

SUMMARY
Kinetoplastids, including Trypanosoma brucei, control gene expression primarily at the posttranscriptional level. Nuclear mRNA export is an important, but understudied, step in this process. The general heterodimeric export factors, Mex67/Mtr2, function in the export of mRNAs and tRNAs in T. brucei, but RNA binding proteins (RBPs) that regulate export processes by controlling the dynamics of Mex67/Mtr2 ribonucleoprotein formation or transport have not been identified. Here, we report that DRBD18, an essential and abundant T. brucei RBP, associates with Mex67/Mtr2 in vivo, likely through its direct interaction with Mtr2. DRBD18 downregulation results in partial accumulation of poly(A)+ mRNA in the nucleus, but has no effect on localization of intron-containing or mature tRNAs. Comprehensive analysis of transcriptomes from whole cell and cytosol in DRBD18 knockdown parasites demonstrates that depletion of DRBD18 leads to impairment of nuclear export of a subset of mRNAs. CLIP experiments reveal association of DRBD18 with several of these mRNAs. Moreover, DRBD18 knockdown leads to a partial accumulation of the Mex67/Mtr2 export receptors in the nucleus. Taken together, the current study supports a model in which DRBD18 regulates the selective nuclear export of mRNAs by promoting the mobilization of export competent mRNPs to the cytosol through the nuclear pore complex.
Keywords: Trypanosome, RNA binding protein, mRNA export, RNAseq, FISH, Nucleoporin
Graphical Abstract
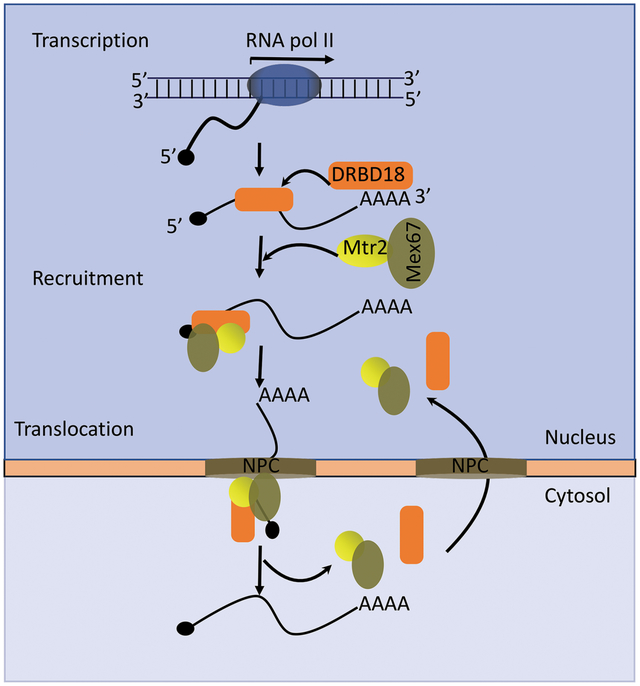
1. INTRODUCTION
Trypanosoma brucei species are early diverged unicellular eukaryotic parasites causing African Sleeping Sickness in humans and Nagana in domestic animals (Keating et al., 2015, Büscher et al., 2017). They are members of the family of Kinetoplastida, which includes other parasites such as Trypanosoma cruzi, the causative agent of Chagas disease, and various species of Leishmania that cause leishmaniases. These parasites all have complex life cycles that alternate between an insect vector and the mammalian host. In T. brucei, two well studied life cycle stages are the procyclic form (PF), which is found in the tsetse fly vector and the bloodstream form (BF), present in the blood of the infected animal host (Matthews, 2005). T. brucei exhibits several unique biological features, such as a condensed bipartite mitochondrial genome with its unique replication machinery, uridine insertion/deletion RNA editing of mitochondrial mRNAs, as well as a complex mechanism to evade the host immune system by frequent changes of variant surface glycoproteins (Jensen & Englund, 2012, Verner et al., 2015, Bangs, 2018, Zimmer et al., 2018, Aphasizheva et al., 2020). The process of transcription in these parasites is also unusual. Most protein coding genes are transcribed by RNA polymerase II to produce long polycistronic units which are co-transcriptionally processed to monocistronic mRNAs. Individual mRNAs are produced by 5’ trans-splicing that adds a 39-nt long spliced leader RNA to the 5′ end of each monocistronic RNA and 3’ addition of a poly(A) tail (Michaeli, 2011). Due to the absence of promoters upstream of individual genes, gene regulation in T. brucei takes place primarily at the posttranscriptional level (Clayton, 2019).
Posttranscriptional gene expression in eukaryotes involves multiple steps including processing of nascent transcripts, nuclear export of mature mRNAs, and regulation of both mRNA decay and translational efficiency. mRNA export machineries have been extensively studied in the model organism Saccharomyces cerevisiae and later in metazoan species (Köhler & Hurt, 2007, Wickramasinghe & Laskey, 2015). In the established universal model of mRNA export, pre-mRNA is co-transcriptionally assembled to a messenger ribonucleoprotein (mRNP) by the TREX complex, followed by mRNA processing and maturation. Adaptor proteins that exhibit RNA binding activity recruit the heterodimeric export receptor Mex67/Mtr2 to the mRNP and promote export through the nuclear pore complex (NPC) (Tutucci & Stutz, 2011). In trypanosomes, homologues of Mex67 and Mtr2 interact and function as nuclear export receptors for both mRNA and tRNA (Schwede et al., 2009, Dostalova et al., 2013, Hegedűsová et al., 2019). Nevertheless, the domain organization of Mex67 in T. brucei is quite different than that of its yeast and metazoan orthologues. Like yeast or metazoan Mex67, T. brucei Mex67 contains both leucine-rich repeat (LRR) and an NTF2-like domains; however, it lacks an RNA recognition motif (RRM) and a ubiquitin-associated domain (UBA) (Dostalova et al., 2013, Rink & Williams, 2019). Moreover, T. brucei Mex67 contains a CCCH zinc finger (ZC3H) motif in its N-terminal region, which is essential for mRNA export (Dostalova et al., 2013). Other aspects of the nuclear export machinery also differ in T. brucei compared to other well studied systems. Unlike yeast or metazoans, trypanosomatids contain no recognizable components of the TREX complex, with the exception of the DEAD box helicase, Sub2 (Serpeloni et al., 2011a, Dostalova et al., 2013). Recent studies suggest that active transcription is necessary to initiate mRNA export in T. brucei, although neither the completion of transcription nor splicing appear to be essential for export (Goos et al., 2018).
RNA binding proteins (RBPs) play major roles in gene expression and development in trypanosomes. In some cases, RBPs have been shown to bind specific cis-acting regulatory elements present in the 3’-untranslated regions (3’-UTRs) of mRNAs to modulate mRNA stability or translation (Kolev et al., 2014, Clayton, 2019). RBPs are classified into different groups based on the presence of distinct RNA binding domains, with RNA recognition motif (RRM) and ZC3H-containing proteins being the most common RBPs in trypanosomes (Kolev et al., 2014). Examples of the impacts of RBPs in T. brucei include the critical roles in developmental gene regulation played by RRM-containing RBPs, RBP10 and RBP6, and REG9.1 (Kolev et al., 2012, Mugo & Clayton, 2017, Rico et al., 2017). With regard to ZC3H proteins, ZC3H11 mediates gene expression during heat shock (Droll et al., 2013), and ZC3H20 binds specifically to a subset of mRNAs expressed in PFs (Liu et al., 2020).
DRBD18 is an essential and abundant an RRM-containing RBP in T. brucei (Lott et al., 2015). Our previous studies demonstrated that depletion of DRBD18 causes extensive transcriptome rearrangement in PF T. brucei through both stabilization or destabilization of distinct subsets of mRNAs. The molecular functions of DRBD18 in mRNA stabilization or destabilization and protein-protein interactions are markedly controlled by methylation of arginine residues present between its two RRM domains. Towards an understanding of the biochemical mechanisms of DRBD18 function, mass spectrometry analysis of Tandem Affinity Purified (TAP)-tagged DRBD18 revealed the interaction of DRBD18 with numerous proteins involved in RNA biology, including the general mRNA export receptors Mex67 and Mtr2, and the Sub2 RNA helicase (Lott et al., 2015). Previous immunofluorescence analysis revealed a strong perinuclear concentration of DRBD18 that, together with its interaction with Mex67/Mtr2, is consistent with a role for DRBD18 in nucleocytosolic mRNA transport (Lott et al., 2015). In the present study, we confirm the in vivo association between DRBD18 and Mex67/Mtr2 and demonstrate that DRBD18 and Mtr2 directly interact in vitro. High throughput sequencing and immunofluorescence studies show that DRBD18 promotes the export of a subset of mRNAs from nucleus to cytosol. At the same time, DRBD18 also facilitates the export of a fraction of Mex67/Mtr2 from the nucleus. In contrast, DRBD18 does not share a function with Mex67/Mtr2 in the export of intron-containing or mature tRNAs. The mRNA export machinery is not well characterized in T. brucei due to the absence of many conserved export factors. The current study is the first to document a T. brucei RBP that targets the mRNA export machinery.
2. RESULTS
2.1. DRBD18 interacts with export receptor proteins Mex67 and Mtr2 in vivo
Our previous mass spectrometry analysis suggested that DRBD18 associates with several RNA processing complexes, including the nuclear export factors, Mex67 and Mtr2 (Lott et al., 2015). These two factors are evolutionarily conserved and typically form a heterodimer, although distinct functions for Mex67 and Mtr2 have also been reported (Braun et al., 2002, Dostalova et al., 2013, Hegedűsová et al., 2019). To explore whether DRBD18 plays a role in Mex67/Mtr2 mediated RNA export, we began by confirming the interactions of Mex67 and Mtr2 with DRBD18 in vivo. For this purpose, we generated PF T. brucei cells expressing HA-tagged Mtr2 from its genomic locus. Immunoprecipitation (IP) using anti-HA antibody in the aforementioned cell line confirmed that Mtr2 associates with DRBD18, as well as with Mex67 as expected, while both are absent in a control IP (Fig. 1A). As DRBD18 is an RNA binding protein (Lott et al., 2015), we next asked whether the DRBD18-Mtr2 interaction is impacted by RNA by performing co-IPs using HA-Mtr2 tagged T. brucei cell lysate treated either with an RNase cocktail or with RNase inhibitor (Figs. 1B and 1C). DRBD18 consistently displayed an increased interaction with Mtr2 in the absence of RNA, whereas the interaction between Mex67 and Mtr2 was unaffected by RNA. To further probe the RNA dependence of DRBD18-Mex67/Mtr2 interaction, we immunoprecipitated DRBD18 with anti-DRBD18 antibody from the HA-Mtr2 tagged T. brucei cell lysate in presence and absence of RNase, and demonstrated increased co-immunoprecipitation of both Mex67 and Mtr2 with DRBD18 in RNase treated samples (Figs. 1D and 1E). Thus, we conclude that DRBD18 associates with Mex67/Mtr2 in vivo, and that this interaction is strengthened in the absence of RNA.
