

Abstract
Objective:
A cardinal feature of Marfan syndrome is thoracic aortic aneurysm (TAA). The contribution of the renin angiotensin system via angiotensin II (AngII) receptor type 1a (AT1aR) to TAA progression remains controversial because the beneficial effects of angiotensin receptor blockers have been ascribed to off-target effects. This study used genetic and pharmacologic modes of attenuating angiotensin receptor and ligand, respectively, to determine their roles on TAA in mice with fibrillin-1 haploinsufficiency (Fbn1C1041G/+).
Approach and Results:
TAA in Fbn1C1041G/+ mice was found to be strikingly sexual dimorphic. Males displayed aortic dilation over 12 months while aortic dilation in Fbn1C1041G/+ females did not differ significantly from wild type mice. To determine the role of AT1aR, Fbn1C1041G/+ mice that were either +/+ or −/− for AT1aR were generated. AT1aR deletion reduced expansion of ascending aorta and aortic root diameter from 1 to 12 months of age in males. Medial thickening and elastin fragmentation were attenuated. An antisense oligonucleotide against angiotensinogen (AGT-ASO) was administered to male Fbn1C1041G/+ mice to determine the effects of AngII depletion. AGT-ASO administration attenuated dilation of the ascending aorta and aortic root and reduced extracellular remodeling. Aortic transcriptome analyses identified potential targets by which inhibition of the renin angiotensin system reduced aortic dilation in Fbn1C1041G/+ mice.
Conclusions:
Deletion of AT1aR or inhibition of AngII production exerted similar effects in attenuating pathology in the proximal thoracic aorta of male Fbn1C1041G/+ mice. Inhibition of the renin angiotensin system attenuated dysregulation of genes within the aorta related to pathology of Fbn1C1041G/+ mice.
Keywords: Marfan syndrome, Thoracic Aortic Aneurysm, Renin Angiotensin System, Angiotensin II, Angiotensinogen
Graphic Abstract
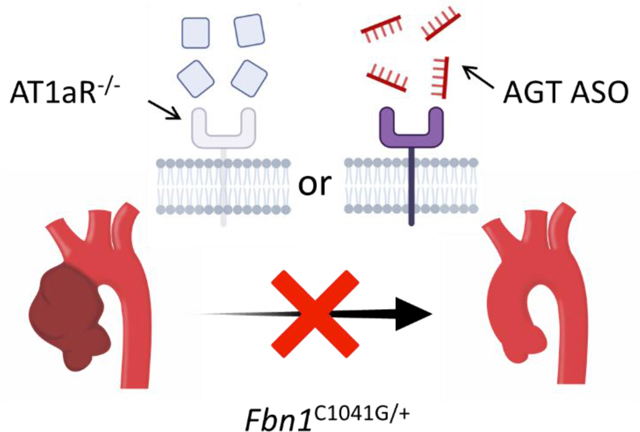
Introduction
Marfan syndrome is an autosomal dominant genetic disorder associated with thoracic aortic aneurysm (TAA) that enhance the risk for aortic rupture due to loss of aortic integrity. 1 The disease is caused by mutations in fibrillin 1 (FBN1 in humans, Fbn1 in mice); a protein incorporated into the microfibrils that decorate elastic fibers. 2 To gain insight into the mechanisms of the disease, mice have been developed with a heterozygous expression of the C1041G mutation of the mouse fibrillin 1 protein, which is analogous to the C1039Y mutation in humans.3 These mice have a haploinsufficiency of Fbn1 and mimic some pathologies present in patients with Marfan syndrome including progressive expansion of the proximal thoracic aorta.
The renin angiotensin system has been invoked as a mediator of TAA in patients with Marfan syndrome.4 Experimental evidence for the role of the renin angiotensin system has been based predominantly on the observation that losartan inhibits aortic pathology in mice. This was demonstrated initially in Fbn1C1041G/+ mice administered losartan starting at the prenatal phase of life.5 Additionally, it has been consistently demonstrated that losartan reduces aortic expansion in many other mouse models of TAA.6–11 However, losartan has many well-characterized effects independent of AT1 receptor antagonism which potentially could compromise interpretation as a pharmacological tool to specifically study AT1 receptors.12 Indeed, the benefit of losartan in inhibiting aortic root dilation in Fbn1C1041G/+ mice has been attributed to effects such as TGF-β antagonism or nitric oxide synthase stimulation.5, 6 To overcome the limitations of pharmacological approaches, there is a critical need to determine the role of AT1aR using genetic deletion to specifically ascribe a function of AT1 receptors in general and AT1aR specifically.
Additionally, it is unclear whether deletion of AT1 receptors has similar effects on TAA compared to depletion of its activating ligands. Previous studies have relied on use of angiotensin converting enzyme inhibitors such as enalapril. These studies demonstrated that enalapril did not affect TAA or dilated cardiomyopathy in Marfan syndrome model mice.8, 13 However, enalapril and other angiotensin converting enzyme inhibitors have multiple effects on the cardiovascular system including modulation of the kinin-kallikrein system, necessitating a study of the effects of angiotensin ligand depletion alone. Indeed, the protective effect of enalapril on hypertension has been attributed to kinin inhibition rather than angiotensin converting enzyme inhibition.14 Therefore, we aimed to define if there was a differential effect of AT1 receptor deletion and specific depletion of angiotensinogen, the sole precursor of angiotensin receptor ligands, on Marfan syndrome associated TAA.
The aim of the present study was to define the contribution of the renin angiotensin system to the progressive expansion of the proximal thoracic aorta in Fbn1C1041G/+ mice. Aortic diameters were measured for a 1-year interval using a standardized ultrasound protocol.15 In accord with current guidelines, the study was performed in both sexes of these mice. These studies demonstrated a strong sexual dimorphism with greater expansion in Fbn1C1041G/+ males and minimal progressive expansion in Fbn1C1041G/+ females. The role of AT1aR was determined subsequently using male mice with global AT1aR deletion. The role of angiotensin ligands was determined using an angiotensinogen antisense oligonucleotide (AGT ASO) that causes a protracted depletion of the unique precursor of AngII. Finally, transcriptome analyses were performed using ascending aortas to understand potential mechanisms underlying the attenuation of TAA by AGT ASO in Fbn1C1041G/+ mice.
