

Abstract
In anticancer drug discovery, multi-targeting compounds have been beneficial due to their advantages over single-targeting compounds. For instance, VEGFR-2 has a crucial role in angiogenesis and cancer management, whereas HDACs are well-known regulators of epigenetics and have been known to contribute significantly to angiogenesis and carcinogenesis. Herein, we have reported nineteen novel VEGFR-2 and HDAC dual-targeting analogs containing diaryl-pyrazoline thiazolidinediones and their in vitro and in vivo biological evaluation. In particular, the most promising compound 14c has emerged as a dual inhibitor of VEGFR-2 and HDAC. It demonstrated anti-angiogenic activity by inhibiting in vitro HUVEC proliferation, migration, and tube formation. Moreover, an in vivo CAM assay showed that 14c repressed new capillary formation in CAMs. In particular, 14c exhibited cytotoxicity potential on different cancer cell lines such as MCF-7, K562, A549, and HT-29. Additionally, 14c demonstrated significant potency and selectivity against HDAC4 in the sub-micromolar range. To materialize the hypothesis, we also performed molecular docking on the crystal structures of both VEGFR-2 (PDB ID: 1YWN) and HDAC4 (PDB-ID: 4CBY), which corroborated the designing and biological activity. The results indicated that compound 14c could be a potential lead to develop more optimized multi-target analogs with enhanced potency and selectivity.
In anticancer drug discovery, multi-targeting compounds have been beneficial due to their advantages over single-targeting compounds.
Introduction
Cancer is the most dreaded disease and a major contributor of death worldwide. To battle against cancer, it is imperative to develop an agent with target specificity and potency. Clinical observations suggest that the use of a single-targeting agent might fail to produce the desired therapeutic effects due to development of resistance to the target or mutation.1 Moreover, the progression of cancer relies on multiple receptors or signaling pathways, and thus, an alternative strategy would be to develop multi-targeting agents.2 Angiogenesis is known to be involved in cancer progression;3,4 a variety of factors are involved in regulation of angiogenesis, in which vascular endothelial growth factors (VEGFs) and their kinase receptors remain the most prominent ones. Tumor cells secrete VEGFs under various conditions such as oncogenic mutations, inflammatory stimuli, hypoxia, stromal and inflammatory cells etc. VEGFs interact with their respective VEGF receptors (VEGFRs) which initiates the critical downstream signaling pathway that brings about tumor angiogenesis; thus, targeting VEGFR would be a reasonable therapeutic approach.5 There are many FDA-approved agents that target VEGFRs (sorafenib, sunitinib, pazopanib, vandetanib, axitinib, regorafenib etc.). However, drug resistance is the major concern associated with the monotherapy of these agents;6,7 hence, over decades researchers have been directing the focus on the potential treatment of resistance.8 Various dual-targeting agents have been reported in which one target is VEGFR-2 along with other therapeutic targets.2,9–12 For example, in 2011, using a multitarget approach, cabozantinib was developed as a dual inhibitor of VEGFR-2 and c-Met and used for the treatment of renal cell carcinoma.13–15
Though genetics is crucial in cancer progression, epigenetics has become equally important in this field. In the process of epigenetic alterations, histone deacetylases (HDACs) play a key role and therefore become therapeutic targets for anticancer drug discovery.16,17 HDACs are subdivided into three zinc-dependent classes. Considering only human enzymes, class I contains HDACs 1, 2, 3 and 8; class II is subdivided into class IIa consisting of HDACs 4, 5, 7 and 9 and class IIb containing HDAC6 and HDAC10; class IV has only one member, HDAC11. The zinc-dependent HDACs have in common a sequentially and structurally highly conserved active site including a catalytic zinc ion. For the sake of completeness, HDAC class III has also to be mentioned. This special class consists of 7 human members, also named sirtuins, which exploit a different NAD+-dependent deacetylation mechanism. Overexpression of HDACs can cause various types of cancers, e.g., colorectal, pancreatic, lung, breast and colon as well as leukemia and hepatocellular carcinoma.18–22 There is emerging evidence that class IIa HDACs and particularly HDAC4 are involved in cancer.23,24 One of the best investigated pathways which controls the proliferation of mammary epithelial cells is the MEF2-HDAC axis. Consequently, targeting class IIa HDACs was suggested as a therapeutic strategy for the treatment of breast cancer.25 Currently, there are five HDAC inhibitors clinically approved as novel antitumor drugs: vorinostat (SAHA), panobinostat (LBH-589), belinostat (PXD-101), romidepsin (FK228) and chidamide. However, their monotherapy fails to display any significant effectiveness against solid tumors1 and they are associated with various side effects such as thrombocytopenia, leukopenia, diarrhoea, fatigue and potential mutagenicity.26,27 Mutagenicity is suspected to be associated with hydroxamate zinc binding groups (ZBGs),28–31 the mechanism being the Lossen rearrangement, a reaction in which hydroxamates are transformed into the corresponding isocyanate.29 Thus, the best alternative strategy to circumvent mutagenicity would be the development of agents containing non-hydroxamate ZBGs. In addition, there are several mechanisms by which HDAC inhibitors also exhibit anti-angiogenesis activity such as: (1) hypoxia causes expression of HIF-1α which initiates the transcription of downstream pathways such as overexpression of VEGF,32 HDAC inhibitors cause suppression of HIF-1α by initiating the expression of noncoding RNAs that target HIF-1α33 and by regulating the transforming growth factor-β (TGF-β) pathway,34–36 leading to anti-angiogenic effects; (2) regulating the acetylation of many non-histone proteins such as P53 which is known to promote degradation of HIF-1α;37,38 (3) tie2-expressing macrophages (TEMs) are known to promote angiogenesis and HDAC inhibitors suppress the M2 polarization of TEMs, thus causing anti-angiogenesis;39,40 (4) arresting the cell cycle; and (5) apoptosis.41 Interestingly, one of the mechanisms by which HDACs exert their antitumor effects is downregulation of VEGF32,42 and suppression of neovascularization through alteration of genes directly involved in angiogenesis.38,43–45 Thus, using a multitarget approach, selecting VEGFR-2 and HDAC as therapeutic targets for the development of novel dual-targeting agents would offer benefits such as reduced side effects and increased potency.
Results and discussion
Designing
The main challenge in designing dual VEGFR-2 and HDAC inhibitors was to achieve a unique framework which satisfied the binding requirements of both targets. In our previous reports, a distinct pharmacophoric drug design was used to achieve both classes of compounds.46,47 Thus, based on our previous experience with both targets, a multi-targeting strategy was developed by considering VEGFR-2 and HDAC pharmacophore properties.
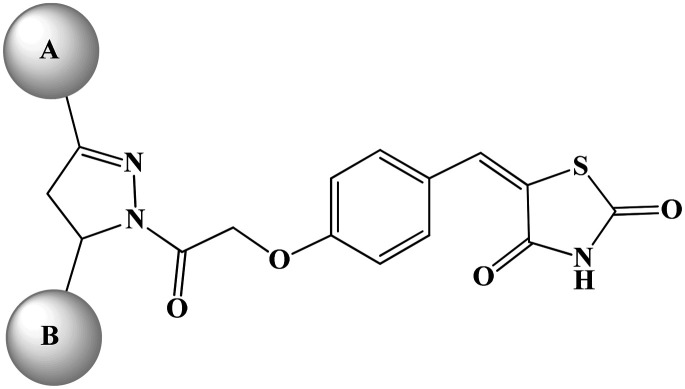
The pharmacophoric requirement of VEGFR-2 inhibitors are (a) a “hinge-binding” moiety that interacts with the ATP pocket, (b) a “linker” usually comprising 3–5 bond lengths and a hydrogen bond acceptor donor system and (c) a hydrophobic moiety occupying the allosteric site.48 With these design considerations, we had previously reported 5-benzylidene-2,4-thiazolidinedione analogs as VEGFR-2 inhibitors (Fig. 1;47 VEGFR-2 IC50 – 0.5 μM). It was hypothesized that a bulky hydrophobic moiety was found to be essential to fill the large empty pocket of the DFG motif;49,50 thus, we modified the 5-benzylidene-2,4-thiazolidinedione analogs by replacing the terminal aryl group with the bulkier substituted diaryl-pyrazoline (ring A and ring B) and retained the remaining structural features to occlude the basic VEGFR-2 inhibitory activity. Since, pyrazoline-containing compounds have been reported to possess excellent VEGFR-2 inhibition capability51–55 and antiproliferative potential,56–59 they were thought to impart antiangiogenic activity to the molecule.
Fig. 1. Rationale of designing novel VEGFR-2 and HDAC dual inhibitors.

