

Abstract
The X protein of hepatitis B virus (HBV) is a transcriptional activator which is required for infection and may play an important role in HBV-associated hepatocarcinogenesis. It has been suggested that X acts as a nuclear coactivator or stimulates several signal transduction pathways by acting in the cytoplasm. One of these pathways leads to the nuclear translocation of NF-κB. A recent report indicates that X activates NF-κB by acting on two cytoplasmic inhibitors of this family of transcription factors: IκBα and the precursor/inhibitor p105. We demonstrate here that X directly interacts with IκBα, which is able to transport it to the nucleus by a piggyback mechanism. This transport requires a region of IκBα (the second ankyrin repeat) which has been demonstrated to be involved in its nuclear import following NF-κB activation. Using deletion mutants, we showed that amino acids 249 to 253 of IκBα (located in the C-terminal part of the sixth ankyrin repeat) play a critical role in the interaction with X. This small region overlaps one of the domains of IκBα mediating the interaction with the p50 and p65 subunits of NF-κB and is also close to the nuclear export sequence of IκBα, therefore providing a potential explanation for the nuclear accumulation of IκBα with X. This association can also be observed upon the induction of endogenous IκBα by tumor necrosis factor alpha (TNF-α) treatment of Chang cells expressing X. In accordance with this observation, band shift analysis indicates that X induces a sustained NF-κB activation following TNF-α treatment, probably by preventing the reassociation of newly synthesized nuclear IκBα with DNA-bound NF-κB complexes.
Hepatitis B virus (HBV) belongs to the family Hepadnaviridae, whose members replicate their genomes by reverse transcription, and causes both acute and chronic infections of the liver. The gene encoding the X protein is highly conserved among all mammalian hepatitis viruses and is expressed at low levels during acute and chronic hepatitis (5, 25, 52, 68). The function of X in the viral life cycle is still enigmatic. Previous studies showed that in cell cultures transfected with viral DNA, viral replication does not depend on a functional X protein. However, X is required for infectivity in vivo (8, 71) and thus may play an essential role during early steps of viral infection. In addition, several lines of evidence have implicated X in the regulation of liver cell proliferation and viability during liver carcinogenesis (10, 16, 22, 30, 38, 52, 55). X shows transcriptional activity, but the relevance of this property in the viral life cycle is still unclear (32, 43, 68). X has been shown to activate a wide variety of cellular and viral genes in trans, including HBV enhancers; human immunodeficiency virus long terminal repeats; class II and III promoters; the proto-oncogenes c-jun, c-fos, and c-myc; and, recently, cytokines such as tumor necrosis factor alpha (TNF-α) (33). The mechanism(s) by which X activates gene transcription in trans is only partially understood. The findings that X by itself does not bind to double-stranded DNA and that genes stimulated by X lack any obvious consensus sequences suggest that X stimulates transcription presumably by interacting with cellular proteins and/or components of signal transduction pathways (14, 23). The transactivation function of X has been shown to involve both direct interaction with transcriptional factors, such as RPB5 and RMP of RNA polymerases (14), TATA-binding protein (40, 61), and ATF/CREB (65), and activation of signal transduction pathways, such as Ras/Raf/MAP kinase (4), protein kinase C (29), Jak1-STAT signaling (34), and NF-κB (9, 35, 47, 51, 58). Although X seems to act in the nucleus to activate transcription from certain promoters, the great majority of X is cytosolic and is likely to act from this compartment to activate pathways leading to the activation of promoters bearing AP-1, NF-AT, or NF-κB sites (9, 32, 48, 51, 52). We focus here on the mechanisms involved in X-induced NF-κB activation.
Members of the Rel/NF-κB family of transcription factors play important roles in immune, inflammatory, and apoptotic responses, through the induction of the expression of numerous cellular and viral genes (3, 36, 60). NF-κB activity is composed of homo- or heterodimers of related proteins that share a conserved DNA-binding and dimerization domain called the Rel homology domain. In most cell types, NF-κB is sequestered in the cytoplasm bound to inhibitory proteins called IκBα, IκBβ, and IκBɛ. In response to diverse stimuli, including inflammatory cytokines and mitogens, as well as several viral proteins, active NF-κB is translocated to the nucleus as a result of the proteolytic degradation of IκB proteins. This mechanism has been best studied for the IκBα inhibitor and demonstrated to involve phosphorylation on two specific serine residues followed by ubiquitination and degradation by the 26S proteasome (6, 7, 42, 56, 64). More recently, a specific protein kinase activity responsible for the phosphorylation of IκBα has been identified as a large multisubunit complex, and two kinase subunits (IKK1/α and IKK2/β) as well as a structural component (NEMO or IKKγ) have been cloned (12, 37, 41, 44, 66, 67, 70). While the process leading to the degradation of the IκB proteins is relatively well understood, the mechanism by which a variety of distinct signals are transduced to their common targets, the IκB proteins, remains to be elucidated. This is particularly true for the viral proteins which are known to activate NF-κB, including human T-cell leukemia virus 1 Tax, Epstein-Barr virus LMP1, and HBV X. LMP1 has been shown to act like a constitutive TNF-like receptor (15). Concerning Tax, the situation is less clear, despite a number of studies suggesting that this molecule might interact with several members of the NF-κB or IκB family. More recently, it has been shown that Tax can interact directly with the IKK complex or with one of the putative upstream kinases (11, 21, 59, 69). In contrast, NF-κB activation by X has been much less studied: two recent reports indicate that the transient expression of X induces the degradation of two NF-κB cytoplasmic inhibitors, IκBα and the p105 precursor of the p50 NF-κB subunit (9, 51). While the role of the IKK complex in X-induced NF-κB activation will be the subject of a separate study (61a), we demonstrate here that X interacts with IκBα and IκBβɛ but not IκBβ and that the interaction between X and IκBα results in the nuclear colocalization of these two molecules.
We also show that IκBα is responsible for transporting X to the nucleus; we have mapped the residues necessary for the interaction between these two proteins to amino acids 249 to 253 of IκBα. This region overlaps the recently identified contact points between IκBα and the p50 and p65 subunits of NF-κB and is also close to the nuclear export sequence (NES) of IκBα, therefore providing a potential explanation for the nuclear accumulation of IκBα that is associated with X. The observation that transfected X also associates with newly synthesized endogenous IκBα following TNF treatment (but not with endogenous IκBα in nonstimulated cells) suggests that under these conditions, X could induce a sustained NF-κB activation by preventing the reassociation of newly synthesized nuclear IκBα with DNA-bound NF-κB complexes. Indeed, band shift analysis indicated that X induced a sustained NF-κB activation following TNF-α treatment.
MATERIALS AND METHODS
Cell culture and reagents.
Chang cells and 293T cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum, penicillin, and streptomycin (Life Technologies Inc., Cergy Pontoise, France).
Antisera.
The antisera used were as follows. Anti-X (kindly provided by S. Urban) is a polyclonal rabbit antiserum raised against a fusion protein between X and glutathione S-transferase (GST). IκBα polyclonal antiserum 52008 was raised against recombinant human IκBα (62). Anti-IκBβ serum 37015 is a polyclonal rabbit antiserum generated against a GST fusion protein encompassing amino acids 258 to 360 of mouse IκBβ (62). Anti-IκBɛ serum M121 was purchased from Santa Cruz. Anti-green fluorescence protein (GFP) monoclonal antibody is from Clontech. Anti-hemagglutinin is murine monoclonal antibody 12CA5.
Plasmids and constructs.