Figure 1. DRBD18 interacts with the general export receptor proteins Mex67 and Mtr2 in vivo.
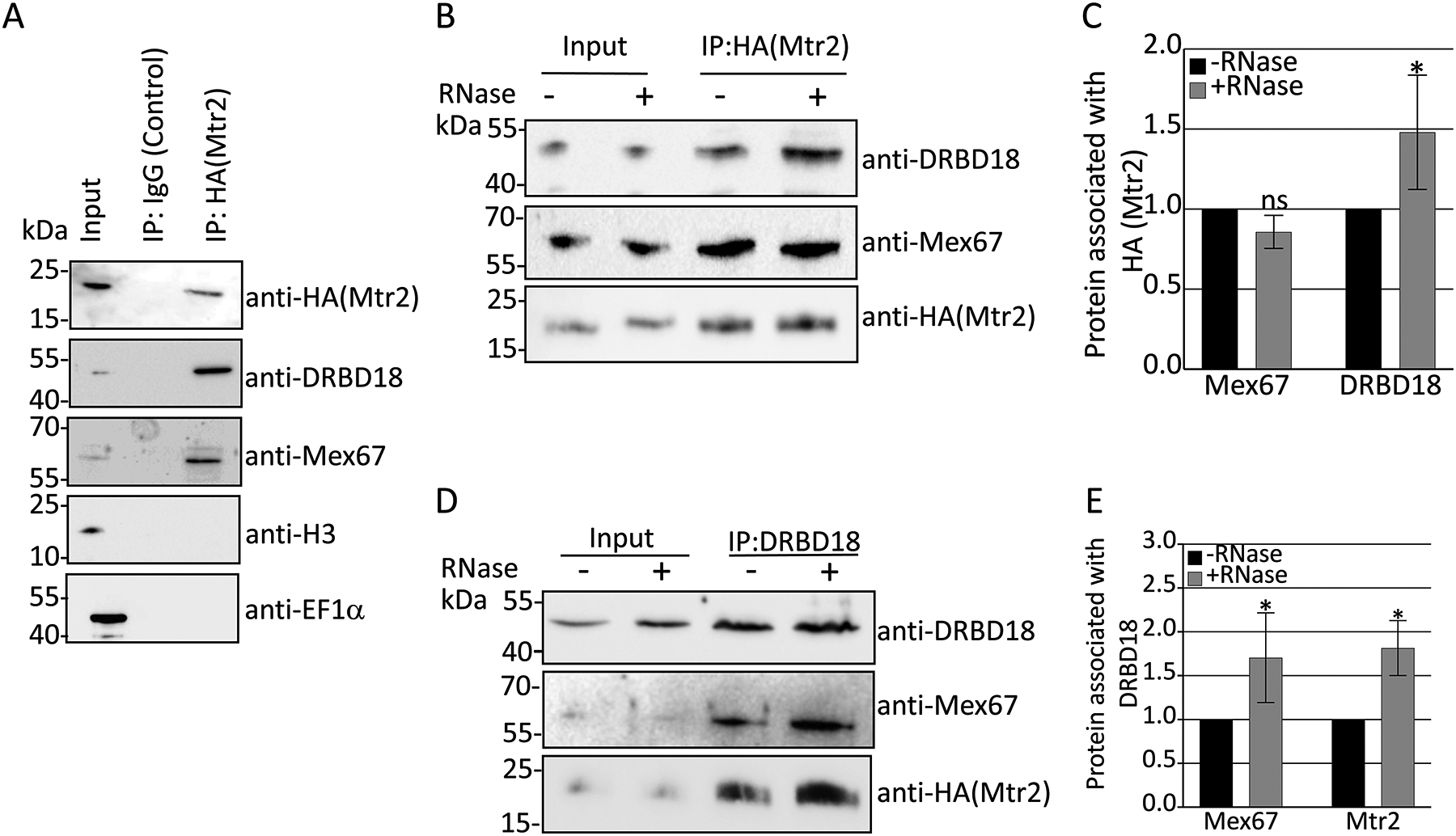
(A) Immunoprecipitations (IP) of T. brucei cells expressing 3XHA-tagged Mtr2 from its genomic locus were carried out using anti-HA beads or IgG Sepharose beads (Control). Bound proteins were eluted using 100 mM glycine and analyzed by Western blot with anti-HA, anti-DRBD18, anti-Mex67, anti-Histone H3 (H3), and anti-EF1α antibodies. Migration of molecular weight standards (kDa) is indicated on the left. Two biological replicate experiments, each with two technical replicates, were performed, and a representative experiment is shown. (B) HA-Mtr2 and associated proteins were immunoprecipitated from T. brucei cell extracts that were treated with either an RNase cocktail (+RNase) or RNase inhibitor (−RNase). Proteins were eluted and Western blots performed as in (A). Two biological replicate experiments, each with two technical replicates, were performed, and a representative experiment is shown. (C) Quantification of band intensities of the experiment shown in (B). Protein levels were normalized to amount of HA-Mtr2 for a given IP. The normalized protein levels from the +RNase IP were then compared to that of the −RNase IP (which was set to 1) to calculate the protein associated with HA-Mtr2. Bar graphs represent the average and standard deviation (SD) of two biological replicates, each with two technical replicates. (D) DRBD18 was immunoprecipitated from HA-Mtr2 expressing cells with specific antibodies. Proteins were eluted and Western blots with anti-HA and anti-Mex67 antibodies were performed as in (A). Two biological replicate experiments, each with two technical replicates, were performed, and a representative experiment is shown. (E) Quantification of band intensities from the experiment shown in (D). Bar graphs represent the average and standard deviation (SD) of two biological replicates, each with two technical replicates. Significance was determined by unpaired t-test with Welch’s correction. * indicates p < 0.05.
2.2. DRBD18 directly interacts with Mtr2 in vitro
Having demonstrated the in vivo DRBD18-Mex67/Mtr2 interaction, we next asked if DRBD18 interacts directly with the export receptors. To this end, we expressed and purified recombinant His-Mtr2, GST-DRBD18, and GST in E. coli as described in Experimental Procedures. Purity of the recombinant proteins was analyzed by SDS-PAGE analysis (Fig. 2A). Next, we performed GST-pulldown assays using GST-DRBD18 as bait protein, GST as a negative control, and purified His-Mtr2 as prey protein. For this purpose, we incubated equimolar amounts of purified His-tagged Mtr2 with the immobilized GST or GST-DRBD18. Western blot analysis showed that Mtr2 is pulled down by the GST-tagged DRBD18 but not by GST, indicating that DRBD18 interacts directly with Mtr2 (Fig. 2B). These data collectively demonstrate that DRBD18 interacts with Mtr2 through a protein-protein interaction.
Figure 2. In vitro interaction of DRBD18 with Mex67 and Mtr2.
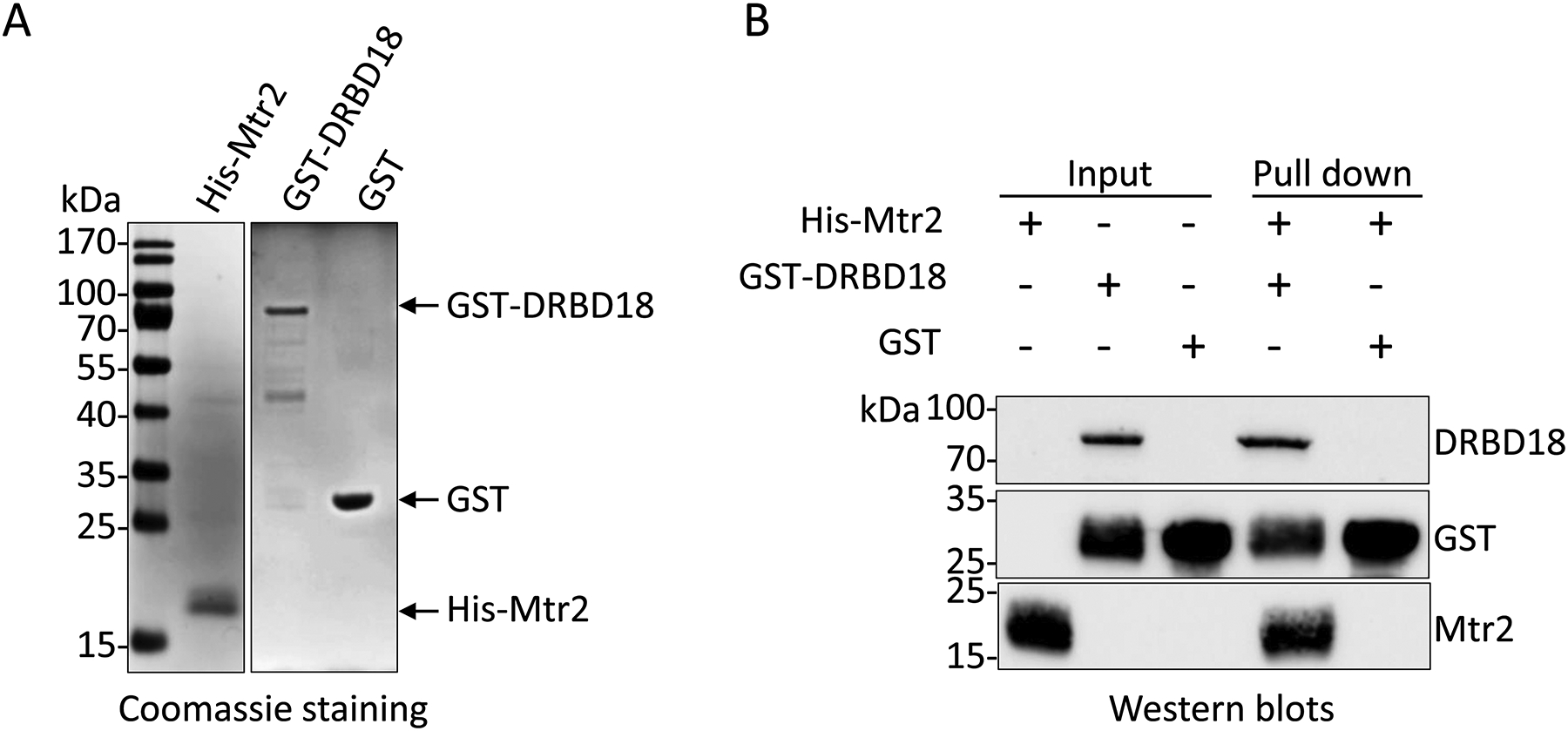
(A) Recombinant His-tagged Mtr2, GST-tagged DRBD18, and GST were expressed, purified by affinity chromatography, and analyzed by 12.5% SDS-PAGE followed by Coomassie brilliant blue staining. Migration of molecular weight standards (kDa) is indicated on the left. (B) Recombinant GST-DRBD18 or GST were immobilized on glutathione-Sepharose 4B beads, followed by incubation with His-Mtr2 for GST pulldown assays. Western blot analyses were performed using anti-His and anti-GST antibodies to detect His-Mtr2 and GST or GST-DRBD18, respectively. Purified His-Mtr2 (in lane 1), and GST-DRBD18 or GST on glutathione-Sepharose 4B beads incubated without His-Mtr2 were used as inputs for the experiments (in lane 2 and 3 respectively). Two biological replicates, each with two technical replicates, were performed, and a representative experiment is shown.
