

Abstract
Amyotrophic lateral sclerosis (ALS) is an adult-onset neurodegenerative disease with progressive motor neuron death, where patients usually die within 5 years of diagnosis. Previously, we showed that the C-boutons, which are large cholinergic synapses to motor neurons that modulate motor neuron activity, are necessary for behavioral compensation in mSOD1G93A mice, a mouse model for ALS. We reasoned that, since the C-boutons likely increase the excitability of surviving motor neurons to compensate for motor neuron loss during ALS disease progression, then amplitude modulation through the C-boutons likely increases motor neuron stress and worsens disease progression. By comparing male and female mSOD1G93A mice to mSOD1G93A mice with genetically silenced C-boutons [mSOD1G93A; Dbx1::cre; ChATfl/fl (mSOD1G93A/Coff)], we show that the C-boutons do not influence the humane end point of mSOD1G93A mice; however, our histologic analysis shows that C-bouton silencing significantly improves fast-twitch muscle innervation over time. Using immunohistology, we also show that the C-boutons are active in a task-dependent manner, and that symptomatic mSOD1G93A mice show significantly higher C-bouton activity than wild-type mice during low-intensity walking. Last, by using behavioral analysis, we provide evidence that C-bouton silencing in combination with swimming is beneficial for the behavioral capabilities of mSOD1G93A mice. Our observations suggest that manipulating the C-boutons in combination with a modulatory-targeted training program may therefore be beneficial for ALS patients and could result in improved mobility and quality of life.
SIGNIFICANCE STATEMENT Despite decades of research on amyotrophic lateral sclerosis (ALS), there have been little improvements in treatments and therapies. We sought to better understand how the activation of C-boutons, which are large cholinergic modulatory synapses on motor neurons, change and affect the disease as it progresses. When these C-boutons are genetically silenced and exercises designed to otherwise activate the C-boutons are frequently performed in ALS model mice, the mice perform better than their untreated counterparts over time. C-bouton-targeted therapies could therefore be beneficial for ALS patients and could result in improved mobility and quality of life.
Keywords: amyotrophic lateral sclerosis, behavior, C-boutons, genetics, mice, Sod1G93A
Introduction
Amyotrophic lateral sclerosis (ALS) is an adult-onset neurodegenerative disease of progressive motor neuron death. Over time, patients with the disease lose their ability to move, talk, eat, and breathe. There is currently no cure for ALS, and patients usually die within 5 years of the disease being diagnosed (Cleveland and Rothstein, 2001; Robberecht and Philips, 2013). The glutamate release inhibitor riluzole is the most common drug used to treat ALS patients; however, it only extends a patient's life span by ∼2–3 months (Rothstein, 1996; Miller et al., 2012). Despite extensive knowledge of the relationship between ALS and the motor neurons, edaravone has been the only other effective therapy developed for ALS treatment (Yoshino, 2019).
Factors that regulate motor neuron activity likely play a significant role in ALS disease progression and could provide new drug targets for ALS disease treatment. The C-boutons, for instance, are large cholinergic synapses to motor neurons that modulate motor neuron activity (Conradi and Skoglund, 1960; Zagoraiou et al., 2009). The C-boutons originate from V0c interneurons that express the Dbx1 and Pitx2 transcription factors near the central canal (Zagoraiou et al., 2009). Changes in the C-boutons have been observed in both ALS patients and in transgenic model mice that carry the mutant form of superoxide dismutase 1 (mSOD1G93A; Hegedus et al., 2007, 2008; Gordon et al., 2010), suggesting that they play a role in ALS disease progression (Nagao et al., 1998; Pullen and Athanasiou, 2009; Herron and Miles, 2012; Milan et al., 2015; Dukkipati et al., 2017). Previously, we showed that these C-boutons are necessary for behavioral compensation in mSOD1G93A mice (Landoni et al., 2019); however, it is still unclear whether these C-boutons influence motor neuron health and ALS disease progression. We hypothesized that, since the C-boutons increase the excitability of surviving motor neurons to compensate for motor neuron loss during ALS disease progression, then amplitude modulation through the C-boutons likely increases motor neuron stress and worsens disease progression.
In this study, we compare mSOD1G93A mice and mSOD1G93A mice that had their C-boutons genetically silenced [mSOD1G93A; Dbx1::cre; ChATfl/fl (mSOD1G93A/Coff)]. We show that, although the C-boutons do not influence the humane end point of mSOD1G93A mice, silencing the C-boutons significantly improves fast-twitch muscle innervation. By combining behavioral training with c-Fos immunohistology, we also found that the C-boutons are active in a task-dependent manner and that they are more active in mSOD1G93A mice than wild-type mice during slow walking, confirming that the C-boutons do indeed compensate for motor neuron loss by modulating the surviving neurons. Last, we demonstrate that C-bouton silencing combined with swimming is beneficial for the behavioral capabilities of mSOD1G93A mice. Manipulating the C-boutons in combination with a modulatory-targeted training program may therefore be beneficial for ALS patients.
Materials and Methods
All experiments were performed according to the Canadian Council on Animal Care guidelines and were approved by the Dalhousie University Committee on Laboratory Animals. The mice were housed on a 12 h light/dark cycle (light on from 07:00 A.M. to 7:00 P.M.) with access to laboratory chow and water ad libitum.
Animals.
Both male and female mice were used for all experiments. The following three mouse lines were used for experiments: wild-type (WT) C57BL/6 mice; mSOD1G93A mice (The Jackson Laboratory); mSOD1G93A; Dbx1::cre; ChATfl/fl (mSOD1G93A/Coff) mice, in which the C-boutons of mSOD1G93A mice are genetically silenced [the details of this mouse line were previously described (Zagoraiou et al., 2009); Dbx1::cre mice were obtained from the laboratory of Dr. Rob Brownstone at Dalhousie University, Brain Repair Center (Bielle et al., 2005); and ChATfl/fl mice were obtained from The Jackson Laboratory].
The humane end point of mSOD1G93A mice was determined by Dalhousie University animal care staff based on the health and ability of the mice, or whether the mice had lost 15% of their peak weight. Otherwise, behavioral mice were followed up until postnatal day 130 (P130) as this is when mSOD1G93A mice could no longer swim without significant intervention.
We verified the copy numbers in our mSOD1G93A and mSOD1G93A/Coff mice using quantitative PCR (qPCR). Experimental mSOD1G93A/Coff mice experienced only a 0.022 ± 0.174-fold decrease in mSOD1G93A gene expression relative to the mSOD1G93A mice, indicating that there was no difference in gene expression between these two groups of mice (Extended Data Fig. 5-1A,B).
Behavioral experiments.
Behavioral mice were divided into swimming and resting groups. Beginning at P30 to P33, swimming mice swam 3 d/week on Mondays, Wednesdays, and Fridays. The mice swam freely in a 30 × 3 × 7 cubic inch swimming pool with a starting temperature of 38°C and an end temperature of 31°C. The mice alternated between swimming for 4 min and resting for 1 min for half an hour. While resting, the mice were held in a heated paper towel that was replaced every 10 min. To ensure that the mice swam as much as possible, the back of the necks of the mice were poked with mouse holding forceps (catalog #NC1239918, Thermo Fisher Scientific) whenever the mice stopped swimming. Swimming mice were returned to their cages and placed on a heating pad for 15–30 min after swimming. Resting mice remained in their cages and did not swim. Experiments were stopped at P130 when mSOD1G93A swimming mice could no longer swim.
The swimming performance of the mice was scored based on the average number of prodding/interventions required per lap: score 10, no intervention; score 8, <2 prods per lap; score 6, 2–3 prods per lap; score 4, 4–5 prods per lap; score 2, >5 prods per lap; score 0, drowning behavior or early termination of experiment. Mice were placed between these scores (9, 7, 5, 3, 1) if there was high variability in performance and they could not easily be placed in one score or another.
Once a week and before swimming, both resting and swimming mice underwent behavioral recordings for their weight, maximum walking speed, balance on a rotarod, and grip force. Recordings were performed on either Mondays or Fridays depending on when the mice were born. The swimming mice were given at least a 2 h break after the behavioral recordings before swimming. Swimming performance did not fluctuate on the days that behavioral recordings were also recorded (Extended Data Fig. 5-1C–F).
The maximum speed of the mice was determined using a 25 × 6 × 16 cm3 treadmill (model 802, Zoological Institute, University of Cologne, Cologne, Germany). The treadmill speed was set to 0.1 m/s for the first trial and was gradually increased until the mouse lagged on the back wall of the treadmill. The speed was then reduced to the nearest 0.05 m/s interval, and the mice were given a short break before starting the next trial at the new initial speed. Trials were repeated until the start speed of the next trial was the same as the start speed of the previous trial. This speed was then recorded as the maximum. In cases where the mice could not start at a speed of 0.1 m/s, the speed was instead started at 0.05 m/s. If the mice could not walk at the initial speed for a trial, their maximum speed was recorded as the next lowest 0.05 m/s interval.
The performance of the mice on a rotating rod was determined using a commercial Rota Rod (Ugo Basile). The rotarod was programmed to start at 5 rpm and accelerate to 30 rpm over 120 s. The Rota Rod then remained at 30 rpm until the maximum time of 180 s. Three trials were performed for each mouse in which their time to fall was recorded. The best time of the three trials was used for analysis.
The grip force of the mice was determined using a grip strength apparatus (model GT3, BIOSEB) with the straight bar attachment. A single trial consisted of pulling the mice from the bar 5 to 13 times, depending on how well the mice behaved. The maximum force of the trial was then recorded. Five trials were conducted for each mouse, and the average of the five trials was used for analysis.
C-Fos experiments.
To induce c-Fos expression in interneurons, mice rested, walked, or swam. Resting mice were perfused immediately from their cages. Walking mice walked at 0.15 m/s on the same 25 × 6 × 16 cm3 treadmill described above. The mice alternated between walking for 4 min and resting for 1 min for 1 h. The mice remained on the motionless treadmill while resting. Swimming mice swam under the same protocol described above, but for 1 h instead of half an hour, with a final water temperature of 28°C. One hour after walking or swimming, the mice were perfused.
Tissue preparation.
The mice were deeply anesthetized with Euthanyl (100 µl sodium pentobarbital, 100 mg/kg, i.p.; Bimeda-MTC) and transcardially perfused with saline followed by 4% paraformaldehyde (PFA). Both legs were placed in PBS, while the vertebral column was postfixed in PFA overnight. The gastrocnemius (GS), soleus (Sol), and tibialis anterior (TA) muscles were then dissected, weighed, and cryoprotected in 30% sucrose overnight, while the spinal cord was dissected and cryoprotected overnight. The muscles, L2, L3-4, and L5 segments of the spinal cord were frozen in Optimal Cutting Temperature compound (Tissue-Tek, Sakura Finetek) and stored at −80°C. The muscles were longitudinally sectioned at 40 µm, and the spinal segments were transversely sectioned at 30 µm using a cryostat (model CM 3050S, Leica).
Muscle immunofluorescence.
Red fluorescent α-bungarotoxin conjugated to tetramethylrhodamine (BTX; 1:200, 5 µg/ml; catalog #T1175, Thermo Fisher Scientific) was used to label the nicotinic acetylcholine receptors on the postsynaptic site of the neuromuscular junction (NMJ), while goat anti-VAChT primary (1:1000; catalog #ABN100, Millipore Sigma) and donkey anti-goat secondary (1:1000; catalog #A32814, Thermo Fisher Scientific) were used to label the presynaptic axon terminals. NMJs were considered innervated if ≥20% of BTX-labeled postsynaptic terminals were colabeled with VAChT+ axon terminals, or denervated if <20% of BTX-labeled postsynaptic terminals were colabeled with VAChT+ axon terminals. NMJs were counted using a microscope (model DM LB2, Leica). Approximately 16 GS, 24 Sol, and 28 TA sections from each leg were used for analysis. Sections were placed on the slides sequentially such that each slide was a representation of the whole muscle. The proportion of innervation was determined for each muscle, and the average innervation between the left and right leg muscles of each mouse was used for analysis. If the muscle of only one leg was collected, then its innervation was used for analysis.