The expression vectors for transfection into Chang cells were obtained by subcloning cDNAs encoding IκBα or its derivative into the plasmid pRc-CMV or pcDNA-3 (Invitrogen). To identify the domains of IκBα required for in vivo interaction with X protein, modified forms of IκBα (αΔC290, αΔC278, αΔC260, αΔC253, and αΔC249) were generated by site-directed mutagenesis with a PCR-based strategy (reference 64 and unpublished data). The deletion of amino acids 268 to 317 of IκBα was obtained by digestion with PvuII, and the resulting construct is referred to as αΔC268. Deletions were confirmed by sequencing. The X-GFP, X-myc, and pcDNA3-X expression vectors have been described (48). IκBα-110A3 was a kind gift from M. Hannink (46). (His)6-IκBα was obtained by cloning a PCR-amplified IκBα cDNA into the pRSETA vector (Invitrogen). Recombinant (His)6-IκBα was purified according to the manufacturer’s instructions. DNA encoding the SV5-tagged version of IκBα sequences for amino acids 68 to 317 was amplified by PCR with the pcDNA-3-IκBα ctag vector (1) as a template and cloned into the BamHI/XbaI restriction sites of eukaryotic expression vector pEGFP-C1 (Clontech), generating pEGFP-IκBα(68-317) ctag.
Generation and purification of bacterial fusion proteins.
DNA constructs allowing the synthesis of GST fusion proteins encompassing human IκBα or X were created by PCR and standard recombinant technology. All PCR-generated fragments were cloned into the pGEX-2T vector (Pharmacia). They were fully sequenced to ensure that they contained no mutation. For the production of fusion proteins, bacterial cultures were grown to mid-log phase and then stimulated with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). After 4 h of additional growth, the bacteria were harvested in a solution containing 50 mM Tris-HCl (pH 8.0), 1% Nonidet P-40 (NP-40), 150 mM NaCl, 2 mM EDTA (pH 8.0), 10% glycerol supplemented with protease inhibitors (10 μg each of leupeptin, aprotinin, N-tosyl-l-phenylalanine chloromethyl ketone [TPCK], Nα-p-tosyl-l-lysine chloromethyl ketone [TLCK], and phenylmethylsulfonyl fluoride per ml), and phosphatase inhibitors (100 mM sodium fluoride and 2 mM sodium orthovanadate). Fusion proteins were immobilized on glutathione coupled to agarose beads (Sigma).
In vitro translation of pcDNA-3-X was performed with wheat germ extracts according to the manufacturer’s instructions (Promega).
Cell transfection.
Transfection of Chang cells was carried out by the calcium phosphate coprecipitation technique. A total of 10 μg of DNA per 10-cm plate was used.
Preparation of nuclear extracts and electrophoretic mobility shift assay.
Transfected 293T cells were pelleted and solubilized for 5 min at 4°C in EMSA I buffer (50 mM Tris-HCl [pH 7.9], 10 mM KCl, 1 mM EDTA, 0.2% NP-40, 10% glycerol) supplemented with protease inhibitors (10 μg each of leupeptin, aprotinin, TPCK, TLCK, and phenylmethylsulfonyl fluoride per ml) and phosphatase inhibitors (100 mM sodium fluoride and 2 mM sodium orthovanadate). The lysates were centrifuged at 6,500 × g for 3 min. The pelleted nuclei were washed extensively with the same buffer without NP-40 and then incubated for 20 min at 4°C with EMSA II buffer containing 400 mM NaCl, 20% glycerol, 20 mM HEPES (pH 7.9), 10 mM KCl, 1 mM EDTA, and protease and phosphatase inhibitors. The extracts were centrifuged at 14,000 × g for 10 min, and supernatants were used for the mobility shift assay. The following partially double-stranded oligonucleotide probe, KBF1, was used for the mobility shift assays:
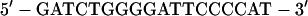 |
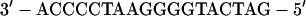 |
Mobility shift assays were performed in a total volume of 20 μl in the following buffer: 4% Ficoll, 20 mM HEPES (pH 7.5), 70 mM NaCl, 2 mM dithiothreitol, 100 μg of bovine serum albumin per ml, and 0.01% NP-40. Each reaction [mixtures also contained 1 μl of the probe, end labelled with 32P, and 1 μg of poly(dI-dC) (Pharmacia)] was initiated by the addition of 10 μg of nuclear extract, and mixtures were allowed to incubate at room temperature for 20 min prior to electrophoretic analysis on a 5% native polyacrylamide gel in 0.5× Tris-borate-EDTA buffer.
Immunoprecipitations and immunoblots.
Cells were lysed by adding 250 μl of 1× Chris buffer (50 mM Tris [pH 8.0], 0.5% NP-40, 200 mM NaCl, and 0.1 mM EDTA) as well as the protease and phosphatase inhibitors described above to 5 × 105 cells. Specific polypeptides were then recovered by immunoprecipitation from equivalent amounts of cellular proteins, using one of the following antibodies: anti-IκBα, anti-IκBβ, anti-IκBɛ, anti-X, or anti-GFP. Immune complexes were collected with Staphylococcus aureus protein A (Pansorbin; Calbiochem) or protein G-Sepharose (Sigma). Immunoprecipitates were then washed three times in lysis buffer and resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Subsequent immunoblot analyses were performed according to a previously described protocol. Anti-GFP monoclonal antibody was used at a 1/500 dilution. Anti-IκBα, anti-IκBβ, anti-IκBɛ, and anti-X were used at a 1/1,000 dilution for enhanced chemiluminescence (ECL) or 1/200 for 125I-protein A, as indicated in the figure legends. Proteins transferred to Immobilon membranes (Millipore) were revealed either with the Amersham ECL system (for immunoblotting of mouse monoclonal immunoprecipitates or for direct immunoblotting of total cell extracts) or by incubation with 125I-protein A (Amersham) (for immunoblotting of rabbit polyclonal immunoprecipitates).
In vitro binding assays.
In vitro-translated X or recombinant (His)6-IκBα was added to immobilized GST fusion proteins in 1× Chris buffer. To avoid the formation of mixed disulfide bonds, the buffer was supplemented with 0.5 mM dithiothreitol. The mixtures were incubated at 4°C for an hour with gentle rotation. The agarose beads were washed three times with 1 ml of 1× Chris buffer. Bound proteins were subsequently eluted with sample buffer, boiled, and resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
Indirect immunofluorescence.
Our procedure has been described previously (48). Briefly, Chang cells were seeded onto coverslips and transfected. After 30 to 40 h, the cells were either fixed in 3% paraformaldehyde, rinsed, and permeabilized in phosphate-buffered saline containing 0.2% Triton or fixed and permeabilized with methanol and acetone, blocked with goat serum, and incubated with primary antibody at a 1/100 dilution for 1 h at room temperature. The secondary antibody was anti-rabbit immunoglobulin G conjugated with Texas red (Vector Laboratory) or fluorescein isothiocyanate (Sanofi Diagnostics Pasteur) at a 1:60 dilution. We used also Texas red-conjugated anti-mouse immunoglobulin G at the same dilution (Sigma). After being washed in phosphate-buffered saline, coverslips were mounted in Mowiol, and cells were examined with an epifluorescence microscope (Zeiss). Alternatively, confocal laser scanning microscopy and immunofluorescence analysis were performed with a TCS4D confocal microscope based on an immunofluorescence microscope interfaced with a mixed-gas (argon-krypton) laser (Leica Laser Technic). Fluorescence acquisition was performed with the 488- and 568-nm lasers to excite the fluorescein isothiocyanate and Texas red dyes, respectively, with a 100× oil immersion PL APO objective.
