

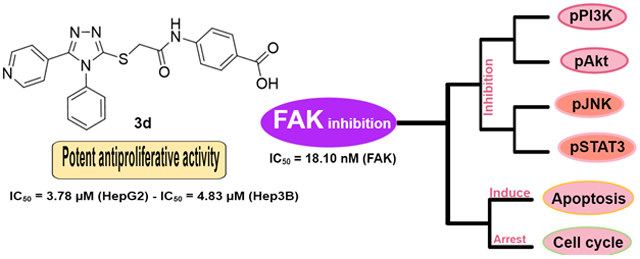
Abstract
Small molecule inhibitors of the focal adhesion kinase are regarded as promising tools in our armamentarium for treating cancer. Here, we identified four 1,2,4-triazole derivatives that inhibit FAK kinase significantly and evaluated their therapeutic potential. Most tested compounds revealed potent antiproliferative activity in HepG2 and Hep3B liver cancer cells, in which 3c and 3d were the most potent (IC50 range; 2.88~4.83 μM). Compound 3d possessed significant FAK inhibitory activity with IC50 value of 18.10 nM better than the reference GSK-2256098 (IC50 = 22.14 nM). The preliminary mechanism investigation by Western blot analysis showed that both 3c and 3d repressed FAK phosphorylation comparable to GSK-2256098 in HepG2 cells. As a result of FAK inhibition, 3c and 3d inhibited the pro-survival pathways by decreasing the phosphorylation levels of PI3K, Akt, JNK, and STAT3 proteins. This effect led to apoptosis induction and cell cycle arrest. Taken together, these results indicate that 3d could serve as a potent preclinical candidate for the treatment of cancers.
Keywords: FAK inhibitors; Synthesis; 5-pyridinyl-1,2,4-triazoles; Anticancer activity; Docking study
Graphical Abstract
Focal adhesion kinase (FAK), a non-receptor tyrosine kinase, is a critical enzyme in the signaling pathway and is overexpressed in different solid tumors such as liver cancer, breast cancer, colon cancer, etc 1. FAK plays a prominent role in cell survival, migration, adhesion, and proliferation 2. Also, FAK consists of three different domains, including an N terminal FERM (four-point-one, ezrin, radixin, moesin) domain, a catalytic kinase domain (central), and a C-terminal focal adhesion targeting (FAT) domain. These domains contain tyrosine phosphorylation sites that regulate the molecular functions and the catalytic activity of FAK 3. Ample evidence indicates that FAK potentiates cancer progression and that inhibiting the kinase is a promising therapeutic strategy 3, 4. Consequently, FAK is recognized as an important therapeutic target for the treatment of cancer.
FAK inhibition influences other essential proteins such as Akt and Signal transducer and activator of transcription 3 (STAT3) 5, 6. Akt plays a fundamental role in regulating numerous cellular functions such as proliferation, survival, metabolism, and growth, while STAT3 is crucial for cell growth and is activated mostly in solid tumors (the same as FAK). In this respect, the inhibition of Akt and STAT3 phosphorylation leads to remarkable anticancer activity. Interestingly, the FAK inhibition results in suppressing the phosphorylation of both Akt and STAT3 5, 7, 8.
Recently, numerous efforts devoted to developing potent FAK inhibitors to treat cancers 4, 9, 10. Most of the developed inhibitors targeted the ATP binding site of FAK and were designed to bind to the essential amino acids in the active site, such as Cys502 in the kinase hinge and Asp564 of the DFG motif. TAE226 is one of the earliest discovered FAK inhibitors with potent antiproliferative activity (Figure 1) 11. Despite its superior potency, TAE226 did not enter clinical trials due to its effect on glucose metabolism 12. Four FAK inhibitors; PND-1186, PF-562271, CEP37440, and GSK-2256098, finished phase I clinical trials, whereas defactinib is the only compound that completed phase II clinical trials (Figure 1) 4. The main pharmacophore of FAK inhibitors comprises diphenyl five/six-membered nitrogenous heterocyclic rings.
