

Abstract
The COVID-19 pandemic has clearly brought the healthcare systems worldwide to a breaking point, along with devastating socioeconomic consequences. The SARS-CoV-2 virus, which causes the disease, uses RNA capping to evade the human immune system. Nonstructural protein (nsp) 14 is one of the 16 nsps in SARS-CoV-2 and catalyzes the methylation of the viral RNA at N7-guanosine in the cap formation process. To discover small-molecule inhibitors of nsp14 methyltransferase (MTase) activity, we developed and employed a radiometric MTase assay to screen a library of 161 in-house synthesized S-adenosylmethionine (SAM) competitive MTase inhibitors and SAM analogs. Among six identified screening hits, SS148 inhibited nsp14 MTase activity with an IC50 value of 70 ± 6 nM and was selective against 20 human protein lysine MTases, indicating significant differences in SAM binding sites. Interestingly, DS0464 with an IC50 value of 1.1 ± 0.2 µM showed a bisubstrate competitive inhibitor mechanism of action. DS0464 was also selective against 28 out of 33 RNA, DNA, and protein MTases. The structure–activity relationship provided by these compounds should guide the optimization of selective bisubstrate nsp14 inhibitors and may provide a path toward a novel class of antivirals against COVID-19, and possibly other coronaviruses.
Keywords: nsp14, SARS-CoV-2, COVID-19, coronavirus
Introduction
COVID-19, a severe acute respiratory syndrome in humans, is caused by SARS-CoV-2. It first surfaced in December 20191 and soon became a pandemic with more than 140 million confirmed cases and more than 3.1 million deaths reported worldwide at the time of this writing (https://www.who.int/emergencies/diseases/novel-coronavirus-2019). SARS-CoV-2 belongs to the Coronaviridae family of viruses,2 , 3 which also includes the severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle Eastern respiratory syndrome coronavirus (MERS-CoV), which caused the SARS and MERS epidemics in 2002 and 2012, respectively.4 , 5 SARS-CoV-2 contains a nonsegmented, positive-sense 30 kb RNA that consists of 14 open reading frames (ORFs),6 encoding 16 nonstructural proteins (nsps) and four main structural and accessory proteins.7 The 16 nsps (referred to as nsp1 to nsp16) are more conserved among coronaviruses compared with the structural and accessory proteins.8 These nsps in coronaviruses form a replicase–transcriptase complex and are essential for the transcription and replication of the virus.9 Among these, nsp14 and nsp16 are RNA methyltransferases (MTases) involved in RNA capping.10
Nsp14 is a bifunctional protein with a C-terminal MTase domain catalyzing N7- guanosine methylation and an N-terminal exoribonuclease domain (Suppl. Fig. S1). In the replicase–transcriptase complex of coronaviruses, nsp14 functions as an exoribonuclease and is involved in maintaining the fidelity of coronavirus RNA synthesis.11 Nsp14 in complex with nsp10 can function as a proofreading exoribonuclease and removes 3′-end mismatched nucleotides from dsRNA.12 Breaking this interaction between nsp10 and nsp14 results in a decrease in virus replication fidelity.13 Besides nsp10, nsp14 also interacts with the nsp7-nsp8-nsp12 complex, where the exonuclease function of nsp14 decreases the incidence of mismatched nucleotides14 by erasing the mutated nucleotides.11
While complex formation between nsp10 and nsp14 is required for enhanced exoribonuclease activity, the MT activity of nsp14 is independent of nsp10-nsp14 complex formation.15 , 16 The nsp14 S-adenosylmethionine (SAM)-dependent MTase activity is essential for viral mRNA capping.16 , 17 The cap1 structure at the 5′-end of viral RNA helps in masking the virus from the host immune system.18 , 19 The cap (GpppN) structure in nascent RNA of coronaviruses is formed by nsp1320 , 21 and a guanylyltransferase (GTase). Nsp14 methylates this cap structure at the N7 position of the guanosine, forming a cap-O (N7mGpppN).17 Nsp16 further 2′-O-methylates the product of the nsp14 MTase activity, completing the capping process (N7mGpppNm).16 , 22
Nsp14 is conserved among the seven coronaviruses known to infect humans to date (Suppl. Fig. S2).23 The SARS-CoV-2 nsp14 overall amino acid sequence shows 95.1%, 62.7%, 57.8%, 58.5%, 52.9%, and 53.7% identity with nsp14 from SARS-CoV, MERS-CoV, OC43, HKU1, 299E, and NL63, respectively. This suggests the possibility of SARS-CoV-2 nsp14 inhibitors also inhibiting nsp14 MTase activity of other coronaviruses. Such pan inhibitors would be priceless for developing pan antiviral therapeutics for COVID-19 that would also be effective on future coronaviruses that may jump to humans. In this study, we first developed a radiometric high-throughput activity assay for SARS-CoV-2 nsp14 MTase activity and screened a library of 161 SAM competitive MTase inhibitors we previously synthesized and SAM analogs. We identified six reproducible hits that we mapped on the active site of SARS-CoV-2 nsp14. The data show for the first time a clear path toward the development of potent and selective bisubstrate nsp14 inhibitors that may lead to a novel class of therapeutics for COVID-19 and possibly other coronaviruses to come.