Spinal cord immunofluorescence.
Goat anti-ChAT (choline acetyltransferase) primary (1:250; catalog #AB144P, Millipore Sigma) and donkey anti-goat secondary (1:1000; catalog #A32814, Thermo Fisher Scientific) were used to putatively label the V0c interneurons, while rabbit anti-c-Fos primary antibody (1:1000; catalog #226003, Synaptic Systems) and donkey anti-rabbit secondary antibody (1:1000; catalog #A32794, Thermo Fisher Scientific) were used to label active interneurons. The putative V0c interneurons were counted as active if there was overlap between ChAT+ interneurons and c-Fos+ nuclei, or inactive if only ChAT+ interneurons were present. Sections were counted using a microscope (model DM LB2, Leica). All sections collected per mouse were used for analysis (200 ± 20 sections).
Experimental design and statistical analysis.
The number and sex of mice used for the behavioral experiments are as follows: WT rest three male (M) and one female (F); mSOD1G93A rest 2 M and 3F; mSOD1G93A swim 3 M and 2 F; mSOD1G93A/Coff swim 4 M and 7 F. The mice used for the behavioral experiments were mutually exclusive from those used during the c-Fos experiments. The experimental unit for all data presented is the mouse. For the behavioral data, the groups were statistically compared on a week-by-week basis. Male and female mice are separated for data presentation in all instances, though for the purposes of statistical analysis were pooled together. Differences between male and female mice were assessed at P130 for the mSOD1G93A/Coff swim mice for their weight and all three motor measurements. Significant differences between the male and female mice of this group are noted in text.
Power analyses were performed to determine the size and sufficiency of our sample sizes. Unless otherwise noted, all statistical tests involving two groups were performed using a t test, and all tests involving more than two groups were performed using a one-way ANOVA with a Tukey post hoc test. Whenever possible, tissue from multiple mouse lines were blinded and counted together. For spinal cord immunofluorescence, the samples were blinded among resting, walking, and swimming mice of the same mouse line. Immunofluorescent images were obtained using a laser-scanning confocal microscope (model LSM 710, Zeiss). Bar graphs are presented as the mean ± SD. Boxplots display lower and upper extremes, lower and upper quartiles, and medians. Significance was labeled as follows: *p < 0.05; **p < 0.01; ***p < 0.001.
Excel 2016 (Microsoft) and Graph Pad Prism 6.07 (GraphPad Software) were used for statistical analysis. Excel 2016, Graph Pad Prism 6.07, and Illustrator CS6 (Adobe) were used for data presentation.
Results
C-Boutons do not influence the humane end point but worsen fast-twitch muscle innervation in mSOD1G93A mice
Given that the C-boutons are necessary for behavioral compensation in mSOD1G93A mice (Landoni et al., 2019), we wanted to know whether the C-boutons influence overall disease progression. To do this, we compared the ages of mSOD1G93A and mSOD1G93A/Coff mice at their humane end points. Histologic assessment demonstrated widespread C-boutons in WT mice (Fig. 1A), whereas there was a total absence of ChAT at the sites of the C-boutons in the mSOD1G93A/Coff mice (Fig. 1B). qPCR also revealed that SOD1G93A gene copy expression was consistent between mSOD1G93A and mSOD1G93A/Coff mice (Extended Data Fig. 5-1A,B). The mean age at humane end point was 156.4 ± 7.3 d for the mSOD1G93A mice and 152.5 ± 7.9 d for the mSOD1G93A/Coff mice, which are in line with previously reported mSOD1G93A mice end points (Heiman-Patterson et al., 2011). We found no significant difference between the humane end points of these groups (ANOVA, p > 0.05; Fig. 1C), suggesting that the C-boutons do not significantly influence the humane end point of mSOD1G93A mice.
Figure 1.
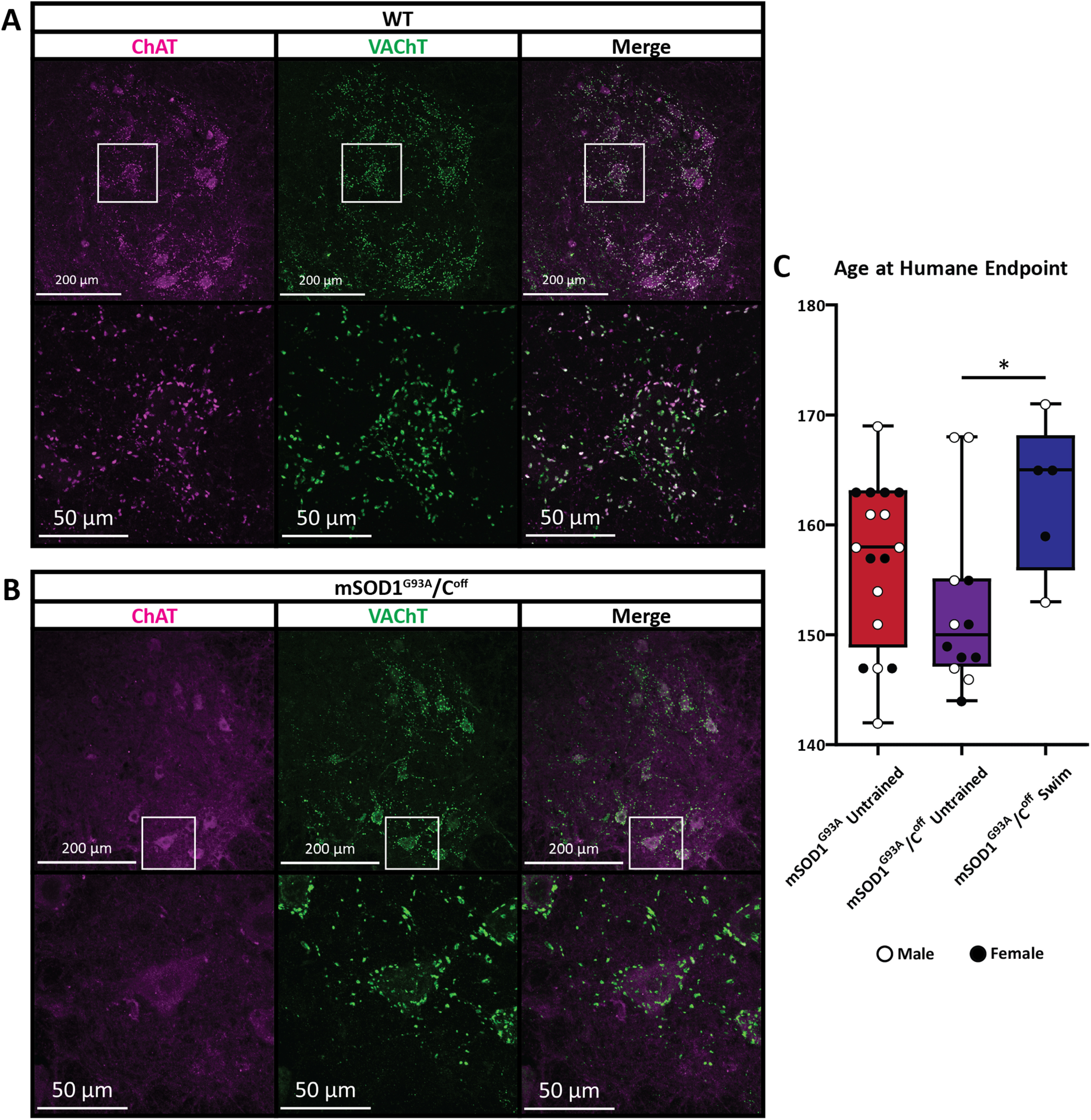
C-bouton silencing and humane end points of mSOD1G93A/Coff mice. A, B, The C-boutons are present in wild-type mice (A), but were completely silenced in mSOD1G93A/Coff mice (B), as shown by the absence of ChAT+ synapses to the motor neurons. C, The humane end points of mSOD1G93A and mSOD1G93A/Coff mice were determined by the Dalhousie University animal care staff based on the health and ability of the mice, or if the mice lost 15% of their peak weight. There were no significant differences in the age at the humane end point between untrained mSOD1G93A and mSOD1G93A/Coff mice (ANOVA, p > 0.05); however, mSOD1G93A/Coff mice that frequently swam reached the humane end point significantly later than their untrained mSOD1G93A/Coff counterparts (ANOVA with Tukey's test, p < 0.05). Boxplot displays lower and upper extremes, lower and upper quartiles, and median.
Despite these results, previous evidence we collected suggests that mSOD1G93A/Coff mice reach symptomatic stages of the disease sooner than mSOD1G93A mice (Landoni et al., 2019). This prompted us to investigate possible differences in muscle denervation between mSOD1G93A and mSOD1G93A/Coff mice, as silencing the C-boutons could worsen muscle innervation. We examined the GS, Sol, and TA muscle innervation of WT, mSOD1G93A, and mSOD1G93A/Coff mice across their life span (Fig. 2A). As expected, WT mouse muscle innervation remained near 100% for all muscles and ages examined. In contrast, the mSOD1G93A mice experienced a gradual decrease in endplate innervation beginning at approximately P45 for GS and TA, and P60 for the Sol muscles. Denervation progressed the fastest in the GS and TA as indicated by the steeper slope of the regression line (Fig. 2B,C; GS slope, −0.0048; TA slope, −0.0049), with Sol denervation progressing the slowest (Fig. 2D; slope, −0.0038). There was also a significant difference in the slope of the GS and TA when comparing the WT and mSOD1G93A mice, but not in the Sol (ANOVA: GS, p = 0.0003; TA, p = 0.0098; Sol, p = 0.106). These data support previous findings that denervation occurs sooner and faster in muscles with a higher proportion of type IIa and IIb fibers (GS and TA) relative to muscles made mostly of type I fibers (Sol; Kawamura et al., 1981; Frey et al., 2000; Hegedus et al., 2007).
Figure 2.
