

Abstract
Deregulations in gut microbiota may play a role in vascular and bone disease in chronic kidney disease (CKD). As glomerular filtration rate declines, the colon becomes more important as a site of excretion of urea and uric acid, and an increased bacterial proteolytic fermentation alters the gut microbial balance. A diet with limited amounts of fibre, as well as certain medications (eg phosphate binders, iron supplementation, antibiotics) further contribute to changes in gut microbiota composition among CKD patients. At the same time, both vascular calcification and bone disease are common in patients with advanced kidney disease. This narrative review describes emerging evidence on gut dysbiosis, vascular calcification, bone demineralization and their interrelationship termed the ‘gut‐bone‐vascular axis’ in progressive CKD. The role of diet, gut microbial metabolites (ie indoxyl sulphate, p‐cresyl sulphate, trimethylamine N‐oxide (TMAO) and short‐chain fatty acids (SCFA)), vitamin K deficiency, inflammatory cytokines and their impact on both bone health and vascular calcification are discussed. This framework may open up novel preventive and therapeutic approaches targeting the microbiome in an attempt to improve cardiovascular and bone health in CKD.
Keywords: bone, chronic kidney disease, gut microbiota, vascular calcification
1. INTRODUCTION
The human gut microbiome is composed of trillions of microorganisms including bacteria, fungi, viruses and archaea, which co‐exist in a balanced relationship with their host in healthy state.1, 2 Among others, the microbiome protects against pathogens, modulates the immune system and regulates endogenous metabolism of nutrients, in concert with lifestyle‐related factors.1 The advent of microbial DNA sequencing technologies has allowed profiling of diverse microbial communities in the gut, providing evidence that although its composition displays huge inter‐individual variation, there is a common pattern.3 The human gut microbiota is composed of more than 1000 different microbial species with particular taxonomic characteristics.3 The prevailing phyla composing the usual gut microbiota are Firmicutes, Bacteroidetes, Actinobacteria, Proteobacteria, Fusobacteria and Verrucomicrobia, with the two first corresponding to 90% of gut microbiota.2 Although gut microbiota composition is considered generally stable at short‐term, fluctuations and acute changes may occur both at short‐ and long‐term after exposition to disruptive factors such as diet, medication, ageing and pathological conditions.2, 3
Emerging evidence implicates alterations in gut microbiota (ie gut dysbiosis) to play a central role in the pathogenesis of many diseases including chronic kidney disease (CKD).4 CKD is a noteworthy global public health issue with rising prevalence that affects over 850 million individuals worldwide.5 Common causes of CKD include diabetes, hypertension, glomerulopathies, tubulo‐interstitial nephritis secondary to infections or exposure to nephrotoxic agents, among others.6 As kidney function declines, CKD is characterized by metabolic complications including anaemia, metabolic acidosis and abnormalities in bone and mineral metabolism.7 Abnormalities in bone and mineral metabolism not only lead to an increased risk of fractures,8 but have also been associated with an increased risk of vascular calcification, cardiovascular complications and mortality.9 Uraemia also promotes an imbalance in the intestinal microbiota, resulting in an increased production of toxins and deleterious effects, such as vascular and renal disease progression.10
Although vascular calcification and bone disorders in CKD patients are generally considered to be driven by shared pathophysiological mechanisms, it has only recently been appreciated that gut microbiota may be a major third component influencing both vasculature and bone. This review aims to describe emerging evidence on abnormalities in the vascular system, bone, and gut and their interrelationship termed the ‘gut‐bone‐vascular axis’ in progressive CKD.
2. VASCULAR AND BONE ABNORMALITIES IN CKD
2.1. Vascular calcification
Along with the decline in GFR, a systemic change in mineral metabolism takes place in patients with progressive CKD. These changes are collectively termed Chronic Kidney Disease‐Mineral and Bone Disorder (CKD‐MBD). CKD‐MBD is characterized by one or more abnormalities in circulating minerals or their regulating hormones (eg calcium, phosphorus, PTH and vitamin D), bone abnormalities and vascular calcification.11
Chronic kidney disease is accompanied by an excessively high cardiovascular mortality risk, which may be at least in part be attributed to vascular calcification in the context of CKD‐MBD. Extensive deregulations in bone and mineral metabolism may be observed even in very young dialysis patients and contribute to detrimental outcomes.12 Vascular calcification is an active and highly regulated cellular process, defined by the deposition of calcium‐phosphate crystals within the intima and media layers of the vasculature and/or heart valves.13 Besides well‐known traditional risk factors for vascular calcification such as age, male gender, diabetes, dyslipidaemia, hypertension, smoking and inflammation, vascular calcification in CKD patients (since early stages until after renal replacement therapies) is additionally driven by deregulations in mineral metabolism,14, 15, 16, 17 and phenotypic changes in vascular smooth muscle cells (VSMC) represent the initial step in this pathological process, namely osteochondrogenic differentiation and apoptosis of VSMC.18 Multifaceted intricate mechanisms in CKD‐induced vascular calcification also comprise the instability and liberation of matrix extracellular vesicles containing calcium and phosphate from bone and VSMC, and elastin (most abundant protein in media wall) degradation, promoting calcium deposition.19 Moreover, an unbalanced environment brought by the mineral deregulations impairs the effects of vascular calcification inhibitors (eg pyrophosphate, adenosine, matrix Gla protein, osteopontin, fetuin‐A, osteoprotegerin, bone morphogenetic protein‐7, magnesium, vitamin K, klotho), while enhancing the action of promoters (eg inflammatory cytokines, oxidative stress, uremic toxins, osteocalcin, osteonectin, bone morphogenetic protein‐2, Runx2, secondary calciprotein particles).19, 20, 21, 22, 23 Deregulations in many of these factors have been identified and linked with vascular calcification and adverse clinical outcomes.16, 24, 25 A recent study with CKD patients with different levels of renal function has demonstrated an augmented expression of alkaline phosphatase and Runx2 in VSMC of CKD arteries, an indicative of osteogenic differentiation.26 The role of Wnt inhibitors such as sclerostin and DKK1 (dickkopf‐1) remains controversial.27, 28 Interestingly, many of the above‐mentioned factors are not only involved in vascular calcification, but also regulate bone metabolism, and are implicated in bone abnormalities in CKD.
2.2. Bone disease in chronic kidney disease
Chronic kidney disease is characterized by a strongly elevated facture risk, with dialysis patients exhibiting a risk ranging from 1.5 to 8‐fold in comparison with the general population.29 Between 1992 and 2009, there has been an increase of 50.2% in the incidence of hip and vertebral fractures and of 40.6% in arm and leg fractures requiring hospitalization among haemodialysis patients in the United States.30 A more recent epidemiological study has disclosed an inverse association between estimated glomerular filtration rate (eGFR) and fracture incidence with a hazard ratio of 1.25 (95% CI 1.05‐1.49) for eGFR <60 mL/min/1.73 m2 and 1.65 (95% CI 1.14‐2.37) for eGFR <45 mL/min/1.73 m2.31 Furthermore, the effect of decreased eGFR on fracture incidence was more noticeable in younger and male patients.31 Moreover, haemodialysis patients presenting with a fracture had higher unadjusted rates of death (3.7‐fold) and death/rehospitalization (4.0‐fold) dialysis patients without a fracture.29 The increased risk of fracture has also been observed among CKD patients after kidney transplantation.32, 33 Over the first 3 years following transplantation, the risk of a hip fracture is 34% higher than in dialysis patients, and over the first 5 years post‐transplant one‐fourth will have a fracture.33 A recent meta‐analysis has disclosed an overall fracture incidence rate of 8.95 per 1,000 person‐years among dialysis patients and kidney transplant recipients (KTR).32
2.3. Vascular calcification and bone disease in CKD: Shared aetiology?
The parallel development of bone disease and vascular calcification in CKD patients and the overlapping factors driving these deregulations implicate a shared aetiology, at least in part. A large body of evidence has already documented the coexistence of bone disease and vascular calcification in CKD patients.34, 35, 36 Nevertheless, the mechanisms connecting abnormalities in bone and vasculature remain incompletely understood. Since bone matrix is rich in regulatory factors that are also active in the vasculature, such as osteopontin and MGP, they may be released into the circulation during bone resorption.37 Cells with both osteoblastic and osteoclastic potential have been described in vascular tissues, and bone‐related proteins have been identified in calcified arterial lesions.38 Alternatively, causal factors driving both vascular calcification and bone loss in CKD may exist, supported by the several risk factors common in both disorders, including vitamin K and D abnormalities, chronic inflammation, dyslipidaemia, ageing, hyperparathyroidism, hyperhomocysteinemia, oestrogen deficiency and oxidative stress.37 As discussed in the 2006 KDIGO guidelines, the term CKD‐MBD should not solely be used to describe the syndrome of biochemical and bone abnormalities observed in CKD, but also encompasses the extra‐skeletal calcification occurring in these patients resulting from derangement of complex systems biology involving the kidney, skeleton and cardiovascular system.39
Although a causal relationship has not been conclusively established, CKD‐MBD, and particularly hyperphosphatemia, represents one of the major triggers for vascular calcification.19 Hyperphosphatemia stimulates both PTH and fibroblast growth factor‐23 (FGF23) synthesis, subsequently inducing phosphaturia through an effect on type II renal sodium‐dependent phosphate transporters.18, 19 However, abnormal levels of PTH and FGF23 may be associated with vascular calcification even before the development of hyperphosphatemia. High levels of PTH and FGF23 in CKD promote bone resorption, which may further increase serum calcium‐phosphate product in a vicious circle.40 When vascular smooth muscle cells (VSMC) are exposed to a calcium and phosphorus‐rich environment they undergo osteogenic differentiation becoming bone‐formative cells, namely osteoblast/chondrocyte‐like cells.18 These cells lose their contractile properties but produce a collagen matrix and form calcium‐ and phosphorus‐rich matrix vesicles, which in turn may initiate the mineralization of the vascular wall.18
At the same time, a dynamic bone disease, characterized by markedly reduced bone formation rate and low PTH (low bone remodelling), may also be associated with ectopic calcification of vessels valves and heart.41 It seems that osteochondrogenic differentiation of VSMC depends on higher expression of type III sodium‐dependent phosphate transporters (Pit‐1 and Pit‐2) hence contributing to vascular calcification in CKD‐MBD. There is experimental evidence that Klotho, a co‐receptor relevant for canonical FGF23 signalling, confers anti‐calcification effects by inducing phosphaturia, preserving GFR and even through a direct effect on soft tissues including the vascular smooth muscle.23 Thus, deregulations in bone and mineral metabolism may induce both bone disease and vascular calcification in CKD. At the same time, additional factors such as chronic inflammation, which is common in CKD, and also loss of Klotho 42 may adversely affect both bone and vasculature.