RESULTS
Interaction between X and IκBα.
Expression of the HBV X protein results in NF-κB activation by an incompletely characterized mechanism, possibly involving phosphorylation of the IκBα molecules by the recently characterized IKK complex. As Tax, another viral protein known to activate NF-κB, has been suggested to directly interact with members of the NF-κB and IκB families, the possibility that X could directly interact with IκB molecules in vivo and in vitro was evaluated.
To assay for a possible interaction between X and IκBα, we cotransfected X-GFP and IκBα expression vectors in Chang cells and carried out coimmunoprecipitations. As depicted in Fig. 1A, precipitation of IκBα followed by immunoblotting for X demonstrated the association between these proteins (lanes 12 and 15). Anti-GFP blotting detected X-GFP only in the presence of IκBα, but not when X-GFP, IκBα, or IκBβ was expressed alone (Fig. 1A, top, lanes 9 through 11) or when X-GFP was coexpressed with IκBβ (lane 13). No association could be seen when normal rabbit serum (NRS) was used for immunoprecipitation (lane 1). We also confirmed that the interaction of X-GFP with IκBα was not mediated by GFP (lane 14). In the bottom panel of Fig. 1A, an anti-IκBα immunoblot of IκBα immunoprecipitates verified in parallel the level of endogenous and exogenous IκBα. The reverse experiment (the immunoprecipitation of X-GFP with anti-GFP followed by immunoblotting with anti-IκBα) confirmed the interaction between X-GFP and IκBα (Fig. 1B, lane 5). Importantly, a direct IκBα immunoblot of the corresponding cellular lysates (lanes 6 through 9) indicated that the augmented presence of IκBα in the anti-GFP immunoprecipitate (lane 5) was not due to a change in its cellular abundance.
FIG. 1.

X associates with IκBα but not with IκBβ or IκBɛ. (A) Chang cells were transfected with the indicated cDNAs (α, IκBα; β, IκBβ). After 30 h, lysates were immunoprecipitated (IP) with NRS, anti-X, or anti-IκBα. Immunoblotting was performed with anti-GFP monoclonal antibody followed by ECL (top) or anti-IκBα polyclonal antibody followed by incubation with 125I-protein A (bottom). (B) After transfection with the indicated plasmids, IκBα was detected by immunoblotting of total cell lysates (lanes 6 to 9) or anti-hemagglutinin (HA) (as a nonrelevant control antibody [lane 1]) or anti-GFP (to detect X-GFP [lanes 2 to 5]) immunoprecipitates. (C) (Left) Binding of 35S-labelled in vitro-translated X to a GST-IκBα column. A total of 0 (lane 1), 1 (lane 2), 5 (lane 3), or 10 (lane 4) μl of the translated product was incubated with the bacterially produced GST fusion protein. After extensive washing, bound 35S-X was detected by autoradiography. One microliter of the wheat germ extract translation product was run in parallel (lane 5). (Right) Binding of (His)6-IκBα (His-α) to GST-X. One microliter (50 ng) of (His)6-IκBα was incubated with GST-X (lane 1) or GST (lane 2). After extensive washing, the level of IκBα was determined by immunoblotting. To quantify the interaction, we loaded 1.5 ng of (His)6-IκBα on the gel (lane 3). (D) Chang cells were transfected with the indicated cDNAs. Lysates were immunoprecipitated (IP) with NRS or immunopurified anti-IκBβ and immunoblotted with anti-GFP or anti-IκBβ. (E) Same as panel D, but with IκBɛ. Cell extracts were immunoprecipitated with NRS (lane 1) or immunopurified anti-IκBɛ (lanes 2 to 7) before immunoblotting with anti-GFP (top). To evaluate the expression of X-GFP in total lysates, we performed an immunoblot with an anti-GFP antibody.
The specificity of this interaction was then examined in vitro with GST pull-down experiments: GST fusion proteins encompassing IκBα or X were produced in bacteria, immobilized on agarose-glutathione beads, and used to detect, respectively, their association with in vitro-translated 35S-labelled X (Fig. 1C, left) or (His)6-IκBα (right). The addition of increasing amounts of 35S-labelled X protein resulted in a parallel augmentation of its interaction with GST-IκBα fusion protein (Fig. 1C, left, lanes 2 and 3), with a plateau for the highest concentration (left, lane 4). Recombinant (His)6-IκBα was incubated with equivalent amounts of immobilized GST or GST-X (Fig. 1C, right) and found to be associated only with GST-X, demonstrating specific association between IκBα and X (right, lane 1).
The finding that IκBα associates with X raised the possibility that the two other NF-κB inhibitors, IκBβ and IκBɛ, could also interact with X. To examine this possibility, Chang cells were transfected with plasmids encoding these proteins and/or X-GFP. Lysates were subjected to immunoprecipitation with NRS as a negative control, immunopurified anti-IκBβ antiserum (Fig. 1D), or anti-IκBɛ antiserum (Fig. 1E), and the presence of X-GFP was determined by anti-GFP immunoblotting. These experiments did not demonstrate any significant interaction between X and IκBβ (Fig. 1D, lane 7); however, when the cells were transfected with X or cotransfected with X and IκBɛ, we observed an association of X with either endogenous (lane 3) or transfected (lane 7) IκBɛ. The abundance of IκBβ was also measured by anti-IκBβ immunoblotting (Fig. 1D, bottom) to ensure that it was efficiently expressed and immunoprecipitated. As the bottom part of Fig. 1E shows, a direct GFP immunoblot of the corresponding cellular lysates (lanes 6 through 9) indicated that the augmented presence of X-GFP in the anti-IκBɛ immunoprecipitate (lane 7) was not due to a change in its cellular abundance.
In conclusion, these data demonstrate that X and IκBα interact both in vitro and ex vivo. Moreover, this interaction also occurs with IκBɛ but not with IκBβ.
IκBα promotes the import and accumulation of X in the nucleus.
To evaluate whether the association between X and IκBα induces a change in subcellular distribution of these proteins, overexpressed X and IκBα were localized by indirect immunofluorescence. IκBα, detected with an immunopurified anti-IκBα antibody, was localized to the cytoplasm and the nucleus in Chang cells with a diffuse distribution (Fig. 2A). To study the subcellular localization of X, we took advantage of the fusion construct between X and the reporter GFP. As was previously reported, X-GFP showed a discrete granular appearance and was distributed throughout the cytoplasm (Fig. 2B and reference 48). In contrast, when cotransfected with IκBα, X exhibited a nuclear distribution in large structures (Fig. 2D), and double staining indicated that X and IκBα colocalized in these nuclear structures (Fig. 2E). To ensure that fusion with GFP did not alter the nuclear localization of X, we performed immunofluorescence analysis in cells cotransfected with a plasmid carrying a gene encoding a myc-tagged form of X (X-myc) and pEGFP-IκBα(68-317) ctag cDNA. As shown in Fig. 2G, X-myc (in red) exhibited a nuclear distribution that overlaps with nuclear structures observed with GFP-IκBα(68-317) (Fig. 2F). Double staining indicated that X and GFP-IκBα(68-317) colocalized in these nuclear structures (Fig. 2H). Thus, the presence of IκBα induced a relocalization of the majority of X to the nucleus. In agreement with our biochemical results (Fig. 1D), we could not detect any effect of IκBβ on the subcellular localization of X (data not shown).
FIG. 2.

