

Abstract
Duchenne muscular dystrophy (DMD) is a progressive and lethal disease, caused by X‐linked mutations of the dystrophin encoding gene. The lack of dystrophin leads to muscle weakness, degeneration, fibrosis, and progressive loss of skeletal, cardiac, and respiratory muscle function resulting in premature death due to the cardiac and respiratory failure. There is no cure for DMD and current therapies neither cure nor arrest disease progression. Thus, there is an urgent need to develop new approaches and safer therapies for DMD patients. We have previously reported functional improvements which correlated with increased dystrophin expression following transplantation of dystrophin expressing chimeric (DEC) cells of myoblast origin to the mdx mouse models of DMD. In this study, we demonstrated that systemic‐intraosseous transplantation of DEC human cells derived from myoblasts of normal and DMD‐affected donors, increased dystrophin expression in cardiac, respiratory, and skeletal muscles of the mdx/scid mouse model of DMD. DEC transplant correlated with preservation of ejection fraction and fractional shortening on echocardiography, improved respiratory function on plethysmography, and improved strength and function of the limb skeletal muscles. Enhanced function was associated with improved muscle histopathology, revealing reduced mdx pathology, fibrosis, decreased inflammation, and preserved muscle morphology and architecture. Our findings confirm that DECs generate a systemic protective effect in DMD‐affected target organs. Therefore, DECs represents a novel therapeutic approach with the potential to preserve or enhance multiorgan function of the skeletal, cardiac, and respiratory muscles critical for the well‐being of DMD patients.
Keywords: cardiac protection/function, cellular therapy, DEC therapy, Duchenne muscular dystrophy, dystrophin expressing chimeric cells, muscle regeneration, myoblasts, pulmonary protection/function
Systemic‐intraosseous transplantation of human dystrophin expressing chimeric (DEC) cells (0.5 × 106 and 1 × 106) results in significant increase in dystrophin expression in heart, diaphragm, and gastrocnemius muscle (GM) which correlates with the reduction of centrally nucleated fibers in diaphragm and GM indicating reduced muscle pathology and further confirming beneficial effect of DEC therapy at 90 days after systemic transplant to mdx/scid mouse model of DMD.

Significance statement.
Duchenne muscular dystrophy (DMD) is a lethal disease, caused by X‐linked mutations of dystrophin encoding gene. Currently there is no cure for DMD. A novel approach was developed and dystrophin expressing chimeric (DEC) human cells derived from myoblasts of normal and DMD‐affected donors for treatment of DMD was created. The efficacy of DEC therapy after systemic‐intraosseous transplant to mdx mice model of DMD was tested. This study confirmed that DECs improve the function and reduce the pathology in DMD‐affected target organs. Therefore, DECs represent a novel therapeutic approach with the potential to ameliorate the function of cardiac, respiratory, and skeletal muscles critical for well‐being of DMD patients.
1. INTRODUCTION
Duchenne muscular dystrophy (DMD) affects about 1 in 3500 to 5000 live male births and represents the most severe congenital form of muscular dystrophy caused by mutations in the dystrophin encoding gene. Dystrophin is a vital structural link between the extracellular matrix and the cytoskeletal proteins and plays an essential role in several important biochemical extracellular signaling pathways. Dystrophin deficiency clinically manifests as skeletal, cardiac, and respiratory muscle weakness as myofibrils undergo damage, inflammation, and fibrosis. This results in the devastating multiorgan failure, first manifested by the gradual loss of skeletal muscle strength and motor function affecting the ability to walk, followed by the development of cardiomyopathy, deterioration of cardiac function corresponding with progressive respiratory failure, all leading to the premature death. Currently, there is no cure for DMD patients.1
Since DMD is a chronic, progressive disease and the age of onset of clinical manifestations and the subsequent organ failure varies between DMD patients depending on gene mutation, this represents a significant challenge for the currently tested DMD therapies.
Several potential gene therapies aiming at dystrophin restoration, such as exon skipping, microdystrophin gene delivery via adeno‐associated viruses (AAV),2 and gene editing therapies using clustered regularly interspaced short palindromic repeats (CRISPR) system,3, 4, 5 have their limitations and safety concerns. The CRISPR therapies are targeting only very specific and limited populations of DMD patients. The microdystrophin AAV delivery therapies show promising results2; however, the AAV9 approach still poses a risk of an adverse immune response, which is often dose‐related.6 Moreover, the efficacy of concomitant dystrophin restoration in all affected muscles including limb skeletal muscles, diaphragm as well as cardiac muscle to the clinically relevant levels has been limited using these gene therapy approaches.3, 4, 5, 7, 8, 9, 10, 11, 12
Additionally, the past clinical trials testing gene‐based therapies reported improvements in functional outcomes measured by standard motor tests, with relatively limited evidence of concomitant improvement of cardiac or pulmonary function during the course of therapy.11, 12, 13 In contrast, current clinical trials using systemic delivery of microdystrophin are reporting promising results and improvements in the skeletal muscles as well as cardiac function.2, 14, 15, 16
Relatively low levels of dystrophin expression are reported in some of the clinical trials.17 In contrast, cell‐based therapies are organ‐specific and are either targeting skeletal muscles by local intramuscular injections of the allogenic myoblasts (MBs) to the selected skeletal muscle groups18, 19 or are organ‐specific and target cardiac muscle via intracoronary infusion of cardiosphere‐derived cells to prevent the development of cardiomyopathy and progression to cardiac failure.20 Pulmonary function in DMD patients is maintained by standard supportive measures including administration of prednisone, glucocorticosteroids, idebenone, or deflazacort.21, 22
There are limited reports providing evidence that currently tested gene therapies are leading to functional improvements in all major target organs responsible for the progressive decline of the DMD patients.20, 23
Cell‐based therapies of either autologous or allogenic stem cell origin have shown promising results as an alternative method for DMD treatment, however limited or short‐term cell engraftment, allogenic immune response, and side effects of supportive immunosuppressive therapy challenged routine clinical application of stem cells for the treatment of DMD patients.18, 19, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34 Thus, there is an urgent need for the development of novel therapies and approaches which will elicit systemic effects irrespective of gene mutation, the stage of the disease or the patient's age.
Based on our 20 years of experience with the application of chimerism‐based strategies for tolerance induction in bone marrow and vascularized composite allograft transplantation,35, 36, 37, 38, 39, 40 we have addressed the limitations of current stem cell therapies, by introducing chimeric cells as a novel therapeutic approach for DMD.
Hence, we applied the concept of cell fusion technology to create human chimeric cell lines of MB origin as a novel, dystrophin delivery platform, presenting the allogeneic donor in the context of “self” in order to minimize the immune response and eliminate the need for immunosuppression.41, 42, 43
We have previously reported in both the mdx and mdx/scid mouse models of DMD that local transplantation of DEC cells significantly improved dystrophin expression which correlated with significant improvement of muscle strength and function.41, 42 Moreover, systemic intraosseous DEC transplantation confirmed protective effect on cardiac function by the rebound effect on the ejection fraction (EF) and fractional shortening (FS).43
In the current study, we aimed to demonstrate that systemic‐intraosseous transplantation of human DEC cells will elicit target organ‐oriented systemic effect and will protect the DMD‐affected cardiac, respiratory, and skeletal muscles from progressive functional decline.
First, we have created human DEC cell lines via PEG‐mediated fusion of MBs from normal and DMD‐affected donors. Next, the long‐term efficacy of engraftment and systemic effect of human DEC cells was assessed at 90 days after intraosseous transplant to the mdx/scid mouse model of DMD. After DEC transplant, we have addressed the problem of multiorgan failure observed in DMD patients1, 44, 45 by assessment of functional outcomes, histology, and mdx muscle pathology in the most affected by DMD progression organs, including cardiac, respiratory, and skeletal muscles.
We have also posed the question, if systemic DEC transplant will delay or slow progression of cardiac and respiratory failure via synchronized stabilization of skeletal muscle's function, leading to the maintenance of cardiac and respiratory function at the baseline levels, thus changing the dynamics of DMD progression.
In this study, we have confirmed that systemic intraosseous transplant of human DEC cells restored dystrophin expression and improved mdx pathology which correlated with the amelioration of cardiac, respiratory, and skeletal muscle's function. This study introduces DEC as a potential drug candidate for the treatment of DMD with protective systemic effect in the DMD‐affected organs.
2. MATERIALS AND METHODS
2.1. Mice and animal care
This study was approved by the Institutional Animal Care and Use Committee of University of Illinois at Chicago, which is accredited by the American Association for the Accreditation of Laboratory Animal Care. All animals received humane care in compliance with the “Principles of Laboratory Animal Care” formulated by the National Society for Medical Research and the Guide for the Care and Use of Laboratory Animal Resources. Six‐ to 8‐week‐old male mdx/scid mice—animal model for DMD (B10ScSn.Cg‐Prkdcscid Dmdmdx/J, stock number 018018) with respective background wild‐type (WT) mice (C57BL/10ScSnJ, stock number 000476)—were purchased from Jackson Laboratories (Mount Desert Island, Maine). Animals were kept in pathogen‐free environment on light/dark cycle. Prior to study initiation, aged matched male mdx/scid mice were ear‐tagged and randomized into experimental groups: vehicle (n = 5, 60 μL phosphate‐buffered saline [PBS]), fused MBN/MBDMD DEC (n = 13) 0.5 × 106 in 60 μL PBS, and fused MBN/MBDMD DEC n = 5, 1 × 106 in 60 μL PBS).