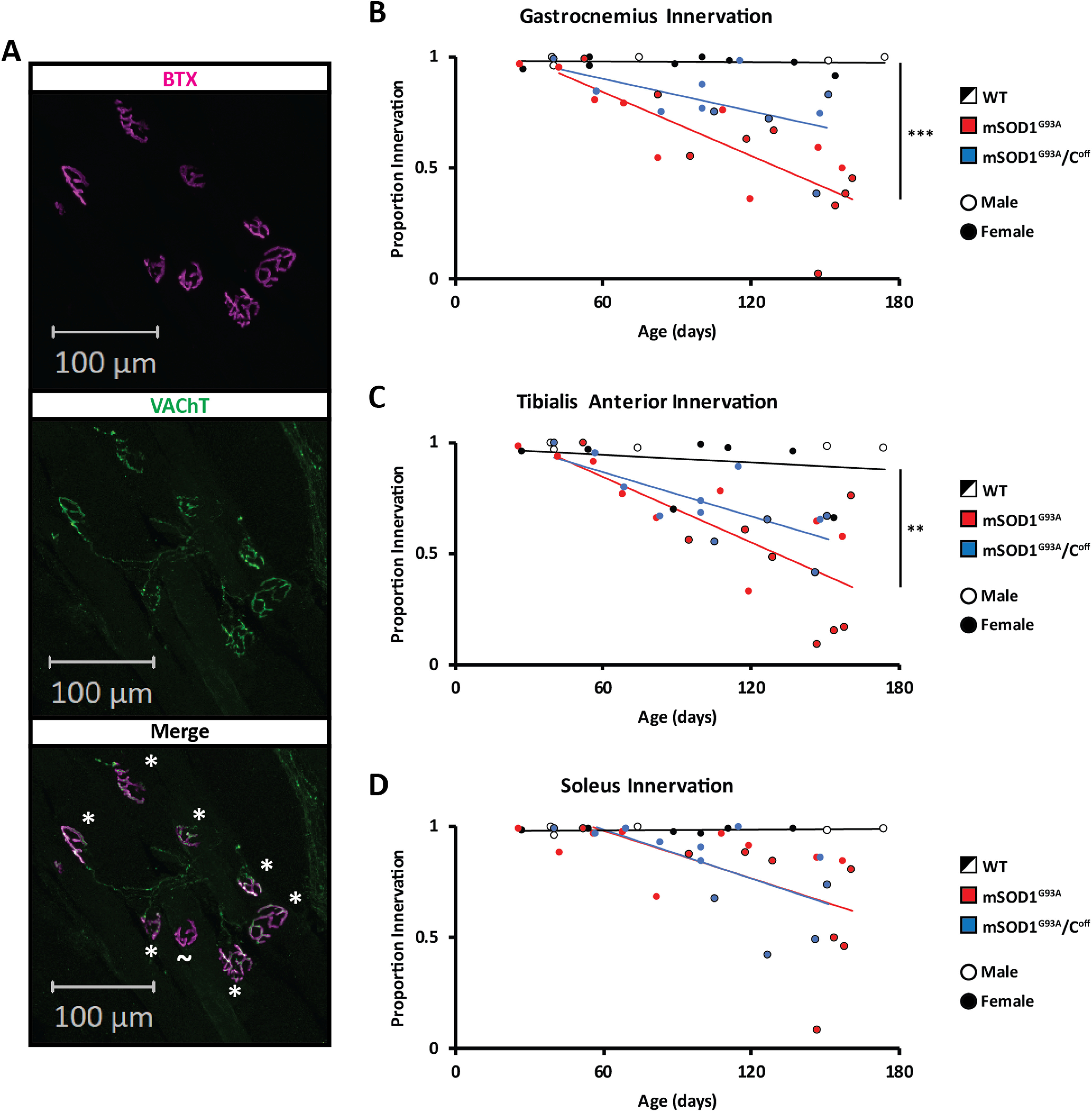
Muscle innervation in WT, mSOD1G93A, and mSOD1G93A/Coff mice. A, Muscle innervation was calculated based on the number of BTX+ postsynaptic terminals that did and did not have VAChT+ presynaptic terminals (* and ∼, respectively). B–D, The proportion of innervated NMJs was calculated for the GS (B), TA (C), and Sol (D) of WT, mSOD1G93A, and mSOD1G93A/Coff mice. NMJs were considered innervated if ≥20% of BTX-labeled postsynaptic terminals were colabeled with VAChT+ axon terminals or were denervated if <20% of BTX-labeled postsynaptic terminals were colabeled with VAChT+ axon terminals. The regression lines were calculated based on the beginning of denervation in the mSOD1G93A and mSOD1G93A/Coff mice, which we estimated to be approximately P45 for the GS and TA, and P60 for the Sol. Regression lines were compared with a linear regression comparison (ANOVA) using the StatistiXL extension in Excel. **p < 0.01, ***p < 0.001.
To investigate the effect of the C-boutons on muscle denervation, we then calculated the proportion of innervated endplates in mSOD1G93A/Coff mice and compared the results to the WT and mSOD1G93A data. The slope of the regression lines in both the GS (Fig. 2B; −0.0024) and TA (Fig. 2C; −0.0033) was higher in the mSOD1G93A/Coff mice than in the mSOD1G93A mice, though not significantly so (ANOVA: GS, p = 0.213; TA, p = 0.615). However, there was no significant difference between the GS and TA of the WT and mSOD1G93A/Coff mice, indicating that mSOD1G93A/Coff mice experience slower denervation in these muscles (ANOVA: GS, p = 0.196; TA, p = 0.227). The regression of the Sol muscle in both mSOD1G93A and mSOD1G93A/Coff mice was also similar (Fig. 2D; ANOVA: p = 0.977; mSOD1G93A/Coff slope: −0.0041), suggesting that the rate of denervation for the Sol muscle is not affected by the C-boutons. These observations suggest that silencing the C-boutons improves innervation in fast-twitch leg muscles such as the GS and TA, but not in slow-twitch leg muscles such as the Sol.
C-Bouton activity is upregulated during walking in symptomatic mSOD1G93A mice
Given that the C-boutons are detrimental to muscle innervation, we wanted to know whether frequent activation of the C-boutons would exacerbate behavioral deterioration in mSOD1G93A mice. To do this, we first wanted to confirm that the C-boutons are indeed active in a task-dependent manner, such that swimming elicits high C-bouton activation, as previous evidence suggests (Zagoraiou et al., 2009). We thus counted the proportion of active V0c interneurons that give rise to the C-boutons (Zagoraiou et al., 2009) using antibody staining against c-Fos following resting, slow walking at 0.15 m/s, and free swimming in WT mice (Fig. 3A–E; see Materials and Methods). We observed either clear c-Fos nuclear staining (Fos+) or no c-Fos staining (Fos–) in cholinergic neurons in the region of V0c neurons (Fig. 3A).
Figure 3.
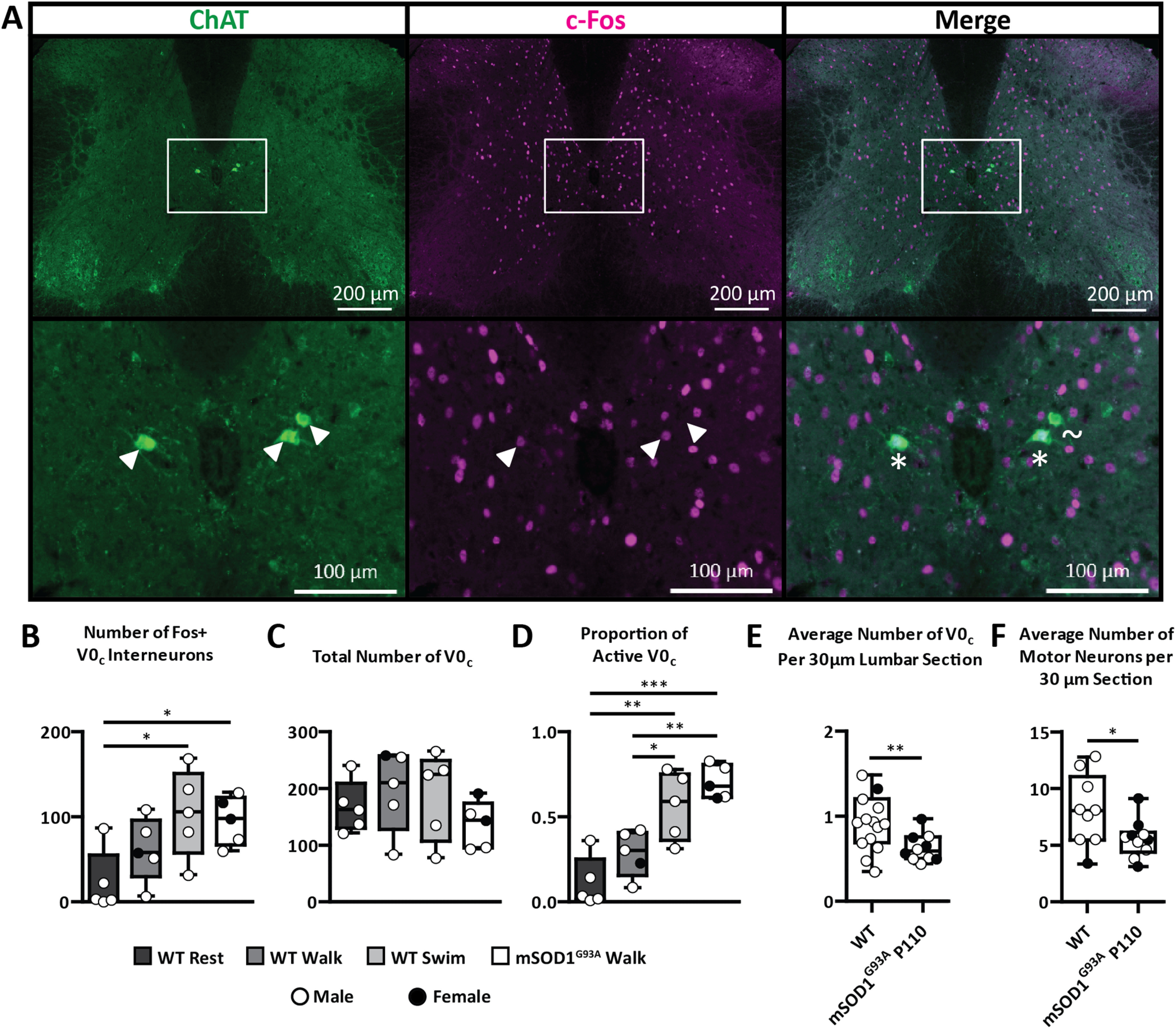
The number and proportion of active V0c interneurons following training in WT and mSOD1G93A mice. A, The number and proportion of active V0c interneurons following exercise were determined in WT and mSOD1G93A mice based on whether ChAT-labeled V0c interneurons did or did not contain c-Fos+ antibody staining in the nucleus (* and ∼, respectively). B–D, The number of Fos+ (B) and the total number of V0c (C) interneurons were used to calculate the proportion of active V0c interneurons in resting, walking, and swimming WT and mSOD1G93A mice (D). As task intensity increases, V0c interneuron activity also increases. There is also a significant increase in the proportion of active V0c interneurons during walking in symptomatic mSOD1G93A mice relative to WT mice. E, After standardizing for the number of spinal cord sections counted, there was a significant difference between the number of V0c interneurons in WT and mSOD1G93A mice at P110 (Student's t test, p = 0.0056). F, There was also a significant reduction in the number of motor neurons in mSOD1G93A mice at P110 relative to WT mice (Student's t test, p = 0.0388). Boxplots display lower and upper extremes, lower and upper quartiles, and median. White arrowheads are used to indicate the putative V0c and their location. *p < 0.05, **p < 0.01, ***p < 0.001.
The number of Fos– and Fos+ (Fig. 3B) V0c interneurons were counted and used to determine the total number (Fig. 3C) and proportion of active V0c interneurons (Fig. 3D) in the WT and mSOD1G93A mice. On average, only 11 ± 15% of all V0c neurons were active in resting WT mice. After 1 h of slow walking, the proportion of active V0c interneurons was 29 ± 14% in WT mice, which was not statistically significant (ANOVA, p > 0.05). In contrast, 56 ± 20% of V0c interneurons were active in WT mice after 1 h of free swimming. This was significantly higher than after resting (ANOVA, p < 0.01) and walking (ANOVA, p < 0.05). This observation provides direct evidence that V0c activity is modulated in a task-dependent manner, supporting the conclusion that C-boutons are involved in task-dependent amplitude modulation (Zagoraiou et al., 2009).
Given that the C-boutons maintain motor behavior by compensating for motor neuron loss (Landoni et al., 2019), we reasoned that the C-boutons must be more active in symptomatic mSOD1G93A mice than in WT mice during low-intensity exercise. To investigate this, we put symptomatic mSOD1G93A mice through the same walking experiment (Fig. 3). There were no immediate differences in the total number of V0c interneurons between any of the groups (ANOVA, p > 0.05; Fig. 3C). Proportionally, while the WT walk mice showed c-Fos expression in 29 ± 14% of the V0c interneurons, symptomatic walking mSOD1G93A mice showed a significantly higher proportion of c-Fos expression in the V0c interneurons at 70 ± 10% (ANOVA, p < 0.01; Fig. 3D). This upregulation supports our previous hypothesis that the C-boutons are responsible for behavioral compensation during disease progression in mSOD1G93A mice (Landoni et al., 2019).
