. Furthermore, Relish mutant flies have a downregulation of Gst genes, suggesting a role for Relish in mediating the antioxidant response (104). Conversely, if tissue injury is such that high levels of ROS are produced (e.g., oxidative stress, which is known to occur with chronic inflammation), there is sustained JNK activation, which likely contributes to inflammation-induced apoptosis as has been observed in mammalian models (105-107). In addition to oxidative stress, JNK signaling is negatively regulated by Relish. Activation of Relish attenuates JNK activity by promoting the proteasomal degradation of dTAK1 (108). In the absence of Relish activation, JNK signaling is sustained, resulting in a pro-apoptotic response. Although it has not been directly evaluated in TBI, such a mechanism could account for enhanced survival observed in Relish-deficient flies (66).
Figure 2.

Immune signaling pathways in Drosophila that are activated in response to CNS injury. The red line indicates negative interactions between the indicated pathways. Created with BioRender.com
Several genes that regulate activity in the JAK/STAT pathway, including Socs36E and Tep2, are induced following injury, which suggests that the JAK/STAT signaling pathway is activated in flies as has been reported in mammals (Figure 2) (36, 60, 109). The JAK/STAT pathway is implicated in regenerative inflammatory signaling in response to TBI. Tep2 is a member of the thioester-containing protein (TEP) family of genes, which are complement-like proteins that share homology with the α2-macroglobulin/complement C3 family. Drosophila expresses a single STAT protein, Stat92E, that, although not directly evaluated in dTBI models, is a critical element in the axonal response to injury (see below). In addition, Stat92E, in association with dAP-1, regulates the production of AMPs (110). The AP-1/JNK and JAK/STAT pathways interact to negatively regulate Relish activation, which plays an important role in terminating immune responses. Specifically, dAP-1 (Jra) and Stat92E combine with the Drosophila HMG protein (a homolog of mammalian HMGB1, the prototypic DAMP), dorsal switch protein (Dsp1), and histone deacetylase to inhibit the transcription of the diverse immune effector genes activated by Relish (110). This is important to avoid the potentially negative effects that would occur with prolonged activation of the innate immune response (e.g., chronic inflammation). Whether this regulatory mechanism is operative after dTBI in insects or mammals is not known.
4.3. Axonal degeneration and regeneration
Axonal degeneration in the CNS and the peripheral nervous system have been observed in Drosophila and mammalian models of TBI and neurodegenerative disease. The response to axotomy in Drosophila resembles the responses observed in mammalian models. Specifically, within minutes, the formation of retraction bulbs and filopodial sprouts is observed in the proximal axonal fragments, with vesiculation and fragmentation apparent in the distal segments (111). These characteristic features of Wallerian degeneration indicate that this is a conserved evolutionary process. Similar to the limited regeneration observed in mammalian CNS axons, the sprouting response in Drosophila is transient with only limited regrowth of axons.
The clearance of axonal debris generated in response to axotomy is a prerequisite for axonal regeneration. Several studies have aimed at identifying the signaling pathways that promote clearance of axonal debris in the CNS. Early work indicated a requirement for Draper in the clearance of axonal debris in the CNS (Figure 2) (112). Later studies identified ensheathing glia as the predominant CNS phagocytic cells expressing draper following axotomy and established that Draper-mediated engulfment of axonal debris requires dCed-6, Src42a, and Shark (113, 114). DAMPs bind to Draper and activate the dJNK pathway and the JAK/STAT pathway (115) and JNK signaling and Stat92E in glial cells are both required for upregulation of Draper after axotomy (116). In addition, Eiger/TNF signaling via dTRAF6 activates Dorsal/NF-κB to induce proliferation of ensheathing glia following stab injury of the ventral nerve cord (117), and transcriptional reporters for JNK pathway signaling to dAP-1 are increased in glia after axonal injury (116). Collectively, these data provide several mechanisms by which activation of JNK and Jak/STAT signaling in response to axotomy can provide cross-regulation of NF-κB signaling pathways that regulate the innate immune response to injury.
Axonal regenerative responses in Drosophila can be enhanced by activating several signaling pathways, including PKA, JNK, and the JAK/STAT pathway (111). Similar to cAMP-based improvements in axonal regeneration observed in mammals (118, 119), increased PKA activity enhanced axonal sprouting in flies (111). The JNK pathway also plays a key role in axonal regeneration after injury. Activation of JNK signaling by overexpression of a constitutively active form of hemipterous, the Drosophila homolog of mitogen-activated protein kinase kinase 7 (MAP2K7), increased CNS axonal regeneration after injury, an effect that was blocked by overexpression of a dominant-negative allele of basket, the Drosophila homolog of JNK (111). Larvae from adult Drosophila that were previously given a closed head TBI exhibited axonal and dendritic regeneration via activation of the dual leucine zipper kinase (DLK) and the JNK pathway (120). While the mechanisms of action are not yet well understood, researchers hypothesize that inactivation/activation of these pathways is mediated in part by the enzymatic and cellular pathway changes that occur post-TBI.
4.4. Cell death
Cell death after TBI results from mechanically induced primary injury and secondary neuroinflammation. While apoptosis, necroptosis, pyroptosis, and necrosis are all implicated in TBI patients and mammalian models, cell death reported in fly TBI models is primarily limited to brain vacuole formation, where the contributions of apoptosis, necroptosis, or necrosis are not clear (56, 68, 121-124). Drosophila studies have advanced our understanding of conserved apoptotic pathways (Figure 3) (125, 126). Interestingly, while intrinsic apoptosis machinery in flies and mammals comprises many of the same components and mechanisms of regulation, its contribution to cell death has been primarily observed during development. Whether the lack of evidence of apoptosis in dTBI is due to its limited contribution to cell death in the fly model or to scarcity of research conducted in this area remains to be determined. If the latter, powerful genetic manipulation/expression tools and highly conserved apoptotic homologs are untapped resources. For example, the fly adaptor protein Dark is a functional homolog of mammalian intrinsic pathway-mediated cytoplasmic apaf-1 (mediated by NLR signaling in mammals) that promotes the formation of apoptosomes in conjunction with the initiator caspase Dronc (mammalian homolog of caspase-9; Table 1) (127-129). This subsequently activates effector caspases DrICE (caspase 3 homolog) and Dcp-1 (127-131) promotes apoptosis. Whether cytochrome c is involved in fly apoptosome formation is unclear (132-136). Dark shares cytochrome c interacting domains WD40 and CARD, suggesting that it may (127, 128, 133); however, fly apoptosome formation can occur in the apparent absence of cytochrome c (132-134).
Figure 3. Intrinsic apoptosis in Drosophila.

The scheme above depicts interactions of Drosophila apoptosis players and their mammalian (in grey font) orthologs and homologs. Dashed arrow (⇢) indicates translocation. Created with BioRender.com
As in mammals, fly mitochondria swell in response to stress (72), suggesting that mitochondrial membrane permeabilization may be involved in initiating intrinsic apoptotic signaling. Apoptosis regulation also seems to be conserved as flies express homologs of apoptosis regulating proteins including anti-apoptotic protein Buffy, and pro-apoptotic Debcl, which are Bcl-2 and Bok homologs, respectively. Interestingly, Buffy appears to bind to both endoplasmic reticulum and mitochondrial membranes to suppress the unfolded protein response, while Debcl functions more like Bok by binding to mitochondrial membranes (131, 137-141). Drosophila inhibitor of apoptosis (IAP) protein, Diap1, regulates DrICE caspase activity via ubiquitination and competitive binding (142), and dOmi, a fly pro-apoptotic protein that functions like its mammalian counterpart, HtrA2/Omi in its inhibition of IAPs (135, 136, 143). Diap2 may also provide protection against inflammation as Imd pathway components are also Diap2 ubiquitination substrates (144-146). Conversely, IAP antagonist Hid transcription can be triggered by JNK signaling upon Eiger binding of its receptor Grindelwald (GRND) (147, 148). GRND is the fly homolog of mammalian TNF-α receptor, and its endogenous agonist Eiger is the fly homolog of TNF-alpha. It is important to note that developmental apoptosis pathways may differ from degenerative ones; where only the former is well characterized. Reaper, Hid, Grim, and Sickle are functional homologs to pro-apoptotic proteins SMAC/DIABLO in that they inhibit Diap1; however, unlike their mammalian IAP antagonist homologs, Reaper, Hid, Grim, and Sickle activities are transcriptionally regulated (149). Reaper, Hid, Grim bind to mitochondria to promote membrane permeabilization during developmental apoptosis (150).
While flies have fewer orthologs of mammalian necroptosis proteins, functional studies suggest homologous pathways that signal necrotic cell death exist in flies. Indeed, there are numerous reports of changes in innate immune protein levels following dTBI; thus, necrosis or necroptosis may be primary contributors to cell death. For instance, as discussed above, Drosophila Imd and PGRP-LC pathways share functional homology with mammalian RIP1/TNFR1 and RIPK2 nucleotide-binding oligomerization domain 1/2 (NOD1/2) mammalian necroptotic pathways, respectively (151-154). Fly necroptosis appears to be mediated via Imd and JNK signaling pathways. Additionally, inhibition of target of rapamycin (TOR, the Drosophila ortholog of mammalian TOR) increases lifespan via downregulation of conserved substrate cyclic AMP response element-binding protein coactivator transducer of regulated CREB activity (TORC) (155). Flies harbor a single isoform of TORC (crtc) that seems to perform the functions of mammalian TORC1 and TORC2 (155, 156). Although not yet demonstrated in flies, disrupted TOR activity may trigger inflammatory pathways resulting from decreased autophagy (157-159).
5. Improving translatability of Drosophila in TBI research
The heterogeneity of TBI involves multiple entities that influence pathogenesis and rate of disease progression. Indeed, several factors, including age, injury location, injury severity, previous head injuries, genetics, sex, drug abuse, comorbid psychological disorders, and sub-optimal post-morbid conditions contribute to continuing or late-onset TBI symptoms (160). Current TBI classification is primarily based on duration of loss of consciousness, GCS score, and length of post-traumatic amnesia, along with several neuroimaging measures used for diagnosis and prognosis; however, the ongoing pathophysiological processes are not considered. The present situation surrounding the management of the secondary injury phase of TBI reflects the current lack of success for therapeutic intervention. Animal TBI models have helped to reproduce clinically relevant injuries and generate behavioral, functional, and structural consequences of human TBI. These models provide assessments of acute neurological responses and evaluation of chronic behavioral morbidities.
Despite the encouraging discoveries that have paved the way for understanding the consequences of TBI, the full translational benefit has not been achieved. An unanswered question as to how and why TBI leads to persistent morbidity can be partially attributed to an incomplete understanding of the self-propagating secondary injury mechanisms driving chronic pathophysiological discord long after TBI. Moreover, it is unclear how these injury mechanisms further confer interindividual vulnerability toward various neurological deficits. While animal models are essential to translational progress, the roles of the intact nervous system, presence of vascular, and immunologic components contribute unaccounted complexity to result interpretation. Some of the challenges to modeling TBI in rodents and larger mammals include longer study duration, drug development, drug delivery, vivarium maintenance, sample size, and trained personnel, which contributes to costly experiments (albeit less expensive than clinical trials). In addition, technical aspects such as the requirement of pre-injury surgical procedures and the use of anesthetics, which are not representative of “real-life” human TBI, can influence some outcome measures. Other factors include over-optimistic pre-clinical interpretations, lack of inclusion of both sexes, the absence of valid biomarkers, and the use of inbred animals that lack genetic diversity possessed by the human population (161). Therefore, utilizing multiple TBI models that serve as an excellent in vivo system to delineate secondary injury mechanisms that underlie TBI pathology likely sets the stage to substantially improve our understanding of the pathogenesis and promote translational confidence.
5.1. Validity in Drosophila
As discussed previously, great progress has been made with fly TBI models to recapitulate important neurological, biochemical, pathological, and behavioral analogies of human brain injury. There are several acute and subacute pathological and behavioral phenotypes that fly TBI models collectively display that are remarkably similar to key features of human TBI, including axonal shearing and motor deficits possessing face validity. For example, injured flies exhibit temporary suppression of reflexes causing loss of the righting reflex and start moving after a delay, the duration of which was found to be associated with the injury severity (Figure 4)(68, 162). The flies remain momentarily disoriented and often demonstrate problems with balance, including circling, ataxia, incoordination, and loss of equilibrium, similar to difficulties in coordination, posture and steadiness of movement observed in humans after TBI (Figure 5; Movie S1-S4). In addition, TBI has been associated with the development of epilepsy, including the occurrence of early seizures (162). Similarly, flies subjected to TBI exhibit spontaneous seizures involving twitches and uncontrolled jumps (68). Another observation from our lab using the HIT model is the temporary splaying of wings. The splaying occurs in the absence of overt damage to the wings, and wing position returns to normal within minutes to hours post-injury. The splaying of wings has similarities to the ‘fencing response’ noticed as temporary abnormal flexion or extension posturing of the extremities in rodent studies of TBI (163) and tonic posturing after sport-related human head injuries. The fencing response occurs without convulsions, immediately after injury, and during a loss of consciousness. Similar to the Parachute and Moro reflexes in human infants (in response to rotation or startle – infants arms extend or flex as if to break the fall), the response is thought to be a phylogenetically conserved spinal reflex involving activation of vestibular nuclei (164). In mice, Math1 gives rise to spinocerebellar and cuneocerebellar systems involved in fine motor control, proprioception, and posture (165). While this requires further evaluation, the ato homolog of Math1 is conserved across species and involved in the development of Drosophila chordotonal organs, which are associated with the joints of wings, legs, and the second antennal segment and thought to be involved in proprioception and required for geotaxis (166, 167). The rest-activity cycle in Drosophila also has apparent similarities with human sleep-wake rhythm and has been particularly useful to study post-traumatic sleep changes (66).