Our above designed structural framework also fits perfectly into the pharmacophoric requirements of HDAC inhibitors. Typical HDAC inhibitors share a broad pharmacophore as a surface recognition cap (SRC) which binds with amino acids of the active site, a hydrophobic linker which occupies the active site channel, and a zinc-binding group (ZBG) which chelates the catalytic zinc ion.16 The diaryl pyrazoline moiety of our designed molecule was assumed to occupy the cap portion of HDAC, as recent literature highlights pyrazolines as an appropriate surface recognition motif in HDAC inhibitor design.60–63 Additionally, the literature reports non-classical HDAC inhibitors containing a cyclic ring as a linker rather than a straight hydrocarbon chain;16,64–66 thus, the central phenyl ring was assumed as a cyclic linker. Several recently reported HDAC inhibitors, the so-called non-hydroxamates, have been an interesting part of the HDAC inhibitors' discovery. In this regard, several non-hydroxamic ZBGs have been identified and reported. On a similar line, in our previously reported HDAC inhibitors, we had incorporated thiazolidinedione (TZD) as a non-hydroxamate ZBG which was found to be an effective antitumor agent (Fig. 1; Mohan et al. 2012);46,66–69 hence, the TZD portion of the currently designed series was assumed as ZBG.
This rational design led to a unique structural framework that could be considered in accordance with the pharmacophoric model of both the targets (Fig. 1). The substituted diaryl-pyrazoline group would correspond to the hydrophobic tail of VEGFR-2 and CAP for HDAC. The central phenyl ring with a C O–CH2–O– framework would be the hydrophobic linker for VEGFR-2 and will also function as a cyclic linker of HDAC. The terminal TZD ring would be the hinge binding moiety for VEGFR-2 and ZBG for HDAC. Therefore, we were fairly confident that these compounds could have the capacity to target simultaneously VEGFR-2 and HDAC.
Chemistry
All the final compounds were synthesized by Schemes 1 and 2. The detailed procedure and spectral observations are presented in the Materials and methods. The purity of all the final compounds, which was >95%, was confirmed by HPLC. All the intermediates were structurally confirmed by 1H NMR and FTIR spectroscopy, and the final compounds by 1H NMR, 13C NMR, FTIR and mass spectrometry. Scheme 1 outlines the synthesis of variously substituted chalcones (3a, 3b, 3d–3g, 3j, 3k, 6a–6g, 6j, 9c, 9e, and 9g), chloroacetylated pyrazolines (4a, 4b, 4d–4g, 4j, 4k, 7a–7g, 7j, 10c, 10e, and 10g) and 5-(4-hydroxybenzylidene)-2,4-thiazolidenedione (12). The first step was the synthesis of chalcones (3a, 3b, 3d–3g, 3j, 3k, 6a–6g, 6j, 9c, 9e, and 9g) which occurred via the renowned Claisen–Schmidt condensation reaction, where different acetophenones (1, 5 and 8) were reacted with substituted aromatic aldehydes (2a–2g, 2j, and 2k) in basic medium to obtain the respective chalcones. The second step involved synthesis of chloroacetylated pyrazoline intermediates (4a, 4b, 4d–4g, 4j, 4k, 7a–7g, 7j, 10c, 10e, and 10g), which proceeded via refluxing different chalcones (3a, 3b, 3d–3g, 3j, 3k, 6a–6g, 6j, 9c, 9e, and 9g) with hydrazine hydrate and chloroacetyl chloride in chloroform. The third step was Knoevenagel condensation of 2,4-thiazolidinedione (11) with 2-hydroxybenzaldehyde in the presence of piperidine benzoate in toluene to afford 5-(4-hydroxybenzylidene)-2,4-thiazolidenedione (12). Scheme 2 describes the synthesis of the final compounds (13a, 13b, 13d–13g, 13j, 13k, 14a–14g, 14j, 15c, 15e, and 15g). Reaction of chloroacetylated pyrazolines (4a, 4b, 4d–4g, 4j, 4k, 7a–7g, 7j, 10c, 10e, and 10g) and 5-(4-hydroxybenzylidene)-2,4-thiazolidenedione (12) in DMF and K2CO3 by stirring at RT for 24 h afforded the final compounds (13a, 13b, 13d–13g, 13j, 13k, 14a–14g, 14j, 15c, 15e, and 15g).
Scheme 1. Synthesis of intermediates. Reagents and conditions: (a) aqueous NaOH, EtOH, RT, 5–6 h; (b) NH2NH2·H2O, CHCl3, 80 °C, 12 h, then K2CO3, ClCH2COCl, rt, 12 h; (c) 4-hydroxybenzaldehyde, piperidine benzoate, toluene, reflux, 4 h.

Scheme 2. Synthesis of final compounds. Reagents and conditions: (d) K2CO3, DMF, stirred at RT, 24 h.

In vitro HDAC enzyme inhibition assay on HDAC4 and HDAC8
To evaluate the HDAC inhibitory potential of all the newly synthesized compounds, an initial in vitro screening was performed at 50 μM concentration on HDAC4 (class II) and HDAC8 (class I) and all the compounds showed satisfactory inhibition against both isoforms (Table 1). Compounds with less than 50% residual enzyme activity were evaluated further to determine their inhibitory concentration (IC50); the best compound 13j showed activity in the sub-micromolar range against HDAC4. Most of the analogs were essentially inactive (IC50 >50 μM) or exhibited 2-digit micromolar IC50 values against HDAC8. “Cpd16” and “Cpd31” were used as HDAC4 selective reference inhibitors, and the TZD analogs exhibited less potency against HDAC4 than the reference inhibitors.70,71 In contrast to these reference compounds, the presently reported TZD inhibitors did not contain the zinc-chelating hydroxamate warhead, which mostly contributes to the affinity of classical HDAC inhibitors. Therefore, sub-micromolar activities without typical zinc-chelating groups appear very promising for the development of HDAC inhibitors with presumably less unwanted toxic effects in later pharmaceutical applications. It is noteworthy that the overall results suggest that the novel diaryl-pyrazoline TZDs exhibit more selectivity and potency towards HDAC4 as compared to HDAC8. Based on the in vitro HDAC enzyme inhibition assay results, representative compounds with the most potent inhibitory activity were selected to further determine their anti-angiogenesis capability.
Preliminary screening and IC50 determination of pyrazoline-based TZD on HDAC4 and HDAC8 isoforms.
| Sr. no. | Compounds | Residual HDAC4 activity at 50 μM (%) | Residual HDAC8 activity at 50 μM (%) | HDAC4 IC50 (μM) | HDAC8 IC50 (μM) |
|---|---|---|---|---|---|
| 1 | 13a | 11 ± 2 | 39 ± 1 | 1.6 ± 0.2 | 20 ± 1 |
| 2 | 13b | 14 ± 1 | 27 ± 3 | 1.2 ± 0.3 | 29 ± 1 |
| 3 | 13d | 12 ± 1 | 8.2 ± 0.2 | 1.0 ± 0.2 | 16 ± 1 |
| 4 | 13e | 2.5 ± 0.5 | 26 ± 2 | 1.7 ± 0.4 | 26 ± 1 |
| 5 | 13f | 17 ± 1 | 33 ± 3 | 2.2 ± 0.2 | 34 ± 10 |
| 6 | 13g | 4.6 ± 0.2 | 36 ± 2 | 1.2 ± 0.6 | 31 ± 1 |
| 7 | 13j | 5.9 ± 0.3 | 9.4 ± 1.6 | 0.7 ± 0.3 | 10 ± 1 |
| 8 | 13k | 7.0 ± 0.6 | 21 ± 2 | 1.2 ± 2.0 | 35 ± 1 |
| 9 | 14a | 10 ± 1 | 58 ± 1 | 4.2 ± 0.2 | >50 |
| 10 | 14b | 3.3 ± 1.3 | 28 ± 3 | 1.1 ± 0.9 | >50 |
| 11 | 14c | 36 ± 2 | 45 ± 9 | 0.88 ± 4 | >50 |
| 12 | 14d | 9.2 ± 1.5 | 81 ± 3 | 1.3 ± 3 | >50 |
| 13 | 14e | 3.3 ± 0. | 61 ± 12 | 1.4 ± 0.4 | >50 |
| 14 | 14f | 10 ± 2 | 63 ± 4 | 1.6 ± 0.5 | >50 |
| 15 | 14g | 10 ± 1 | 81 ± 6 | 1.8 ± 0.4 | >50 |
| 16 | 14j | 13 ± 2 | 22 ± 3 | 0.8 ± 0.2 | 25 ± 7 |
| 17 | 15c | 22 ± 3 | 100 ± 8 | 15 ± 2 | >50 |
| 18 | 15e | 10 ± 4 | 69 ± 5 | 1.5 ± 0.7 | >50 |
| 19 | 15g | 9.4 ± 1.8 | 69 ± 3 | 1.5 ± 0.2 | >50 |
| 20 | Cpd 16 (ref. 72) | — | — | 0.039 | 6.7 |
| 21 | Cpd 31 (ref. 73) | — | — | 0.02 | 0.36 |
HDAC selectivity profiling on a panel of HDACs
From the results of the primary evaluation on HDAC4 and HDAC8, compounds 13j, 14c, and 14j were selected for further screening on a panel of HDACs (Table 2) to establish their selectivity profiling on different HDAC isoforms. The assay revealed that 13j and 14j were active towards most of the HDAC isoforms; whereas 14c was more selective towards HDAC4 and its IC50 was found to be in the sub-micromolar range (Fig. 2). Thus, 14c could be used as a lead for the development of more optimized molecules and rigorous structural modifications could provide analogs with enhanced potency and efficacy towards HDAC4 isoforms in the future.