2.4. Inflammation: Driving bone disease and calcification in CKD?
In CKD, the presence of so‐called uremic micro‐inflammation has been recognized as an essential pathophysiological constituent, that plays an important role as a contributor to both vascular calcification and bone disease.43 A large cohort study has disclosed that biomarkers of inflammation such as IL‐1β (interleukin 1 beta), IL‐6 (interleukin 6) and TNF‐α (tumour necrosis factor alpha) were inversely associated with eGFR (estimated glomerular filtration rate) and cystatin‐C and positively with albuminuria,44 indicating a role for inflammation on CKD development and progression. The mechanisms linking inflammation with vascular calcification are complex and multifactorial. A great amount of inflammatory mediators, such as oxidative stress, carbonyl stress, C‐reactive protein and cytokines may directly stimulate vascular calcification. Moreover, inflammation decreases the calcification inhibitor fetuin‐A.45 Importantly, the pro‐inflammatory cytokines TNF‐α, receptor activator of NF‐κB ligand (RANKL) and interleukins 1 and 17 also have adverse effects on bone.46 TNF exerts an effect on osteoclastogenesis by acting directly on osteoclast precursors while IL‐6 can upregulate RANKL and thus indirectly support osteoclast formation via the interaction with mesenchymal cells.46 A histomorphometric study performed by Viaene et al47 in end‐stage kidney disease (ESKD) patients showed that inflammatory markers IL‐6 and TNF‐ α were independently associated, respectively, with aortic calcification and low bone area, emphasizing the role of micro‐inflammation in the bone‐vascular axis in CKD. The source of chronic inflammation remains unclear in many CKD patients, and its aetiology may be multifactorial. Accumulating evidence points towards the gastrointestinal tract as a major source of inflammation in CKD.48
3. GUT MICROBIOTA IN CHRONIC KIDNEY DISEASE
Experimental and clinical evidence points towards a distinct intestinal microflora composition in CKD.4 It is well known that a healthy, balanced colonic microbiota is primarily composed of saccharolytic bacteria (ie carbohydrates fermenting bacteria).3 In the context of a reduced GFR, the intestine is exposed to uraemia. The prolonged exposure to high concentrations of urea causes overgrowth in urease containing bacterial families,49 accounting for greater ammonia generation and further increased intestinal pH. Moreover, in ESKD, there is also an increased concentration of uric acid and oxalate secretion in the gastrointestinal (GI) tract since colon becomes their main route of excretion,4 accounting for an increasing abundance of uricase producing microbes.49 The aforementioned increased proliferation of proteolytic bacterial species (ie uricase and urease containing species) confers gut dysbiosis in CKD. A recent systematic review showed that patients in any stage of CKD, ranging from early GFR decline to end‐stage kidney disease, presented with substantial differences in gut microbiota composition, compared to healthy individuals.50 However, some differences in microbiota composition within the CKD population can also be discerned. In early CKD, some beneficial microbial families such as Ruminococcaceae and Lachnospiraceae, which are enriched in the general population and capable of degrading polysaccharides, were observed.50 In advanced CKD, families associated with the production of uremic toxins are considerably more common, whereas families linked with production of SCFAs are markedly reduced.50 These findings suggest that the microbiota in early CKD may be closer to healthy individuals, whereas in advanced CKD dysbiosis is more common.
Gut dysbiosis promoted by the uraemic milieu also accounts for the loss of gut barrier integrity, which in turn allows the translocation of living bacteria, endotoxin molecules (lipopolysaccharides), and gut‐derived uremic toxins into the systemic circulation.51 The increased exposure of the host to these components may activate innate immunity and systemic inflammation, increasing the ratio of the intestinal T helper 17 (Th17) cells over T regulatory (Treg) cells, creating a vicious circle.52, 53 Moreover, the aforementioned proteolytic bacterial species amplified in CKD produce uraemic toxins generated through protein fermentation in the intestine,54 including p‐cresyl sulphate (P‐CS), indoxyl sulphate (IS) and trimethylamine N‐oxide (TMAO),54 all of which have pro‐inflammatory effects.54 Alterations in microbiota composition also comprise the depletion of protective microbial species including the ones capable of producing anti‐inflammatory and cytoprotective molecules such as short‐chain fatty acids (SCFA).49
In addition to uraemia, other aspects that further contribute to the changes in gut microbiota composition in CKD patients include diet and medications.55 Protein restriction is a major component of the ‘renal diet’, which not only aims to preserve renal function, but also to reduce uremic toxin production.55 Moreover, in order to avoid hyperkalemia and oxalate overload, dietary recommendations include limiting the consumption of fruits and vegetables, food groups that are also rich in fibre, hence interfering with symbiosis in GI tract.56 Instead of focussing on specific nutrients, some authors have investigated the potential role of healthy dietary patterns, such as vegetarian and Mediterranean diet, upon gut microbiota composition in CKD.57, 58 A cross‐sectional study has disclosed that vegetarian haemodialysis patients presented lower levels of IS and P‐CS when compared to omnivorous patients.57 The higher fibre and reduced amount of animal protein of a vegetarian diet may account for such beneficial effects. The Mediterranean diet is characterized by providing a favourable ratio of protein‐to‐carbohydrate intake for gut microbiota, since it is mainly composed of whole grains, nuts, fruits and vegetables, with modest intake of fish and dairy products and low intake of red meat. Although this dietary pattern did not reduce uraemic toxin production in CKD patients,58 it was able to increase healthy gut microbiota constituents and SCFA production in healthy subjects.55 Chronic intestinal constipation, another frequent condition in CKD, might also influence the intestinal microflora.59, 60
Chronic kidney disease and its frequent comorbidities leads to polypharmacy (defined as the continuous use of five or more medications per day) in 80% of patients, of whom 20% use >10 different medications per day.61 Many drugs have documented impact on gut microbiota composition.62, 63, 64, 65, 66, 67, 68, 69 For example, phosphate binders can affect gut microbiota by impairing vitamin and phosphate absorption.62 Besides affecting microbiota, calcium‐based phosphate binders may further increase the risk of extra‐skeletal calcifications by inducing a rise in serum calcium levels or reducing intestinal absorption of vitamin K2.62 A recent review has reported adverse effects of oral iron supplementation on gut microbiota composition, gut metabolome and intestinal health, which in turn may result in increased production of uremic toxins in CKD.64 The frequent use of antibiotics by CKD patients does not only impact the target pathogen but also commensal gut bacteria.63 Proton‐pump inhibitors (PPI), another class of medications commonly used by CKD patients, have been associated with important changes in gut microbiome because of long‐term reduction of gastric acid secretion and hypochlorhydria.65, 66, 67 Finally, immunosuppression therapy after kidney transplantation can induce profound alterations in gut microbiota composition.65, 68
Interestingly, deregulations in gut microbiota may contribute to vascular and bone disease in CKD patients (the ‘gut‐vascular‐bone axis’), among others through a metabolic shift from a predominant saccharolytic to a proteolytic fermentation pattern, an altered intestinal barrier function and impaired vitamin K status.69
4. GUT‐VASCULAR‐BONE AXIS IN CKD
Several factors may connect gut abnormalities with vascular calcification and bone disease in CKD patients; these include uraemic toxins, TMAO, vitamin K and SCFA.
4.1. Uremic toxins
The uremic toxins P‐CS and IS originate from intestinal microbial metabolization of tyrosine/phenylalanine and tryptophan, respectively.10 After absorption by colonic mucosa, these compounds are conjugated with sulphate in the liver to IS and P‐CS, which are either bound to albumin or circulate in free forms.10, 54 In CKD, the augmented microbial production and increased intestinal permeability both contribute to progressive accumulation of serum uremic toxins.54, 70 Conventional haemodialysis is not able to efficiently remove IS due to its high binding affinity to albumin in plasma.71 Higher levels of both P‐CS and IS are associated with accelerated CKD progression and cardiovascular disease.72, 73 In CKD patients, plasma IS concentrations can be 100‐fold higher than healthy individuals.74 This disproportionate amount of plasmatic uremic toxins prompts the production of free radicals in both renal cells and VSMC through oxidative stress and inflammation, causing tissue injury.75 IS induces the expression of osteoblast‐specific proteins and aortic wall calcification and thickening in Dahl salt‐sensitive hypertensive rats.76 In line, IS has been shown to promote a phenotypic switch of vascular smooth muscular cells (VSMC) from a contractile to an osteogenic phenotype.77 An experimental study has demonstrated that a concentration of IS comparable to that observed in ESKD patients was able to cause VSMC proliferation in rats.78 In vitro, P‐CS may act as a pro‐osteogenic and procalcific toxin.79 In a more recent elegant in vivo study, long‐term exposure to IS and P‐CS significantly increased calcification of the aorta and peripheral arteries of CKD rats.80 Jing et al 81 demonstrated that serum P‐CS triggers monocyte‐endothelial cell interaction in vitro through increased production of reactive oxygen species. Additionally, in vivo, increased P‐CS levels promoted atherogenesis in 5/6‐nephrectomized apoEL/L mice, compared with controls.81 Moreover, the observed activation of inflammation and coagulation signalling pathways in aorta preceding the calcification, implicated these signalling pathways in toxin‐induced arterial calcification.80
Only a handful of clinical studies have investigated the association between uremic toxins and vascular calcification in CKD. Barreto et al73 have disclosed a positive association between IS serum levels and pulse wave velocity and aortic calcification in different stages of CKD, with IS being a powerful predictor of overall and cardiovascular mortality. Subsequently, the same group demonstrated an association between P‐CS and vascular calcification in CKD patients, suggesting P‐CS levels as predictors of overall and cardiovascular death.72 Rossi et al found progressively increased levels of total and free serum IS and P‐CS in more advanced stages of CKD.82 Furthermore, total and free serum IS and P‐CS were independently associated with structural and functional markers of cardiovascular disease, such as carotid intima‐media thickness (cIMT), and endothelial function.82 Among haemodialysis (HD) patients, PC‐S levels were associated with high carotid‐femoral pulse wave velocity (PWV) and seemed to be predictors of higher arterial stiffness in ESKD.83 In line, a study in HD patients demonstrated increased levels of serum P‐CS in patients with carotid atherosclerotic plaque (CAP), and reported a positive correlation with higher total plaque area.81 Moreover, P‐CS levels were independently associated with the incidence and progression of CAP, and promoted induction of inflammatory factors and adhesion molecule expression in endothelial cells and macrophages.81 Finally, a large cohort study of children with CKD also found significant associations between a higher IS level and higher cIMT and progression of PWV, independent of other risk factors.84 It is worth mentioning that all these data derive from observational studies, requiring further confirmation in prospective intervention studies in humans.