Figure 4.

Phenotypic similarity of mammalian/vertebrate and Drosophila acute response to diffuse TBI. Created with BioRender.com
Figure 5.

TBI-induced Drosophila phenotypes. Sham and injured flies are shown trapped in the end of a standard food vial as part of a HIT device. Normal upright positioning and wing placement are depicted in uninjured flies (left). After TBI, Drosophila can become incapacitated, inverted, and their wings splayed (right, black arrows).
To shed light on TBI pathogenesis, it is necessary for a thorough understanding of the behavioral consequences that emerge longitudinally after injury. Such outcomes can be exploited to test therapeutic approaches capable of modulating multiple downstream secondary injury pathways to promote behavioral recovery. The fly has been utilized for studying various simple and complex behaviors such as locomotion, sleep patterns, circadian rhythm, courtship, and mating relevant to several human diseases. Flies can be visually scored, timed, and measured by negative geotaxis and visual closed-loop flight (56, 63, 101, 104, 168). Locomotor disabilities are commonly observed after TBI in humans and animal models, and the HIT model has been shown capable of reproducing movement deficits with decreased spontaneous locomotor activity (101). Testing for negative geotaxis reveals a loss of 50% more of normal progression toward light. When using a dark/light box exploration, flies, which normally are phototaxic, tend to stay in the dark side of the box, indicating photosensitivity after TBI (169) or increased anxiety-like behavior. Motor disturbances similar to those observed in humans after TBI have been reported in flies using the test for climbing behavior in response to gentle mechanical stimulus. Chronic phenotypic behavior deficits detected in flies have the potential to be evaluated for circuit-related disruption, where altered neurotransmission and maladaptive circuit reorganization contribute to the persistence of symptoms (170). For instance, sleep/wake cycles impairment was observed out to 5 days post-injury (171), and aggressive behavior has been observed in brain-injured flies and offspring (121), where early modulation of secondary injury cascades could improve behavioral deficits. Further, there is the potential to test for other phenotypic TBI symptoms, including depression and anxiety-like behavior, diapause, equilibrium, grooming (indicative of stress), learning and memory, phototaxis (circadian rhythms, sensory sensitivity, or anxiety), response to novel and familiar stimuli, and visual discrimination (indicative of TBI-induced optic neuropathy). Collectively, these endpoints suggest Drosophila carries a good face validity to longitudinally study TBI pathophysiology associated with TBI-like symptoms.
5.2. Systemic effects
It is now recognized that TBI compromises systemic physiology causing substantial changes in the functioning of peripheral organs. Several recent clinical and preclinical reports have supported the importance of brain-peripheral interactions mediated by several direct and indirect pathways involving microbiome, hypothalamic-pituitary-adrenal (HPA) functioning, immune signaling, neurotransmitters, hormones, and other neuropeptides. There is now emerging evidence that the influence of TBI on the systemic organs reverts on the brain and contributes to exacerbating the pathogenesis of TBI. Experimental data have implicated the role of bidirectional interaction in promoting secondary post-injury neuroinflammation, particularly associated with changes to systemic immunity. Other findings have indicated that alterations to the systemic immune system promote changes in immune signaling within the brain, and enhanced peripheral inflammation has been found in several neuropsychiatric disorders. Post-TBI infections and the initiation of the inflammatory response can continue chronically and promote neurodegeneration (172, 173). Among several comorbidities, gastrointestinal (GI) disturbances have been reported in TBI patients, including intestinal barrier dysfunction, changes in gut motility, and gastric emptying (174, 175). It has been reported that intestinal barrier dysfunction is associated with mortality in Drosophila after TBI (59). It appears that alterations of genes involved in tissue barrier function and homeostasis were primarily responsible for a higher risk of death after TBI (176). From the metabolic standpoint, TBI contributes to metabolic dysfunction characterized in peripheral organs, which can promote secondary brain damage.
Fly TBI models have expanded the animal findings to include changes in metabolic components after TBI. Indeed, findings have indicated that post-injury metabolic derangements with glucose dysregulation promotes mortality (176). In addition, transcriptomic analysis of w1118 flies injured with the HIT device showed conserved long intron sequences in genes involved in mitochondrial energy metabolism (70, 177). As gene regulatory mechanisms have emerged as important determinants of cellular functions such as metabolism, these results support the utility of the fly TBI model to uncover translationally relevant physiological processes and provide more mechanistic evidence for secondary cellular injury. Overall, continued research on this frontier will facilitate more important systemic changes on the pathophysiological outcomes of TBI.
5.3. Challenges in modeling TBI in flies
Several common challenges remain inherent to the modeling of TBI in flies and require consideration in interpretation. Lacking a conserved closed cardiovascular and respiratory system in Drosophila, complications and contributions from TBI-induced elevated intracranial pressure, arterial blood pressure, irregular breathing, and reduced heart rate are not a factor of outcomes. Inherent differences in the vascular milieu potentially limit the utility of flies to understand vascular injury that promotes local hypoxia and ischemia, secondary hemorrhage that has negative consequences on cerebrovascular autoregulatory function, and metabolic function. Also, differences in the immune strategies between flies and humans offer limited insights into the contribution of the adaptive immune system. A thorough understanding of the adaptive immune responses role in injury, repair, and restoration of tissue homeostasis requires the use of a mammalian system to integrate the information derived from molecular entities and pathways studied in flies. While these factors differentiate fly versus mammalian experiments, they also provide opportunities to isolate relevant pathways without the influence of cardiovascular alterations and adaptive immunity.
5.4. Gaps in knowledge
Despite a small brain, small body size, and the presence of an exoskeleton, Drosophila have an intact sophisticated and well understood nervous system and innate immune system. Several fly models of TBI have been developed and modified over the years for direct relevance to human TBI. The remarkable progress made in recapitulating principal features of TBI in flies is supported by the understanding that cellular pathways involved in TBI pathogenesis are present in both flies and humans. To address how acute TBI-induced injuries can transition to persisting and late-onset morbidities relevant to human post-TBI symptoms, multiple fly TBI models have been developed and show conserved behavioral and secondary cellular changes that are analogous to human TBI. Fly TBI models use mechanical force and a continuum of injury severities to induce brain trauma, similar to human TBI and mammalian models.
6. Strategies for using Drosophila in TBI research
The mechanics and methods of injury associated with primary injuries are well documented and can be replicated in animal models. Secondary injuries after TBI are challenging to study, as mammalian models have redundant signaling pathway proteins, interactions between innate and adaptive immunity, complex local circuits, and higher-order interconnected networks. Mammals also have a substantially longer lifespan, complicating essential longitudinal evaluations required for a temporal and comprehensive understanding of beneficial vs. detrimental downstream cascades. Drosophilae are gaining traction as a model for TBI research as novel insights into the causative mechanisms involved in secondary injuries can be better studied due to the relatively simple brain, control of gene expression, and conserved brain function and neurotransmitters (71, 178). Drosophila melanogaster models of AD, PD, and ALS are already established and contributing useful information on neurodegenerative diseases (reviewed in (179, 180)). Genetically modulating gene expression to isolate targets implicated in playing a role in secondary injury is relatively simple in flies due to the lack of redundant signaling pathway proteins. Findings can then be validated in vertebrate animal models that are physiologically more similar to humans.
6.1. Utility of Drosophila as a model for TBI and neurodegenerative disease
While etiologies of neurodegenerative diseases are largely unknown, in rare cases, disease-causing mutations are inherited. Observing effects of disease-causing mutations offers clues to pathophysiology, but complex neuroanatomy and genetic redundancy complicate brain injury studies in mammals. The relatively small fly brain controls shared fundamental behaviors that are facilitated by activity of functionally homologous structures and conserved neurotransmitters, receptors and signaling pathways (181-183). Flies have orthologs or homologs of many human genes implicated in neurodegenerative diseases including, APP (Appl), MAPT/TAU (tau), ATM (tefu), BACE1 (bace), LRRK2 (Lrrk2), PRKN (park), PINK1 (Pink1), DJ1 (DJ-1alpha, dj-1beta), TDP-43 (TBPH), SOD1 (Sod2), Frederick’s ataxia-associated FXN (fh), and spinal cerebellar ataxia-associated ATXN1 (atx-1), (184-195). The ability to control gene expression, and the ease of conducting high throughput measurements of stereotypical behaviors make Drosophila an attractive model for elucidating contributions of conserved signaling pathways to neuropathology (71, 178). Fly TBI studies also provide additional insight into putative neuroprotective compound screening.
Drosophila models of AD, PD, ALS, and ataxia-telangiectasia implicate aberrant mitochondrial dynamics, oxidative stress, inflammation, disrupted protein turnover, altered autophagy events, and apoptosis in their pathology, all of which are consistent with TBI pathology (72, 76, 179, 180, 196-206). For example, tau phosphorylation increases under cell stress in Drosophila, although it does not seem prone to aggregation in flies (64). A variety of genetic reporters of oxidative stress are available, including MitoTimer and redox-sensing GFPs (roGFPs) that can be targeted to the cytosol or mitochondria (207-210). Fluorescent markers like mitoGFP and mCherry-atg8a can be used to observe mitochondrial dynamics and autophagy initiation, respectively; the two can be used in conjunction to quantify autophagosome-to-mitochondria recruitment as an initial event in mitophagy (72, 209). Available at the Bloomington Drosophila Stock Center in Bloomington, IN, are fly stocks that harbor genetic markers of innate immunity/inflammation, including those for circulating macrophages (hemocytes; no. 30140), CD36 member Cr12 (no. 25041), nitric oxide synthase (no. 76766), toll receptor-ligand spaetzle (no. 83269), and anti-microbial peptides (no. 36558; https://bdsc.indiana.edu/index.html). A wide variety of mutant stocks and stocks harboring RNAi, CRISPRCas9 machinery, or overexpressing constructs for a wide variety of genes involved in neurodegenerative disease studies are also available. Protocols measuring deficits in fly behaviors associated with neurodegeneration include assessments for memory, sleep behavior, aggression, courtship, gut dysbiosis, lifespan, and mobility (211-216).
6.2. Sex, development, and peripheral effects after TBI
Since the use of Drosophila as a model organism for TBI is relatively novel, many gaps in knowledge are ripe for exploration. Historically, clinical and preclinical neurotrauma research did not include females or treat them as an independent variable. Recent reports indicate profound sex differences in neuroinflammation, response to treatment, fertility, and long-term morbidity; where valuable information can be discovered using flies (217). Clinical and preclinical studies indicate that estrogen has neuroprotective and anti-inflammatory effects, with evidence that females may have a higher tolerance to stress and injury. Flies express estrogen related receptors and synthesize the ligand, ecdysone, from their diet (218), indicating several approaches to evaluating the lineal roles of this receptor-mediated pathway in TBI-induced secondary injury cascades. TBI in adult women can lead to an increased risk of preterm labor/delivery and their infants developing neurologic disorders. Flies exposed to TBI (HIT method) have decreased pupation, longer larval length, faster pupae formation, and metamorphosis rates (63). Offspring from brain-injured females also had decreased negative geotaxis and implications of impaired social interaction (63). Together, these data indicate that the secondary injury cascades activated after TBI during pregnancy can directly impact first-generation offspring in flies, mice, and humans (219). TBI in 0-7 days post-eclosion Drosophila caused intestinal barrier permeability associated with secondary damage to the septate and tight junctions. Permeability was demonstrated by increased passage of bacteria and dietary glucose across the intestinal barrier, which activated the innate immune response and was associated with a higher incidence of death within 24 hours of injury. Antibiotic administration decreased the number of endogenous bacteria, but did not improve survival after TBI, indicating that increased mortality is not directly due to increased permeability to bacteria, but primary or other associated secondary injury cascades (59).
7. Concluding remarks
TBI in Drosophila provides the opportunity to isolate and evaluate specific secondary injury cascades for a deeper understanding of the development of chronic neurological deficits. Injuries in flies recapitulate pathological pathways associated with oxidative stress, innate neuroinflammatory response, axonal degeneration/regeneration, and cell death, providing feasibility and essential groundwork for future research (Figure 6). Loss of responsiveness, discoordination, disorientation, wing splay, and post-TBI activity indicate good face validity compared to rodents and clinical observations. Full body and head-specific injury models and established behavioral paradigms similar to those currently used in rodent models are optimized and available for the longitudinal evaluation of behaviorally relevant circuit function. Genetically modifying Drosophila serves as a complementary tool to isolate and modulate pathophysiological mechanisms and assess the impact on gene expression, molecular function, and behavioral outcomes. High throughput compound screening is feasible due to rapid reproduction (breeding), short lifespan, and affordability of the flies to identify therapeutic compounds to test in mammalian TBI models.