Figure 1:

Chemical structures of different six-membered FAK inhibitors
Several five-membered heterocyclic FAK inhibitors, such as oxadiazoles (VII, VIII) 13, 14, triazoles (IX, X) 15, 16, and thiadiazoles (XI, XII) 17, 18 revealed potent FAK inhibitory and antiproliferative activity (Figure 2). The structural features analysis displayed that the diphenyl substitution at ring A and ring B on the five-membered heterocyclic ring is fundamental for the activity. Several compounds bearing diphenyl dioxine, substituted phenyl, or pyridinyl in ring A possessed significant anti-FAK and antiproliferative activity, whereas ring B was separated from the heterocyclic ring by a linker containing thioacetamide, butanamide, thioacetohydrazide, or amide.
Figure 2:

Chemical structures of various five-membered FAK inhibitors
The 1,2,4-triazole and pyridine rings are well-known moieties present in several anticancer agents 13, 19-21. The 1,2,4-triazole is an important bioisostere to various bonds and heterocyclic ring and possesses H-bond accepting properties, strong dipole moment, and pi electron-deficient aromaticity 22. Besides, both 1,2,4-triazole and pyridine moieties display considerable stability towards chemical and metabolic degradation.
Based on the abovementioned findings and as a probe for the investigation of more potent FAK inhibitors, we report herein the synthesis and biological investigation of 5-pyridinyl-1,2,4-triazole derivatives as potential FAK inhibitors. Pyridine and benzoic acid were investigated as ring A and ring B, respectively. The 1,2,4-triazole (five-membered ring) was connected to ring B via thioacetamide moiety.
The synthesis of derivatives 3a-d was accomplished by treating isonicotinic acid hydrazide with the appropriate isothiocyanate (methyl, ethyl, allyl, or phenyl) at 70 °C (Scheme 1). The resulting thiosemicarbazide was then reacted with 2N NaOH and acidified with HCl to yield the 1,2,4-triazole-3-thiol derivatives 1a-d, whereas 4-(2-bromoacetamido) benzoic acid 2 was obtained by reacting p-aminobenzoic acid with bromoacetylbromide. The target compounds 3a-d were obtained through reacting 1a-d with 2 in the presence of triethylamine in 93-96% yield at 70 °C. The structures of the target compounds were checked by 1HNMR, 13CNMR, mass spectroscopy, and further confirmed by elemental analyses. The purity of compound 3d was fatherly checked by HPLC analysis using a linear gradient of methanol: water mixture at a rate of 1.0 mL/min leading to elution of hybrid 3d as a single peak after 4.79 min at λmax of 220 nm (supporting information) 23.
Scheme 1:
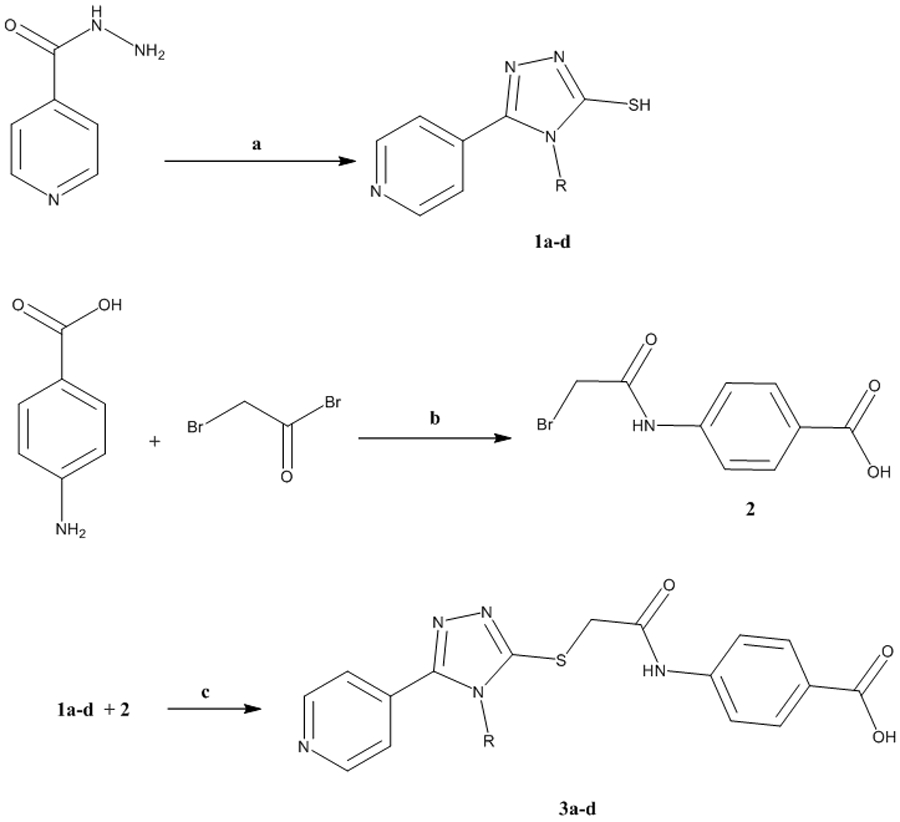
Synthetic approach of the target 1,2,4-triazole derivatives 3a-d
a) (i) RNCS, EtOH, 70 °C, 4h, (ii) 2N NaOH, 3h, (iii) HCl; b) K2CO3, 24h; c) TEA, DMF, 70 °C, 10h.