Methods
Tabulated data, including aortic measurements by ultrasonography and read counts of RNA sequencing, are available in Supplemental Excel File I-V.
Mice
Studies were performed in accordance with recommendations for design and reporting of animal aortopathy studies.16, 17 Studies were performed using littermate controls. Mice and genealogy were tracked with Mosaic Vivarium Laboratory Animal Management Software (Virtual Chemistry). Male and female AT1aR deleted (AT1aR-/-) (stock #002682) and Fbn1C1041G/+ (stock #012885) mice were obtained from The Jackson Laboratory. Male AT1aR heterozygous (AT1aR+/−) x Fbn1C1041G/+ were bred with female AT1aR+/− x fibrillin-1 wild type (Fbn1+/+) mice to generate four experimental groups per sex: male and female AT1aR wild type x Fbn1+/+ (WT), AT1aR-/- x Fbn1+/+ (AT1aR-/-), AT1aR+/+ x Fbn1C1041G/+ (Fbn1C1041G/+), and AT1aR-/- x Fbn1C1041G/+ mice. Littermates were separated by sex and genotypes and were randomized when housing mice after weaning. For AGT ASO experiments, 2-month-old male Fbn1C1041G/+ mice were procured from The Jackson Laboratory and randomized into experimental groups using a random number generator. Mice were checked daily for health, and necropsy was performed to adjudicate cause of death. Mice were housed up to 5 per cage and maintained on a 14:10 hour light:dark cycle. Mice were fed Teklad Irradiated Global 18% Protein Rodent Diet # 2918 ad libitum and allowed ad libitum access to water via a Lixit system. Bedding was provided by P.J. Murphy (Coarse SaniChip) and changed weekly during the study. Cotton pads were provided as enrichment. The room temperature was maintained at 21°C and room humidity was maintained at 50%. All protocols were approved by University of Kentucky IACUC.
Genotyping
Mice were genotyped twice using tail tissue. Group allocation was based on genotyping performed after weaning at postnatal day 28. The genotype of mice was confirmed using tissue acquired at the termination of each study. AT1aR deletion was assayed using forward primer (5’-AAATGGCCCTTAACTCTTCTACTG-3’) and reverse primer (5’-ATTAGGAAAGGGAAC AGGAAGC-3’) covering a neo cassette that disrupts AT1aR spanning bps 110–635. The neo cassette removed approximately 0.5 kb and inserted approximately 1 kb of neo gene. AT1aR+/+ generated a 631 bp product. AT1aR−/− generated a ~1.1 kbp product. Fbn1C1041G/+ was assayed using forward primer (5’-CTCATCATTTTTGGCCAGTTG-3’) and reverse primer (5’-GCACTTGATGCACATTCA CA-3’) covering a single loxP intronic sequence within intron 24 which is not present in wild type mice. The protocol used was as described by The Jackson Laboratory. Fbn1+/+ generates a 164 bp product. Fbn1C1041G/+ generates a 212 bp product. Post-termination validation genotyping was performed by Transnetyx.
Antisense Oligonucleotides
Scrambled control ASO (5’-GGCTACTACGCCGTCA-3’) and AGT ASO (5’-ATCATTTATTCTCGGT-3’) were provided by Ionis Pharmaceuticals. ASOs were 3–10-3 2’ – 4’ constrained ethyl gapmers that have demonstrated improved potency and tolerability versus locked nucleic acid or 2’-O-methoxyethyl gapmers.18 Mice were randomized to study group using a random number generator. Two-month-old male Fbn1C1041G/+ mice were administered control ASO or AGT ASO (80 mg/kg) subcutaneously at day 1 and 3 of study. Mice were maintained on subcutaneous control ASO or AGT ASO (40 mg/kg) every 7 days for the remainder of the study.
Ultrasound Measurements
Ultrasonography was performed by standardized protocols as described previously.15, 19 Briefly, mice were anesthetized using inhaled isoflurane (2–3% vol/vol) and maintained at a heart rate of 450–550 beats per minute during image capture to reduce anesthesia exposure and maintain consistent heart rate between animals (Somnosuite, Kent Scientific). The order by which mice were subject to ultrasound was randomized. Ultrasound images were captured in the right parasternal view using a Vevo 3100 system with a 40 MHz transducer (Visualsonics). Images captured were standardized according to two anatomical landmarks: the innominate artery branch point and aortic valves. The largest luminal ascending aortic diameter between the sinotubular junction and the innominate artery were measured in end-diastole over three cardiac cycles by two independent observers.
Measurement of in situ Aortic Diameters
Mice were terminated by overdose of ketamine:xylazine followed by cardiac puncture and saline perfusion. The order in which mice were terminated was randomized. Aortas were dissected away from surrounding tissue and Optimal Cutting Temperature Compound (Sakura Finetek) was introduced into the left ventricle to maintain aortic patency.20 A black plastic sheet was inserted beneath the aorta and heart to increase contrast and facilitate visualization of aortic borders. Aortas were imaged using a Nikon SMZ800 stereoscope and measurements were recorded using NIS-Elements AR 4.51 software (Nikon Instruments Inc.). Ascending aortic diameters were measured at the largest width perpendicular to the vessel.
Histology
Mice were ranked according to their ascending aortic diameter by ultrasound, and the median five per group were selected for histology. Tissue sections (10 μm) were acquired from the aortic root to the aortic arch at 100 μm intervals using a cryostat. The section corresponding to a region of maximal dilation between the sinotubular junction and the arch was analyzed. Elastin fragmentation was visualized by Verhoeff elastin staining under 20x magnification and images from three high powered fields (40x) per section were recorded for analysis. Individual data were represented as the mean of three high power fields. Fragmentation was defined as the presence of discernable breaks of continuous elastic lamina. Medial thickness was measured at the greatest thickness from inner to external elastic laminae in 3 images using NIS-Elements AR software. Measurements were verified by an independent investigator who was blinded to sample identification. More detailed descriptions of these protocols are available on protocol.io dx.doi.org/10.17504/protocols.io.be9mjh46 and dx.doi.org/10.17504/protocols.io.be9sjh6e
AGT Western Blotting
Reducing buffer (Bio-Rad 161–0737 and Sigma M7522) and plasma (0.3 μL) from mice administered control or AGT ASO were heated to 95°C for 5 minutes. Samples were resolved by SDS-PAGE (10% wt/vol; Bio-Rad 456–8033). Proteins were transferred to PVDF membranes via a Trans-blot system (Bio-Rad 170–4256). Total proteins were detected by Ponceau S. Membranes were blocked by a solution of powdered milk (5% wt/vol; Bio-Rad 170–6404) in TBS-T (0.1% wt/vol). Membranes were then incubated with antibodies against AGT (0.1 μg/mL; IBL 28101) for 1 hour at room temperature then with an HRP-conjugated goat-antirabbit IgG (0.2 μg/mL; Vector Pi-1000). Membranes were incubated with Clarity Max ECL (Bio-Rad 1705064) and signals were visualized on a ChemiDoc MP system. Blots were quantified using Bio-Rad CFX software.