Material and Methods
Reagents
S-Adenosylhomocysteine (SAH) and sinefungin were purchased from Sigma-Aldrich (St. Louis, MO). S-adenosyl-l-methionine, 3H-SAM, and the 384-well FlashPlate PLUS coated with streptavidin and scintillant (cat. SMP410A001PK) were purchased from PerkinElmer (Waltham, MA). Biotin-labeled single-stranded RNA (5′ GpppACCCCCCCCC-Biotin 3′), referred to as RNA substrate from here on, was custom synthesized by TriLink BioTechnologies (San Diego, CA). All RNA solutions were prepared by solubilizing in nuclease-free water in the presence of RNAseOUT recombinant ribonuclease inhibitor (Thermo Fisher, Waltham, MA; cat. 10777019) at a final concentration of 0.4 U/µL. The D- (+) Biotin, [3H(G)], referred to as 3H-biotin, was purchased from PerkinElmer. The biotinylated single- and double-stranded miR-145 substrates for BCDIN3D and METTL3-14 were synthesized by Integrated DNA Technologies (Boston, MA). All peptide substrates were synthesized by Tufts (Boston, MA) or GenScript (Piscataway, NJ). All other reagents were from Sigma-Aldrich.
All compounds tested in vitro were ≥95% pure, except JL27-56A1, which was 86% pure at the time of screening. Purity was determined by analytical high-performance liquid chromatography (HPLC) on an Agilent 1100 series instrument equipped with a Phenomenex KINETEX column (50.0 mm × 4.6 mm, C18, 2.6 μM) at 25 °C. A linear gradient starting from 5% acetonitrile and 95% water to 95% acetonitrile and 5% water over 4 min, followed by elution at 95% acetonitrile and 5% water, was employed. Formic acid (0.1%) was added to all solvents.
Protein Expression and Purification
The expression and purification of SARS-CoV-2 nsp14 are provided as supplemental data.
Radiometric Assay Development
Assay Optimization
The MTase activity of nsp14 was measured using a radiometric assay. The transfer of the 3H-methyl group from 3H-SAM to the RNA substrate (5′ GpppACCCCCCCCC-Biotin 3′) was monitored using a scintillation proximity assay (SPA). Unless stated otherwise, all reactions were carried out in a 20 µL final volume and in triplicate at room temperature. For assay optimization, several concentrations of nsp14 (0.5 nM to 1 µM) were tested by mixing 1 µM 3H-SAM and 1 µM RNA in 20 µL of buffer. The reaction was stopped after 30 min by adding 20 µL of 7.5 M guanidium chloride and 20 µL of 20 mM Tris HCl, pH 8. The overall assay mixture was then transferred to a 384-well FlashPlate (SPA plate) coated with streptavidin/scintillant. After 3 h, the amount of methylated RNA formed was quantified using a TopCount (counts per minute [CPM]). To test additives and buffers, nsp14 (1.5 nM) was mixed with 50 nM RNA and 250 nM 3H-SAM and the reaction was stopped after 30 min. Each buffer (Tris, HEPES, and BTP) was tested at 20 mM at pH 7.5. The effect of pH was evaluated in Tris, pH 6.5–9.5. The effect of additives was monitored individually by titrating the reaction mixture with varying concentrations of DTT (from 0.1 mM to 100 mM), MgCl2 (from 0.1 mM to 100 mM), Triton X-100 (from 0.01% to 10%), and DMSO (from 1.25% to 10%) and comparing their relative activity to the samples with no additive.
Kinetic Characterization
The MTase activity of nsp14 was determined using the optimum buffer conditions identified (20 mM Tris HCl, pH 7.5, 250 µM MgCl2, 5 mM DTT, and 0.01% Triton X-100). The kinetic parameters (K m app and k cat app) of nsp14 were determined using a series of reactions containing nsp14 at a saturating concentration of one substrate (1 µM RNA or 1 µM 3H-SAM) and varying concentrations of the others (from 15.6 nM to 2000 nM for 3H-SAM and from 7.8 nM to 1000 nM for RNA). Reactions were stopped at various time points (2, 5, 10, 15, 20, and 30 min). Initial velocities of the reaction were calculated from the linear portion of the reaction curves (see Suppl. Fig. S4) and were plotted as a function of each substrate concentration (3H-SAM and RNA) to determine their K m app values using the Michaelis–Menten equation. The maximum velocity (V max) obtained from the Michaelis–Menten plot was used to calculate the apparent turnover number (k cat app) in each experiment. To determine the optimum reaction time for screening, linearity of the methylation reaction over time was monitored. The reaction mixture containing 1.5 nM nsp14, 250 nM 3H-SAM, 50 nM RNA in 20 mM Tris HCl, pH 7.5, 250 µM MgCl2, 5 mM DTT, and 0.01% Triton X-100 was incubated for various durations (0, 2, 5, 10, 15, 20, and 30 min), and the amount of methylated RNA was quantified over time.
IC50 Determination
Compounds were tested at various concentrations from 12 nM to 200 µM final concentration to determine their half-maximal inhibitory concentration (IC50) values. Potent compounds (SS148, SAH, and WZ16) were tested from 3 nM to 50 µM. The final DMSO concentration was 2%. The final reaction mixture consisted of 1.5 nM nsp14, 250 nM 3H-SAM, 50 nM RNA in 20 mM Tris HCl, pH 7.5, 250 µM MgCl2, 5 mM DTT, and 0.01% Triton X-100. The reaction time was 20 min. Data were fitted to the four-parameter logistic equation using GraphPad Prism 8.
Z′-Factor Determination
To evaluate the effectiveness of the nsp14 assay for screening purposes, the Z′ factor was determined by running 96 different reactions in the presence or absence of 1 µM sinefungin, a known MTase inhibitor. The final reaction consisted of 1.5 nM nsp14, 250 nM 3H-SAM, 50 nM RNA in 20 mM Tris HCl, pH 7.5, 250 µM MgCl2, 5 mM DTT, and 0.01% Triton X-100. Reactions were stopped after 20 min. The Z′ factor was calculated as described by Zhang et al.24
Screening
Nsp14 was screened against the in-house library of 161 compounds at 50 µM in 1% DMSO. Compounds with inhibition of more than 75% were selected as screening hits for further analysis. The hits were tested for assay signal quenching at 50 µM. The signal was generated using 0.1 µM 3H-biotin in a 384-well FlashPlate that is coated with streptavidin/scintillant. Compounds that did not quench the signal were tested by dose-response against nsp14 at various concentrations from 12 nM to 200 µM final concentration to determine their IC50 values.