Although little is known on the effects of uraemic toxins on bone health in CKD patients, emerging evidence indicates that IS may inhibit osteoclast differentiation and PTH signalling.85, 86 Moreover, several in vitro studies have disclosed that IS jeopardizes the differentiation of mesenchymal stem cells to osteoblasts, inhibit osteoblast proliferation, bone mineralization, alkaline phosphatase activity and expression of bone formation‐related genes.87, 88 A study in parathyroidectomized rats demonstrated that IS‐induced low bone turnover may be due to mechanisms unrelated to skeletal resistance to PTH.89 Treatment of mesenchymal stem cells (MSC) with high concentrations of uremic toxins, namely P‐CS or IS, during osteogenic differentiation promoted down‐regulation of collagen type I, reduction in alkaline phosphatase activity and impairment of MSC mineralization.90 These results reinforce that uraemic toxins negatively influence osteogenesis. The resistance to PTH effects on bone in CKD setting is well recognized, although the underlying mechanisms have not been fully elucidated yet. In line, Nii‐Kono et al 91 demonstrated that higher serum IS promotes PTH resistance in osteoblasts. In an experimental model of CKD, IS and other uremic toxins changed bone composition.92 In haemodialysis patients, IS was inversely correlated with alkaline phosphatase and bone‐specific alkaline phosphatase, irrespective of intact PTH, suggesting that IS may be implicated in skeletal PTH resistance in uraemia.93 Furthermore, increased levels of IS prompt 24‐hydroxylase (CYP24A1) activity, causing 25‐hydroxyvitamin D and active vitamin D degradation, thereby reducing calcitriol (1,25‐dihydroxycholecalciferol) levels.94
Altogether, these findings suggest that patients with CKD may develop low‐turnover bone disease by a direct action of IS on both osteoblast and osteoclast precursors to suppress bone formation and bone resorption. However, further studies are needed to elucidate the mechanisms of action of uraemic toxins in bone disease progression in uraemia.
4.2. Trimethylamine‐N‐oxide
Trimethylamine N‐oxide is derived from metabolization of trimethylamine (TMA)‐containing substrates, such as L‐carnitine, choline and betaine, by intestinal bacteria.95 These compounds are found in a variety of foods, including those of animal origin such as red meat, eggs, milk, fish, poultry and shellfish, and those of plant origin, such as green vegetables, whole grains, spinach and beets.95 Bacterially produced TMA is efficiently absorbed into the circulation and metabolized by the hepatic enzyme flavin monooxygenase to form TMAO.95
In previous large clinical studies, high serum levels of TMAO have already been associated with cardiovascular diseases,96, 97, 98 besides promoting atherosclerosis in rodents.99 It is well established that CKD patients, with different levels of renal function, have increased production and serum levels of TMAO due to altered gut microbiota and reduced renal function.98, 100, 101 A recent well‐designed study involving haemodialysis patients showed higher TMAO levels in patients with aortic calcification, compared with patients without calcification, and a positive association between serum TMAO and aortic calcification scores.102 In cultured VSMCs, TMAO promoted calcium/phosphate‐induced calcification, and in an animal CKD model, TMAO promoted vascular calcification.102 Furthermore, reducing TMAO levels by using antibiotics ameliorated vascular calcification in CKD rats.102 The authors have also demonstrated that TMAO promotes vascular calcification and osteogenic differentiation of VSMCs in the context of CKD via NLRP3 (nucleotide‐binding domain, leucine‐rich‐containing family, pyrin domain‐containing‐3) and NF‐κB (nuclear factor κB) signalling pathways.102
Recent studies investigated the role of TMAO in bone health and in age‐related osteoporosis 103 and type 2 diabetes,104 but data in CKD patients are currently lacking. Interestingly, TMAO treatment in vitro promoted adipogenic differentiation and inhibited the osteogenic differentiation of bone marrow mesenchymal stem cells (BMSCs) by upregulating the NF‐κB signalling pathway.105 Such observations highlight the importance of investigating the role of gut microbiota of CKD patients on serum levels of TMAO and whether it impacts on vascular calcification and bone health.
4.3. Vitamin K
Vitamin K, a fat‐soluble vitamin, plays a role in bone metabolism and vascular calcification and its deficiency is common in CKD.106 Subclinical vitamin K deficiency is also observed in CKD patients,107, 108, 109 further supporting an increased risk vascular calcification and bone demineralization.110 It exists in two main forms: vitamin K1 (phylloquinone, PK), synthesized by plants and found in green leafy vegetables; and vitamin K2 (menaquinone‐n, MK), mainly synthesized by bacteria, can be found in yogurts and other fermented foods besides being produced by gut microbes.111 Bacterial MK present in the colon are mainly MK‐10 and MK‐11, synthesized by Bacteroides, MK‐8 by Enterobacteria, MK‐7 by Veillonella and MK‐6 by Eubacterium lentum.112 Usually, up to 90% of total vitamin K intake comes from PK, with 10 to 25% deriving from MK.113 Yet, some authors consider that even small amounts of vitamin K2 derived from intestinal bacteria can have a significant impact on health.114
Diets poor in potassium (reduced leafy green vegetables rich in K1) and low in phosphate (reduced dairy products rich in K2) normally prescribed for CKD patients, thus represent a potential cause of vitamin K deficiency.56 Besides dietary factors and the use of medications such as vitamin K antagonists and phosphate binders,107, 109, 115 the altered gut microbiota composition in CKD patients may also contribute to worsening of vitamin K deficiency in this population. A recent study in patients with Crohn's disease has shown a reduction in the diversity of gut bacteria in vitamin K‐deficient patients.114 A well‐designed experimental study has shown that mice with disrupted microbiota displayed modified abundance of microbial genes responsible for the synthesis of vitamin K.116 In addition, the disruption of gut microbiome was associated with decreased crystallinity and impaired bone tissue strength.116 The potential role of gut microbiota composition in relationship with vitamin K status in the CKD setting remains to be addressed.
Vitamin K‐dependent proteins (VKDP), including osteocalcin (OC) and matrix Gla‐protein (MGP), are important regulators of bone mineralization.117 OC is exclusively synthesized by osteoblasts and binds to calcium ions and hydroxyapatite crystals exerting regulatory effects on bone mineral matrix.117 MGP is produced by VSMC and chondrocytes to prevent ectopic calcification.106, 117 Reduced serum levels of carboxylated MGP appear to be a noticeably contributor to the development and progression of vascular calcification.118 Furthermore, by stimulating the xenobiotic receptor on osteoblasts, vitamin K can alter bone mineralization processes.119 Given that vitamin K is essential to functionalize a series of proteins including OC, the most abundant non‐collagenous protein in bone matrix,119 the lack of matrix‐bound OC in bone turns it more fragile and susceptible to fracture.119 Reduced intake of vitamin K and functional vitamin K deficiency, as determined by circulating biomarkers, such as dephosphorylated‐uncarboxylated MGP (dp‐ucMGP) or undercarboxilated OC (unOC), are associated with low BMD and increased risk of fractures in general population 120 as well as in CKD patients.121 An observational study which evaluated the impact of vitamin K upon vascular calcification and bone health in haemodialysis patients revealed that vitamin K1 deficiency was the strongest predictor of vertebral fractures, while MK4 deficiency was a predictor of aortic calcification and MK7 deficiency a predictor of iliac calcification.121 Several studies have found positive associations between vascular calcification and vitamin K deficiency.110, 122, 123 A recent cohort study with ESKD patients has disclosed that dp‐ucMGP was strongly associated with coronary artery calcium (CAC) and aortic valve calcium; however, a stepwise regression analysis revealed that dp‐ucMGP was not an independent determinant of vascular calcification. At the same time, a randomized clinical trial has shown that 12 months of vitamin K2 supplementation did not improve vascular stiffness in non‐dialysis CKD patients, and this was similar in an updated meta‐analysis.124 These findings suggest a complex and yet unclarified interconnection between vitamin K deficiency, bone disease and vascular calcification. Six randomized clinical trials to investigate the effects of vitamin K supplementation upon vascular calcification in haemodialysis patients are ongoing.125
4.4. Short‐chain fatty acids
Short‐chain fatty acids are the products of bacterial fermentation of non‐digestible carbohydrates in the colon, such as dietary fibres and resistant starches.126 SCFA account for 2 to 10% of the total energy consumption in humans and serve as energy source for the colonic epithelial cells and the microbiota.126 The most abundant SCFA are acetate, propionate and butyrate, mainly produced by Firmicutes and Bacteroides, and rapidly absorbed by the intestinal epithelium through specific transporters or diffusion.3, 126 The benefits of SCFA are not limited to the intestine, where they are produced.127 SCFA seems to contribute to the improvement of vascular phenotypes.75 Some studies have shown immunomodulatory capacities of SCFA. Inflammatory cells such as neutrophils, macrophages, dendritic cells and T cells are responsive to SCFA treatment, in line with their anti‐inflammatory effects in a wide range of inflammatory diseases, and reduction of vascular calcification,128 although studies in this field are still limited.