Aortic mRNA Sequencing and Transcriptome Analyses
Ascending aortas were harvested from male wild type or Fbn1C1041G/+ mice at 7 to 12 weeks of age. Additional male Fbn1C1041G/+ mice were administered control or AGT ASO for 2 weeks. After euthanasia using a ketamine:xylazine cocktail, the thoracic cavity was cut open and saline (8 mL) was perfused via the left ventricle. After removing periaortic tissues, ascending aortas were harvested and endothelial cells were removed using a cotton swab. Four aortic samples were pooled in a tube with RNAlater solution (#AM7020, Invitrogen). Subsequently, RNA was extracted by a RNeasy Fibrous Tissue Mini Kit (#74704, Qiagen). mRNA sequencing was performed with a blinded mode of identification by Novogene according to their contract research organization protocols. Briefly, a library was generated from total mRNA which was subsequently sequenced on Illumina platform. Quality control including validation via error rate distribution, G-C content distribution, and data filtering were performed. Alignment and read assignment were performed to allow transcript quantification and differential gene expression analysis.
Sequencing libraries were generated using a NEGNext Ultra™ RNA Library Prep Kit for Illumina (NEB) from mRNA samples. cDNA libraries were then sequenced by a Next Generation sequencer, NovaSeq 6000 (Illumina), in a pair-end fashion to reach more than 1,500,000 reads. FASTQ sequence data were mapped to mouse genome mm10 using STAR (v2.6.1d, mismatch=2).
Statistics
Statistical analyses were performed using R (ver 3.6.2) and Sigmaplot (ver 14.0). For transcriptome data, edgeR package (ver 3.28.1) was used on R. Visualizations were made with BioRender and Adobe Illustrator. Equal variance and normality of data determined whether non-linear, logarithmic transformation was performed and whether parametric or non-parametric tests were used. A linear mixed effect model with a random intercept and slopes was used to compare data obtained sequentially. For data at a single timepoint, Two-way analysis of variance (ANOVA) or Student’s t-test was performed for parametric comparisons; Holm-Sidak was used for post-hoc tests. Kruskal-Wallis or Rank Sum was performed for non-parametric comparisons with Dunn’s method for post hoc tests. Data are represented as individual data points, mean ± standard error of mean (SEM), or median and interquartile range. P<0.05 was considered as significant. Precise P values are described in Supplemental Excel File I.
Results
Progression of Aortic Dimensions was Sexually Dimorphic in Fbn1C1041G/+ Mice
In initial studies, the progression of aortic diameters over a 12-month interval was determined in both male and female wild type and Fbn1C1041G/+ mice. Because TAA in Fbn1C1041G/+ mice had variable manifestations within the proximal thoracic aorta (Supplemental Figure I), several parameters were measured. These included the ascending aortic diameter, aortic root diameter, and ascending aortic length.
In wild type mice, there was no statistical difference in aortic root diameters ascending aorta diameters, and ascending aortic length between female and male through 1 to 12 months of age (Figure 1A-C). Compared to male wild type mice, male Fbn1C1041G/+ mice exhibited significantly increased luminal expansions in aortic roots, ascending aortas, and ascending aortic lengths over 12 months. In contrast, female Fbn1C1041G/+ mice did not demonstrate significant luminal dilatations in aortic roots, ascending aortas, and ascending aortic lengths over 12 months (Figure 1A-C). Since female Fbn1C1041G/+ mice had no significant differences in the progression of aortic dimensions compared to their wild type littermates, most subsequent experiments only used male mice.
Figure 1: TAA in Fbn1C1041G/+ mice is sexually dimorphic.

Sequential ultrasound measurements of the A, ascending aorta, B, aortic root, and C, aortic length in diastole from 1 month to 12 months of age of male and female wild type (WT) and Fbn1C1041G/+ mice.
Aortic parameters were compared using a linear mixed effect model with a random intercept and slopes. * p<0.05, *** p<0.001 Male wild type vs Fbn1C1041G/+ mice. † p<0.05, †† p<0.01, male Fbn1C1041G/+ vs female Fbn1C1041G/+ mice (n=9–15/group).
AT1aR Deletion Attenuated Aortic Dilation in Male Fbn1C1041G/+ Mice
To study the effects of AT1aR on aortic dilation in Fbn1C1041G/+ mice, Fbn1C1041G/+ mice that were either AT1aR+/+ or AT1aR−/− were generated. Fbn1C1041G/+ mice were also compared against Fbn1+/+ mice that were also either AT1aR+/+ or AT1aR-/−. Aortic dimensions were measured using ultrasound images acquired from a right parasternal view at diastole (Figure 2A). Images were acquired from every mouse at the stated intervals up to 12 months of age, with no deaths of any cause occurring during the study.
Figure 2: AT1aR deletion attenuated ascending aortic dilation in male Fbn1C1041G/+ mice.

A, Representative ultrasound images of the thoracic aorta in male wild type, AT1aR-/-, Fbn1C1041G/+, and AT1aR-/- x Fbn1C1041G/+ mice. Green bar=1 mm. Sequential ultrasound measurements of the B, ascending aorta C, aortic root and D, aortic length. ** p<0.01, *** p<0.001 of male wild type vs Fbn1C1041G/+ mice; †† p<0.01, ††† p<0.001 of male Fbn1C1041G/+ vs AT1aR-/- x Fbn1C1041G/+ mice. n=11–15/group. E) Representative in situ images of the thoracic aorta. F) Measurement of in situ aortic dimensions taken at the maximal aortic diameter. ** p<0.01; *** p<0.001. (n=10–15/group).