Mechanism of Action
The mechanism of action (MOA) of DS0464 was investigated by determining the IC50 values of the compound for nsp14 at the saturating concentration of one substrate (250 nM RNA or 1.25 µM 3H-SAM) and varying concentrations of the others (from 62.5 nM to 1.25 µM for 3H-SAM and from 12.5 nM to 250 nM for RNA).
Selectivity Assays
Selectivity assays were performed as previously described.25 Briefly, compounds were tested at 50 µM in duplicate using radiometric assays. IC50 values were determined for compounds with higher than a 50% inhibitory effect, as described above.
Surface Plasmon Resonance
KD values for initial screening hits for nsp14 were determined by surface plasmon resonance (SPR) using a Biacore T200 from GE Healthcare (Chicago, IL). N-terminally biotinylated nsp14 (amino acids 1–527) and C-terminally biotinylated SETD3 (amino acids 1–605, as control) were coupled on a CM5 SPR Sensor chip (GE Healthcare). Compounds were injected into the sensitized chip at five concentrations (0.6, 1.9, 5.6, 16.7, and 50 µM) plus 0.5% DMSO at 50 µL/min, using HBS-EP Plus buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 2 mM EDTA, and 0.005% Tween-20) and 0.5% DMSO. Compounds with lower IC50 values (SS148, SAH, WZ16, and JL27-56A1) were injected at the following concentrations: 0.06, 0.19, 0.56, 1.67, and 5 µM. The contact time was 60 s and the disassociation time was 120 s. Buffer alone (plus 0.5% DMSO) was used for blank injections, and buffers containing 0.4%–0.6% DMSO were used for buffer corrections.
Modeling
DS0464 was docked into the MTase active site of SARS-CoV nsp14 (Protein Data Bank [PDB] code 5C8S) with ICM (Molsoft, San Diego, CA). PDB coordinates were loaded, side chains that were missing due to poor electron density were built and energy minimized with a biased-probability Monte Carlo simulation, hydrogens were added, and rotameric states of hydroxyl groups, terminal amides, and histidine side chains were optimized in the internal coordinate space. Ligand docking was conducted with fully flexible ligand and a grid representation of the protein with a Monte Carlo-based energy minimization.26
Results
Assay Development and Optimization for High-Throughput Screening
Developing therapeutics for COVID-19 and other coronaviruses requires reliable high-throughput screening assays. Radiometric assays have been widely used for developing potent substrate and SAM competitive inhibitors for human MTases within the last decade.25 Using biotinylated RNA substrate, we developed a radiometric nsp14 MTase activity assay with 3H-SAM as a methyl donor ( Fig. 1). Nsp14 MTase activity was evaluated in various buffers, pH values, and additives (Suppl. Fig. S3). The highest MTase activity was observed in Tris HCl buffer at pH 7.5. No significant effect was observed for DTT up to 10 mM, and it was included in the assay reaction mixture at 5 mM to maintain reducing conditions. Triton X-100 at 0.01% was added to the reaction buffer to minimize the binding of proteins and compounds to plates. DMSO at concentrations up to 10% had little effect on nsp14 MTase activity. However, MgCl2 was only tolerated at concentrations below 1 mM (Suppl. Fig. S3). The assay optimization resulted in selecting 20 mM Tris HCl, pH 7.5, 250 µM MgCl2, 5 mM DTT, and 0.01% Triton X-100 for testing nsp14 MTase activity and determining its kinetic parameters.
Figure 1.
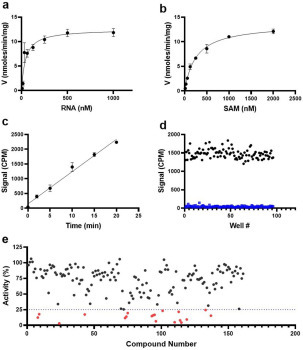
Kinetic characterization and screening of nsp14 MTase activity. The optimized radiometric MTase assay was used to determine the Kmapp and kcatapp values for (a) RNA and (b) SAM at saturating concentrations of SAM and RNA, respectively. (c) The linearity of the initial velocity of the nsp14 MTase reaction was tested at Km for both substrates. (d) The Z′ factor was determined at the same conditions as in c. Nsp14 activity was assessed in the absence (100% activity) ( ) and presence (
) and presence ( ) of 1 µM sinefungin. (e) Nsp14 was screened against a library of SAM analogs. A collection of 161 SAM competitive inhibitors and analogs was tested at 50 µM for inhibition of nsp14 activity under conditions described for Z′-factor determination. Compounds with higher than 75% inhibition (
) of 1 µM sinefungin. (e) Nsp14 was screened against a library of SAM analogs. A collection of 161 SAM competitive inhibitors and analogs was tested at 50 µM for inhibition of nsp14 activity under conditions described for Z′-factor determination. Compounds with higher than 75% inhibition ( ) were selected for follow-up experiments. Values in a, b, and c are presented as the mean ± standard deviation of three independent experiments (n = 3). Values in d and e are from single screening.
) were selected for follow-up experiments. Values in a, b, and c are presented as the mean ± standard deviation of three independent experiments (n = 3). Values in d and e are from single screening.