The synthesized carboxylic acid derivatives 3a-d were evaluated for their antiproliferative activity on HepG2 and Hep3B liver cancer cell lines. These cells were selected deliberately because they profoundly express FAK protein 24, 25. Doxorubicin and GSK2256098 were used as a positive control. As illustrated in Table 1, the derivatives 3a-d possessed a distinctive potency on both tested cells; compounds 3c and 3d showed potent antiproliferative activity in HepG2 cells with IC50 values of 4.72 and 3.78 μM, respectively. Besides, 3a revealed moderate antiproliferative activity in HepG2 cells (IC50 = 8.83 μM). Despite this promising activity of the prepared compounds on HepG2 cells, the derivative 3b did not possess notable activity and showed 20-folds lower antiproliferative activity than Doxorubicin. On Hep3B cells, compounds 3a, 3c, and 3d showed more potent antiproliferative activity than Doxorubicin and GSK2256098. The most active derivatives 3c and 3d were tested for their FAK inhibitory activity, using GSK2256098 as a positive control. Interestingly, both tested compounds revealed strong FAK inhibitory activity in low nanomolar concentrations (Table 1). It is worth mentioning that 3d possessed comparable FAK inhibitory activity to the reference GSK2256098 with IC50 value of 18.10 nM.
Table 1:
Concentration producing 50% growth inhibition (IC50, μM±SEM) of the carboxylic acid derivatives 3a-d on HepG2 and Hep3B liver cancer cells
| Cpd | R | Antiproliferative activity, μM | FAK inhibitory activity, nM |
|
|---|---|---|---|---|
| HepG2 | Hep3B | |||
| 3a | Methyl | 8.83±0.52 | 3.87±0.23 | N.T. |
| 3b | Ethyl | 40.97±2.4 | 17.94±1.05 | N.T. |
| 3c | Allyl | 4.72±0.28 | 2.88±0.17 | 36.37±1.8 |
| 3d | Phenyl | 3.78±0.22 | 4.83±0.28 | 18.10±0.92 |
| Doxorubicin | - | 1.99±0.12 | 7.21±0.42 | N.T. |
| GSK2256098 | - | 3.24±0.57 | 6.91±1.02 | 22.14±1.12 |
Moreover, the immunoblotting analysis was performed in HepG2 cells for compounds 3c and 3d, whereas GSK-2256098 was used as a positive control. The obtained results revealed that both 3c and 3d displayed a substantial ability to repress FAK phosphorylation comparable to the reference GSK-2256098 at 4 μM concentration (Figure 3A and 3B). As FAK plays an important role in the activation of different pro-survival mechanisms, including PI3K/Akt and JNK/STAT3 in the liver cancer cells, we tested the effect of 3c and 3d on the PI3K/Akt and JNK/STAT3 pathways using western blotting 26. We found that 3c and 3d significantly repressed the phosphorylated PI3K, Akt, JNK, and STAT3 (Figure 3A, 3C-F) in HepG2 cell line. These degrees of suppression of the signaling pathways were positively correlated with the observed antiproliferative activity. We hypothesized that such suppression of the PI3K/Akt and JNK/STAT3 signaling pathways might alter cell survival and lead to apoptosis. Therefore, we examined the effect of 3d on cell cycle progression in HepG2 cells using its IC50 concentration (3.78 μM), we observed that compound 3d was able to arrest the cell cycle at sub G1 and S phases (41.61% and 54.11%, respectively), (Figure 4). Meanwhile, 3d revealed a substantial ability to induce apoptosis by dissipating and dispersing cellular integrity leading to increasing the early and late apoptotic cells by 4.98% and 23.62%, respectively (Figure 5).