There is a reduction in the number of V0c interneurons in symptomatic mSOD1G93A mice, which correlates with motor neuron loss
Recent evidence suggests that spinal interneurons are lost during ALS disease progression (Salamatina et al., 2020). Moreover, disruption to the cholinergic system can lead to the phagoptosis, and thus loss, of the V0c interneurons (Jiang et al., 2018). Determining whether mSOD1G93A mice also experience a loss of V0c interneurons is therefore beneficial for understanding the relationship between the C-boutons and ALS and could reveal a natural mechanism by which to target the C-boutons therapeutically. We thus compared the number of V0c interneurons in WT and middle stage mSOD1G93A mice aged P110. Since there were differences in the number of spinal cord sections counted per mouse, the number of V0c interneurons we counted was normalized to the number of sections counted per mouse (Fig. 3E). The mean number of V0c interneurons per 30 µm section was 0.93 ± 0.32 in WT mice and 0.63 ± 0.16 in symptomatic mSOD1G93A mice, representing a 32.3% reduction in V0c interneurons. An F test (Snedecor and Cochran, 1989) determined that there was a significant difference in variance between these two groups (p = 0.0446). A t test was thus performed with Welch's correction, which revealed a significant reduction in V0c interneurons in the mSOD1G93A mice relative to the age-matched WT mice (p = 0.0056).
We were then interested to know what the relationship was between V0c interneuron loss and motor neuron loss. We thus counted and compared the number of motor neurons in the same WT and mSOD1G93A mice (Fig. 3F). The mean number of motor neurons per 30 µm section was 8.09 ± 3.12 in WT mice and 5.56 ± 1.65 in symptomatic mSOD1G93A mice, representing a 31.2% reduction, which was statistically significant (t test, p = 0.0388). V0c and motor neuron loss is therefore tightly correlated at the middle stage of the disease, given their similar percentage loss (32.3% and 31.2%, respectively). This correlation could also further indicate a neurologic relationship or connection between V0c and motor neuron loss.
Training toward amplitude modulation may worsen overall ALS progression
Since we found the C-boutons to be most active during swimming, and since the C-boutons were previously found to be involved in the task-dependent modulation of motor neurons (Zagoraiou et al., 2009), we sought to determine whether frequent activation of the C-boutons via swimming would exacerbate behavioral deterioration in mSOD1G93A mice. We trained mSOD1G93A mice toward amplitude modulation via frequent swimming (mSOD1G93A swim) and compared their weight (Fig. 4) and behavioral capabilities (Fig. 5) over time to those in resting WT and mSOD1G93A mice (WT rest and mSOD1G93A rest, respectively; see Materials and Methods). At P130, the mSOD1G93A swim mice rarely swam and spent most of the training time floating (Extended Data Fig. 5-1G). P130 was thus chosen to be the end age for these experiments.
Figure 4.
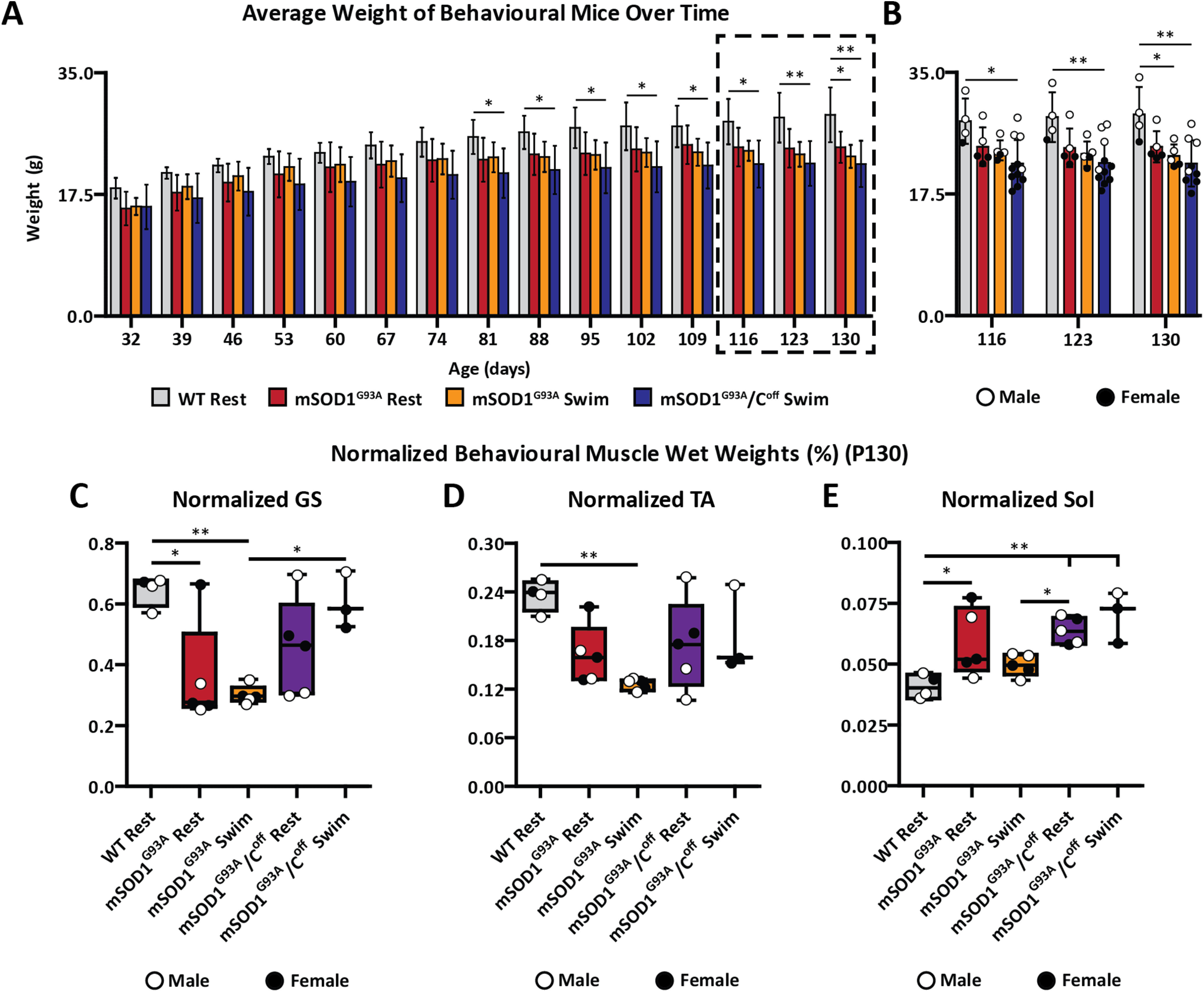
Training toward amplitude modulation in the absence of C-boutons significantly worsens gross weight in mSOD1G93A mice, but significantly improves GS muscle weights. A, B, mSOD1G93A swim mice weighed significantly less than WT mice at P130 (A), whereas the mSOD1G93A/Coff swim mice weighed significantly less than WT mice beginning at approximately P81 (B). C–E, C-bouton silencing paired with swimming resulted in mSOD1G93A/Coff GS muscles that made up a significantly higher proportion of the total mouse weight than mSOD1G93A swimming mice (C), but not for the TA (D) or Sol (E). Bar graphs display the mean ± SD. Boxplots display lower and upper extremes, lower and upper quartiles, and median. *p < 0.05, **p < 0.01.
Figure 5.
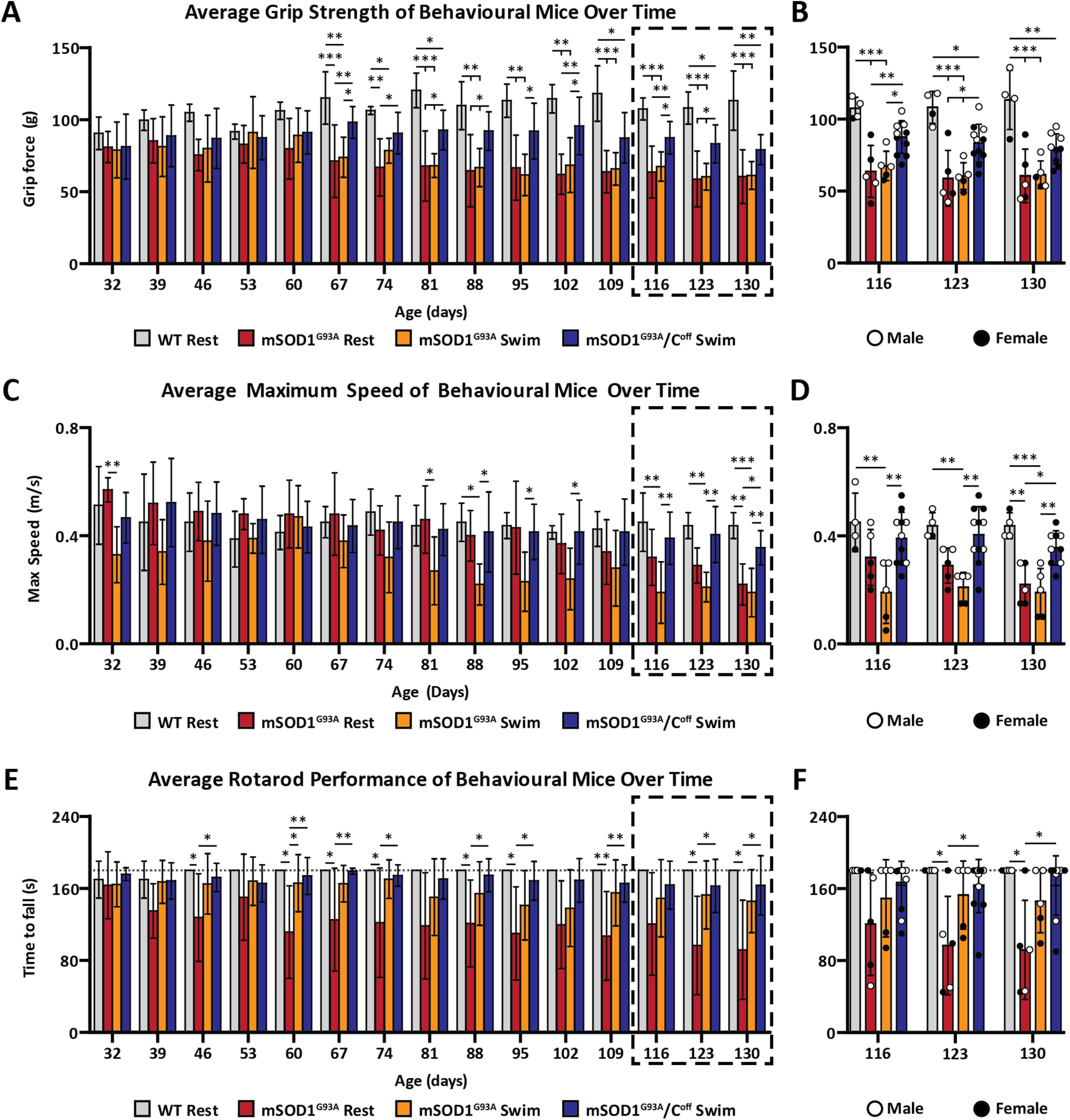
Training toward amplitude modulation in the absence of C-boutons significantly improves behavioral performance in mSOD1G93A mice. A, B, WT mice are significantly stronger than mSOD1G93A mice beginning at approximately P67 but are only significantly stronger than mSOD1G93A/Coff swim mice beginning at P109. mSOD1G93A/Coff mice also perform significantly better than their mSOD1G93A counterparts beginning at P67, though this difference is not present at P130. C, D, mSOD1G93A/Coff swim mice are significantly faster than their mSOD1G93A counterparts beginning at approximately P116 and never perform significantly worse than WT mice at all ages examined. E, F, mSOD1G93A/Coff swim mice outperformed the mSOD1G93A rest mice on the rotarod beginning at approximately P123. See Extended Data Figure 5-1 for data regarding qPCR results and swim performance. Bar graphs display the mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001.
qPCR results and swim performance. A, qPCR revealed tight overlap between the SOD1G93A gene amplification sigmoidal curves of the mSOD1G93A and mSOD1G93A/Coff mice (left cluster) relative to the housekeeping gene (right cluster). B, 2^ΔΔCt was calculated to be 0.978 ± 0.174 for the mSOD1G93A/Coff mice relative to the mSOD1G93A mice, translating to a 0.022 ± 0.174-fold decrease in SOD1G93A gene expression and indicating that there was no difference in gene expression between these two groups of mice. C–G, The swim performance of the mice was scored based on the average number of prodding/interventions required per lap: score 10, no intervention; score 8, <2 prods per lap; score 6, 2–3 prods per lap; score 4, 4–5 prods per lap; score 2, >5 prods per lap; score 0, drowning behavior or early termination of experiment. Mice were placed between these scores (9, 7, 5, 3, 1) if there was high variability in performance and they could not easily be placed in one score or another other. An example of male and female performance is given for the mSOD1G93A mice (C, D) and mSOD1G93A/Coff mice (E, F), where the red dots indicate days that motor performance measurements were obtained at least 2 h prior to swimming. G, Average swim performance of the mSOD1G93A and mSOD1G93A/Coff mice. mSOD1G93A/Coff mice required consistently and significantly fewer interventions than their mSOD1G93A counterparts beginning at P120. Bar graphs display the mean ± SD. p < 0.05, p < 0.01, p < 0.001. Download Figure 5-1, TIF file (12.4MB, tif) .