2.2. Creation of human DEC cells
2.2.1. Cell culture
Normal human MBs were purchased from Lonza Bioscience (Mapleton, Illinois), and DMD‐affected MBs were purchased from Axol Bioscience Ltd. (Little Chesterford, UK). MBs were cultured in Skeletal Muscle Cell Growth Medium‐2 (Lonza Clonetics, Mapleton, Illinois) supplemented with (Lonza Clonetics): human epidermal growth factor, fetal bovine serum (FBS), dexamethasone, gentamicin/amphotericin B. Upon reaching 60% to 70% confluence, MBs were harvested using 0.25% trypsin/EDTA (Sigma‐Aldrich, St. Louis, Missouri). Enzymatic activity was inhibited with serum‐supplemented culture media. Human MB were harvested between passages 3 and 7, which are optimal for ex vivo cell fusion procedure.
2.2.2. Cell fusion
After harvesting, staining, and viability assessment with 0.4% Trypan Blue (Gibco‐Thermo Fisher Scientific, Waltham, Massachusetts), parent MBs (MBN and MBDMD) were washed in serum‐free media supplemented with antibiotics (1% Antibiotic‐Antimycotic solution, Gibco‐Thermo Fisher Scientific). Then, cell fusion was performed. As previously described.36, 37, 38 parent MBs (MBN and MBDMD) were fluorescently labeled using PKH26 or PKH67 (Sigma‐Aldrich) membrane dyes according to manufacturer's instructions. Prior to fusion, parent cells were mixed, washed, and fusion was performed using 1.46 g/mL PEG solution (PEG 4000, EMD) containing 16% DMSO (Sigma).36, 37, 38 Fused cells were transferred to fluorescently activated cells sorting (FACS) buffer containing 5% HEPES, 1% EDTA, and 5% FBS. Finally, cells presenting double (PKH26/PKH67) staining were selected via FACS (MoFlow Astrios, Beckman Coulter, San Jose, California) and used for in vitro analysis or transplanted to mdx/scid mice.
2.3. Systemic‐intraosseous DEC transplantation
Mice were anesthetized with 2% isoflurane inhalation along with 1 mL/kg buprenorphine subcutaneous injection. DEC intraosseous transplant was performed as previously reported.35, 42, 46, 47 Briefly, 5 mm incision was made in lateral‐mid thigh and muscles were separated to expose femur. DECs were transferred in 60 μL volume of sterile PBS to tuberculin syringe (BD). A 25G needle was used to aspirate 60 μL of bone marrow followed by DEC cells injection directly into the femur. Bone wax was applied to injection site, next muscles were reapproximated and wound was closed with 5‐0 nylon sutures. Animals recovered in a heated environment with postoperative monitoring and returned to the colony.
2.4. Histological and immunofluorescence (IF) analysis
For histological analysis of heart, diaphragm and gastrocnemius muscle (GM) paraffin blocks were cut at 5 μm nonconsecutive transverse cross‐sections. Samples were deparaffinized and subsequently stained with H&E and mounted (Poly‐Mount, PolySciences Inc, Warrington, Pennsylvania) to analyze muscle structure and to quantify centrally nucleated fibers (CNFs) which were normalized to the total number of nuclei in the region of interest. Fiber diameters were assessed by minimal Feret's diameter using ImageJ measurement plug‐ins. Average number of fibers falling into 5 μm increments was normalized to the total number of fibers and expressed as a percentage. Inflammatory foci were analyzed on H&E sections by counting total number of small (5‐9 inflammatory cells) and large (group of 10 or more inflammatory cells) within representative images. The number of foci counts were normalized to area unit (number of foci/mm2). To analyze fibrosis, gastrocnemius and diaphragm sections were stained with trichrome stain kit for visualization of collagenous fibrotic tissue. For cardiac muscle fibrosis, cross‐sections of heart samples were stained with Picro‐Sirius red kit specific for cardiac muscle (Abcam, cat. 245887, Cambridge, Massachusetts). The digital images were acquired (BX51/IX70 Olympus Japan) and processed using ImageJ (NIH). Pixels corresponding to the area stained in blue (for trichrome) or red (for Picro‐Sirius) indicating collagenous areas for fibrosis assessment were normalized to the total pixel area of the tissue in image, and results were expressed as a percentage of fibrotic area vs total tissue area.
For IF, OCT frozen sections of heart, diaphragm, and gastrocnemius were fixed with ice‐cold acetone for 10 minutes and blocked for the unspecific binding with 10% normal goat serum for 30 minutes in 4°C. Next, specimens were incubated with mouse monoclonal anti‐human dystrophin primary antibody (abcam15277, 1:50, Abcam), followed by incubation with a goat anti‐mouse secondary conjugate AlexaFluor‐647 antibody (1:400, Thermo Fisher Scientific). Nuclear counter‐staining was performed using 4′,6‐diamidino‐2‐phenylindole (DAPI; Abcam, cat. 104139). A Zeiss Meta confocal microscope with ZEN software (Carl Zeiss, Oberkochen, Germany) and Leica DM4000B microscope were used for fluorescence signal detection and analysis.
2.5. Assessment of cardiac muscle function
2.5.1. Echocardiography
Echocardiography was performed as previously described.43 Mice were anesthetized with isoflurane and placed on ultrasound stage. Paws were taped to the electrocardiograph electrodes to monitor heart rate (maintained at 350‐450 bmp). Assessments were made at day 0, 30, and 90 after intraosseous DEC transplant. M‐mode echocardiographic images were obtained from the parasternal long axis view through the center of left ventricle.48, 49, 50
2.6. Assessment of respiratory muscle function
2.6.1. Plethysmography
Whole body plethysmography was applied for the assessment of respiratory function as described elsewhere.51 Small Animal Plethysmography setup (Buxco/DSI, St. Paul MN) using FinePointe (Buxco/DSI) was applied. The following parameters were calculated: enhanced pause (PENH), expiratory time (Te), tidal volume (Tv), and breathing frequency (F).
2.7. Assessment of skeletal muscle's function
2.7.1. Grip strength test
Mice motor function was monitored weekly up to 90‐day endpoint with a grip meter (Digital Force Gauge, HL‐50) as described previously.41, 42 This test allows for forelimb force measurements, providing information on muscle strength of DEC‐injected vs control mice.
2.7.2. Ex vivo muscle force test
After animal euthanasia, the contractile and passive properties of the GM were measured ex vivo using Aurora Scientific test system as described previously.42 After whole GM dissection, the Achilles tendon and proximal pole of muscle were attached to the force transducer. Muscle force was measured after establishing optimal length through a standardized stimuli pattern until reaching maximal wave and maximal strain. Results are reported as force/muscle weight (g/g).
2.8. Statistical analysis
Data are expressed as mean ± SEM (standard error of the mean). GraphPrism software was used to perform statistical analysis. Two‐tailed Student t tests or one‐way analysis of variance with Tukey post hoc test for group comparisons were used to define statistical significance. Results were considered statistically significant for P < .05.
3. RESULTS
3.1. Intraosseous DEC transplantation results in dystrophin expression and reduced pathology of cardiac, respiratory, and skeletal muscles
We have previously reported that systemic‐intraosseous transplant of murine DEC cells resulted in the restoration of dystrophin expression which correlated with the protection of cardiac function in the mdx mice.43 In the current study, to test the therapeutic clinical potential of DEC, we created human DEC cell line and tested functional efficacy of engraftment after systemic‐intraosseous transplant of two doses of human DEC cells (0.5 × 106 or 1 × 106) to the mdx/scid mice (Figure 1A). IF staining confirmed long‐term engraftment and dystrophin expression in the selected target organs of heart, diaphragm, and GM of mdx/scid‐injected mice at 90 days after systemic transplant of human DEC (Figure 1B; Figure S1).78 Restoration of dystrophin was dose‐dependent and reveled significantly increased expression in heart (16.44% ± 0.94%; Figure 1C), diaphragm (17.83% ± 1.11%; Figure 1D), and GM (13.00% ± 0.66%; Figure 1E) of DEC‐injected compared to the vehicle‐injected mdx/scid controls.
FIGURE 1.