Selectivity profile of pyrazoline-based TZD inhibitors against a panel of human HDACs. IC50 values are means, and errors were obtained from statistical analysis of non-linear regression fits to sets of 10 data points.
| IC50 (μM) | |||||||
|---|---|---|---|---|---|---|---|
| Compound | HDAC1 | HDAC2 | HDAC3 | HDAC4 | HDAC6 | HDAC7 | HDAC8 |
| 13j | 4.9 | 13 | 9.1 | 0.7 ± 0.3 | 39 | >50 | 10 ± 1.0 |
| 14c | >50 | >50 | >50 | 0.88 ± 4 | 7.6 | >50 | >50 |
| 14j | 11 | 21 | 11 | 0.8 ± 0.2 | 13 | 6.7 | 25 ± 7.0 |
Fig. 2. Dose–response curve of compounds 13j, 14c, and 14f on different HDAC isoforms.

Thermal shift assay on HDAC4
Thermal shift is used to demonstrate target engagement (TE) by a ligand. Binding of a ligand to the target protein can shift the melting temperature of the protein to the higher or lower side as compared to free protein, thus producing a thermal shift. Test compounds with HDAC4 inhibitory potential (13j and 14j) were subjected to this assay with the HDAC4 isoform to examine the thermal stabilization of the ligands. The melting temperature of free HDAC4 (DMSO control) was 57.3 °C, and those of the protein in the presence of the test compounds were 56.7 °C and 57.3 °C for 13j and 14j, respectively (Fig. 3). These minor changes in comparison with the melting point of the unbound enzyme were within the error range of the method. Altogether, even though the compounds were able to inhibit HDAC4 in sub-micromolar concentrations, they contribute nothing or only to a minor degree to protein stabilization.
Fig. 3. Thermal stabilization of 3 μM HDAC4 upon binding of 100 μM 13j and 14j.

Cytotoxicity assessment on HUVEC proliferation
Human umbilical vein endothelial cells (HUVECs) are the most commonly used human endothelial cells for in vitro angiogenesis assays.74,75 In the development of sprouting angiogenesis, endothelial cells undergo proliferation, which is related to survival of new vessels; thus, the effects of anti-angiogenic agents on the proliferation of HUVECs could be measured by the MTT assay,74–77 in which the number of viable cells is determined. Nine compounds with the highest HDAC4 inhibition potential (13b, 13d, 13j, 14a, 14c–14e, 14j, and 15g) were selected to determine their in vitro cytotoxicity effects on HUVEC proliferation. STS was used as a positive control. Five different concentrations (10, 1, 0.1, 0.01, and 0.001 μM) of test compounds as well as the positive control were used to determine their inhibition potential. Results showed that compounds 13b, 14c, 14d and 14j exhibited satisfactory inhibition effects on HUVEC proliferation (IC50 <10 μM), while 13d, 13j, 14a, 14e and 15g displayed poor inhibition (>10 μM). Moreover, 14c showed inhibitory activity comparable to that of STS (Table 3); thus, we assume that it may have potential anti-angiogenic activity.
IC50 of HUVECs by compounds 13b, 13d, 13j, 14a, 14c–14e, 14j, and 15g.
| ||||
|---|---|---|---|---|
| Sr. no. | Code | IC50a (μM) | A ring | B ring |
| 1 | 13b | 2 | Phenyl | 2-Cl phenyl |
| 2 | 13d | >10 | Phenyl | 4-CF3 phenyl |
| 3 | 13j | >10 | Phenyl | 3,4-DiCl phenyl |
| 4 | 14a | >10 | 4-F phenyl | Phenyl |
| 5 | 14c | 0.7 | 4-F phenyl | 2-Furyl |
| 6 | 14d | 6 | 4-F phenyl | 4-CF3 phenyl |
| 7 | 14e | >10 | 4-F phenyl | 4-F phenyl |
| 8 | 14j | 2 | 4-F phenyl | 3,4-DiCl phenyl |
| 9 | 15g | >10 | 2,4-DiF phenyl | 4-Tolyl |
| 10 | STS | 0.5 | — | — |
Assays were performed in replicate (n ≥ 2).
VEGFR-2 inhibition assay
Test compounds with inhibitory capability on HUVEC proliferation (13a, 14c, 14d, and 14j) were selected to determine their effects on the activity of VEGFR-2 phosphorylation, which is known to activate other signalling cascades associated to endothelial cells and bring about proliferation, migration, invasion, and cell differentiation.78,79 An in vitro cell-based ELISA method was used in which all four test compounds along with the positive control STS were preliminarily tested at 10 μM concentration against pVEGFR-2. The test compounds with noticeable structural differences displayed poor to good % VEGFR-2 inhibition, wherein 14c showed >50% inhibition, and the IC50 of 14c further confirmed its pVEGFR-2 inhibition potential. Structurally, 14c contains 4-fluorophenyl at ring A and 2-furyl at ring B, while the other two compounds with 4-fluorophenyl at ring A (14d and 14j) and other substituents at ring B (14d: 4-trifluoromethylphenyl; 14j: 3,4-dichlorophenyl) displayed <50% inhibition (Table 4). However, compound 14j exhibited a remarkable decrease in % VEGFR-2 inhibitory activity as compared to 14c and 14d, which suggested that dichlorophenyl at ring B is of least preference as compared to the other substituents.
VEGFR-2 inhibitory activity of 13a, 14c, 14d, and 14j.
| Sr. no. | Code | % Inhibitiona (at 10 μM) | IC50 (μM) |
|---|---|---|---|
| 1 | 13b | 48.95 | NDb |
| 2 | 14c | 56.66 | 5 |
| 3 | 14d | 48.71 | NDb |
| 4 | 14j | 22.36 | NDb |
| 5 | STSc | 87.85 | 0.5 |
Assays were performed in replicate (n ≥ 2).
Not determined.
STS represents staurosporine.
Molecular docking study
A complete docking study was performed to predict the binding pose of the ligand and determine the putative molecular determinants of protein–ligand binding. The docking procedure was validated by redocking of the ligand in the crystal structure of VEGFR-2 (PDB-ID: 1YWN) and HDAC4 (PDB-ID: 4CBY).70 The ligands were docked into the protein receptor which yielded an almost perfect overlap with the X-ray pose (ESI†). Thus, by assuming that all parameters were well configurated for docking of pyrazoline TZD analogs, the same crystal structures were used throughout the docking study. The GBVI/WSA dG docking score of the redocked ligand was −11.2 on VEGFR-2 and −13.6 on HDAC4 (ESI†). Docking of the complementary stereoisomers of 14c and 14j showed similar binding poses of (S)-14c, (R)-14j and (S)-14j having the same absolute configuration at the pyrazole ring (ESI†). The binding mode of (S)-14c to VEGFR-2 resembles that of previously reported TZD analogs.47 The TZD warhead forms two hydrogen bonds with L838 and A1048. Multiple hydrophobic pi-alkyl and alkyl interactions contribute further to affinity (Fig. 4).
Fig. 4. Docking of (S)-14c into VEGFR-2. (A) 3D binding pose showing hydrogen bonds (dotted green lines) to L838 and A1048. (B) Detailed 2D protein–ligand interactions.

Furthermore, the docking study of compounds 14c and 14j with HDAC4 gained more insight into the mode of molecular interaction. The results for the two enantiomers of these analogs show rather similar docking scores, which were considerably lower than that obtained from redocking the reference compound “Cpd 31” into the crystal structure of its complex with the catalytic domain of HDAC4 (Fig. 5).71 This appears realistic, since “Cpd 31” is about 45-fold more active against HDAC4 than 13d and 14c (Table 1). Looking closer at the binding pose of (S)-14c, it was revealed that the TZD group was able to replace the hydroxamate warhead of classic HDAC inhibitors, since the TZD group not only interacted with the catalytic zinc ion in the catalytic site but also showed amide-pi stacking with G811 and pi-alkyl interaction with L943 (Fig. 5). The binding was also strengthened by parallel pi-stacking interactions with F871. The branched pyrazoline group serving as capping group was largely exposed to the surrounding solvent but showed an amide-pi interaction with F870. Therefore, it is not unexpected that different substitution patterns at the aromatic rings of the capping group have no big influence on the enzyme inhibitory activity (ESI†). Thus, the docking results confirmed that the pyrazoline TZD analogs of this study were biologically active against both target proteins VEGFR-2 and HDAC4.