Naturally, the WT mice gradually gained weight over time, beginning with an average weight of 18.4 ± 1.5 g at P32 and ending with an average weight of 29.0 ± 4.0 g at P130 (Fig. 4A,B). Beginning at P32, the mSOD1G93A rest mice weighed, on average, 15.5 ± 2.4 g, a weight that rose sharply until approximately P60 (21.4 ± 3.6 g), after which it increased slowly until P130 (24.7 ± 2.4 g). At P32, the mSOD1G93A swim mice weighed, on average, 15.8 ± 1.2 g, a weight that sharply rose until approximately P60 (21.8 ± 2.5 g). The weight of the mSOD1G93A swim mice did not rise as quickly as the mSOD1G93A rest mice after P60, however, and these mice even experienced a small decline in weight beginning at P116 (Fig. 4B). This resulted in a significantly lower weight than the WT mice at P130 (23.0 ± 1.6 g; ANOVA, p < 0.05. Since there were no significant differences between the WT and mSOD1G93A rest mice, these findings suggest that training toward amplitude modulation may worsen the health of mSOD1G93A mice at middle to late stages of the disease.
To further investigate the influence of training toward amplitude modulation on mSOD1G93A mice, all the behavioral mice were perfused at approximately P130, and their muscles were dissected and weighed (Fig. 4C–E). Given that the mSOD1G93A swim mice weighed significantly less than the WT rest mice at P130, it is possible that the raw muscle weights underestimate the true differences between these groups. To account for this, we normalized the weight of each muscle to the weight of the mice they were obtained from. After normalizing, we observed significant differences between the GS of the WT and mSOD1G93A swim mice and between the WT and mSOD1G93A rest mice (ANOVA: p < 0.01 and p < 0.05, respectively; WT rest, 0.646 ± 0.051%; mSOD1G93A rest, 0.361 ± 0.173%; mSOD1G93A swim, 0.302 ± 0.030%; Fig. 4C). There was also a significant difference in the normalized weight of the TA of the WT and mSOD1G93A swim mice (ANOVA, p < 0.01), but not in that of the WT rest and mSOD1G93A rest mice (ANOVA, p > 0.05), indicating that frequent swimming brought the weight of the TA further from that of a healthy mouse (WT rest, 0.236 ± 0.019%; mSOD1G93A rest, 0.162 ± 0.037%; mSOD1G93A swim, 0.126 ± 0.007%; Fig. 4D). Interestingly, the normalized weight of the Sol in the mSOD1G93A rest mice was significantly higher than that of the WT mice (WT rest, 0.041 ± 0.005%; mSOD1G93A rest, 0.059 ± 0.014%; mSOD1G93A swim, 0.050 ± 0.004%; ANOVA, p < 0.05; Fig. 4E). Given that the Sol is known to be resistant to ALS disease progression (Gurney et al., 1994; Frey et al., 2000; Hegedus et al., 2007; Valdez et al., 2012), this finding is behaviorally insignificant and an artifact resulting from normalization. Overall, these findings indicate that training toward amplitude modulation likely worsened the weights of the GS and TA.
Training toward amplitude modulation in the absence of the C-boutons improves fast-twitch muscle weight during ALS progression
To verify that the differences we observed between the WT and mSOD1G93A rest and swim mice were indeed the result of higher C-bouton activity, we put mSOD1G93A/Coff mice through the same swimming experiments (mSOD1G93A/Coff swim; Figs. 4, 5). Remarkably, the mSOD1G93A/Coff swim mice performed much better than expected. Of the 11 mice used, 2 were stopped at P123 and used for other experiments (not included here). The remaining nine mice continued swimming until P130 and could all swim on their own with minimal encouragement despite lacking C-bouton modulation. Consistently significant improvements in swimming performance over their mSOD1G93A counterparts began at P120 (Student's t test; p < 0.001) and continued until P130. By comparison, all five mSOD1G93A swim mice spent most of the training time floating at P130, despite frequent encouragement (Extended Data Fig. 5-1C–G, Movie 1).
The mSOD1G93A swim mice spent most of their time floating at P130, whereas the mSOD1G93A/Coff mice swam with minimal encouragement. A, B, A male mSOD1G93A swim mouse (A) and a male mSOD1G93A/Coff swim mouse (B) at P130 demonstrating the difference in quality of swimming between these strains. Videos were captured at 500 frames/s with 4× playback speed. The distance between the calibration dots measures 1.5 cm.
The mSOD1G93A/Coff swim mice tended to gain weight slower than their mSOD1G93A counterparts (Fig. 4A,B). At P32, the mSOD1G93A/Coff swim mice weighed 15.7 ± 3.2 g and increased steadily in weight until P130 (21.9 ± 3.3 g). Because the mSOD1G93A/Coff swim mice gained weight slower, there were significant differences when compared with the WT rest mice beginning as early as P81 (ANOVA, p < 0.05). There were, however, no significant differences in the weight of any of the mSOD1G93A groups at any age (ANOVA, p > 0.05). Unsurprisingly, the male mSOD1G93A/Coff mice weighed significantly more than their female counterparts at P130 (Student's t test, p = 0.0103). These findings indicate that C-bouton silencing, in combination with swimming, worsened the gross weight of mSOD1G93A mice, and that male and female mSOD1G93A/Coff mice display similar weight differences to those of WT and mSOD1G93A mice previously reported (Oliván et al., 2015).
Since the mSOD1G93A/Coff swim mice weighed significantly less than the WT mice as early as P81, we wanted to know whether their muscles also weighed less. The mSOD1G93A/Coff swim mice were therefore perfused, and their muscles were dissected and weighed following the final experiments at P130 (Fig. 4C–E). As before, we normalized the weight of the mSOD1G93A/Coff swim muscles to the weight of the mice they were dissected from. After normalizing, the GS of the mSOD1G93A/Coff swim mice made up significantly more of the weight of mice than those of the mSOD1G93A swim mice (ANOVA, p < 0.05) and was similar to that of the WT rest mice (ANOVA, p > 0.05; WT rest, 0.646 ± 0.051%; mSOD1G93A rest, 0.361 ± 0.173%; mSOD1G93A swim, 0.302 ± 0.030%; mSOD1G93A/Coff swim, 0.606 ± 0.094%; Fig. 4C). No differences were observed between the TA of the mSOD1G93A/Coff swim mice and that of any other group (ANOVA, p > 0.05; WT rest, 0.236 ± 0.019%; mSOD1G93A rest, 0.162 ± 0.037%; mSOD1G93A swim, 0.126 ± 0.007%; mSOD1G93A/Coff swim, 0.187 ± 0.054%; Fig. 4D). Though the Sol of the mSOD1G93A/Coff swim mice weighed the most after normalizing and significantly more than that of the WT mice (ANOVA, p < 0.01), this is again behaviorally insignificant because of the nature of the Sol muscle (WT rest, 0.041 ± 0.005%; mSOD1G93A rest, 0.059 ± 0.014%; mSOD1G93A swim, 0.050 ± 0.004%; mSOD1G93A/Coff swim, 0.070 ± 0.010%; ANOVA, p > 0.05; Fig. 4E). These findings suggest that training toward amplitude modulation alone lead to lower fast-twitch muscle weights in the mSOD1G93A mice, and that training toward amplitude modulation in the absence of C-boutons not only rescued this reduction but improved the weights of fast-twitch muscles in mSOD1G93A mice.
Given that mSOD1G93A/Coff mice in general have higher fast-twitch innervation over time relative to mSOD1G93A mice, we wanted to verify that this finding was the result of both training and C-bouton silencing, rather than C-bouton silencing alone. Last, we perfused untrained mSOD1G93A/Coff mice at P130 (mSOD1G93A/Coff rest) and dissected and weighed their muscles. There were no significant differences between the normalized weights of the GS (Fig. 4C) and TA (Fig. 4D) of the mSOD1G93A/Coff rest mice relative to the other groups (ANOVA, p > 0.05; GS: mSOD1G93A/Coff rest, 0.454 ± 0.162%; mSOD1G93A/Coff swim, 0.606 ± 0.094%; TA: mSOD1G93A/Coff rest, 0.174 ± 0.056%; mSOD1G93A/Coff swim, 0.187 ± 0.054%). As expected, the weight of the Sol was significantly higher in the mSOD1G93A/Coff rest mice relative to the WT mice (ANOVA, p < 0.01), though behaviorally insignificant (mSOD1G93A/Coff rest, 0.064 ± 0.005%; mSOD1G93A/Coff swim, 0.070 ± 0.011%). Since the mSOD1G93A/Coff swim mice, but not the mSOD1G93A/Coff rest mice, had significantly higher GS weights than the mSOD1G93A swim mice, these findings suggest that training toward amplitude modulation has a minor detriment to fast-twitch muscle weights in mSOD1G93A mice, but when combined with C-bouton silencing has a beneficial effect on fast-twitch muscle weight, specifically regarding the GS.
Training toward amplitude modulation alters mSOD1G93A mouse behavior
To determine whether training toward amplitude modulation exacerbates the behavioral deterioration of mSOD1G93A mice, we put the WT rest, mSOD1G93A rest, and mSOD1G93A swim mice through weekly tests of their grip strength, maximum speed, and rotarod performance (Fig. 5A–F). Unsurprisingly, the grip strength of the WT mice gradually increased over time, while that of the mSOD1G93A rest and swim mice gradually decreased (Fig. 5A,B). Though both mSOD1G93A groups began performing significantly worse than the WT mice beginning at only P67, we found no significant differences between the grip forces of the mSOD1G93A rest and swim mice at any age (ANOVA, p > 0.05). Both mSOD1G93A groups performed equally well at P32 (mSOD1G93A rest, 81.0 ± 10.8 g; mSOD1G93A swim, 79.0 ± 19.3 g) and decreased at a near-identical rate until P130 (mSOD1G93A rest, 60.5 ± 18.7 g; mSOD1G93A swim, 61.3 ± 9.7 g), suggesting that training toward amplitude modulation did not influence the strength of the mSOD1G93A mice.