Kinetic Parameters of SARS CoV-2 Nsp14 MTase Activity
Using optimized assay conditions, we assessed the linearity of initial velocities (activity vs time) at various concentrations of RNA at a fixed SAM concentration (1 µM) (Suppl. Fig. S4a) and at various concentrations of SAM at a fixed RNA concentration (1 µM) (Suppl. Fig. S4b). The slopes of the linear initial velocities were calculated and used to determine kinetic parameters for nsp14 MTase activity. Apparent K m (K m app) values of 43 ± 15 nM and 257 ± 20 nM were determined for RNA and SAM, respectively. The calculated k cat app values from the two sets of experiments, 48 ± 4 h–1 (RNA) (Fig. 1a) and 52 ± 1 h–1 (SAM) (Fig. 1b), were reasonably close. The addition of nsp10 at various molar ratios from 1 (nsp10):1 (nsp14) to 20 (nsp10):1 (nsp14) did not have any significant effect on nsp14 MTase activity (Suppl. Fig. S5).
Assay Amenability to High-Throughput Screening
In small-molecule screening campaigns, typically the assays are performed at K m of the substrates to allow potential inhibitors to compete with the substrates and allow their binding to be detected. However, the activity of the enzyme should be linear during the assay period. As the radiometric MTase assays are endpoint assays, lack of linearity may mask inhibition of some compounds. Testing the activity of nsp14 at 50 nM RNA and 250 nM SAM indicated that the assay can be run for at least 20 min while maintaining the linearity (Fig. 1c). To determine the reproducibility of such conditions for high-throughput screening, the assay was performed in the presence (1 µM) and absence of sinefungin, a pan MTase inhibitor that inhibits nsp14 MTase activity with an IC50 value of 0.019 ± 0.01 µM (Suppl. Fig. S6). A Z′ factor of 0.69 was calculated for nsp14 screening, indicating suitability of the assay for high-throughput screening (Fig. 1d).
Screening SARS CoV-2 nsp14 against a Collection of Potential SAM Competitive Inhibitors
An in-house library of 161 SAM competitive MTase inhibitors and SAM analogs was screened against nsp14 at 50 µM, and 19 compounds were identified that inhibited nsp14 MTase activity more than 75% (Fig. 1e). Compounds that interfered with the readout signal or were not reproducible were eliminated. The remaining six compounds ( Fig. 2) inhibited nsp14 MTase activity with IC50 values ranging from 70 nM to 95 µM ( Table 1, Fig. 3). We also tested these compounds for binding to nsp14 by SPR. Binding was detected for all hits and K D values were calculated for all except WZ16, with its relatively linear fitting, which was not reliable for accurate K D calculation (Table 1, Suppl. Fig. S7).
Figure 2.
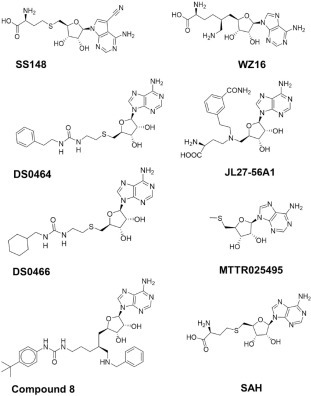
Structures of nsp14 screening hits.
Table 1.
Confirmation and Selectivity of nsp14 Screening Hits.
| Compound | SPR KD (µM) of Nsp14 | IC50 (µM)a | |||||
|---|---|---|---|---|---|---|---|
| Nsp 14 | G9a | BCDIN3D | ALKBH5 | SETD3 | METTL3-14 | ||
| SS148 | 0.05 | 0.07 ± 0.006 | NI | 0.03 ± 0.002 | NI | NI | 0.9 |
| WZ16 | NA | 0.19 ± 0.04 | NI | 1.7 | NI | 3.4 | 3.4 |
| DS0464 | 1.6 | 1.1 ± 0.2 | NI | 46 ± 9 | NI | NI | NI |
| DS0466 | 2.4 | 3.4 ± 1.4 | NI | >50 | NI | NI | NI |
| JL27-56A1 | 0.2 | 0.26 ± 0.13 | NI | 19 | NI | 23 | 10 |
| Compound 8 | 18.9 | 95 ± 6 | NI | NI | NI | NI | NI |
| SAH | 0.14 | 0.13 ± 0.03 | NA | NA | NA | NA | NA |
| MTTR025495 | 6.2 | 17 ± 2 | NA | NA | NA | NA | NA |
The screening hits were tested for binding to nsp14 by SPR and for inhibition of MTase activity of nsp14 and selected MTases by activity assays. All values are from experiments presented in Figure 3andSupplemental Figures S7, S10, S11, S12, S13, and S14. NA, not assessed; NI, no inhibition. aIC50 values are presented as the mean ± standard deviation (n = 3).
Figure 3.
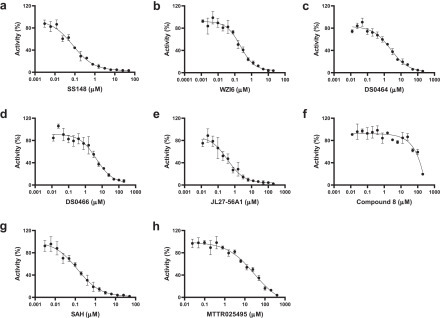
Dose–response curves of nsp14 screening hits. The IC50 values were determined for (a) SS148 (0.07 ± 0.006 µM), (b) WZ16 (0.19 ± 0.04 µM), (c) DS0464 (1.1 ± 0.2 µM), (d) DS0466 (3.4 ± 1.4 µM), (e) JL27-56A1 (0.26 ± 0.13 µM), (f) compound 8 (95 ± 6 µM), (g) SAH (0.13 ± 0.03 µM), and (h) MTTR025495 (17 ± 2 µM) by testing their inhibitory effects ranging from 3 nM to 200 µM final concentration. IC50 values are also presented in Table 1. All values are presented as the mean ± standard deviation of three independent experiments (n = 3).
