

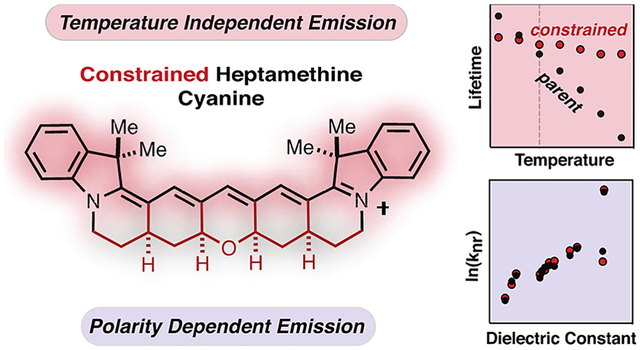
Abstract
Appending conformationally restraining ring systems to the cyanine chromophore creates exceptionally bright fluorophores in the visible range. Here, we report the application of this strategy in the near-infrared range through the preparation of the first restrained heptamethine indocyanine. Time-resolved absorption spectroscopy and fluorescence correlation spectroscopy verify that, unlike the corresponding parent unrestrained variant, the restrained molecule is not subject to photoisomerization. Notably, however, the room-temperature emission efficiency and the fluorescence lifetime of the restrained cyanine are not extended relative to the parent cyanine, even in viscous solvents. Thus, in contrast to prior reports, the photoisomerization of heptamethine cyanines does not contribute significantly to the excited-state chemistry of these molecules. We also find that the fluorescence lifetime of the restrained heptamethine cyanine is temperature-insensitive and significantly extended at moderately elevated temperatures relative to the parent cyanine. Finally, computational studies have been used to evaluate the impact of the conformational restraint on atomic and orbital structure across the cyanine series. These studies clarify the role of photoisomerization in the heptamethine cyanine scaffold and demonstrate the dramatic effect of restraint on the temperature sensitivity of these dyes.
Graphical Abstract
INTRODUCTION
Optical imaging in the near-infrared (NIR) range benefits from reduced autofluorescence and enhanced tissue penetration.1 Improving the photon output of NIR fluorescent probes would be an enabling advance for many imaging applications. This is challenging because, as observed across multiple scaffolds, strategies that shift absorbance maxima toward longer wavelengths result in decreased fluorescence quantum yield (ΦF).2–4 While increased internal conversion as the S0–S1 energy gap is decreased is a major element of this issue, and chemical features also have dramatic effects on the emissive properties of NIR dyes. As the magnitude of ΦF results from the interplay of multiple dye- and solvent-dependent pathways, it is important to clearly define the link between structure and emission efficiency.
Cyanine dyes are broadly used fluorescent probes with applications that span single-molecule to bulk-tissue imaging.5,6 Unique to the cyanine dye class, two-carbon homologation extends the absorbance maxima in roughly 100 nm increments. The indocarbocyanines, including the broadly utilized Cy3, Cy5, and Cy7 series, form the chemical backbone of many invaluable fluorescent reagents.7 While these dyes exhibit exceptionally high absorbance cross sections, emission can compete with a trans-to-cis photoisomerization pathway limiting photon output.5,6,9 Fusing a polycyclic ring system to the polymethine chromophore to block even the possibility of photoisomerization is an appealing strategy. This approach was implemented first on the trimethine cyanines by Waggoner et al.8 and, more recently, by us on the synthetically challenging pentamethine series.10,11 The resulting dyes are dramatically brighter with the tri- and pentamethine derivatives exhibiting ~8× and ~4× greater room-temperature ΦF values, respectively (Figure 1A).10,11 The improved brightness of these restrained dyes is consistent with various spectroscopic observations suggesting that photoisomerization is a major component of the excited-state chemistry of these molecules.5,12
Figure 1.
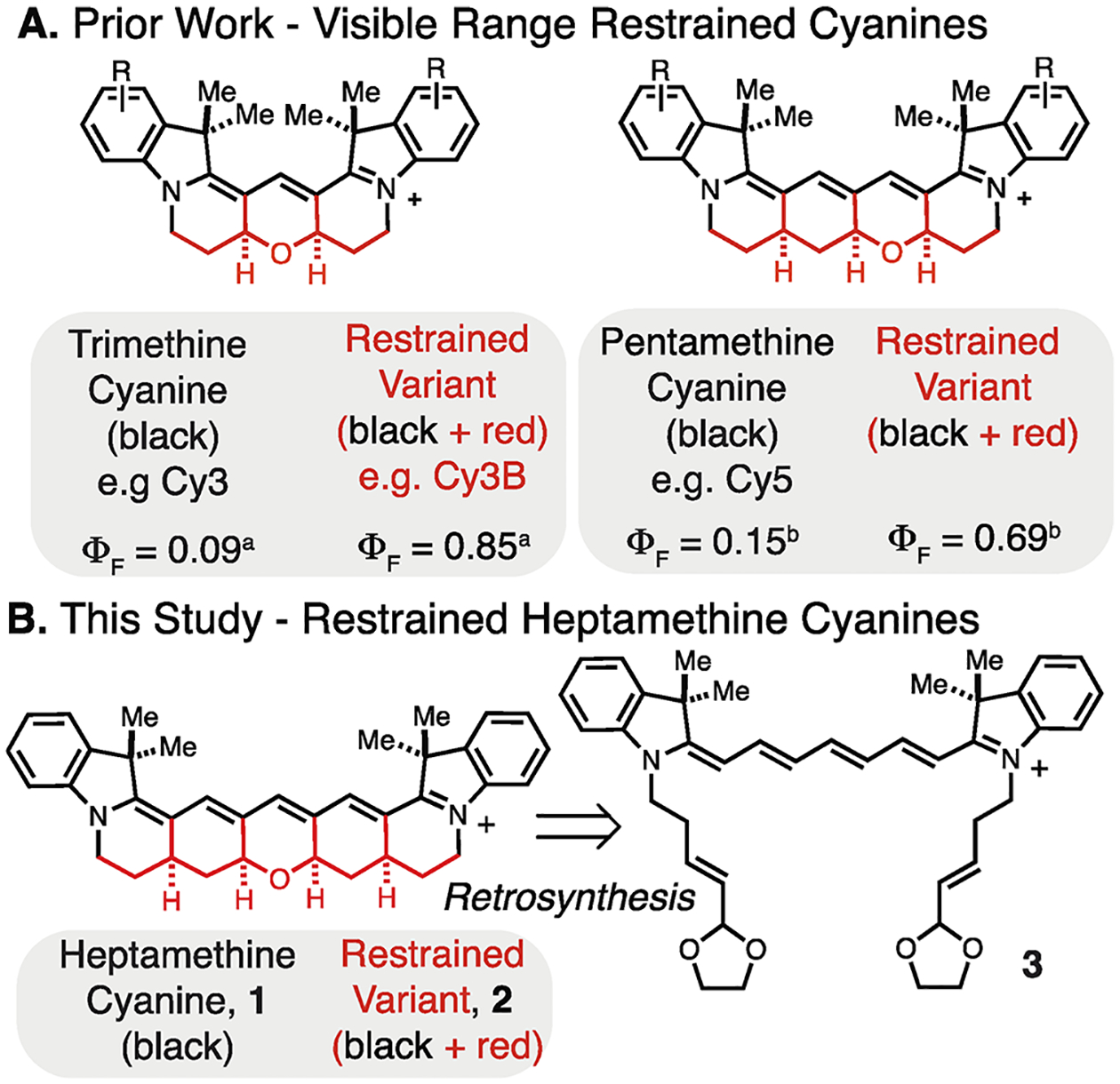
Prior restrained cyanines (A) and this study (B). aΦF values in H2O.10,17,18bΦF values in MeOH11.