Male wild type mice had modest increases in diameters of the ascending aorta (Figure 2B), aortic root (Figure 2C), and lengths of the ascending aorta (Figure 2D) during the 12 months of observation. These increases were not significantly different from increases in AT1aR−/− mice. Meanwhile, Fbn1C1041G/+ mice exhibited significant dilatations in ascending and root diameters and aortic lengths (Figure 2B-D), as expected. It is of note that systemic AT1aR deletion attenuated the dilatation of ascending aortic diameters and aortic lengths in Fbn1C1041G/+ mice. Direct measurements of the maximal aortic diameter on in situ aortas also demonstrated a significant attenuation of aortic dilatation in AT1aR−/− x Fbn1C1041G/+ mice (Figure 2E, F).
At 1 month of age, male Fbn1C1041G/+ mice had increased diameters of ascending aorta, aortic root, and ascending length compared to wild type mice. At this early age, deletion of AT1aR had no effect on aortic dimensions (Supplemental Figure IIA, C, E). Consistent with previous publications,21 body weight and systolic blood pressure were not correlated with ascending aortic dimensions in mice (Supplemental Figure IIIA-C).
Female wild type and Fbn1C1041G/+ mice that were either AT1aR +/+ or −/− were generated, and aortic dimensions were measured from 1 to 12 months of age. As noted above, beyond the initial differences at 1 month of age, progressive changes in aortic dimensions were not different between wild type and Fbn1C1041G/+ female mice. Aortic dimensions were not different between female wild type and AT1aR−/− or between female Fbn1C1041G/+ and AT1aR−/− x Fbn1C1041G/+ mice (Supplemental Figure IVA-C).
AT1aR Deletion Attenuated Aortic Medial Pathology in Male Fbn1C1041G/+ Mice
To determine if AT1aR deletion impacted the structure of the aortic media, histological characteristics were determined in aortic tissues acquired at 12 months of age. Since the most dramatic differences in changes of dimensions described above were in the ascending aorta, this region was selected for tissue characterization using our validated and reproducible method (Supplemental Figure VA, B). Ascending aortic tissues from wild type mice had elastic fibers with minimal fragmentation (Figure 3A). Neither the extent of fragmentation nor medial thickness were altered by the absence of AT1aR in wild type mice (Figure 3B, C). In contrast, Fbn1C1041G/+ x AT1aR+/+ mice had extensive fragmentation of elastic fibers and marked medial thickening. Deletion of AT1aR in these mice significantly reduced elastin fragmentation and medial thickening.
Figure 3: AT1aR deletion attenuated medial remodeling in male Fbn1C1041G/+ mice.

A, Representative images of Verhoeff’s elastin staining in ascending aortic sections from male WT, AT1aR-/-, Fbn1C1041G/+, and AT1aR-/- x Fbn1C1041G/+ mice. Green bar=100 μm. B, Number of breaks per high powered field detected in aortic sections. C, Medial thickness as measured by the distance between the inner elastic lamina and external elastic lamina in aortic sections. * p<0.05, *** p<0.001. (n=5/group).
Depletion of Plasma AGT Concentrations by AGT ASO Attenuated Aortic Pathology in Male Fbn1C1041G/+ Mice
We have demonstrated previously that administration of AGT ASO markedly reduces plasma concentration of AGT and attenuates AngII responses in mice.22, 23 Using ASO against the same target as previous publications, male Fbn1C1041G/+ mice received a loading dose (80 mg/kg) of either AGT or control ASO on day 1 and day 4 of the study. Starting on day 7, mice received a maintenance dose (40 mg/kg) every 7 days for 6 months. (Figure 4A). Mice tolerated the ASO well and displayed minimal toxicity after administration of loading doses (Supplemental Figure VI) AGT ASO effectively depleted AGT in plasma (Figure 4B, Supplemental Figure VII).
Figure 4: AGT ASOs depleted AGT and attenuated TAA in male Fbn1C1041G/+ mice.

A, Study design and administration schedule of ASOs in male Fbn1C1041G/+ mice. A loading dose of control ASO or AGT ASO (80 mg/kg) was administered day 1 and 4 of study. Maintenance doses of control ASO or AGT ASO (40 mg/kg) was administered every 7 days. B, Western blot of plasma AGT and total plasma protein in 8-month-old male Fbn1C1041G/+ mice administered either control ASO or AGT ASOs. Blot represents one of two experiments. *** p<0.001 (n=6/group). Representative C, ultrasound and D, in situ images of aortas from 8-month-old Fbn1C1041G/+ mice administered either control ASO or AGT ASO. Green bar=1 mm. Sequential ultrasound measurements of the E, ascending aorta, F, aortic root, and G, aortic length in diastole from 2 months to 8 months of age in male Fbn1C1041G/+ mice dosed with either control ASO or AGT ASO.** p<0.01, *** p<0.001. (n=8–10/group).
Aortic dimensions were acquired starting at 2 months of age, and every month for a further 6 months using the same process described above (Figure 4C) with in situ aortic measurements at termination confirming the ultrasound measurement. (Figure 4D). Although aortic root diameters were not different significantly (Figure 4F), AGT depletion achieved by the ASO administration led to statistically significant difference of diameters of ascending aorta diameter (Figure 4E) and length of ascending aorta (Figure 4G) in male Fbn1C1041G/+ mice after 6 months of ASO administration.
To determine whether AGT ASO impacted aortic medial structure, histology was performed on ascending aortic tissue. Consistent with our previous observation, we detected aortic medial remodeling in 8-month-old male Fbn1C1041G/+ mice administered control ASO (Figure 5A). Compared to male Fbn1C1041G/+ mice administered control ASO, male Fbn1C1041G/+ mice administered AGT ASO exhibited less elastin fragmentation and medial thickening (Figure 5B, C).
Figure 5: AGT ASOs attenuated medial remodeling in male Fbn1C1041G/+ mice.