Docking and Modeling
This limited and focused screening exercise provided important insights on the structural chemistry of SARS CoV-2 nsp14 inhibition (Table 1). First, we noted that the demethylated cofactor S-adenosyl-l-homocysteine is a very potent inhibitor of nsp14 (IC50 = 130 ± 30 nM; K D = 140 nM). Decorating the adenine scaffold with a nitrile group at position 7 improved the potency (SS148 IC50 = 70 ± 6 nM, K D = 50 nM), while installing an ethylamine in place of the sulfur group of SAH had little effect on potency (WZ16: IC50 = 190 ± 40 nM). Deleting the amino acid end of SAH resulted in a 130-fold loss in SARS-CoV-2 nsp14 inhibition (MTTR025495 IC50 = 17 ± 2 µM) (Table 1, Fig. 3). Interestingly, compound DS0464, where the amino acid moiety is replaced with a physicochemically more favorable phenyl-ethyl-urea, retains significant inhibitory activity (IC50 = 1.1 ± 0.2 µM). Since all residues lining SAH and the substrate RNA cap GpppA in the SARS-CoV nsp14 structure are conserved in SARS-CoV-2 (Suppl. Fig. S8), the SARS-CoV structure was used to dock DS0464 ( Fig. 4). The model revealed a possible arrangement where the adenosine end of the inhibitor overlays with the adenosine of the bound cofactor, and the terminal phenyl group recapitulates stacking interactions observed between the guanine ring of the RNA cap and surrounding residues (Y420, F426, F506, and N386) (Fig. 4). Such a binding mode suggests an MOA where DS0464 behaves as a bisubstrate inhibitor. This possibility was further tested by performing MOA studies with DS0464, which revealed that DS0464 competes against both SAM and RNA and can act as a bifunctional inhibitor ( Fig. 5, Suppl. Fig. S9).
Figure 4.
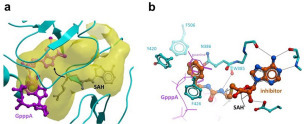
Docking model of DS0464. (a) A large and enclosed binding pocket (yellow) in the MTase domain of nsp14 accommodates both the methyl-donating cofactor (gray; SAH is shown instead) and the methyl-accepting RNA cap GpppA (purple) (PDB code 5C8S). Black arrow, site of methyl transfer. (b) The docked inhibitor DS0464 occupies the cofactor site and extends into the substrate binding site.
Figure 5.
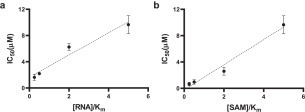
MOA of DS0464. IC50 values were determined for DS0464 at (a) varying concentrations of RNA at 1.25 µM SAM and (b) varying concentrations of SAM at 250 nM RNA. DS0464 competes against both RNA and SAM. All values are presented as the mean ± standard deviation of three independent experiments (n = 3).
Selectivity of Screening Hits
The selectivity of all six compounds was tested against the human RNA MTase BCDIN3D and the METTL3-METTL14 complex (METTL3-14), the RNA demethylase ALKBH5, and protein lysine MTases G9a and SETD3 (Table 1, Suppl. Figs. S10–S14). Interestingly, none of the compounds inhibited G9a or ALKBH5 activities, indicating some level of selectivity. SS148 and DS0464 potently inhibited BCDIN3D with IC50 values of 0.03 ± 0.002 and 46 ± 9 µM, respectively, but not G9a, SETD3, or ALKBH5 (Table 1). G9a and SETD3 are SET domain MTases that are structurally distinct from class I MTases such as nsp14, BCDIN3D, or METTL3-METTL14, which could explain the observed specificity profile.27 For instance, the channel separating the substrate and cofactor binding sites is wide in nsp14 but narrow in G9a. Additionally, a cavity that can accommodate the nitrile group of SS148 in nsp14 and BCDIN3D is absent in G9a, in agreement with the obtained IC50 values ( Fig. 6). To further characterize our nsp14 inhibitors, the selectivity of SS148 and DS0464 was evaluated against a larger panel of lysine, arginine, DNA, and RNA MTases ( Table 2, Fig. 7). As expected, while SS148 inhibited arginine, DNA, and RNA MTases (all class I MTases), it did not inhibit any of the 20 SET domain lysine MTases (Fig. 7, Table 2, Suppl. Fig. S15). DS0464 was even more selective and inhibited only PRMT4, PRMT5, PRMT7, DOT1L, and BCDIN3D, but none of the protein lysine MTases tested (Table 2, Suppl. Fig. S16).
Figure 6.
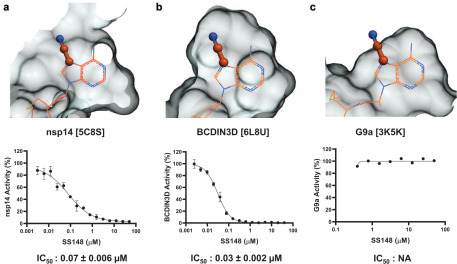
Structural determinant for SS148 selectivity. A nitrile group (stick) at position 7 of the cofactor adenine ring is compatible with the crystal structure of SAH (wire) bound to (a) nsp14 and (b) BCDIN3D but not (c) G9a. The Van der Waals surface of the cofactor binding pocket is shown as gray mesh. Dose–response curves for SS148 against these proteins with their IC50 values are also shown.
Table 2.
Selectivity of SS148 and DS0464.
| Methyltransferase | IC50 (µM)a | |
|---|---|---|
| SS148 | DS0464 | |
| PRMT1 | 3 ± 0.07 | NI |
| PRMT3 | 2 ± 0.07 | NI |
| PRMT4 | 1 ± 0.1 | 10 ± 0.8 |
| PRMT5 | 9 ± 2 | 13 ± 1.2 |
| PRMT6 | 1 ± 0.2 | NI |
| PRMT7 | 0.3 ± 0.03 | 14 ± 1.3 |
| PRMT8 | 2 ± 0.05 | NI |
| PRMT9 | 39 ± 4 | NI |
| DOT1L | 0.1 ± 0.009 | 7 ± 0.2 |
| DNMT1 | 2 ± 0.3 | NI |
| DNMT3A/3L | 0.1 ± 0.002 | NI |
| DNMT3B/3L | 0.03 ± 0.006 | NI |
| METTL3-METTL14 | 0.9 ± 0.1 | NI |
| SETD3 | NI | NI |
| BCDIN3D | 0.03 ± 0.002 | 46 ± 9 |
| Nsp14 | 0.07 ± 0.006 | 1.1 ± 0.2 |
NI, no inhibition.