Colocalization of X and IκBα in the nucleus. Chang cells were transfected with plasmids expressing IκBα and/or X-GFP and fixed 30 h after transfection. The cellular distribution of X was visualized by the reporter GFP, and IκBα was visualized by immunostaining with purified anti-IκBα antibody followed by Texas red-conjugated anti-rabbit antibody. (A) Cells transfected with IκBα alone and stained with anti-IκBα. (B) Cells transfected with X-GFP alone and subjected to immunofluorescent detection of GFP. (C to E) Cells cotransfected with IκBα and X-GFP and stained for IκBα (C), GFP (D), or both (E); areas of coincidence of red and green fluorescence (yellow) indicate overlapping distributions of X and IκBα. Panel E shows fluorescence superimposed on a Nomarski image. (F to H) Cells cotransfected with pEGFP-IκBα(68-317) and X-myc were stained for GFP and X-myc. The localization of GFP-IκBα(68-317) is visualized by its green fluorescence (F), whereas X-myc is detected in red (G). Panel H shows fluorescence superimposed on a Nomarski image.
To get insight into the mechanism responsible for X nuclear localization, X was coexpressed in Chang cells with IκBα-110A3. This variant of IκBα is mutated in the second ankyrin repeat, a region responsible for the nuclear import of IκBα, and therefore remains predominantly cytoplasmic when ectopically expressed (46, 57). The subcellular distribution of IκBα-110A3 and X was determined by indirect immunofluorescence. X was found exclusively in the cytoplasm. As previously reported, IκBα-110A3 displayed a diffuse cytoplasmic localization (Fig. 3A) and was also found in cytoplasmic dots containing X (Fig. 3B and C). Coimmunoprecipitation experiments confirmed that X interacts with IκBα-110A3 (data not shown).
FIG. 3.

Colocalization of X and mutant IκBα-110A3 in the cytoplasm. Chang cells were cotransfected with plasmids expressing IκBα-110A3 and X-GFP and fixed 30 h after transfection. IκBα-110A3 was visualized by immunostaining with purified anti-IκBα antibody followed by Texas red-conjugated anti-rabbit antibody (A). The subcellular distribution of X was visualized by the reporter GFP (B). The distribution of both fluorochromes, superimposed on a Nomarski image, is shown in panel C. Areas of coincidence of red and green fluorescence (yellow) indicate overlapping distributions of X and IκBα-110A3.
These data indicate that a mutation which impairs the ability of IκBα to translocate to the nucleus is sufficient to prevent the nuclear accumulation of X. Together, these data show that IκBα is able to transport X to the nucleus by a piggyback mechanism.
Mapping of the domain for interaction of IκBα with X.
To analyze the site of binding of IκBα to X, we employed both wild-type IκBα (α wt) and a series of C-terminal-deletion mutants of IκBα termed αΔC290, αΔC278, αΔC268, αΔC253, and αΔC249. All these proteins were efficiently immunoprecipitated by the polyclonal anti-IκBα antibody (Fig. 4, bottom). When X was coexpressed in vivo with these proteins, it could associate with mutants αΔC290, αΔC278, αΔC268 (Fig. 4, left, lanes 3 to 5), and αΔC253 (Fig. 4, right, lane 3), but binding was hardly visible with mutant αΔ249 (Fig. 4, right, lane 4). Parallel immunoblotting with GFP confirmed that X-GFP was normally expressed in each immunoprecipitate. We confirmed these results by the immunofluorescence of cotransfected cells (Fig. 5). IκBα mutants αΔC260 and αΔC253 colocalized with X in nuclear structures (Fig. 5A through C and D through F, respectively), but mutant αΔC249 showed no colocalization with X and no evidence of punctate structures, while X remained fully cytoplasmic (Fig. 5G through I).
FIG. 4.

Structural requirement for IκBα-X interaction. Chang cells were transiently transfected with X-GFP expression vector (lanes 1) or cotransfected with X-GFP and plasmids encoding various mutants of IκBα: α wt, αΔC290, αΔC278, αΔC268, αΔC253, and αΔ249. Cell lysates were immunoprecipitated with anti-IκBα antiserum and analyzed by immunoblotting with a monoclonal anti-GFP antibody or a polyclonal anti-IκBα antibody. αΔC268 migrates more slowly than αΔC278 because it contains a few extra C-terminal amino acids derived from the cloning vector (this extra sequence does not interfere with its activity [data not shown]).
FIG. 5.

Structural requirement for IκBα-X colocalization. Chang cells were cotransfected with X-GFP and plasmids expressing either αΔC260 (A to C), αΔC253 (D to F), or αΔC249 (G to I). Cells were analysed by immunofluorescence with immunopurified anti-IκBα antibody (A, D, and G) or directly tested for GFP fluorescence (B, E, and H) and visualized by confocal laser scanning microscopy. Merging of the two stainings is shown in panels C, F, and I.
X interacts with endogenous IκBα following TNF-α treatment.
Because we were not able to detect an association between X and endogenous IκBα (Fig. 1A, lane 9), we decided to evaluate this interaction in cells treated with TNF-α, a situation where free IκBα is resynthesized following degradation (Fig. 6). Chang cells transfected with X-GFP cDNA were subjected to a time course of TNF-α stimulation (Fig. 6, lanes 1 to 7), whereas mock-transfected cells were left untreated (lane 8) or stimulated for 15 min with TNF-α (lane 9). Following these treatments, cell lysates were subjected to immunoprecipitation with an anti-IκBα antiserum, followed by immunoblotting with anti-GFP to evaluate X-GFP–IκBα association (Fig. 6, top) or with anti-IκBα to evaluate the kinetics of IκBα degradation and resynthesis (middle). The anti-IκBα Western blot demonstrates that IκBα was partially degraded at 15 min (this degradation seemed to be more effective in the presence of X; compare Fig. 6, middle, lanes 2 and 9) and was progressively resynthesized from 30 min to 6 h (lanes 3 to 6). Monitoring the interaction between X and IκBα demonstrated that such interaction occurred only when IκBα was resynthesized (Fig. 6, top, lanes 3 to 6). Indeed, despite observing a larger amount of IκBα visible in nontreated cells, we could not detect any association with X-GFP (Fig. 6, top; compare lane 1 with lanes 3 to 6). A parallel Western blotting of total cell lysates with an anti-GFP antibody showed the level of expression of X-GFP. The reduced expression of IκBα and X-GFP after 18 h of TNF-α treatment (Fig. 6, top and middle, lanes 7) could be a consequence of cell apoptosis upon long treatment with TNF-α and X.
FIG. 6.

Association of X with newly synthesized IκBα. Chang cells transfected with X (lanes 1 to 7) or mock transfected (lanes 8 and 9) were stimulated for the indicated periods of time with TNF-α, lysed in 1× Chris buffer, and subjected to anti-IκBα immunoprecipitation followed by anti-GFP or anti-IκBα immunoblotting. The abundance of X-GFP was measured by immunoblotting of lysates with an anti-GFP monoclonal antibody (bottom).
X sustained NF-κB activation following TNF-α treatment.
Our results demonstrate that a region of IκBα critical for interaction with X encompasses amino acids 249 to 253. The recent resolution of the IκBα–NF-κB complexes (27, 28) demonstrates that this region also directly contacts either p50 or p65 (see Discussion). Therefore, it is possible that X might interfere with the reassociation of newly synthesized nuclear IκBα with DNA-bound NF-κB complexes and thus prevent the termination of the activation process.