A, Representative images of Verhoeff’s elastin staining in aortic sections from male Fbn1C1041G/+ mice administered either control ASO or AGT ASO for 6 months. B, Number of breaks per high powered field detected in aortic sections. C, Medial thickness as measured by the distance between the inner elastic lamina and external elastic lamina in aortic sections. * p<0.05, *** p<0.001. (n=5/group). Green bar=100 μm.
Aortic Transcriptomic Changes in Male Fbn1C1041G/+ Mice Are Reversed by Inhibition of AGT Synthesis
To explore potential mechanisms of aortic pathology in male mice, transcriptomic alterations were determined in the ascending aorta of Fbn1C1041G mice. RNA sequencing revealed that there were 490 differentially expressed genes in Fbn1C1041G/+ mice compared to wild type controls (Figure 6A). It is of note that, none of the major components of the renin angiotensin system were differentially altered in ascending aortas of Fbn1C1041G/+ mice (Figure 6B).
Figure 6. Impact of AGT ASO on transcriptomic alteration of the ascending aorta in Fbn1C1041G/+ mice.

A, Volcano plot of the transcriptomes and B, box plots for mRNAs related to the renin angiotensin system in wild type and Fbn1C1041G/+ mice (n=4/group). C, Volcano plot in Fbn1C1041G/+ mice with either control ASO (Control) or AGT-ASO (ASO) injection. n=5/group. D, Heatmap for mRNAs associated with smooth muscle contraction, collagens, metalloproteinases, and inflammation in Fbn1C1041G/+ mice administered control ASO vs AGT ASO. E, Venn’s diagram of the number of differentially expressed genes in each RNAseq. F, Heatmap for mRNAs showing opposite alterations in RNAseq #1 and #2. Color scale indicates Log2 fold changes in each comparison; RNAseq #1 - wild type vs Fbn1C1041G/+ and RNAseq #2 - control vs AGT-ASO in Fbn1C1041G/+ mice.
Next, we determined the effects of depleting AngII through inhibition of AGT synthesis on the aortic mRNA transcriptome in the ascending aorta of male Fbn1C1041G/+ mice. AGT ASO was administered in Fbn1C1041G/+ mice prior to developing profound disease. Differential gene analysis demonstrated a total of 145 genes (Figure 6C). mRNA transcripts associated with proliferation were not significantly altered. (Figure 6D) However, several genes related to smooth muscle cells were decreased by AGT ASO. AGT ASO also downregulated multiple genes related to contraction, collagen, metalloproteinases, and inflammation (Figure 6D) in male Fbn1C1041G/+ mice.
Since the two RNAseq studies identified multiple genes as targets, we further compared the RNAseq datasets to identify possible targets responsible for aortic dilatation in Fbn1C1041G/+ mice. In comparing differentially expressed genes between the first and second RNAseq datasets, 19 genes were common (Figure 6E). Of interest, 16 genes demonstrated opposite responses in the two datasets (Figure 6F). For instance, Igfbp2, Acan, and Rgs17 were highly abundant in Fbn1C1041G/+ compared to wild type mice, whereas it was decreased by AGT ASO. In contrast, Ppp1r1b, Adra1d, and Gm830 were less abundant in Fbn1C1041G/+ mice and AGT ASO increased their abundance. These results indicate that these 16 genes may contribute to the pathophysiology of TAA in Fbn1C1041G/+ mice and contribute to the protective effects of AGT ASO.
Discussion
There have been consistent demonstrations that losartan attenuates aortic pathology in mice with Fbn1 manipulations.5–8, 24–26 However, it has been proposed that losartan may exert these beneficial actions independent of AT1 receptor antagonism.6–8 Additionally, it has been suggested that AngII may not be responsible for cardiovascular pathology in mice with genetically manipulated Fbn1.13 The present study demonstrates that both genetic deletion of AT1aR and techniques to reduce AngII availability led to reduced aortic pathology in male Fbn1C1041G/+ mice. The protective effects of renin angiotensin system inhibition in Marfan syndrome model mice occur by modulating ascending aortic dilation, attenuating medial remodeling, and affecting the aortic transcriptome.
Sexual dimorphism of TAA formation in Marfan syndrome has been reported in humans and mice.27–29 Consistent with previous reports, the present study demonstrated sexual dimorphism of TAA in Fbn1C1041G/+ mice. Although Marfan syndrome is an autosomal dominant disease, retrospective cross-sectional studies demonstrated that male patients develop more severe aortic dilation compared to female patients.27 A previous study reported that androgen accelerates TAA formation with increase of TGF-β signaling in male Fbn1C1041G/+ mice.29 However, it is still unclear how the renin angiotensin system interacts with TAA formation through androgen. Further research is needed to explore mechanisms responsible for sexual dimorphism of TAA in Marfan syndrome.
A previous study have noted that AT1aR deficiency had no effect on expansion of the aortic root at 3 and 6 months of age in Fbn1C1041G/+ mice, whereas losartan had a divergent effect and decreased aortic root expansion in these mice.6 The beneficial effects of losartan were attributed to preservation of endothelial function in an AT1aR independent manner through an alternative VEGFR2/eNOS pathway. The basis for the disparity relative to the present study is not clear, and comparisons are hampered by the paucity of data on the protocol for ultrasound acquisition and on the sex of the mice in each group. However, the present study offers better temporal resolution compared to previous studies of AT1aR deleted Fbn1C1041G/+ mice through sequential measurement of the aorta. The sequential measurement of aortic dimensions over a protracted interval in multiple groups required development of a standardized ultrasound protocol for image acquisition. We have noted previously the variance imparted by the differences acquiring dimension at systole or diastole.15 Given that this excursion can be as much as 0.2 mm, lack of consistency in acquiring data could have a profound effect on data interpretation. The approach used in this study also consistently imaged the aorta from the right parasternal view.30 While this view is optimal for determining dimensions of the ascending aorta, it reduces precision of aortic root measurements. In the present study, there was strenuous adherence to a standardized protocol. This degree of measurement validation enabled acquisition of reliable data while reducing variability between sequential measurements. Use of more robust measurement protocols alongside sequential measurement of the ascending aorta serves to increase discriminatory power of ultrasound measurements and may explain these discordant observations.