Figure 6.

Summary of findings from fly TBI studies to elucidate injury models, secondary injury mechanisms, systemic changes, sex differences, and behaviors (220-223). Created with BioRender.com
Supplementary Material
Highlights.
Traumatic brain injury (TBI)-induced secondary injury cascades are simplified and conserved in flies
Fly TBI models trigger oxidative stress, innate immunity, cell death, and neurodegenerative pathways
Permits the isolation and modulation of pathways responsible for phenotypically similar behavioral deficits in mammals
Short lifespan allows detailed longitudinal tests to identify therapeutic windows and test interventions
Validation of environmental, biological, and genetic factors can guide and accelerate translational TBI research
ACKNOWLEDGEMENTS
We thank Vanessa Moreno for contributing to the review of fly TBI models and the lab's initial testing of HIT device. We would also like to thank Tracy Mackey and Amber Juba for HIT optimization, Andrew Pielage, and Cory Williams for assistance in capturing and editing of videography. We thank Carol A. Haussler for proofreading. This work was supported by Midwestern University’s Biomedical Sciences program to L.M.B. and National Institutes of Health (R01NS100793) and Phoenix Children's Hospital Mission Support to T.C.T.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing financial interests: The authors declare no biomedical financial interests or potential conflicts of interest.
References:
- 1.Dewan MC, Rattani A, Gupta S, Baticulon RE, Hung YC, Punchak M, Agrawal A, Adeleye AO, Shrime MG, Rubiano AM, Rosenfeld JV, Park KB. Estimating the global incidence of traumatic brain injury. Journal of neurosurgery. 2018:1–18. Epub 2018/04/28. doi: 10.3171/2017.10.Jns17352. PubMed PMID: 29701556. [DOI] [PubMed] [Google Scholar]
- 2.de la Tremblaye PB, O'Neil DA, LaPorte MJ, Cheng JP, Beitchman JA, Thomas TC, Bondi CO, Kline AE. Elucidating opportunities and pitfalls in the treatment of experimental traumatic brain injury to optimize and facilitate clinical translation. Neurosci Biobehav Rev. 2018;85:160–75. Epub 2017/06/04. doi: 10.1016/j.neubiorev.2017.05.022. PubMed PMID: 28576511; PMCID: PMC5709241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maas AI, Stocchetti N, Bullock R. Moderate and severe traumatic brain injury in adults. The Lancet Neurology. 2008;7(8):728–41. Epub 2008/07/19. doi: 10.1016/s1474-4422(08)70164-9. PubMed PMID: 18635021. [DOI] [PubMed] [Google Scholar]
- 4.Faul M, Coronado V. Epidemiology of traumatic brain injury. Handbook of clinical neurology. 2015;127:3–13. Epub 2015/02/24. doi: 10.1016/b978-0-444-52892-6.00001-5. PubMed PMID: 25702206. [DOI] [PubMed] [Google Scholar]
- 5.Gardner RC, Yaffe K. Epidemiology of mild traumatic brain injury and neurodegenerative disease. Mol Cell Neurosci. 2015;66(Pt B):75–80. Epub 2015/03/10. doi: 10.1016/j.mcn.2015.03.001. PubMed PMID: 25748121; PMCID: PMC4461453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levin HS, Diaz-Arrastia RR. Diagnosis, prognosis, and clinical management of mild traumatic brain injury. The Lancet Neurology. 2015;14(5):506–17. Epub 2015/03/25. doi: 10.1016/s1474-4422(15)00002-2. PubMed PMID: 25801547. [DOI] [PubMed] [Google Scholar]
- 7.Gupta R, Sen N. Traumatic brain injury: a risk factor for neurodegenerative diseases. Rev Neurosci. 2016;27(1):93–100. Epub 2015/09/10. doi: 10.1515/revneuro-2015-0017. PubMed PMID: 26352199. [DOI] [PubMed] [Google Scholar]
- 8.Krishna G, Beitchman JA, Bromberg CE, Currier Thomas T. Approaches to Monitor Circuit Disruption after Traumatic Brain Injury: Frontiers in Preclinical Research. Int J Mol Sci. 2020;21(2). Epub 2020/01/23. doi: 10.3390/ijms21020588. PubMed PMID: 31963314; PMCID: PMC7014469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelly DF, Martin NA, Kordestani R, Counelis G, Hovda DA, Bergsneider M, McBride DQ, Shalmon E, Herman D, Becker DP. Cerebral blood flow as a predictor of outcome following traumatic brain injury. J Neurosurg. 1997;86(4):633–41. Epub 1997/04/01. doi: 10.3171/jns.1997.86.4.0633. PubMed PMID: 9120627. [DOI] [PubMed] [Google Scholar]
- 10.Soldozy S, Sharifi KA, Desai B, Giraldo D, Yeghyayan M, Liu L, Norat P, Sokolowski JD, Yagmurlu K, Park MS, Tvrdik P, Kalani MYS. Cortical Spreading Depression in the Setting of Traumatic Brain Injury. World Neurosurg. 2020;134:50–7. Epub 2019/10/28. doi: 10.1016/j.wneu.2019.10.048. PubMed PMID: 31655239. [DOI] [PubMed] [Google Scholar]
- 11.Weber JT. Altered calcium signaling following traumatic brain injury. Front Pharmacol. 2012;3:60. Epub 2012/04/21. doi: 10.3389/fphar.2012.00060. PubMed PMID: 22518104; PMCID: PMC3324969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andersen JK. Oxidative stress in neurodegeneration: cause or consequence? Nat Med. 2004;10Suppl:S18–25. Epub 2004/08/10. doi: 10.1038/nrn1434. PubMed PMID: 15298006. [DOI] [PubMed] [Google Scholar]
- 13.Feldmann LK, Le Prieult F, Felzen V, Thal SC, Engelhard K, Behl C, Mittmann T. Proteasome and Autophagy-Mediated Impairment of Late Long-Term Potentiation (l-LTP) after Traumatic Brain Injury in the Somatosensory Cortex of Mice. Int J Mol Sci. 2019;20(12). Epub 2019/06/27. doi: 10.3390/ijms20123048. PubMed PMID: 31234472; PMCID: PMC6627835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarkar C, Zhao Z, Aungst S, Sabirzhanov B, Faden AI, Lipinski MM. Impaired autophagy flux is associated with neuronal cell death after traumatic brain injury. Autophagy. 2014;10(12):2208–22. Epub 2014/12/09. doi: 10.4161/15548627.2014.981787. PubMed PMID: 25484084; PMCID: PMC4502690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lipinski MM, Wu J, Faden AI, Sarkar C. Function and Mechanisms of Autophagy in Brain and Spinal Cord Trauma. Antioxid Redox Signal. 2015;23(6):565–77. Epub 2015/03/27. doi: 10.1089/ars.2015.6306. PubMed PMID: 25808205; PMCID: PMC4545370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu J, Lipinski MM. Autophagy in Neurotrauma: Good, Bad, or Dysregulated. Cells. 2019;8(7). Epub 2019/07/13. doi: 10.3390/cells8070693. PubMed PMID: 31295858; PMCID: PMC6678153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ng SY, Lee AYW. Traumatic Brain Injuries: Pathophysiology and Potential Therapeutic Targets. Front Cell Neurosci. 2019;13:528. Epub 2019/12/13. doi: 10.3389/fncel.2019.00528. PubMed PMID: 31827423; PMCID: PMC6890857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaur S, Sharma N, Nehru B. Anti-inflammatory effects of Ginkgo biloba extract against trimethyltin-induced hippocampal neuronal injury. Inflammopharmacology. 2018;26(1):87–104. Epub 2017/09/18. doi: 10.1007/s10787-017-0396-2. PubMed PMID: 28918573. [DOI] [PubMed] [Google Scholar]
- 19.Kumar A, Loane DJ. Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun. 2012;26(8):1191–201. Epub 2012/06/26. doi: 10.1016/j.bbi.2012.06.008. PubMed PMID: 22728326. [DOI] [PubMed] [Google Scholar]
- 20.Needham EJ, Helmy A, Zanier ER, Jones JL, Coles AJ, Menon DK. The immunological response to traumatic brain injury. Journal of neuroimmunology. 2019;332:112–25. Epub 2019/04/22. doi: 10.1016/j.jneuroim.2019.04.005. PubMed PMID: 31005712. [DOI] [PubMed] [Google Scholar]
- 21.Kelso ML, Gendelman HE. Bridge between neuroimmunity and traumatic brain injury. Curr Pharm Des. 2014;20(26):4284–98. Epub 2013/09/13. PubMed PMID: 24025052; PMCID: PMC4135046. [PMC free article] [PubMed] [Google Scholar]
- 22.Kelley BJ, Farkas O, Lifshitz J, Povlishock JT. Traumatic axonal injury in the perisomatic domain triggers ultrarapid secondary axotomy and Wallerian degeneration. Experimental neurology. 2006;198(2):350–60. Epub 2006/02/02. doi: 10.1016/j.expneurol.2005.12.017. PubMed PMID: 16448652. [DOI] [PubMed] [Google Scholar]
- 23.Hill CS, Coleman MP, Menon DK. Traumatic Axonal Injury: Mechanisms and Translational Opportunities. Trends in neurosciences. 2016;39(5):311–24. Epub 2016/04/05. doi: 10.1016/j.tins.2016.03.002. PubMed PMID: 27040729; PMCID: PMC5405046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gadani SP, Walsh JT, Lukens JR, Kipnis J. Dealing with Danger in the CNS: The Response of the Immune System to Injury. Neuron. 2015;87(1):47–62. Epub 2015/07/04. doi: 10.1016/j.neuron.2015.05.019. PubMed PMID: 26139369; PMCID: PMC4491143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feerick CL, McKernan DP. Understanding the regulation of pattern recognition receptors in inflammatory diseases - a 'Nod' in the right direction. Immunology. 2017;150(3):237–47. Epub 2016/10/06. doi: 10.1111/imm.12677. PubMed PMID: 27706808; PMCID: PMC5290251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clark IA, Vissel B. Broader Insights into Understanding Tumor Necrosis Factor and Neurodegenerative Disease Pathogenesis Infer New Therapeutic Approaches. J Alzheimers Dis. 2021;79(3):931–48. Epub 2021/01/19. doi: 10.3233/JAD-201186. PubMed PMID: 33459706; PMCID: PMC7990436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen J, Wu X, Shao B, Zhao W, Shi W, Zhang S, Ni L, Shen A. Increased expression of TNF receptor-associated factor 6 after rat traumatic brain injury. Cell Mol Neurobiol. 2011;31(2):269–75. Epub 2010/11/13. doi: 10.1007/s10571-010-9617-6. PubMed PMID: 21072581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zulfiqar Z, Shah FA, Shafique S, Alattar A, Ali T, Alvi AM, Rashid S, Li S. Repurposing FDA Approved Drugs as JNK3 Inhibitor for Prevention of Neuroinflammation Induced by MCAO in Rats. J Inflamm Res. 2020;13:1185–205. Epub 2021/01/02. doi: 10.2147/JIR.S284471. PubMed PMID: 33384558; PMCID: PMC7770337. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Hong MY, Cui JZ, Li R, Tian YX, Wang H, Wang HT, Gao JL. [Effect of expression of c-jun N-terminal kinase on neuron autophagy following diffuse brain injury in rats]. Zhonghua Wai Ke Za Zhi. 2012;50(2):166–70. Epub 2012/04/12. PubMed PMID: 22490359. [PubMed] [Google Scholar]
- 30.Luo SY, Li R, Le ZY, Li QL, Chen ZW. Anfibatide protects against rat cerebral ischemia/reperfusion injury via TLR4/JNK/caspase-3 pathway. Eur J Pharmacol. 2017;807:127–37. Epub 2017/04/10. doi: 10.1016/j.ejphar.2017.04.002. PubMed PMID: 28390871. [DOI] [PubMed] [Google Scholar]
- 31.Bramlett HM, Dietrich WD. Long-Term Consequences of Traumatic Brain Injury: Current Status of Potential Mechanisms of Injury and Neurological Outcomes. J Neurotrauma. 2015;32(23):1834–48. Epub 2014/08/27. doi: 10.1089/neu.2014.3352. PubMed PMID: 25158206; PMCID: PMC4677116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simon DW, McGeachy MJ, Bayir H, Clark RS, Loane DJ, Kochanek PM. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol. 2017;13(3):171–91. Epub 2017/02/12. doi: 10.1038/nrneurol.2017.13. PubMed PMID: 28186177; PMCID: PMC5675525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang HY, Jin XB, Lue TF. Three important components in the regeneration of the cavernous nerve: brain-derived neurotrophic factor, vascular endothelial growth factor and the JAK/STAT signaling pathway. Asian J Androl. 2011;13(2):231–5. Epub 2010/12/21. doi: 10.1038/aja.2010.162. PubMed PMID: 21170078; PMCID: PMC3739200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raible DJ, Frey LC, Brooks-Kayal AR. Effects of JAK2-STAT3 signaling after cerebral insults. JAKSTAT. 2014;3:e29510. Epub 2014/08/12. doi: 10.4161/jkst.29510. PubMed PMID: 25105066; PMCID: PMC4124058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen XM, Yu YH, Wang L, Zhao XY, Li JR. Effect of the JAK2/STAT3 signaling pathway on nerve cell apoptosis in rats with white matter injury. Eur Rev Med Pharmacol Sci. 2019;23(1):321–7. Epub 2019/01/19. doi: 10.26355/eurrev_201901_16779. PubMed PMID: 30657573. [DOI] [PubMed] [Google Scholar]