The WT rest mice were consistently capable of locomoting at a maximum speed of at least 0.4 m/s during most of our experiments (Fig. 5C,D). The mSOD1G93A rest mice, however, had an initial maximum speed of 0.57 ± 0.04 m/s at P32, which decreased steadily until P130 (0.22 ± 0.08 m/s). Interestingly, the maximum speed of the mSOD1G93A swim mice was significantly lower than the mSOD1G93A rest mice at P32 (mSOD1G93A rest, 0.57 ± 0.04 m/s; mSOD1G93A swim, 0.33 ± 0.10 m/s; ANOVA, p < 0.01), likely since the mSOD1G93A mice swam at P30, while their first behavioral test was at P32. Their performance nonetheless improved each week until P60 when the maximum speeds of the two groups were nearly identical (mSOD1G93A rest, 0.48 ± 0.13 m/s; mSOD1G93A swim, 0.47 ± 0.12 m/s; ANOVA, p > 0.05). Unlike the mSOD1G93A rest mice, the maximum speed of the mSOD1G93A swim mice decreased rapidly from P60 to P88, after which their maximum speed remained constant until the end of the experiments at P130 (0.19 ± 0.09 m/s). This trend resulted in significant differences between the mSOD1G93A rest and swim groups at P88 (ANOVA, p < 0.05). The mSOD1G93A swim mice also displayed qualitative differences in walking behavior relative to the mSOD1G93A rest mice at P95, most notably the dragging of the backside, which likely influenced these results (Movie 2). Despite this, the rest and swim mice had similar maximum speeds at P130 (mSOD1G93A rest, 0.22 ± 0.08 m/s; mSOD1G93A swim, 0.19 ± 0.09 m/s; ANOVA, p > 0.05). Consistently significant differences between the mSOD1G93A groups and the WT mice only began at P116 (WT rest, 0.45 ± 0.10 m/s; mSOD1G93A rest, 0.32 ± 0.10 m/s; mSOD1G93A swim, 0.19 ± 0.11 m/s). These findings suggest that training toward amplitude modulation impaired the maximum speed of the mSOD1G93A swim mice.
The mSOD1G93A/Coff swim mice never displayed the backside dragging that the mSOD1G93A swim mice did at P95, even when assessed at P129. A–C, Treadmill walking of a male mSOD1G93A rest mouse (A), and swim mouse (B) at P95, and a male mSOD1G93A/Coff swim mouse at P129 (C). Note that the mSOD1G93A swim mouse displays backside dragging at P95 that the mSOD1G93A rest mouse does not. We did not observe this phenotype in the mSOD1G93A/Coff swim mice, even when assessed at P129. Videos were captured at 500 frames/s with 4× playback speed. The distance between the calibration dots measures 1.5 cm.
Unsurprisingly, almost all the WT mice reached the maximum trial time of 180 s on the rotarod every week (Fig. 5E,F). The mSOD1G93A swim mice, however, performed better than the mSOD1G93A rest mice on the rotarod. The mSOD1G93A rest mice lasted 163 ± 37 s on the rotarod at P32, and gradually worsened until P130 (92 ± 55 s). The mSOD1G93A swim performance was like that of the mSOD1G93A rest mice at P32, with a time to fall of 164 ± 25 s. Their performance, however, decreased at a much slower rate than the mSOD1G93A rest mice, with a time of fall of 146 ± 35 s at P130. Despite this, there were no significant differences between these groups at any age because of the variance in the mSOD1G93A rest data (ANOVA, p > 0.05). Only one of the five mSOD1G93A rest mice reached the maximum time of 180 s at P130, whereas two of the five mSOD1G93A swim mice reached maximum time at P130.
Training toward amplitude modulation has a beneficial effect on mSOD1G93A mouse behavior if C-boutons are silenced
To verify that the behavioral differences we observed between the mSOD1G93A rest and swim mice were indeed the result of frequent C-bouton activation, we also recorded the grip force, maximum speed, and rotarod performance of the mSOD1G93A/Coff swim mice over time (Fig. 5A–F). Amazingly, the mSOD1G93A/Coff swim mice performed well above the capabilities of the mSOD1G93A mice. At P32, the grip strength of the mSOD1G93A/Coff swim mice was, on average, 81.2 ± 22.5 g, which improved steadily until P60 (91.1 ± 15.0 g; Fig. 5A,B). At P67, these mice experienced a spike in grip strength (98.1 ± 11.1 g), which was significantly more than the mSOD1G93A rest and swim mice (ANOVA: p < 0.01 and p < 0.05 respectively). This significant improvement lasted until P123, with sporadic changes in statistical confidence (notably, P109 when significance was not present between these groups). Their strength then decreased only slightly by P130 (79.1 ± 10.5). Interestingly, the male mice from this group were significantly stronger than their female counterparts at P130 (Student's t test, p = 0.0078). By comparison, the grip strengths of the mSOD1G93A rest and swim mice were only 60.5 ± 18.7 and 61.3 ± 9.7 g at P130, respectively. Significant differences between the WT and mSOD1G93A/Coff swim began at approximately P109 (ANOVA, p < 0.05). In contrast, significant differences between the WT and mSOD1G93A mice began 42 d earlier at P67. Unexpectedly, these findings suggest that training toward amplitude modulation in the absence of C-boutons significantly improves the strength of SOD1G93A mice.
The maximum speed of the mSOD1G93A/Coff swim mice barely deteriorated over time (Fig. 5C,D). At P32, their maximum speed was like that of the WT mice (WT, 0.51 ± 0.14 m/s; mSOD1G93A/Coff swim, 0.47 ± 0.09 m/s), and only decreased slightly by P130 (0.36 ± 0.06 m/s). No significant differences were observed between the male and female mice of this group at P130 (Student's t test, p > 0.05). It is interesting that none of the nine mSOD1G93A/Coff mice examined at P130 walked below a comfortable walking speed of 0.2 m/s, whereas three of the five mice in each of the mSOD1G93A groups could only walk at or below this speed at P130. Qualitatively, the mSOD1G93A/Coff swim mice did not display the backside dragging to the extent that the mSOD1G93A swim mice did at P95, even when assessed 34 d later at P129 (Movie 2). Since the speed of their mSOD1G93A counterparts deteriorated much faster, the mSOD1G93A/Coff swim mice began performing better than the mSOD1G93A rest mice at P130 (mSOD1G93A rest, 0.22 ± 0.08 m/s; mSOD1G93A/Coff swim, 0.36 ± 0.06 m/s; ANOVA, p < 0.05), with significant improvements over the mSOD1G93A swim mice beginning at P116 (mSOD1G93A swim, 0.19 ± 0.11 m/s; mSOD1G93A/Coff swim, 0.39 ± 0.10 m/s; ANOVA, p < 0.01). Of note is that the mSOD1G93A/Coff swim mice never performed significantly worse than the WT mice. Given that the drop in maximum speed of untrained mSOD1G93A/Coff mice has previously been shown to occur sooner than in mSOD1G93A mice (Landoni et al., 2019), these results suggest that training toward amplitude modulation in the absence of C-boutons helps to maintain locomotor capability over time.
At P32, the mSOD1G93A/Coff swim mice performed similarly to their mSOD1G93A counterparts on the rotarod (mSOD1G93A/Coff swim, 175.8 ± 7.1 s); however, the mSOD1G93A/Coff mice never deteriorated as the mSOD1G93A mice did (Fig. 5E,F). Remarkably, six of the nine mSOD1G93A/Coff swim mice that continued swimming at P130 reached the maximum trial time of 180 s, resulting in a score of 163.3 ± 33.1 s. By comparison, only two of the five mSOD1G93A swim mice, and one of the mSOD1G93A rest mice achieved this. Though there were no significant differences between the mSOD1G93A/Coff swim and mSOD1G93A swim mice at any age (ANOVA, p > 0.05), the mSOD1G93A/Coff swim mice began performing significantly better than the mSOD1G93A rest group at P123 (ANOVA, p < 0.05). Like with their maximum speed, we never observed significant differences between the performance of the WT and mSOD1G93A/Coff mice. Given that most mice in the mSOD1G93A/Coff swim group reached the maximum trial time every week, we were unable to determine whether training toward amplitude modulation in the absence of C-boutons had a significant influence on rotarod performance. We also did not detect significant differences between the male and female mice of this group at P130 (Student's t test; p > 0.05), though this may have been the result of the maximum trial time. Recent evidence suggests that C-bouton silencing alone improves rotarod performance (Konsolaki et al., 2020). In line with this evidence, our findings nonetheless suggest that silencing C-boutons in mSOD1G93A mice significantly improves rotarod performance relative to untrained mSOD1G93A mice.
Training toward amplitude modulation in mSOD1G93A/Coff improves humane end point over untrained mSOD1G93A/Coff mice, but not over untrained mSOD1G93A mice
Given the significant improvement in performance of the mSOD1G93A/Coff swim mice over their mSOD1G93A counterparts, we last wanted to know whether this increase in behavioral performance translated to an increase in life span. Five of the 11 mSOD1G93A/Coff swim mice thus continued swimming beyond P130 until they could no longer swim, and then lived until the humane end point. On average, the mSOD1G93A/Coff mice were able to swim until P150.4 ± 10.9 d, thus being able to swim for ∼20 d more than their mSOD1G93A counterparts. The mSOD1G93A/Coff mice then reached a humane end point ∼12 d later at P162.6 ± 6.8 d (Fig. 1C). This life span was significantly longer than that of the untrained mSOD1G93A/Coff mice by ∼10 d, or a 6.6% longer life span (152.5 ± 7.9 d; ANOVA, p < 0.05), but not of the untrained mSOD1G93A mice (156.4 ± 7.3 d), which was only increased by ∼8 d, or 3.9% (ANOVA; p > 0.05). These findings therefore indicate that training toward amplitude modulation in the absence of C-boutons significantly improves life span over untrained mSOD1G93A mice that lack C-boutons, but not over untrained mSOD1G93A mice with C-boutons.
Discussion
Here, we show that C-bouton silencing, in combination with training toward amplitude modulation, significantly improves life span over untrained mSOD1G93A mice with silenced C-boutons, but not over untrained mSOD1G93A mice. The presence of C-boutons also significantly worsens fast-twitch muscle innervation over time. We also show that the V0c interneurons, and thus C-boutons, are active in a task-dependent manner, and that the V0c interneurons in symptomatic mSOD1G93A mice show a significantly higher proportion of c-Fos activity than those of wild-type mice during low-intensity walking. Lastly, we provide evidence that C-bouton silencing in combination with high-intensity modulatory-targeted training worsens gross weight but improves fast-twitch muscle weight and is beneficial for the behavioral capabilities of mSOD1G93A mice. Our observations suggest that manipulating the C-boutons in combination with a modulatory-targeted training program may therefore be a beneficial therapy for ALS patients.
mSOD1G93A mice experience V0c interneuron loss at middle stage of ALS
Recent evidence regarding V2a interneurons loss in ALS has suggested that the depletion of direct connectivity to motor neurons, or in other words the failure for the V2a interneurons to signal downstream, is what drives V2a loss (Ravits, 2014; Salamatina et al., 2020). The V0c interneuron loss we detected here likely occurs through a similar mechanism. This hypothesis is also supported by our finding that the percentage loss of the V0c and motor neurons are tightly correlated (Fig. 3E,F), indicating that motor neuron loss may, indeed, be a driving factor for V0c loss. Despite conflicting evidence in the literature regarding the number and size of C-boutons on motor neurons over time in ALS (Pullen and Athanasiou, 2009; Herron and Miles, 2012; Gallart-Palau et al., 2014), evidence suggests that the density of C-boutons on surviving motor neurons in mSOD1G93A mice does not reduce over time (Lobsiger et al., 2013; Dukkipati et al., 2017). V0c interneuron loss is thus unlikely to precede the death of the motor neurons they project to. In such a case, the loss of the V0c interneurons would not directly influence disease progression. This hypothesis could also explain the conflicting evidence regarding C-bouton density changes in ALS; if V0c interneurons die because most of their C-boutons synapse onto dead motor neurons, then we would expect some loss in C-bouton density, as the dead V0c interneurons were likely still synapsing onto some surviving motor neurons (Ravits, 2014; Salamatina et al., 2020).