While the results of the present study clearly demonstrate the attenuation of aortic pathology in AT1aR deleted Fbn1C1041G/+, it is unclear which cell type is responsible for these protective effects. A previous study by Galatioto and colleagues compared the impact of AT1aR deletion between endothelial and smooth muscle cells on aortic dilatation in Fbn1mgR/mgR mice.31 Endothelial cell-specific deletion of AT1aR exhibited modest suppression of aortic dilatations, whereas smooth muscle cell-specific AT1aR deletion did not attenuate development of TAA in Fbn1mgR/mgR mice. Cellular specificity can be observed in the AngII infusion model of TAA.32 In this model, smooth muscle cell-specific AT1aR deletion had no effect on TAA, but AT1aR deletion in endothelial cells showed a partial reduction in Ang II-induced TAA formation. However, the effects of cell specific deletion of AT1aR have only represented a small component of the reduction, compared to mice with global AT1R deletion. Further study is needed to identify the responsible cell type exerting the dominant role in TAA attenuation by AT1aR inhibition.
We used an ASO to decrease the synthesis of the unique precursor of all angiotensin peptides to determine whether AT1aR stimulation in aortopathies required AngII as a ligand. This approach is advantageous over the more common mode of reducing AngII production through inhibiting angiotensin-converting enzyme, which regulates other pathways including the kinin-kallikrein system. Additionally, the protracted half-life of ASO leads to persistent inhibition of AGT synthesis and profound reductions in plasma AGT concentrations. Use of this pharmacologic modality also avoids adverse consequences of genetic deletion of the renin angiotensin system components. Previous genetic approaches have included the use of mice with global deficiencies of AGT. However, these mice have several major developmental abnormalities including poor growth and cardiomyopathy.33 Inhibition of AGT synthesis by an ASO reduces plasma concentrations by approximately 90% in the postnatal phase with no observable toxicity as demonstrated in the present study and other reports.22, 34 Therefore, the use of ASO to deplete AGT demonstrated the need for the presence of angiotensin ligands to augment aortic pathology in Fbn1C1041G/+ mice.
Randomized control trials of angiotensin receptor blockers have yielded mixed results in Marfan syndrome associated TAA, in contrast to the consistent results that have been generated using mouse models of the disease.5–8, 24–26 Most of the mouse and human studies have been performed using losartan, which is characterized by a relatively short half-life and surmountable antagonism. The deficiencies of this drug were likely to have been ameliorated in mouse studies by consistent delivery, via osmotic pumps and diet, leading to a persistent inhibition. AT1 receptor antagonists with enhanced pharmacological profiles, such as irbesartan and candesartan, would be preferable to test the role of AT1 receptor inhibition in humans. Indeed, it has been demonstrated recently that irbesartan significantly attenuated aortic root expansion in individuals with Marfan syndrome.35 Conversely, ASO affords chronic and persistent inhibition of AGT synthesis to effect long-term depletion of angiotensin ligands. These durable effects of ASO enables inhibition of AGT synthesis to be tested as a possible approach to reduce TAA in Marfan syndrome.
There is compelling evidence that pharmacological inhibition of AT1aR using losartan attenuates TAA formation in Fbn1C1041G/+ mice. 5, 6, 8 The present study reported protective effects of systemic AT1aR deletion and inhibition of AGT synthesis in Fbn1C1041G/+ mice. However, the molecular mechanism by which the renin angiotensin system contributes to development of TAA in Marfan syndrome remains unclear. In fact, our transcriptomic analysis revealed that abundance of the renin angiotensin system components was not altered in the ascending aorta of Fbn1C1041G/+ mice. Although there was no evidence of local activation of the renin angiotensin system, this RNA sequencing analyses identified multiple genes as targets. Several molecules in these genes have physiological or pathophysiological functions in the cardiovascular system. Aggrecan is a proteoglycan which accumulates in TAA and aortic dissection.36 Connective tissue growth factor is implicated in development of abdominal aortic aneurysms.37 Igfbp2, known to interact with matrix metalloprotease 2 and exert a role in angiogenesis, was identified previously via a transcriptome analysis using aortas from Fbn1mgR/mgR mice.38 Optc is a protein found to stabilize collagen in bone cartilage.39 Ppp1r1b contributes to calcium signaling in cardiac muscle cells.40 It is important to elucidate the precise role of these molecules in the pathophysiology of TAA and would be interesting to investigate the interaction of these molecules to the renin angiotensin system in Fbn1C1041G/+ mice.
In conclusion, our study provided strong evidence that both AT1aR deletion and AGT depletion resulted in significant attenuation of ascending aortic dilation in male Fbn1C1041G/+ mice. These data are consistent with both AT1aR and angiotensin receptor ligands being necessary for TAA progression in male Fbn1C1041G/+ mice and that profound and persistent depletion of either component is sufficient to attenuate TAA.
Supplementary Material
{kind=link}
Highlights.
Profound sexual dimorphism of aortic disease was observed in Fbn1C1041G/+ mice, with female mice being more resistant and male mice being more susceptible.
Inhibition of the AngII-AT1aR axis attenuated aortic pathology in male Fbn1C1041G/+ mice.
Antisense oligonucleotides targeting angiotensinogen diminished plasma angiotensinogen and attenuated thoracic aortic aneurysms.
Transcriptome analyses identified several genes associated with attenuation of aortic dilatation by renin angiotensin system inhibition in Fbn1C1041G/+ mice.
Acknowledgments
We thank the University of Kentucky – Baylor College of Medicine Aortic Research Center investigators for their input in this manuscript.
Sources of Funding
The authors’ research work is supported by the American Heart Association SFRN in Vascular Disease (18SFRN33960163) and the NIH NHLBI under award numbers R01HL133723. JC and MS have been supported by NCATS UL1TR001998 and JC has been supported by NHLBI F30143943. HS was supported by an AHA postdoctoral fellowship (18POST33990468). The content in this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the American Heart Association or the National Institutes of Health.
Non-standard abbreviations:
- TAA
thoracic aortic aneurysm
- AT1aR
angiotensin II receptor type 1a
- Fbn1
fibrillin 1
- AGT
angiotensinogen
- AngII
angiotensin II
- ASO
antisense oligonucleotide
Footnotes
Disclosures
MS, AD, HL, and JC have submitted a patent application for use of antisense oligonucleotides targeted against angiotensinogen in thoracic aneurysmal disease. AM is an employee of Ionis Pharmaceuticals, who provided the control ASO and AGT ASO. Ionis Pharmaceuticals did not provide funding for this study.