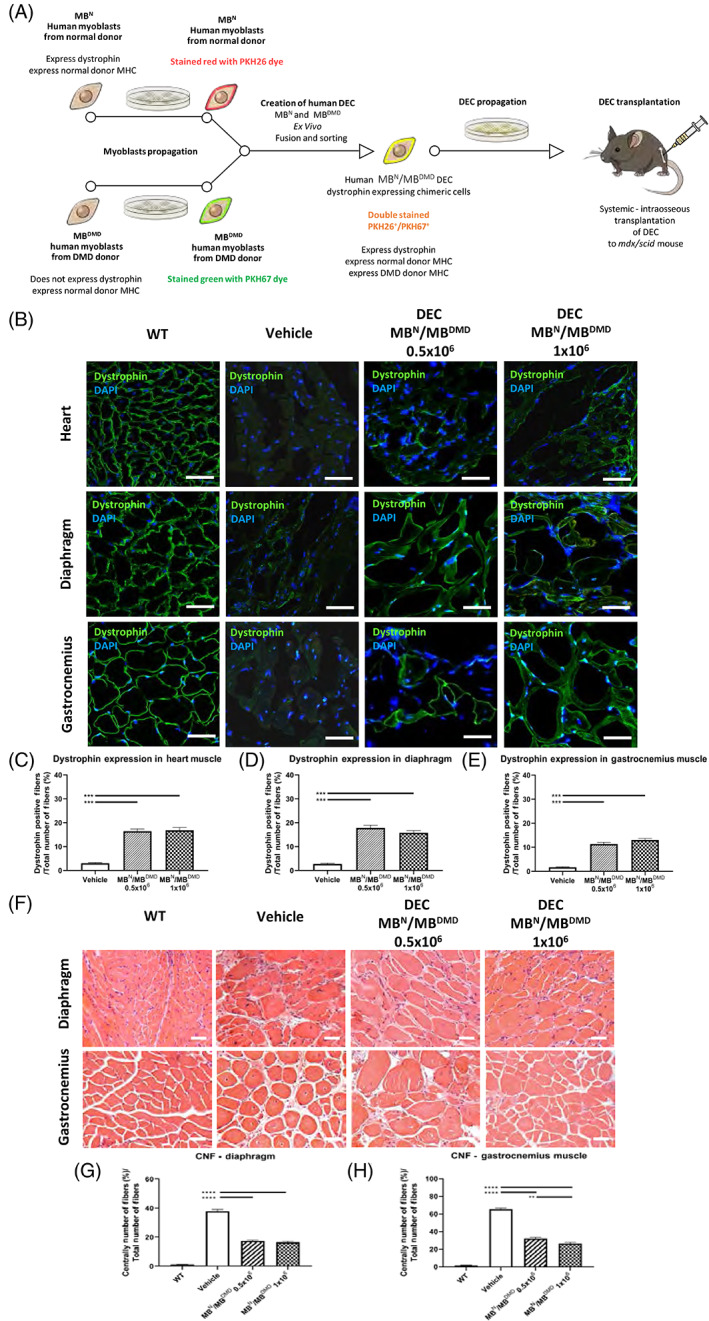
Systemic‐intraosseous transplantation of human DEC (0.5 × 106 and 1 × 106) increases dystrophin expression and improves morphology of cardiac, respiratory, and skeletal muscles at 90 days post‐transplant to mdx/scid mouse. A, Diagram of ex vivo creation and systemic intraosseous transplantation of human DEC cell line to mdx/scid mouse. B, Immunofluorescence images of dystrophin expression in the target organs of heart, diaphragm, and gastrocnemius muscle (GM) after systemic DEC transplant compared to vehicle and WT, magnification 58×; scale bar = 50 μm; for merge: dystrophin (green), DAPI nuclei counterstaining (blue). C‐E, Significant increase of dystrophin expression in the selected target organs: (C) heart, (D) diaphragm, and (E) gastrocnemius muscle of DEC‐injected compared to vehicle‐injected mdx/scid mice n = 3/group, 10 ROI/organ/mouse. Dystrophin‐positive fibers were counted and normalized to the total number of fibers; Two tailed Student's t test. F, H&E‐stained transverse sections of diaphragm and GM in DEC transplant groups compared to vehicle and WT, magnification 20×; scale bar = 100 μm. G,H, Significant decrease of CNF in diaphragm and gastrocnemius muscle of DEC‐injected mdx/scid mice, (G) confirming reduced pathology in the diaphragm and (H) significant dose‐dependent CNF reduction in gastrocnemius muscle, n = 3/ group, 12 ROI/organ/mouse. CNF were counted in the diaphragm (G) and GM (H) samples and normalized to the total number of fibers; One‐way analysis of variance with post hoc Tukey's test. All data are presented as mean ± SEM. CNF, centrally nucleated fibers; DAPI, 4′,6‐diamidino‐2‐phenylindole; DEC, dystrophin expressing chimeric; H&E, hematoxylin and eosin; ROI, region of interest; WT, wild type; **P < .01, ***P < .001, ****P < .0001
To further test the potential beneficial effect of DEC therapy on reducing muscle pathology, we assessed the number of CNFs representing the hallmark of mdx pathology on hematoxylin and eosin (H&E) sections of diaphragm (Figure 1F, upper panel) and GM (Figure 1F, lower panel) and confirmed significant reduction of CNF in the diaphragm of DEC‐injected (0.5 × 106, 17.33% ± 0.74% and 1 × 106, 16.36% ± 0.75%) compared to vehicle‐injected mdx/scid controls (37.78% ± 1.22%; Figure 1G).
There was also significant dose‐dependent (32.07% ± 1.37% for 0.5 × 106 vs 26.44% ± 1.24% for 1 × 106 dose) reduction of CNF in GM muscles of DEC‐injected compared to vehicle injected mdx/scid mice (65.45% ± 1.22%) (Figure 1H).
3.2. Systemic DEC transplant protects cardiac function which correlates with reduced fibrosis, inflammation, and amelioration of EF and FS
Since dilated cardiomyopathy‐dependent heart failure is the major cause of death in DMD patients, we have assessed the systemic effect of DEC therapy on cardiac muscle fibrosis and inflammation. More importantly, we tested the correlation between reduced muscle pathology and cardiac function by echocardiography at baseline, 30, and 90 days after DEC transplant (Figure 2).
FIGURE 2.
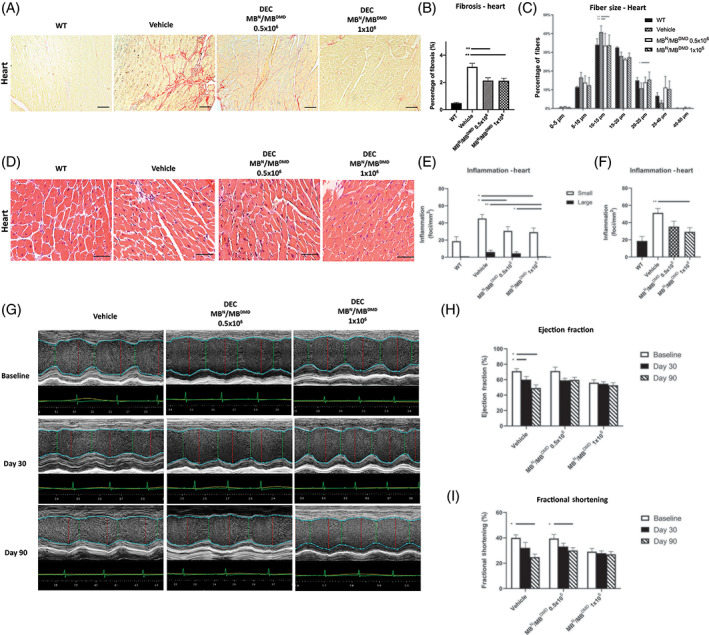
Intraosseous DEC transplant protects cardiac function via reduced fibrosis and inflammation, normalized fiber size, and amelioration of ejection fraction and fractional shortening at 90 days. A, Picro‐Sirius staining of heart cross‐sections assessing fibrosis after DEC therapy compared to vehicle and WT, fibrotic collagenous tissue (red), muscle fibers (yellow), magnification 20×, scale bar = 50 μm, n = 3/group, 12 ROI/organ/mouse. B, Significant decrease of fibrosis after DEC transplant, indicating reduced cardiac pathology. Fibrosis was measured and normalized to total tissue area; two‐tailed Student's t test. C, Feret's diameter measurements reveled rightward shift in fiber size distribution toward WT phenotype, bars represent means ± SEM; unpaired t test. D, H&E heart sections confirmed reduced inflammation in DEC‐injected groups, magnification 40×, scale bars = 100 μm, n = 3/ group, 12 ROI/organ/mouse. E, Number of small foci was reduced in both DEC groups; no large foci were seen in 1 × 106 group. F, Total number of inflammatory foci was reduced in DEC‐injected but not vehicle‐injected mice. G, M‐mode of LV parasternal long axis after DEC transplant. Vehicle‐injected mice demonstrated gross thickening of the posterior LV wall and reduced LV size compared to DEC‐injected mice. H,I, Echocardiography reveled maintenance of EF (H) and FS (I) values over 90 days in both DEC‐injected groups indicating protective DEC effect, while vehicle‐injected mice revealed significant drop of EF and FS, indicating progressive impairment of cardiac function. All data are presented as mean ± SEM. DEC, dystrophin expressing chimeric; EF, ejection fraction; FS, fractional shortening; H&E, hematoxylin and eosin; LV, left ventricle; ROI, region of interest; WT, wild type; *P < .05, **P < .01
Assessment of cardiac fibrosis by Picro‐Sirius staining (Figure 2A) revealed significant reduction in collagenous, fibrotic tissue distribution in DEC‐injected vs vehicle‐injected mdx/scid mice (Figure 2B).