2.3. DRBD18 does not impact Mex67/Mtr2-dependent export of tRNAs
To begin to understand the functional significance of the DRBD18-Mex67/Mtr2 interaction, we first confirmed that DRBD18 does not alter the steady state levels or heterodimerization of Mex67 and Mtr2 (Fig. S1). As these parameters are unaffected by DRBD18 knockdown, the interaction between DRBD18 and Mex67/Mtr2 suggests that DRBD18 could impact some functions of these export factors. It was recently reported that Mex67 and Mtr2 play important and distinct roles in tRNA nuclear export in T. brucei (Hegedűsová et al., 2019). Knockdown of Mtr2 results in nuclear accumulation of all type of tRNAs, while Mex67 knockdown leads to nuclear accumulation of subsets of tRNAs that are modified with queuosine. To investigate the functional role of the DRBD18-Mex67/Mtr2 interaction in tRNA export, we analyzed total RNA from uninduced and induced DRBD18 RNAi cells by Northern hybridization. We first monitored the splicing of tRNATyr, which occurs exclusively in the cytosol in T. brucei and, thus, serves as a measure of the export of immature, intron-containing tRNATyr. We observed no effect of DRBD18 knockdown on this parameter, similar to the lack of Mex67 or Mtr2 impact on export of this immature tRNA (Fig. S2). Following splicing, tRNATyr undergoes retrograde import into the nucleus, at which point it acquires a queuosine modification and is then re-exported. Impairment of this re-export step, which requires both Mex67 and Mtr2, results in aberrantly high queuosine levels on tRNATyr (Hegedűsová et al., 2019). We next asked whether DRBD18 exerts any modulation of the function of Mex67 and Mtr2 in the re-export of tRNATyr by analyzing the level of queuosine modified tRNATyr using boronate affinity gel electrophoresis followed by Northern blot hybridization (Kessler et al., 2018). RNA samples treated with the oxidizing agent, sodium periodate, served as a negative control (Fig. 3A). We detected similar levels of queuosine modified tRNATyr in RNA from uninduced and induced DRBD18 RNAi cells (Fig. 3A), indicating that DRBD18 does not impact the Mex67/Mtr2 mediated re-export of queuosine modified tRNATyr. To further investigate the potential role of DRBD18 in tRNA export, we analyzed mature tRNATyr and tRNAGlu (Figs. 3B and 3C) by fluorescence in situ hybridization (FISH) using specific fluorophore labeled oligonucleotide probes (Table S1). These probes detect spliced tRNA, regardless of its queuosine modification status. Unlike what is observed in Mtr2 or Mex67 knockdowns, we did not observe any nuclear accumulation of mature tRNATyr or tRNAGlu when DRBD18 is knocked down. Collectively, these data show that DRBD18 does not impact the functions of Mex67 or Mtr2 with respect to their roles in the export or re-export of tRNAs.
Figure 3. DRBD18 RNAi does not impact tRNA export.
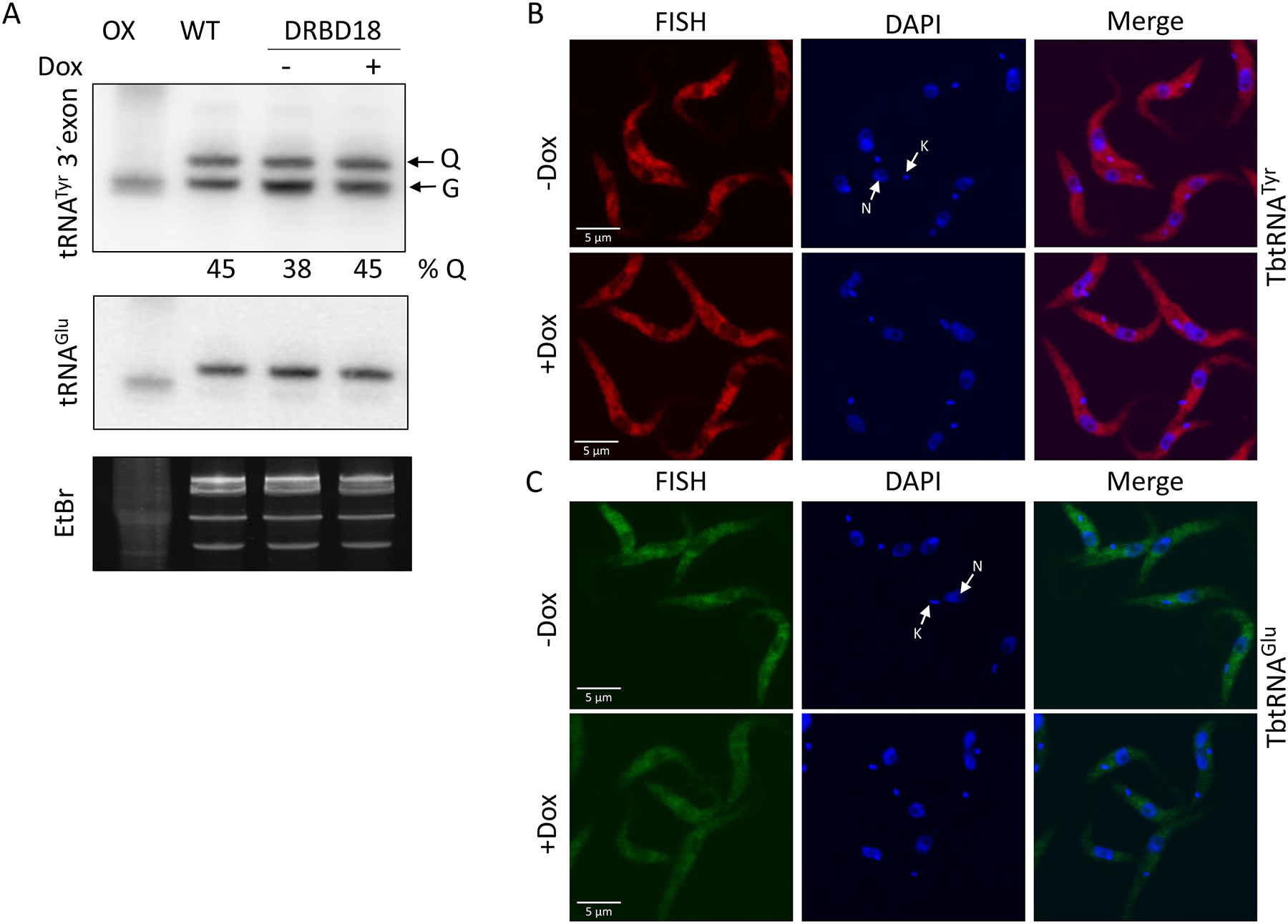
(A)T. brucei cells harboring the DRBD18 RNAi construct were grown for 23 h in the presence (+Dox) or absence (−Dox) of doxycycline. Total RNA was separated by boronate affinity electrophoresis followed by Northern blotting. A probe recognizing the tRNATyr 3’ exon was used to detect queuosine (Q)-modified tRNATyr levels, based on the electrophoretic shift caused by the presence of Q as described elsewhere (Igloi & Kössel, 1985). G indicates unmodified tRNATyr. The levels of Q-modified tRNA are shown below the panel. WT and OX indicate untreated and sodium periodate treated cells as a negative control. A probe against tRNAGlu (middle panel) and the ethidium bromide stained gel (bottom panel) were used as a loading controls. (B and C) Subcellular localization of tRNATyr (red-Cy3) (B) and tRNAGlu (green-AF488) (C) in uninduced (−Dox) or induced (+Dox) DRBD18 RNAi cells were monitored by fluorescence in situ hybridization (FISH). DAPI was used to stain the DNA of the nucleus (N) and the kinetoplast (K). An overlay of the FISH and DAPI signals (Merge) is shown.
2.5. DRBD18 plays role in mRNA export from nucleus to cytosol
Mex67/Mtr2 plays an important role in mRNA transport from nucleus to cytosol in T. brucei (Dostalova et al., 2013), but the mechanisms of export are poorly understood. The interaction of DRBD18 with these export factors (Figs. 1 and 2) suggests that DRBD18 could modulate export of at least some mRNAs. To determine whether DRBD18 exerts any functional role on mRNA export from nucleus to cytosol, we first assessed the subcellular location of poly(A)+ RNA in DRBD18 RNAi uninduced and induced cells by oligo(dT) FISH (Fig. 4A). To quantify the mRNA distribution in the nucleus, we plotted the fluorescence intensities of the FISH and DAPI signals across the nucleus, with overhangs covering the cytosol, across multiple cells (Fig. 4B). In DRBD18 RNAi induced cells, we observed substantial overlap between FISH signals and DAPI signals (lower panel), while uninduced cells exhibited minimum overlap between two fluorophores (upper panel). These observations demonstrate that DRBD18 depletion causes partial accumulation of mRNA in nucleus, thereby indicating a role for DRBD18 in export of mature mRNA from nucleus to cytosol.
Figure 4. DRBD18 RNAi causes partial accumulation of mRNA in the nucleus.