Given the need for brighter NIR fluorophores, improving the photon output of heptamethine cyanines is an important objective. However, the role of photoisomerization was less clear in this case, and several seemingly paradoxical findings were reported. Studies using a benzoxazole heptamethine cyanine observed a strong temperature dependence on emission, which was ascribed to increased photoisomerization with elevated temperature.13 Also consistent with a role for photoisomerization, albeit nonquantitatively, transient spectroscopy studies directly observed the isomerized species, as does as an approach using gas-phase mass spectrometry.14,15 By contrast, a study looking at two highly substituted heptamethine cyanines found that room-temperature emission was independent of solvent viscosity.16 This observation suggests that, at least in these examples, the contribution of photoisomerization to nonradiative singlet-state deactivation is insignificant. Finally, a single theoretical study found that the activation energy for isomerization in the excited state increases with polymethine length, potentially suggesting a less important role of photoisomerization for heptamethine cyanines compared to the shorter variants.10
To address the ambiguity regarding the excited-state chemistry of these broadly used molecules, we have prepared and evaluated the first fully conformationally restrained heptamethine cyanine. We detail the preparation of the indocyanine, 2, through an acid-catalyzed reaction that forms the pentacyclic ring system from an acyclic precursor in a single step (Figure 1B). Time-resolved transient absorption spectroscopy and fluorescence correlation spectroscopy (FCS) both confirm that, unlike the conventional heptamethine cyanine, compound 2 does not undergo excited-state isomerization. However, at ambient temperature, the restrained and parent cyanines exhibit similar ΦF and lifetimes (τF). Moreover, τF does not correlate with solvent viscosity in either molecule. Notably, however, the τF of 2 is nearly insensitive to temperature and at moderately elevated temperatures significantly exceeds that of the highly temperature sensitive unrestrained variant 1. This later observation is attributed to the temperature dependence of internal conversion and not photoisomerization, as suggested previously. Finally, computational studies analyze the impact of chromophore length and conformational restraint on orbital and atomic structures across the tri-, penta-, and heptamethine cyanine series. In total, these studies provide mechanistic insights that clarify the relative contribution of polymethine isomerization and the role of solvent and temperature in the internal conversion kinetics in this important class of fluorescent probes.
RESULTS AND DISCUSSION
In considering the structure of restrained heptamethine cyanines, several possible (poly)-dihydropyran and carbocyclic ring systems are feasible. We first examined the approach shown in reaction 1, which involves the condensation of malonyldialdehyde with compound 4 or its partially cyclized variant to form the restrained cyanine 5. Despite examining a range of protic and Lewis acids with several malonyldialdehyde precursors, no stable polycyclic compounds were isolated. We then developed the retrosynthetic disconnection shown in Figure 1B to compound 3, which only contains a single dihydropyran ring. In this approach, we envisioned carrying out a tandem Michael addition/Prins cyclization cascade to assemble the pentacyclic framework (Figure 1B). In this design, we were motivated by our earlier synthesis of the pentamethine cyanine by the introduction of a carbocyclic ring, and only a single dihydropyran ring was critical to creating a stable molecule.11
The synthesis of 2 was carried out as shown in Scheme 1. We found that previously reported 6 could be converted to 7 in useful yield using the Hoveyda–Grubbs II catalyst.11 Indolenine 7 and cyanine precursor 8 react efficiently to form cyanine 3 using standard cyanine preparative conditions (Ac2O, Et3N, CH2Cl2). The pentacyclization reaction proved difficult to fully optimize but was ultimately found to proceed in a modest, albeit reproducible, yield of 14% through a two-step sequence involving initial formation of a mixture of diastereomers in 1:1 1 M HCl:THF at rt followed by conversion to single, all-syn product 2 using 1:1 0.3 M HCl:MeOH at 60 °C (Figures S1 and S2).
Scheme 1:
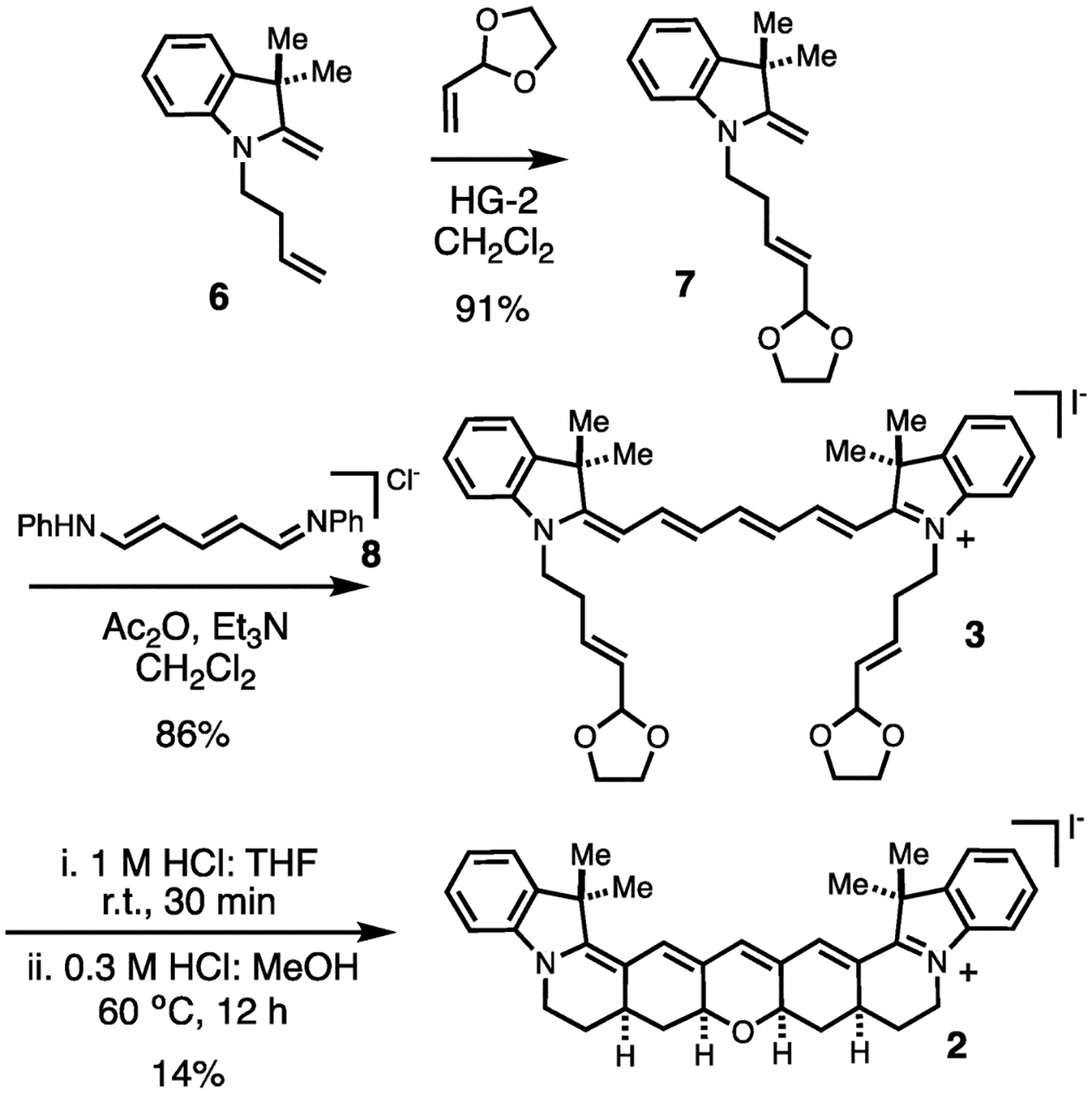
Synthesis of Cyanine 2.
We first evaluated the impact of conformational restraint using spectroscopic methods that directly assess photoisomerization. As an unrestrained comparison for 2, we have used the simplest heptamethine indocyanine hexamethylindotricarbocyanine iodide (HITCI), 1. Transient absorption spectroscopy experiments were carried out with 1 and 2 in ethanol and glycerol at room temperature (Figure 2A and Figure S3A). We were able to observe a transient spectrum around 780 nm with a lifetime of approximately 50 μs for 1 in ethanol, but not in glycerol, that matches the spectrum reported previously for the photoisomer of the same dye in DMSO.19 No transient signals were observed for 2 in either solvent, providing strong evidence that photoisomerization was blocked due to installation of the ring system. We also carried out fluorescence correlation spectroscopy (FCS) measurements, which have been previously used to characterize photoisomerization in Cy5 and other unrestricted cyanines.20,21 While the decay of the conformationally restrained 2 can be fitted with a pure diffusion model (Figure 2B), the decay of the parent heptamethine cyanine 1 shows a component with a small amplitude in the μs timescale not observed for 2 when measured under the same conditions. Fluctuations in the μs timescale were also observed in FCS studies of Cy5 and other unrestricted cyanines, which were attributed to photoisomerization. Although the amplitude of the fast component observed with 1 is quite small, the fact that this component is absent in the FCS decay of the restrained compound 2 suggests that it is indeed due to the formation of a dark isomer. Together, the transient spectroscopy and FCS experiments indicate that photoisomerization occurs in 1 but not in 2. We note that neither transient spectroscopy nor FCS is a suitable method to determine the efficiency of photoisomerization of 1 in a quantitative manner because the absorption cross sections of the isomers are unknown. However, the fact that signals are quite small suggests that the efficiency of photoisomerization may be less than those of other photophysical processes that depopulate the singlet excited state, including fluorescence and internal conversion.