Feret's diameter measurements revealed a rightward shift in fiber size distribution toward WT phenotype in DEC‐injected mice compared to vehicle‐injected controls (Figure 2C).
Inflammatory response assessed on H&E heart sections (Figure 2D) presented significant decreases in small inflammatory foci (30.67 ± 4.94 vs 45.16 ± 4.73 foci/mm2) for both DEC doses (Figure 2E) and significant drop in total number of inflammatory foci in DEC‐injected (29.26 ± 4.83 foci/mm2) compared to vehicle‐injected mice (51.25 ± 5.16 foci/mm2; Figure 2F). Interestingly, there was a lack of large inflammatory infiltrates observed only in DEC‐injected mdx/scid heart sections at both doses (Figure 2E).
M‐mode imaging at 30‐ and 90‐days post‐transplant revealed gross thickening of the posterior left ventricular wall in the control mice and reduced left ventricular chamber size in DEC‐injected mdx/scid mice. The images at 30 and 90 days after intraosseous human DEC transplant confirm a protective effect of DEC therapy on the maintenance of LV function, and morphology in mdx/scid mice injected with two doses of human DEC cells (Figure 2G).
Echocardiography assessments at 90 days after systemic DEC transplant revealed protective effect of DEC therapy on cardiac function, confirmed by maintenance of EF at baseline levels for 1 × 106 DEC dose (baseline −56.11% ± 3.73% vs day 90‐52.74% ± 3.30%; Figure 2H). Similar trend was observed for FS values which were maintained at baseline level (baseline 56.11% ± 3.73% vs day 90‐52.74% ± 3.30%; Figure 2I) over 90 days follow‐up after DEC transplant to mdx/scid mice. In contrast significant drop of EF and FS values was observed in vehicle‐injected mdx/scid controls (Figure 2H,I).
3.3. Intraosseous DEC transplantation results in preservation of respiratory function and amelioration of diaphragm pathology
Since pulmonary fibrosis and respiratory failure is significantly contributing to the premature death of DMD patients, we assessed fibrotic and inflammatory changes in diaphragm following intraosseous DEC transplant (Figure 3A‐D). Furthermore, to evaluate the potential correlation between reduced diaphragm pathology and improved function, we evaluated pulmonary function via plethysmography at the baseline and at 90 days (Figure 3E‐H).
FIGURE 3.
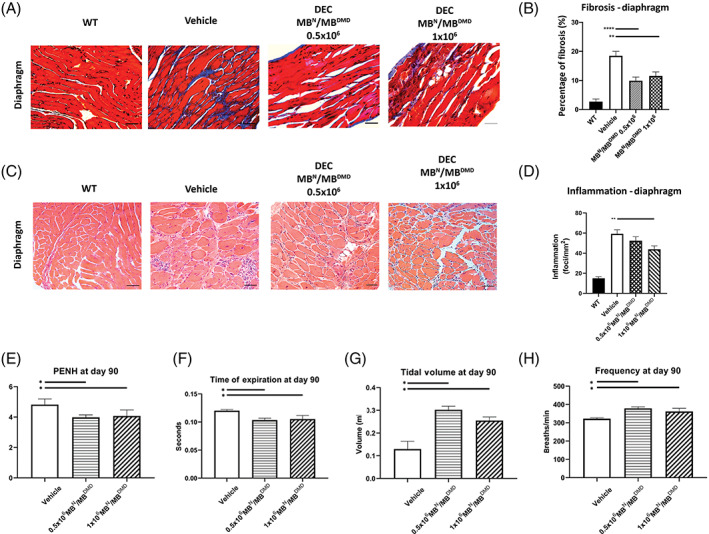
Systemic DEC transplant results in rescue of respiratory function and reduced diaphragm pathology. A, Assessment of fibrosis on Trichrome‐stained transverse sections of diaphragm of mdx/scid mice at 90 day postsystemic intraosseous DEC transplant, compared to vehicle‐injected controls and WT, blue‐stained areas represent collagenous connective tissue (fibrosis) and red‐stained areas represent muscle fibers, magnification 20×, scale bars = 100 μm. B, Significant reduction in percentage of diaphragm fibrosis was observed in DEC‐injected mice compared to vehicle‐injected controls, indicating reduced muscle pathology at 90 days postsystemic DEC transplant. C, H&E‐stained transverse sections of diaphragm confirmed reduced inflammation in diaphragm after administration of 0.5 × 106 and 1 × 106 DEC compared with vehicle controls and WT, magnification 20×, scale bars = 100 μm, n = 3/ group, 12 ROI/organ/mouse. D, The total number of inflammatory foci in diaphragm was significantly reduced in DEC‐injected compared to vehicle‐injected mdx/scid mice. E‐H, Confirmation of reduced mdx‐mediated respiratory disease after DEC transplant assessed by whole body plethysmography: E, significant decrease in PENH (enhanced pause); F, decrease of expiration time along with reduced diaphragm fibrosis indicates amelioration of passive exhalation; G, increase in tidal volume in DEC‐injected mdx/scid mice compared to vehicle‐injected controls indicate more complete contraction of diaphragm and utilization of a larger percentage of lung's capacities; H, significant increase of respiratory frequency was observed in both DEC‐injected groups compared to vehicle‐injected mice. All data are presented as mean ± SEM. DEC, dystrophin expressing chimeric; H&E, hematoxylin and eosin; ROI, region of interest; WT, wild type; *P < .05, **P < .01, ****P < .0001
Assessment of muscle fibrosis by Trichome staining (Figure 3A) revealed significant reduction of interstitial fibrosis observed in the diaphragm of DEC‐injected compared with vehicle‐injected mdx/scid controls: 0.5 × 106 and 1 × 106 doses of DEC (9.90% ± 1.27% and 11.55% ± 0.86%, respectively) when compared to vehicle‐injected controls (18.50% ± 1.53%; Figure 3B).
Reduced fibrosis correlated with reduced inflammation assessed on diaphragm H&E sections (Figure 3C) which revealed significant decrease in total number of inflammatory foci in diaphragm sections of 1 × 106 DEC‐injected (43.87 ± 3.40 foci/mm2) compared to vehicle‐injected (59.25 ± 4.14 foci/mm2) mdx/scid controls assessed at 90 days postsystemic DEC transplant (Figure 3D).
Assessment of pulmonary function via plethysmography confirmed significant reduction in PENH (3.98 ± 0.45; 4.07 ± 0.79) in DEC‐injected mdx/scid mice compared to vehicle‐injected controls (4.82 ± 0.65) at 90 days after intraosseous DEC transplant (Figure 3E). There was a significant decrease of expiration time observed in DEC‐injected mice, likely due to reduced diaphragm fibrosis, indicating amelioration of passive exhalation (Figure 3F). The tidal volume was significantly increased for both, 0.5 × 106 and 1 × 106 DEC doses (0.30 mL ± 0.04 mL and 0.25 ± 0.03 mL, respectively), indicating enhanced contraction of the diaphragm (Figure 3G). Significant increase of respiratory frequency was observed in both DEC‐injected groups (378.95 ± 23.12 breaths/min, and 361.87 ± 34.09 breaths/min for 0.5 × 106 and 1 × 106 doses, respectively) when compared to vehicle‐injected controls (322.49 ± 9.02 breaths/min; Figure 3H). Increased breath frequency in two treatment groups reflects less fibrosis and overall faster inhalation and exhalation rates.
3.4. DEC transplant reduces dystrophic muscle pathology and improves skeletal muscle strength and function
Deterioration of skeletal muscle's function in DMD patients is often the first sign of developing DMD pathology. Thus, we evaluated both the pathological changes in GM muscles as well as the progression of functional deterioration via standard in vivo grip strength and ex vivo Aurora tests (Figure 4).
FIGURE 4.