- 36.Raible DJ, Frey LC, Del Angel YC, Carlsen J, Hund D, Russek SJ, Smith B, Brooks-Kayal AR. JAK/STAT pathway regulation of GABAA receptor expression after differing severities of experimental TBI. Exp Neurol. 2015;271:445–56. Epub 2015/07/15. doi: 10.1016/j.expneurol.2015.07.001. PubMed PMID: 26172316; PMCID: PMC5969808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uryu K, Laurer H, McIntosh T, Praticò D, Martinez D, Leight S, Lee VM, Trojanowski JQ. Repetitive mild brain trauma accelerates Abeta deposition, lipid peroxidation, and cognitive impairment in a transgenic mouse model of Alzheimer amyloidosis. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2002;22(2):446–54. Epub 2002/01/11. doi: 10.1523/jneurosci.22-02-00446.2002. PubMed PMID: 11784789; PMCID: PMC6758680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huber BR, Meabon JS, Martin TJ, Mourad PD, Bennett R, Kraemer BC, Cernak I, Petrie EC, Emery MJ, Swenson ER, Mayer C, Mehic E, Peskind ER, Cook DG. Blast exposure causes early and persistent aberrant phospho- and cleaved-tau expression in a murine model of mild blast-induced traumatic brain injury. Journal of Alzheimer's disease : JAD. 2013;37(2):309–23. Epub 2013/08/21. doi: 10.3233/jad-130182. PubMed PMID: 23948882; PMCID: PMC4126588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tan XL, Wright DK, Liu S, Hovens C, O'Brien TJ, Shultz SR. Sodium selenate, a protein phosphatase 2A activator, mitigates hyperphosphorylated tau and improves repeated mild traumatic brain injury outcomes. Neuropharmacology. 2016;108:382–93. Epub 2016/05/11. doi: 10.1016/j.neuropharm.2016.05.001. PubMed PMID: 27163189. [DOI] [PubMed] [Google Scholar]
- 40.Dias V, Junn E, Mouradian MM. The role of oxidative stress in Parkinson's disease. J Parkinsons Dis. 2013;3(4):461–91. Epub 2013/11/21. doi: 10.3233/JPD-130230. PubMed PMID: 24252804; PMCID: PMC4135313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barrett JP, Henry RJ, Shirey KA, Doran SJ, Makarevich OD, Ritzel RM, Meadows VA, Vogel SN, Faden AI, Stoica BA, Loane DJ. Interferon-β Plays a Detrimental Role in Experimental Traumatic Brain Injury by Enhancing Neuroinflammation That Drives Chronic Neurodegeneration. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2020;40(11):2357–70. Epub 2020/02/08. doi: 10.1523/jneurosci.2516-19.2020. PubMed PMID: 32029532; PMCID: PMC7083281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Loane DJ, Kumar A, Stoica BA, Cabatbat R, Faden AI. Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. J Neuropathol Exp Neurol. 2014;73(1):14–29. Epub 2013/12/18. doi: 10.1097/NEN.0000000000000021. PubMed PMID: 24335533; PMCID: PMC4267248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoffman AN, Paode PR, May HG, Ortiz JB, Kemmou S, Lifshitz J, Conrad CD, Currier Thomas T. Early and Persistent Dendritic Hypertrophy in the Basolateral Amygdala following Experimental Diffuse Traumatic Brain Injury. J Neurotrauma. 2017;34(1):213–9. doi: 10.1089/neu.2015.4339. PubMed PMID: 27306143; PMCID: PMC5198057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beitchman JA, Griffiths DR, Hur Y, Ogle SB, Bromberg CE, Morrison HW, Lifshitz J, Adelson PD, Thomas TC. Experimental Traumatic Brain Injury Induces Chronic Glutamatergic Dysfunction in Amygdala Circuitry Known to Regulate Anxiety-Like Behavior. Frontiers in Neuroscience. 2020;13(1434). doi: 10.3389/fnins.2019.01434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krishna GB, C.E;Currier Thomas T. Circuit reorganizations after diffuse axonal injury: Utility of the whisker barrel circuit. In: Rajadram VP C, editor. Cellular, Molecular, Physiological, and Behavioral Aspects of Traumatic Brain Injury: Elsevier Inc. ; 2022. [Google Scholar]
- 46.Keyvani K, Schallert T. Plasticity-associated molecular and structural events in the injured brain. J Neuropathol Exp Neurol. 2002;61(10):831–40. Epub 2002/10/22. doi: 10.1093/jnen/61.10.831. PubMed PMID: 12387449. [DOI] [PubMed] [Google Scholar]
- 47.Greer JE, McGinn MJ, Povlishock JT. Diffuse traumatic axonal injury in the mouse induces atrophy, c-Jun activation, and axonal outgrowth in the axotomized neuronal population. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31(13):5089–105. Epub 2011/04/01. doi: 10.1523/jneurosci.5103-10.2011. PubMed PMID: 21451046; PMCID: PMC3076099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rowe RK, Rumney BM, May HG, Permana P, Adelson PD, Harman SM, Lifshitz J, Thomas T. Diffuse traumatic brain injury affects chronic corticosterone function in the rat. Endocr Connect. 2016. Epub 2016/06/19. doi: 10.1530/EC-16-0031. PubMed PMID: 27317610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bromberg CE, Condon AM, Ridgway SW, Krishna G, Garcia-Filion PC, Adelson PD, Rowe RK, Thomas TC. Sex-Dependent Pathology in the HPA Axis at a Sub-acute Period After Experimental Traumatic Brain Injury. Front Neurol. 2020;11:946. Epub 2020/10/27. doi: 10.3389/fneur.2020.00946. PubMed PMID: 33101162; PMCID: PMC7554641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thomas TC, Ogle SB, Rumney BM, May HG, Adelson PD, Lifshitz J. Does time heal all wounds? Experimental diffuse traumatic brain injury results in persisting histopathology in the thalamus. Behavioural brain research. 2018;340:137–46. doi: 10.1016/j.bbr.2016.12.038. PubMed PMID: 28042008; PMCID: PMC5491365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sukhina AS, Oatman OJ, Lewis KS, Thomas TC, Brown D, Rowe RK, Adelson PD, Lifshitz J. Failure to Thrive in a 15-month-old with a History of Head Trauma. Pediatr Rev. 2021;42(Suppl 1):S55–S9. Epub 2021/01/03. doi: 10.1542/pir.2018-0069. PubMed PMID: 33386363. [DOI] [PubMed] [Google Scholar]
- 52.Rowe RK, Oritz JB, Currier Thomas T. Mild and Moderate Traumatic Brain Injury and Repeated Stress Affect Corticosterone in the Rat. Neurotrauma Reports. 2020;1(1):113–24. Epub Jan. 2020. doi: 10.1089/neur.2020.0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Faden AI, Barrett JP, Stoica BA, Henry RJ. Bidirectional Brain-Systemic Interactions and Outcomes After TBI. Trends in neurosciences. 2021. Epub 2021/01/27. doi: 10.1016/j.tins.2020.12.004. PubMed PMID: 33495023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Busnelli M, Manzini S, Chiesa G. The Gut Microbiota Affects Host Pathophysiology as an Endocrine Organ: A Focus on Cardiovascular Disease. Nutrients. 2019;12(1). Epub 2020/01/02. doi: 10.3390/nu12010079. PubMed PMID: 31892152; PMCID: PMC7019666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Matthews KA, Kaufman TC, Gelbart WM. Research resources for Drosophila: the expanding universe. Nature reviews Genetics. 2005;6(3):179–93. Epub 2005/03/02. doi: 10.1038/nrg1554. PubMed PMID: 15738962. [DOI] [PubMed] [Google Scholar]
- 56.Katzenberger RJ, Loewen CA, Wassarman DR, Petersen AJ, Ganetzky B, Wassarman DA. A Drosophila model of closed head traumatic brain injury. Proceedings of the National Academy of Sciences. 2013;110(44):E4152–E9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Katzenberger RJ, Loewen CA, Bockstruck RT, Woods MA, Ganetzky B, Wassarman DA. A method to inflict closed head traumatic brain injury in Drosophila. Journal of visualized experiments: JoVE. 2015(100). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Putnam LJ, Willes AM, Kalata BE, Disher ND, Brusich DJ. Expansion of a fly TBI model to four levels of injury severity reveals synergistic effects of repetitive injury for moderate injury conditions. Fly. 2019;13(1-4):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Katzenberger RJ, Chtarbanova S, Rimkus SA, Fischer JA, Kaur G, Seppala JM, Swanson LC, Zajac JE, Ganetzky B, Wassarman DA. Death following traumatic brain injury in Drosophila is associated with intestinal barrier dysfunction. Elite. 2015;4. Epub 2015/03/06. doi: 10.7554/eLife.04790. PubMed PMID: 25742603; PMCID: PMC4377547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Katzenberger RJ, Ganetzky B, Wassarman DA. Age and Diet Affect Genetically Separable Secondary Injuries that Cause Acute Mortality Following Traumatic Brain Injury in Drosophila. Genes, Genomes, Genetics. 2016;6(12):4151–66. Epub 2016/10/19. doi: 10.1534/g3.116.036194. PubMed PMID: 27754853; PMCID: PMC5144983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Swanson LC, Trujillo EA, Thiede GH, Katzenberger RJ, Shishkova E, Coon JJ, Ganetzky B, Wassarman DA. Survival Following Traumatic Brain Injury in Drosophila Is Increased by Heterozygosity for a Mutation of the NF-κB Innate Immune Response Transcription Factor Relish. Genetics. 2020;216(4):1117–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Swanson LC, Rimkus SA, Ganetzky B, Wassarman DA. Loss of the Antimicrobial Peptide Metchnikowin Protects Against Traumatic Brain Injury Outcomes in Drosophila melanogaster. G3 (Bethesda). 2020;10(9):3109–19. Epub 2020/07/08. doi: 10.1534/g3.120.401377. PubMed PMID: 32631949; PMCID: PMC7466987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chauhan V, Chauhan A. Traumatic injury in female Drosophila melanogaster affects the development and induces behavioral abnormalities in the offspring. Behav Brain Funct. 2019;15(1):11. Epub 2019/10/28. doi: 10.1186/s12993-019-0163-1. PubMed PMID: 31653253; PMCID: PMC6815055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barekat A, Gonzalez A, Mauntz RE, Kotzebue RW, Molina B, El-Mecharrafie N, Conner CJ, Garza S, Melkani GC, Joiner WJ, Lipinski MM, Finley KD, Ratliff EP. Using Drosophila as an integrated model to study mild repetitive traumatic brain injury. Sci Rep. 2016;6:25252. Epub 2016/05/05. doi: 10.1038/srep25252. PubMed PMID: 27143646; PMCID: PMC4855207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sun M, Chen LL. A Novel Method to Model Chronic Traumatic Encephalopathy in Drosophila. Journal of visualized experiments : JoVE. 2017(125):55602. doi: 10.3791/55602. PubMed PMID: 28715400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van Alphen B, Stewart S, Iwanaszko M, Xu F, Bang E, Rozenfeld S, Ramakrishnan A, Itoh TQ, Braun RI, Allada R. Glial immune-related pathways as mediators of closed head TBI effects on behavior in Drosophila 2018. doi: 10.1101/422535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Saikumar J, Kim J, Byrns CN, Hemphill M, Meaney DF, Bonini NM. Inducing different severities of traumatic brain injury in Drosophila using a piezoelectric actuator. Nat Protoc. 2021;16(1):263–82. Epub 2020/12/06. doi: 10.1038/s41596-020-00415-y. PubMed PMID: 33277631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Saikumar J, Byrns CN, Hemphill M, Meaney DF, Bonini NM. Dynamic neural and glial responses of a head-specific model for traumatic brain injury in Drosophila. Proc Natl Acad Sci U S A. 2020;117(29):17269–77. Epub 2020/07/03. doi: 10.1073/pnas.2003909117. PubMed PMID: 32611818; PMCID: PMC7382229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97(6): 1634–58. Epub 2006/06/30. doi: 10.1111/j.1471-4159.2006.03907.x. PubMed PMID: 16805774. [DOI] [PubMed] [Google Scholar]