Figure 2.
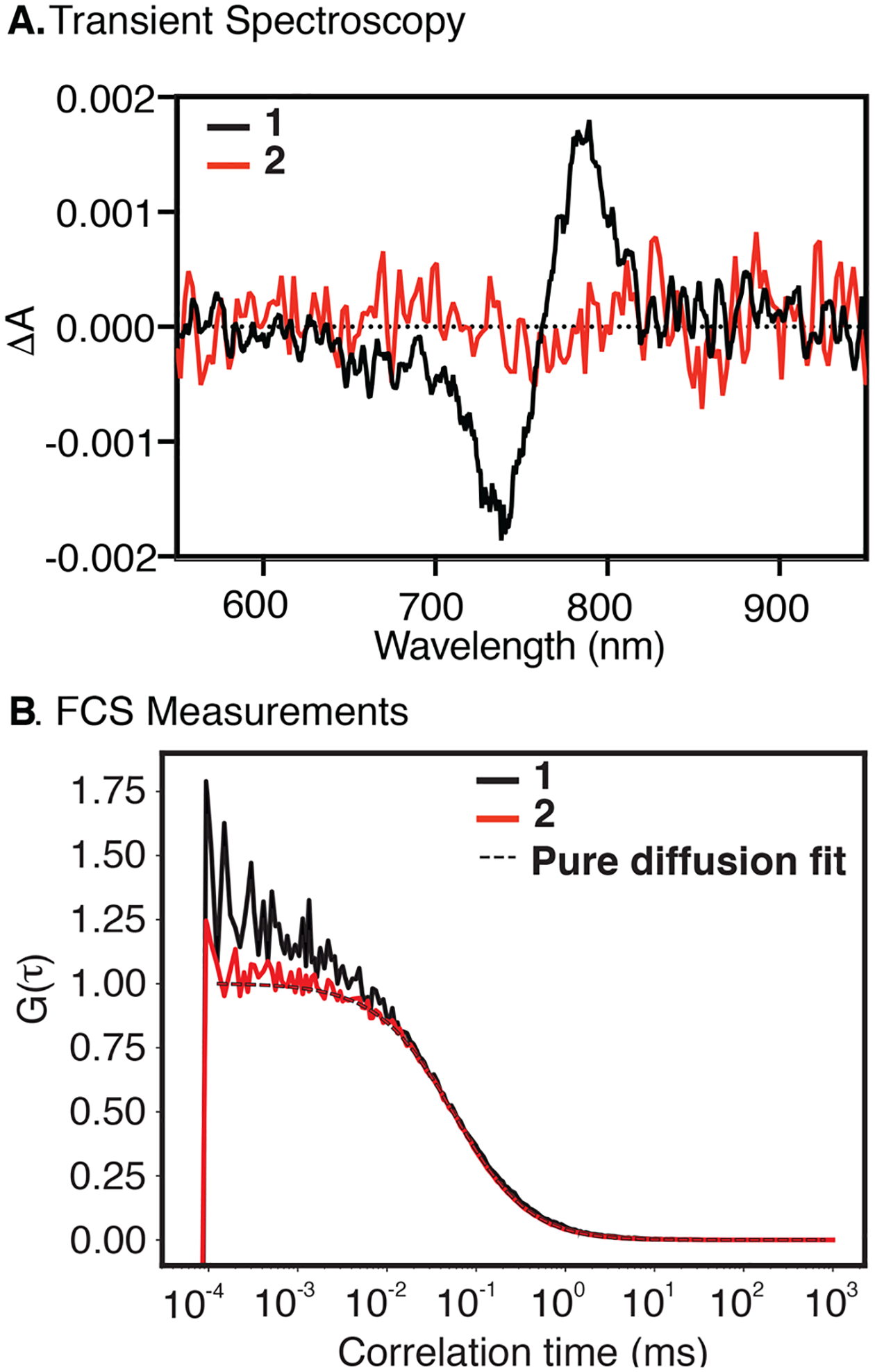
Transient difference spectrum (A, in ethanol) recorded 50 ns after excitation. FCS (B, in aqueous buffer containing ROXS photostabilization system)22 measurements of 1 and 2. The FCS curve of 2 can be described by pure diffusion fit, while in the FCS curve of 1, we observed an additional component in the microsecond timescale which we attribute to a photoisomerization.
We then examined the impact of conformational restraint on the emission properties of 1 and 2. As shown in Table 1 and Table S1, the room-temperature ΦF and τF values are similar between the two dyes (Figures S4 and S5). The differences between the two dyes are maximally a 25% increase for the ΦF of 2 in H2O but typically less than 10% in the other solvents examined. This is in stark contrast to the trimethine and penthamethine members of the family where the restrained cyanines are significantly more fluorescent than their parent compounds. This suggests that, in contrast to the shorter cyanines, isomerization does not play a major role in deactivating the excited state of 1. Indeed, the excited-state lifetime of 1 does not increase with increasing solvent viscosity as has been observed for many trimethine and penthamethine cyanines (Figure S9B and Table S1).23 For example, while the fluorescence lifetime of trimethine indocyanine is ca. 18-fold greater in glycerol than in water, the fluorescence lifetime of 2 is shorter in glycerol (τF = 0.83 ns) than in all measured solvents except water.24
Table 1.
Key Structures and Spectroscopic Properties of 1 and 2
| ||||||
|---|---|---|---|---|---|---|
| compound | solvent | ε (M−1 cm−1) | ΦF | τF(ns) | ||
| 1 | methanol | 740 | 270,000 | 772 | 0.24 | 0.91 |
| glycerol | 751 | 165,000 | 776 | 0.26 | 0.83 | |
| DCM | 756 | 266,000 | 788 | 0.46 | 1.6 | |
| H2Oa | 736 | 107,000 | 771 | 0.064 | 0.37 | |
| D2Oa | 735 | 136,000 | 771 | 0.20 | 0.97 | |
| 2 | methanol | 782 | 265,000 | 803 | 0.29 | 0.93 |
| glycerol | 794 | 134,000 | 808 | 0.28 | 0.83 | |
| DCM | 775 | 303,000 | 811 | 0.40 | 1.7 | |
| H2Oa | 775 | 106,000 | 795 | 0.081 | 0.38 | |
| D2Oa | 770 | 107000 | 791 | 0.18 | 1.2 | |
w/ 5% DMSO.
To analyze the role of solvent in greater detail, the rate constant for nonradiative deactivation (knr) was calculated from ΦF and τF as knr = (1 – ΦF)/τF (Figure S4 and Table S1). Experiments with various trimethine and penthamethine cyanines show an excellent correlation between knr and solvent viscosity. This observation has been interpreted in terms of frictional effects on the photoisomerization reaction of the unrestrained cyanines.23,25 In contrast, knr values do not correlate with solvent viscosity for either 1 or 2 (Figure 3A). Most dramatically, knr values for 1 are almost the same in glycerol (η = 1412 cp) and methanol (η = 0.54 cp), and the differences between 1 and 2 are very small. These observations indicate that the rate-limiting step in the deactivation of the excited state of 1 does not involve large molecular rearrangements. Instead, there is a clear correlation between knr and solvent polarity (Figure 3B), which has not been observed with the shorter cyanine dyes. Values of knr are higher in protic solvents compared to aprotic solvents of the same dielectric constant, suggesting hydrogen-bond-assisted contributions to the nonradiative deactivation process. Differences are particularly striking for τF and ΦF in D2O vs H2O for 1 and 2 (Figure 3B and Table S1), as observed previously by Heilemann et al. for several cyanines.26 Overall, these observations highlight the significant role that internal conversion plays for singlet-state deactivation in the NIR range and the dramatic role of solvent in mediating this process.
Figure 3.