Fig. 5. Docking pose of (S)-14c in the active pocket of HDAC4 (PDB-ID: 4CBY). (A) 3D binding pose showing hydrogen bonds with F871 and Zn2+ interaction. (B) Detailed 2D protein–ligand interactions.

Endothelial cell migration assay
Endothelial cell mobility is significant for tumor angiogenesis which is known to be initiated by different growth factors.80,81 To measure the extent of endothelial cell motility against test compounds, scrape wound healing assay was performed. The migration capability of HUVECs was determined for the most active compound 14c along with positive control STS, which was measured in percent wound healing. As compared to untreated HUVECs, STS-treated HUVECs suppressed the endothelial cell migration to a greater extent (Fig. 6). The inhibitory effect of 14c on the mobility of HUVECs was less than that of STS but greater than that of untreated, evident by percent wound healing. Thus, with the results it was clear that 14c has the potential to interfere with the migration capability of endothelial cells.
Fig. 6. (A–C) Representative images of cellular migration assay in HUVECs after 8 h of untreated, STS (10 μM mL−1), and 14c (10 μM mL−1), respectively. (D) Graphical representation of percent wound healing. Error bars represent SEM, n = 3, *p ≤ 0.01; ***p ≤ 0.0001 (compared to untreated; calculated with unpaired Student's t-test).

Capillary tube formation assay
The process of angiogenesis requires capillary-like tube formation of endothelial cells for ease of blood flow, and it is considered as representative of later stages of angiogenesis. Thus, the tube formation assay is extensively used to determine the in vitro anti-angiogenic effects of test compounds.74–77 Compound 14c exhibited substantial inhibition of HUVEC proliferation, VEGFR-2 phosphorylation, and migration; thus, it was further evaluated to determine its effect on the tube formation capability of HUVECs. After 24 h, STS-treated HUVECs reduced the number of capillaries to a greater extent as compared to untreated which formed hollow capillary-like structures (Fig. 7A and B). However, 14c did not significantly decrease the capillary formation in comparison with STS (Fig. 7C and Table S2† (p = 0.31)). Thus, we could state that 14c has less potential to decrease the tube formation activity of HUVECs as compared to STS.
Fig. 7. (A–C) Images of HUVEC capillary-like tube formation assay after 48 h of untreated, STS (10 μM mL−1), and 14c (10 μM mL−1), respectively. (D) Graphical representation of intersection counts with different treatments. Error bars represent SEM, n = 3, ***p ≤ 0.0001 (STS compared to untreated; calculated with unpaired Student's t-test).

MTT cytotoxicity assay
Considering in vitro results of anti-angiogenic assays, 14c was evaluated for its effects on the cytotoxicity of four different cancer cell lines: MCF-7 (human breast cancer), K562 (leukaemia), A549 (human lung cancer), and HT-29 (human colorectal adenocarcinoma). Results revealed that 14c exhibited moderate to good cytotoxicity on these four cancer cell lines (Table 5). The positive control paclitaxel was several fold more potent than 14c, whereas cisplatin showed variation in activity. HT-29 cell lines appeared to be most affected by 14c followed by A549, MCF-7 and K562 (ESI†). Thus, we assume that 14c has potential cytotoxicity against these cancer cell lines.
Cancer cell viability of 14c.
| Compound | IC50a (μM) | |||
|---|---|---|---|---|
| MCF-7 | K562 | A549 | HT-29 | |
| 14c | 28.41 ± 4.2 | 46.27 ± 1.6 | 19.52 ± 1.44 | 18.84 ± 1.1 |
| Paclitaxel | 0.35 | 0.29 | 0.32 | 0.28 |
| Cisplatin | 10.57 ± 1.1 | 58.4 ± 1.4 | 16.68 ± 1.74 | 10.6 ± 1.2 |
Assays were performed in replicate (n ≥ 2).
In vivo chick chorioallantoic membrane (CAM) assay
The CAMs are easy to access outside the embryo and represent a reliable approach to study the anti-angiogenic effects of compounds in vivo.77 Compound 14c was chosen to evaluate its inhibitory effects on growing CAMs to confirm the in vivo anti-angiogenic activity. After 12 days of implanting sponges loaded with the test compound into the CAMs, the average number of blood vessels was determined. It was observed that untreated CAMs were surrounded by allantoic vessels with newly formed capillaries (Fig. 8A), whereas 14c-treated (Fig. 8B) CAMs demonstrated significant reduction in the number of branching of capillaries, further supporting its anti-angiogenic effects.
Fig. 8. Representative images of untreated (A) and 14c-treated (B) CAMs. (C) Graphical representation of CAM assay. Error bars represent SEM, n = 4, ***p ≤ 0.0001 (STS compared to untreated; calculated with unpaired Student's t-test).

Materials and methods
Chemistry
All the reagents, solvents, chemicals etc. were procured from sources, viz. Sigma Aldrich, S.D. Fine Chem. Ltd., and Himedia, and utilized without any further purification. The reactions were monitored at each step by thin layer chromatography (TLC) using Merck precoated silica gel 60 F-254 plates under short wave UV light (254 nm) to identify the UV absorbing spots for completion of the reaction and also to trace any impurities. All intermediates were purified by a recrystallization method using suitable solvents such as chloroform, methanol etc. All the final molecules were purified by a column chromatography technique on silica gel 60 (60 to 120 mesh) using a suitable combination of different solvents. The melting points of all the intermediates were obtained by using a VEEGO model VMP-DS melting point apparatus and those of the final compounds were obtained using a DSC1 STAR system differential scanning calorimeter from Mettler Toledo. The purity of all final compounds was determined using an Agilent 1200 high-performance liquid chromatography (HPLC) system with the software EZ chrome Elite. The chromatographic column used was HemochromIntsil A31 C18 5U 150 mm × 4.6 mm Sn-B180127, detection at 300 nm. A UV-visible detector was used with a flow rate of 1 mL min−1. The oven temperature was maintained at 30 °C; gradient elution with a run time of 10 min using methanol : formic acid (1%) (formic acid: in 1000 mL double-distilled water 1 mL formic acid was added) in an 80 : 20/90 : 10 ratio. The structures of intermediates were confirmed by FTIR and 1H NMR and that of the final compounds by FTIR, 1H NMR, 13C NMR and mass spectrometry. IR analysis was performed using a JASCO FT/IR-4100 type A spectrometer using a manual sampling method. 1H NMR spectra were recorded using a Bruker Avance 400 MHz spectrometer with DMSO-d6. All shifts are reported in δ (ppm) units relative to the signals for solvent DMSO (δ – 2.50 ppm). All coupling constants (J values) are reported in hertz (Hz). NMR abbreviations are bs, broad singlet; s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; and dd, doublet of doublets. 13C NMR was recorded on a Bruker Avance spectrometer at 100 MHz with DMSO-d6. The mass spectrum was determined on an LC-MS Agilent Technologies 1260 Infinity instrument. All final compounds were synthesized by Schemes 1 and 2. Scheme 1 represents the synthesis of intermediates and Scheme 2 of the final compounds.
Procedure for synthesis of chalcone intermediates
Different chalcones were synthesized using a previously reported procedure82,83 with some minor modifications. In short, to ethanolic NaOH solution (10%, 20 mL), different acetophenones (0.04 mol-acetophenone, 4-fluoroacetophenone, and 2,4-difluoroacetophenone) were added followed by aldehydes (0.04 mol 2a–2g, 2j, and 2k) in an ice bath and stirred at room temperature (RT) for 5–6 h. The solid precipitate was filtered and washed with cold water. It was then recrystallized with ethanol to obtain the appropriate chalcones (3a, 3b, 3d–3g, 3j, 3k, 6a–6g, 6j, 9c, 9e, and 9g) (Scheme 1).
Procedure for synthesis of chloroacetylated pyrazoline intermediates
Various pyrazole derivatives were synthesized and previously reported by us.82,83 With minor modifications in the previous procedure, we synthesized pyrazoline derivatives from their respective chalcones. To a solution of chalcones (3a, 3b, 3d–3g, 3j, 3k, 6a–6g, 6j, 9c, 9e, and 9g) (0.02 mol) in chloroform, hydrazine hydrate (0.04 mol) was added and refluxed for 12 h. In this reaction mixture K2CO3 (0.05 mol) was added and stirred for 15 min followed by chloroacetyl chloride (0.03 mol) with stirring under an ice bath and stirred at RT. After 12 h the reaction was stopped, and the chloroform layer was washed with water to remove excess K2CO3 and evaporated to obtain the solid. The crude was purified by extracting with diethyl ether to obtain the appropriate chloroacetylated pyrazoline intermediates (4a, 4b, 4d–4g, 4j, 4k, 7a–7g, 7j, 10c, 10e, and 10g) (Scheme 1).