To test this possibility, nuclear NF-κB DNA-binding activity was assayed in 293T cells treated or not with TNF-α in the presence or absence of transfected X-GFP (Fig. 7). DNA-binding activity was not detected in unstimulated 293T cells (Fig. 7, lane 1) but was strongly induced after the addition of TNF-α for 30 min (lane 2). When the cytokine was removed for an additional 120-min chase period, NF-κB activity was completely abolished (lane 3) as a consequence of IκBα resynthesis, nuclear import, and NF-κB withdrawal. The transient expression of the X-GFP plasmid in 293T cells led to some NF-κB activation (lane 4), although it was at a much lower level than activation following TNF-α treatment (lane 2). This activation was potentiated by treatment with TNF-α (lane 5). Interestingly, during the chase period, X sustained NF-κB DNA-binding activity (lane 6) to a level higher than that observed either with X alone (lane 4) or after the chase period in the absence of X (lane 3).
FIG. 7.

X prolongs TNF-α-induced NF-κB activation. 293T cells either mock transfected (lanes 1 to 3) or transfected with GFP-X (lanes 4 to 6) for 30 h were either left untreated (lanes 1 and 4), treated for 30 min with TNF-α (lanes 2 and 5), or treated with TNF-α, followed by extensive washing and a 120-min chase in culture medium (lanes 4 and 6). Nuclear extracts were prepared, and band shift assays were performed as indicated in Materials and Methods.
DISCUSSION
A body of evidence points to X as an important regulator of HBV genome expression, infection, and proliferation and viability in liver cells. This has been correlated by several studies suggesting that X can bind cellular proteins, such as P53, DNA repair proteins, proteasome subunits, and Jak1 (17, 26, 34, 49, 54). The actual specificity and in vivo relevance of such findings are, however, still debated.
One example of the potential interaction between X and cellular metabolism is the capacity of X to activate the NF-κB signaling pathway. A recent report indicates that X activates this pathway by acting on two cytoplasmic inhibitors of the NF-κB family of transcription factors, IκBα and the precursor/inhibitor p105 (51). Our results provide an additional mechanism which in part accounts for the activation of NF-κB by demonstrating that X directly interacts with the IκBα inhibitor. A structure-function analysis of IκBα has allowed definition of several functional domains: a core domain of six ankyrin repeats; an N-terminal region containing two serine and two lysine residues involved in signal-induced phosphorylation and ubiquitination, respectively; and a C-terminal PEST domain. More recently, a region responsible for the nuclear import of IκBα has been identified in its second ankyrin repeat (46, 57), and a leucine-rich NES (from amino acids 266 to 281) in the C-terminal part of the sixth ankyrin repeat has been characterized (1, 19). The current understanding of the roles of these two sequences is as follows: following signaling and degradation of the IκB inhibitors, IκBα molecules are quickly resynthesized and translocated to the nucleus. This translocation requires the presence of the second ankyrin repeat. Once in the nucleus, these newly synthesized free IκBα molecules dissociate NF-κB complexes from DNA and transport them back to the cytoplasm (1). This step requires the presence of the NES; this sequence is homologous to the nuclear export signal found in human immunodeficiency virus Rev and the inhibitor of protein kinase A and is specifically recognized by the nuclear protein CRM1, which promotes the export from the nucleus to the cytoplasm (18, 20, 39, 50). Therefore, the localization of IκBα is the result of a dynamic equilibrium between nuclear import and export, also controlled by the accessibility of two sequences, the one in the second ankyrin repeat and the NES.
We demonstrate here that the X protein directly interacts with a region of the IκBα inhibitor located towards the N-terminal part of the sixth ankyrin repeat. Interestingly, while X has been shown to be essentially cytoplasmic, the cotransfection of X and IκBα results in the nuclear colocalization of the two molecules. This localization is dependent on the presence of a functional nuclear import region in IκBα, demonstrating that it is not due to unmasking of a cryptic NLS sequence in X. In order to characterize the nuclear localization of X-IκBα complexes, we performed double indirect immunofluorescence studies using antibodies specifically recognizing different punctate structures in the nucleus, such as SC-35 (a spliceosome assembly factor), SP-10, SM-1, and PML (31). However, the distribution pattern of these proteins was distinctly different from that of X (data not shown).
These results raise two questions: (i) why are the two molecules localized to the nucleus, and (ii) what is the physiological implication of this phenomenon?
A deletion analysis demonstrated that the region encompassing amino acids 249 to 253 of IκBα is critical for the interaction with X. A deletion mutant of IκBα containing amino acids 1 to 249 showed no association or colocalization with X, while a construct containing amino acids 1 to 253 exhibited a behavior similar to that of the wild-type molecule. The localization of this region is interesting in view of the recent resolution of the structure of a p50-p65-IκBα complex (27, 28). The domain for interaction between IκBα and X is located in the N-terminal region of the sixth ankyrin repeat, while the NES is in the C-terminal part of this repeat. The structure shows that these two regions are not far from each other, and it is conceivable that interaction between IκBα and X might interfere with the accessibility of the NES to the export machinery. As the subcellular localization of IκBα is the result of an equilibrium between import (controlled by the second ankyrin repeat) and export (controlled by the NES), masking of the latter sequence would provide an explanation for the nuclear colocalization and accumulation of the two molecules. A similar situation has recently been described (45): complexes between the v-rel oncogene product and IκBα are localized to the cytoplasm, but the treatment of cells with leptomycin, a specific inhibitor of CRM1-mediated nuclear export, results in nuclear relocalization of the v-rel–IκBα complex, indicating that continuous nuclear export is required for the cytoplasmic retention of this complex. Experiments are in progress to determine whether interaction between X and IκBα prevents access to the IκBα NES and, in particular, prevents binding of CRM1 to the NES.
Another important piece of information provided by the crystal structure concerns the contact points between IκBα and p50 or p65 (27, 28). Amino acids 249, 251, 255, 256, 258, 259, and 260 of IκBα directly contact either p50 or p65. Therefore, it is likely that the interaction of IκBα with X would interfere with IκBα’s interaction with NF-κB. One possibility is that X might be able to dissociate preformed NF-κB–IκBα complexes and therefore induce a sustained nuclear localization of NF-κB complexes. However, we have been unable to demonstrate dissociation of NF-κB–IκBα complexes by recombinant X (data not shown); this is in agreement with the observation that X is unable to interact with endogenous IκBα in nonstimulated cells (Fig. 6). Another possibility is that X associates and translocates to the nucleus with newly synthesized free IκBα. Free IκBα has been observed in cells treated with a NF-κB-activating stimulus, such as TNF-α: following stimulation, newly synthesized IκBα translocates to the nucleus, where it dissociates NF-κB complexes from DNA and takes them back to the cytoplasm. Figure 6 indicates that newly synthesized endogenous IκBα indeed associates with X. Therefore, this association might prevent IκBα from interacting with DNA-bound NF-κB complexes and lead to sustained NF-κB activation. This hypothesis was strengthened by the observation that in the presence of X, the complete disappearance of nuclear NF-κB DNA-binding activity which had been observed after TNF-α treatment following a chase period (2) could no longer be observed, and sustained NF-κB activity was detected (Fig. 7).
X interacts with a region of IκBα which is well conserved among other IκBs; however, interaction could be observed with IκBɛ but not with IκBβ. The significance of the association with IκBɛ is currently unknown. IκBɛ has not been demonstrated to shuttle between the cytoplasm and nucleus, and the association between X and endogenous IκBɛ in nonstimulated cells (contrary to what is observed with IκBα) suggests that this might fulfill a different function. Further experiments are required to clarify this point.