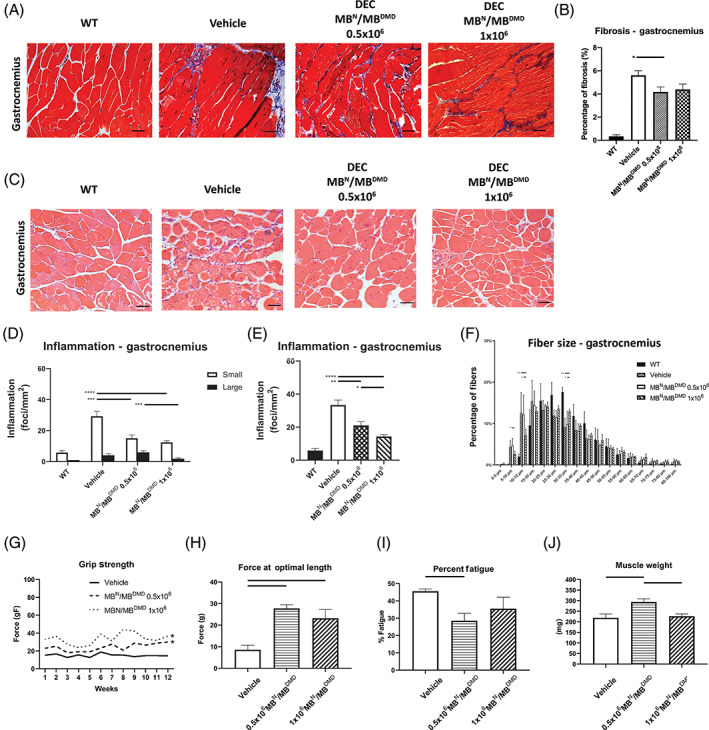
DEC transplant improves skeletal muscle's function via reduced mdx pathology in mdx/scid mice at 90 days. A, Reduced fibrosis in Trichrome‐stained transverse sections of gastrocnemius muscles (GM) after DEC transplant. B, Significant decrease in percentage of GM fibrosis in DEC‐injected mdx/scid mice, indicating reduced muscle pathology, magnification 20×, scale bar = 50 μm, n = 3/group, 12 ROI/organ/mouse. Fibrotic area was measured and normalized to the total tissue area; two‐tailed Student's t test; C, H&E‐stained transverse sections of gastrocnemius confirm reduced inflammation in DEC‐injected mice, magnification 20×, scale bars = 100 μm, n = 3/ group, 12 ROI/organ/mouse. D, The number of small foci decreased significantly in mdx/scid mice injected with either DEC dose; significant dose‐dependent reduction of large inflammatory foci was observed in DEC‐injected mice. E, Significant dose‐dependent decrease in the total number of foci was seen in both DEC groups, n = 3/group, 12 ROI/organ/mouse. F, Feret's diameter measurements reveled rightward shift in fiber size distribution toward larger fibers in both DEC‐injected cohorts with significant difference observed for fiber size increment of 30 to 35 μm. One‐way analysis of variance with post hoc Tukey's test, magnification 20×, scale bar = 50 μm. G, Measurements of grip strength revealed significantly increased muscle force in DEC‐injected mice, two‐tailed Student's t test. H, Improved muscle force confirmed by ex vivo Aurora test in DEC‐injected mice. I, Reduced muscle fatigue was confirmed in 1 × 106 DEC group. J, GM weight increased in DEC‐injected mdx/scid mice (*P < .05). *P < .05, **P < .01, ***P < .001, ****P < .0001. All data are presented as mean ± SEM. DEC, dystrophin expressing chimeric; H&E, hematoxylin and eosin; ROI, region of interest
GM fibrosis assessed by Trichome staining (Figure 4A) revealed reduced fibrosis for both DEC‐injected doses (0.5 × 106 and 1 × 106) compared to vehicle‐injected mdx/scid controls (Figure 4B).
Assessment of small and large inflammatory foci on H&E gastrocnemius specimens (Figure 4C) presented a significant dose‐dependent decrease in the large inflammatory foci seen in DEC‐injected mice (Figure 4D). Moreover, we observed dose‐dependent decrease of total number of inflammatory foci for both doses (0.5 × 106 and 1 × 106) in DEC‐injected (21.08 ± 2.25 foci/mm2, and 14.34 ± 1.22 foci/mm2, respectively) compared to vehicle‐injected mdx/scid controls (33.38 ± 2.99 foci/mm2; Figure 4E).
Feret's diameter measurement in GM revealed benefit of DEC therapy by an increase in diameters and a shift in fiber size distribution toward larger fiber size of 30 to 35 μm in DEC‐injected compared to vehicle‐injected mdx/scid mice, further confirming improved fiber homogeneity and architecture (Figure 4F). Functional testing confirmed improved performance in grip strength, which revealed dose‐dependent 2.53‐fold increase in 1 × 106 DEC‐injected compared to vehicle‐injected mice (Figure 4G). This is correlated with improved muscle strength assessed by ex vivo Aurora test which revealed significantly increased muscle force at optimal length in both 0.5 × 106 and 1 × 106 DEC therapy groups (29.78 ± 1.67 and 23.22 ± 4.04 g, respectively) when compared to vehicle‐injected controls (8.60 ± 2.04 g; Figure 4H). Furthermore, significantly lower fatigue values were seen in DEC‐injected compared to the vehicle‐injected mdx/scid controls (Figure 4I). These data were in line with increased muscle mass in DEC‐injected mdx/scid mice (Figure 4J).
4. DISCUSSION
Despite significant scientific and clinical efforts to introduce new DMD therapies,52, 53, 54, 55 currently there is no cure for DMD. One of the approaches of regenerative medicine is to enable the correction of genetic diseases via cellular replacement therapies.56
The emerging MB and satellite stem cell‐based and gene therapies show promising results in clinical trials.4 However, limited engraftment, low cell survival, and the need for immunosuppression to prevent cell rejection precluded their routine clinical applications.
Recent studies applying genetically modified autologous MBs25 and highly proliferative stem cell populations52, 54 received attention; however, their routine clinical applications are unclear due to safety concern of the fate of genetically modified cells including: off‐target mutations, sensitization, and potential carcinogenic transformation.57, 58, 59 Recent reports on application of mesoangioblasts in experimental models and clinical trials are encouraging, however bear the inherent problems such as low efficacy of donor cell engraftment and lack of functional improvements.17
Other approaches include testing of induced pluripotent stem (iPS) cells as the potential therapeutic option for muscular dystrophies. However, the major challenge is to achieve satisfactory dedifferentiation of iPS toward MB lineage.56 Thus, there is an urgent need to develop new, more effective strategies for DMD patients. To address this unmet need and to develop a novel cell‐based therapy for DMD with unique prerequisites including, MBs phenotype, a low immune profile and tolerogenic properties, we applied our ex vivo cell fusion technology, which was established for the creation of chimeric cells of hematopoietic, mesenchymal, and MB stem cell lineages.35, 41, 42, 43, 60, 61 We developed novel DEC cell lines via ex vivo fusion of normal and dystrophin deficient MBs. These DEC lines were successfully tested in the preclinical models of mdx and mdx/scid mice.41, 42, 43 We confirmed increase in dystrophin expression which correlated with significant improvement of muscle strength and function at 90 days after DEC transplant.42
In a search of the “perfect” donor cell for skeletal muscle regeneration, researchers agreed that the cell candidate should be easily accessible, able to expand, engraft, preserve myogenic phenotype, and display a high survival rate.62, 63, 64 Moreover, low immunogenic profile is also critical for long‐term engraftment without the need for immunosuppressive therapy. Our DEC cells fulfill the prerequisites of the preferred MB therapy candidates, since DEC are of MB origin, are easily accessible and expandable, preserve myogenic phenotype in culture, and are characterized by long‐term engraftment without the need of supportive immunosuppression.41, 42, 43
However, in order to elicit therapeutic effect, the most important factors responsible for efficacy of MB‐based therapy include long‐term MB engraftment and the route of MB administration. Most of the experimental and clinical studies testing MB therapeutic effect in DMD were based on local intramuscular MBs delivery.18, 30 This resulted only in transient engraftment without long‐term therapeutic effects. Recent studies on stem cell‐based therapies confirmed the crucial role of delivery routes on the efficacy of therapeutic effect.17, 20 To enhance cell engraftment and systemic therapeutic effect, the intracardiac, intravenous, and intra‐arterial routs of delivery were tested for cardiomyocytes, MSC, iPS, and mesoangioblasts with different success rates.17, 20, 65, 66, 67 To address this issue, based on our experience and established protocol for intraosseous delivery of hematopoietic, MSCn and chimeric cells which confirmed higher engraftment rates and efficacy compared to intravenous cell delivery,35, 46, 47 we tested potential systemic effect of DEC therapy following intraosseous administration. We confirmed the protection of cardiac function which correlated with increased dystrophin expression at 90 days following intraosseous DEC transplant to mdx mice.43 In the current study to assess potential clinical applications of DEC, we created and tested efficacy of human DEC cell line and confirmed our hypothesis that intraosseous DEC transplant will result in the engraftment to the selected, DMD‐affected target organs leading to amelioration of cardiac, pulmonary, and skeletal muscle's function in the mdx/scid mouse model of DMD. Our findings of multiorgan systemic effect observed after intraosseous DEC transplant are confirmed by other investigators showing higher engraftment rate and overall safety and efficacy following intrabone transplant of cord blood and MSC in both the adult and pediatric patients' population.68, 69, 70, 71, 72, 73 In this study, the long‐term systemic effect of DEC transplant in the selected organs of heart, diaphragm, and GM was confirmed by histology, IF as well as by functional tests specific for these organs. IF analysis revealed significant dose‐dependent restoration of dystrophin expression in the heart, diaphragm, and GM of DEC‐injected mdx/scid mice compared to vehicle‐injected controls at 90 days post‐DEC transplant.