References
- 1.Milewicz DM, Dietz HC, Miller DC. Treatment of aortic disease in patients with marfan syndrome. Circulation. 2005;111:e150–157 [DOI] [PubMed] [Google Scholar]
- 2.Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, Puffenberger EG, Hamosh A, Nanthakumar EJ, Curristin SM, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–339 [DOI] [PubMed] [Google Scholar]
- 3.Judge DP, Biery NJ, Keene DR, Geubtner J, Myers L, Huso DL, Sakai LY, Dietz HC. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of marfan syndrome. J Clin Invest. 2004;114:172–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz HC, 3rd. Angiotensin ii blockade and aortic-root dilation in marfan’s syndrome. N Engl J Med. 2008;358:2787–2795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C, Podowski M, Neptune ER, Halushka MK, Bedja D, Gabrielson K, Rifkin DB, Carta L, Ramirez F, Huso DL, Dietz HC. Losartan, an at1 antagonist, prevents aortic aneurysm in a mouse model of marfan syndrome. Science. 2006;312:117–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sellers SL, Milad N, Chan R, Mielnik M, Jermilova U, Huang PL, de Crom R, Hirota JA, Hogg JC, Sandor GG, Van Breemen C, Esfandiarei M, Seidman MA, Bernatchez P. Inhibition of marfan syndrome aortic root dilation by losartan: Role of angiotensin ii receptor type 1-independent activation of endothelial function. Am J Pathol. 2018;188:574–585 [DOI] [PubMed] [Google Scholar]
- 7.Cook JR, Clayton NP, Carta L, Galatioto J, Chiu E, Smaldone S, Nelson CA, Cheng SH, Wentworth BM, Ramirez F. Dimorphic effects of transforming growth factor-beta signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for marfan syndrome. Arterioscler Thromb Vasc Biol. 2015;35:911–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Habashi JP, Doyle JJ, Holm TM, Aziz H, Schoenhoff F, Bedja D, Chen Y, Modiri AN, Judge DP, Dietz HC. Angiotensin ii type 2 receptor signaling attenuates aortic aneurysm in mice through erk antagonism. Science. 2011;332:361–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuang SQ, Geng L, Prakash SK, Cao JM, Guo S, Villamizar C, Kwartler CS, Peters AM, Brasier AR, Milewicz DM. Aortic remodeling after transverse aortic constriction in mice is attenuated with at1 receptor blockade. Arterioscler Thromb Vasc Biol. 2013;33:2172–2179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gallo EM, Loch DC, Habashi JP, Calderon JF, Chen Y, Bedja D, van Erp C, Gerber EE, Parker SJ, Sauls K, Judge DP, Cooke SK, Lindsay ME, Rouf R, Myers L, ap Rhys CM, Kent KC, Norris RA, Huso DL, Dietz HC. Angiotensin ii-dependent tgf-beta signaling contributes to loeys-dietz syndrome vascular pathogenesis. J Clin Invest. 2014;124:448–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramnath NW, Hawinkels LJ, van Heijningen PM, te Riet L, Paauwe M, Vermeij M, Danser AH, Kanaar R, ten Dijke P, Essers J. Fibulin-4 deficiency increases tgf-beta signalling in aortic smooth muscle cells due to elevated tgf-beta2 levels. Sci Rep. 2015;5:16872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sadoshima J. Novel at(1) receptor-independent functions of losartan. Circ Res. 2002;90:754–756 [DOI] [PubMed] [Google Scholar]
- 13.Cook JR, Carta L, Benard L, Chemaly ER, Chiu E, Rao SK, Hampton TG, Yurchenco P, Gen TACRC, Costa KD, Hajjar RJ, Ramirez F. Abnormal muscle mechanosignaling triggers cardiomyopathy in mice with marfan syndrome. J Clin Invest. 2014;124:1329–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elmarakby AA, Morsing P, Pollock DM. Enalapril attenuates endothelin-1-induced hypertension via increased kinin survival. Am J Physiol Heart Circ Physiol. 2003;284:H1899–1903 [DOI] [PubMed] [Google Scholar]
- 15.Chen JZ, Sawada H, Moorleghen JJ, Weiland M, Daugherty A, Sheppard MB. Aortic strain correlates with elastin fragmentation in fibrillin-1 hypomorphic mice. Circ Rep. 2019;1:199–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robinet P, Milewicz DM, Cassis LA, Leeper NJ, Lu HS, Smith JD. Consideration of sex differences in design and reporting of experimental arterial pathology studies-statement from atvb council. Arterioscler Thromb Vasc Biol. 2018;38:292–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daugherty A, Tall AR, Daemen M, Falk E, Fisher EA, Garcia-Cardena G, Lusis AJ, Owens APr, Rosenfeld ME, Virmani R. Recommendation on design, execution, and reporting of animal atherosclerosis studies: A scientific statement from the american heart association. Arterioscler Thromb Vasc Biol. 2017;37:e131–e157 [DOI] [PubMed] [Google Scholar]
- 18.Seth PP, Siwkowski A, Allerson CR, Vasquez G, Lee S, Prakash TP, Wancewicz EV, Witchell D, Swayze EE. Short antisense oligonucleotides with novel 2’−4’ conformationaly restricted nucleoside analogues show improved potency without increased toxicity in animals. J Med Chem. 2009;52:10–13 [DOI] [PubMed] [Google Scholar]