Procedure for synthesis of 5-(4-hydroxybenzylidene)-2,4-thiazolidenedione (12)
Intermediate 12 was synthesized using a previously reported procedure,84,85 in which 2,4-thiazolidinedione (4.68 g, 0.04 mmol) and 4-hydroxy benzaldehyde (4.88 g, 0.04 mmol) were dispersed in toluene followed by a catalytic amount of piperidinium benzoate and this reaction mixture was refluxed in a Dean Stark apparatus for 4 h. The reaction mixture was cooled to RT and the solid precipitated out was collected by vacuum filtration to obtain 12 (Scheme 1). Yellow solid. Yield 11 g (89%). M. P. 300–302 °C.
Procedure for synthesis of final compounds
The target compounds were synthesized using a similar synthetic route mentioned previously.66,85 5-(4-Hydroxybenzylidene)-2,4-thiazolidenedione (12) (1.0 mol) and potassium carbonate (K2CO3) (1.5 mol) were added to dimethyl formamide (DMF) (10 mL) and stirred for 5 min at RT. To this solution chloroacetylated pyrazolines (4a, 4b, 4d–4g, 4j, 4k, 7a–7g, 7j, 10c, 10e, and 10g) (1.5 mol) were added and stirred for 24 h at RT. The reaction was quenched by adding water (50 mL); the precipitated solid was collected by filtration and washed with water. The crude was purified by column chromatography with hexane : ethyl acetate (1 : 1 to 1.5 : 0.5) solvent to obtain the final products (13a, 13b, 13d–13g, 13j, 13k, 14a–14g, 14j, 15c, 15e, and 15g) (Scheme 2).
The ESI† contains all the characterization data.
HDAC enzyme inhibition assay
Recombinant HDAC1, 2, 3, 6 and 7 were purchased from BPS Bioscience. Recombinant HDAC8 was produced as described recently.86 In short, HDAC8 was produced in E. coli (BL21) DE3 pLysS cells using a pET14b vector containing codon-optimized human HDAC8. The expression of recombinant cHDAC4 was performed according to another recently published procedure.87 Recombinant cHDAC4 was expressed in E. coli (BL21) DE3 pLysS using a pET14b vector (Novagen, EMD Millipore) containing the codon-optimized catalytic domain of human HDAC4, fused to an N-terminal His6-SUMO tag and a C-terminal SII tag and auto-induction medium. Enzyme activity assays were performed in a two-step procedure as described in detail previously.87 The fluorogenic activity assay relies on the transformation of Boc-Lys(trifluoroacetyl)-AMC (Bachem) as substrate for HDAC4, 7 and 8 and Boc-Lys(acetyl)-AMC as substrate for HDAC1, 2, 3 and 6. Afterwards, the deacetylated substrates are converted into a fluorescent product by trypsin.
Thermal shift assay of HDAC4
HDAC4 was tested in the absence (DMSO control) and in the presence of the compounds. For the thermal shift assay, 3 μM HDAC4 was mixed with 100 μM of compound (dissolved in DMSO) and incubated for 15 min at 30 °C. Ten aliquots of this mix were transferred into PCR strips (25 μl per well) and further incubated for 60 min at 30 °C in a PCR cycler (T Gradient, Biometra). Each indicated temperature was held for 10 min followed by a temperature increase of 2 °C. After 9.5 min, a sample from the respective well was stored on ice. After the sample collection for all indicated temperatures, the samples were centrifuged at 4 °C and 18 000g for 15 min. After centrifugation the supernatant was treated with Laemmli buffer and denatured via heat followed by SDS-PAGE. By using ImageJ program, the relative band intensity at each indicated temperature was calculated, and the GraphPad Prism program was used for plotting intensities against respective temperatures and fitting the data to a logistic function.88 The melting point of the protein is the x-value of the point of infliction, which is the IC50 value of the logistic function.
Antiproliferative assay on HUVECs
HUVECs were procured from Vinod Nursing Home, Bhopal, India; umbilical cord cells were isolated by collagenase treatment of the umbilical cord and HUVECs were further cultured and maintained for all further experiments. [(3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide)] or MTT, a pale-yellow substrate, is known to be cleaved by living cells to form a dark blue formazan product. The process involves active mitochondria, and freshly dead cells do not cleave a significant amount of MTT. Hence, the amount of MTT cleaved is in direct proportion with the number of viable cells; this is quantified by colorimetric methods. Test compounds and positive control staurosporine (STS) were solubilized in DMSO and diluted with complete medium to obtain 5 different ranges of test concentrations (10, 1, 0.1, 0.01, and 0.001 μM). DMSO concentration was maintained at <0.1% in all the samples. HUVECs maintained in suitable environments were seeded in 96-well plates and treated with different concentrations of all test compounds and incubated at 37 °C in 5% CO2 for 96 h. MTT reagents were added to the well plates and kept for incubation for 4 h. The formazan products (dark blue) formed by the living cells were dissolved in DMSO under a safety cabinet and read at 550 nm. The % inhibitions were calculated and IC50 values for test compounds were determined by plotting the different concentrations used.
VEGFR-2 inhibition assay
HUVECs were seeded at 4000–5000 cells per well by using DMEM and 10% FCS for 12 h. Post-incubation, the cells were incubated in serum for 24 h. To the test compound and STS (10 μM), serum was added and incubated for 30 min at 37 °C. The medium was removed, and cells were fixed with 4% formaldehyde in PBS. The cells were washed thrice with PBS. The cells were incubated with anti-phospho VEGF Tyr 1175 antibody (1 : 1000) for 1 h. The cells were washed thrice with PBS T and incubated with secondary antibody labelled with HRP (1 : 5000) for 1 h. The cells were washed and incubated with TMB. The reaction was immobilized with 2N H2SO4 and read at 450 nM. The % inhibition was calculated by normalizing with the control.
Migration assay
HUVECs were grown to 90% confluency in 6-well plates in DMEM and 10% FCS. A scratch was made with a sterile pipette tip to create a wound. Test compound 14c and positive control STS 10 μM were added immediately and the cells were imaged. The cells were splashed with 1 mL of DMEM and then with 2 mL of DMEM. They were further incubated at 37 °C in a 5% CO2 atmosphere. Images were acquired again after 8 h. Cell migration was determined by wound healing (%), which was the distance the cells migrated starting from the original wound margin. Wounds were measured using ImageJ software and % wound closure was calculated and recorded.
Capillary tube formation assay
In order to perform capillary-like tube formation assay, HUVECs were trypsinized at 80% confluency and seeded in a 6-well plate at 50 000 cells and DMEM + 10% FCS per well coated with collagen gel. Test compound 14c and STS in 10 μM concentrations were added at 3 h post seeding and PBS with 0.01% DMSO was used as a control. The plates were incubated at 37 °C in 5% CO2 for 48 h. The intersections of the tubes were measured in a given field and recorded.
MTT cytotoxicity assay
In vitro antiproliferative assays were conducted using a procedure previously described.66,89–91 All the cell lines (MCF-7, K562, A549, and HT-29) were obtained from National Centre for Cell Sciences (NCCS), Pune. To summarize, the cells were seeded in 96-well flat-bottom micro plates and maintained at 37 °C in 95% humidity and 5% CO2 overnight. Different concentrations (100, 75, 50, 25, 10, 2.5 μm ml−1) of test compound 14c and positive controls (paclitaxel and cisplatin) were treated. The cells were incubated for 48 h. The wells were washed twice with PBS, 20 μL of MTT staining solution was added to each well and the plate was incubated at 37 °C. After 4 h, 100 μL of DMSO was added to each well to dissolve the formazan crystals, and absorbance was recorded at 570 nm using a microplate reader. The IC50 of compounds was calculated by using Graph Pad Prism Version 5.1.
Formula:Surviving cells (%) = Mean OD of test compound/Mean OD of Negative control × 100
Molecular docking
Preparation, visualization of structural data and molecular docking were performed using MOE 2019 software (Chemical Computing Group ULC, Canada). The crystal structures of VEGFR-2 (PDB-ID: 1YWN) and HDAC4 (PDB-ID: 4CBY) were obtained from the RCSB Protein Data Bank. The structure files were loaded into the program and subjected to structure preparation including 3D protonation for subsequent docking. The partial charges of all protein and ligand atoms were calculated using the implemented Amber14 force field. Molecular docking was performed choosing the triangle matcher for placement of the ligand in the binding site and ranked with the London dG scoring function. The best 50 poses were passed to the refinement and energy minimization in the pocket using the induced fit method and then rescored using the GBVI/WSA dG scoring function. The protein–ligand complexes were subsequently energy minimized within a radius of 10 Å around the ligand using the Amber14 force field.
CAM assay
Test sample preparation
Test sample 14c was submitted in airtight glass vials and stored at 4 °C in a light-controlled environment. A 10 or 1 μg μl−1 solution of test samples was prepared in PBS and sterilized by passing through a syringe filter (0.22 μm). hVEGF (SIGMA) 50 ng μl−1 was prepared in sterile PBS.
Grafting
Gelatin sponges (Abogel) were cut in approximately 2 mm3 pieces and loaded with 2 μl of a 1 : 1 mixture of test substance solution and VEGF solution. The graft was placed on the CAM.