- 70.Sen A, Gurdziel K, Liu J, Qu W, Nuga OO, Burl RB, Hüttemann M, Pique-Regi R, Ruden D. Smooth, an hnRNP-L homolog, might decrease mitochondrial metabolism by post-transcriptional regulation of isocitrate dehydrogenase (Idh) and other metabolic genes in the sub-acute phase of traumatic brain injury. Frontiers in genetics. 2017;8:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shah EJ, Gurdziel K, Ruden DM. Mammalian models of traumatic brain injury and a place for drosophila in TBI research. Frontiers in Neuroscience. 2019;13. Epub 2019/05/21. doi: 10.3389/fnins.2019.00409. PubMed PMID: 31105519; PMCID: PMC6499071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cackovic J, Gutierrez-Luke S, Call GB, Juba A, O'Brien S, Jun CH, Buhlman LM. Vulnerable Parkin Loss-of-Function Drosophila Dopaminergic Neurons Have Advanced Mitochondrial Aging, Mitochondrial Network Loss and Transiently Reduced Autophagosome Recruitment. Frontiers in cellular neuroscience. 2018;12:39. Epub 2018/03/03. doi: 10.3389/fncel.2018.00039. PubMed PMID: 29497364; PMCID: PMC5818410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Martín-Maestro P, Gargini R, García E, Perry G, Avila J, García-Escudero V. Slower Dynamics and Aged Mitochondria in Sporadic Alzheimer's Disease. Oxid Med Cell Longev. 2017;2017:9302761-. Epub 2017/10/19. doi: 10.1155/2017/9302761. PubMed PMID: 29201274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.D'Amico E, Factor-Litvak P, Santella RM, Mitsumoto H. Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free radical biology & medicine. 2013;65:509–27. Epub 2013/06/21. doi: 10.1016/j.freeradbiomed.2013.06.029. PubMed PMID: 23797033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cioffi F, Adam RHI, Broersen K. Molecular Mechanisms and Genetics of Oxidative Stress in Alzheimer's Disease. Journal of Alzheimer's disease : JAD. 2019;72(4):981–1017. doi: 10.3233/JAD-190863. PubMed PMID: 31744008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Whitworth AJ, Theodore DA, Greene JC, Beneš H, Wes PD, Pallanck LJ. Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson's disease. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(22):8024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10Suppl:S10–7. Epub 2004/07/24. doi: 10.1038/nm1066. PubMed PMID: 15272267. [DOI] [PubMed] [Google Scholar]
- 78.Ding H, Wang X, Wang H, Zhu L, Wang Q, Jia Y, Wei W, Zhou C, Wu H, Ding K. Nrf2-ARE signaling provides neuroprotection in traumatic brain injury via modulation of the ubiquitin proteasome system. Neurochem Int. 2017;111:32–44. Epub 2017/05/04. doi: 10.1016/j.neuint.2017.04.016. PubMed PMID: 28465088. [DOI] [PubMed] [Google Scholar]
- 79.Anderson EN, Gochenaur L, Singh A, Grant R, Patel K, Watkins S, Wu JY, Pandey UB. Traumatic injury induces stress granule formation and enhances motor dysfunctions in ALS/FTD models. Hum Mol Genet. 2018;27(8):1366–81. Epub 2018/02/13. doi: 10.1093/hmg/ddy047. PubMed PMID: 29432563; PMCID: PMC6455923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cruz-Haces M, Tang J, Acosta G, Fernandez J, Shi R. Pathological correlations between traumatic brain injury and chronic neurodegenerative diseases. Translational neurodegeneration. 2017;6(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jassam YN, Izzy S, Whalen M, McGavern DB, El Khoury J. Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift. Neuron. 2017;95(6):1246–65. Epub 2017/09/15. doi: 10.1016/j.neuron.2017.07.010. PubMed PMID: 28910616; PMCID: PMC5678753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Witcher KG, Eiferman DS, Godbout JP. Priming the inflammatory pump of the CNS after traumatic brain injury. Trends in neurosciences. 2015;38(10):609–20. Epub 2015/10/08. doi: 10.1016/j.tins.2015.08.002. PubMed PMID: 26442695; PMCID: PMC4617563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–60. Epub 1998/05/23. doi: 10.1146/annurev.immunol.16.1.225. PubMed PMID: 9597130. [DOI] [PubMed] [Google Scholar]
- 84.Lye SH, Chtarbanova S. Drosophila as a Model to Study Brain Innate Immunity in Health and Disease. Int J Mol Sci. 2018;19(12). Epub 2018/12/14. doi: 10.3390/ijms19123922. PubMed PMID: 30544507; PMCID: PMC6321579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stuart LM, Ezekowitz RA. Phagocytosis and comparative innate immunity: learning on the fly. Nat Rev Immunol. 2008;8(2):131–41. Epub 2008/01/26. doi: 10.1038/nri2240. PubMed PMID: 18219310. [DOI] [PubMed] [Google Scholar]
- 86.Georgel P, Naitza S, Kappler C, Ferrandon D, Zachary D, Swimmer C, Kopczynski C, Duyk G, Reichhart JM, Hoffmann JA. Drosophila immune deficiency (IMD) is a death domain protein that activates antibacterial defense and can promote apoptosis. Dev Cell. 2001;1(4):503–14. Epub 2001/11/13. doi: 10.1016/s1534-5807(01)00059-4. PubMed PMID: 11703941. [DOI] [PubMed] [Google Scholar]
- 87.Ferrandon D, Imler JL, Hetru C, Hoffmann JA. The Drosophila systemic immune response: sensing and signalling during bacterial and fungal infections. Nat Rev Immunol. 2007;7(11):862–74. Epub 2007/10/20. doi: 10.1038/nri2194. PubMed PMID: 17948019. [DOI] [PubMed] [Google Scholar]
- 88.Silverman N, Maniatis T. NF-kappaB signaling pathways in mammalian and insect innate immunity. Genes & development. 2001;15(18):2321–42. Epub 2001/09/20. doi: 10.1101/gad.909001. PubMed PMID: 11562344. [DOI] [PubMed] [Google Scholar]
- 89.Hancock RE, Haney EF, Gill EE. The immunology of host defence peptides: beyond antimicrobial activity. Nat Rev Immunol. 2016;16(5):321–34. Epub 2016/04/19. doi: 10.1038/nri.2016.29. PubMed PMID: 27087664. [DOI] [PubMed] [Google Scholar]
- 90.Imler JL, Bulet P. Antimicrobial peptides in Drosophila: structures, activities and gene regulation. Chem Immunol Allergy. 2005;86:1–21. Epub 2005/06/25. doi: 10.1159/000086648. PubMed PMID: 15976485. [DOI] [PubMed] [Google Scholar]
- 91.Lazzaro BP, Zasloff M, Rolff J. Antimicrobial peptides: Application informed by evolution. Science. 2020;368(6490). Epub 2020/05/02. doi: 10.1126/science.aau5480. PubMed PMID: 32355003; PMCID: PMC8097767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lemaitre B, Hoffmann J. The host defense of Drosophila melanogaster. Annu Rev Immunol. 2007;25:697–743. Epub 2007/01/05. doi: 10.1146/annurev.immunol.25.022106.141615. PubMed PMID: 17201680. [DOI] [PubMed] [Google Scholar]
- 93.Tanji T, Hu X, Weber AN, Ip YT. Toll and IMD pathways synergistically activate an innate immune response in Drosophila melanogaster. Mol Cell Biol. 2007;27(12):4578–88. Epub 2007/04/18. doi: 10.1128/MCB.01814-06. PubMed PMID: 17438142; PMCID: PMC1900069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cao Y, Chtarbanova S, Petersen AJ, Ganetzky B. Dnr1 mutations cause neurodegeneration in Drosophila by activating the innate immune response in the brain. Proc Natl Acad Sci U S A. 2013;110(19):E1752–60. Epub 2013/04/25. doi: 10.1073/pnas.1306220110. PubMed PMID: 23613578; PMCID: PMC3651420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chinchore Y, Gerber GF, Dolph PJ. Alternative pathway of cell death in Drosophila mediated by NF-kappaB transcription factor Relish. Proc Natl Acad Sci U S A. 2012;109(10):E605–12. Epub 2012/02/14. doi: 10.1073/pnas.1110666109. PubMed PMID: 22328149; PMCID: PMC3309745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kounatidis I, Chtarbanova S, Cao Y, Hayne M, Jayanth D, Ganetzky B, Ligoxygakis P. NF-kappaB Immunity in the Brain Determines Fly Lifespan in Healthy Aging and Age-Related Neurodegeneration. Cell reports. 2017;19(4):836–48. Epub 2017/04/27. doi: 10.1016/j.celrep.2017.04.007. PubMed PMID: 28445733; PMCID: PMC5413584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Petersen AJ, Rimkus SA, Wassarman DA. ATM kinase inhibition in glial cells activates the innate immune response and causes neurodegeneration in Drosophila. Proc Natl Acad Sci U S A. 2012;109(11):E656–64. Epub 2012/02/23. doi: 10.1073/pnas.1110470109. PubMed PMID: 22355133; PMCID: PMC3306708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kono H, Onda A, Yanagida T. Molecular determinants of sterile inflammation. Curr Opin Immunol. 2014;26:147–56. Epub 2014/02/22. doi: 10.1016/j.coi.2013.12.004. PubMed PMID: 24556412. [DOI] [PubMed] [Google Scholar]
- 99.Kozlov A, Koch R, Nagoshi E. Nitric oxide mediates neuro-glial interaction that shapes Drosophila circadian behavior. PLoS Genet. 2020;16(6):e1008312. Epub 2020/07/01. doi: 10.1371/journal.pgen.1008312. PubMed PMID: 32598344; PMCID: PMC7367490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Simon DW, McGeachy MJ, Bayir H, Clark RSB, Loane DJ, Kochanek PM. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol. 2017;13(9):572. Epub 2017/08/05. doi: 10.1038/nrneurol.2017.116. PubMed PMID: 28776601. [DOI] [PubMed] [Google Scholar]
- 101.Shah EJ, Gurdziel K, Ruden DM. Drosophila Exhibit Divergent Sex-Based Responses in Transcription and Motor Function After Traumatic Brain Injury. Front Neurol. 2020;11:511. Epub 2020/07/09. doi: 10.3389/fneur.2020.00511. PubMed PMID: 32636795; PMCID: PMC7316956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sanuki R, Tanaka T, Suzuki F, Ibaraki K, Takano T. Normal aging hyperactivates innate immunity and reduces the medical efficacy of minocycline in brain injury. Brain Behav Immun. 2019;80:427–38. Epub 2019/04/16. doi: 10.1016/j.bbi.2019.04.023. PubMed PMID: 30986429. [DOI] [PubMed] [Google Scholar]
- 103.Vidal S, Khush RS, Leulier F, Tzou P, Nakamura M, Lemaitre B. Mutations in the Drosophila dTAK1 gene reveal a conserved function for MAPKKKs in the control of rel/NF-kappaB-dependent innate immune responses. Genes & development. 2001;15(15):1900–12. Epub 2001/08/04. doi: 10.1101/gad.203301. PubMed PMID: 11485985; PMCID: PMC524699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Swanson LC, Trujillo EA, Thiede GH, Katzenberger RJ, Shishkova E, Coon JJ, Ganetzky B, Wassarman DA. Survival Following Traumatic Brain Injury in Drosophila Is Increased by Heterozygosity for a Mutation of the NF-kappaB Innate Immune Response Transcription Factor Relish. Genetics. 2020;216(4):1117–36. Epub 2020/10/29. doi: 10.1534/genetics.120.303776. PubMed PMID: 33109529; PMCID: PMC7768241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wilhelm M, Kukekov NV, Schmit TL, Biagas KV, Sproul AA, Gire S, Maes ME, Xu Z, Greene LA. Sh3rf2/POSHER protein promotes cell survival by ring-mediated proteasomal degradation of the c-Jun N-terminal kinase scaffold POSH (Plenty of SH3s) protein. The Journal of biological chemistry. 2012;287(3):2247–56. Epub 2011/12/01. doi: 10.1074/jbc.M111.269431. PubMed PMID: 22128169; PMCID: PMC3265902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxidants & redox signaling. 2014;20(7):1126–67. Epub 2013/09/03. doi: 10.1089/ars.2012.5149. PubMed PMID: 23991888; PMCID: PMC3929010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Meier P, Banreti A. Tissue Repair: How to Inflame Your Neighbours. Current biology : CB. 2016;26(5):R192–4. Epub 2016/03/10. doi: 10.1016/j.cub.2016.01.033. PubMed PMID: 26954436. [DOI] [PubMed] [Google Scholar]
- 108.Park JM, Brady H, Ruocco MG, Sun H, Williams D, Lee SJ, Kato T Jr., Richards N, Chan K, Mercurio F, Karin M, Wasserman SA. Targeting of TAK1 by the NF-kappa B protein Relish regulates the JNK-mediated immune response in Drosophila. Genes & development. 2004;18(5):584–94. Epub 2004/03/24. doi: 10.1101/gad.1168104. PubMed PMID: 15037551; PMCID: PMC374239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhao JB, Zhang Y, Li GZ, Su XF, Hang CH. Activation of JAK2/STAT pathway in cerebral cortex after experimental traumatic brain injury of rats. Neuroscience letters. 2011;498(2):147–52. Epub 2011/05/21. doi: 10.1016/j.neulet.2011.05.001. PubMed PMID: 21596098. [DOI] [PubMed] [Google Scholar]