Figure 3:
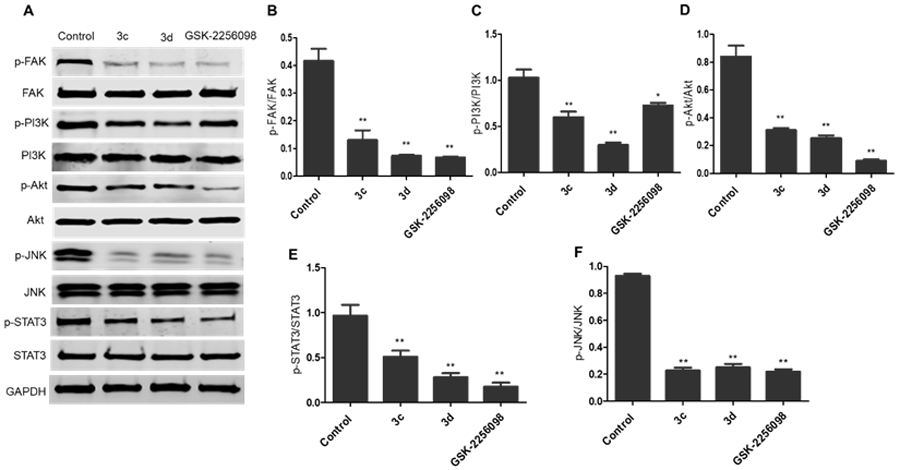
The 1,2,4-triazole derivatives 3c and 3d inhibited FAK phosphorylation and its downstream cascades. A) Western blotting analysis showed that levels of p-FAK, p-PI3K, p-Akt, p-JNK, and p-STAT3 were downregulated in HepG2 cells treated with 3c and 3d, DMSO served as a negative control, and GAPDH served as a loading control; B-F) The relative protein expression levels of p-FAK, p-PI3K, p-Akt, p-JNK, and p-STAT3, compared to their total form FAK, PI3K, Akt, JNK, and STAT3, respectively, were quantified by Image J. The values are the means ± SEM of three different experiments. *, P < 0.05, ** and P < 0.01 indicate a significant difference compared with DMSO negative control treatment.
Figure 4:

Compound 3d arrested the cell cycle at sub G1 and S phases in HepG2 cells. Representative cell cycle distribution data are shown by flow cytometric analysis upon incubation with 3d or DMSO as a negative control for 24 h.
Figure 5:

Apoptotic effect of compound 3d on HepG2 cells. Representative cytograms of apoptotic HepG2 cells stimulated with 3d or DMSO as a negative control for 24 h.
Compound 3d was docked into the ATP binding site of FAK kinase (PDB code: 2JKK) to elucidate its binding mode 11. The native ligand (TAE226) was re-docked into the active site to validate the docking study (RMDS = 1.3813). Compound 3d showed two possible binding modes inside the ATP binding site of FAK, and both modes revealed essential interactions inside the active site. Regarding the 1st mode, N1 of the 1,2,4-triazole made a hydrogen bond with Cys502 of the kinase hinge (Figure 6A). Meanwhile, the oxygen of the amide group showed two hydrogen bonds with Asp564 of the DFG motif of the activation loop and Gly563 amino acids. The sulfur atom formed a hydrogen bond with Glu506 amino acid and the 1,2,4-triazole-3-thiol moiety pointed toward the gatekeeper amino acid Met499. Also, the pyridine ring formed H…π interaction with Ile428 amino acid. On the other hand, the 2nd mode revealed an essential hydrogen bond between the nitrogen of the pyridine moiety and Cys502 of the kinase hinge (Figure 6B). Also, both pyridine and 1,2,4-triazole rings formed H…π contacts with Leu501 and Leu567 amino acids, respectively, whereas N2 of the latter ring showed a hydrogen bond with Gly429 amino acid. The presence of the phenyl group helped in the formation of several π interactions and was in high proximity to Asp564 of the DFG motif. Moreover, the carboxylic acid formed a hydrogen bond with Ser568. Both obtained poses are reasonable and showed several essential interactions. However, the 2nd pose might be more realistic because it rationalized the role of both the nitrogen in the pyridine moiety and the phenyl group through the formation of different electrostatic interactions.