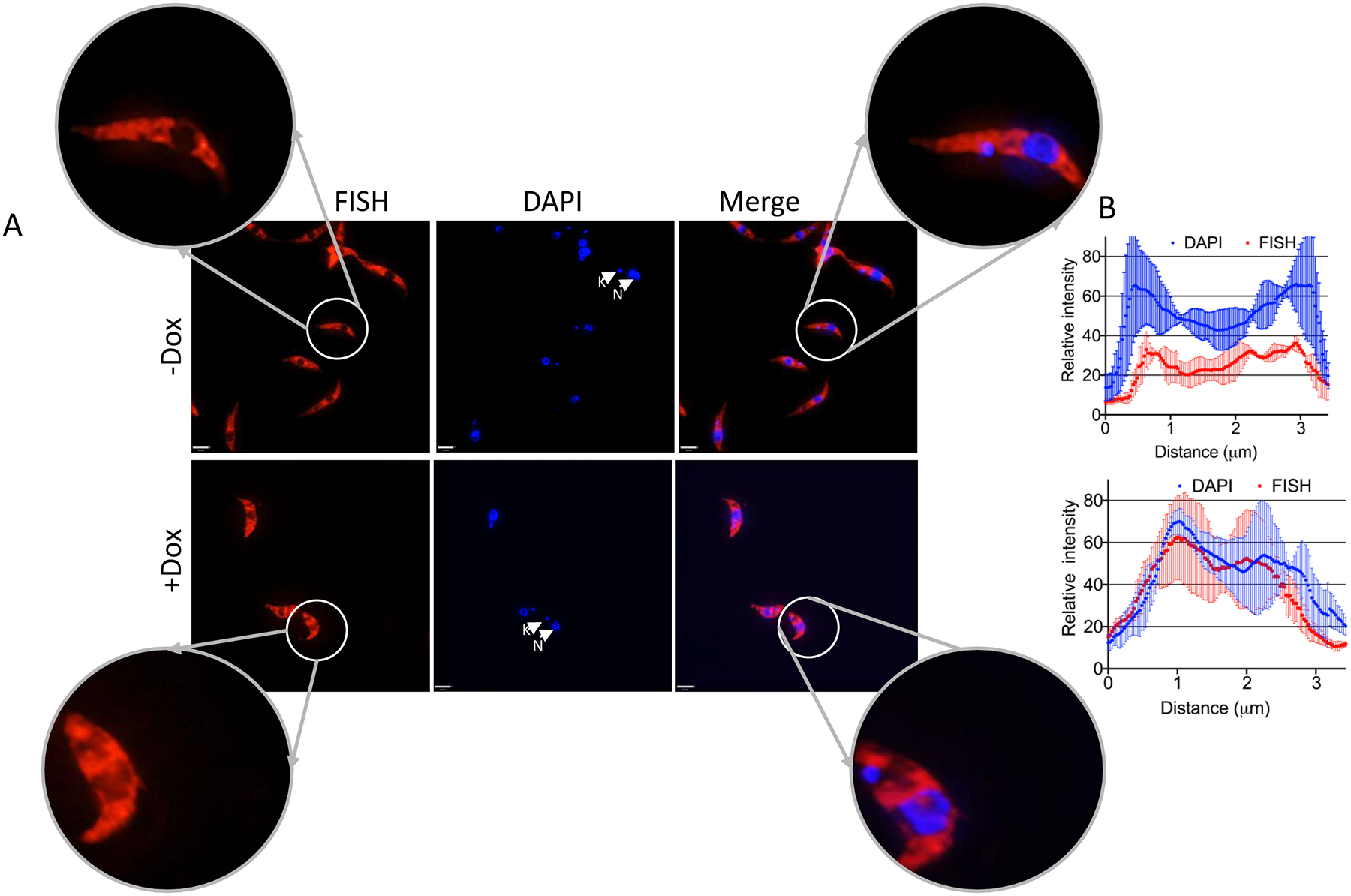
PF T. brucei cells harboring the DRBD18 RNAi construct were used to assess the relative abundance of mRNA in the nucleus upon depletion of DRBD18. (A) Subcellular localization of total mRNA in uninduced (−Dox) or induced (+Dox) DRBD18 RNAi cells was monitored by fluorescence in situ hybridization (FISH) using fluorescently labelled oligo(dT). DAPI was used to stain the DNA of the nucleus (N) and the kinetoplast (K). An overlay of the FISH and DAPI signals (Merge) is shown. Bars, 3.2 μm. Images of individual cells were enlarged to enhance visualization of the nucleus. (B) Quantification of the fluorescence intensities of FISH (red-Alexa 594) and DAPI (blue) fluorophores were calculated on a line drawn across the nucleus, with overhangs covering the cytosol, using ImageJ (NIH) software. Two biological replicate experiments were performed. The relative intensities represent the average ± SD from six randomly selected cells (three from each replicate).
2.6. DRBD18 regulates gene expression by promoting the export of a subset of mRNAs
The partial nuclear retention of mRNA in DRBD18 depleted cells (Fig. 4) suggests that DRBD18 impacts nuclear export of only a subset of mRNAs. To identify a cohort of mRNAs whose export is affected by DRBD18, we carried out Illumina NextSeq analysis of total RNA and cytosolic RNA from uninduced and induced DRBD18 RNAi cells. We identified mRNAs whose abundance is unchanged in whole cell RNA, but decreased in cytosolic RNA upon DRBD18 RNAi, as an indication of decreased nuclear export (Müller-McNicoll et al., 2016, Rehwinkel et al., 2004, Wickramasinghe et al., 2014). The purity of subcellular fractionation and the depletion of DRBD18 by RNAi induction were confirmed by Western blot analysis using antibodies against cytosolic and nuclear marker proteins and anti-DRBD18 antibody, respectively (Fig. S3). In whole cell RNA, we identified 259 transcripts that decreased in abundance and 378 that increased upon DRBD18 knockdown (1.5 fold change; corrected p-value <0.05) (Table S2), similar to our previously published findings (Lott et al., 2015). In the cytosolic fraction, we identified 216 transcripts with decreased abundance and 593 transcripts with increased abundance (1.5 fold change; corrected p-value <0.05) upon DRBD18 knockdown (Table S3, Fig. S4). Next, we focused on the set of transcripts whose nuclear export was apparently promoted by DRBD18, consistent with the partial nuclear accumulation of poly(A)+ RNA that we observed by FISH (Fig. 5). To this end, we identified mRNAs reduced in cytosol and unchanged in the whole cell upon DRBD18 RNAi (Fig. 5A; Fig. S4). Of the 174 transcripts fitting these parameters (corrected p-value <0.05; Fig. 5A), 95 exhibited a fold decrease in the cytosol of >1.5 fold (Fig. 5A, blue). These 95 transcripts comprise the list of potential DRBD18 facilitated export targets (Table S4). No significantly enriched GO terms or nucleotide motifs were apparent in this mRNA set. To validate the relative expression of the transcripts in cytosol and whole cell in DRBD18 RNAi induced or uninduced cells, we performed qRT-PCR analyses on a subset of these transcripts. By qRT-PCR, we detected either no decrease or small decreases in the transcripts in whole cell RNA samples as expected (Figs. 5B and S5), and we confirmed significantly larger decreases in the cytosol for seven of the transcripts tested (Fig. 5B). An additional four transcripts showed no significant differences between whole cell and cytosol, although some appeared to trend in this direction (Fig. S5). If the nuclear export of these transcripts is directly promoted by DRBD18, we would expect them to associate with DRBD18 in vivo. To test this, we performed cross-linking immunoprecipitation (CLIP) experiments in which we measured the enrichment of the transcripts in Fig. 5B using anti-DRBD18 antibodies relative to a non-specific antibody control. Indeed, all seven transcripts were enriched, from 2- to 17-fold, while a negative control transcript (Tb927.7.7400; Table S2) was not (Fig. 5C). The reduced abundance of specific mRNAs in cytosol compared to that in the whole cell RNA population upon DRBD18 depletion is consistent with a function for DRBD18 in the export of these mRNAs. Moreover, DRBD18 binds differentially transported mRNAs in vivo, suggesting it plays a direct role in promoting the nucleocytosolic transport of a subset of mRNAs.
Figure 5. DRBD18 RNAi inhibits export of a subset of mRNAs from nucleus to cytosol and associates with differentially transported mRNAs.
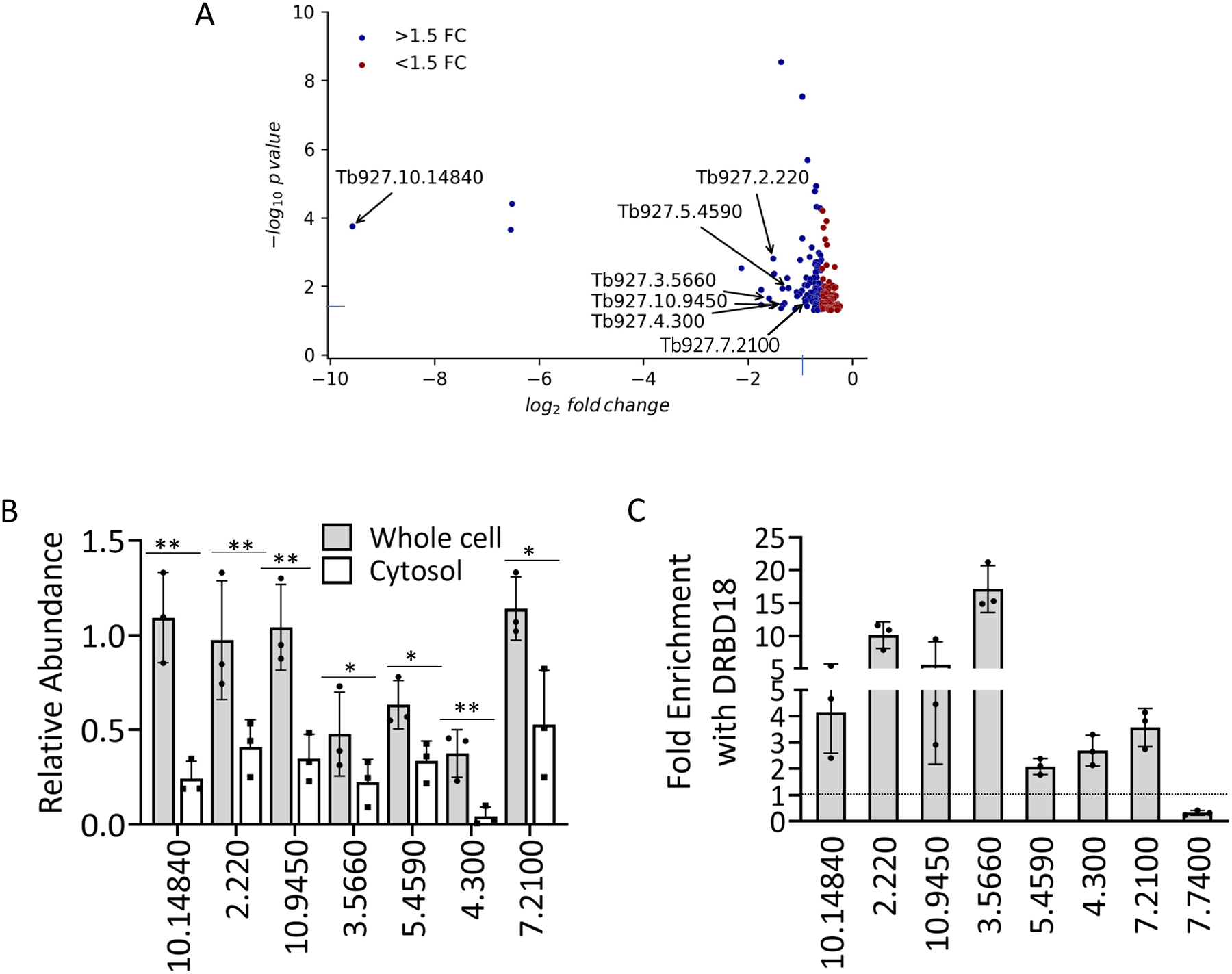
(A) Volcano plot (generated from RNAseq data) of transcripts whose abundance is decreased in cytosol but unchanged in whole cell RNA (corrected p-value <0.05) upon DRBD18 RNAi. Transcripts with fold change (FC) >1.5 are colored blue, and those with FC <1.5 are red. Transcripts analyzed in Fig. 5B are indicated by their TriTrypDB numbers. (B) Representative transcripts, identified by the last four to seven digits of their TriTrypDB numbers, were quantified by qRT-PCR in whole cell and cytosolic fractions. Relative abundance represents RNA levels in induced (+Dox) cells compared to levels in uninduced (−Dox) cells. RNA levels were normalized to 18S rRNA. Values represent the mean of three biological replicates, each with three technical replicates. Significance was determined by unpaired t-test. **p < 0.01; *p < 0.05. (C) Enrichment of transcripts shown in (B) in anti-DRBD18 immunoprecipitations relative to a non-specific antibody control was measured by UV Cross-Linking ImmunoPrecipitation (CLIP). The 7.7400 transcript serves as a negative control. Values represent the mean of three biological replicates, each with three qRT-PCR technical replicates. A value of 1 (dotted line) indicates no enrichment.