Recent emerging evidence supports beneficial effects of SCFA on bone health.129, 130 Mice treated with SCFA for 8 weeks showed significantly increased bone mass and decreased bone resorption.129 In the same study, the authors also investigated the influence of a fibre‐rich diet, which increased SCFA levels in the cecum and serum, upon bone health,129 and concluded that the diet helped to further increase bone mass. Zhou et al130 have reported a direct association between dietary fibre consumption and BMD mainly in men and among participants with lower genetically determined gut microbiota‐derived SCFA propionate.
As aforementioned, the gut microbiota of CKD patients is characterized by a higher abundance of bacterial species containing proteases compared to species containing enzymes that degrade fibre and generate SCFA,49 besides a low fibre intake due to renal dietary restrictions.56 Serum butyrate levels were threefold lower in Chinese CKD patients compared to healthy controls.131 Moreover, an inverse correlation between butyrate and renal function has been reported.131 In contrast, Terpstra et al132 could not find a reduction in butyrate‐producing species nor in butyrate production capacity in a sample of Dutch CKD patients, although total SCFA was not measured. Thus, although SCFA could be another link in the gut‐bone‐vascular axis, the relevance in CKD patients deserves further study.
5. THERAPEUTIC INTERVENTIONS
Individualized approaches pursuing the re‐establishment of a symbiotic status of the gut microbiota in CKD may benefit the gut‐vascular‐bone axis in CKD, hence helping to mitigate vascular calcification and bone mineral disease. These targets may be accomplished through dietary modifications, mainly by increasing fibre intake, administrating probiotics, prebiotics or symbiotics and through the supplementation of vitamin K, when necessary.
Probiotics are defined by the Food and Agriculture Organization of the United Nations (FAO) and the World Health Organization (WHO) as ‘living organisms that provide health benefits in the host when consumed in the appropriate quantity’. To date, there are no studies that directly investigate the effects of probiotics, prebiotics or symbiotics supplementation upon vascular calcification and bone health in CKD patients. However, a recent randomized controlled trial with non‐dialysis CKD patients has shown that a three‐month supplementation with fructooligosaccharides (FOS) did not affect arterial stiffness, but lowered circulating levels of IL‐6 and preserved endothelial function in patients with less damaged endothelium.133
Furthermore, some studies indicate decreased uremic toxin production in response to probiotics in experimental and clinical CKD, with ameliorated biomarkers of inflammation and oxidative stress.134 On the other hand, Borges et al135 have found in a recent randomized, double‐blind trial in haemodialysis patients a significant increase in IS plasma levels after three months of probiotic supplementation, which could be a consequence of increased permeability of the gut barrier. These findings highlight that probiotic therapy should be further explored but used with caution in CKD patients. Prebiotics, non‐digestible carbohydrates that act stimulating the growth and activity of beneficial bacteria in the colon, have also been associated with reduction of uremic toxins and inflammatory markers in haemodialysis patients.136, 137 On the other hand, in non‐dialysis‐dependent CKD patients, supplementation of FOS for three months did not lead to changes in uremic toxins levels.138 Likewise, symbiotics, which are defined as the combination of both prebiotics and probiotics, reduced levels of serum P‐CS in CKD patients.134
Interestingly, AST‐120, an orally administered spherical carbon adsorbent approved for clinical use in CKD patients in Japan and Asia, was able to inhibit the hepatic synthesis of indoxyl sulphate by blocking the gastrointestinal absorption of its biochemical precursor indole in an experimental study.139 Moreover, serum P‐CS levels were significantly reduced by AST‐120 in an animal model.140 More recently, mice with adenine‐induced kidney damage that were treated with AST‐120 did not have p‐cresol in faecal content.141 In addition, AST‐120 reduced the abundance of Erysipelotrichaceae uncultured and Clostridium sensu stricto, species that are involved in p‐cresol production.141 These preclinical studies set the stage for studies in humans. An initial study reported that AST‐120 decreased serum levels of indoxyl sulphate in a dose‐dependent manner in patients with moderate to severe CKD.142 This was followed by a more recent large, multinational, randomized clinical trial that unfortunately could not show a beneficial effect of AST‐120 on the progression of CKD.143 An ongoing clinical trial aims to study the effects of oral absorbent and probiotics on vascular function of CKD patients.144 Overall, although some studies show promising effects of interventions aiming to promote gut health on intermediate endpoints, other studies show null results or even safety signals. This highlights the need for further studies to find efficient interventions, to identify relevant specific target (sub‐)populations, and to demonstrate effects on clinically relevant (cardiovascular and bone) outcomes.
6. CONCLUSIONS AND FUTURE PERSPECTIVES
In conclusion, CKD and gut dysbiosis share abnormalities in factors that promote or inhibit vascular calcification, as summarized in Table 1. Although a clear interplay exists between gut microbiota, bone health and vascular calcification (Figure 1), most studies to date focus on associations between two of these factors, but did not analyse all factors together. Therefore, there is still much to learn regarding the pathways that link gut microbiota to both bone and vascular health. To reach this aim, large, well‐designed cohort studies addressing microbiome composition in relation to, preferably, both vascular calcification and bone disease in the setting of CKD are warranted.
TABLE 1.
Potential factors acting as promoters or inhibitors of vascular calcification in the context of CKD and gut dysbiosis
| CKD | Gut dysbiosis | |
|---|---|---|
| VC promoters | TMAO 101 | TMAO 144 |
| Inflammatory cytokines 45, 47 | Inflammatory cytokines 145 | |
| Oxidative stress 13 | Oxidative stress 146 | |
| Indoxyl‐sulphate 72, 77, 81, 83 | ||
| P‐cresyl‐sulphate 71, 81, 82 | ||
| BMPs 13 | ||
| Secondary CPPs 43 | ||
| Serum calcium 13, 19 | ||
| Serum phosphate 13, 19 | ||
| Serum Ca‐P product 13, 19 | ||
| Serum PTH 13, 41 | ||
| Serum FGF‐23 13 | ||
| Runx2 19 | ||
| VC inhibitors | Vitamin K 120 | Vitamin K 110, 121, 122 |
| SCFA 74 | SCFA 127 | |
| Klotho 13, 19 | ||
| Osteoprotegerin 13, 19 | ||
| Osteopontin 19 | ||
| MGP 43 | ||
| Pyrophosphate 19 | ||
| Fetuin‐A 13, 19 |
Abbreviations: BMP, bone morphogenetic protein; Ca, calcium; CKD, chronic kidney disease; CPP, calciprotein particles; FGF‐23, fibroblast growth factor‐23; MGP, matrix Gla‐protein; P, phosphorus; PTH, parathyroid hormone; SCFA, short‐chain fatty acids; TMAO, trimethylamine‐N‐oxide; VC, vascular calcification.
FIGURE 1.
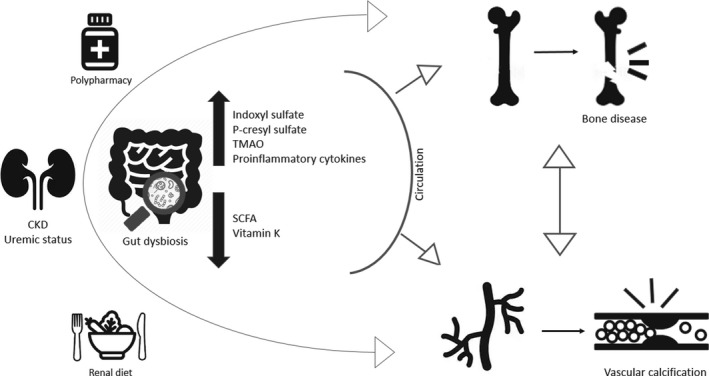
The uraemic milieu is one of the main contributors for gut dysbiosis in CKD. As renal function declines, the colon replaces the kidney as the primary site of excretion of urea and uric acid, which become alternative substrates for the gut bacteria, instead of indigestible complex carbohydrates. Besides, renal diet (limited intake of fibre‐rich foods, eg fruits and vegetables) and polypharmacy (ie phosphate binders, antibiotics, iron supplementation, PPI, immunosuppressants) may account for altered gut dysbiosis in CKD setting. The hypothesis upon gut‐vascular‐bone axis in CKD revolves around the augmented exposure of the referred tissues to uremic toxins, such as indoxyl sulphate (IS) and p‐cresyl sulphate (P‐CS), TMAO (Trimethylamine‐N‐oxide) and pro‐inflammatory cytokines given the presence of a disrupted intestinal barrier. In addition, the reduced production of SCFA (short‐chain fatty acids) and the deficiency of vitamin K might also exacerbate this mechanism
Another major outstanding question, as also addressed in this review, is whether interventions targeting the microbiome improve cardiovascular and bone health in CKD. The difficulties regarding such studies rely among others on the lack of feasible and practical methods of evaluating changes in gut microbiome over time in a clinical trial. While it is important to address dietary and/or pro/prebiotic interventions modulating gut microbiota in trials in CKD patients aiming to assess their impact on gut metabolites (uremic toxins, TMAO, SCFA), it would be even more important to focus on markers of bone/vascular disease like fracture risk and CAC. Moreover, since current data seem to indicate heterogenic efficacy of interventions targeting gut health regarding intermediate outcomes such as uremic toxins, it would be highly relevant to identify subgroups of patients that are potentially more susceptible to positive treatment response. Although there may be a long road ahead, ultimately, correcting derangements in gut microbiota may be a promising approach to improve both bone and cardiovascular health in CKD patients.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
FGR conducted the research and wrote the manuscript. MSO discussed and enriched the manuscript. IPH, SJLB and MHB supervised and reviewed the manuscript.