- 110.Kim LK, Choi UY, Cho HS, Lee JS, Lee WB, Kim J, Jeong K, Shim J, Kim-Ha J, Kim YJ. Down-regulation of NF-kappaB target genes by the AP-1 and STAT complex during the innate immune response in Drosophila. PLoS Biol. 2007;5(9):e238. Epub 2007/09/07. doi: 10.1371/journal.pbio.0050238. PubMed PMID: 17803358; PMCID: PMC1964775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ayaz D, Leyssen M, Koch M, Yan J, Srahna M, Sheeba V, Fogle KJ, Holmes TC, Hassan BA. Axonal injury and regeneration in the adult brain of Drosophila. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28(23):6010–21. Epub 2008/06/06. doi: 10.1523/JNEUROSCI.0101-08.2008. PubMed PMID: 18524906; PMCID: PMC2693324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.MacDonald JM, Beach MG, Porpiglia E, Sheehan AE, Watts RJ, Freeman MR. The Drosophila cell corpse engulfment receptor Draper mediates glial clearance of severed axons. Neuron. 2006;50(6):869–81. Epub 2006/06/15. doi: 10.1016/j.neuron.2006.04.028. PubMed PMID: 16772169. [DOI] [PubMed] [Google Scholar]
- 113.Doherty J, Logan MA, Tasdemir OE, Freeman MR. Ensheathing glia function as phagocytes in the adult Drosophila brain. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29(15):4768–81. Epub 2009/04/17. doi: 10.1523/JNEUROSCI.5951-08.2009. PubMed PMID: 19369546; PMCID: PMC2674269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Doherty J, Sheehan AE, Bradshaw R, Fox AN, Lu TY, Freeman MR. PI3K signaling and Stat92E converge to modulate glial responsiveness to axonal injury. PLoS Biol. 2014;12(11):e1001985. Epub 2014/11/05. doi: 10.1371/journal.pbio.1001985. PubMed PMID: 25369313; PMCID: PMC4219656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Freeman MR. Drosophila Central Nervous System Glia. Cold Spring Harb Perspect Biol. 2015;7(11). Epub 2015/02/28. doi: 10.1101/cshperspect.a020552. PubMed PMID: 25722465; PMCID: PMC4632667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Macdonald JM, Doherty J, Hackett R, Freeman MR. The c-Jun kinase signaling cascade promotes glial engulfment activity through activation of draper and phagocytic function. Cell death and differentiation. 2013;20(9):1140–8. Epub 2013/04/27. doi: 10.1038/cdd.2013.30. PubMed PMID: 23618811; PMCID: PMC3741495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kato K, Forero MG, Fenton JC, Hidalgo A. The glial regenerative response to central nervous system injury is enabled by pros-notch and pros-NFkappaB feedback. PLoS Biol. 2011;9(8):e1001133. Epub 2011/09/14. doi: 10.1371/journal.pbio.1001133. PubMed PMID: 21912512; PMCID: PMC3166069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pearse DD, Pereira FC, Marcillo AE, Bates ML, Berrocal YA, Filbin MT, Bunge MB. cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat Med. 2004;10(6):610–6. Epub 2004/05/25. doi: 10.1038/nm1056. PubMed PMID: 15156204. [DOI] [PubMed] [Google Scholar]
- 119.Stiles TL, Kapiloff MS, Goldberg JL. The role of soluble adenylyl cyclase in neurite outgrowth. Biochimica et biophysica acta. 2014;1842(12 Pt B):2561–8. Epub 2014/07/30. doi: 10.1016/j.bbadis.2014.07.012. PubMed PMID: 25064589; PMCID: PMC4262618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Stone MC, Albertson RM, Chen L, Rolls MM. Dendrite injury triggers DLK-independent regeneration. Cell reports. 2014;6(2):247–53. Epub 2014/01/15. doi: 10.1016/j.celrep.2013.12.022. PubMed PMID: 24412365; PMCID: PMC3954604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lee DC, Vali K, Baldwin SR, Divino JN, Feliciano JL, Fequiere JR, Fernandez MA, Frageau JC, Longo FK, Madhoun SS, Mingione VP, O'Toole TR, Ruiz MG, Tanner GR. Dietary Supplementation With the Ketogenic Diet Metabolite Beta-Hydroxybutyrate Ameliorates Post-TBI Aggression in Young-Adult Male Drosophila. Front Neurosci. 2019;13:1140. Epub 2019/11/19. doi: 10.3389/fnins.2019.01140. PubMed PMID: 31736687; PMCID: PMC6833482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hill CS, Sreedharan J, Loreto A, Menon DK, Coleman MP. Loss of highwire Protects Against the Deleterious Effects of Traumatic Brain Injury in Drosophila Melanogaster. Front Neurol. 2020;11:401. Epub 2020/06/02. doi: 10.3389/fneur.2020.00401. PubMed PMID: 32477254; PMCID: PMC7235382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Miñambres E, Ballesteros MA, Mayorga M, Marin MJ, Muñoz P, Figols J, López-Hoyos M. Cerebral apoptosis in severe traumatic brain injury patients: an in vitro, in vivo, and postmortem study. J Neurotrauma. 2008;25(6):581–91. Epub 2008/03/28. doi: 10.1089/neu.2007.0398. PubMed PMID: 18363508. [DOI] [PubMed] [Google Scholar]
- 124.Liu T, Zhao D-x, Cui H, Chen L, Bao Y-h, Wang Y, Jiang J-y. Therapeutic hypothermia attenuates tissue damage and cytokine expression after traumatic brain injury by inhibiting necroptosis in the rat. Scientific reports. 2016;6:24547-. doi: 10.1038/srep24547. PubMed PMID: 27080932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.White K, Grether ME, Abrams JM, Young L, Farrell K, Steller H. Genetic control of programmed cell death in Drosophila. Science. 1994;264(5159):677–83. Epub 1994/04/29. doi: 10.1126/science.8171319. PubMed PMID: 8171319. [DOI] [PubMed] [Google Scholar]
- 126.Cashio P, Lee TV, Bergmann A. Genetic control of programmed cell death in Drosophila melanogaster. Semin Cell Dev Biol. 2005;16(2):225–35. Epub 2005/03/31. doi: 10.1016/j.semcdb.2005.01.002. PubMed PMID: 15797833. [DOI] [PubMed] [Google Scholar]
- 127.Kanuka H, Sawamoto K, Inohara N, Matsuno K, Okano H, Miura M. Control of the cell death pathway by Dapaf-1, a Drosophila Apaf-1/CED-4-related caspase activator. Mol Cell. 1999;4(5):757–69. Epub 2000/01/05. doi: 10.1016/s1097-2765(00)80386-x. PubMed PMID: 10619023. [DOI] [PubMed] [Google Scholar]
- 128.Rodriguez A, Oliver H, Zou H, Chen P, Wang X, Abrams JM. Dark is a Drosophila homologue of Apaf-1/CED-4 and functions in an evolutionarily conserved death pathway. Nature cell biology. 1999;1(5):272–9. Epub 1999/11/13. doi: 10.1038/12984. PubMed PMID: 10559939. [DOI] [PubMed] [Google Scholar]
- 129.Zhou L, Song Z, Tittel J, Steller H. HAC-1, a Drosophila homolog of APAF-1 and CED-4 functions in developmental and radiation-induced apoptosis. Mol Cell. 1999;4(5):745–55. Epub 2000/01/05. doi: 10.1016/s1097-2765(00)80385-8. PubMed PMID: 10619022. [DOI] [PubMed] [Google Scholar]
- 130.Muro I, Berry DL, Huh JR, Chen CH, Huang H, Yoo SJ, Guo M, Baehrecke EH, Hay BA. The Drosophila caspase Ice is important for many apoptotic cell deaths and for spermatid individualization, a nonapoptotic process. Development. 2006;133(17):3305–15. Epub 2006/08/05. doi: 10.1242/dev.02495. PubMed PMID: 16887831. [DOI] [PubMed] [Google Scholar]
- 131.Quinn L, Coombe M, Mills K, Daish T, Colussi P, Kumar S, Richardson H. Buffy, a Drosophila Bcl-2 protein, has anti-apoptotic and cell cycle inhibitory functions. The EMBO journal. 2003;22(14):3568–79. doi: 10.1093/emboj/cdg355. PubMed PMID: 12853472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Varkey J, Chen P, Jemmerson R, Abrams JM. Altered cytochrome c display precedes apoptotic cell death in Drosophila. J Cell Biol. 1999;144(4):701–10. Epub 1999/02/26. doi: 10.1083/jcb.144.4.701. PubMed PMID: 10037791; PMCID: PMC2132929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dorstyn L, Read S, Cakouros D, Huh JR, Hay BA, Kumar S. The role of cytochrome c in caspase activation in Drosophila melanogaster cells. J Cell Biol. 2002;156(6):1089–98. Epub 2002/03/20. doi: 10.1083/jcb.200111107. PubMed PMID: 11901173; PMCID: PMC2173478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zimmermann KC, Ricci JE, Droin NM, Green DR. The role of ARK in stress-induced apoptosis in Drosophila cells. J Cell Biol. 2002;156(6):1077–87. Epub 2002/03/20. doi: 10.1083/jcb.20112068. PubMed PMID: 11901172; PMCID: PMC2173462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Challa M, Malladi S, Pellock BJ, Dresnek D, Varadarajan S, Yin YW, White K, Bratton SB. Drosophila Omi, a mitochondrial-localized IAP antagonist and proapoptotic serine protease. The EMBO journal. 2007;26(13):3144–56. Epub 2007/06/09. doi: 10.1038/sj.emboj.7601745. PubMed PMID: 17557079; PMCID: PMC1914093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Igaki T, Suzuki Y, Tokushige N, Aonuma H, Takahashi R, Miura M. Evolution of mitochondrial cell death pathway: Proapoptotic role of HtrA2/Omi in Drosophila. Biochemical and biophysical research communications. 2007;356(4):993–7. Epub 2007/04/03. doi: 10.1016/j.bbrc.2007.03.079. PubMed PMID: 17397804. [DOI] [PubMed] [Google Scholar]
- 137.Galindo KA, Lu WJ, Park JH, Abrams JM. The Bax/Bak ortholog in Drosophila, Debcl, exerts limited control over programmed cell death. Development. 2009;136(2):275–83. Epub 2008/12/18. doi: 10.1242/dev.019042. PubMed PMID: 19088092; PMCID: PMC2685970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Brachmann CB, Jassim OW, Wachsmuth BD, Cagan RL. The Drosophila bcl-2 family member dBorg-1 functions in the apoptotic response to UV-irradiation. Current biology : CB. 2000;10(9):547–50. Epub 2000/05/10. doi: 10.1016/s0960-9822(00)00474-7. PubMed PMID: 10801447. [DOI] [PubMed] [Google Scholar]
- 139.Colussi PA, Quinn LM, Huang DC, Coombe M, Read SH, Richardson H, Kumar S. Debcl, a proapoptotic Bcl-2 homologue, is a component of the Drosophila melanogaster cell death machinery. J Cell Biol. 2000;148(4):703–14. Epub 2000/02/23. doi: 10.1083/jcb.148.4.703. PubMed PMID: 10684252; PMCID: PMC2169366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Igaki T, Kanuka H, Inohara N, Sawamoto K, Núñez G, Okano H, Miura M. Drob-1, a Drosophila member of the Bcl-2/CED-9 family that promotes cell death. Proc Natl Acad Sci U S A. 2000;97(2):662–7. Epub 2000/01/19. doi: 10.1073/pnas.97.2.662. PubMed PMID: 10639136; PMCID: PMC15387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zhang H, Huang Q, Ke N, Matsuyama S, Hammock B, Godzik A, Reed JC. Drosophila pro-apoptotic Bcl-2/Bax homologue reveals evolutionary conservation of cell death mechanisms. The Journal of biological chemistry. 2000;275(35):27303–6. Epub 2000/05/16. doi: 10.1074/jbc.M002846200. PubMed PMID: 10811653. [DOI] [PubMed] [Google Scholar]
- 142.O'Riordan MX, Bauler LD, Scott FL, Duckett CS. Inhibitor of apoptosis proteins in eukaryotic evolution and development: a model of thematic conservation. Dev Cell. 2008;15(4):497–508. Epub 2008/10/16. doi: 10.1016/j.devcel.2008.09.012. PubMed PMID: 18854135; PMCID: PMC2676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Khan FS, Fujioka M, Datta P, Fernandes-Alnemri T, Jaynes JB, Alnemri ES. The interaction of DIAP1 with dOmi/HtrA2 regulates cell death in Drosophila. Cell death and differentiation. 2008;15(6):1073–83. Epub 2008/02/15. doi: 10.1038/cdd.2008.19. PubMed PMID: 18259196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Huh JR, Foe I, Muro I, Chen CH, Seol JH, Yoo SJ, Guo M, Park JM, Hay BA. The Drosophila inhibitor of apoptosis (IAP) DIAP2 is dispensable for cell survival, required for the innate immune response to gram-negative bacterial infection, and can be negatively regulated by the reaper/hid/grim family of IAP-binding apoptosis inducers. The Journal of biological chemistry. 2007;282(3):2056–68. Epub 2006/10/28. doi: 10.1074/jbc.M608051200. PubMed PMID: 17068333. [DOI] [PubMed] [Google Scholar]