Eggs
Fertile hen eggs were procured from a hatchery and were cleaned and decontaminated using alcohol. 1 ml of albumin was removed using a syringe and incubated for 8 days. Grafts were placed on developing CAMs and further incubated to day 12. On day 12, CAMs were fixed with formaldehyde and dissected.
Imaging
Fixed CAMs were observed and scored under constant illumination and magnification under a stereomicroscope by two independent experts.
Statistical analysis
Data were analyzed on MS Excel 2007.
Conclusions
In the anticancer drug discovery field, single-targeting agents have failed to show the desired pharmacological effects due to several factors such as intrinsic and acquired resistance. Detailed cellular reports suggest that cancer cells obey Darwin's law of evolution and follow alternative pathways for their survival. Thus, the new strategy to combat drug resistance would be to develop multi-targeting agents endowed with anti-tumor efficacy. Angiogenesis is the fundamental process significant for overall survival of solid tumors; therefore, the search for anti-angiogenic agents has become the primary line of investigation in this field. The epigenetic modulator histone deacetylase has been known to contribute significantly in the progression and metastasis of different cancers. Thus, selecting VEGFR-2 and HDAC would be a rational multi-target approach for the drug discovery of novel chemical weapons to combat cancer.
Herein, we have rationally designed, synthesized, purified, and structurally characterized diaryl-pyrazoline TZD derivatives (nineteen compounds). All the synthetic molecules were primarily screened at 50 μM on two different HDAC isoforms (HDAC4 and HDAC8), which revealed their selectivity towards class II HDAC isoform, i.e., HDAC4, as compared to class I HDAC8. All compounds displayed excellent inhibitory activity on HDAC4 (<10 μM) and only a few showed HDAC8 inhibition. Moreover, 13d, 13j and 14c exhibited uncompromised HDAC4 IC50 (<1 μM), which suggests that placing a pyrazoline scaffold at the terminal portion and TZD at the ZBG site of the framework was beneficial in enhancing the HDAC potency of the compounds. Based on the results obtained by this study, nine molecules with the best HDAC inhibitory activity were selected to further evaluate their detailed anti-angiogenic capability. These synthesized derivatives were evaluated to determine their effects on HUVEC proliferation, among which 13d, 14c, 14d, and 14j demonstrated inhibitory activity at <10 μM, indicating their anti-angiogenic potential. These compounds were further screened at 10 μM on VEGFR-2 which revealed the best compound 14c with >50% VEGFR-2 inhibition and IC50 of 5 μM. Furthermore, 14c also possessed an inhibition potential of endothelial cell migration. It was screened on four different cancer cell lines (MCF-7, K562, A549 and HT-29), and it was worth noting that it exhibited anti-proliferative activity on all four cell lines. The anti-angiogenic capability of compound 14c was further confirmed by an in vivo CAM assay which demonstrated significant reduction in the number of capillary branching. Additionally, molecular docking was performed to determine the mechanism of binding mode of 14c at the VEGFR-2 (PDB ID: 1YWN) and HDAC4 (PDB-ID: 4CBY) receptor sites. Docking results showed essential binding interactions such as F1045 and D1044 on VEGFR-2, and H842 and F871 on HDAC4.
The results obtained were in agreement with our hypothesis that diaryl-pyrazoline thiazolidinedione analog 14c holds both anti-angiogenic and HDAC inhibitory activity. To conclude, this study unfolds that 14c has the potential to simultaneously target both VEGFR-2 and HDAC4. Based on these findings, we put forward that 14c could be used as a lead for the discovery of more optimized dual inhibitors.
Conflicts of interest
There are no conflicts of interest to declare.
Supplementary Material
Acknowledgments
This research work was supported by an “Indo-ASEAN Collaborative Research Project Grant” from the ASEAN-India S&T Development Fund (AISTDF), Department of Science and Technology (DST), Government of India, Project Reference Number IMRC/AISTDF/CRD/2018/000001 (to CSR); the National Medical Research Council of Singapore (to APK); and the National Medical Research Council of Singapore and the Singapore Ministry of Education under its Research Centres of Excellence Initiative to the Cancer Science Institute of Singapore, National University of Singapore (to APK). We also acknowledge funding through the LOEWE priority program TRABITA, State of Hesse, Germany (to FJMA).
Electronic supplementary information (ESI) available. See DOI: 10.1039/d1md00125f
References
- Yang F. Zhao N. Ge D. Chen Y. RSC Adv. 2019;9:19571–19583. doi: 10.1039/C9RA02985K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu R. Sun Y. Sheng W. Liao D. Eur. J. Med. Chem. 2017;136:195–211. doi: 10.1016/j.ejmech.2017.05.016. [DOI] [PubMed] [Google Scholar]
- Carmeliet P. Jain R. K. Nature. 2011;473:298. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerbel R. S. N. Engl. J. Med. 2008;358:2039–2049. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara N. Oncology. 2005;69(Suppl 3):11–16. doi: 10.1159/000088479. [DOI] [PubMed] [Google Scholar]
- Bergers G. Hanahan D. Nat. Rev. Cancer. 2008;8:592. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gacche R. N. Meshram R. J. Biochim. Biophys. Acta, Rev. Cancer. 2014;1846:161–179. doi: 10.1016/j.bbcan.2014.05.002. [DOI] [PubMed] [Google Scholar]
- Peng F.-W. Liu D.-K. Zhang Q.-W. Xu Y.-G. Shi L. Expert Opin. Ther. Pat. 2017;27:987–1004. doi: 10.1080/13543776.2017.1344215. [DOI] [PubMed] [Google Scholar]
- Li Y. Tan C. Gao C. Zhang C. Luan X. Chen X. Liu H. Chen Y. Jiang Y. Bioorg. Med. Chem. 2011;19:4529–4535. doi: 10.1016/j.bmc.2011.06.022. [DOI] [PubMed] [Google Scholar]
- Raghavendra N. M. Pingili D. Kadasi S. Mettu A. Prasad S. V. U. M. Eur. J. Med. Chem. 2018;143:1277–1300. doi: 10.1016/j.ejmech.2017.10.021. [DOI] [PubMed] [Google Scholar]
- Smith B. D. Kaufman M. D. Leary C. B. Turner B. A. Wise S. C. Ahn Y. M. Booth R. J. Caldwell T. M. Ensinger C. L. Hood M. M. Mol. Cancer Ther. 2015;14:2023–2034. doi: 10.1158/1535-7163.MCT-14-1105. [DOI] [PubMed] [Google Scholar]
- Zang J. Liang X. Huang Y. Jia Y. Li X. Xu W. Chou C. J. Zhang Y. J. Med. Chem. 2018;61:5304–5322. doi: 10.1021/acs.jmedchem.8b00384. [DOI] [PubMed] [Google Scholar]
- Kurzrock R. Sherman S. I. Ball D. W. Forastiere A. A. Cohen R. B. Mehra R. Pfister D. G. Cohen E. E. W. Janisch L. Nauling F. Hong D. S. Ng C. S. Ye L. Gagel R. F. Frye J. Müller T. Ratain M. J. Salgia R. J. Clin. Oncol. 2011;29:2660–2666. doi: 10.1200/JCO.2010.32.4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyseng-Williamson K. A. Drugs Ther. Perspect. 2018;34:457–465. doi: 10.1007/s40267-018-0547-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S. Quinn D. Dorff T. OncoTargets Ther. 2016;9:5825–5837. doi: 10.2147/OTT.S97397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller T. A. Witter D. J. Belvedere S. J. Med. Chem. 2003;46:5097–5116. doi: 10.1021/jm0303094. [DOI] [PubMed] [Google Scholar]