Moreover, assessment of CNFs representing the hallmark of mdx pathology74 revealed significant dose‐dependent decrease of CNF in the diaphragm and GMs of DEC‐injected mdx/scid mice indicating reduced dystrophic pathology and further confirming protective systemic effect of DEC therapy.
Histological analysis of cross‐sections of heart, diaphragm, and GM revealed reduced muscle fibrosis and inflammation as evidenced by the reduced total number of inflammatory foci in DEC‐injected compared to vehicle‐injected mdx/scid controls. Interestingly, there was a lack of large inflammatory foci observed in the higher (1 × 106) DEC dose group. We have confirmed that the beneficial effects observed on histological assessments and morphological improvements showing reduced DMD‐pathology, correlated with functional improvements in the three selected target organs of heart, diaphragm, and GM, as summarized below.
First, we assessed cardiac function after DEC transplant, since cardiomyopathy‐dependent heart failure is the major cause of death in DMD patients.75, 76 Our echocardiography data revealed maintenance of EF and FS values at 90 days postintraosseous DEC transplant to the mdx/scid mice. In contrast, significant drops of EF and FS were seen in the vehicle‐injected controls. These functional outcomes correlated with restoration of dystrophin expression, reduced central nucleation, muscle fibrosis, and inflammation leading to amelioration of mdx pathology, thus confirming the protective systemic effect of DEC therapy.
The second organ which we selected to test the efficacy of systemic DEC transplant was the diaphragm, since respiratory failure contributes significantly to the premature death of DMD patients.21 We have assessed pulmonary function via whole body plethysmography at 90 days after systemic DEC transplant and found significant dose‐dependent improvement in several functional parameters including the PENH, increased tidal volume indicating more complete contraction of a diaphragm, thus utilizing a larger percentage of the lungs capacity. Moreover, significant increase of respiratory frequency was observed, as wells as decrease of the expiration time due to reduced diaphragm fibrosis indicating amelioration of passive exhalation. These functional improvements correlated with the restoration of dystrophin expression and improved muscle pathology as evidenced by reduced number of CNF as well as reduced fibrosis and inflammation at 90 days after systemic DEC transplant to the mdx/scid mice, confirming protective effect of DEC therapy on respiratory function.
Finally, to further assess the systemic effect of DEC therapy, we selected GM as the canonical skeletal muscle, since deterioration of skeletal muscle's function in DMD patients represent the early signs of the disease. Standard functional tests of grip strength confirmed beneficial effect of DEC therapy which correlated with significant dose‐dependent improvement of muscle strength and tolerance to fatigue in GM of systemically DEC‐injected compared to the vehicle injected controls. These functional GM improvements correlated with restoration of dystrophin expression and significant dose‐dependent reduction of inflammation confirming the anti‐inflammatory effect of systemic DEC therapy. Moreover, reduced dystrophic pathology was confirmed by reduced GM fibrosis and improved fiber homogeneity assessed by Feret's diameter measurements further confirming improved fiber architecture in the DEC‐injected mice indicative of improved dystrophic phenotype after intraosseous DEC transplant.
It should be addressed that testing DEC cell therapy in the immunocompromised animal model allows for the assessment of human cell engraftment and efficacy, but limits the evaluation of the potential immune response. However, we have proven in the immunocompetent mdx mouse model that intramuscular DEC transplant resulted in increased dystrophin expression which correlated with significant improvement of muscle function, whereas intraosseous DEC injection confirmed protection of cardiac function without evidence of side effect and without the need for immunosuppression.42, 43 Moreover, mdx/scid mouse model of DMD allows to test human cell line created from normal and DMD‐affected human donors according to the same manufacturing protocol which will be used in the clinical scenario and this is one of the regulatory requirements stating that preclinical efficacy studies should be testing the same type of cell‐based therapy which will be applied in the clinical trials.77
5. CONCLUSIONS
In this study, we have addressed the problem of multiorgan failure observed in the DMD patients by assessing morphological, pathological, and functional changes in the cardiac, respiratory, and skeletal muscles representing the most frequently affected organs in DMD patients. We confirmed that systemic‐intraosseous transplant of DEC cells resulted in increased dystrophin expression, reduction of mdx pathology, and amelioration of cardiac, pulmonary, and skeletal muscle's function. Therefore, the DEC therapy represents a novel promising strategy for the prevention of cardiomyopathy and stabilization of musculoskeletal and pulmonary function in DMD.
To the best of our knowledge, this is the first report confirming protective effect of DEC therapy on the selected, DMD‐targeted organs of heart, diaphragm, and skeletal muscle after systemic‐intraosseous transplantation.
CONFLICT OF INTEREST
M.S. is CMO and shareholder of Dystrogen Therapeutics SA, the company holds a license for DEC Therapy. M.S. received research from the University of Illinois Chancellor's Innovation Fund (CIF) Proof of Concept Award. M.S. is the inventor on the patent application filed by University of Illinois at Chicago related to chimeric cell therapy for Duchenne muscular dystrophy (WO/2016/201182). M.S. is also a consultant for Cleveland Clinic and received honoraria from MMI SpA, Axogen. A.H. is the advisor to the Dystrogen Therapeutics. The other authors declared no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
M.S.: conception and design, data analysis and interpretation, manuscript writing, final manuscript approval, financial support; P.L.: collection and assembly of data, data analysis, manuscript writing; M.H., J.C.: collection of data; M.S., J.S., A.H.: collection and assembly of data, data analysis. All authors edited and approved the manuscript.
Supporting information
Supplemental Figure S1 Immunofluorescence images of dystrophin expression in the target organ of gastrocnemius muscle (GM) at 90 days after systemic DEC transplant. (A) Costameric staining with the dystrophin antibody. The dystrophin glycoprotein complex has been demonstrated to be in register with the sarcomere proteins.78 The costamer pattern is observed in these slightly tangential sections (white arrows) which identifies true dystrophin staining. (B) An additional example of a tangential cross section of GM showing costameric staining (see white arrows), magnification 58x, scale bar 50 μm; for merge: dystrophin (green), DAPI nuclei counterstaining (blue). Abbreviations: DEC‐dystrophin expressing chimeric, GM, gastrocnemius muscle.
ACKNOWLEDGMENTS
The authors thank Sonia Brodowska, MSc, Katarzyna Budzynska, MD, Kristina Perchishena, and Luke Mittelstaedt for technical support, Maria Sikorska, PhD, for Figure 1A design, and Flow Cytometry Staff (Dr. Ganesh Balaji, Wei Feng), Fluorescence Imaging Core (Dr. Peter Toth, Dr. Ke Ma), Cardiovascular Research Core (Dr. Jiwang Chen, Maricela Castellon), Research Histology and Tissue Imaging Core (Dr. Maria Sverdlov), Preclinical Imaging Core (Dr. Weiguo Li, Jin Gao) for technical assistance with data acquisition and sample processing. This work was supported by the University of Illinois Chancellor's Innovation Fund (CIF) Proof of Concept Award. ADS research fellowship was supported by The Kosciuszko Foundation.