Figure 6:

3D representation of 3d B (cyan) within the ATP binding site of FAK. A) Proposed binding mode 1; B) Proposed binding mode 2 (PDB code: 2JKK)
The prediction of Lipinski and ADME properties is one of the significant and widely identified parameters used to predict the oral bioavailability of active therapeutics 27, 28. Compounds that obey all or three of these parameters: molecular weight (MW) ≤ 500, number of hydrogen bond acceptors (HBA) ≤ 10, number of hydrogen bond donors (HBD) ≤ 5, and octanol-water partition coefficient (iLogP) ≤ 5, will most probably have acceptable oral bioavailability. Therefore, Lipinski violations and drug-likeness properties of the synthesized 1,2,4-triazoles were calculated using the SwissADME online predictor. All the prepared derivatives showed no Lipinski violations and showed acceptable topological polar surface area (TPSA) and bioavailability scores (F). Fortunately, none of the prepared derivatives was classified as Pan-Assay Interference Compounds (PAINS). In other words, these compounds are predicted to interact specifically with the ATP binding site of FAK instead of binding to random targets, and hence, likely to have fewer off-target effects.
In conclusion, the 5-pyridinyl-1,2,4-triazole derivatives 3a-d were synthesized and evaluated for their antiproliferative activity in two hepatic cancer cell lines (HepG2 and Hep3B). Compounds 3c and 3d emerged as potent antiproliferative agents against both cell lines tested. The top potent derivatives 3c and 3d were assessed for their anti-FAK activity and revealed remarkable potency at nanomolar concentrations. Furthermore, the study of FAK phosphorylation inhibition in HepG2 cells revealed that both 3c and 3d repress the phosphorylation of FAK significantly. Preferably, compound 3d decreased the phosphorylation of PI3K, Akt, JNK, and STAT3. Moreover, 3d induced apoptosis and arrested preG1 and S phases of the cell cycle. In support of this study, molecular docking analysis revealed that 3d showed good fitting within the ATP binding site of FAK. Also, the obtained ADME properties were in an acceptable range. From the above data, it could be assumed that the synthesized 5-pyridinyl-1,2,4-triazole carboxylic acid derivatives (3a-d) are a set of promising compounds for the development of potent antiproliferative agents as well as FAK inhibitors.
Supplementary Material
Acknowledgements
A.A.A was supported by an NIH-funded Cancer Therapeutics Training Program (CT2, T32 CA121938) and P.G by the NIH (CA238042, CA100768 and CA160911). The authors gratefully acknowledge Radwa Taher (Assistant lecturer, Department of Pharmacognosy, Faculty of Pharmacy, Deraya University) and Fatma A. Abo Elsoud (Assistant lecturer, Department of organic chemistry, Faculty of Dentistry, Deraya University) for their efforts and help in running HPLC analysis for compound 3d.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have influenced the work reported in this paper.