- 145.Kleino A, Valanne S, Ulvila J, Kallio J, Myllymäki H, Enwald H, Stöven S, Poidevin M, Ueda R, Hultmark D, Lemaitre B, Rämet M. Inhibitor of apoptosis 2 and TAK1-binding protein are components of the Drosophila Imd pathway. The EMBO journal. 2005;24(19):3423–34. Epub 2005/09/16. doi: 10.1038/sj.emboj.7600807. PubMed PMID: 16163390; PMCID: PMC1276168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Valanne S, Kleino A, Myllymäki H, Vuoristo J, Rämet M. Iap2 is required for a sustained response in the Drosophila Imd pathway. Dev Comp Immunol. 2007;31(10):991–1001. Epub 2007/03/09. doi: 10.1016/j.dci.2007.01.004. PubMed PMID: 17343912. [DOI] [PubMed] [Google Scholar]
- 147.Fogarty CE, Diwanji N, Lindblad JL, Tare M, Amcheslavsky A, Makhijani K, Brückner K, Fan Y, Bergmann A. Extracellular Reactive Oxygen Species Drive Apoptosis-Induced Proliferation via Drosophila Macrophages. Current biology : CB. 2016;26(5):575–84. Epub 2016/02/24. doi: 10.1016/j.cub.2015.12.064. PubMed PMID: 26898463; PMCID: PMC4765900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Biteau B, Karpac J, Hwangbo D, Jasper H. Regulation of Drosophila lifespan by JNK signaling. Exp Gerontol. 2011;46(5):349–54. Epub 2010/11/25. doi: 10.1016/j.exger.2010.11.003. PubMed PMID: 21111799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Oberst A, Bender C, Green DR. Living with death: the evolution of the mitochondrial pathway of apoptosis in animals. Cell death and differentiation. 2008;15(7):1139–46. Epub 2008/05/03. doi: 10.1038/cdd.2008.65. PubMed PMID: 18451868; PMCID: PMC2612587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kornbluth S, White K. Apoptosis in Drosophila: neither fish nor fowl (nor man, nor worm). J Cell Sci. 2005;118(Pt 9):1779–87. Epub 2005/04/30. doi: 10.1242/jcs.02377. PubMed PMID: 15860727. [DOI] [PubMed] [Google Scholar]
- 151.Dondelinger Y, Hulpiau P, Saeys Y, Bertrand MJM, Vandenabeele P. An evolutionary perspective on the necroptotic pathway. Trends Cell Biol. 2016;26(10):721–32. Epub 2016/07/03. doi: 10.1016/j.tcb.2016.06.004. PubMed PMID: 27368376. [DOI] [PubMed] [Google Scholar]
- 152.Kanda H, Igaki T, Okano H, Miura M. Conserved metabolic energy production pathways govern Eiger/TNF-induced nonapoptotic cell death. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(47):18977–82. Epub 2011/11/07. doi: 10.1073/pnas.1103242108. PubMed PMID: 22065747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Li M, Sun S, Priest J, Bi X, Fan Y. Characterization of TNF-induced cell death in Drosophila reveals caspase- and JNK-dependent necrosis and its role in tumor suppression. Cell death & disease. 2019;10(8):613-. doi: 10.1038/s41419-019-1862-0. PubMed PMID: 31409797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Obata F, Kuranaga E, Tomioka K, Ming M, Takeishi A, Chen C-H, Soga T, Miura M. Necrosis-driven systemic immune response alters SAM metabolism through the FOXO-GNMT axis. Cell reports. 2014;7(3):821–33. [DOI] [PubMed] [Google Scholar]
- 155.Bjedov I, Toivonen JM, Kerr F, Slack C, Jacobson J, Foley A, Partridge L. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 2010;11(1):35–46. Epub 2010/01/16. doi: 10.1016/j.cmet.2009.11.010. PubMed PMID: 20074526; PMCID: PMC2824086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wang B, Goode J, Best J, Meltzer J, Schilman PE, Chen J, Garza D, Thomas JB, Montminy M. The insulin-regulated CREB coactivator TORC promotes stress resistance in Drosophila. Cell Metab. 2008;7(5):434–44. Epub 2008/05/08. doi: 10.1016/j.cmet.2008.02.010. PubMed PMID: 18460334; PMCID: PMC3704161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. Regulation of TORC1 by Rag GTPases in nutrient response. Nature cell biology. 2008;10(8):935–45. Epub 2008/07/08. doi: 10.1038/ncb1753. PubMed PMID: 18604198; PMCID: PMC2711503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Royce GH, Brown-Borg HM, Deepa SS. The potential role of necroptosis in inflammaging and aging. Geroscience. 2019;41(6):795–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Xu T, Nicolson S, Denton D, Kumar S. Distinct requirements of Autophagy-related genes in programmed cell death. Cell death and differentiation. 2015;22(11):1792–802. Epub 2015/04/18. doi: 10.1038/cdd.2015.28. PubMed PMID: 25882046; PMCID: PMC4648326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ryan LM, Warden DL. Post concussion syndrome. International review of psychiatry (Abingdon, England). 2003;15(4):310–6. Epub 2004/07/28. doi: 10.1080/09540260310001606692. PubMed PMID: 15276952. [DOI] [PubMed] [Google Scholar]
- 161.Loane DJ, Faden AI. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci. 2010;31(12):596–604. Epub 2010/11/03. doi: 10.1016/j.tips.2010.09.005. PubMed PMID: 21035878; PMCID: PMC2999630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Yeh CC, Chen TL, Hu CJ, Chiu WT, Liao CC. Risk of epilepsy after traumatic brain injury: a retrospective population-based cohort study. Journal of neurology, neurosurgery, and psychiatry. 2013;84(4):441–5. Epub 2012/11/03. doi: 10.1136/jnnp-2012-302547. PubMed PMID: 23117492. [DOI] [PubMed] [Google Scholar]
- 163.Hosseini AH, Lifshitz J. Brain injury forces of moderate magnitude elicit the fencing response. Medicine and science in sports and exercise. 2009;41(9):1687–97. Epub 2009/08/07. doi: 10.1249/MSS.0b013e31819fcd1b. PubMed PMID: 19657303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Futagi Y, Toribe Y, Suzuki Y. The grasp reflex and moro reflex in infants: hierarchy of primitive reflex responses. Int J Pediatr. 2012;2012:191562. Epub 2012/07/11. doi: 10.1155/2012/191562. PubMed PMID: 22778756; PMCID: PMC3384944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Bermingham NA, Hassan BA, Wang VY, Fernandez M, Banfi S, Bellen HJ, Fritzsch B, Zoghbi HY. Proprioceptor pathway development is dependent on Math1. Neuron. 2001;30(2):411–22. Epub 2001/06/08. doi: 10.1016/s0896-6273(01)00305-1. PubMed PMID: 11395003. [DOI] [PubMed] [Google Scholar]
- 166.Hassan BA, Bermingham NA, He Y, Sun Y, Jan YN, Zoghbi HY, Bellen HJ. atonal regulates neurite arborization but does not act as a proneural gene in the Drosophila brain. Neuron. 2000;25(3):549–61. Epub 2000/04/25. doi: 10.1016/s0896-6273(00)81059-4. PubMed PMID: 10774724. [DOI] [PubMed] [Google Scholar]
- 167.Field LH, Matheson T. Chordotonal organs of insects. Advances in insect physiology. 1998;27:1–228. [Google Scholar]
- 168.Sherman A, Dickinson MH. A comparison of visual and haltere-mediated equilibrium reflexes in the fruit fly Drosophila melanogaster. The Journal of experimental biology. 2003;206(Pt 2):295–302. Epub 2002/12/13. doi: 10.1242/jeb.00075. PubMed PMID: 12477899. [DOI] [PubMed] [Google Scholar]
- 169.Lateef S, Holman A, Carpenter J, James J. Can Therapeutic Hypothermia Diminish the Impact of Traumatic Brain Injury in Drosophila melanogaster? J Exp Neurosci. 2019;13:1179069518824852. Epub 2019/02/09. doi: 10.1177/1179069518824852. PubMed PMID: 30733630; PMCID: PMC6343440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Krishna G, Bromberg C, Connell EC, Mian E, Hu C, Lifshitz J, Adelson PD, Thomas TC. Traumatic Brain Injury-Induced Sex-Dependent Changes in Late-Onset Sensory Hypersensitivity and Glutamate Neurotransmission. Front Neurol. 2020;11:749. Epub 2020/08/28. doi: 10.3389/fneur.2020.00749. PubMed PMID: 32849211; PMCID: PMC7419702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kempf J, Werth E, Kaiser PR, Bassetti CL, Baumann CR. Sleep-wake disturbances 3 years after traumatic brain injury. Journal of neurology, neurosurgery, and psychiatry. 2010;81(12):1402–5. Epub 2010/10/05. doi: 10.1136/jnnp.2009.201913. PubMed PMID: 20884672. [DOI] [PubMed] [Google Scholar]
- 172.Sharma R, Shultz SR, Robinson MJ, Belli A, Hibbs ML, O'Brien TJ, Semple BD. Infections after a traumatic brain injury: The complex interplay between the immune and neurological systems. Brain Behav Immun. 2019;79:63–74. Epub 2019/04/29. doi: 10.1016/j.bbi.2019.04.034. PubMed PMID: 31029794. [DOI] [PubMed] [Google Scholar]
- 173.Puntambekar SS, Saber M, Lamb BT, Kokiko-Cochran ON. Cellular players that shape evolving pathology and neurodegeneration following traumatic brain injury. Brain Behav Immun. 2018;71:9–17. Epub 2018/03/31. doi: 10.1016/j.bbi.2018.03.033. PubMed PMID: 29601944. [DOI] [PubMed] [Google Scholar]
- 174.Kao CH, ChangLai SP, Chieng PU, Yen TC. Gastric emptying in head-injured patients. The American journal of gastroenterology. 1998;93(7):1108–12. Epub 1998/07/22. doi: 10.1111/j.1572-0241.1998.00338.x. PubMed PMID: 9672339. [DOI] [PubMed] [Google Scholar]
- 175.Faries PL, Simon RJ, Martella AT, Lee MJ, Machiedo GW. Intestinal permeability correlates with severity of injury in trauma patients. The Journal of trauma. 1998;44(6):1031–5; discussion 5-6. Epub 1998/06/24. doi: 10.1097/00005373-199806000-00016. PubMed PMID: 9637159. [DOI] [PubMed] [Google Scholar]
- 176.Katzenberger RJ, Ganetzky B, Wassarman DA. The gut reaction to traumatic brain injury. Fly (Austin). 2015;9(2):68–74. Epub 2015/08/21. doi: 10.1080/19336934.2015.1085623. PubMed PMID: 26291482; PMCID: PMC5019014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sen A, Gurdziel K, Liu J, Qu W, Nuga OO, Burl RB, Huttemann M, Pique-Regi R, Ruden DM. Smooth, an hnRNP-L Homolog, Might Decrease Mitochondrial Metabolism by Post-Transcriptional Regulation of Isocitrate Dehydrogenase (Idh) and Other Metabolic Genes in the Sub-Acute Phase of Traumatic Brain Injury. Front Genet. 2017;8:175. Epub 2017/12/01. doi: 10.3389/fgene.2017.00175. PubMed PMID: 29187863; PMCID: PMC5694756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Leyssen M, Hassan BA. A fruitfly's guide to keeping the brain wired. EMBO Reports 2007. p. 46–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Pandey UB, Nichols CD. Human disease models in Drosophila melanogaster and the role of the fly in therapeutic drug discovery. Pharmacol Rev. 2011;63(2):411–36. Epub 03/17. doi: 10.1124/pr.110.003293. PubMed PMID: 21415126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lessing D, Bonini NM. Maintaining the Brain: Insight into Human Neurodegeneration From Drosophila Mutants. Nature reviews Genetics. 2009;10(6):359. doi: 10.1038/nrg2563. PubMed PMID: 19434080; PMCID: 2820605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Strausfeld NJ, Hirth F. Deep Homology of Arthropod Central Complex and Vertebrate Basal Ganglia. Science. 2013;340(6129):157–61. [DOI] [PubMed] [Google Scholar]
- 182.Okano H, Okabe M, Taguchi A, Sawamoto K. Evolutionarily conserved mechanisms regulating neural development: lessons from the development of Drosophila peripheral nervous systems. Hum Cell. 1997;10(3):139–50. Epub 1998/01/22. PubMed PMID: 9436033. [PubMed] [Google Scholar]
- 183.Kendler KS, Greenspan RJ. The nature of genetic influences on behavior: lessons from "simpler" organisms. Am J Psychiatry. 2006;163(10):1683–94. Epub 2006/10/03. doi: 10.1176/ajp.2006.163.10.1683. PubMed PMID: 17012675. [DOI] [PubMed] [Google Scholar]
- 184.Heidary G, Fortini ME. Identification and characterization of the Drosophila tau homolog. Mechanisms of development. 2001;108(1-2):171–8. [DOI] [PubMed] [Google Scholar]