Siemionow M, Langa P, Harasymczuk M, et al. Human dystrophin expressing chimeric (DEC) cell therapy ameliorates cardiac, respiratory, and skeletal muscle's function in Duchenne muscular dystrophy. STEM CELLS Transl Med. 2021;10(10):1406‐1418. 10.1002/sctm.21-0054
Funding information The Kosciuszko Foundation; University of Illinois Chancellor's Innovation Fund (CIF) Proof of Concept Award
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1.Van Ruiten HJ, Marini Bettolo C, Cheetham T, et al. Why are some patients with Duchenne muscular dystrophy dying young: an analysis of causes of death in North East England. Eur J Paediatr Neurol. 2016;20(6):904‐909. [DOI] [PubMed] [Google Scholar]
- 2.Duan D. Micro‐dystrophin gene therapy goes systemic in Duchenne muscular dystrophy patients. Hum Gene Ther. 2018;29(7):733‐736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Long C, McAnally JR, Shelton JM, et al. Prevention of muscular dystrophy in mice by CRISPR/Cas9‐mediated editing of germline DNA. Science. 2014;345(6201):1184‐1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gee P, Xu H, Hotta A. Cellular reprogramming, genome editing, and alternative CRISPR Cas9 technologies for precise gene therapy of Duchenne muscular dystrophy. Stem Cells Int. 2017;2017:8765154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doetschman T, Georgieva T. Gene editing with CRISPR/Cas9 RNA‐directed nuclease. Circ Res. 2017;120(5):876‐894. [DOI] [PubMed] [Google Scholar]
- 6.Colella P, Ronzitti G, Mingozzi F. Emerging issues in AAV‐mediated in vivo gene therapy. Mol Ther Methods Clin Dev. 2017;8:87‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nelson CE, Hakim CH, Ousterout DG, et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351(6271):403‐407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malik V, Rodino‐Klapac LR, Viollet L, et al. Gentamicin‐induced readthrough of stop codons in Duchenne muscular dystrophy. Ann Neurol. 2010;67(6):771‐780. [DOI] [PubMed] [Google Scholar]
- 9.Kinali M, Arechavala‐Gomeza V, Feng L, et al. Local restoration of dystrophin expression with the morpholino oligomer AVI‐4658 in Duchenne muscular dystrophy: a single‐blind, placebo‐controlled, dose‐escalation, proof‐of‐concept study. Lancet Neurol. 2009;8(10):918‐928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jearawiriyapaisarn N, Moulton HM, Sazani P, et al. Long‐term improvement in mdx cardiomyopathy after therapy with peptide‐conjugated morpholino oligomers. Cardiovasc Res. 2010;85(3):444‐453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cirak S, Arechavala‐Gomeza V, Guglieri M, et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open‐label, phase 2, dose‐escalation study. Lancet. 2011;378(9791):595‐605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goemans NM, Tulinius M, van den Hauwe M, et al. Long‐term efficacy, safety, and pharmacokinetics of drisapersen in Duchenne muscular dystrophy: results from an open‐label extension study. PLoS One. 2016;11(9):e0161955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakamura A. Moving towards successful exon‐skipping therapy for Duchenne muscular dystrophy. J Hum Genet. 2017;62(10):871‐876. [DOI] [PubMed] [Google Scholar]
- 14.Systemic gene delivery clinical trial for Duchenne muscular dystrophy. 2020. https://www.clinicaltrials.gov/ct2/show/NCT03375164
- 15.Mendell JR, Sahenk Z, Lehman K, et al. Assessment of systemic delivery of rAAVrh74.MHCK7. Micro‐dystrophin in children with Duchenne muscular dystrophy: a nonrandomized controlled trial. JAMA Neurol. 2020;77(9):1122‐1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilcocks Willcocks RJ, Forbes SC, Walter GA, et al. Assessment of rAAVrh.74.MHCK7.Micro‐dystrophin gene therapy using magnetic resonance imaging in children with Duchenne muscular dystrophy. JAMA Netw Open. 2021;4(1):e2031851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cossu G, Previtali SC, Napolitano S, et al. Intra‐arterial transplantation of HLA‐matched donor mesoangioblasts in Duchenne muscular dystrophy. EMBO Mol Med. 2016;8(12):1470‐1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gussoni E, Bennett RR, Muskiewicz KR, et al. Long‐term persistence of donor nuclei in a Duchenne muscular dystrophy patient receiving bone marrow transplantation. J Clin Invest. 2002;110(6):807‐814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skuk D, Tremblay JP. Cell therapy in muscular dystrophies: many promises in mice and dogs, few facts in patients. Expert Opin Biol Ther. 2015;15(9):1307‐1319. [DOI] [PubMed] [Google Scholar]
- 20.Taylor M, Jefferies J, Byrne B, et al. Cardiac and skeletal muscle effects in the randomized HOPE‐Duchenne trial. Neurology. 2019;92(8):e866‐e878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buyse GM, Goemans N, van den Hauwe M, et al. Effects of glucocorticoids and idebenone on respiratory function in patients with Duchenne muscular dystrophy. Pediatr Pulmonol. 2013;48(9):912‐920. [DOI] [PubMed] [Google Scholar]
- 22.McDonald CM, Sajeev G, Yao Z, et al. Deflazacort vs prednisone treatment for Duchenne muscular dystrophy: a meta‐analysis of disease progression rates in recent multicenter clinical trials. Muscle Nerve. 2020;61(1):26‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alfano LN, Charleston JS, Connolly AM, et al. Long‐term treatment with eteplirsen in nonambulatory patients with Duchenne muscular dystrophy. Medicine (Baltimore). 2019;98(26):e15858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goudenege S, Lebel C, Huot NB, et al. Myoblasts derived from normal hESCs and dystrophic hiPSCs efficiently fuse with existing muscle fibers following transplantation. Mol Ther. 2012;20(11):2153‐2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meng J, Counsell JR, Reza M, et al. Autologous skeletal muscle derived cells expressing a novel functional dystrophin provide a potential therapy for Duchenne muscular dystrophy. Sci Rep. 2016;6:19750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nitahara‐Kasahara Y, Hayashita‐Kinoh H, Ohshima‐Hosoyama S, et al. Long‐term engraftment of multipotent mesenchymal stromal cells that differentiate to form myogenic cells in dogs with Duchenne muscular dystrophy. Mol Ther. 2012;20(1):168‐177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Noviello M, Tedesco FS, Bondanza A, et al. Inflammation converts human mesoangioblasts into targets of alloreactive immune responses: implications for allogeneic cell therapy of DMD. Mol Ther. 2014;22(7):1342‐1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palmieri B, Tremblay JP, Daniele L. Past, present and future of myoblast transplantation in the treatment of Duchenne muscular dystrophy. Pediatr Transplant. 2010;14(7):813‐819. [DOI] [PubMed] [Google Scholar]
- 29.Sitzia C, Farini A, Jardim L, et al. Adaptive immune response impairs the efficacy of autologous transplantation of engineered stem cells in dystrophic dogs. Mol Ther. 2016;24(11):1949‐1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Skuk D, Tremblay JP. Confirmation of donor‐derived dystrophin in a Duchenne muscular dystrophy patient allotransplanted with normal myoblasts. Muscle Nerve. 2016;54(5):979‐981. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y, Zhu Y, Li Y, et al. Long‐term engraftment of myogenic progenitors from adipose‐derived stem cells and muscle regeneration in dystrophic mice. Hum Mol Genet. 2015;24(21):6029‐6040. [DOI] [PubMed] [Google Scholar]
- 32.Rajput BS, Chakrabarti SK, Dongare VS, et al. Human umbilical cord mesenchymal stem cells in the treatment of Duchenne muscular dystrophy: safety and feasibility study in India. J Stem Cells. 2015;10(2):141‐156. [PubMed] [Google Scholar]
- 33.Skuk D, Goulet M, Roy B, et al. First test of a "high‐density injection" protocol for myogenic cell transplantation throughout large volumes of muscles in a Duchenne muscular dystrophy patient: eighteen months follow‐up. Neuromuscul Disord. 2007;17(1):38‐46. [DOI] [PubMed] [Google Scholar]
- 34.Torrente Y, Belicchi M, Marchesi C, et al. Autologous transplantation of muscle‐derived CD133+ stem cells in Duchenne muscle patients. Cell Transplant. 2007;16(6):563‐577. [DOI] [PubMed] [Google Scholar]
- 35.Hivelin M, Klimczak A, Cwykiel J, et al. Immunomodulatory effects of different cellular therapies of bone marrow origin on chimerism induction and maintenance across MHC barriers in a face allotransplantation model. Arch Immunol Ther Exp (Warsz). 2016;64(4):299‐310. [DOI] [PubMed] [Google Scholar]
- 36.Siemionow M, Rampazzo A, Gharb BB, et al. The reversed paradigm of chimerism induction: donor conditioning with recipient‐derived bone marrow cells as a novel approach for tolerance induction in vascularized composite allotransplantation. Microsurgery. 2016;36(8):676‐683. [DOI] [PubMed] [Google Scholar]
- 37.Siemionow M, Papay F, Alam D, et al. Near‐total human face transplantation for a severely disfigured patient in the USA. Lancet. 2009;374(9685):203‐209. [DOI] [PubMed] [Google Scholar]
- 38.Siemionow MZ, Papay F, Djohan R, et al. First U.S. near‐total human face transplantation: a paradigm shift for massive complex injuries. Plast Reconstr Surg. 2010;125(1):111‐122. [DOI] [PubMed] [Google Scholar]
- 39.Siemionow M, Demir Y, Mukherjee A, et al. Development and maintenance of donor‐specific chimerism in semi‐allogenic and fully major histocompatibility complex mismatched facial allograft transplants. Transplantation. 2005;79(5):558‐567. [DOI] [PubMed] [Google Scholar]
- 40.Siemionow MZ, Klimczak A, Unal S. Different routes of donor‐derived hematopoietic stem cell transplantation for donor‐specific chimerism induction across MHC barrier. Transplant Proc. 2005;37(1):62‐64. [DOI] [PubMed] [Google Scholar]