Rodrigues FG, Ormanji MS, Heilberg IP, Bakker SJL, de Borst MH. Interplay between gut microbiota, bone health and vascular calcification in chronic kidney disease. Eur J Clin Invest. 2021;51:e13588. 10.1111/eci.13588
REFERENCES
- 1.Eckburg PB, Bik EM, Bernstein CN, et al. Diversity of the human intestinal microbial flora. Science (80‐). 2005;308(5728):1635‐1638. 10.1126/science.1110591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rinninella E, Raoul P, Cintoni M, et al. What is the healthy gut microbiota composition? A changing ecosystem across age, environment, diet, and diseases. Microorganisms. 2019;7(1):14. 10.3390/microorganisms7010014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jandhyala SM, Talukdar R, Subramanyam C, Vuyyuru H, Sasikala M, Nageshwar RD. Role of the normal gut microbiota. World J Gastroenterol. 2015;21(29):8787‐8803. 10.3748/wjg.v21.i29.8787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vaziri ND, Wong J, Pahl M, et al. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013;83(2):308‐315. 10.1038/ki.2012.345 [DOI] [PubMed] [Google Scholar]
- 5.Jager KJ, Kovesdy C, Langham R, Rosenberg M, Jha V, Zoccali C. A single number for advocacy and communication‐worldwide more than 850 million individuals have kidney diseases. Nephrol Dial Transplant. 2019;34(11):1803‐1805. 10.1093/ndt/gfz174 [DOI] [PubMed] [Google Scholar]
- 6.Chen TK, Knicely DH, Grams ME. Chronic kidney disease diagnosis and management a review. JAMA. 2019;322(12):1294‐1304. 10.1001/jama.2019.14745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levey AS, Coresh J. Chronic kidney disease. Lancet. 2012;379(9811):165‐180. 10.1016/S0140-6736(11)60178-5 [DOI] [PubMed] [Google Scholar]
- 8.Graciolli FG, Neves KR, Barreto F, et al. The complexity of chronic kidney disease–mineral and bone disorder across stages of chronic kidney disease. Kidney Int. 2017;91(6):1436‐1446. 10.1016/j.kint.2016.12.029 [DOI] [PubMed] [Google Scholar]
- 9.Bangalore S, Maron DJ, O’Brien SM, et al. Management of coronary disease in patients with advanced kidney disease. N Engl J Med. 2020;382(17):1608‐1618. 10.1056/NEJMoa1915925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vanholder R, Schepers E, Pletinck A, Nagler EV, Glorieux G. The uremic toxicity of indoxyl sulfate and p‐cresyl sulfate: a systematic review. J Am Soc Nephrol. 2014;25(9):1897‐1907. 10.1681/ASN.2013101062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bover J, Ureña‐Torres P, Mateu S, et al. Evidence in chronic kidney disease–mineral and bone disorder guidelines: is it time to treat or time to wait? Clin Kidney J. 2020;13(4):513‐521. 10.1093/ckj/sfz187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hénaut L, Chillon JM, Kamel S, Massy ZA. Updates on the mechanisms and the care of cardiovascular calcification in chronic kidney disease. Semin Nephrol. 2018;38(3):233‐250. 10.1016/j.semnephrol.2018.02.004 [DOI] [PubMed] [Google Scholar]
- 13.Evenepoel P, Opdebeeck B, David K, D’Haese PC. Bone‐vascular axis in chronic kidney disease. Adv Chronic Kidney Dis. 2019;26(6):472‐483. 10.1053/j.ackd.2019.09.006 [DOI] [PubMed] [Google Scholar]
- 14.Ammirati AL, Dalboni MA, Cendoroglo M, et al. The progression and impact of vascular calcification in peritoneal dialysis patients. Perit Dial Int. 2007;27(3):340‐346. 10.1177/089686080702700325 [DOI] [PubMed] [Google Scholar]
- 15.Budoff MJ, Rader DJ, Reilly MP, et al. Relationship of estimated GFR and coronary artery calcification in the CRIC (Chronic Renal Insufficiency Cohort) study. Am J Kidney Dis. 2011;58(4):519‐526. 10.1053/j.ajkd.2011.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keyzer CA, De Borst MH, Van Den Berg E, et al. Calcification propensity and survival among renal transplant recipients. J Am Soc Nephrol. 2016;27(1):239‐248. 10.1681/ASN.2014070670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sotomayor CG, te Velde‐Keyzer CA, de Borst MH, Navis GJ, Bakker SJL. Lifestyle, Inflammation, and vascular calcification in kidney transplant recipients: perspectives on long‐term outcomes. J Clin Med. 2020;9(6):1911. 10.3390/jcm9061911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paloian NJ, Giachelli CM. A current understanding of vascular calcification in CKD. Am J Physiol ‐ Ren Physiol. 2014;307(8):F891‐F900. 10.1152/ajprenal.00163.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamada S, Giachelli CM. Vascular Calcification in CKD‐MBD: roles for phosphate, FGF23, and Klotho. Bone. 2017;100:87‐93. 10.1016/j.bone.2016.11.012.Vascular [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vervloet M, Cozzolino M. Vascular calcification in chronic kidney disease: different bricks in the wall? Kidney Int. 2017;91(4):808‐817. 10.1016/j.kint.2016.09.024 [DOI] [PubMed] [Google Scholar]
- 21.Ter Braake AD, Eelderink C, Zeper LW, et al. Calciprotein particle inhibition explains magnesium‐mediated protection against vascular calcification. Nephrol Dial Transplant. 2020;35(5):765‐773. 10.1093/ndt/gfz190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salam S, Gallagher O, Gossiel F, Paggiosi M, Eastell R, Khwaja A. Vascular calcification relationship to vascular biomarkers and bone metabolism in advanced chronic kidney disease. Bone. 2021;143:115699. 10.1016/j.bone.2020.115699 [DOI] [PubMed] [Google Scholar]
- 23.Hu MC, Shi M, Zhang J, et al. Klotho deficiency causes vascular calcification in chronic kidney disease. J Am Soc Nephrol. 2011;22(1):124‐136. 10.1681/ASN.2009121311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keyzer CA, Vermeer C, Joosten MM, et al. Vitamin K status and mortality after kidney transplantation: a cohort study. Am J Kidney Dis. 2015;65(3):474‐483. 10.1053/j.ajkd.2014.09.014 [DOI] [PubMed] [Google Scholar]
- 25.Chen J, Budoff MJ, Reilly MP, et al. Coronary artery calcification and risk of cardiovascular disease and death among patients with chronic kidney disease. JAMA Cardiol. 2017;2(6):635. 10.1001/jamacardio.2017.0363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang JF, Liu SH, Lu KC, et al. Uremic vascular calcification is correlated with oxidative elastic lamina injury, contractile smooth muscle cell loss, osteogenesis, and apoptosis: the human pathobiological evidence. Front Med. 2020;7:1‐12. 10.3389/fmed.2020.00078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klingenschmid G, Tschiderer L, Himmler G, et al. Associations of serum dickkopf‐1 and sclerostin with cardiovascular events: results from the prospective Bruneck study. J Am Heart Assoc. 2020;9(6):e014816. 10.1161/JAHA.119.014816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brandenburg VM, Kramann R, Koos R, et al. Relationship between sclerostin and cardiovascular calcification in hemodialysis patients: a cross‐sectional study. BMC Nephrol. 2013;14(1):219. 10.1186/1471-2369-14-219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tentori F, McCullough K, Kilpatrick RD, et al. High rates of death and hospitalization follow bone fracture among hemodialysis patients. Kidney Int. 2014;85(1):166‐173. 10.1038/ki.2013.279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wagner J, Jhaveri KD, Rosen L, Sunday S, Mathew AT, Fishbane S. Increased bone fractures among elderly United States hemodialysis patients. Nephrol Dial Transplant. 2014;29(1):146‐151. 10.1093/ndt/gft352 [DOI] [PubMed] [Google Scholar]
- 31.Desbiens LC, Goupil R, Madore F, Mac‐Way F. Incidence of fractures in middle‐aged individuals with early chronic kidney disease: a population‐based analysis of CARTaGENE. Nephrol Dial Transplant. 2020;35(10):1712‐1721. 10.1093/ndt/gfz259 [DOI] [PubMed] [Google Scholar]
- 32.Tan J, Li Y, Wu Z, Zhao J. Risk of hip fracture in patients on dialysis or kidney transplant: a meta‐analysis of 14 cohort studies. Ther Clin Risk Manag. 2018;14:1747‐1755. 10.2147/TCRM.S171970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ball AM, Gillen DL, Weiss NS, et al. Risk of hip fracture among dialysis and renal transplant recipients. JAMA ‐ J Am Med Assoc. 2002;288(23):3014‐3018. [DOI] [PubMed] [Google Scholar]
- 34.Barreto DV, de Barreto FC, de Carvalho AB, et al. Association of changes in bone remodeling and coronary calcification in hemodialysis patients: a prospective study. Am J Kidney Dis. 2008;52(6):1139‐1150. 10.1053/j.ajkd.2008.06.024 [DOI] [PubMed] [Google Scholar]
- 35.Rodríguez‐García M, Gómez‐Alonso C, Naves‐Díaz M, et al. Vascular calcifications, vertebral fractures and mortality in haemodialysis patients. Nephrol Dial Transplant. 2009;24(1):239‐246. 10.1093/ndt/gfn466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sotomayor CG, Benjamens S, Gomes‐Neto AW, et al. Bone mineral density and aortic calcification. Transplantation. 2020. 10.1097/tp.0000000000003226 [DOI] [PubMed] [Google Scholar]