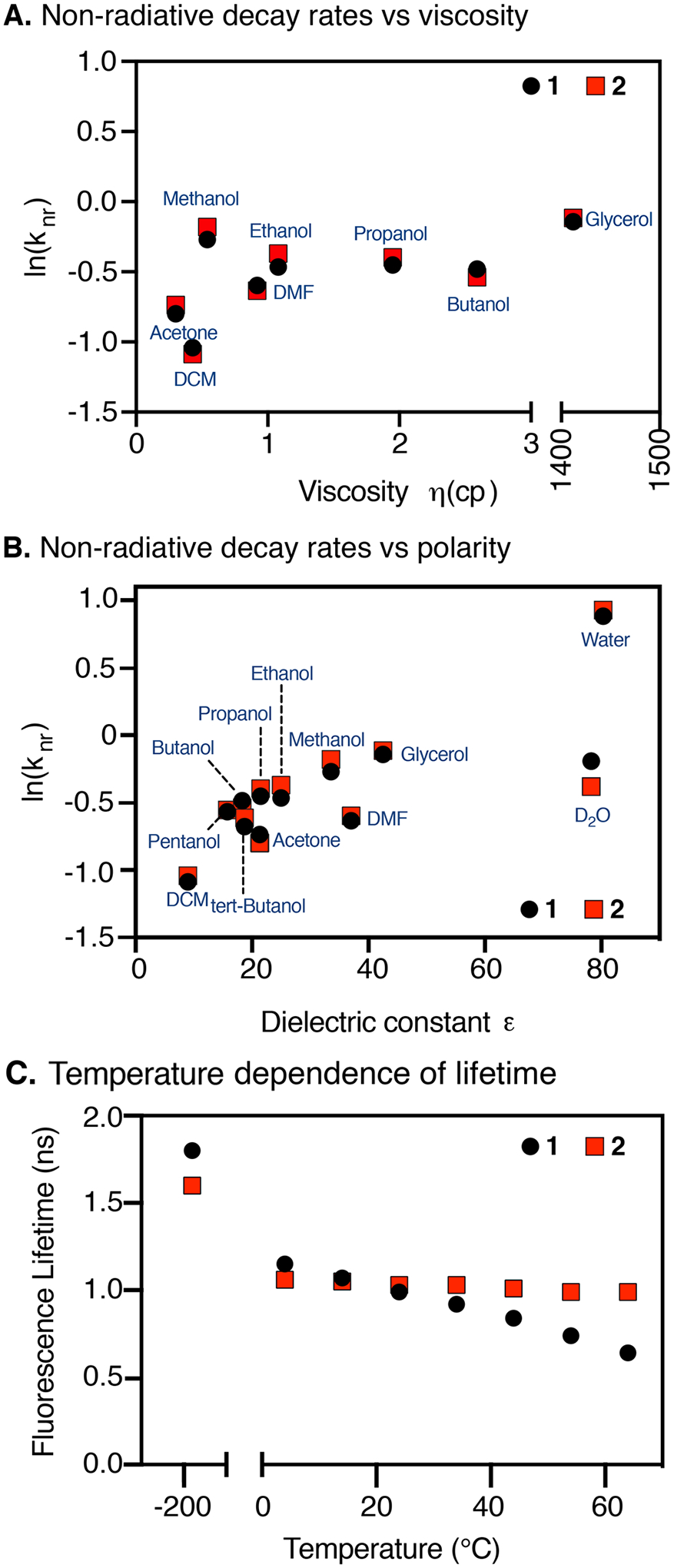
Relationship between ln(knr) vs (A) viscosity and (B) dielectric constant. (C) Temperature dependence of fluorescence lifetimes of 1 and 2 (in EtOH). Note the break in the x-axis of panel (A) to accommodate the glycerol data point.
We then examined the role of temperature in the photon output of these dyes. Temperature-dependent lifetime measurements of 1 and 2 in ethanol are shown in Figure 3C (and Figure S5). The τF of 2 is fairly insensitive to temperature, but the τF of 1 decreases with increasing temperature. Interestingly, this observation is similar to that made in the trimethine and pentamethine series where this observation was linked to the photoisomerization of these dyes. In fact, this dependence has been used to calculate the activation energy of photoisomerization.27 However, in the case of 1, this interpretation is inconsistent with the observation made above that τF and knr are independent of solvent viscosity. To resolve this apparent disconnect, we suggest the observed differences between 1 and 2 are due to temperature effects on the internal conversion rate of 1, which are minimized in the conformationally restrained compound, 2. Thus, conformational restraint, which rigidifies the heptamethine cyanine chromophore, also limits the impact of increased temperature on internal conversion. It is interesting to note that the τF measured at 77 K is lower for 2 than for 1, (1.6 vs 1.8 ns, respectively), indicating that conformational restriction has a negative impact on the intrinsic radiative lifetime of the dye. This rationalizes the observation that the two dyes have very similar lifetimes at room temperature despite the fact that knr increases more steeply with temperature for 1 than for 2.
These experimental results prompted us to carry out the first computational analysis seeking to compare restrained and conventional cyanines. Full details and data are provided in the Supporting Information, but in outline, we have used time-dependent density functional theory28,29 with the B3LYP functional30–32 (TDDFT-B3LYP) and occupation restricted multiple active space with second-order perturbation theory (ORMAS-PT2).33,34 Solvent water effects were included via the polarizable continuum model (PCM),35–37 and the GAMESS package was used for all calculations.38,39 We note that TDDFT methods have been used broadly to predict various aspects of cyanine chemistry despite a systematic overestimation of orbital energies.40,41 In earlier efforts, we found that ORMAS-PT2-PCM accurately modeled modified heptamethine cyanine absorbance energies,42 and we sought to further compare the two methods in these studies. We found that both methods predict absorbance and emission energies (Table S2 and Figure S6) that match exactly the experimentally observed trends, viz., (1) trimethine > pentamethine > heptamethine and (2) conventional > restrained. Overall, ORMAS-PT2-PCM absorbance energies are closer to experimental than TDDFT-B3LYP-PCM (mean unsigned errors (MUEs) are 0.143 and 0.377 eV, respectively), but surprisingly, emission predictions are even more accurate for both (MUEs = 0.078 and 0.057, respectively). It is not clear why computed emission energies are more reliable; however, we note that the error differences cause the TDDFT-B3LYPPCM Stokes shifts to be less accurate than ORMAS-PT2-PCM. Nonetheless, both methods predict the correct trends, and importantly (for discussion below), TDDFT singlet excited states represent almost pure single electron jumps from the highest occupied molecular orbital to the lowest unoccupied molecular orbital, i.e., HOMO → LUMO (see the Supporting Information).
We then examined the orbital structure to understand why there is a bathochromic shift upon cyanine restraint and why its magnitude increases when going from trimethine to pentamethine to heptamethine (Table S2). As shown in Figure 4A, the B3LYP-PCM HOMOs and LUMOs appear mostly similar for the representative heptamethine case, albeit with a notable difference. Specifically, in the HOMO of the restrained variant, there is additional orbital density on the CH σ-bonds adjacent to the unsaturated chromophore. However, when examining the orbital energies (Figure 4B), we find that HOMOs become destabilized when going from parent to restrained and that this destabilization gets larger with increasing cyanine length. LUMO energies decrease with cyanine length and increase slightly when going from conventional to restrained, but these changes are smaller than for the HOMOs. These orbital energy changes serve to explain the bathochromic shift of the restrained structures. In examining the impact of restraint on the structures of these compounds, we observed a clear trend in which the bond lengths of the core chromophore are slightly lengthened upon constraint (up to 0.008 Å) due to a compression of the bond angles (up to 4°) (Figure S7). The magnitude of these effects correlates with chromophore length, being greatest in the heptamethine series.
Figure 4.