- 41.Siemionow M, Cwykiel J, Heydemann A, et al. Creation of dystrophin expressing chimeric cells of myoblast origin as a novel stem cell based therapy for Duchenne muscular dystrophy. Stem Cell Rev. 2018;14(2):189‐199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Siemionow M, Cwykiel J, Heydemann A, et al. Dystrophin expressing chimeric (DEC) human cells provide a potential therapy for Duchenne muscular dystrophy. Stem Cell Rev. 2018;14(3):370‐384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siemionow M, Malik M, Langa P, et al. Cardiac protection after systemic transplant of dystrophin expressing chimeric (DEC) cells to the mdx mouse model of Duchenne muscular dystrophy. Stem Cell Rev Rep. 2019;15(6):827‐841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang M, Zhao R, Yu T, et al. Sudden cardiac death of Duchenne muscular dystrophy with NT‐proBNP in pericardial fluid as a useful biomarker for diagnosis of the cause of death: a case report. Forensic Sci Res. 2020;5(2):165‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wasilewska E, Malgorzewicz S, Sobierajska‐Rek A, et al. Transition from childhood to adulthood in patients with Duchenne muscular dystrophy. Medicina (Kaunas). 2020;56(9):426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Siemionow M, Zielinski M, Ozmen S, et al. Intraosseus transplantation of donor‐derived hematopoietic stem and progenitor cells induces donor‐specific chimerism and extends composite tissue allograft survival. Transplant Proc. 2005;37(5):2303‐2308. [DOI] [PubMed] [Google Scholar]
- 47.Ozmen S, Ulusal BG, Ulusal AE, et al. Trafficking of donor‐derived bone marrow correlates with chimerism and extension of composite allograft survival across MHC barrier. Transplant Proc. 2006;38(5):1625‐1633. [DOI] [PubMed] [Google Scholar]
- 48.Fayssoil A, Renault G, Guerchet N, et al. Cardiac characterization of mdx mice using high‐resolution doppler echocardiography. J Ultrasound Med. 2013;32(5):757‐761. [DOI] [PubMed] [Google Scholar]
- 49.Bondoc AB, Detombe S, Dunmore‐Buyze J, et al. Application of 3‐d echocardiography and gated micro‐computed tomography to assess cardiomyopathy in a mouse model of Duchenne muscular dystrophy. Ultrasound Med Biol. 2014;40(12):2857‐2867. [DOI] [PubMed] [Google Scholar]
- 50.Bauer R, Straub V, Blain A, et al. Contrasting effects of steroids and angiotensin‐converting‐enzyme inhibitors in a mouse model of dystrophin‐deficient cardiomyopathy. Eur J Heart Fail. 2009;11(5):463‐471. [DOI] [PubMed] [Google Scholar]
- 51.Roberts NW, Holley‐Cuthrell J, Gonzalez‐Vega M, et al. Biochemical and functional comparisons of mdx and Sgcg(−/−) muscular dystrophy mouse models. Biomed Res Int. 2015;2015:131436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Muir LA, Murry CE, Chamberlain JS. Prosurvival factors improve functional engraftment of myogenically converted dermal cells into dystrophic skeletal muscle. Stem Cells Dev. 2016;20(20):1559‐1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boldrin L, Morgan JE. Modulation of the host skeletal muscle niche for donor satellite cell grafting. Methods Mol Biol. 2013;1035:179‐190. [DOI] [PubMed] [Google Scholar]
- 54.Pelatti MV, Gomes JP, Vieira NM, et al. Transplantation of human adipose mesenchymal stem cells in non‐immunosuppressed GRMD dogs is a safe procedure. Stem Cell Rev. 2016;12(4):448‐453. [DOI] [PubMed] [Google Scholar]
- 55.Li HL, Fujimoto N, Sasakawa N, et al. Precise correction of the dystrophin gene in Duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR‐Cas9. Stem Cell Rep. 2015;4(1):143‐154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Abujarour R, Bennett M, Valamehr B, et al. Myogenic differentiation of muscular dystrophy‐specific induced pluripotent stem cells for use in drug discovery. Stem Cells Translational Medicine. 2014;3(2):149‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Long C, Amoasii L, Mireault AA, et al. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2016;351(6271):400‐403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huard J, Mu X, Lu A. Evolving paradigms in clinical pharmacology and therapeutics for the treatment of Duchenne muscular dystrophy. Clin Pharmacol Ther. 2016;100(2):142‐146. [DOI] [PubMed] [Google Scholar]
- 59.Kosicki M, Tomberg K, Bradley A. Repair of double‐strand breaks induced by CRISPR‐Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol. 2018;36(8):765‐771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Siemionow M, Madajka M, Cwykiel J. Application of cell‐based therapies in facial transplantation. Ann Plast Surg. 2012;69(5):575‐579. [DOI] [PubMed] [Google Scholar]
- 61.Cwykiel J, Siemionow MZ. Cellular therapy models: ex vivo chimera model. In: Siemionow MZ, ed. Plastic and Reconstructive Surgery: Experimental Models and Research Design. London, UK: Springer‐Verlag Ltd; 2015;593‐603. [Google Scholar]
- 62.Tedesco FS, Cossu G. Stem cell therapies for muscle disorders. Curr Opin Neurol. 2012;25(5):597‐603. [DOI] [PubMed] [Google Scholar]
- 63.Meregalli M, Farini A, Sitzia C, et al. Advancements in stem cells treatment of skeletal muscle wasting. Front Physiol. 2014;5:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meng J, Muntoni F, Morgan JE. Stem cells to treat muscular dystrophies ‐ where are we? Neuromuscul Disord. 2011;21(1):4‐12. [DOI] [PubMed] [Google Scholar]
- 65.Chun JL, O'Brien R, Song MH, et al. Injection of vessel‐derived stem cells prevents dilated cardiomyopathy and promotes angiogenesis and endogenous cardiac stem cell proliferation in mdx/utrn‐/‐ but not aged mdx mouse models for Duchenne muscular dystrophy. Stem Cells Translational Medicine. 2013;2(1):68‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Markert CD, Atala A, Cann JK, et al. Mesenchymal stem cells: emerging therapy for Duchenne muscular dystrophy. PM R. 2009;1(6):547‐559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Biressi S, Filareto A, Rando TA. Stem cell therapy for muscular dystrophies. J Clin Invest. 2020;130(11):5652‐5664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bonifazi F, Dan E, Labopin M, et al. Intrabone transplant provides full stemness of cord blood stem cells with fast hematopoietic recovery and low GVHD rate: results from a prospective study. Bone Marrow Transplant. 2019;54(5):717‐725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Frassoni F, Gualandi F, Podesta M, et al. Direct intrabone transplant of unrelated cord‐blood cells in acute leukaemia: a phase I/II study. Lancet Oncol. 2008;9(9):831‐839. https://pubmed.ncbi.nlm.nih.gov/18693069/ [DOI] [PubMed] [Google Scholar]
- 70.Goto T, Murata M, Terakura S, et al. Phase I study of cord blood transplantation with intrabone marrow injection of mesenchymal stem cells: a clinical study protocol. Medicine (Baltimore). 2018;97(17):e0449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee H, Park JB, Lee S, et al. Intra‐osseous injection of donor mesenchymal stem cell (MSC) into the bone marrow in living donor kidney transplantation; a pilot study. J Transl Med. 2013;11:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Marktel S, Scaramuzza S, Cicalese MP, et al. Intrabone hematopoietic stem cell gene therapy for adult and pediatric patients affected by transfusion‐dependent ß‐thalassemia. Nat Med. 2019;25(2):234‐241. [DOI] [PubMed] [Google Scholar]
- 73.Doring M, Kluba T, Cabanillas Stanchi KM, et al. Longtime outcome after intraosseous application of autologous mesenchymal stromal cells in pediatric patients and young adults with avascular necrosis after steroid or chemotherapy. Stem Cells Dev. 2020;29(13):811‐822. https://pubmed.ncbi.nlm.nih.gov/32295491/ [DOI] [PubMed] [Google Scholar]
- 74.Yu L, Zhang X, Yang Y, et al. Small‐molecule activation of lysosomal TRP channels ameliorates Duchenne muscular dystrophy in mouse models. Sci Adv. 2020;6(6):eaaz2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hagenbuch SC, Gottliebson WM, Wansapura J, et al. Detection of progressive cardiac dysfunction by serial evaluation of circumferential strain in patients with Duchenne muscular dystrophy. Am J Cardiol. 2010;105(10):1451‐1455. [DOI] [PubMed] [Google Scholar]
- 76.Zito C, Longobardo L, Citro R, et al. Ten years of 2D longitudinal strain for early myocardial dysfunction detection: a clinical overview. Biomed Res Int. 2018;2018:8979407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tappenbeck N, Schroder HM, Niebergall‐Roth E, et al. In vivo safety profile and biodistribution of GMP‐manufactured human skin‐derived ABCB5‐positive mesenchymal stromal cells for use in clinical trials. Cytotherapy. 2019;21(5):546‐560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gumerson JD, Michele DE. The dystrophin‐glycoprotein complex in the prevention of muscle damage. J Biomed Biotechnol. 2011;2011:210797. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1 Immunofluorescence images of dystrophin expression in the target organ of gastrocnemius muscle (GM) at 90 days after systemic DEC transplant. (A) Costameric staining with the dystrophin antibody. The dystrophin glycoprotein complex has been demonstrated to be in register with the sarcomere proteins.78 The costamer pattern is observed in these slightly tangential sections (white arrows) which identifies true dystrophin staining. (B) An additional example of a tangential cross section of GM showing costameric staining (see white arrows), magnification 58x, scale bar 50 μm; for merge: dystrophin (green), DAPI nuclei counterstaining (blue). Abbreviations: DEC‐dystrophin expressing chimeric, GM, gastrocnemius muscle.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.