- Mottamal M. Zheng S. Huang T. L. Wang G. Molecules. 2015;20:3898–3941. doi: 10.3390/molecules20033898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. Y. Jun J. A. Jeong K. J. Heo H. J. Sohn J. S. Lee H. Y. Park C. G. Kang J. Oncol. Rep. 2011;25:1677–1681. doi: 10.3892/or.2011.1236. [DOI] [PubMed] [Google Scholar]
- Nakagawa M. Oda Y. Eguchi T. Aishima S.-I. Yao T. Hosoi F. Basaki Y. Ono M. Kuwano M. Tanaka M. Tsuneyoshi M. Oncol. Rep. 2007;18:769–774. [PubMed] [Google Scholar]
- Singh A. Patel P. Jageshwar Patel V. K. Jain D. K. Kamal M. Rajak H. Curr. Cancer Drug Targets. 2018;18:720–736. doi: 10.2174/1568009617666170630124643. [DOI] [PubMed] [Google Scholar]
- Lernoux M. Schnekenburger M. Dicato M. Diederich M. Biochem. Pharmacol. 2020;173:113698. doi: 10.1016/j.bcp.2019.113698. [DOI] [PubMed] [Google Scholar]
- Kim H. S. Shen Q. Nam S. W. J. Korean Med. Sci. 2015;30:1375–1380. doi: 10.3346/jkms.2015.30.10.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asfaha Y. Schrenk C. Alves Avelar L. A. Hamacher A. Pflieger M. Kassack M. U. Kurz T. Bioorg. Med. Chem. 2019;27:115087. doi: 10.1016/j.bmc.2019.115087. [DOI] [PubMed] [Google Scholar]
- Guerriero J. L. Sotayo A. Ponichtera H. E. Castrillon J. A. Pourzia A. L. Schad S. Johnson S. F. Carrasco R. D. Lazo S. Bronson R. T. Davis S. P. Lobera M. Nolan M. A. Letai A. Nature. 2017;543:428–432. doi: 10.1038/nature21409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clocchiatti A. Di Giorgio E. Viviani G. Streuli C. Sgorbissa A. Picco R. Cutano V. Brancolini C. J. Cell Sci. 2015;128:3961–3976. doi: 10.1242/jcs.170357. [DOI] [PubMed] [Google Scholar]
- Eckschlager T. Plch J. Stiborova M. Hrabeta J. Int. J. Mol. Sci. 2017;18:1414. doi: 10.3390/ijms18071414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian S. Bates S. E. Wright J. J. Espinoza-Delgado I. Piekarz R. L. Pharmaceuticals. 2010;3:2751–2767. doi: 10.3390/ph3092751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goracci L. Deschamps N. Randazzo G. M. Petit C. Dos Santos Passos C. Carrupt P.-A. Simões-Pires C. Nurisso A. Sci. Rep. 2016;6:29086. doi: 10.1038/srep29086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S. Kozikowski A. P. ChemMedChem. 2016;11:15–21. doi: 10.1002/cmdc.201500486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T. Miyata N. Curr. Med. Chem. 2005;12:2867–2880. doi: 10.2174/092986705774454706. [DOI] [PubMed] [Google Scholar]
- Kerr J. S. Galloway S. Lagrutta A. Armstrong M. Miller T. Richon V. M. Andrews P. A. Int. J. Toxicol. 2010;29:3–19. doi: 10.1177/1091581809352111. [DOI] [PubMed] [Google Scholar]
- Deng B. Luo Q. Halim A. Liu Q. Zhang B. Song G. DNA Cell Biol. 2020;39:167–176. doi: 10.1089/dna.2019.4877. [DOI] [PubMed] [Google Scholar]
- Choudhry H. Harris A. L. McIntyre A. Mol. Aspects Med. 2016;47–48:35–53. doi: 10.1016/j.mam.2016.01.003. [DOI] [PubMed] [Google Scholar]
- Cook K. M. Figg W. D. Ca-Cancer J. Clin. 2010;60:222–243. doi: 10.3322/caac.20075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich M. Gerbeth L. Gerling M. Rosenthal R. Steiger K. Weidinger C. Keye J. Wu H. Schmidt F. Weichert W. Siegmund B. Glauben R. Mucosal Immunol. 2019;12:656–667. doi: 10.1038/s41385-019-0135-7. [DOI] [PubMed] [Google Scholar]
- Wanjare M. Kusuma S. Gerecht S. Biotechnol. J. 2013;8:434–447. doi: 10.1002/biot.201200199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W. Roeder R. G. Cell. 1997;90:595–606. doi: 10.1016/S0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- Kim M. S. Kwon H. J. Lee Y. M. Baek J. H. Jang J.-E. Lee S.-W. Moon E.-J. Kim H.-S. Lee S.-K. Chung H. Y. Nat. Med. 2001;7:437. doi: 10.1038/86507. [DOI] [PubMed] [Google Scholar]
- Chen L. Li J. Wang F. Dai C. Wu F. Liu X. Li T. Glauben R. Zhang Y. Nie G. He Y. Qin Z. Cancer Res. 2016;76:6828–6838. doi: 10.1158/0008-5472.CAN-16-1114. [DOI] [PubMed] [Google Scholar]
- Pucci F. Venneri M. A. Biziato D. Nonis A. Moi D. Sica A. Di Serio C. Naldini L. De Palma M. Blood. 2009;114:901–914. doi: 10.1182/blood-2009-01-200931. [DOI] [PubMed] [Google Scholar]
- Bassett S. A. Barnett M. P. Nutrients. 2014;6:4273–4301. doi: 10.3390/nu6104273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrgovic I. Doll M. Pinter A. Kaufmann R. Kippenberger S. Meissner M. Exp. Dermatol. 2017;26:194–201. doi: 10.1111/exd.13159. [DOI] [PubMed] [Google Scholar]
- Kwon H. J. Kim M. S. Kim M. J. Nakajima H. Kim K.-W. Int. J. Cancer. 2002;97:290–296. doi: 10.1002/ijc.1602. [DOI] [PubMed] [Google Scholar]
- Michaelis M. Michaelis U. R. Fleming I. Suhan T. Cinatl J. Blaheta R. A. Hoffmann K. Kotchetkov R. Busse R. Nau H. Mol. Pharmacol. 2004;65:520–527. doi: 10.1124/mol.65.3.520. [DOI] [PubMed] [Google Scholar]
- Qian D. Z. Wang X. Kachhap S. K. Kato Y. Wei Y. Zhang L. Atadja P. Pili R. Cancer Res. 2004;64:6626–6634. doi: 10.1158/0008-5472.CAN-04-0540. [DOI] [PubMed] [Google Scholar]
- Mohan R. Sharma A. K. Gupta S. Ramaa C. S. Med. Chem. Res. 2012;21:1156–1165. doi: 10.1007/s00044-011-9623-3. [DOI] [Google Scholar]
- Bhanushali U. Rajendran S. Sarma K. Kulkarni P. Chatti K. Chatterjee S. Ramaa C. S. Bioorg. Chem. 2016;67:139–147. doi: 10.1016/j.bioorg.2016.06.006. [DOI] [PubMed] [Google Scholar]
- McTigue M. Murray B. W. Chen J. H. Deng Y.-L. Solowiej J. Kania R. S. Proc. Natl. Acad. Sci. U. S. A. 2012;109:18281–18289. doi: 10.1073/pnas.1207759109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhanushali U. Rajendran S. Sarma K. Kulkarni P. Chatti K. Chatterjee S. Ramaa C. S. Bioorg. Chem. 2016;67:139–147. doi: 10.1016/j.bioorg.2016.06.006. [DOI] [PubMed] [Google Scholar]
- Vijayan R. S. K. He P. Modi V. Duong-Ly K. C. Ma H. Peterson J. R. Dunbrack Jr R. L. Levy R. M. J. Med. Chem. 2014;58:466–479. doi: 10.1021/jm501603h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasiotis K. M. Tzanetou E. N. Haroutounian S. A. Front. Chem. 2014;2(78) doi: 10.3389/fchem.2014.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto N. Sakai N. Hirayama T. Miwa K. Oguro Y. Oki H. Okada K. Takagi T. Iwata H. Awazu Y. Bioorg. Med. Chem. 2013;21:2333–2345. doi: 10.1016/j.bmc.2013.01.074. [DOI] [PubMed] [Google Scholar]
- Wang M. Xu S. Lei H. Wang C. Xiao Z. Jia S. Zhi J. Zheng P. Zhu W. Bioorg. Med. Chem. 2017;25:5754–5763. doi: 10.1016/j.bmc.2017.09.003. [DOI] [PubMed] [Google Scholar]
- Mainolfi N. Karki R. Liu F. Anderson K. ACS Med. Chem. Lett. 2016;7:363–367. doi: 10.1021/acsmedchemlett.5b00486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L.-L. Li G.-B. Ma S. Zou C. Zhou S. Sun Q.-Z. Cheng C. Chen X. Wang L.-J. Feng S. Li L.-L. Yang S.-Y. J. Med. Chem. 2013;56:1641–1655. doi: 10.1021/jm301537p. [DOI] [PubMed] [Google Scholar]
- Sharma A. Pathan T. Mohan R. Ramaa C. S. Lett. Drug Des. Discovery. 2011;8:843–849. doi: 10.2174/157018011797200812. [DOI] [Google Scholar]
- Havrylyuk D. Zimenkovsky B. Vasylenko O. Gzella A. Lesyk R. J. Med. Chem. 2012;55:8630–8641. doi: 10.1021/jm300789g. [DOI] [PubMed] [Google Scholar]