- 37.Demer LL, Tintut Y. Vascular calcification: pathobiology of a multifaceted disease. Circulation. 2008;117(22):2938‐2948. 10.1161/CIRCULATIONAHA.107.743161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boström K, Watson KE, Horn S, Wortham C, Herman IM, Demer LL. Bone morphogenetic protein expression in human atherosclerotic lesions. J Clin Invest. 1993;91(4):1800‐1809. 10.1172/JCI116391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kidney Disease: Improving Global Outcomes (KDIGO) CKD‐MBD Work Group . KDIGO clinical practice guidelines for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease‐mineral and bone disorder (CKD‐MBD). Kidney Int Suppl. 2009;113:S1‐130. 10.1038/ki.2009.188 [DOI] [PubMed] [Google Scholar]
- 40.Cozzolino M, Dusso AS, Slatopolsky E. Role of calcium‐phosphate product and bone‐associated proteins on vascular calcification in renal failure. J Am Soc Nephrol. 2001;12(11):2511‐2516. [DOI] [PubMed] [Google Scholar]
- 41.Martola L, Barany P, Stenvinkel P. Why do dialysis patients develop a heart of stone and bone of China? Blood Purif. 2005;23(3):203‐210. 10.1159/000084890 [DOI] [PubMed] [Google Scholar]
- 42.Henao Agudelo J, Baia L, Ormanji M, et al. Fish oil supplementation reduces inflammation but does not restore renal function and klotho expression in an adenine‐induced CKD model. Nutrients. 2018;10(9):1283. 10.3390/nu10091283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Viegas C, Araújo N, Marreiros C, Simes D. The interplay between mineral metabolism, vascular calcification and inflammation in Chronic Kidney Disease (CKD): Challenging old concepts with new facts. Aging (Albany NY). 2019;11(12):4274‐4299. 10.18632/aging.102046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gupta J, Mitra N, Kanetsky PA, et al. Association between albuminuria, kidney function, and inflammatory biomarker profile in CKD in CRIC. Clin J Am Soc Nephrol. 2012;7(12):1938‐1946. 10.2215/CJN.03500412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moe SM, Chen NX. Inflammation and vascular calcification. Blood Purif. 2005;23(1):64‐71. 10.1159/000082013 [DOI] [PubMed] [Google Scholar]
- 46.Schett G. Effects of inflammatory and anti‐inflammatory cytokines on the bone. Eur J Clin Invest. 2011;41(12):1361‐1366. 10.1111/j.1365-2362.2011.02545.x [DOI] [PubMed] [Google Scholar]
- 47.Viaene L, Behets GJ, Heye S, et al. Inflammation and the bone‐vascular axis in end‐stage renal disease. Osteoporos Int. 2016;27(2):489‐497. 10.1007/s00198-015-3233-8 [DOI] [PubMed] [Google Scholar]
- 48.Lau WL, Kalantar‐Zadeh K, Vaziri ND. The gut as a source of inflammation in chronic kidney disease. Nephron. 2015;130(2):92‐98. 10.1159/000381990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wong J, Piceno YM, DeSantis TZ, Pahl M, Andersen GL, Vaziri ND. Expansion of urease‐ and uricase‐containing, indole‐ and p‐cresol‐forming and contraction of short‐chain fatty acid‐producing intestinal microbiota in ESRD. Am J Nephrol. 2014;39(3):230‐237. 10.1159/000360010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chung SY, Barnes JL, Astroth KS. Gastrointestinal microbiota in patients with chronic kidney disease: a systematic review. Adv Nutr. 2019;10(5):888‐901. 10.1093/advances/nmz028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Magnusson M, Magnusson KE, Sundqvist T, Denneberg T. Impaired intestinal barrier function measured by differently sized polyethylene glycols in patients with chronic renal failure. Gut. 1991;32(7):754‐759. 10.1136/gut.32.7.754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anders HJ, Andersen K, Stecher B. The intestinal microbiota, a leaky gut, and abnormal immunity in kidney disease. Kidney Int. 2013;83(6):1010‐1016. 10.1038/ki.2012.440 [DOI] [PubMed] [Google Scholar]
- 53.Onal EM, Afsar B, Covic A, Vaziri ND, Kanbay M. Gut microbiota and inflammation in chronic kidney disease and their roles in the development of cardiovascular disease. Hypertens Res. 2019;42(2):123‐140. 10.1038/s41440-018-0144-z [DOI] [PubMed] [Google Scholar]
- 54.Evenepoel P, Meijers BKI, Bammens BRM, Verbeke K. Uremic toxins originating from colonic microbial metabolism. Kidney Int. 2009;76:S12‐S19. 10.1038/ki.2009.402 [DOI] [PubMed] [Google Scholar]
- 55.Mafra D, Borges N, Alvarenga L, et al. Dietary components that may influence the disturbed gut microbiota in chronic kidney disease. Nutrients. 2019;11(3):1‐23. 10.3390/nu11030496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cupisti A, D’Alessandro C, Gesualdo L, et al. Non‐traditional aspects of renal diets: Focus on fiber, alkali and Vitamin K1 intake. Nutrients. 2017;9(5):1‐15. 10.3390/nu9050444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kandouz S, Mohamed AS, Zheng Y, Sandeman S, Davenport A. Reduced protein bound uraemic toxins in vegetarian kidney failure patients treated by haemodiafiltration. Hemodial Int. 2016;20(4):610‐617. 10.1111/hdi.12414 [DOI] [PubMed] [Google Scholar]
- 58.Pignanelli M, Just C, Bogiatzi C, et al. Mediterranean diet score: Associations with metabolic products of the intestinal microbiome, carotid plaque burden, and renal function. Nutrients. 2018;10(6):779. 10.3390/nu10060779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ramos CI, Armani RG, Canziani ME, et al. Bowel habits and the association with uremic toxins in non–dialysis‐dependent chronic kidney disease patients. J Ren Nutr. 2020;30(1):31‐35. 10.1053/j.jrn.2019.02.004 [DOI] [PubMed] [Google Scholar]
- 60.Sumida K, Yamagata K, Kovesdy CP. Constipation in CKD. Kidney Int Rep. 2020;5(2):121‐134. 10.1016/j.ekir.2019.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schmidt IM, Hübner S, Nadal J, et al. Patterns of medication use and the burden of polypharmacy in patients with chronic kidney disease: The German Chronic Kidney Disease study. Clin Kidney J. 2019;12(5):663‐672. 10.1093/ckj/sfz046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rahbar Saadat Y, Niknafs B, Hosseiniyan Khatibi SM, et al. Gut microbiota; an overlooked effect of phosphate binders. Eur J Pharmacol. 2020;868: 10.1016/j.ejphar.2019.172892 [DOI] [PubMed] [Google Scholar]
- 63.Jernberg C, Löfmark S, Edlund C, Jansson JK. Long‐term impacts of antibiotic exposure on the human intestinal microbiota. Microbiology. 2010;156(11):3216‐3223. 10.1099/mic.0.040618-0 [DOI] [PubMed] [Google Scholar]
- 64.Kortman GAM, Reijnders D, Swinkels DW. Oral iron supplementation: Potential implications for the gut microbiome and metabolome in patients with CKD. Hemodial Int. 2017;21:S28‐S36. 10.1111/hdi.12553 [DOI] [PubMed] [Google Scholar]
- 65.Swarte JC, Douwes RM, Hu S, et al. Characteristics and dysbiosis of the gut microbiome in renal transplant recipients. J Clin Med. 2020;9(2):386. 10.3390/jcm9020386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Imhann F, Bonder MJ, Vich Vila A, et al. Proton pump inhibitors affect the gut microbiome. Gut. 2016;65(5):740‐748. 10.1136/gutjnl-2015-310376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lin Y‐T, Lin T‐Y, Hung S‐C, et al. Anti‐acid drug treatment induces changes in the gut microbiome composition of hemodialysis patients. Microorganisms. 2021;9(2):286. 10.3390/microorganisms9020286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee JR, Muthukumar T, Dadhania D, et al. Gut microbiota and tacrolimus dosing in kidney transplantation. PLoS One. 2015;10(3):e0122399. 10.1371/journal.pone.0122399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Evenepoel P, Dejongh S, Verbeke K. The role of gut dysbiosis in the bone – vascular axis in chronic kidney disease. Toxins. 2020;12(5):1‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Miyamoto Y, Watanabe H, Noguchi T, et al. Organic anion transporters play an important role in the uptake of p‐cresyl sulfate, a uremic toxin, in the kidney. Nephrol Dial Transplant. 2011;26(8):2498‐2502. 10.1093/ndt/gfq785 [DOI] [PubMed] [Google Scholar]
- 71.Meijers BKI, De Loor H, Bammens B, Verbeke K, Vanrenterghem Y, Evenepoel P. P‐Cresyl Sulfate and Indoxyl Sulfate in Hemodialysis Patients. Clin J Am Soc Nephrol. 2009;4(12):1932‐1938. 10.2215/CJN.02940509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liabeuf S, Barreto DV, Barreto FC, et al. Free p‐cresylsulphate is a predictor of mortality in patients at different stages of chronic kidney disease. Nephrol Dial Transplant. 2010;25(4):1183‐1191. 10.1093/ndt/gfp592 [DOI] [PubMed] [Google Scholar]
- 73.Barreto FC, Barreto DV, Liabeuf S, et al. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin J Am Soc Nephrol. 2009;4(10):1551‐1558. 10.2215/CJN.03980609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lano G, Burtey S, Sallée M. Indoxyl sulfate, a uremic endotheliotoxin. Toxins (Basel). 2020;12(4):1‐14. 10.3390/toxins12040229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yin L, Li XX, Ghosh S, Xie C, Chen J, Huang H. Role of gut microbiota‐derived metabolites on vascular calcification in CKD. J Cell Mol Med. 2021;25(3):1332‐1341. 10.1111/jcmm.16230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Adijiang A, Goto S, Uramoto S, Nishijima F, Niwa T. Indoxyl sulphate promotes aortic calcification with expression of osteoblast‐specific proteins in hypertensive rats. Nephrol Dial Transplant. 2008;23(6):1892‐1901. 10.1093/ndt/gfm861 [DOI] [PubMed] [Google Scholar]