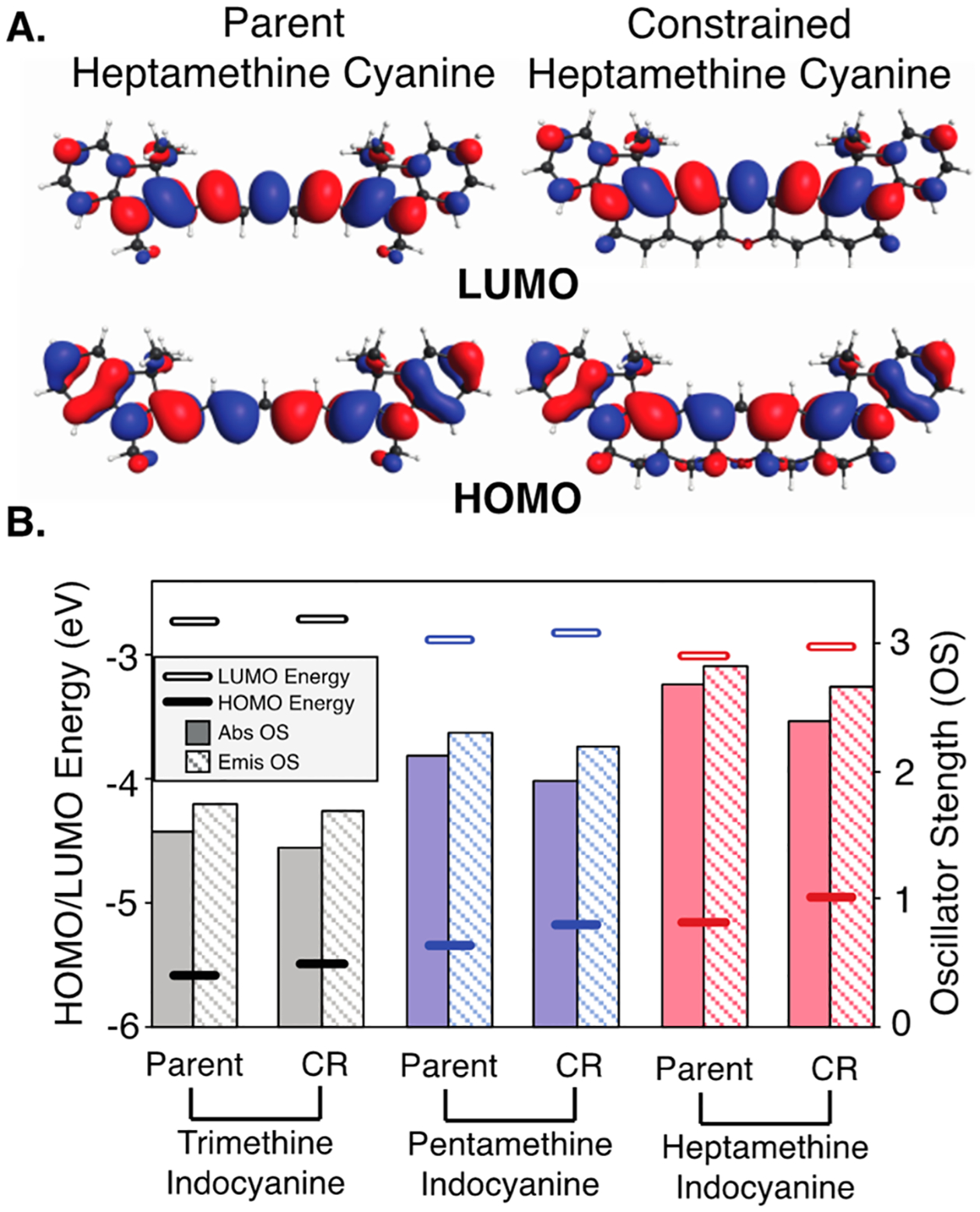
(A) HOMO/LUMO plots for 1 and 2 (contour values = 0.02 (electron/bohr3)1/2); (B) computed B3LYP-PCM HOMO (lower bars) and LUMO (higher bars) energies (in eV, left axis scale) and TDDFT-B3LYP-PCM oscillator strengths (OS) for absorbance (solid columns) and emission (striped columns) (right axis scale). Parent = conventional cyanine; CR = conformationally restrained cyanine.
Finally, we looked at TDDFT-B3LYP-PCM oscillator strengths, which is a dimensionless value to express the quantum probability of the transition between ground and excited states (Figure 4B). Computed absorbance (excitation) and emission (deexcitation) oscillator strengths decrease by about 5–10% upon ring constraint, which may explain why the absorbance cross sections (ε) of the restrained dyes are modestly lower across the series. Additionally, reduced deexcitation oscillator strengths predict lower emission (ΦF) for the restrained dyes relative to unrestrained cyanines. Both of these features likely result, at least in part, from HOMO delocalization onto the CH σ-bonds adjacent to the unsaturated chromophore leading to loss of pure π-symmetry and an energy penalty due to introduced nodes. Of note, the emission oscillator strengths for conventional cyanines do not account for the impact of excited-state isomerization on emission. This explains why the impact of these computational observations is particularly apparent in low-temperature emission comparison of 1 and 2, where isomerization has been prevented in both cases and the τF of 1 exceeds that of 2. In total, these studies demonstrate that conformational restraint impacts not only the capacity for photoisomerization but also imposes notable effects on the chromophore and orbital structure.
CONCLUSIONS
Here, we detail the application of cyanine conformational restraint in the heptamethine scaffold. These studies reveal several features that distinguish the heptamethine cyanines from their trimethine and pentamethine congeners. First, while solvent viscosity strongly correlates with emission for unrestrained tri- and pentamethine cyanines, it does not in either the parent or restrained heptamethine case. This observation serves as conclusive evidence that photoisomerization does not meaningfully impact the room-temperature excited-state chemistry of the broadly used heptamethine cyanine dyes. While solvent viscosity is independent, solvent polarity strongly correlates with heptamethine emission—a trend that is not true for shorter cyanines. This latter observation highlights the outsized role that solvent-mediated pathways, particularly aqueous solvents, have on internal conversion rates of these NIR fluorophores. Finally, despite not impacting photoisomerization, restrained, but not unrestrained, heptamethine cyanines exhibit nearly temperature-independent emission. This indicates that the rigidifying ring system dramatically decreases the temperature dependence of internal conversion. The minimal temperature sensitivity of these dyes may have significant utility in certain contexts.
These studies have significant bearing on the future design of NIR dyes for biological use. As indicated by our computational studies, the placement of CH-bonds adjacent to the polymethine chromophore can impact the HOMO/LUMO orbital structure and oscillator strength. These effects should be considered alongside the standard considerations of unsaturated chromophore design. Furthermore, studies seeking to improve the photon output of NIR dyes, particularly in aqueous biological settings, should be conscious of the magnitude of solvent effects on internal conversion kinetics. One solution is the use of encapsulation strategies using engineered proteins or other organic/inorganic frameworks that serve to alter the local environment. While these have been studied extensively with visible-range fluorophores,43,44 related efforts with NIR fluorophores are likely to be constructive and merit further investigation.
EXPERIMENTAL SECTION
General Materials and Methods.
All commercially obtained reagents were used as received. 2,3,3-Trimethyl indoline, N-[5-(phenylamino)-2,4-pentadienylidene]aniline monohydrochloride, and Hoveyda–Grubbs II catalyst were purchased from Sigma-Aldrich (St. Louis, MO). Flash column chromatography was performed using reversed phase (100 Å, 20–40 micron particle size, RediSep Rf Gold Reversed-phase C18 or C18Aq) on a CombiFlash Rf 200i (Teledyne Isco, Inc., Lincoln, NE). High-resolution LC/MS analyses were conducted on a ThermoFisher LTQ-Orbitrap-XL hybrid mass spectrometer system with an Ion MAX API electrospray ion source in negative ion mode. Analytical LC/MS was performed using a Shimadzu LC/MS-2020 Single Quadrupole utilizing a Kinetex 2.6 μm C18 100 Å (2.1 × 50 mm) column obtained from Phenomenex, Inc. (Torrance, CA). Runs employed a gradient of 0 → 90% MeCN/0.1% aqueous formic acid over 4.5 min at a flow rate of 0.2 mL/min. 1H NMR and 13C NMR spectra were recorded on Bruker spectrometers (at 400 or 500 MHz or at 100 or 125 MHz) and are reported relative to deuterated solvent signals. Data for 1H NMR spectra are reported as follows: chemical shift (δ ppm), multiplicity, coupling constant (Hz), and integration. Data for 13C NMR spectra are reported in terms of chemical shift. Absorbance curves were obtained on a Shimadzu UV-2550 spectrophotometer operated by UVProbe 2.32 software. Data analysis and curve fitting were performed using MS Excel 2011 and GraphPad Prism 7.
Synthesis of the Dioxolane Adduct (7).