How could V0c interneuron loss be occurring? Evidence suggests that the V0c interneurons undergo phagoptosis following neuronal C1q expression when descending projections to the V0c interneurons are disrupted (Brown and Neher, 2012; Jiang et al., 2018). Interestingly, evidence also suggests that C1q-silencing in mSOD1G37R mice results in the loss of C-boutons at the end-stage of ALS (Lobsiger et al., 2013). If the V0c loss we illustrated here is indeed driven by C1q expression in ALS, C1q expression in the V0c interneurons may allow for a specific “kill switch” in V0c interneurons that project to dead motor neurons. As we have shown, C-bouton silencing in combination with modulatory-targeted training results in behavioral improvements in mSOD1G93A mice. Further understanding of the mechanism behind V0c interneuron loss could therefore be crucial in developing treatments and therapies for ALS focused on C-bouton silencing or removal.
Frequent swimming in the absence of C-boutons likely recruits the serotonergic system in mSOD1G93A mice
Here, we demonstrated the relationship between the C-boutons and mSOD1G93A ALS disease progression (Fig. 6). We measured nearly 100% muscle innervation throughout the life span of WT mice and found that the V0c interneurons modulate motor neuron excitability through the C-boutons in a task-dependent manner (Fig. 6A). The motor neurons of mSOD1G93A mice, however, gradually withdraw and die (Chiu et al., 1995; Fischer et al., 2004), altering their interaction with C-boutons (Fig. 6B). At early stages of the disease (approximately P65 to P95), mSOD1G93A mice appear functionally healthy despite reduced muscle innervation (Akay, 2014). Previously, we showed that this behavioral compensation occurs through the C-boutons (Landoni et al., 2019). Here, we show that the V0c interneurons do indeed have upregulated activity during walking at symptomatic ages (Fig. 3D). Further assessment of c-Fos expression in mSOD1G93A mice is required, however, as it is likely that resting mSOD1G93A mice have upregulated V0c interneuron activity relative to resting WT mice. Despite this behavioral compensation, the physical capabilities of mSOD1G93A mice deteriorate during the middle stage of the disease (approximately P95 to P125), likely because of motor neuron loss beyond what the cholinergic system can compensate for or muscle atrophy (Fig. 6B, middle). We also measured a reduction in the number of V0c interneurons that mirrored the percentage loss of motor neurons (Fig. 3E,F). At late stages of the disease (P125+), mSOD1G93A mice become paralyzed because of near-complete muscle denervation (Fig. 6B, right).
Figure 6.
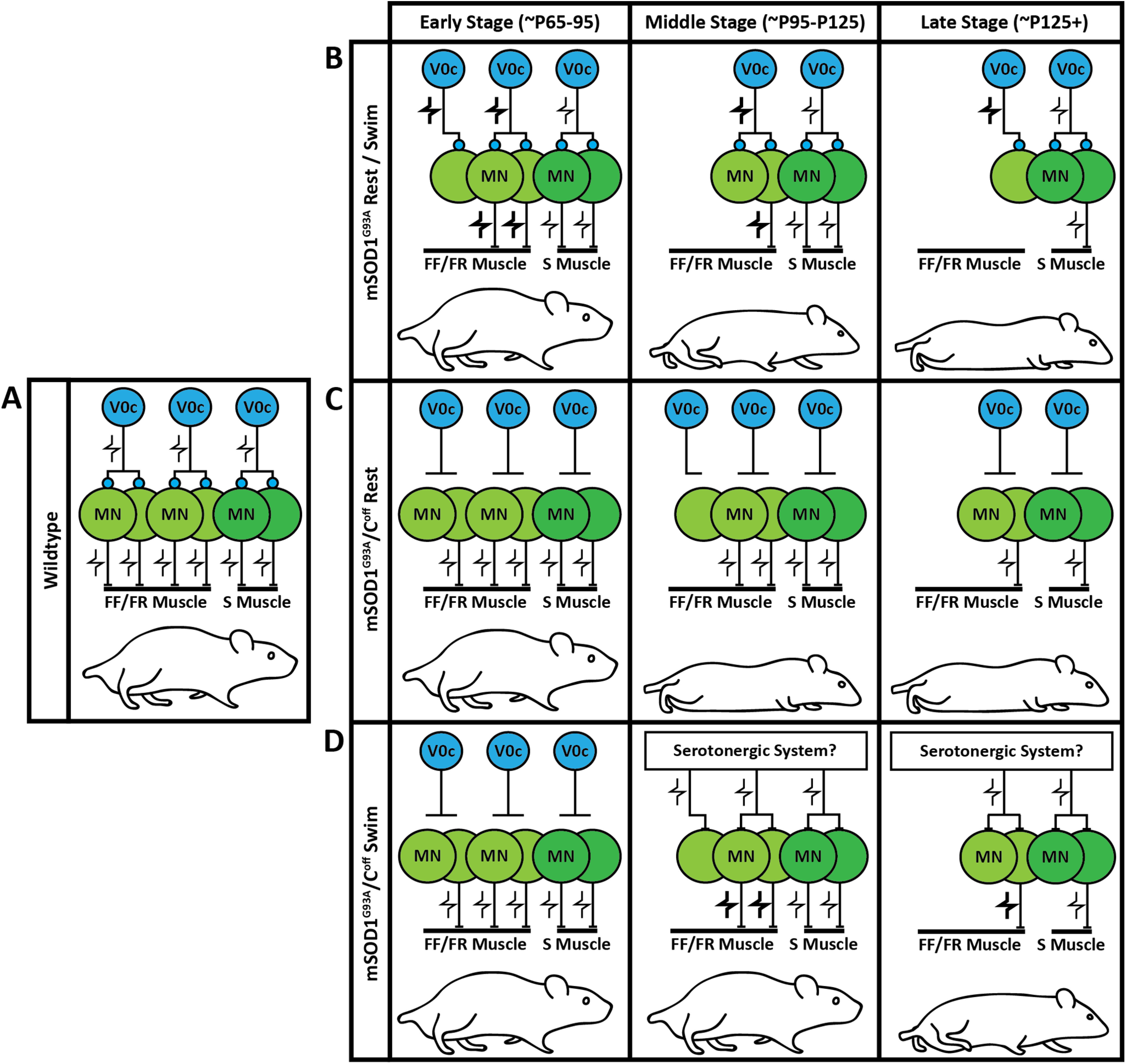
A schematic illustrating the influence of the C-boutons on muscle innervation and behavioral performance under various conditions. A, The interaction between the C-boutons and motor neurons (MNs) is stable in WT mice. Modulation of the motor neurons through the C-boutons occurs in a task-dependent manner. B, As motor neurons die in mSOD1G93A mice, C-bouton activity is upregulated to compensate. This, however, seems to increase motor neuron stress and hastens muscle denervation. The result is a gradual reduction in behavioral performance that eventually leads to paralysis. The V0c interneurons are also lost as the disease progresses. C, Silencing the C-boutons in mSOD1G93A mice improves muscle innervation at the cost of behavioral compensation. This results in an earlier reduction in behavioral performance relative to mSOD1G93A mice (Landoni et al., 2019). D, When C-bouton-silenced mSOD1G93A mice undergo frequent swimming, another modulatory mechanism seems to be recruited that helps to maintain behavioral performance at the middle stage (approximately P95 to P125) and the beginning of the late stage (approximately P125+) of the disease. The mechanism at play is likely the serotonergic system, because of its known gain control of motor neurons and benefit in ALS (Turner et al., 2003; Wei et al., 2014; El Oussini et al., 2016). FF, Fast-fatigable; FR, fast-fatigue resistant; S, slow.
Despite compensating for locomotor behavior, the C-boutons are detrimental to fast-twitch muscle innervation (Fig. 2B–D). At early stages of the disease, mSOD1G93A/Coff mice are functionally healthy, likely driven by higher fast-twitch muscle innervation relative to mSOD1G93A mice (Fig. 6C, left). At the middle stage of the disease, the locomotor capabilities of mSOD1G93A/Coff mice significantly decline as muscle denervation continues in the absence of behavioral compensation (Fig. 6C, middle; Landoni et al., 2019). This mechanism continues during the late stage of the disease when mSOD1G93A/Coff mice become paralyzed despite higher muscle innervation relative to mSOD1G93A mice (Fig. 6C, right). Characterization of innervation in the mSOD1G93A/Coff mice is incomplete, however, and it is unclear whether the improved muscle innervation over mSOD1G93A resulted from improved motor neuron survival or axonal sprouting, and whether there were differences in partial innervation between these groups of mice.
Surprisingly, mSOD1G93A/Coff mice that undergo frequent swimming perform significantly better during motor tests than mSOD1G93A mice by a late stage of the disease (Fig. 6D). It is unlikely that these findings are the result of the improved fast-twitch muscle innervation we observed for two reasons. First, the fast-twitch muscle weights of the mSOD1G93A/Coff swim mice were like those of the unexperimented mSOD1G93A/Coff mice at the late stage of the disease (P130; Fig. 4C–E). Second, the mSOD1G93A/Coff swim mice performed well beyond what we previously observed in untrained mSOD1G93A/Coff mice (Landoni et al., 2019). Another alternative modulatory system must therefore be compensating for the loss of C-bouton modulation in the mSOD1G93A/Coff swim mice to maintain task-dependent amplitude modulation (Fig. 6D, middle, right).
It is likely that the alternative modulatory system is a serotonergic system for three reasons: first, the serotonergic system has been shown to modulate motor neuron excitability by increasing persistent inward currents (Wei et al., 2014). Second, it has also been shown to slow disease progression and improve motor function in ALS (Turner et al., 2003; El Oussini et al., 2016). Last, the V0c interneurons also receive serotonergic input (Zagoraiou et al., 2009); therefore, an interaction exists between the serotonergic and cholinergic modulatory systems. When V0c interneuron output is eliminated in mSOD1G93A mice and the mice are forced to swim, the alternative amplitude modulator, which we think is the serotonergic system (Wei et al., 2014), may be upregulated to compensate for the V0c loss, resulting in the improved motor function we observed. Upregulating the serotonergic system and downregulating the C-boutons along with amplitude modulation could therefore amplify the beneficial effects of both systems. This hypothesis could also explain why we only saw improved life span in our mSOD1G93A/Coff swim mice relative to untrained mSOD1G93A/Coff mice, and not mSOD1G93A mice. To our knowledge, no experiments have been conducted that have amplified the serotonergic system and downregulated the cholinergic system. Further investigation into the relationship among ALS, the serotonergic system, and the cholinergic system is therefore critical in developing treatments and therapies targeting neuromodulation in ALS.
Implications
We have demonstrated that the effects of the C-boutons on mSOD1G93A mice are not strictly beneficial or detrimental. Restricting cholinergic neurotransmission by genetically silencing the C-boutons improves muscle innervation but worsens behavioral performance over time because of the loss of behavioral compensation. Under appropriate conditions, another modulatory system such as the serotonergic system might be recruited in the absence of the C-boutons that are beneficial to behavioral capability. Possible therapies for ALS may thus involve downregulating the C-boutons to improve muscle innervation while upregulating other modulatory systems to maintain behavioral compensation.
Footnotes
This work was supported by the QE II Foundation/BRC (Discovery Grant, 2015), and the ALS Canada (Project Grant, 2017). We thank Brenda Ross for technical assistance and for maintaining the mouse colonies. We also thank Laura Taylor and Igor Tatamikov for assistance with qPCR. In addition, we thank Marwan Ibrahim and Rachel Banks for help with some of the experiments presented in this article. We also thank Dr. Rob Brownstone for his valuable comments on the manuscript.
The authors declare no competing financial interests.