- 77.Muteliefu G, Enomoto A, Jiang P, Takahashi M, Niwa T. Indoxyl sulphate induces oxidative stress and the expression of osteoblast‐specific proteins in vascular smooth muscle cells. Nephrol Dial Transplant. 2009;24(7):2051‐2058. 10.1093/ndt/gfn757 [DOI] [PubMed] [Google Scholar]
- 78.Yamamoto H, Tsuruoka S, Ioka T, et al. Indoxyl sulfate stimulates proliferation of rat vascular smooth muscle cells. Kidney Int. 2006;69(10):1780‐1785. 10.1038/sj.ki.5000340 [DOI] [PubMed] [Google Scholar]
- 79.Chang JF, Hsieh CY, Liou JC, et al. Scavenging intracellular ros attenuates p‐cresyl sulfate‐triggered osteogenesis through mapk signaling pathway and NF‐κB activation in human arterial smooth muscle cells. Toxins (Basel). 2020;12(8):472. 10.3390/toxins12080472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Opdebeeck B, Maudsley S, Azmi A, et al. Indoxyl sulfate and p‐cresyl sulfate promote vascular calcification and associate with glucose intolerance. J Am Soc Nephrol. 2019;30(5):751‐766. 10.1681/ASN.2018060609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jing YJ, Ni JW, Ding FH, et al. P‐Cresyl sulfate is associated with carotid arteriosclerosis in hemodialysis patients and promotes atherogenesis in apoE‐/‐ mice. Kidney Int. 2016;89(2):439‐449. 10.1038/ki.2015.287 [DOI] [PubMed] [Google Scholar]
- 82.Rossi M, Campbell K, Johnson D, et al. Uraemic toxins and cardiovascular disease across the chronic kidney disease spectrum: An observational study. Nutr Metab Cardiovasc Dis. 2014;24(9):1035‐1042. 10.1016/j.numecd.2014.04.006 [DOI] [PubMed] [Google Scholar]
- 83.Lai YH, Wang CH, Kuo CH, Lin YL, Tsai JP, Hsu BG. Serum P‐cresyl sulfate is a predictor of central arterial stiffness in patients on maintenance hemodialysis. Toxins (Basel). 2020;12(1):10. 10.3390/toxins12010010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Holle J, Querfeld U, Kirchner M, et al. Indoxyl sulfate associates with cardiovascular phenotype in children with chronic kidney disease. Pediatr Nephrol. 2019;34(12):2571‐2582. 10.1007/s00467-019-04331-6 [DOI] [PubMed] [Google Scholar]
- 85.Yamamoto S, Fukagawa M. Uremic toxicity and bone in CKD. J Nephrol. 2017;30(5):623‐627. 10.1007/s40620-017-0406-x [DOI] [PubMed] [Google Scholar]
- 86.Mozar A, Louvet L, Godin C, et al. Indoxyl sulphate inhibits osteoclast differentiation and function. Nephrol Dial Transplant. 2012;27(6):2176‐2181. 10.1093/ndt/gfr647 [DOI] [PubMed] [Google Scholar]
- 87.Kim YH, Kwak KA, Gil HW, Song HY, Hong SY. Indoxyl sulfate promotes apoptosis in cultured osteoblast cells. BMC Pharmacol Toxicol. 2013;14:60. 10.1186/2050-6511-14-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Watanabe K, Tominari T, Hirata M, et al. Indoxyl sulfate, a uremic toxin in chronic kidney disease, suppresses both bone formation and bone resorption. FEBS Open Bio. 2017;7(8):1178‐1185. 10.1002/2211-5463.12258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hirata J, Hirai K, Asai H, et al. Indoxyl sulfate exacerbates low bone turnover induced by parathyroidectomy in young adult rats. Bone. 2015;79:252‐258. 10.1016/j.bone.2015.06.010 [DOI] [PubMed] [Google Scholar]
- 90.Kamprom W, Tawonsawatruk T, Oodi SM, Anansilp K. P ‐ cresol and indoxyl sulfate impair osteogenic differentiation by triggering mesenchymal stem. Cell Senescence. 2021;18: 10.7150/ijms.48492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nii‐Kono T, Iwasaki Y, Uchida M, et al. Indoxyl sulfate induces skeletal resistance to parathyroid hormone in cultured osteoblastic cells. Kidney Int. 2007;71(8):738‐743. 10.1038/sj.ki.5002097 [DOI] [PubMed] [Google Scholar]
- 92.Iwasaki Y, Kazama JJ, Yamato H, Shimoda H, Fukagawa M. Accumulated uremic toxins attenuate bone mechanical properties in rats with chronic kidney disease. Bone. 2013;57(2):477‐483. 10.1016/j.bone.2013.07.037 [DOI] [PubMed] [Google Scholar]
- 93.Goto S, Fujii H, Hamada Y, Yoshiya K, Fukagawa M. Association between indoxyl sulfate and skeletal resistance in hemodialysis patients. Ther Apher Dial. 2010;14(4):417‐423. 10.1111/j.1744-9987.2010.00813.x [DOI] [PubMed] [Google Scholar]
- 94.Liu W‐C, Wu C‐C, Hung Y‐M, et al. Pleiotropic effects of vitamin D in chronic kidney disease. Clin Chim Acta. 2016;453:1‐12. 10.1016/j.cca.2015.11.029 [DOI] [PubMed] [Google Scholar]
- 95.Moraes C, Fouque D, Amaral ACF, Mafra D. Trimethylamine N‐oxide from gut microbiota in chronic kidney disease patients: focus on diet. J Ren Nutr. 2015;25(6):459‐465. 10.1053/j.jrn.2015.06.004 [DOI] [PubMed] [Google Scholar]
- 96.Wang Z, Klipfell E, Bennett BJ, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472(7341):57‐65. 10.1038/nature09922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang Z, Tang WHW, Buffa JA, et al. Prognostic value of choline and betaine depends on intestinal microbiota‐generated metabolite trimethylamine‐N‐oxide. Eur Heart J. 2014;35(14):904‐910. 10.1093/eurheartj/ehu002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stubbs JR, House JA, Ocque AJ, et al. Serum Trimethylamine‐N‐Oxide is Elevated in CKD and Correlates with Coronary Atherosclerosis Burden. J Am Soc Nephrol. 2016;27(1):305‐313. 10.1681/ASN.2014111063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Koeth RA, Wang Z, Levison BS, et al. Intestinal microbiota metabolism of l‐carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19(5):576‐585. 10.1038/nm.3145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pelletier CC, Croyal M, Ene L, et al. Elevation of trimethylamine‐N‐oxide in chronic kidney disease: contribution of decreased glomerular filtration rate. Toxins (Basel). 2019;11(11):635. 10.3390/toxins11110635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bain MA, Faull R, Fornasini G, Milne RW, Evans AM. Accumulation of trimethylamine and trimethylamine‐N‐oxide in end‐stage renal disease patients undergoing haemodialysis. Nephrol Dial Transplant. 2006;21(5):1300‐1304. 10.1093/ndt/gfk056 [DOI] [PubMed] [Google Scholar]
- 102.Zhang X, Li Y, Yang P, et al. Trimethylamine‐N‐oxide promotes vascular calcification through activation of NLRP3 (nucleotide‐binding domain, leucine‐rich‐containing family, pyrin domain‐containing‐3) inflammasome and NF‐κB (nuclear factor κb) signals. Arterioscler Thromb Vasc Biol. 2020;3:751‐765. 10.1161/ATVBAHA.119.313414 [DOI] [PubMed] [Google Scholar]
- 103.Li L, Chen B, Zhu R, et al. Fructus Ligustri Lucidi preserves bone quality through the regulation of gut microbiota diversity, oxidative stress, TMAO and Sirt6 levels in aging mice. Aging (Albany NY). 2019;11(21):9348‐9368. 10.18632/aging.102376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhou T, Heianza Y, Chen Y, et al. Circulating gut microbiota metabolite trimethylamine N‐oxide (TMAO) and changes in bone density in response to weight loss diets: The Pounds lost trial. Diabetes Care. 2019;42(8):1365‐1371. 10.2337/dc19-0134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lin H, Liu T, Li X, Gao X, Wu T, Li P. The role of gut microbiota metabolite trimethylamine N‐oxide in functional impairment of bone marrow mesenchymal stem cells in osteoporosis disease. Ann Transl Med. 2020;8(16):1009. 10.21037/atm-20-5307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cozzolino M, Fusaro M, Ciceri P, Gasperoni L, Cianciolo G. The role of vitamin K in vascular calcification. Adv Chronic Kidney Dis. 2019;26(6):437‐444. 10.1053/j.ackd.2019.10.005 [DOI] [PubMed] [Google Scholar]
- 107.Cranenburg ECM, Schurgers LJ, Uiterwijk HH, et al. Vitamin K intake and status are low in hemodialysis patients. Kidney Int. 2012;82(5):605‐610. 10.1038/ki.2012.191 [DOI] [PubMed] [Google Scholar]
- 108.Mansour AG, Hariri E, Daaboul Y, et al. Vitamin K2 supplementation and arterial stiffness among renal transplant recipients—a single‐arm, single‐center clinical trial. J Am Soc Hypertens. 2017;11(9):589‐597. 10.1016/j.jash.2017.07.001 [DOI] [PubMed] [Google Scholar]
- 109.Holden RM, Morton AR, Garland JS, Pavlov A, Day AG, Booth SL. Vitamins K and D status in stages 3–5 chronic kidney disease. Clin J Am Soc Nephrol. 2010;5(4):590‐597. 10.2215/CJN.06420909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schurgers LJ, Barreto DV, Barreto FC, et al. The circulating inactive form of matrix gla protein is a surrogate marker for vascular calcification in chronic kidney disease: a preliminary report. Clin J Am Soc Nephrol. 2010;5(4):568‐575. 10.2215/CJN.07081009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Walther B, Philip Karl J, Booth SL, Boyaval P. Menaquinones, bacteria, and the food supply: The relevance of dairy and fermented food products to vitamin K requirements. Adv Nutr. 2013;4(4):463‐473. 10.3945/an.113.003855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Booth SL, Dallal G, Shea MK, Gundberg C, Peterson JW, Dawson‐Hughes B. Effect of vitamin K supplementation on bone loss in elderly men and women. J Clin Endocrinol Metab. 2008;93(4):1217‐1223. 10.1210/jc.2007-2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shearer M, Newman P. Metabolism and cell biology of vitamin K. Thromb Haemostasis. 2008;100(4):530‐547. [PubMed] [Google Scholar]