References
- 1.Chatzizacharias NA, Kouraklis GP, Theocharis SE. Clinical significance of FAK expression in human neoplasia. Histology and histopathology. 2008. [DOI] [PubMed] [Google Scholar]
- 2.Schaller MD. Cellular functions of FAK kinases: insight into molecular mechanisms and novel functions. Journal of cell science. 2010;123(7): 1007–1013. [DOI] [PubMed] [Google Scholar]
- 3.Lu Y, Sun H. Progress in the Development of Small Molecular Inhibitors of Focal Adhesion Kinase (FAK). Journal of Medicinal Chemistry. 2020;63(23): 14382–14403. [DOI] [PubMed] [Google Scholar]
- 4.Cao F-Y, Zhou X-P, Su J, et al. Chemical Structure Characteristics and Bioactivity of Small Molecule FAK Inhibitors. Anti-Cancer Agents in Medicinal Chemistry (Formerly Current Medicinal Chemistry-Anti-Cancer Agents). 2016;16(8): 934–941. [DOI] [PubMed] [Google Scholar]
- 5.Visavadiya NP, Keasey MP, Razskazovskiy V, et al. Integrin-FAK signaling rapidly and potently promotes mitochondrial function through STAT3. Cell Communication and Signaling. 2016;14(1): 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kong D, Chen F, Sima N. Inhibition of focal adhesion kinase induces apoptosis in bladder cancer cells via Src and the phosphatidylinositol 3-kinase/Akt pathway. Experimental and therapeutic medicine. 2015;10(5): 1725–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fagard R, Metelev V, Souissi I, Baran-Marszak F. STAT3 inhibitors for cancer therapy: Have all roads been explored? Jak-stat. 2013;2(1): e22882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang N, Dai Q, Su X, Fu J, Feng X, Peng J. Role of PI3K/AKT pathway in cancer: the framework of malignant behavior. Molecular Biology Reports. 2020: 1–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dao P, Lietha D, Etheve-Quelquejeu M, Garbay C, Chen H. Synthesis of novel 1, 2, 4-triazine scaffold as FAK inhibitors with antitumor activity. Bioorganic & Medicinal Chemistry Letters. 2017;27(8): 1727–1730. [DOI] [PubMed] [Google Scholar]
- 10.Zificsak CA, Gingrich DE, Breslin HJ, et al. Optimization of a novel kinase inhibitor scaffold for the dual inhibition of JAK2 and FAK kinases. Bioorganic & medicinal chemistry letters. 2012;22(1): 133–137. [DOI] [PubMed] [Google Scholar]
- 11.Lietha D, Eck MJ. Crystal structures of the FAK kinase in complex with TAE226 and related bis-anilino pyrimidine inhibitors reveal a helical DFG conformation. PloS one. 2008;3(11): e3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hao H-f, Takaoka M, Bao X-h, et al. Oral administration of FAK inhibitor TAE226 inhibits the progression of peritoneal dissemination of colorectal cancer. Biochemical and biophysical research communications. 2012;423(4): 744–749. [DOI] [PubMed] [Google Scholar]
- 13.Zhang L-R, Liu Z-J, Zhang H, et al. Synthesis, biological evaluation and molecular docking studies of novel 2-(1, 3, 4-oxadiazol-2-ylthio)-1-phenylethanone derivatives. Bioorganic & medicinal chemistry. 2012;20(11): 3615–3621. [DOI] [PubMed] [Google Scholar]
- 14.Altıntop MD, Sever B, Çiftçi GA, Turan-Zitouni G, Kaplancıklı ZA, Özdemir A. Design, synthesis, in vitro and in silico evaluation of a new series of oxadiazole-based anticancer agents as potential Akt and FAK inhibitors. European journal of medicinal chemistry. 2018;155: 905–924. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y-B, Liu W, Yang Y-S, et al. Synthesis, molecular modeling, and biological evaluation of 1, 2, 4-triazole derivatives containing pyridine as potential anti-tumor agents. Medicinal Chemistry Research. 2013;22(7): 3193–3203. [Google Scholar]
- 16.Jia Y, Si L, Lin R, et al. Thiophenol-formaldehyde triazole causes apoptosis induction in ovary cancer cells and prevents tumor growth formation in mice model. European Journal of Medicinal Chemistry. 2019;172: 62–70. [DOI] [PubMed] [Google Scholar]