2.7. DRBD18 levels impact the relative abundance of export receptors in nucleus and cytosol mRNA
One mechanism by which DRBD18 may promote export of selected mRNAs is by facilitating the nuclear export of the Mex67/Mtr2-DRBD18 mRNP. To determine whether DRBD18 impacts the transport of Mex67/Mtr2 from nucleus to cytosol, we fractionated DRBD18 RNAi induced and uninduced cells into nuclear and cytosolic fractions, and validated the fractionation by western blot analysis of histone H3 and EF1α, respectively (Figs. 6A and 6B). We then blotted for Mex67 and Mtr2 in both fractions and normalized their expression to the expression of histone H3 (nucleus) or EF1α (cytosol). We detected a significant and reproducible approximately two-fold increase in nuclear Mex67 and Mtr2 in cells depleted of DRBD18. We did not detect significant concomitant changes in Mtr2 or Mex67 level in cytosolic fraction of DRBD18 RNAi induced and uninduced cells, possibly due to the greater abundance of Mex67/Mtr2 in the cytosol in our assay system. These data indicate that DRBD18 modestly impacts the dynamics of export receptor trafficking between the two subcellular compartments, consistent with its ability to promote the export of a subset of mRNAs.
Figure 6. DRBD18 RNAi causes partial accumulation of export receptors in the nucleus.
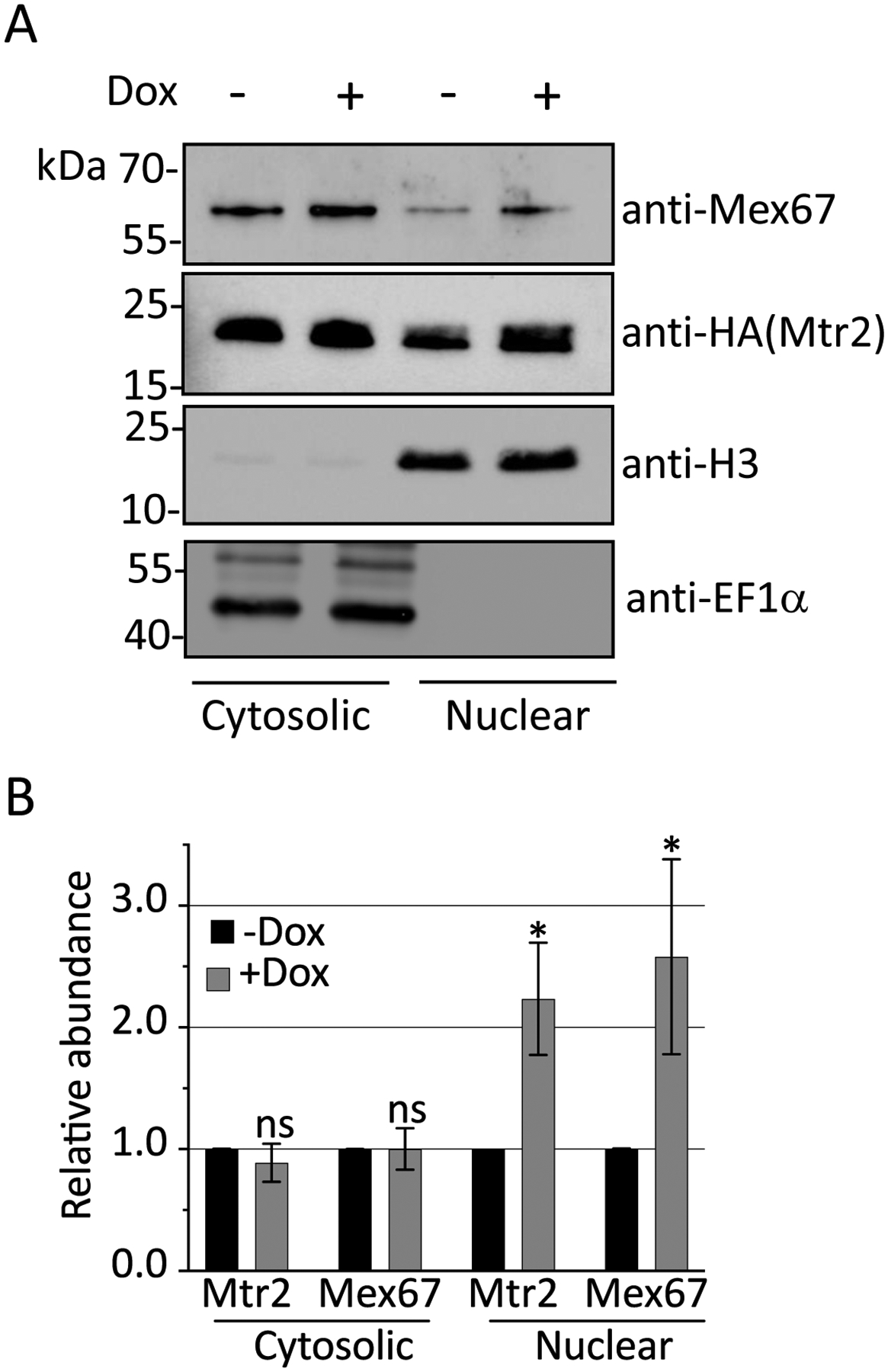
(A) The relative abundance of Mtr2 and Mex67 in nuclear and cytosolic fractions in uninduced (-Dox) or induced (+Dox) DRBD18 RNAi cells were analyzed by subcellular fractionation followed by Western blot analysis. Anti-Histone H3 (H3) and anti-EF1α antibodies were used as loading controls for nuclear and cytosolic fractions, respectively. (B) Quantification of Western blots in (A). Relative abundance of Mex67 or Mtr2 in nuclear and cytosolic fractions was normalized to the expression of Histone H3 and EF1α, respectively. The normalized protein expression in nuclear and cytosolic fractions from the +Dox were then compared to that of the −Dox (which was set to 1) to calculate the relative abundances in the DRBD18 knockdown. Bar graphs represent the average and standard deviation (SD) of four samples (two biological replicates, each with two technical replicates). Significance was determined by unpaired t-test with Welch’s correction. *p < 0.05 and ns = non-significant.
3. DISCUSSION
Gene expression in kinetoplastids, including T. brucei, is modulated primarily at the posttranscriptional level, with RBPs constituting the key gene regulatory factors (Clayton, 2019). While RBPs and cis-acting sequences that modulate mRNA stability and translation have been identified in T. brucei, no proteins outside the basal nuclear export machinery have been reported to control mRNA export (Schwede et al., 2009, Dostalova et al., 2013). Here, we show that the abundant and essential T. brucei RBP, DRBD18, promotes the nucleocytosolic transport of a subset of mRNAs. While nuclear mRNA export was originally thought to be constitutive, more recent studies reveal that nuclear export of mRNAs can be highly selective, and this process is now established as an important level of gene regulation (Wickramasinghe & Laskey, 2015). In mammals, processes such as DNA repair, maintenance of pluripotency, stress responses, and cell proliferation can be regulated by selective nuclear export promoted by distinct proteins. We demonstrate here that T. brucei DRBD18 binds the general nuclear export receptor, Mex67/Mtr2, likely through a direct interaction with Mtr2, and facilitates the export of a subset of mRNAs. Thus, DRBD18 is the first reported nuclear mRNA export specificity factor in trypanosomes.
Bulk nuclear mRNA export is facilitated in yeast and mammals by the TREX complex, which binds to Mex67 and increases its affinity for mRNA. With the exception of the Sub2 RNA helicase, homologues of TREX complex components are absent in T. brucei (Serpeloni et al., 2011b); however, DRBD18 clearly does not fulfill the bulk export role of a TREX component, as it impacts the export of only a few hundred mRNAs. Indeed, it has been suggested that T. brucei Mex67 may not utilize a homolog of the TREX complex to increase bulk mRNA binding due to the presence of a kinetoplastid-specific ZC3H domain at its N terminus (Dostalova et al., 2013). DRBD18 may act as an adaptor protein that promotes association of a select subset of mRNAs with Mex67/Mtr2. Alternatively, or in addition, DRBD18 may function as a nuclear export chaperone. The nuclear envelope defines the physical barrier between nucleoplasm and cytosol, and communication between the two compartments is mediated by cylindrical macromolecular NPCs. Rate limiting steps in mRNA nuclear export are diffusion of mRNA loaded Mex67/Mtr2 through the nucleoplasm and loading onto NPC, the latter of which requires the activity of chaperones that direct cargo to the NPC (Scott et al., 2019). Chaperoning of Mex67/Mtr2 to the NPC has been proposed as a good target for regulation of the export rate of specific subsets of mRNAs (Scott et al., 2019).