IκBα is not the only inhibitor which has been demonstrated to be present in the nucleus: an underphosphorylated form of IκBβ has indeed been demonstrated to translocate to the nucleus in association with NF-κB complexes, but it has been postulated to induce a prolonged NF-κB activation. The specific association of X with IκBα and the postulated mechanism described above are consistent with the observation that IκBα is the only IκB known to date to exhibit an inhibitory function in the nucleus.
The subcellular localization of X is still debated. Most studies have suggested a mainly cytoplasmic localization (13, 48, 53), and members of our groups have, in particular, demonstrated the colocalization of X with proteasome in this compartment (48). Some studies have suggested, however, that X might also show a nuclear localization with the nuclear and cytoplasmic X modulating the cellular signal transduction networks differently (13). Thus, nuclear X might directly interact with the transcription machinery and act as a coactivator (14, 24). Our data should help to reconcile such apparently contradictory findings. We indeed demonstrate that TNF-α stimulation leads to the nuclear localization of X through its binding to IκBα. Inflammation is a hallmark of chronic hepatitis and is associated with cytokine synthesis. Collectively, the results suggest a model whereby X, despite its major cytoplasmic localization, might transiently interact with the nuclear machinery.
While it is likely that this unusual interaction per se cannot be solely responsible for NF-κB activation (61a), especially in view of the fact that X is unable by itself to dissociate preformed NF-κB–IκBα complexes, it is likely to play a role in the establishment of a prolonged NF-κB response. We are currently addressing this question by trying to isolate mutants of IκBα that no longer interact with X but keep their NF-κB inhibitory functions; by reintroducing these molecules into IκBα −/− fibroblasts and determining the parameters of X-induced NF-κB activation on both wild-type and mutant IκBα backgrounds, we will be able to assign a role to this interaction.
ACKNOWLEDGMENTS
R.W. and H.S. contributed equally to this work.
We thank S. Whiteside for the gift of the C-terminal-deletion mutants of IκBα. We are grateful to R. Hellio for technical assistance with confocal microscopy. We thank S. Urban for the gift of anti-X antibody. We thank M. Hannink for the gift of the IκBα-110A3 mutant.
This research was sponsored in part by grants from ARC, ANRS, and the Ligue Nationale contre le Cancer and by Biomed contract 97-2567 and TMR contract 960026 to A.I.; grants from INSERM, EU, ARC, the Ligue Nationale contre le Cancer, and CNAM to C.B.; and grants from ARC and the Ligue Nationale contre le Cancer to C.D. R.W. is a recipient of a long-term fellowship from SIDACTION. H.S. is a recipient of a fellowship from DFG. C.G. is a recipient of a fellowship from FRM.
REFERENCES
- 1.Arenzana-Seisdedos F, Turpin P, Rodriguez M, Thomas D, Hay R T, Virelizier J L, Dargemont C. Nuclear localization of IκBα promotes active transport of NF-κB from the nucleus to the cytoplasm. J Cell Sci. 1997;110:369–378. doi: 10.1242/jcs.110.3.369. [DOI] [PubMed] [Google Scholar]
- 2.Arenzana-Seisdedos F, Thompson J, Rodriguez M S, Bachelerie F, Thomas D, Hay R T. Inducible nuclear expression of newly synthesized IκBα negatively regulates DNA-binding and transcriptional activities of NF-κB. Mol Cell Biol. 1995;15:2689–2696. doi: 10.1128/mcb.15.5.2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baldwin A S. The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 4.Benn J, Schneider R J. Hepatitis B virus HBx protein activates Ras-GTP complex formation and establishes a Ras, Raf, MAP kinase signaling cascade. Proc Natl Acad Sci USA. 1994;91:10350–10354. doi: 10.1073/pnas.91.22.10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bréchot C. Molecular mechanisms of hepatitis B and C viruses related liver carcinogenesis. In: Rizzetto M, Purcell R H, Gerin J L, Verme G, editors. Viral hepatitis and liver disease. Rome, Italy: Edizioni Minerva Medica; 1997. pp. 490–508. [Google Scholar]
- 6.Brockman J A, Scherer D C, McKinsey T A, Hall S M, Qi X, Lee W Y, Ballard D W. Coupling of a signal response domain in IκBα to multiple pathways for NF-κB activation. Mol Cell Biol. 1995;15:2809–2818. doi: 10.1128/mcb.15.5.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Control of IκBα proteolysis by site-specific, signal-induced phosphorylation. Science. 1995;267:1485–1488. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- 8.Chen H-S, Kaneko S, Girones R, Anderson R W, Hornbuckle W E, Tennant B C, Cote P J, Gerin J L, Purcell R H, Miller R H. The woodchuck hepatitis virus X gene is important for establishment of virus infection in woodchucks. J Virol. 1993;67:1218–1226. doi: 10.1128/jvi.67.3.1218-1226.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chirillo P, Falco M, Puri P L, Artini M, Balsano C, Levrero M, Natoli G. Hepatitis B virus pX activates NF-κB-dependent transcription through a Raf-independent pathway. J Virol. 1996;70:641–646. doi: 10.1128/jvi.70.1.641-646.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chirillo P, Pagano S, Natoli G, Puri P L, Burgio V L, Balsano C, Levrero M. The hepatitis B virus X gene induces p53-mediated programmed cell death. Proc Natl Acad Sci USA. 1997;94:8162–8167. doi: 10.1073/pnas.94.15.8162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chu Z L, DiDonato J A, Hawiger J, Ballard D W. The Tax oncoprotein of human T-cell leukemia virus type 1 associates with and persistently activates IκB kinases containing IKKα and IKKβ. J Biol Chem. 1998;273:15891–15894. doi: 10.1074/jbc.273.26.15891. [DOI] [PubMed] [Google Scholar]
- 12.DiDonato J A, Hayakawa M, Rothwarf D M, Zandi E, Karin M. A cytokine-responsive IκB kinase that activates the transcription factor NF-κB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 13.Doria M, Klein N, Lucito R, Schneider R J. The hepatitis B virus HBx protein is a dual specificity cytoplasmic activator of Ras and nuclear activator of transcription factors. EMBO J. 1995;14:4747–4757. doi: 10.1002/j.1460-2075.1995.tb00156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dorjsuren D, Lin Y, Wei W, Yamashita T, Nomura T, Hayashi N, Murakami S. RMP, a novel RNA polymerase II subunit 5-interacting protein, counteracts transactivation by hepatitis B virus X protein. Mol Cell Biol. 1998;18:7546–7555. doi: 10.1128/mcb.18.12.7546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eliopoulos A G, Rickinson A B. Epstein-Barr virus: LMP1 masquerades as an active receptor. Curr Biol. 1998;8:196–198. doi: 10.1016/s0960-9822(98)70123-x. [DOI] [PubMed] [Google Scholar]