- 17.Sun J, Yang Y-S, Li W, et al. Synthesis, biological evaluation and molecular docking studies of 1, 3, 4-thiadiazole derivatives containing 1, 4-benzodioxan as potential antitumor agents. Bioorganic & medicinal chemistry letters. 2011;21(20): 6116–6121. [DOI] [PubMed] [Google Scholar]
- 18.Yang X-H, Xiang L, Li X, et al. Synthesis, biological evaluation, and molecular docking studies of 1, 3, 4-thiadiazol-2-amide derivatives as novel anticancer agents. Bioorganic & medicinal chemistry. 2012;20(9): 2789–2795. [DOI] [PubMed] [Google Scholar]
- 19.Mustafa M, Anwar S, Elgamal F, Ahmed ER, Aly OM. Potent combretastatin A-4 analogs containing 1, 2, 4-triazole: Synthesis, antiproliferative, anti-tubulin activity, and docking study. European journal of medicinal chemistry. 2019;183: 111697. [DOI] [PubMed] [Google Scholar]
- 20.Mustafa M, Abdelhamid D, Abdelhafez EM, Ibrahim MA, Gamal-Eldeen AM, Aly OM. Synthesis, antiproliferative, anti-tubulin activity, and docking study of new 1, 2, 4-triazoles as potential combretastatin analogues. European journal of medicinal chemistry. 2017;141: 293–305. [DOI] [PubMed] [Google Scholar]
- 21.Altaf AA, Shahzad A, Gul Z, et al. A review on the medicinal importance of pyridine derivatives. J Drug Des Med Chem. 2015;1(1): 1. [Google Scholar]
- 22.Malik MS, Ahmed SA, Althagafi II, Ansari MA, Kamal A. Application of triazoles as bioisosteres and linkers in the development of microtubule targeting agents. RSC Medicinal Chemistry. 2020;11(3): 327–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Compound 3d: Yellow powder (0.62 g ym-CHM, CDCl3) (ppm): 4.24(s, 2H, S-CH2), 7.25(d, 2H, J=4.81 Hz, Ar-H), 7.47(d, 2H, J=7.21 Hz, Ar-H), 7.54-7.60(m, 3H, Ar-H),7.65(d, 2H, J=8.81 Hz, Ar-H), 7.87(d, 2H, J=8.81 Hz, Ar-H), 8.52(d, 2H, J=5.61 Hz, Ar-H), 10.67(s, 1H, NH), 12.74(s, 1H, OH); 13CNMR (100 MHz, CDCl3, δ ppm): 37.36, 118.94, 122.05, 125.97, 128.08, 130.79, 131.04, 131.14, 133.81, 134.32, 143.31, 150.66, 152.84, 153.50, 166.50, 167.42; Anal. Calcd for C22H17N5O3S: C, 61.24; H, 3.97; N, 16.23; S, 7.43. Found: C, 61.32; H, 4.12; N, 16.45; S, 7.61; ESI-MS: [M-H]-, 430.4, found: 430.3; HPLC-analysis: linear gradient of water-methanol (20:80) for 10 min, followed by linear gradient to 50% methanol within 30 min, at a flow rate of 1 ml/min, Rt = 3.26 min.
- 24.Zheng X, Liu W, Xiang J, et al. Collagen I promotes hepatocellular carcinoma cell proliferation by regulating integrin β1/FAK signaling pathway in nonalcoholic fatty liver. Oncotarget. 2017;8(56): 95586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Azzariti A, Mancarella S, Porcelli L, et al. Hepatic stellate cells induce hepatocellular carcinoma cell resistance to sorafenib through the laminin-332/α3 integrin axis recovery of focal adhesion kinase ubiquitination. Hepatology. 2016;64(6): 2103–2117. [DOI] [PubMed] [Google Scholar]
- 26.Xia H, Nho RS, Kahm J, Kleidon J, Henke CA. Focal adhesion kinase is upstream of phosphatidylinositol 3-kinase/Akt in regulating fibroblast survival in response to contraction of type I collagen matrices via a beta 1 integrin viability signaling pathway. J Biol Chem. 2004;279(31): 33024–33034. [DOI] [PubMed] [Google Scholar]
- 27.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced drug delivery reviews. 1997;23(1-3): 3–25. [DOI] [PubMed] [Google Scholar]
- 28.Mustafa M, Mostafa YA. A facile synthesis, drug-likeness, and in silico molecular docking of certain new azidosulfonamide–chalcones and their in vitro antimicrobial activity. Monatshefte für Chemie-Chemical Monthly. 2020;151: 417–427. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.