IC50 values are presented as the mean ± standard deviation (n = 3).
Figure 7.

Selectivity of SS148 and DS0464. Both SS148 and DS0464 were screened for selectivity against 33 human RNA, DNA, and protein MTases at 50 µM. All experiments were performed in duplicate. When significant inhibition was observed, IC50 values were determined and are presented in Table 2.
Discussion
The frequent emergence in the last two decades of novel coronaviruses as human pathogens, highlighted by the current COVID-19 pandemic, urgently needs to be addressed, preferably with pan coronavirus drugs. Nsp14 is an essential MTase in RNA cap formation that is required for protecting viral RNA and proper replication of coronaviruses. Therefore, targeting nsp14 MTase activity would be a viable option toward developing antiviral therapeutics.28 MTases are druggable.29 In the last decade, a significant number of selective and cell-active small molecules (chemical probes) have been discovered for human MTases,25 , 30 , 31 and some are in clinical trials for various cancers.30 , 31
Key to a successful discovery campaign of such chemical probes is the availability of reliable screening methods that could enable medium- to high-throughput screening with low false-positive and false-negative rates. Various assays including mass spectrometry,32, 33, 34 fluorescence,35 and radiometric assays25 have been used for screening libraries of compounds. Mass spectrometry-based assays require more expensive instrumentation and expertise. Fluorescence assays can be performed in any lab; however, many fluorescent compounds in chemical libraries may increase the background and lead to high numbers of false positives to triage following screening large libraries. Radiometric assays are typically more reliable and have fewer false positives, leading to identifying more reliable screening hits.25 In this study, we have developed a radiometric assay for nsp14 and employed this assay for screening a small library of selected SAM competitive inhibitors and analogs.
Targeting the SAM binding site has successfully led to the discovery of chemical probes for human MTases such as DOT1L,36 , 37 EZH2/EZH1,38 and SMYD2.39 Synthesis of adenine dinucleoside SAM analogs as specific inhibitors of SARS-CoV (SARS) nsp14 RNA cap guanine-N7-MTase activity has also been recently reported.40 SS148 (nsp14 IC50 = 70 ± 6 nM) was reported as a DOT1L inhibitor with a nitrile as a nontraditional replacement for heavy halogen atoms.41 This is consistent with the selectivity of SS148 against all other protein lysine methyltransferases (PKMTs) due to narrower active sites that could not fit the added nitrile group (Fig. 4). Keeping this substitution in designing future nsp14 inhibitors will provide selectivity against PKMTs. WZ1642 and compound 8 are PRMT4 (CARM1) inhibitors. Although compound 8 is a relatively weak nsp14 inhibitor (IC50 = 95 ± 6 µM), it is likely cell permeable (US20180305391A1). JL27-56A1 is known to modestly inhibit nicotinamide N-methyltransferase (NNMT).43 , 44 Optimization of these series for NNMT resulted in the discovery of MS2734, a bisubstrate inhibitor. DS0464 and DS0466 were originally designed for DOT1L inhibition.36 DS0464, with replacement of the amino acid moiety with a physicochemically more favorable phenyl-ethyl-urea, retains significant nsp14 inhibitory activity (IC50 = 1.1 ± 0.2 µM). Structural modeling and kinetic data both suggest a mechanism of inhibition consistent with bisubstrate competition. These data reveal a path to developing a series of bifunctional inhibitors to increase the potency of nsp14 SAM competitive inhibitors. Interestingly, DS0464 was selective against 28 out of 33 human RNA, DNA, and protein MTases, indicating a path to developing selective bisubstrate inhibitors toward the discovery of therapeutics for COVID-19 and closely related coronaviruses. In this study, we developed a radiometric assay for nsp14 MTase activity, determined the kinetic parameters, and optimized the assay for high-throughput screening. Through limited screening of SAM competitive inhibitors, we identified six confirmed hits that we used to probe the active site of nsp14. Our study revealed a path toward developing selective bisubstrate inhibitors for nsp14.
Acknowledgments
We thank Dr. Aled Edwards and Dr. Cheryl Arrowsmith for continued support, and Brian Wilson for performing compound quality control.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by University of Toronto COVID-19 Action Initiative-2020 and COVID-19 Mitacs Accelerate postdoctoral awards to A.K.Y. and S.P.; U.S. National Institutes of Health grant R35GM131858 to M.L.; and NIH/NIAID contract HHSN272201700060C to K.J.F.S. The Structural Genomics Consortium is a registered charity (no. 1097737) that receives funds from AbbVie, Bayer AG, Boehringer Ingelheim, Canada Foundation for Innovation, Genentech, Genome Canada through Ontario Genomics Institute (OGI-196), EU/EFPIA/OICR/McGill/KTH/Diamond Innovative Medicines Initiative 2 Joint Undertaking (EUbOPEN grant 875510), Janssen, Merck KGaA (aka EMD in Canada and the United States), Pfizer, Takeda, and Wellcome (106169/ZZ14/Z).