To a solution of 6 (300 mg, 0.83 mmol, 1.0 equiv.), prepared according to ref 11 and confirmed by NMR, and Hoveyda–Grubbs II catalyst (50 mg, 0.082 mmol, 0.10 equiv.) in anhydrous DCM (4 mL, 0.2 M) was added 2-vinyl-1,3-dioxolane (420 μL, 4.13 mmol, 5 equiv.) under argon after degassing with vacuum. The reaction was refluxed in an oil bath at 40 °C for 20 h and reapplied to static vacuum/argon every 30 min for the first 4 h. The progress of the reaction was monitored by LC/MS, and upon completion, the reaction was cooled to room temperature and quenched with saturated aqueous NaHCO3 (4 mL). The biphasic mixture was then separated, extracted with DCM (3 × 30 mL), dried (Na2SO4), and concentrated in vacuo. The crude reaction mixture was then purified by normal-phase column chromatography (40 g silica column, 0–50% Hex/EtOAc) to afford the desired product 7 (215 mg, 0.75 mmol, 91% yield) as a pale-pink oil. 1H NMR (500 MHz, CDCl3) δ 7.13 (td, J = 7.7, 1.2 Hz, 1H), 7.11–7.08 (m, 1H),6.77 (td, J = 7.4, 1.1 Hz, 1H), 6.54 (d, J = 7.8 Hz, 1H), 5.98 (dt, J =15.2, 6.9 Hz, 1H), 5.60 (ddt, J = 15.5, 6.2, 1.4 Hz, 1H), 5.22 (d, J =6.2 Hz, 1H), 4.02–3.93 (m, 2H), 3.92–3.86 (m, 4H), 3.62–3.56 (m, 2H), 2.44 (ddd, J = 14.3, 7.2, 1.3 Hz, 2H), 1.35 (s, 6H). 13C{1H} NMR (126 MHz, CDCl3) δ 161.2, 145.5, 137.5, 133.8, 128.4, 127.5, 121.9, 118.4, 105.1, 103.7, 73.4, 64.9, 44.2, 41.3, 30.1, 28.5. HRMS (ESI) m/z: [M + H]+ Calcd for C18H23NO2 286.1802; Found 286.1813.
Synthesis of the Bisindole Cyanine Adduct (3).
To a solution of 7 (300 mg, 1.1 mmol, 3 equiv.), N-[5-(phenylamino)-2,4-pentadienylidene]aniline monohydrochloride (100 mg, 0.35 mmol, 1.0 equiv.), and Et3N (250 μL, 1.8 mmol, 5 equiv.) in anhydrous DCM (1.75 mL, 0.2 M) was added Ac2O (100 μL, 1.052 mmol, 3 equiv.). The reaction mixture was left to stir at room temperature for 12 h and then heated to 40 °C for an additional 3 h. The progress of the reaction was monitored by LC/MS, and upon completion, the reaction was cooled to room temperature and quenched with NaHCO3 (2 mL). The biphasic mixture was then separated, extracted over DCM (3 × 15 mL), and concentrated in vacuo. The crude reaction mixture was then redissolved in a 1:1 mixture of DCM (20 mL) and 1 M NaI (20 mL) and stirred vigorously over 2 h. Next, the DCM layer was separated, washed with H2O (10 mL), dried (Na2SO4), and concentrated in vacuo. The crude reaction mixture was purified via normal-phase column chromatography (24 g silica column, 0–10% DCM/MeOH) to yield 3 (230 mg, 0.302 mmol, 86% yield) as a dark blue solid. 1H NMR (500 MHz, MeOD) δ 7.93 (t, J =13.1 Hz, 2H), 7.46 (dd, J = 7.5, 1.1 Hz, 2H), 7.39 (td, J = 7.8, 1.2 Hz, 2H), 7.27 (d, J = 7.9 Hz, 2H), 7.23 (t, J = 7.4 Hz, 2H), 6.58 (t, J =12.7 Hz, 2H), 6.30 (d, J = 13.6 Hz, 2H), 5.96 (dt, J = 15.0, 7.3 Hz, 2H), 5.47 (dd, J = 15.9, 6.3 Hz, 2H), 5.08 (d, J = 5.8 Hz, 2H), 4.20 (t, J = 6.9 Hz, 4H), 3.84–3.79 (m, 4H), 3.79–3.75 (m, 4H), 2.66–2.53 (m, 4H), 1.69 (s, 12H). 13C{1H} NMR (126 MHz, MeOD) δ 173.4, 157.7, 153.0, 143.6, 142.5, 132.3, 131.9, 129.7, 127.2, 126.0, 123.4, 112.0, 105.4, 104.2, 100.1, 65.8, 50.4, 43.8, 31.2, 28.1. HRMS (ESI) m/z: [M]+ Calcd for C41H49N2O4 (M)+ 633.3687; Found 633.3669.
Synthesis of a Diastereomeric Mixture of the Constrained Cyanine.
To a dark blue solution of 3 (50 mg, 0.069 mmol, 1.0 equiv.) in THF (1 mL) was added 1 M HCl (1 mL). Upon addition of acid, the reaction mixture immediately turned dark green. The reaction mixture was left to stir at room temperature, and the progress was monitored by LC/MS analysis. Significant product formation was seen in 30 min, after which time the reaction mixture was quenched with saturated aqueous NaHCO3 (1 mL), extracted over DCM (3 × 15 mL), dried (Na2SO4), and concentrated in vacuo. The crude reaction mixture was then redissolved in a 1:1 mixture of DCM (15 mL) and 1 M NaI (15 mL) and stirred vigorously over 2 h. Next, the DCM layer was separated, washed with H2O (10 mL), dried (Na2SO4), and concentrated in vacuo. The concentrate was purified via normal-phase column chromatography (4 g silica column, 0–10% DCM/MeOH) to yield a diastereomeric mixture of the desired product (14 mg, 0.021 mmol, 31% yield) as a dark green solid.
Equilibration of a Diastereomeric Mixture Yielding the Single Diastereomer (2).
To a solution of the diastereomeric mixture from the previous reaction (15 mg, 0.023 mmol, 1 equiv.) in MeOH (1.5 mL) was added aqueous HCl (0.3 M, 4.5 mL). The reaction was heated to 60 °C in an oil bath and left to stir overnight. After 2 h, the reaction was quenched with NaHCO3 (4.5 mL), extracted over DCM (3 × 30 mL), dried (Na2SO4), and concentrated in vacuo. The crude reaction mixture was then redissolved in a 1:1 mixture of DCM (15 mL) and 1 M NaI (15 mL) and stir vigorously over 2 h. Next, the DCM layer was separated, washed with H2O (10 mL), dried (Na2SO4), and concentrated in vacuo. The concentrate was then purified via normal-phase column chromatography (4 g silica column, 0–10% DCM/MeOH) to yield a single diastereomer 2 (6.9 mg, 0.015 mmol, 46% yield) as a dark green solid. 1H NMR (500 MHz, MeOD) δ 7.50 (s, 2H), 7.47 (d, J = 6.9 Hz, 2H), 7.39 (td, J =7.8, 1.2 Hz, 2H), 7.24 (t, J = 7.4 Hz, 2H), 7.22 (s, 1H), 7.21 (m, 2H),4.58 (dd, J = 11.4, 4.8 Hz, 2H), 4.28 (dd, J = 13.7, 4.1 Hz, 2H), 3.80 (td, J = 13.2, 4.0 Hz, 2H), 2.80 (tt, J = 12.4, 4.1 Hz, 2H), 2.44 (dt, J =11.4, 4.3 Hz, 2H), 2.40–2.32 (m, 2H), 1.76–1.73 (m, 2H), 1.70 (d, J = 4.5 Hz, 12H), 1.45 (q, J = 11.7 Hz, 2H). 13C{1H} NMR (126 MHz, MeOD) δ 167.9, 143.6, 143.1, 142.5, 141.3, 132.7, 129.7, 126.1, 123.3, 115.4, 111.1, 73.8, 50.0, 44.1, 36.5, 32.6, 28.8, 28.2, 27.7. HRMS (ESI) m/z: [M+] Calcd for C37H39N2O 527.3057; Found 527.3043.
Absorption and Steady-State Fluorescence Spectroscopy.
Absorption spectra were measured using a Shimadzu UV-1700 PharmaSpec UV–vis spectrophotometer. Fluorescence emission spectra for absolute quantum yield determinations were recorded on a PTI QuantaMaster steady-state spectrofluorometer operated by FelixGX 4.2.2 software, with 4 nm excitation and emission slit widths and a 0.1 s integration rate. The absolute quantum yields (ϕF) of 1 and 2 in DCM were measured using an integrating sphere Quantaurus-QY spectrometer (model C11374) from Hamamatsu. Measurements were carried out using dilute samples (A < 0.1), and self-absorption corrections were performed using the instrument’s software.
Fluorescence emission spectra for relative quantum yield determinations were measured on a Nanolog fluorometer (Horiba Jobin Yvon). Fluorescence spectra were corrected for wavelength-dependent detector responsivity using correction curves determined with a calibrated light source. Relative fluorescence quantum yields were determined for 1 and 2 in a variety of solvents using the corresponding compounds in DCM as reference (ϕF = 0.455 and ϕF = 0.401 for 1 and 2 in DCM, respectively).