References
- Akay T (2014) Long-term measurement of muscle denervation and locomotor behavior in individual wild-type and ALS model mice. J Neurophysiol 111:694–703. 10.1152/jn.00507.2013 [DOI] [PubMed] [Google Scholar]
- Bielle F, Griveau A, Narboux-Nême N, Vigneau S, Sigrist M, Arber S, Wassef M, Pierani A (2005) Multiple origins of Cajal-Retzius cells at the borders of the developing pallium. Nat Neurosci 8:1002–1012. 10.1038/nn1511 [DOI] [PubMed] [Google Scholar]
- Brown GC, Neher JJ (2012) Eaten alive! Cell death by primary phagocytosis: “phagoptosis”. Trends Biochem Sci 37:325–332. 10.1016/j.tibs.2012.05.002 [DOI] [PubMed] [Google Scholar]
- Chiu AY, Zhai P, Dal Canto MC, Peters TM, Kwon YW, Prattis SM, Gurney ME (1995) Age-dependent penetrance of disease in a transgenic mouse model of familial amyotrophic lateral sclerosis. Mol Cell Neurosci 6:349–362. 10.1006/mcne.1995.1027 [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Rothstein JD (2001) From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci 2:806–819. 10.1038/35097565 [DOI] [PubMed] [Google Scholar]
- Conradi S, Skoglund S (1960) Observations on the ultrastructure and distribution of neuronal and glial elements on the motoneuron surface in the lumbosacral spinal cord of the cat during postnatal development. Acta Physiol Scand 333:5–52. [PubMed] [Google Scholar]
- Dukkipati SS, Chihi A, Wang Y, Elbasiouny SM (2017) Experimental design and data analysis issues contribute to inconsistent results of C-bouton changes in amyotrophic lateral sclerosis. eNeuro 4:ENEURO.0281-16.2016. 10.1523/ENEURO.0281-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Oussini H, Bayer H, Scekic-Zahirovic J, Vercruysse P, Sinniger J, Dirrig-Grosch S, Dieterlé S, Echaniz-Laguna A, Larmet Y, Müller K, Weishaupt JH, Thal DR, van Rheenen W, van Eijk K, Lawson R, Monassier L, Maroteaux L, Roumier A, Wong PC, van den Berg LH, et al. (2016) Serotonin 2B receptor slows disease progression and prevents degeneration of spinal cord mononuclear phagocytes in amyotrophic lateral sclerosis. Acta Neuropathol 131:465–480. 10.1007/s00401-016-1534-4 [DOI] [PubMed] [Google Scholar]
- Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD (2004) Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol 185:232–240. 10.1016/j.expneurol.2003.10.004 [DOI] [PubMed] [Google Scholar]
- Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P (2000) Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci 20:2534–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallart-Palau X, Tarabal O, Casanovas A, Sábado J, Correa FJ, Hereu M, Piedrafita L, Calderó J, Esquerda JE (2014) Neuregulin-1 is concentrated in the postsynaptic subsurface cistern of C-bouton inputs to α-motoneurons and altered during motoneuron diseases. FASEB J 28:3618–3632. 10.1096/fj.13-248583 [DOI] [PubMed] [Google Scholar]
- Gordon T, Tyreman N, Li S, Putman CT, Hegedus J (2010) Functional over-load saves motor units in the SOD1-G93A transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 37:412–422. 10.1016/j.nbd.2009.10.021 [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, Chen W, Zhai P, Sufit RL, Siddique T (1994) Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264:1772–1775. 10.1126/science.8209258 [DOI] [PubMed] [Google Scholar]
- Hegedus J, Putman CT, Gordon T (2007) Time course of preferential motor unit loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 28:154–164. 10.1016/j.nbd.2007.07.003 [DOI] [PubMed] [Google Scholar]
- Hegedus J, Putman CT, Tyreman N, Gordon T (2008) Preferential motor unit loss in the SOD1 G93A transgenic mouse model of amyotrophic lateral sclerosis. J Physiol 586:3337–3351. 10.1113/jphysiol.2007.149286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman-Patterson TD, Sher RB, Blankenhorn EA, Alexander G, Deitch JS, Kunst CB, Maragakis N, Cox G (2011) Effect of genetic background on phenotype variability in transgenic mouse models of amyotrophic lateral sclerosis: a window of opportunity in the search for genetic modifiers. Amyotroph Lateral Scler 12:79–86. 10.3109/17482968.2010.550626 [DOI] [PubMed] [Google Scholar]
- Herron LR, Miles GB (2012) Gender-specific perturbations in modulatory inputs to motoneurons in a mouse model of amyotrophic lateral sclerosis. Neuroscience 226:313–323. 10.1016/j.neuroscience.2012.09.031 [DOI] [PubMed] [Google Scholar]
- Jiang YQ, Sarkar A, Am A, Martin JH (2018) Transneuronal downregulation of the premotor cholinergic system after corticospinal tract loss. J Neurosci 38:8329–8344. 10.1523/JNEUROSCI.3410-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura Y, Dyck PJ, Shimono M, Okazaki H, Tateishi J, Doi H (1981) Morphometric comparison of the vulnerability of peripheral motor and sensory neurons in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 40:667–675. 10.1097/00005072-198111000-00008 [DOI] [PubMed] [Google Scholar]
- Konsolaki E, Koropouli E, Tsape E, Pothakos K, Zagoraiou L (2020) Genetic inactivation of cholinergic C bouton output improves motor performance but not survival in a mouse model of amyotrophic lateral sclerosis. Neuroscience 450:71–80. 10.1016/j.neuroscience.2020.07.047 [DOI] [PubMed] [Google Scholar]
- Landoni LM, Myles JR, Wells TL, Mayer WP, Akay T (2019) Cholinergic modulation of motor neurons through the C-boutons are necessary for the locomotor compensation for severe motor neuron loss during amyotrophic lateral sclerosis disease progression. Behav Brain Res 369:111914. 10.1016/j.bbr.2019.111914 [DOI] [PubMed] [Google Scholar]
- Lobsiger CS, Boillée S, Pozniak C, Khan AM, McAlonis-Downes M, Lewcock JW, Cleveland DW (2013) C1q induction and global complement pathway activation do not contribute to ALS toxicity in mutant SOD1 mice. Proc Natl Acad Sci U S A 110:E4385–E4392. 10.1073/pnas.1318309110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milan L, Courtand G, Cardoit L, Masmejean F, Barrière G, Cazalets JR, Garret M, Bertrand SS (2015) Age-related changes in pre- and postsynaptic partners of the cholinergic C-boutons in wild-type and SOD1G93A lumbar motoneurons. PLoS One 10:E0135525. 10.1371/journal.pone.0135525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RG, Mitchell JD, Moore DH (2012) Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev 3:CD001447. [DOI] [PubMed] [Google Scholar]
- Nagao M, Misawa H, Kato S, Hirai S (1998) Loss of cholinergic synapses on the spinal motor neurons of amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 57:329–333. 10.1097/00005072-199804000-00004 [DOI] [PubMed] [Google Scholar]
- Oliván S, Calvo AC, Rando A, Muñoz MJ, Zaragoza P, Osta R (2015) Comparative study of behavioural tests in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Exp Anim 64:147–153. 10.1538/expanim.14-0077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullen AH, Athanasiou D (2009) Increase in presynaptic territory of C-terminals on lumbar motoneurons of G93A SOD1 mice during disease progression. Eur J Neurosci 29:551–555 561. 10.1111/j.1460-9568.2008.06602.x [DOI] [PubMed] [Google Scholar]
- Ravits J (2014) Focality, stochasticity and neuroanatomic propagation in ALS pathogenesis. Exp Neurol 262:121–126. 10.1016/j.expneurol.2014.07.021 [DOI] [PubMed] [Google Scholar]
- Robberecht W, Philips T (2013) The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci 14:248–264. 10.1038/nrn3430 [DOI] [PubMed] [Google Scholar]
- Rothstein JD (1996) Therapeutic horizons for amyotrophic lateral sclerosis. Curr Opin Neurobiol 6:679–687. 10.1016/s0959-4388(96)80103-6 [DOI] [PubMed] [Google Scholar]
- Salamatina A, Yang JH, Brenner-Morton S, Bikoff JB, Fang L, Kintner CR, Jessell TM, Sweeney LB (2020) Differential loss of spinal interneurons in a mouse model of ALS. Neuroscience 450:81–95. 10.1016/j.neuroscience.2020.08.011 [DOI] [PubMed] [Google Scholar]
- Snedecor GW, Cochran WG (1989) Statistical methods, Ed 8. Ames, IA: Iowa State UP. [Google Scholar]
- Turner BJ, Lopes EC, Cheema SS (2003) The serotonin precursor 5-hydroxytryptophan delays neuromuscular disease in murine familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 4:171–176. 10.1080/14660820310009389 [DOI] [PubMed] [Google Scholar]
- Valdez G, Tapia JC, Lichtman JW, Fox MA, Sanes JR (2012) Shared resistance to aging and ALS in neuromuscular junctions of specific muscles. PLoS One 7:e34640. 10.1371/journal.pone.0034640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei K, Glaser JI, Deng L, Thompson CK, Stevenson IH, Wang Q, Hornby TG, Heckman CJ, Kording KP (2014) Serotonin affects movement gain control in the spinal cord. J Neurosci 34:12690–12700. 10.1523/JNEUROSCI.1855-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino H (2019) Edaravone for the treatment of amyotrophic lateral sclerosis. Expert Rev Neurother 19:185–193. 10.1080/14737175.2019.1581610 [DOI] [PubMed] [Google Scholar]
- Zagoraiou L, Akay T, Martin JF, Brownstone RM, Jessell TM, Miles GB (2009) A cluster of cholinergic premotor interneurons modulates mouse locomotor activity. Neuron 64:645–662. 10.1016/j.neuron.2009.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
qPCR results and swim performance. A, qPCR revealed tight overlap between the SOD1G93A gene amplification sigmoidal curves of the mSOD1G93A and mSOD1G93A/Coff mice (left cluster) relative to the housekeeping gene (right cluster). B, 2^ΔΔCt was calculated to be 0.978 ± 0.174 for the mSOD1G93A/Coff mice relative to the mSOD1G93A mice, translating to a 0.022 ± 0.174-fold decrease in SOD1G93A gene expression and indicating that there was no difference in gene expression between these two groups of mice. C–G, The swim performance of the mice was scored based on the average number of prodding/interventions required per lap: score 10, no intervention; score 8, <2 prods per lap; score 6, 2–3 prods per lap; score 4, 4–5 prods per lap; score 2, >5 prods per lap; score 0, drowning behavior or early termination of experiment. Mice were placed between these scores (9, 7, 5, 3, 1) if there was high variability in performance and they could not easily be placed in one score or another other. An example of male and female performance is given for the mSOD1G93A mice (C, D) and mSOD1G93A/Coff mice (E, F), where the red dots indicate days that motor performance measurements were obtained at least 2 h prior to swimming. G, Average swim performance of the mSOD1G93A and mSOD1G93A/Coff mice. mSOD1G93A/Coff mice required consistently and significantly fewer interventions than their mSOD1G93A counterparts beginning at P120. Bar graphs display the mean ± SD. p < 0.05, p < 0.01, p < 0.001. Download Figure 5-1, TIF file (12.4MB, tif) .
The mSOD1G93A swim mice spent most of their time floating at P130, whereas the mSOD1G93A/Coff mice swam with minimal encouragement. A, B, A male mSOD1G93A swim mouse (A) and a male mSOD1G93A/Coff swim mouse (B) at P130 demonstrating the difference in quality of swimming between these strains. Videos were captured at 500 frames/s with 4× playback speed. The distance between the calibration dots measures 1.5 cm.
The mSOD1G93A/Coff swim mice never displayed the backside dragging that the mSOD1G93A swim mice did at P95, even when assessed at P129. A–C, Treadmill walking of a male mSOD1G93A rest mouse (A), and swim mouse (B) at P95, and a male mSOD1G93A/Coff swim mouse at P129 (C). Note that the mSOD1G93A swim mouse displays backside dragging at P95 that the mSOD1G93A rest mouse does not. We did not observe this phenotype in the mSOD1G93A/Coff swim mice, even when assessed at P129. Videos were captured at 500 frames/s with 4× playback speed. The distance between the calibration dots measures 1.5 cm.