Supplemental Material
Supplemental Material
References
- 1.Zhu N., Zhang D., Wang W., et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adams M.J., Lefkowitz E.J., King A.M., et al. Ratification Vote on Taxonomic Proposals to the International Committee on Taxonomy of Viruses (2016) Arch. Virol. 2016;161:2921–2949. doi: 10.1007/s00705-016-2977-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coronaviridae Study Group of the International Committee on Taxonomy of Viruses The Species Severe Acute Respiratory Syndrome-Related Coronavirus: Classifying 2019-nCoV and Naming It SARS-CoV-2. Nat. Microbiol. 2020;5:536–544. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan-Yeung M., Xu R.H. SARS: Epidemiology. Respirology. 2003;8(Suppl.):S9–S14. doi: 10.1046/j.1440-1843.2003.00518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zaki A.M., van Boheemen S., Bestebroer T.M., et al. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 2012;367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 6.Chan J.F., Kok K.H., Zhu Z., et al. Genomic Characterization of the 2019 Novel Human-Pathogenic Coronavirus Isolated from a patient with Atypical Pneumonia after Visiting Wuhan. Emerg. Microbes Infect. 2020;9:221–236. doi: 10.1080/22221751.2020.1719902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Boheemen S, de Graaf M., Lauber C., et al. Genomic Characterization of a Newly Discovered Coronavirus Associated with Acute Respiratory Distress Syndrome in Humans. mBio. 2012;3:e00473-12. doi: 10.1128/mBio.00473-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y., Liu Q., Guo D. Emerging Coronaviruses: Genome Structure, Replication, and Pathogenesis. J. Med. Virol. 2020;92:418–423. doi: 10.1002/jmv.25681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baez-Santos Y.M., St John S.E., Mesecar A.D. The SARS-Coronavirus Papain-Like Protease: Structure, Function and Inhibition by Designed Antiviral Compounds. Antiviral Res. 2015;115:21–38. doi: 10.1016/j.antiviral.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Y., Guo D. Molecular Mechanisms of Coronavirus RNA Capping and Methylation. Virol. Sin. 2016;31:3–11. doi: 10.1007/s12250-016-3726-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eckerle L.D., Becker M.M., Halpin R.A., et al. Infidelity of SARS-CoV Nsp14-Exonuclease Mutant Virus Replication Is Revealed by Complete Genome Sequencing. PLoS Pathog. 2010;6:e1000896. doi: 10.1371/journal.ppat.1000896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bouvet M., Imbert I., Subissi L., et al. RNA 3′-End Mismatch Excision by the Severe Acute Respiratory Syndrome Coronavirus Nonstructural Protein Nsp10/Nsp14 Exoribonuclease Complex. Proc. Natl. Acad. Sci. U.S.A. 2012;109:9372–9377. doi: 10.1073/pnas.1201130109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith E.C., Case J.B., Blanc H., et al. Mutations in Coronavirus Nonstructural Protein 10 Decrease Virus Replication Fidelity. J. Virol. 2015;89:6418–6426. doi: 10.1128/JVI.00110-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Subissi L., Posthuma C.C., Collet A., et al. One Severe Acute Respiratory Syndrome Coronavirus Protein Complex Integrates Processive RNA Polymerase and Exonuclease Activities. Proc. Natl. Acad. Sci. U.S.A. 2014;111:E3900–E3909. doi: 10.1073/pnas.1323705111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Decroly E., Debarnot C., Ferron F., et al. Crystal Structure and Functional Analysis of the SARS-Coronavirus RNA Cap 2′-O-Methyltransferase Nsp10/Nsp16 Complex. PLoS Pathog. 2011;7:e1002059. doi: 10.1371/journal.ppat.1002059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bouvet M., Debarnot C., Imbert I., et al. In Vitro Reconstitution of SARS-Coronavirus mRNA Cap Methylation. PLoS Pathog. 2010;6:e1000863. doi: 10.1371/journal.ppat.1000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y., Cai H., Pan J., et al. Functional Screen Reveals SARS Coronavirus Nonstructural Protein Nsp14 as a Novel Cap N7 Methyltransferase. Proc. Natl. Acad. Sci. U.S.A. 2009;106:3484–3489. doi: 10.1073/pnas.0808790106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marcotrigiano J., Gingras A.C., Sonenberg N., et al. Cocrystal Structure of the Messenger RNA 5′ Cap-Binding Protein (eIF4E) Bound to 7-Methyl-GDP. Cell. 1997;89:951–961. doi: 10.1016/s0092-8674(00)80280-9. [DOI] [PubMed] [Google Scholar]
- 19.Decroly E., Ferron F., Lescar J., et al. Conventional and Unconventional Mechanisms for Capping Viral mRNA. Nat. Rev. Microbiol. 2011;10:51–65. doi: 10.1038/nrmicro2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ivanov K.A., Ziebuhr J. Human Coronavirus 229E Nonstructural Protein 13: Characterization of Duplex-Unwinding, Nucleoside Triphosphatase, and RNA 5′-Triphosphatase Activities. J. Virol. 2004;78:7833–7838. doi: 10.1128/JVI.78.14.7833-7838.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ivanov K.A., Thiel V., Dobbe J.C., et al. Multiple Enzymatic Activities Associated with Severe Acute Respiratory Syndrome Coronavirus Helicase. J. Virol. 2004;78:5619–5632. doi: 10.1128/JVI.78.11.5619-5632.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Decroly E., Imbert I., Coutard B., et al. Coronavirus Nonstructural Protein 16 Is a Cap-0 Binding Enzyme Possessing (Nucleoside-2′O)-Methyltransferase Activity. J. Virol. 