- 185.Luo LQ, Martin-Morris LE, White K. Identification, secretion, and neural expression of APPL, a Drosophila protein similar to human amyloid protein precursor. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1990;10(12):3849–61. Epub 1990/12/01. doi: 10.1523/jneurosci.10-12-03849.1990. PubMed PMID: 2125311; PMCID: PMC6570036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Carmine-Simmen K, Proctor T, Tschäpe J, Poeck B, Triphan T, Strauss R, Kretzschmar D. Neurotoxic effects induced by the Drosophila amyloid-beta peptide suggest a conserved toxic function. Neurobiology of disease. 2009;33(2):274–81. Epub 2008/11/08. doi: 10.1016/j.nbd.2008.10.014. PubMed PMID: 19049874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Liu Z, Wang X, Yu Y, Li X, Wang T, Jiang H, Ren Q, Jiao Y, Sawa A, Moran T, Ross CA, Montell C, Smith WW. A Drosophila model for LRRK2-linked parkinsonism. Proc Natl Acad Sci U S A. 2008;105(7):2693–8. Epub 2008/02/09. doi: 10.1073/pnas.0708452105. PubMed PMID: 18258746; PMCID: PMC2268198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Fortini ME, Skupski MP, Boguski MS, Hariharan IK. A survey of human disease gene counterparts in the Drosophila genome. J Cell Biol. 2000;150(2):F23–30. Epub 2000/07/26. doi: 10.1083/jcb.150.2.f23. PubMed PMID: 10908582; PMCID: PMC2180233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Clark I, Dodson M, Jiang C, Cao J, Huh J, Seol J, Yoo S, Hay B, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441(7097):1162–6. [DOI] [PubMed] [Google Scholar]
- 190.Bandyopadhyay S, Cookson MR. Evolutionary and functional relationships within the DJ1 superfamily. BMC Evol Biol. 2004;4:6-. doi: 10.1186/1471-2148-4-6. PubMed PMID: 15070401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Hazelett DJ, Chang J-C, Lakeland DL, Morton DB. Comparison of parallel high-throughput RNA sequencing between knockout of TDP-43 and its overexpression reveals primarily nonreciprocal and nonoverlapping gene expression changes in the central nervous system of Drosophila. G3 (Bethesda). 2012;2(7):789–802. Epub 2012/07/01. doi: 10.1534/g3.112.002998. PubMed PMID: 22870402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Tsuda H, Jafar-Nejad H, Patel AJ, Sun Y, Chen HK, Rose MF, Venken KJ, Botas J, Orr HT, Bellen HJ, Zoghbi HY. The AXH domain of Ataxin-1 mediates neurodegeneration through its interaction with Gfi-1/Senseless proteins. Cell. 2005;122(4):633–44. Epub 2005/08/27. doi: 10.1016/j.cell.2005.06.012. PubMed PMID: 16122429. [DOI] [PubMed] [Google Scholar]
- 193.Cañizares J, Blanca JM, Navarro JA, Monrós E, Palau F, Moltó MD. dfh is a Drosophila homolog of the Friedreich's ataxia disease gene. Gene. 2000;256(1-2):35–42. Epub 2000/10/31. doi: 10.1016/s0378-1119(00)00343-7. PubMed PMID: 11054533. [DOI] [PubMed] [Google Scholar]
- 194.Lee YM, Misra HP, Ayala FJ. Superoxide dismutase in Drosophila melanogaster: biochemical and structural characterization of allozyme variants. Proc Natl Acad Sci U S A. 1981;78(11):7052–5. Epub 1981/11/01. doi: 10.1073/pnas.78.11.7052. PubMed PMID: 6273906; PMCID: PMC349192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Satterfield TF, Pallanck LJ. Ataxin-2 and its Drosophila homolog, ATX2, physically assemble with polyribosomes. Hum Mol Genet. 2006;15(16):2523–32. Epub 2006/07/13. doi: 10.1093/hmg/ddl173. PubMed PMID: 16835262. [DOI] [PubMed] [Google Scholar]
- 196.Bolus H, Crocker K, Boekhoff-Falk G, Chtarbanova S. Modeling Neurodegenerative Disorders in Drosophila melanogaster. Int J Mol Sci. 2020;21(9). Epub 2020/05/03. doi: 10.3390/ijms21093055. PubMed PMID: 32357532; PMCID: PMC7246467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, Burman JL, Li Y, Zhang Z, Narendra DP, Cai H, Borsche M, Klein C, Youle RJ. Parkin and PINK1 mitigate STING-induced inflammation. Nature. 2018;561(7722):258–62. Epub 2018/08/24. doi: 10.1038/s41586-018-0448-9. PubMed PMID: 30135585; PMCID: PMC7362342. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 198.Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nature reviews Neuroscience. 2015;16(6):358–72. Epub 2015/05/21. doi: 10.1038/nrn3880. PubMed PMID: 25991443. [DOI] [PubMed] [Google Scholar]
- 199.Zare-Shahabadi A, Masliah E, Johnson GV, Rezaei N. Autophagy in Alzheimer's disease. Rev Neurosci. 2015;26(4):385–95. Epub 2015/04/15. doi: 10.1515/revneuro-2014-0076. PubMed PMID: 25870960; PMCID: PMC5039008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Cao J, Zhong MB, Toro CA, Zhang L, Cai D. Endo-lysosomal pathway and ubiquitin-proteasome system dysfunction in Alzheimer's disease pathogenesis. Neuroscience letters. 2019;703:68–78. Epub 2019/03/21. doi: 10.1016/j.neulet.2019.03.016. PubMed PMID: 30890471; PMCID: PMC6760990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Butterfield DA, Halliwell B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nature reviews Neuroscience. 2019;20(3):148–60. Epub 2019/02/10. doi: 10.1038/s41583-019-0132-6. PubMed PMID: 30737462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Singh A, Kukreti R, Saso L, Kukreti S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules. 2019;24(8). Epub 2019/04/25. doi: 10.3390/molecules24081583. PubMed PMID: 31013638; PMCID: PMC6514564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.McCauley ME, Baloh RH. Inflammation in ALS/FTD pathogenesis. Acta Neuropathol. 2019;137(5):715–30. Epub 2018/11/23. doi: 10.1007/s00401-018-1933-9. PubMed PMID: 30465257; PMCID: PMC6482122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Evans CS, Holzbaur ELF. Autophagy and mitophagy in ALS. Neurobiology of disease. 2019;122:35–40. Epub 2018/07/10. doi: 10.1016/j.nbd.2018.07.005. PubMed PMID: 29981842; PMCID: PMC6366665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Sullivan R, Yau WY, O'Connor E, Houlden H. Spinocerebellar ataxia: an update. Journal of neurology. 2019;266(2):533–44. Epub 2018/10/03. doi: 10.1007/s00415-018-9076-4. PubMed PMID: 30284037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Nachun D, Gao F, Isaacs C, Strawser C, Yang Z, Dokuru D, Van Berlo V, Sears R, Farmer J, Perlman S, Lynch DR, Coppola G. Peripheral blood gene expression reveals an inflammatory transcriptomic signature in Friedreich's ataxia patients. Human molecular genetics. 2018;27(17):2965–77. doi: 10.1093/hmg/ddy198. PubMed PMID: 29790959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Gutscher M, Pauleau A-L, Marty L, Brach T, Wabnitz GH, Samstag Y, Meyer AJ, Dick TP. Real-time imaging of the intracellular glutathione redox potential. Nature methods. 2008;5(6):553. [DOI] [PubMed] [Google Scholar]
- 208.Gutscher M, Sobotta MC, Wabnitz GH, Ballikaya S, Meyer AJ, Samstag Y, Dick TP. Proximity-based protein thiol oxidation by H2O2-scavenging peroxidases. Journal of Biological Chemistry. 2009:jbc. M109. 059246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Laker RC, Xu P, Ryall KA, Sujkowski A, Kenwood BM, Chain KH, Zhang M, Royal MA, Hoehn KL, Driscoll M, Adler PN, Wessells RJ, Saucerman JJ, Yan Z. A novel MitoTimer reporter gene for mitochondrial content, structure, stress, and damage in vivo. The Journal of biological chemistry. 2014;289(17):12005–15. Epub 2014/03/18. doi: 10.1074/jbc.M113.530527. PubMed PMID: 24644293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Terskikh A, Fradkov A, Ermakova G, Zaraisky A, Tan P, Kajava AV, Zhao X, Lukyanov S, Matz M, Kim S, Weissman I, Siebert P. "Fluorescent timer": protein that changes color with time. Science. 2000;290(5496):1585–8. Epub 2000/11/25. doi: 10.1126/science.290.5496.1585. PubMed PMID: 11090358. [DOI] [PubMed] [Google Scholar]
- 211.Hendricks JC, Finn SM, Panckeri KA, Chavkin J, Williams JA, Sehgal A, Pack AI. Rest in Drosophila is a sleep-like state. Neuron. 2000;25(1):129–38. Epub 2000/03/09. doi: 10.1016/s0896-6273(00)80877-6. PubMed PMID: 10707978. [DOI] [PubMed] [Google Scholar]
- 212.Shaw PJ, Cirelli C, Greenspan RJ, Tononi G. Correlates of sleep and waking in Drosophila melanogaster. Science. 2000;287(5459):1834–7. Epub 2000/03/10. doi: 10.1126/science.287.5459.1834. PubMed PMID: 10710313. [DOI] [PubMed] [Google Scholar]
- 213.Certel SJ, Kravitz EA. Scoring and analyzing aggression in Drosophila. Cold Spring Harb Protoc. 2012;2012(3):319–25. Epub 2012/03/03. doi: 10.1101/pdb.prot068130. PubMed PMID: 22383642. [DOI] [PubMed] [Google Scholar]
- 214.Goodwin SF, O'Dell KM. The best laid plans: analyzing courtship defects in Drosophila. Cold Spring Harb Protoc. 2012;2012(11):1140–5. Epub 2012/11/03. doi: 10.1101/pdb.prot071647. PubMed PMID: 23118354. [DOI] [PubMed] [Google Scholar]
- 215.Kietz C, Pollari V, Meinander A. Generating Germ-Free Drosophila to Study Gut-Microbe Interactions: Protocol to Rear Drosophila Under Axenic Conditions. Curr Protoc Toxicol. 2018;77(1):e52. Epub 2018/06/23. doi: 10.1002/cptx.52. PubMed PMID: 29933523. [DOI] [PubMed] [Google Scholar]
- 216.Pfeiffenberger C, Lear BC, Keegan KP, Allada R. Locomotor activity level monitoring using the Drosophila Activity Monitoring (DAM) System. Cold Spring Harb Protoc. 2010;2010(11):pdb.prot5518. Epub 2010/11/03. doi: 10.1101/pdb.prot5518. PubMed PMID: 21041391. [DOI] [PubMed] [Google Scholar]
- 217.Currier Thomas T, Bromberg C, Krishna G. Female sex in experimental traumatic brain injury research: forging a path forward. Neural regeneration research. 2022;17(3):550–2. doi: 10.4103/1673-5374.316602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Tennessen JM, Baker KD, Lam G, Evans J, Thummel CS. The Drosophila estrogen-related receptor directs a metabolic switch that supports developmental growth. Cell Metab. 2011;13(2):139–48. Epub 2011/02/03. doi: 10.1016/j.cmet.2011.01.005. PubMed PMID: 21284981; PMCID: PMC3072597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Saber M, Ortiz JB, Rojas-Valencia LM, Ma X, Tallent BR, Adelson PD, Rowe RK, Qiu S, Lifshitz J. Mice born to mothers with gravida traumatic brain injury have distorted brain circuitry and altered immune responses. J Neurotrauma. 2021. Epub 2021/06/23. doi: 10.1089/neu.2021.0048. PubMed PMID: 34155930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Anderson EN, Morera AA, Kour S, Cherry JD, Ramesh N, Gleixner A, Schwartz JC, Ebmeier C, Old W, Donnelly CJ, Cheng JP, Kline AE, Kofler J, Stein TD, Pandey UB. Traumatic injury compromises nucleocytoplasmic transport and leads to TDP-43 pathology. Elife. 2021;10. Epub 2021/06/02. doi: 10.7554/eLife.67587. PubMed PMID: 34060470; PMCID: PMC8169113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Molina B, Mastroianni J, Suarez E, Soni B, Forsberg E, Finley K. Treatment with Bacterial Biologics Promotes Healthy Aging and Traumatic Brain Injury Responses in Adult Drosophila, Modeling the Gut-Brain Axis and Inflammation Responses. Cells. 2021;10(4). Epub 2021/05/01. doi: 10.3390/cells10040900. PubMed PMID: 33919883; PMCID: PMC8070821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Shah EJ, Gurdziel K, Ruden DM. Sex-Differences in Traumatic Brain Injury in the Absence of Tau in Drosophila. Genes (Basel). 2021;12(6). Epub 2021/07/03. doi: 10.3390/genes12060917. PubMed PMID: 34198629; PMCID: PMC8232113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Shah EJ, Huttemann M, Sanderson TH, Gurdziel K, Ruden DM. Inhibiting Mitochondrial Cytochrome c Oxidase Downregulates Gene Transcription After Traumatic Brain Injury in Drosophila. Front Physiol. 2021;12:628777. Epub 2021/04/02. doi: 10.3389/fphys.2021.628777. PubMed PMID: 33790803; PMCID: PMC8005633. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.