- 16.Elmore L W, Hancock A R, Chang S-F, Wang X W, Chang S, Callahan C P, Geller D A, Will H, Harris C C. Hepatitis B virus X protein and p53 tumor suppressor interactions in the modulation of apoptosis. Proc Natl Acad Sci USA. 1997;94:14707–14712. doi: 10.1073/pnas.94.26.14707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fischer M, Runkel L, Schaller H. HBx protein of hepatitis B virus interacts with the C-terminal portion of a novel human proteasome alpha-subunit. Virus Genes. 1995;10:99–102. doi: 10.1007/BF01724303. [DOI] [PubMed] [Google Scholar]
- 18.Fornerod M, Ohno M, Yoshida M, Mattaj I W. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 1997;90:1051–1060. doi: 10.1016/s0092-8674(00)80371-2. [DOI] [PubMed] [Google Scholar]
- 19.Fritz C C, Green M R. HIV Rev uses a conserved cellular protein export pathway for the nucleocytoplasmic transport of viral RNAs. Curr Biol. 1996;6:848–854. doi: 10.1016/s0960-9822(02)00608-5. [DOI] [PubMed] [Google Scholar]
- 20.Fukuda M, Asano S, Nakamura T, Adachi M, Yoshida M, Yanagida M, Nishida E. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature. 1997;390:308–311. doi: 10.1038/36894. [DOI] [PubMed] [Google Scholar]
- 21.Geleziunas R, Ferrell S, Lin X, Mu Y, Cunningham E T, Jr, Grant M, Connelly M A, Hambor J E, Marcu K B, Greene W C. Human T-cell leukemia virus type 1 Tax induction of NF-κB involves activation of the IκB kinase α (IKKα) and IKKβ cellular kinases. Mol Cell Biol. 1998;18:5157–5165. doi: 10.1128/mcb.18.9.5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gottlob K, Pagano S, Levrero M, Graessmann A. Hepatitis B virus X protein transcription activation domains are neither required nor sufficient for cell transformation. Cancer Res. 1998;58:3566–3570. [PubMed] [Google Scholar]
- 23.Haviv I, Matza Y, Shaul Y. pX, the HBV-encoded coactivator, suppresses the phenotypes of TBP and TAFII250 mutants. Genes Dev. 1998;15:1217–1226. doi: 10.1101/gad.12.8.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haviv I, Shamay M, Doitsh G, Shaul Y. Hepatitis B virus pX targets TFIIB in transcription coactivation. Mol Cell Biol. 1998;18:1562–1569. doi: 10.1128/mcb.18.3.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hildt E, Hofschneider P, Urban S. The role of hepatitis B virus (HBV) in the development of hepatocellular carcinoma. Virology. 1996;7:333–347. [Google Scholar]
- 26.Huang J, Kwong J, Sun E C-Y, Liang T J. Proteasome complex as a potential cellular target of hepatitis B virus X protein. J Virol. 1996;70:5582–5591. doi: 10.1128/jvi.70.8.5582-5591.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huxford T, Huang D B, Malek S, Ghosh G. The crystal structure of the IκBα/NF-κB complex reveals mechanisms of NF-κB inactivation. Cell. 1998;95:759–770. doi: 10.1016/s0092-8674(00)81699-2. [DOI] [PubMed] [Google Scholar]
- 28.Jacobs M D, Harrison S C. Structure of an IκBα/NF-κB complex. Cell. 1998;95:749–758. doi: 10.1016/s0092-8674(00)81698-0. [DOI] [PubMed] [Google Scholar]
- 29.Kekule A S, Lauer U, Weiss L, Luber B, Hofschneider P H. Hepatitis B virus transactivator HBx uses a tumour promoter signalling pathway. Nature. 1993;361:742–745. doi: 10.1038/361742a0. [DOI] [PubMed] [Google Scholar]
- 30.Kim H, Lee H, Yun Y. X-gene product of hepatitis B virus induces apoptosis in liver cells. J Biol Chem. 1998;273:381–385. doi: 10.1074/jbc.273.1.381. [DOI] [PubMed] [Google Scholar]
- 31.Lamond A I, Earnshaw W C. Structure and function in the nucleus. Science. 1998;280:547–553. doi: 10.1126/science.280.5363.547. [DOI] [PubMed] [Google Scholar]
- 32.Lara-Pezzi E, Armesilla A L, Majano P L, Redondo J M, Lopez-Cabrera M. The hepatitis B virus X protein activates nuclear factor of activated T cells (NF-AT) by a cyclosporin A-sensitive pathway. EMBO J. 1998;17:7066–7077. doi: 10.1093/emboj/17.23.7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lara-Pezzi E, Majano P L, Gomez-Gonzalo M, Garcia-Monzon C, Moreno-Otero R, Levrero M, Lopez-Cabrera M. The hepatitis B virus X protein up-regulates tumor necrosis factor alpha gene expression in hepatocytes. Hepatology. 1998;28:1013–1021. doi: 10.1002/hep.510280416. [DOI] [PubMed] [Google Scholar]
- 34.Lee Y H, Yun Y D. HBx protein of hepatitis B virus activates Jak1-Stat signaling. J Biol Chem. 1998;273:25510–25515. doi: 10.1074/jbc.273.39.25510. [DOI] [PubMed] [Google Scholar]
- 35.Mahe Y, Mukaida N, Kuno K, Akiyama M, Ikeda N, Matsushima K, Murakami S. Hepatitis B virus X protein transactivates human interleukin-8 gene through acting on nuclear factor κB and CCAAT/enhancer-binding protein-like cis-elements. J Biol Chem. 1991;266:13759–13763. [PubMed] [Google Scholar]
- 36.May M J, Ghosh S. Signal transduction through NF-κB. Immunol Today. 1998;19:80–88. doi: 10.1016/s0167-5699(97)01197-3. [DOI] [PubMed] [Google Scholar]
- 37.Mercurio F, Zhu H Y, Murray B W, Shevchenko A, Bennett B L, Li J W, Young D B, Barbosa M, Mann M. IKK-1 and IKK-2—cytokine-activated IκB kinases essential for NF-κB activation. Science. 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- 38.Oguey D, Dumenco L, Pierce R, Fausto N. Analysis of tumorigenicity of the X gene of hepatitis B virus in a nontransformed hepatocyte cell line and the effects of cotransfection with a murine p53 mutant equivalent to human codon 249. Hepatology. 1996;24:1024–1033. doi: 10.1002/hep.510240508. [DOI] [PubMed] [Google Scholar]
- 39.Ossareh-Nazari B, Bachelerie F, Dargemont C. Evidence for a role of CRM1 in signal-mediated nuclear protein export. Science. 1997;278:141–144. doi: 10.1126/science.278.5335.141. [DOI] [PubMed] [Google Scholar]
- 40.Quadri I, Maguire H, Siddiqui A. Hepatitis B virus transactivator protein X interacts with the TATA-binding protein. Proc Natl Acad Sci USA. 1995;92:1003–1007. doi: 10.1073/pnas.92.4.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Regnier C H, Song H Y, Gao X, Goeddel D V, Cao Z D, Rothe M. Identification and characterization of an IκB kinase. Cell. 1997;90:373–383. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- 42.Rodriguez M S, Michalopoulos I, Arenzana-Seisdedos F, Hay R T. Inducible degradation of IκBα in vitro and in vivo requires the acidic C-terminal domain of the protein. Mol Cell Biol. 1995;15:2413–2419. doi: 10.1128/mcb.15.5.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rossner M T. Hepatitis B virus X-gene product: a promiscuous transcriptional activator. J Med Virol. 1992;36:101–117. doi: 10.1002/jmv.1890360207. [DOI] [PubMed] [Google Scholar]
- 44.Rothwarf D M, Zandi E, Natoli G, Karin M. IKK-γ is an essential regulatory subunit of the IκB kinase complex. Nature. 1998;395:297–300. doi: 10.1038/26261. [DOI] [PubMed] [Google Scholar]