- 19.Sawada H, Chen JZ, Wright BC, Sheppard MB, Lu HS, Daugherty A. Heterogeneity of aortic smooth muscle cells: A determinant for regional characteristics of thoracic aortic aneurysms? J Transl Int Med. 2018;6:93–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohno-Urabe S, Kukida M, Franklin MK, Katsumata Y, Su W, Gong MC, Lu HS, Daugherty A, Sawada H. Authentication of in situ measurements for thoracic aortic aneurysms in mice. Arterioscler Thromb Vasc Biol. 2021;41:2117–2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rateri DL, Davis FM, Balakrishnan A, Howatt DA, Moorleghen JJ, O’Connor WN, Charnigo R, Cassis LA, Daugherty A. Angiotensin ii induces region-specific medial disruption during evolution of ascending aortic aneurysms. Am J Pathol. 2014;184:2586–2595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu H, Wu C, Howatt DA, Balakrishnan A, Moorleghen JJ, Chen X, Zhao M, Graham MJ, Mullick AE, Crooke RM, Feldman DL, Cassis LA, Vander Kooi CW, Daugherty A. Angiotensinogen exerts effects independent of angiotensin ii. Arterioscler Thromb Vasc Biol. 2016;36:256–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu CH, Wang Y, Ma M, Mullick AE, Crooke RM, Graham MJ, Daugherty A, Lu HS. Antisense oligonucleotides targeting angiotensinogen: Insights from animal studies. Biosci Rep. 2019;39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhatt AB, Buck JS, Zuflacht JP, Milian J, Kadivar S, Gauvreau K, Singh MN, Creager MA. Distinct effects of losartan and atenolol on vascular stiffness in marfan syndrome. Vasc Med. 2015;20:317–325 [DOI] [PubMed] [Google Scholar]
- 25.Yang HH, Kim JM, Chum E, van Breemen C, Chung AW. Long-term effects of losartan on structure and function of the thoracic aorta in a mouse model of marfan syndrome. Br J Pharmacol. 2009;158:1503–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hibender S, Franken R, van Roomen C, Ter Braake A, van der Made I, Schermer EE, Gunst Q, van den Hoff MJ, Lutgens E, Pinto YM, Groenink M, Zwinderman AH, Mulder BJ, de Vries CJ, de Waard V. Resveratrol inhibits aortic root dilatation in the fbn1c1039g/+ marfan mouse model. Arterioscler Thromb Vasc Biol. 2016;36:1618–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roman MJ, Devereux RB, Preiss LR, Asch FM, Eagle KA, Holmes KW, LeMaire SA, Maslen CL, Milewicz DM, Morris SA, Prakash SK, Pyeritz RE, Ravekes WJ, Shohet RV, Song HK, Weinsaft JW, Gen TACI. Associations of age and sex with marfan phenotype: The national heart, lung, and blood institute gentac (genetically triggered thoracic aortic aneurysms and cardiovascular conditions) registry. Circ Cardiovasc Genet. 2017;10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Renard M, Muino-Mosquera L, Manalo EC, Tufa S, Carlson EJ, Keene DR, De Backer J, Sakai LY. Sex, pregnancy and aortic disease in marfan syndrome. PLoS One. 2017;12:e0181166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tashima Y, He H, Cui JZ, Pedroza AJ, Nakamura K, Yokoyama N, Iosef C, Burdon G, Koyano T, Yamaguchi A, Fischbein MP. Androgens accentuate tgf-beta dependent erk/smad activation during thoracic aortic aneurysm formation in marfan syndrome male mice. J Am Heart Assoc. 2020;9:e015773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sawada H, Chen JZ, Wright BC, Moorleghen JJ, Lu HS, Daugherty A. Ultrasound imaging of the thoracic and abdominal aorta in mice to determine aneurysm dimensions. J Vis Exp. 2019:10.3791/59013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galatioto J, Caescu CI, Hansen J, Cook JR, Miramontes I, Iyengar R, Ramirez F. Cell type-specific contributions of the angiotensin ii type 1a receptor to aorta homeostasis and aneurysmal disease-brief report. Arterioscler Thromb Vasc Biol. 2018;38:588–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rateri DL, Moorleghen JJ, Knight V, Balakrishnan A, Howatt DA, Cassis LA, Daugherty A. Depletion of endothelial or smooth muscle cell-specific angiotensin ii type 1a receptors does not influence aortic aneurysms or atherosclerosis in ldl receptor deficient mice. PLoS One. 2012;7:e51483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ding Y, Stec DE, Sigmund CD. Genetic evidence that lethality in angiotensinogen-deficient mice is due to loss of systemic but not renal angiotensinogen. J Biol Chem. 2001;276:7431–7436 [DOI] [PubMed] [Google Scholar]
- 34.Ye F, Wang Y, Wu C, Howatt DA, Wu CH, Balakrishnan A, Mullick AE, Graham MJ, Danser AHJ, Wang J, Daugherty A, Lu HS. Angiotensinogen and megalin interactions contribute to atherosclerosis. Arterioscler Thromb Vasc Biol. 2019;39:150–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mullen M, Jin XY, Child A, Stuart AG, Dodd M, Aragon-Martin JA, Gaze D, Kiotsekoglou A, Yuan L, Hu J, Foley C, Van Dyck L, Knight R, Clayton T, Swan L, Thomson JDR, Erdem G, Crossman D, Flather M. Irbesartan in marfan syndrome (aims): A double-blind, placebo-controlled randomised trial. Lancet. 2020;394:2263–2270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cikach FS, Koch CD, Mead TJ, Galatioto J, Willard BB, Emerton KB, Eagleton MJ, Blackstone EH, Ramirez F, Roselli EE, Apte SS. Massive aggrecan and versican accumulation in thoracic aortic aneurysm and dissection. JCI Insight. 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sachdeva J, Mahajan A, Cheng J, Baeten JT, Lilly B, Kuivaniemi H, Hans CP. Smooth muscle cell-specific notch1 haploinsufficiency restricts the progression of abdominal aortic aneurysm by modulating ctgf expression. PLoS One. 2017;12:e0178538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bhushan R, Altinbas L, Jager M, Zaradzki M, Lehmann D, Timmermann B, Clayton NP, Zhu Y, Kallenbach K, Kararigas G, Robinson PN. An integrative systems approach identifies novel candidates in marfan syndrome-related pathophysiology. J Cell Mol Med. 2019;23:2526–2535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farran A, Valverde-Franco G, Tio L, Lussier B, Fahmi H, Pelletier JP, Bishop PN, Monfort J, Martel-Pelletier J. In vivo effect of opticin deficiency in cartilage in a surgically induced mouse model of osteoarthritis. Sci Rep. 2018;8:457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kang X, Zhao Y, Van Arsdell G, Nelson SF, Touma M. Ppp1r1b-lncrna inhibits prc2 at myogenic regulatory genes to promote cardiac and skeletal muscle development in mouse and human. RNA. 2020;26:481–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.