One clue as to how DRBD18 might facilitate export of mRNAs bound by the Mex67/Mtr2-DRBD18 mRNP lies in the reported interactions of Mex67 and DRBD18 with NPC components. The NPC consist of multiple copies of ~30 proteins termed nucleoporins (Nups), of which T. brucei contains two major classes: phenylalanine-glycine (FG) repeats Nups and core scaffold Nups (Fig. 7) (Obado et al., 2016). The core scaffold, which is comprised of two inner ring and two outer ring structural components interacts with the nuclear envelope. FG Nups are disordered proteins responsible for the selective permeability of the NPC to nucleocytosolic transport, playing vital roles in the transport of the soluble transport receptors through hydrophobic interactions (Strambio-De-Castillia et al., 2010). Comprehensive proteomic and interactome analysis of T. brucei NPC by Obado, et al. (Obado et al., 2016) revealed both similarities and differences in the NPC protein composition in this excavate compared to that of the opisthokonts (humans and yeast). Further, affinity capture of T. brucei Mex67 revealed its association with many components of the NPC (Obado et al., 2016). Under high stringency, Mex67 interactions were observed predominantly with the less evolutionarily conserved outer ring and FG Nups. In particular, the retention of FG Nups comprising the Nup76 complex (Nups 76, 140, and 149; see Fig. 7) under high stringency implicates the Nup76 complex as part of the mRNA export docking platform. Previous Tandem Affinity Purification (TAP) of DRBD18-TAP from PF T. brucei also returned several NPC components (Lott et al., 2015). Remarkably, these studies identified as DRBD18 interacting NPC proteins almost exclusively those that also showed high stringency interactions with Mex67, including components of the Nup76 complex (Nups140 and 149) and the outer ring (Nups 158, 132, 109, 89, and 82). Interaction of DRBD18 with the Nup76 complex docking platform may increase the frequency and/or affinity with which the Mex67/Mtr2 complex interacts with the NPC, thereby facilitating export of mRNAs bound to the Mex67/Mtr2-DRBD18 complex (Tetenbaum-Novatt & Rout, 2010). Future studies will be needed to address the precise mechanism by which DRBD18 promotes the export of a distinct mRNA subset.
Figure 7. Model of the role of DRBD18 in mRNA export.
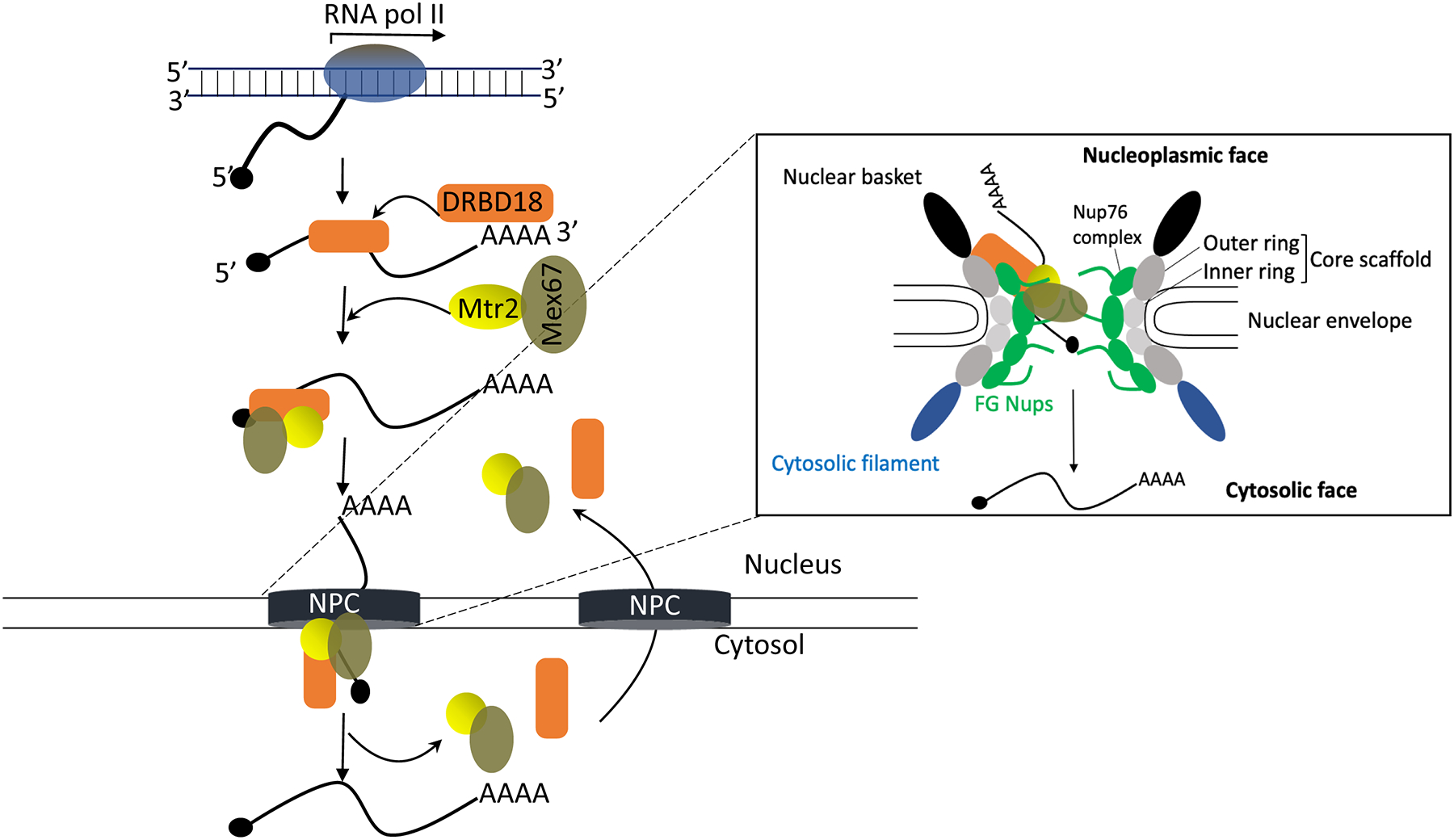
A model summarizing the potential role of DRBD18 in mRNA export from nucleus to cytosol through the Nuclear Pore Complex (NPC; see text for details). Inset shows the detailed architecture of NPC and its structural components (Nups). Core scaffold Nups, dark grey (outer ring) and light grey (inner ring); FG Nups, green; nuclear basket, black; cytosolic filament, blue. The NPC has left to right symmetry, and specific features are labeled on only one side for clarity, although they are present on both sides. DRBD18, orange; Mtr2, yellow; Mex67, olive green. DRBD18 may promote association of select mRNAs with Mex67/Mtr2 and likely acts as a chaperone that facilitates passage of the Mex67/Mtr2-DRBD18 mRNP across the NPC due to its interactions with the outer ring and Nup76 complex.
While DRBD18 clearly facilitates the export of a subset of mRNAs as described here, we also identified numerous mRNAs whose export may be inhibited by DRBD18. Over 500 mRNAs are increased in the cytosol upon DRBD18 knockdown, and approximately 200 of these mRNAs are not increased in whole cell RNA pools (Fig. S4). Thus, it is likely that DRBD18 has dual and opposing roles in mRNA nuclear export. In model organisms, posttranslational modifications of the nuclear export machinery impact numerous steps in this process (Tutucci & Stutz, 2011, Howard & Sanford, 2015, Wickramasinghe & Laskey, 2015). One potential mechanism for regulation of disparate functions of DRBD18 in mRNA export in T. brucei is the methylation of arginine residues that lie between the two DRBD18 RRM domains (Lott et al., 2015). We previously demonstrated that arginine methylation promotes the ability of DRBD18 to stabilize mRNAs, while it inhibits the DRBD18 mediated destabilization of other transcripts. With regard to Mex67/Mtr2, DRBD18 TAP returned increased numbers of Mex67 peptides with hypomethylated DRBD18, but increased Mtr2 peptides with methylmimic DRBD18, so the impact of methylation on the DRBD18-Mex67/Mtr2 interaction is unresolved. Additionally, with a few exceptions, most Nups appeared to bind equivalently to hypomethylated and methylmimic DRBD18 (Lott et al., 2015). It has been suggested that selective mRNA nuclear export in mammals is controlled by combinatorial binding of distinct RBP combinations to a given transcript (Wickramasinghe & Laskey, 2015). Interestingly, TAP of hypomethylated and methylmimic DRBD18 identified several RRM and ZC3H RBPs that were differentially bound to DRBD18 in RNase-treated extracts depending on its methylation status (Lott et al., 2015). This finding suggests that these DRBD18 associated RBPs could contribute to a combinatorial effect on nuclear mRNA export in T. brucei with distinct impacts based on the methylation status of DRBD18.
Nuclear mRNA export interfaces with other RNA processing and gene regulatory events (Howard & Sanford, 2015, Wickramasinghe & Laskey, 2015). One such process is nuclear mRNA decay. Nuclear retained mRNAs are generally turned over, and the balance between mRNA export kinetics and decay can be a key determinant of gene expression (Meola & Jensen, 2017, Tudek et al., 2019). In yeast, experimental inhibition of nuclear export leads to the degradation of protein coding transcripts (Tudek et al., 2018). In light of these findings, our approach to identifying transcripts whose nuclear export is promoted by DRBD18 may have been conservative. Because we identified only those mRNAs whose abundance was statistically unchanged in the whole cell RNA pool, but decreased in cytosol, we would not have identified those mRNAs whose abundance was significantly decreased by decay upon nuclear retention. Thus, the impact of DRBD18 in promoting mRNA export may be broader than reported here. Nuclear mRNA export can also be coupled to transcript specific translation by nucleocytosolic shuttling proteins, such as some SR proteins (Maslon et al., 2014, Howard & Sanford, 2015, Brugiolo et al., 2017). DRBD18 localizes to both nucleus and cytosol, with a perinuclear concentration (Fig. S3) (Lott et al., 2015). This localization pattern, together with its interaction with numerous NPC components, suggests that DRBD18 may undergo nucleocytosolic shuttling. Interestingly, DRBD18-TAP purification revealed the protein’s interaction with ribosomes and translation initiation factors (Lott et al., 2015). In addition, Klein, et al. reported association of DRBD18 with bloodstream form T. brucei polysomes (Klein et al., 2015). Thus, one possible scenario entails sequence specific coupling of mRNA nuclear export and translation by DRBD18 to enhance gene expression. Together, our data suggest that DRBD18 is a multifunctional gene regulatory protein with the potential to integrate mRNA export, stability, and translation.