Fluorescence quantum yields (ϕF) were determined as , where the subscript R refers to the standard used as reference (i.e., the compounds dissolved in DCM), I represents the integrated corrected fluorescence spectrum, A is the absorbance of the solution at the excitation wavelength used to determine I, and n is the refractive index of the solvent. Absorbances at excitation wavelengths (680 nm for 1 and 725 nm for 2) were kept around A ~0.05 in all fluorescence experiments to avoid inner filter artifacts. To improve the precision in the determination of A for the calculation of ϕF, absorbances were measured for solutions displaying A ~0.5 at the shoulder (i.e., excitation wavelength) of the spectrum, and precise 1:10 dilutions were performed before the fluorescence determinations.
Time-Resolved Fluorescence Spectroscopy.
A time-correlated single-photon counting (TCSPC) setup used a Fianium super-continuum laser (SC-450–4-PP) operating at 20 MHz for the excitation light. The excitation wavelength was set at 700 nm for all the lifetime measurements. The emission wavelengths were set at 775 and 800 nm for 1 and 2 lifetime measurements, respectively. The detector was a Hamamatsu MCP-PMT R3809U-50 photomultiplier tube, and data was recorded with a Becker and Hickl TCSPC Timing Card (B&H SPC830). The instrument response function (IRF) was measured by scattering excitation light off a 3% Ludox solution. For the temperature-based experiment, the temperature was controlled using a water circulator and was measured inside the cuvette with a thermocouple. Intensity decays were fitted using ASUFIT, a MATLAB-based program developed at ASU (http://www.public.asu.edu/~laserweb/asufit).
Transient Absorption Measurements.
Transient absorption spectra were measured using an EOS nanosecond-to-millisecond transient absorption spectrometer in the range of 500–1000 nm. The excitation source consists of a KHz regeneratively amplified Ti:S laser and two optical parametric amplifiers (Spectra Physics). The excitation wavelength was set at 750 nm for these experiments. The measurement time window was set at 100 μs. Initial spectra were averaged as mentioned in the legends of the graph. Intensity decays at specific wavelengths were fitted using the ASUFIT.
FCS Measurements.
FCS measurements were carried out on a custom-built confocal microscope based on an Olympus IX-71. The laser (LDH 760 D-C, PicoQuant GmbH, Germany) was cleaned up using a 740/40 bandpass filter (Thorlabs GmbH) and focused onto the sample by an oil-immersion objective (UPLSAPO100XO, NA 1.40, Olympus Deutschland GmbH). A dichroic beam splitter (Quad Line Beamsplitter zt488/543/635/730rpc, Chroma Technology Corporation) separated the excitation from the emission light. The emission light was focused on a 50 μm pinhole (Thorlabs GmbH) and sent through a bandpass filter (810/80, Thorlabs GmbH). Emission light was collected by an avalanche photodiode (SPCM-AQRH-14-TR, Excelitas Technologies GmbH & Co. KG) and registered by a single-photon counting module (HydraHarp 400, PicoQuant GmbH). A commercial software package was used to record time traces and to calculate and fit correlation curves (SymphoTime 64, PicoQuant GmbH). We employed the built-in FLCS correction to suppress the detector after pulsing effects.
Samples were diluted to nanomolar concentrations and measured in a reducing and oxidizing buffer system with enzymatic oxygen removal consisting of 90% buffer A (100 mM MgCl2, 40 mM Tris, 2 mM Trolox/Troloxquinone, 1% w/v glucose) and 10% buffer B (glucose oxidase (1 mg/mL), 0.4% (v/v) catalase (50 μg/mL), 30% glycerol, 12.5 mM KCl in 50 mM TRIS). A laser power of 25 μW and a repetition rate of 80 MHz were used. The confocal volume was placed 10 μm into the solution. Each sample was measured for 30 min.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research. M.J.S. thanks Dr. Joseph Barchi, NCI-CCR, for NMR assistance and Dr. James Kelley, NCI-CCR, for mass spectrometric analysis. Dr. Luke Lavis, Janelia Research Campus, Howard Hughes Medical Institute, is acknowledged for assistance with fluorescence quantum yield determination. This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. M.L. acknowledges use of the Ultra-Fast Laser Spectroscopy Facility at ASU. P.T. acknowledges support by the DFG (excellence cluster e-conversion). V.G. thanks the support of the Alexander von Humboldt Foundation.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.0c00236.
NMR characterization of 1, 2, and other compounds; photophysical measurements of 1 and 2; computational methods and results (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.joc.0c00236
The authors declare no competing financial interest.
Contributor Information
Nikita Kumari, School of Molecular Sciences and The Biodesign Institute at Arizona State University, Tempe, Arizona 85287, United States.
Joseph Ivanic, Advanced Biomedical Computational Science, Frederick National Laboratory for Cancer Research, Frederick, Maryland 21702, United States.
Marcia Levitus, School of Molecular Sciences and The Biodesign Institute at Arizona State University, Tempe, Arizona 85287, United States.
Martin J. Schnermann, Chemical Biology Laboratory, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Frederick, Maryland 21702, United States
REFERENCES
- (1).Hilderbrand SA; Weissleder R Near-infrared fluorescence: application to in vivo molecular imaging. Curr. Opin. Chem. Biol 2010, 14, 71–79. [DOI] [PubMed] [Google Scholar]
- (2).Zhou X; Lai R; Beck JR; Li H; Stains CI Nebraska Red: a phosphinate-based near-infrared fluorophore scaffold for chemical biology applications. Chem. Commun 2016, 52, 12290–12293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Liu J; Sun Y-Q; Zhang H; Shi H; Shi Y; Guo W Sulfone-Rhodamines: A New Class of Near-Infrared Fluorescent Dyes for Bioimaging. ACS Appl. Mater. Interfaces 2016, 8, 22953–22962. [DOI] [PubMed] [Google Scholar]
- (4).Cosco ED; Caram JR; Bruns OT; Franke D; Day RA; Farr EP; Bawendi MG; Sletten EM Flavylium Polymethine Fluorophores for Near- and Shortwave Infrared Imaging. Angew. Chem., Int. Ed 2017, 56, 13126–13129. [DOI] [PubMed] [Google Scholar]
- (5).(a) Levitus M; Ranjit S Cyanine dyes in biophysical research: the photophysics of polymethine fluorescent dyes in biomolecular environments. Q. Rev. Biophys 2011, 44, 123–151. [DOI] [PubMed] [Google Scholar]; (b) Stennett EMS; Ciuba MA; Levitus M Photophysical processes in single molecule organic fluorescent probes. Chem. Soc. Rev 2014, 43, 1057–1075. [DOI] [PubMed] [Google Scholar]
- (6).(a) Gorka AP; Nani RR; Schnermann MJ Cyanine polyene reactivity: scope and biomedical applications. Org. Biomol. Chem 2015, 13, 7584–7598. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gorka AP; Schnermann MJ Harnessing cyanine photooxidation: from slowing photobleaching to near-IR uncaging. Curr. Opin. Chem. Biol 2016, 33, 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gorka AP; Nani RR; Schnermann MJ Harnessing Cyanine Reactivity for Optical Imaging and Drug Delivery. Acc. Chem. Res 2018, 51, 3226–3235. [DOI] [PubMed] [Google Scholar]
- (7).(a) Mujumdar RB; Ernst LA; Mujumdar SR; Lewis CJ; Waggoner AS Cyanine dye labeling reagents: sulfoindocyanine succinimidyl esters. Bioconjugate Chem. 1993, 4, 105–111. [DOI] [PubMed] [Google Scholar]; (b) Mujumdar SR; Mujumdar RB; Grant CM; Waggoner AS Cyanine-labeling reagents: Sulfobenzindocyanine succinimidyl esters. Bioconjugate Chem. 1996, 7, 356–362. [DOI] [PubMed] [Google Scholar]
- (8).Waggoner AS; Mujumdar RB Rigidized trimethine cyanine dyes. U.S. PatentPCT/US1998/02666, 2007.