- Fahmy H. Khalifa N. Ismail M. El-Sahrawy H. Nossier E. Molecules. 2016;21:271. doi: 10.3390/molecules21030271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitulescu G. Draghici C. Olaru O. Int. J. Mol. Sci. 2013;14:21805–21818. doi: 10.3390/ijms141121805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelarapu R. Holzle D. L. Velaparthi S. Bai H. Brunsteiner M. Blond S. Y. Petukhov P. A. J. Med. Chem. 2011;54:4350–4364. doi: 10.1021/jm2001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J. Niu Q. Liu J. Bao Y. Yang J. Luan S. Fan Y. Liu D. Zhao L. Bioorg. Med. Chem. Lett. 2016;26:375–379. doi: 10.1016/j.bmcl.2015.12.007. [DOI] [PubMed] [Google Scholar]
- Yang J. Cheng G. Xu Q. Luan S. Wang S. Liu D. Zhao L. Bioorg. Med. Chem. 2018;26:1418–1425. doi: 10.1016/j.bmc.2017.08.029. [DOI] [PubMed] [Google Scholar]
- Zagni C. Citarella A. Oussama M. Rescifina A. Maugeri A. Navarra M. Scala A. Piperno A. Micale N. Int. J. Mol. Sci. 2019;20:945. doi: 10.3390/ijms20040945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hieu D. T. Anh D. T. Tuan N. M. Hai P.-T. Huong L.-T.-T. Kim J. Kang J. S. Vu T. K. Dung P. T. P. Han S.-B. Nam N.-H. Hoa N.-D. Bioorg. Chem. 2018;76:258–267. doi: 10.1016/j.bioorg.2017.12.007. [DOI] [PubMed] [Google Scholar]
- Minucci S. Pelicci P. G. Nat. Rev. Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- Tilekar K. Upadhyay N. Jänsch N. Schweipert M. Mrowka P. Meyer-Almes F. J. Ramaa C. S. Bioorg. Chem. 2020;95:103522. doi: 10.1016/j.bioorg.2019.103522. [DOI] [PubMed] [Google Scholar]
- Upadhyay N. Tilekar K. Jänsch N. Schweipert M. Hess J. D. Henze Macias L. Mrowka P. Aguilera R. J. Choe J. Meyer-Almes F.-J. Rama C. S. Bioorg. Chem. 2020:103934. doi: 10.1016/j.bioorg.2020.103934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilekar K. Hess J. D. Upadhyay N. Bianco A. L. Schweipert M. Laghezza A. Loiodice F. Meyer-Almes F.-J. Aguilera R. J. Lavecchia A. C S R. J. Med. Chem. 2021;64:6949–6971. doi: 10.1021/acs.jmedchem.1c00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilekar K. Upadhyay N. Hess J. D. Macias L. H. Mrowka P. Aguilera R. J. Meyer-Almes F.-J. Iancu C. V. Choe J.-Y. Ramaa C. S. Eur. J. Med. Chem. 2020;202:112603. doi: 10.1016/j.ejmech.2020.112603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier P. Smil D. V. Wahhab A. Leit S. Rahil J. Li Z. Déziel R. Besterman J. M. Bioorg. Med. Chem. Lett. 2009;19:5684–5688. doi: 10.1016/j.bmcl.2009.08.010. [DOI] [PubMed] [Google Scholar]
- Bürli R. W. Luckhurst C. A. Aziz O. Matthews K. L. Yates D. Lyons Kathy. A. Beconi M. McAllister G. Breccia P. Stott A. J. Penrose S. D. Wall M. Lamers M. Leonard P. Müller I. Richardson C. M. Jarvis R. Stones L. Hughes S. Wishart G. Haughan A. F. O'Connell C. Mead T. McNeil H. Vann J. Mangette J. Maillard M. Beaumont V. Munoz-Sanjuan I. Dominguez C. J. Med. Chem. 2013;56:9934–9954. doi: 10.1021/jm4011884. [DOI] [PubMed] [Google Scholar]
- Luckhurst C. A. Aziz O. Beaumont V. Bürli R. W. Breccia P. Maillard M. C. Haughan A. F. Lamers M. Leonard P. Matthews K. L. Raphy G. Stott A. J. Munoz-Sanjuan I. Thomas B. Wall M. Wishart G. Yates D. Dominguez C. Bioorg. Med. Chem. Lett. 2019;29:83–88. doi: 10.1016/j.bmcl.2018.11.009. [DOI] [PubMed] [Google Scholar]
- Bürli R. W. Luckhurst C. A. Aziz O. Matthews K. L. Yates D. Lyons Kathy. A. Beconi M. McAllister G. Breccia P. Stott A. J. Penrose S. D. Wall M. Lamers M. Leonard P. Müller I. Richardson C. M. Jarvis R. Stones L. Hughes S. Wishart G. Haughan A. F. O'Connell C. Mead T. McNeil H. Vann J. Mangette J. Maillard M. Beaumont V. Munoz-Sanjuan I. Dominguez C. J. Med. Chem. 2013;56:9934–9954. doi: 10.1021/jm4011884. [DOI] [PubMed] [Google Scholar]
- Adel M. Serya R. A. T. Lasheen D. S. Abouzid K. A. M. Bioorg. Chem. 2018;81:612–629. doi: 10.1016/j.bioorg.2018.09.001. [DOI] [PubMed] [Google Scholar]
- Park H.-J. Zhang Y. Georgescu S. P. Johnson K. L. Kong D. Galper J. B. Stem Cell Rev. 2006;2:93–101. doi: 10.1007/s12015-006-0015-x. [DOI] [PubMed] [Google Scholar]
- Goodwin A. M. Microvasc. Res. 2007;74:172–183. doi: 10.1016/j.mvr.2007.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staton C. A. Reed M. W. R. Brown N. J. Int. J. Exp. Pathol. 2009;90:195–221. doi: 10.1111/j.1365-2613.2008.00633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. Y. Park J. A. Kim Y. Noh M. Park S. Lie E. Kim E. Kim Y. Kwon Y. FASEB J. 2019;33:9842–9857. doi: 10.1096/fj.201802516RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q. Lu S. Li T. Yu L. Zhang Y. Zeng H. Qian X. Bi J. Lin Y. J. Exp. Clin. Cancer Res. 2019;38:173. doi: 10.1186/s13046-019-1156-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. Sun X. Wang Z. Chen L. Li D. Zhou J. Liu M. PLoS One. 2012;7:e36389. doi: 10.1371/journal.pone.0036389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z. Zhu C. An B. Chen Y. He X. Qian L. Lan L. Li S. OncoTargets Ther. 2018;11:2937–2944. doi: 10.2147/OTT.S157949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilekar K. Upadhyay N. Meyer-Almes F. Loiodice F. Anisimova N. Y. Spirina T. S. Sokolova D. V. Smirnova G. B. Choe J. Pokrovsky V. S. Lavecchia A. Ramaa C. S. ChemMedChem. 2020;15:1813–1825. doi: 10.1002/cmdc.202000458. [DOI] [PubMed] [Google Scholar]
- Upadhyay N. Tilekar K. Loiodice F. Anisimova N. Yu. Spirina T. S. Sokolova D. V. Smirnova G. B. Choe J. Meyer-Almes F.-J. Pokrovsky V. S. Lavecchia A. Ramaa C. S. Bioorg. Chem. 2020:104527. doi: 10.1016/j.bioorg.2020.104527. [DOI] [PubMed] [Google Scholar]
- Bhanushali U. Rajendran S. Sarma K. Kulkarni P. Chatti K. Chatterjee S. Ramaa C. S. Bioorg. Chem. 2016;67:139–147. doi: 10.1016/j.bioorg.2016.06.006. [DOI] [PubMed] [Google Scholar]
- Patil V. Tilekar K. Mehendale-Munj S. Mohan R. Ramaa C. S. Eur. J. Med. Chem. 2010;45:4539–4544. doi: 10.1016/j.ejmech.2010.07.014. [DOI] [PubMed] [Google Scholar]
- Jänsch N. Meyners C. Muth M. Kopranovic A. Witt O. Oehme I. Meyer-Almes F.-J. Redox Biol. 2019;20:60–67. doi: 10.1016/j.redox.2018.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff B. Jänsch N. Sugiarto W. O. Frühschulz S. Lang M. Altintas R. Oehme I. Meyer-Almes F.-J. Eur. J. Med. Chem. 2019;184:111756. doi: 10.1016/j.ejmech.2019.111756. [DOI] [PubMed] [Google Scholar]
- Volund A. Biometrics. 1978;34:357. doi: 10.2307/2530598. [DOI] [PubMed] [Google Scholar]
- Peram M. R. Jalalpure S. Kumbar V. Patil S. Joshi S. Bhat K. Diwan P. J. Liposome Res. 2019:1–21. doi: 10.1080/08982104.2018.1556292. [DOI] [PubMed] [Google Scholar]
- Upadhyay N. Tilekar K. Jänsch N. Schweipert M. Hess J. D. Henze Macias L. Mrowka P. Aguilera R. J. Choe J. Meyer-Almes F.-J. Rama C. S. Bioorg. Chem. 2020:103934. doi: 10.1016/j.bioorg.2020.103934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilekar K. Upadhyay N. Schweipert M. Hess J. D. Macias L. H. Mrowka P. Meyer-Almes F.-J. Aguilera R. J. Iancu C. V. Choe J.-Y. Ramaa C. S. Eur. J. Pharm. Sci. 2020;154:105512. doi: 10.1016/j.ejps.2020.105512. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.