4. EXPERIMENTAL PROCEDURES
4.1. Cell line generation and culture
PF T. brucei strain 29–13 and all cell lines derived from this strain were grown at 27°C in SM medium supplemented with 10% fetal bovine serum and containing hygromycin (50μg ml−1) and G418 (15μg ml−1). Cells harboring the doxycycline inducible DRBD18 (Tb927.11.14090) RNA interference (RNAi) constructs described previously in (Lott et al., 2015) were grown with the addition of phleomycin (2.5μg ml−1). To induce RNAi, the clonal cell line was incubated for 19 h (high throughput sequencing and qRT-PCR) or 23 h (all other experiments) with 4μg ml−1 doxycycline. In situ tagging of TbMtr2 with a triple hemagglutinin (3XHA) tag was performed by the long primer PCR method, using the pPOTv6 plasmid according to the published protocol by (Dean et al., 2015). Primers are listed in Table S1. PCR products were transfected into T. brucei for genomic integration, and transfectants were selected with blasticidin (20μg ml−1).
4.2. Immunoprecipitation and Western blot
Approximately 1.5 × 109 cells were collected, washed with PBS, and resuspended in lysis buffer (50 mM HEPES (pH 7.4), 3 mM MgCl2, 150 mM KCl, 150 mM sucrose, 0.5% Tween-20, EDTA-free protease inhibitor cocktail (Roche, USA), and 1mM DTT). Cells were lysed by sonication (Sonic Dismembrator, Fisher Scientific, USA) three times for 15s at 30s intervals on ice at 60% amplitude, and clarified by centrifugation at 15,000×g for 30 min at 4°C. For immunoprecipitation with anti-HA antibody, clarified lysates were incubated with agarose immobilized rabbit anti-HA beads (ICL Laboratories, USA) for 2 h at 4°C. As a negative control, lysates were incubated with IgG Sepharose beads (GE Healthcare, USA) for 2 h at 4°C. Bound proteins were eluted with 100 mM glycine (pH 2.5) and the solution neutralized using 1M Tris buffer (pH 7.5). Proteins were electrophoresed on a 12.5% SDS-PAGE gel and transferred to nitrocellulose membrane. Membranes were blocked with 5% non-fat dry milk in Tris buffered saline with Tween-20 (TBST) and probed with polyclonal antibodies against DRBD18 (Lott et al., 2015), p22 (Hayman et al., 2001) and Mex67 (a kind gift from Prof. Mark Carrington, University of Cambridge, UK). Commercially available antibodies against HA-epitope (Mouse monoclonal, Thermo Fisher Scientific), Histone H3 (Rabbit polyclonal, Abcam), and EF1α (Mouse monoclonal, Santa Cruz Biotechnology) were also used. Blots were washed with TBST and subsequently probed with secondary antibodies, either goat anti-rabbit HRP or goat anti-mouse HRP. Signals were detected using an ECL preparation as recommended by the manufacturer (Thermo Fisher Scientific, USA), visualized on a Chemi Doc MP imaging system (BioRad), and quantified using BioRad Image Lab software.
4.3. Protein expression and GST pulldown assay
Expression of recombinant GST-tagged DRBD18 was carried out as described earlier (Lott et al., 2015). For His-Mtr2 (Tb927.7.5760), the ORF was cloned into the MCS-1 of pETDuet-1vector (with MSC2 left empty), with frame shift correction by insertion of single nucleotide before the start codon (Kafková et al., 2017) and was purified using immobilized metal affinity chromatography (IMAC) as described earlier (Klebanov-Akopyan et al., 2018). For GST pulldown assays, all recombinant plasmids including pGEX4T-1, were transformed into Escherichia coli BL21 strain, and protein expression was induced by addition of 0.5 mM isopropyl 1-thio-D-galactopyranoside (IPTG) and subsequent growth at 22°C overnight. Cells expressing GST-DRBD18 or GST were harvested and resuspended in lysis buffer (50 mM Tris-HCl (pH 8.0), 250 mM NaCl, 5 mM EDTA, 10 mM DTT, and 1mM PMSF) and lysed by four cycles of maximum-power sonication bursts of 30 s each at 4°C. Triton X-100 was added to a final concentration of 1%, followed by a 30 min centrifugation at 15,000 ×g and 4°C. The supernatant was incubated with Glutathione-Sepharose 4B beads (GE) at 4°C for 2 h. The column was extensively washed with ice cold lysis buffer. Equal amounts of purified His-Mtr2 were added and incubated for another 2 h at 4°C with the immobilized GST or GST fusion protein. The columns were washed with ice cold wash buffer. The beads were boiled with SDS-PAGE Laemmli buffer and analyzed by Western blot using anti-Histidine (anti-Mouse, Sigma Aldrich) and anti-GST antibodies (anti-Rabbit, Sigma Aldrich).
4.4. RNA isolation from nuclear-cytosolic fractionation
Nuclear-cytosolic fractionation was performed in triplicate as described previously in Dostalova, et al. (Dostalova et al., 2013). Briefly, 2.5 × 108 PF cells were harvested, washed in phosphate buffer saline (PBS), and resuspended in PBS. Nonidet-P40 was added to final concentration of 0.1%, the cell suspension was mixed thoroughly, and nuclei were pelleted by centrifugation at 2300×g for 1 min. Supernatant was considered as cytosolic fraction. To validate the fractionation and DRBD18 knockdown, samples corresponding to same cell equivalents from whole cells and supernatant (cytosolic) fractions were subjected to SDS-PAGE and Western blot analysis with anti-DRBD18, anti-histone H3, and anti-EF1α antibodies. For RNA isolation, Trizol reagent (Invitrogen) was added to the cytosolic fraction and this suspension frozen at −80°C. Total RNA was isolated from whole cell and cytosolic fractions using Trizol reagent and phenol chloroform extraction followed by DNase-I treatment (DNA-free DNase kit; Ambion). The RNA was further purified using phenol chloroform extraction. Purified RNA samples were submitted for RNA sequencing and also kept for qRT-PCR analysis.
4.5. RNA library preparation, sequencing, and bioinformatic analysis
Prior to library preparation, total RNA was quality checked using an Agilent Fragment Analyzer to assess quality and Qubit Fluorescence (Invitrogen) to measure concentration. RNA libraries were prepared following the New England Biolabs NEXT UltraII Directional RNA library kit using polyA+ magnetic beads. Following library preparation, concentration and quality control, final libraries were pooled to 10 nM and the concentration of the pool was determined using the Kapa Biosystems Universal qPCR kit. After dilution and denaturing, the pooled library was loaded onto a NextSeq 500 high output flow cell (PE75) for sequencing. Sequencing quality was assessed using FastQC and MultiQC, and low-quality bases were removed using TrimGalore, a cutadapt wrapper (Andrews, 2010, Krueger, 2015, Ewels et al., 2016, Martin, 2011). Ribosomal RNA (rRNA) reads were removed with SortMeRNA prior to alignment with Spliced Transcripts Alignment to a Reference (STAR) against the TREU927 T. brucei reference genome, downloaded from TriTrypDB v41 (Kopylova et al., 2012, Dobin et al., 2013, Aslett et al., 2010). Transcript abundances were estimated with RNA-Seq by Expectation Maximization (RSEM) (Li & Dewey, 2011). Differential abundances were identified using differential expression analysis for sequence count data (DESeq2) (Love et al., 2014).
The sequencing data used in this study has been deposited in Sequence Read Archive, accession number GSE158584.
4.6. qRT-PCR analysis
Reverse transcription (RT) was performed with 1 μg of DNase I-treated RNA following standard procedures with random hexamer primers and iScript reverse transcriptase (BioRad). Quantitative reverse transcription PCR (qRT-PCR) reactions were performed using primer pairs targeted at specific transcripts (Table S1). Amplification was performed using a CFX Connect Real Time System (Bio Rad), and data were analyzed using BioRad CFX Manager 3.1. Results were analyzed using the Bio-Rad CFX Manager 3.1 software. RNA levels were normalized to 18S rRNA using the standard curve method.
4.7. Fluorescence in situ hybridization (FISH)
For tRNA FISH analysis, cells were harvested, fixed and permeabilized as described (Hegedűsová et al., 2019) and then pre-hybridized for 2h with hybridization solution (2% BSA, 5X Denhardt’s solution, 4X SSC, 5% dextran sulphate, 35% deionized formamide, 10 U ml−1 RNase inhibitor). The slides were then incubated overnight at room temperature in a humid chamber in the presence of 10 ng μl−1 fluorophore labeled oligonucleotide probes (Supplementary Table S1), in the hybridization solution. Slides were washed as described earlier (Hegedűsová et al., 2019) and mounted with mounting medium supplemented DAPI. Images were taken with confocal microscope Olympus Fluo ViewTM FV1000.
mRNA FISH analysis was performed as previously described (Dostalova et al., 2013). Briefly, 2.5 × 107 cells were harvested by centrifugation, washed, resuspended in PBS, and allowed to adhere to poly-L-lysine coated slides for 30 min. Cells were fixed with 4% paraformaldehyde followed by a 10 min treatment with 25mM ammonium chloride. Fixed cells were permeabilized for 1 h in blocking buffer (PBS containing 0.5% saponin, 2% BSA and 10U ml−1 RNase inhibitor). Permeable cells were pre-hybridized for 2 h with hybridization solution (2% BSA, 5X Denhardt’s solution, 4X SSC, 5% dextran sulphate, 35% formamide, 0.5 μg μl−1 tRNA, 10 U ml−1 RNase inhibitor). Alexa 594-labelled oligo-(dT)30 (2 ng μl−1 in hybridization solution; Invitrogen, USA) was hybridized overnight at room temperature. Slides were then washed for 10 min, once with 50μl of 4X SSC with 35% formamide followed by one wash each with 2X SSC and 1X SSC. Labelled cells were mounted with 4′,6-diamino-2-phenylindole dihydrochloride (DAPI) fluoromount-G (Southern Biotech, Birmingham, AL) for visualization. Images were taken with a fluorescent microscope Zeiss Axioimager M2 stand equipped with a rear-mounted excitation filter wheel. mRNA FISH data were quantified using ImageJ (NIH) software as described previously (Chatterjee et al., 2017). Six cells (three from each replicate) were randomly selected and their fluorescence intensities were measured using Image J with a plot profile analysis of the various points at a regular interval along a line drawn across the nucleus with overhangs covering the cytosol (Hegedűsová et al., 2019). The results are expressed as the average value of relative fluorescence intensity ± S.D.
4.8. Denaturing gel electrophoresis and Northern hybridization
Total RNA was isolated using Trizol reagent and phenol chloroform extraction method as described previously (Lott et al., 2015). Boronate affinity electrophoresis was performed as described previously (Kessler et al., 2018). In brief, 5 μg of RNA was resolved by denaturing gel electrophoresis (8% acrylamide, 7 M urea), electroblotted to Zeta probe® (Bio-Rad) membranes, and UV cross-linked (1200 uJ ×100). Oxidation control RNA was deacylated and treated with sodium periodate and then the reaction was quenched by addition of 2.5 mM glucose. The membranes were probed with oligonucleotides radiolabeled with γ32 P-dATP (Supplementary Table S1). Northern hybridization was performed according to the manufacturer’s instructions (Bio-Rad). Subsequently, the membranes were exposed overnight to a Phosphorimager screen and analyzed using Typhoon™ 9410 scanner and Image Quant TL software (GE Healthcare) (Hegedűsová et al., 2019).