- (9).Sanchez-Galvez A; Hunt P; Robb MA; Olivucci M; Vreven T; Schlegel HB Ultrafast radiationless deactivation of organic dyes: evidence for a two-state two-mode pathway in polymethine cyanines. J. Am. Chem. Soc 2000, 122, 2911–2924. [Google Scholar]
- (10).Cooper M; Ebner A; Briggs M; Burrows M; Gardner N; Richardson R; West R Cy3B™: Improving the performance of cyanine dyes. J. Fluoresc 2004, 14, 145–150. [DOI] [PubMed] [Google Scholar]
- (11).Michie MS; Götz R; Franke C; Bowler M; Kumari N; Magidson V; Levitus M; Loncarek J; Sauer M; Schnermann MJ Cyanine Conformational Restraint in the Far-Red Range. J. Am. Chem. Soc 2017, 139, 12406–12409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Chibisov AK; Zakharova GV; Görner H Effects of substituents in the polymethine chain on the photoprocesses in indodicarbocyanine dyes. J. Chem. Soc., Faraday Trans 1996, 92, 4917–4925. [Google Scholar]
- (13).Aramendia PF; Negri RM; San Roman E Temperature Dependence of Fluorescence and Photoisomerization in Symmetric Carbocyanines. Influence of Medium Viscosity and Molecular Structure. J. Phys. Chem 1994, 98, 3165–3173. [Google Scholar]
- (14).Fouassier J-P; Lougnot D-J; Faure J Transient Absorptions in a Polymethine Laser Dye. Chem. Phys. Lett 1975, 35, 189–194. [Google Scholar]
- (15).Adamson BD; Coughlan NJA; Continetti RE; Bieske EJ Changing the shape of molecular ions: photoisomerization action spectroscopy in the gas phase. Phys. Chem. Chem. Phys 2013, 15, 9540–9548. [DOI] [PubMed] [Google Scholar]
- (16).Soper SA; Mattingly QL Steady-State and Picosecond Laser Fluorescence Studies of Nonradiative Pathways in Tricarbocyanine Dyes - Implications to the Design of near-Ir Fluorochromes with High Fluorescence Efficiencies. J. Am. Chem. Soc 1994, 116, 3744–3752. [Google Scholar]
- (17).Waggoner AS; Mujumdar RB Rigidized trimethine cyanine dyes, PCT/US1998/02666, 2007.
- (18).Sanborn ME; Connolly BK; Gurunathan K; Levitus M Fluorescence Properties and Photophysics of the Sulfoindocyanine Cy3 Linked Covalently to DNA. J. Phys. Chem. B 2007, 111, 11064–11074. [DOI] [PubMed] [Google Scholar]
- (19).Fouassier J-P; Lougnot D-J; Faure J Transient Lifetime Measurements in 1,1′-Diethyl-4,4’-Carbocyanine Iodide (Cryptocya-nine, DCI) Using a Train of Picosecond Pulses. Chem. Phys. Lett 1975, 30, 448–450. [Google Scholar]
- (20).Buschmann V; Weston KD; Sauer M Spectroscopic study and evaluation of red-absorbing fluorescent dyes. Bioconjugate Chem. 2003, 14, 195–204. [DOI] [PubMed] [Google Scholar]
- (21).Widengren J; Schwille P Characterization of photoinduced isomerization and back-isomerization of the cyanine dye Cy5 by fluorescence correlation spectroscopy. J. Phys. Chem. A 2000, 104, 6416–6428. [Google Scholar]
- (22).Vogelsang J; Kasper R; Steinhauer C; Person B; Heilemann M; Sauer M; Tinnefeld P A Reducing and Oxidizing System Minimizes Photobleaching and Blinking of Fluorescent Dyes. Angew. Chem., Int. Ed. Engl 2008, 47, 5465–5469. [DOI] [PubMed] [Google Scholar]
- (23).Korppi-Tommola JEI; Hakkarainen A; Hukka T; Subbi J An Isomerization Reaction of a Cyanine Dye in n-Alcohols: Microscopic Friction and an Excited-State Barrier Crossing. J. Phys. Chem 1991, 95, 8482–8491. [Google Scholar]
- (24).Kumari N; Ciuba MA; Levitus M Photophysical properties of the hemicyanine Dy-630 and its potential as a single-molecule fluorescent probe for biophysical applications. Methods Appl. Fluoresc 2019, 8, No. 015004. [DOI] [PubMed] [Google Scholar]
- (25).Sundstroem V; Gillbro T Viscosity-Dependent Isomerization Yields of Some Cyanine Dyes - a Picosecond Laser Spectroscopy Study. J. Phys. Chem 1982, 86, 1788–1794. [Google Scholar]
- (26).Klehs K; Spahn C; Endesfelder U; Lee SF; Fürstenberg A; Heilemann M Increasing the brightness of cyanine fluorophores for single-molecule and superresolution imaging. ChemPhysChem 2014, 15, 637–641. [DOI] [PubMed] [Google Scholar]
- (27).Åkesson E; Sundström V; Gillbro T Solvent-dependent barrier heights of excited-state photoisomerization reactions. Chem. Phys. Lett 1985, 121, 513–522. [Google Scholar]
- (28).Kohn W; Sham LJ Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev 1965, 140, 1133–1138. [Google Scholar]
- (29).Gross EKU; Kohn W Time-Dependent Density-Functional theory. Adv. Quantum Chem 1990, 21, 255–291. [Google Scholar]
- (30).Becke AD Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar]
- (31).Stephens PJ; Devlin FJ; Chabalowski CF; Frisch MJ Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem 1994, 98, 11623–11627. [Google Scholar]
- (32).Hertwig RH; Koch W On the Parameterization of the Local Correlation Functional. What is Becke-3-LYP? Chem. Phys. Lett 1997, 268, 345–351. [Google Scholar]
- (33).Ivanic J Direct Configuration Interaction and Multiconfigurational Self-Consistent-Field Method for Multiple Active Spaces with Variable Occupations. I. Method. J. Chem. Phys 2003, 119, 9364–9376. [Google Scholar]
- (34).Roskop L; Gordon MS Quasi-Degenerate Second-Order Perturbation Theory for Occupation Restricted Multiple Active Space Self-Consistent-Field Reference Functions. J. Chem. Phys 2011, 135, No. 044101. [DOI] [PubMed] [Google Scholar]
- (35).Miertuš S; Scrocco E; Tomasi J Electrostatic Interaction of a Solute with a Continuum. A direct utilizaion of Ab Initio Molecular Potentials for the Prevision of Solvent Effects. J. Chem. Phys 1981, 55, 117–129. [Google Scholar]
- (36).Li H; Jensen JH Improving the Efficiency and Convergence of Geometry Optimization with the Polarizable Continuum Model: New Energy Gradients and Molecular Surface Tessellation. J. Comput. Chem 2004, 25, 1449–1462. [DOI] [PubMed] [Google Scholar]
- (37).Wang Y; Li H Excited State Geometry of Photoactive Yellow Protein Chromophore: A Combined Conductorlike Polarizable Continuum Model and Time-Dependent Density Functional Study. J. Chem. Phys 2010, 133, No. 034108. [DOI] [PubMed] [Google Scholar]
- (38).Schmidt MW; Baldridge KK; Boatz JA; Elbert ST; Gordon MS; Jensen JH; Koseki S; Matsunaga N; Nguyen KA; Su S; Windus TL; Dupuis M; Montgomery JA Jr. The General Atomic and Molecular Electronic Structure System. J. Comput. Chem 1993, 14, 1347–1363. See http://www.msg.ameslab.gov/advances/updatepaper.html for a current list of features [Google Scholar]
- (39).Gordon MS; Schmidt MW Advances in electronic structure theory. In Theory and Applications of Computational Chemistry: the first forty years; Dykstra CE; Frenking G; Kim KS; Scuseria GE, Eds.; Elsevier: Amsterdam; 2005. [Google Scholar]
- (40).Champagne B; Guillaume M; Zutterman F TDDFT investigation of the optical properties of cyanine dyes. Chem. Phys. Lett 2006, 425, 105–109. [Google Scholar]
- (41).Le Guennic B; Jacquemin D Taking up the cyanine challenge with quantum tools. Acc. Chem. Res 2015, 48, 530–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Nani RR; Gorka AP; Nagaya T; Yamamoto T; Ivanic J; Kobayashi H; Schnermann MJ In Vivo Activation of Duocarmycin-Antibody Conjugates by Near-Infrared Light. ACS Cent. Sci 2017, 3, 329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Szent-Gyorgyi C; Schmidt BF; Creeger Y; Fisher GW; Zakel KL; Adler S; Fitzpatrick JAJ; Woolford CA; Yan Q; Vasilev KV; Berget PB; Bruchez MP; Jarvik JW; Waggoner A Fluorogen-activating single-chain antibodies for imaging cell surface proteins. Nat. Biotechnol 2008, 26, 235–240. [DOI] [PubMed] [Google Scholar]
- (44).Smith BD; Gassensmith JJ; Arunkumar E Dye Encapsulation. in Molecular Encapsulation: Reactions in Constrained Systems; Brinker UH, Mieussert J-L, Wiley: New York, 2010, Ch.11., 309–325. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.