- 114.Wagatsuma K, Yamada S, Ao M, et al. Diversity of gut microbiota affecting serum level of undercarboxylated osteocalcin in patients with crohn’s disease. Nutrients. 2019;11(7):1541. 10.3390/nu11071541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jansz TT, Neradova A, Van Ballegooijen AJ, et al. The role of kidney transplantation and phosphate binder use in vitamin K status. PLoS One. 2018;13(8):1‐13. 10.1371/journal.pone.0203157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Guss JD, Taylor E, Rouse Z, et al. The microbial metagenome and bone tissue composition in mice with microbiome‐induced reductions in bone strength. Bone. 2019;127:146‐154. 10.1016/j.bone.2019.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Akbari S, Rasouli‐Ghahroudi AA. Vitamin K and bone metabolism: a review of the latest evidence in preclinical studies. Biomed Res Int. 2018;2018:1‐8. 10.1155/2018/4629383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kurnatowska I, Grzelak P, Masajtis‐Zagajewska A, et al. Effect of Vitamin K2 on progression of atherosclerosis and vascular calcification in nondialyzed patients with chronic kidney disease stages 3–5. Pol Arch Med Wewn. 2015;125(9):631‐640. [DOI] [PubMed] [Google Scholar]
- 119.Atkins GJ, Welldon KJ, Wijenayaka AR, Bonewald LF, Findlay DM. Vitamin K promotes mineralization, osteoblast‐to‐osteocyte transition, and an anticatabolic phenotype by ‐carboxylation‐dependent and ‐independent mechanisms. Am J Physiol Cell Physiol. 2009;25:1358‐1367. 10.1152/ajpcell.00216.2009 [DOI] [PubMed] [Google Scholar]
- 120.Cockayne S, Adamson J, Lanham‐new S, Shearer MJ, Gilbody S, Torgerson DJ. Vitamin K and the prevention of fractures. Arch Intern Med. 2006;166(12):1256‐1261. [DOI] [PubMed] [Google Scholar]
- 121.Fusaro M, Noale M, Viola V, et al. Vitamin K, vertebral fractures, vascular calcifications, and mortality: vitamin K Italian (VIKI) dialysis study. JBMR. 2012;27(11):2271‐2278. 10.1002/jbmr.1677 [DOI] [PubMed] [Google Scholar]
- 122.Thamratnopkoon S, Susantitaphong P, Tumkosit M, et al. Correlations of plasma desphosphorylated uncarboxylated matrix gla protein with vascular calcification and vascular stiffness in chronic kidney disease. Nephron. 2017;135(3):167‐172. 10.1159/000453368 [DOI] [PubMed] [Google Scholar]
- 123.Delanaye P, Krzesinski J‐M, Warling X, et al. Dephosphorylated‐uncarboxylated Matrix Gla protein concentration is predictive of vitamin K status and is correlated with vascular calcification in a cohort of hemodialysis patients. BMC Nephrol. 2014;15(1):145. 10.1186/1471-2369-15-145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Witham MD, Lees JS, White M, et al. Vitamin K supplementation to improve vascular stiffness in CKD: the K4Kidneys randomized controlled trial. J Am Soc Nephrol. 2020;1‐12. 10.1681/ASN.2020020225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Caluwe R, Verbeke F, De Vriese AS. Evaluation of vitamin K status and rationale for vitamin K supplementation in dialysis patients. Nephrol Dial Transpl. 2020;35:23‐33. 10.1093/ndt/gfy373 [DOI] [PubMed] [Google Scholar]
- 126.Den Besten G, Van Eunen K, Groen AK, Venema K, Reijngoud DJ, Bakker BM. The role of short‐chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res. 2013;54(9):2325‐2340. 10.1194/jlr.R036012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Felizardo R, Watanabe IK, Dardi P, Rossoni LV, Câmara N. The interplay among gut microbiota, hypertension and kidney diseases: The role of short‐chain fatty acids. Pharmacol Res. 2019;141:366‐377. 10.1016/j.phrs.2019.01.019 [DOI] [PubMed] [Google Scholar]
- 128.Vinolo MAR, Rodrigues HG, Nachbar RT, Curi R. Regulation of inflammation by short chain fatty acids. Nutrients. 2011;3(10):858‐876. 10.3390/nu3100858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lucas S, Omata Y, Hofmann J, et al. Short‐chain fatty acids regulate systemic bone mass and protect from pathological bone loss. Nat Commun. 2018;9(1):1‐10. 10.1038/s41467-017-02490-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhou T, Wang M, Ma H, Li X, Heianza Y, Qi L. Dietary fiber, genetic variations of gut microbiota‐derived short‐chain fatty acids, and bone health in UK biobank. J Clin Endocrinol Metab. 2021;106(1):201‐210. 10.1210/clinem/dgaa740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wang S, Lv D, Jiang S, et al. Quantitative reduction in short‐chain fatty acids, especially butyrate, contributes to the progression of chronic kidney disease. Clin Sci. 2019;133(17):1857‐1870. 10.1042/CS20190171 [DOI] [PubMed] [Google Scholar]
- 132.Terpstra ML, Sinnige MJ, Peters‐sengers H, Remmerswaal EBM, Geerlings SE, Bemelman FJ. Butyrate production in patients with end‐stage renal disease. Int J Nephrol Renov Dis. 2019;12:87‐101. 10.2147/IJNRD.S200297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Armani RG, Carvalho AB, Ramos CI, et al. Effect of fructooligosaccharide on endothelial function in CKD patients: a randomized controlled trial. Nephrol Dial Transplant. 2021;1‐7. 10.1093/ndt/gfaa335 [DOI] [PubMed] [Google Scholar]
- 134.Armani RG, Ramezani A, Yasir A, Sharama S, Canziani MEF, Raj DS. Gut microbiome in chronic kidney disease. Curr Hypertens Rep. 2017;19(4):29. 10.1007/s11906-017-0727-0 [DOI] [PubMed] [Google Scholar]
- 135.Borges NA, Carmo FL, Stockler‐Pinto MB, et al. Probiotic supplementation in chronic kidney disease: a double‐blind, randomized placebo‐controlled trial. J Ren Nutr. 2018;28(1):28‐36. 10.1053/j.jrn.2017.06.010 [DOI] [PubMed] [Google Scholar]
- 136.Esgalhado M, Kemp JA, Azevedo R, et al. Could resistant starch supplementation improve inflammatory and oxidative stress biomarkers and uremic toxins levels in hemodialysis patients? A pilot randomized controlled trial. Food Funct. 2018;9(12):6508‐6516. 10.1039/c8fo01876f [DOI] [PubMed] [Google Scholar]
- 137.Hill E, Sapa H, Negrea L, et al. Effect of Oat b ‐glucan supplementation on chronic kidney disease : a feasibility study. J Ren Nutr. 2019;1‐8. 10.1053/j.jrn.2019.06.012 [DOI] [PubMed] [Google Scholar]
- 138.Ramos CI, Armani RG, Canziani MEF, et al. Effect of prebiotic (fructooligosaccharide) on uremic toxins of chronic kidney disease patients: A randomized controlled trial. Nephrol Dial Transplant. 2019;34(11):1876‐1884. 10.1093/ndt/gfy171 [DOI] [PubMed] [Google Scholar]
- 139.Niwa T, Yazawa T, Ise M, et al. Inhibitory effect of oral sorbent on accumulation of albumin‐bound indoxyl sulfate in serum of experimental uremic rats. Nephron. 1991;57(1):84‐88. 10.1159/000186222 [DOI] [PubMed] [Google Scholar]
- 140.Sato E, Saigusa D, Mishima E, et al. Impact of the oral adsorbent AST‐120 on organ‐specific accumulation of uremic toxins: LC‐MS/MS and MS imaging techniques. Toxins (Basel). 2018;10(1):19. 10.3390/toxins10010019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sato E, Hosomi K, Sekimoto A, et al. Effects of the oral adsorbent AST‐120 on fecal p‐cresol and indole levels and on the gut microbiota composition. Biochem Biophys Res Commun. 2020;525(3):773‐779. 10.1016/j.bbrc.2020.02.141 [DOI] [PubMed] [Google Scholar]
- 142.Schulman G, Agarwal R, Acharya M, Berl T, Blumenthal S, Kopyt N. A multicenter, randomized, double‐blind, placebo‐controlled, dose‐ranging study of AST‐120 (Kremezin) in patients with moderate to severe CKD. Am J Kidney Dis. 2006;47(4):565‐577. 10.1053/j.ajkd.2005.12.036 [DOI] [PubMed] [Google Scholar]
- 143.Schulman G, Berl T, Beck GJ, Remuzzi G, Ritz E. Randomized Placebo‐Controlled EPPIC Trials of AST‐120 in CKD. J Am Soc Nephrol. 2015;1732‐1746. 10.1681/ASN.2014010042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.National Library of Medicine U . Oral Absorbent and Probiotics in CKD Patients With PAD on Gut Microbiota, IncRNA, Metabolome, and Vascular Function. ClinicalTrials.Gov Identifier (NCT Number): NCT04788914.; 2021.
- 145.Witkowski M, Weeks TL, Hazen SL. Gut microbiota and cardiovascular disease. Circ Res. 2020;127(4):553‐570. 10.1161/CIRCRESAHA.120.316242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Vasquez EC, Pereira TMC, Campos-Toimil M, Baldo MP, Peotta VA. Gut microbiota, diet, and chronic diseases: The role played by oxidative stress. Oxid Med Cell Longev. 2019;2019:1‐3. 10.1155/2019/7092032 [DOI] [PMC free article] [PubMed] [Google Scholar]