- 45.Sachdev S, Hannink M. Loss of IκBα-mediated control over nuclear import and DNA binding enables oncogenic activation of c-Rel. Mol Cell Biol. 1998;18:5445–5456. doi: 10.1128/mcb.18.9.5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sachdev S, Hoffmann A, Hannink M. Nuclear localization of IκBα is mediated by the second ankyrin repeat: the IκBα ankyrin repeats define a novel class of cis-acting nuclear import sequences. Mol Cell Biol. 1998;18:2524–2534. doi: 10.1128/mcb.18.5.2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Siddiqui A, Gaynor R, Srinivasan A, Mapoles J, Farr R W. Trans-activation of viral enhancers including long terminal repeat of the human immunodeficiency virus by the hepatitis B virus X protein. Virology. 1989;169:479–484. doi: 10.1016/0042-6822(89)90177-3. [DOI] [PubMed] [Google Scholar]
- 48.Sirma H, Weil R, Rosmorduc O, Urban S, Israël A, Kremsdorf D, Bréchot C. Cytosol is the prime compartment of hepatitis B virus where it colocalizes with the proteasome. Oncogene. 1998;16:2051–2063. doi: 10.1038/sj.onc.1201737. [DOI] [PubMed] [Google Scholar]
- 49.Sitterlin D, Lee T-H, Prigent S, Tiollais P, Butel J S, Transy C. Interaction of the UV-damaged DNA-binding protein with hepatitis B virus X protein is conserved among mammalian hepadnaviruses and restricted to transactivation-proficient X-insertion mutants. J Virol. 1997;71:6194–6199. doi: 10.1128/jvi.71.8.6194-6199.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stade K, Ford C S, Guthrie C, Weis K. Exportin 1 (Crm1p) is an essential nuclear export factor. Cell. 1997;90:1041–1050. doi: 10.1016/s0092-8674(00)80370-0. [DOI] [PubMed] [Google Scholar]
- 51.Su F, Schneider R J. Hepatitis B virus HBx protein activates transcription factor NF-κB by acting on multiple cytoplasmic inhibitors of rel-related proteins. J Virol. 1996;70:4558–4566. doi: 10.1128/jvi.70.7.4558-4566.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Su F, Schneider R J. Hepatitis B virus HBx protein sensitizes cells to apoptotic killing by tumor necrosis factor α. Proc Natl Acad Sci USA. 1997;94:8744–8749. doi: 10.1073/pnas.94.16.8744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Su Q, Shroder C, Hofmann W, Otto G, Pichlmayr R, Bannasch P. Expression of hepatitis B virus X protein in HBV-infected human livers and hepatocellular carcinomas. Hepatology. 1998;27:1109–1120. doi: 10.1002/hep.510270428. [DOI] [PubMed] [Google Scholar]
- 54.Takada S, Kaneniwa N, Tsuchida N, Koike K. Cytoplasmic retention of the p53 tumor suppressor gene product is observed in the hepatitis B virus X gene-transfected cells. Oncogene. 1997;15:1895–1901. doi: 10.1038/sj.onc.1201369. [DOI] [PubMed] [Google Scholar]
- 55.Terradillos O, Pollicino T, Lecoeur H, Tripodi M, Gougeon M, Tiollais P, Buendia M. p-53-independent apoptotic effects of the hepatitis B virus HBx protein in vivo and in vitro. Oncogene. 1998;17:2115–2123. doi: 10.1038/sj.onc.1202432. [DOI] [PubMed] [Google Scholar]
- 56.Traenckner E B, Baeuerle P A. Appearance of apparently ubiquitin-conjugated IκBα during its phosphorylation-induced degradation in intact cells. J Cell Sci Suppl. 1995;19:79–84. doi: 10.1242/jcs.1995.supplement_19.11. [DOI] [PubMed] [Google Scholar]
- 57.Turpin P, Hay R T, Dargemont C. Characterization of IκBα nuclear import pathway. J Biol Chem, 1999;274:6804–6812. doi: 10.1074/jbc.274.10.6804. [DOI] [PubMed] [Google Scholar]
- 58.Twu J S, Chu K, Robinson W S. Hepatitis B virus X gene activates κ B-like enhancer sequences in the long terminal repeat of human immunodeficiency virus 1. Proc Natl Acad Sci USA. 1989;86:5168–5172. doi: 10.1073/pnas.86.13.5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Uhlik M, Good L, Xiao G T, Harhaj E W, Zandi E, Karin M, Sun S C. NF-κB-inducing kinase and IκB kinases participate in human T-cell leukemia virus I Tax-mediated NF-κB activation. J Biol Chem. 1998;273:21132–21136. doi: 10.1074/jbc.273.33.21132. [DOI] [PubMed] [Google Scholar]
- 60.Verma I M, Stevenson J K, Schwarz E M, Van Antwerp D, Miyamoto S. Rel/NF-κB/IκB family: intimate tales of association and dissociation. Genes Dev. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- 61.Wang H-D, Trivedi A, Johnson D L. Regulation of RNA polymerase I-dependent promoters by the hepatitis B virus X protein via activated Ras and TATA-binding protein. Mol Cell Biol. 1998;18:7086–7094. doi: 10.1128/mcb.18.12.7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61a.Weil, R. Unpublished data.
- 62.Weil R, Laurent-Winter C, Israël A. Regulation of IκBβ degradation—similarities to and differences from IκBα. J Biol Chem. 1997;272:9942–9949. doi: 10.1074/jbc.272.15.9942. [DOI] [PubMed] [Google Scholar]
- 63.Whiteside S T, Epinat J C, Rice N R, Israël A. IκBɛ, a novel member of the IκB family, controls relA and c-rel NF-κB activity. EMBO J. 1997;16:1413–1426. doi: 10.1093/emboj/16.6.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Whiteside S T, Ernst M K, LeBail O, Laurent-Winter C, Rice N, Israël A. N- and C-terminal sequences control degradation of MAD3/IκBα in response to inducers of NF-κB activity. Mol Cell Biol. 1995;15:5339–5345. doi: 10.1128/mcb.15.10.5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Williams J S, Andrisani O M. The hepatitis B virus X protein targets the basic region-leucine zipper domain of CREB. Proc Natl Acad Sci USA. 1995;92:3819–3823. doi: 10.1073/pnas.92.9.3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Woronicz J D, Gao X, Cao Z, Rothe M, Goeddel D V. IκB kinase-β: NF-κB activation and complex formation with IκB kinase-α and NIK. Science. 1997;278:866–869. doi: 10.1126/science.278.5339.866. [DOI] [PubMed] [Google Scholar]
- 67.Yamaoka S, Courtois G, Bessia C, Whiteside S T, Weil R, Agou F, Kirk H E, Kay R J, Israël A. Complementation cloning of NEMO, a component of the IκB kinase complex essential for NF-κB activation. Cell. 1998;93:1231–1240. doi: 10.1016/s0092-8674(00)81466-x. [DOI] [PubMed] [Google Scholar]
- 68.Yen T. Hepadna viral X protein: review of recent progress. J Biomed Sci. 1996;3:20–30. doi: 10.1007/BF02253575. [DOI] [PubMed] [Google Scholar]
- 69.Yin M J, Christerson L B, Yamamoto Y, Kwak Y T, Xu S, Mercurio F, Barbosa M, Cobb M H, Gaynor R B. HTLV-I Tax protein binds to MEKK1 to stimulate IκB kinase activity and NF-κB activation. Cell. 1998;93:875–884. doi: 10.1016/s0092-8674(00)81447-6. [DOI] [PubMed] [Google Scholar]
- 70.Zandi E, Rothwarf D M, Delhase M, Hayakawa M, Karin M. The IκB kinase complex (IKK) contains two kinase subunits, IKK-α and IKK-β, necessary for IκB phosphorylation and NF-κB activation. Cell. 1997;91:243–252. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]
- 71.Zoulim F, Saputelli J, Seeger C. Woodchuck hepatitis virus X protein is required for viral infection in vivo. J Virol. 1994;68:2026–2030. doi: 10.1128/jvi.68.3.2026-2030.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]