2008;82:8071–8084. doi: 10.1128/JVI.00407-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corman V.M., Muth D., Niemeyer D., et al. Hosts and Sources of Endemic Human Coronaviruses. Adv. Virus Res. 2018;100:163–188. doi: 10.1016/bs.aivir.2018.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang J.H., Chung T.D., Oldenburg K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 25.Scheer S., Ackloo S., Medina T.S., et al. A Chemical Biology Toolbox to Study Protein Methyltransferases and Epigenetic Signaling. Nat. Commun. 2019;10:19. doi: 10.1038/s41467-018-07905-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neves M.A., Totrov M., Abagyan R. Docking and Scoring with ICM: The Benchmarking Results and Strategies for Improvement. J. Comput. Aided Mol. Des. 2012;26:675–686. doi: 10.1007/s10822-012-9547-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Campagna-Slater V., Mok M.W., Nguyen K.T., et al. Structural Chemistry of the Histone Methyltransferases Cofactor Binding Site. J. Chem. Inf. Model. 2011;51:612–623. doi: 10.1021/ci100479z. [DOI] [PubMed] [Google Scholar]
- 28.Ferron F., Decroly E., Selisko B., et al. The Viral RNA Capping Machinery as a Target for Antiviral Drugs. Antiviral Res. 2012;96:21–31. doi: 10.1016/j.antiviral.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferreira de, Freitas R., Ivanochko D., Schapira M. Methyltransferase Inhibitors: Competing with, or Exploiting the Bound Cofactor. Molecules. 2019;24:4492. doi: 10.3390/molecules24244492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stein E.M., Garcia-Manero G., Rizzieri D.A., et al. The DOT1L Inhibitor Pinometostat Reduces H3K79 Methylation and Has Modest Clinical Activity in Adult Acute Leukemia. Blood. 2018;131:2661–2669. doi: 10.1182/blood-2017-12-818948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gounder M., Schoffski P., Jones R.L., et al. Tazemetostat in Advanced Epithelioid Sarcoma with Loss of INI1/SMARCB1: An International, Open-Label, Phase 2 Basket Study. Lancet Oncol. 2020;21:1423–1432. doi: 10.1016/S1470-2045(20)30451-4. [DOI] [PubMed] [Google Scholar]
- 32.Rohman M., Wingfield J. High-Throughput Screening Using Mass Spectrometry within Drug Discovery. Methods Mol. Biol. 2016;1439:47–63. doi: 10.1007/978-1-4939-3673-1_3. [DOI] [PubMed] [Google Scholar]
- 33.Haslam C., Hellicar J., Dunn A., et al. The Evolution of MALDI-TOF Mass Spectrometry toward Ultra-High-Throughput Screening: 1536-Well Format and Beyond. J. Biomol. Screen. 2016;21:176–186. doi: 10.1177/1087057115608605. [DOI] [PubMed] [Google Scholar]
- 34.Winter M., Ries R., Kleiner C., et al. Automated MALDI Target Preparation Concept: Providing Ultra-High-Throughput Mass Spectrometry-Based Screening for Drug Discovery. SLAS Technol. 2019;24:209–221. doi: 10.1177/2472630318791981. [DOI] [PubMed] [Google Scholar]
- 35.Janzen W.P. Screening Technologies for Small Molecule Discovery: The State of the Art. Chem. Biol. 2014;21:1162–1170. doi: 10.1016/j.chembiol.2014.07.015. [DOI] [PubMed] [Google Scholar]
- 36.Yu W., Chory E.J., Wernimont A.K., et al. Catalytic Site Remodelling of the DOT1L Methyltransferase by Selective Inhibitors. Nat. Commun. 2012;3:1288. doi: 10.1038/ncomms2304. [DOI] [PubMed] [Google Scholar]
- 37.Daigle S.R., Olhava E.J., Therkelsen C.A., et al. Selective Killing of Mixed Lineage Leukemia Cells by a Potent Small-Molecule DOT1L Inhibitor. Cancer Cell. 2011;20:53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Konze K.D., Ma A., Li F., et al. An Orally Bioavailable Chemical Probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chem. Biol. 2013;8:1324–1334. doi: 10.1021/cb400133j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taylor A.P., Swewczyk M., Kennedy S., et al. Selective, Small-Molecule Co-Factor Binding Site Inhibition of a Su(var)3-9, Enhancer of Zeste, Trithorax Domain Containing Lysine Methyltransferase. J. Med. Chem. 2019;62:7669–7683. doi: 10.1021/acs.jmedchem.9b00112. [DOI] [PubMed] [Google Scholar]
- 40.Ahmed-Belkacem R., Sutto-Ortiz P., Guiraud M., et al. Synthesis of Adenine Dinucleosides SAM Analogs as Specific Inhibitors of SARS-CoV Nsp14 RNA Cap Guanine-N7-Methyltransferase. Eur. J. Med. Chem. 2020;201:112557. doi: 10.1016/j.ejmech.2020.112557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spurr S.S., Bayle E.D., Yu W., et al. New Small Molecule Inhibitors of Histone Methyl Transferase DOT1L with a Nitrile as a Non-Traditional Replacement for Heavy Halogen Atoms. Bioorg. Med. Chem. Lett. 2016;26:4518–4522. doi: 10.1016/j.bmcl.2016.07.041. [DOI] [PubMed] [Google Scholar]
- 42.Cai X.C., Zhang T., Kim E.J., et al. A Chemical Probe of CARM1 Alters Epigenetic Plasticity against Breast Cancer Cell Invasion. Elife. 2019;8:e47110. doi: 10.7554/eLife.47110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Babault N., Allali-Hassani A., Li F., et al. Discovery of Bisubstrate Inhibitors of Nicotinamide N-Methyltransferase (NNMT) J. Med. Chem. 2018;61:1541–1551. doi: 10.1021/acs.jmedchem.7b01422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hong S., Moreno-Navarrete J.M., Wei X., et al. Nicotinamide N-Methyltransferase Regulates Hepatic Nutrient Metabolism through Sirt1 Protein Stabilization. Nat. Med. 2015;21:887–894. doi: 10.1038/nm.3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material