4.9. Cross-linking immunoprecipitation (CLIP)
CLIP was carried out with minor modifications of a previously published method (Mugo & Erben, 2020). Briefly, procyclic form cells (1×107 cells ml−1) were harvested and washed once with cold 1X PBS buffer (pH 7.4). Cells were resuspended in 25 ml of SM medium without FBS to a concentration of ~5 ×109 cells ml−1, transferred to a 100 × 15 mm Petri dish, placed on ice, and UV irradiated at 400 mJ/cm2 in a Stratalinker 1800 (Stratagene). Cells were pelleted, washed with PBS, snap frozen in liquid N2, and stored in −80°C until use. Cells were resuspended in 4 ml of lysis buffer (Tris-HCl (pH 7.5), 150mM NaCl, 0.1 %NP40, and 1% Triton X-100) and then lysed by passing 15–20 times through a 21 gauge needle. Cell lysate was cleared by centrifugation at 18000 rpm for 30 minutes at 4 °C. Supernatant was collected, and the NaCl concentration adjusted to 150 mM. Crosslinked DRBD18-RNA complexes were immunopurified from cellular extracts using anti-DRBD18 antibodies (Lott et al., 2015) attached to Protein A fast flow beads (GE Healthcare); anti-Ty1 antibody attached to Protein A fast flow beads served as the control. Captured protein-RNA complexes were washed with wash buffer (Tris-HCl (pH 7.5), 150mM NaCl, 0.1 %NP40), and 5% of the beads were taken from each sample and used for Western blot was performed to confirm the pulldown of DRBD18. Beads were treated with DNase 1 (Sigma) followed by proteinase K (Roche). RNA was extracted with phenol/chloroform, and cDNA was prepared using gene specific primers (Table S1) and qRT-PCR was performed as described above. Fold change was calculated as described previously (McAdams et al., 2018).
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Kaylen Lott for assistance in the early stages of this work, Mark Carrington for providing anti-Mex67 antibodies, and Brianna Tylec for assistance with figures. This work was supported by National Institutes of Health R01 AI141557 to LKR and Czech Science Foundation [20-11585S to ZP]; ERDF/ESF project Centre for Research of Pathogenicity and Virulence of Parasites [CZ.02.1.01/0.0/0.0/16_019/0000759 to ZP].
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- Andrews S, (2010) FastQC: a quality control tool for high throughput sequence data. In.: Babraham Bioinformatics, Babraham Institute, Cambridge, United Kingdom. [Google Scholar]
- Aphasizheva I, Alfonzo J, Carnes J, Cestari I, Cruz-Reyes J, Göringer HU, Hajduk S, Lukeš J, Madison-Antenucci S, Maslov DA, McDermott SM, Ochsenreiter T, Read LK, Salavati R, Schnaufer A, Schneider A, Simpson L, Stuart K, Yurchenko V, Zhou ZH, Zíková A, Zhang L, Zimmer S, and Aphasizhev R (2020) Lexis and grammar of mitochondrial RNA processing in trypanosomes. Trends Parasitol 36: 337–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslett M, Aurrecoechea C, Berriman M, Brestelli J, Brunk BP, Carrington M, Depledge DP, Fischer S, Gajria B, and Gao X (2010) TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucl Acids Res 38: D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangs JD (2018) Evolution of antigenic variation in African trypanosomes: Variant surface glycoprotein expression, structure, and function. Bioessays 40: e1800181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun IC, Herold A, Rode M, and Izaurralde E (2002) Nuclear export of mRNA by TAP/NXF1 requires two nucleoporin-binding sites but not p15. Mol Cell Biol 22: 5405–5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugiolo M, Botti V, Liu N, Müller-McNicoll M, and Neugebauer KM (2017) Fractionation iCLIP detects persistent SR protein binding to conserved, retained introns in chromatin, nucleoplasm and cytoplasm. Nucl Acids Res 45: 10452–10465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büscher P, Cecchi G, Jamonneau V, and Priotto G (2017) Human African trypanosomiasis. Lancet 390: 2397–2409. [DOI] [PubMed] [Google Scholar]
- Chatterjee K, Majumder S, Wan Y, Shah V, Wu J, Huang H-Y, and Hopper AK (2017) Sharing the load: Mex67–Mtr2 cofunctions with Los1 in primary tRNA nuclear export. Genes Devel 31: 2186–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton C (2019) Regulation of gene expression in trypanosomatids: living with polycistronic transcription. Open Biol 9: 190072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean S, Sunter J, Wheeler RJ, Hodkinson I, Gluenz E, and Gull K (2015) A toolkit enabling efficient, scalable and reproducible gene tagging in trypanosomatids. Open Biol 5: 140197–140197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostalova A, Käser S, Cristodero M, and Schimanski B (2013) The nuclear mRNA export receptor Mex67-Mtr2 of Trypanosoma brucei contains a unique and essential zinc finger motif. Mol Microbiol 88: 728–739. [DOI] [PubMed] [Google Scholar]
- Droll D, Minia I, Fadda A, Singh A, Stewart M, Queiroz R, and Clayton C (2013) Post-transcriptional regulation of the trypanosome heat shock response by a zinc finger protein. PLoS Pathog 9: e1003286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewels P, Magnusson M, Lundin S, and Käller M (2016) MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32: 3047–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goos C, Dejung M, Wehman AM, M-Natus E, Schmidt J, Sunter J, Engstler M, Butter F, and Kramer S (2018) Trypanosomes can initiate nuclear export co-transcriptionally. Nucl Acids Res 47: 266–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayman ML, Miller MM, Chandler DM, Goulah CC, and Read LK (2001) The trypanosome homolog of human p32 interacts with RBP16 and stimulates its gRNA binding activity. Nucl Acids Res 29: 5216–5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedűsová E, Kulkarni S, Burgman B, Alfonzo JD, and Paris Z (2019) The general mRNA exporters Mex67 and Mtr2 play distinct roles in nuclear export of tRNAs in Trypanosoma brucei. Nucl Acids Res 47: 8620–8631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard JM, and Sanford JR (2015) The RNAissance family: SR proteins as multifaceted regulators of gene expression. Wiley Interdiscip Rev RNA 6: 93–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igloi GL, and Kössel H (1985) Affinity electrophoresis for monitoring terminal phosphorylation and the presence of queuosine in RNA. Application of polyacrylamide containing a covalently bound boronic acid. Nucl Acids Res 13: 6881–6898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen RE, and Englund PT (2012) Network news: the replication of kinetoplast DNA. Annu Rev Microbiol 66: 473–491. [DOI] [PubMed] [Google Scholar]
- Kafková L, Debler EW, Fisk JC, Jain K, Clarke SG, and Read LK (2017) The major protein arginine methyltransferase in Trypanosoma brucei functions as an enzyme-p complex. J Biol Chem 292: 2089–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating J, Yukich JO, Sutherland CS, Woods G, and Tediosi F (2015) Human African trypanosomiasis prevention, treatment and control costs: A systematic review. Acta Tropica 150: 4–13. [DOI] [PubMed] [Google Scholar]
- Kessler AC, Kulkarni SS, Paulines MJ, Rubio MAT, Limbach PA, Paris Z, and Alfonzo JD (2018) Retrograde nuclear transport from the cytoplasm is required for tRNA(Tyr) maturation in T. brucei. RNA Biol 15: 528–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebanov-Akopyan O, Mishra A, Glousker G, Tzfati Y, and Shlomai J (2018) Trypanosoma brucei UMSBP2 is a single-stranded telomeric DNA binding protein essential for chromosome end protection. Nucl Acids Res 46: 7757–7771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C, Terrao M, Inchaustegui Gil D, and Clayton C (2015) Polysomes of Trypanosoma brucei: Association with initiation factors and RNA-binding proteins. PLoS One 10: e0135973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler A, and Hurt E (2007) Exporting RNA from the nucleus to the cytoplasm. Nat Rev Mol Cell Biol 8: 761–773. [DOI] [PubMed] [Google Scholar]
- Kolev NG, Ramey-Butler K, Cross GA, Ullu E, and Tschudi C (2012) Developmental progression to infectivity in Trypanosoma brucei triggered by an RNA-binding protein. Science 338: 1352–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolev NG, Ullu E, and Tschudi C (2014) The emerging role of RNA-binding proteins in the life cycle of Trypanosoma brucei. Cell Microbiol 16: 482–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopylova E, Noé L, and Touzet H (2012) SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28: 3211–3217. [DOI] [PubMed] [Google Scholar]
- Krueger F (2015) Trim galore. https://github.com/FelixKrueger/TrimGalore
- Li B, and Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformat 12: 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Kamanyi Marucha K, and Clayton C (2020) The zinc finger proteins ZC3H20 and ZC3H21 stabilise mRNAs encoding membrane proteins and mitochondrial proteins in insect-form Trypanosoma brucei. Mol Microbiol 113: 430–451. [DOI] [PubMed] [Google Scholar]
- Lott K, Mukhopadhyay S, Li J, Wang J, Yao J, Sun Y, Qu J, and Read LK (2015) Arginine methylation of DRBD18 differentially impacts its opposing effects on the trypanosome transcriptome. Nucl Acids Res 43: 5501–5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. journal 17: 10–12. [Google Scholar]
- Maslon MM, Heras SR, Bellora N, Eyras E, and Cáceres JF (2014) The translational landscape of the splicing factor SRSF1 and its role in mitosis. Elife 3: e02028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews KR (2005) The developmental cell biology of Trypanosoma brucei. J Cell Sci 118: 283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAdams NM, Simpson RM, Chen R, Sun Y, and Read LK (2018) MRB7260 is essential for productive protein-RNA interactions within the RNA editing substrate binding complex during trypanosome RNA editing. RNA 24: 540–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meola N, and Jensen TH (2017) Targeting the nuclear RNA exosome: Poly(A) binding proteins enter the stage. RNA Biol 14: 820–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaeli S (2011) Trans-splicing in trypanosomes: machinery and its impact on the parasite transcriptome. Future Microbiol 6: 459–474. [DOI] [PubMed] [Google Scholar]
- Mugo E, and Clayton C (2017) Expression of the RNA-binding protein RBP10 promotes the bloodstream-form differentiation state in Trypanosoma brucei. PLoS Pathog 13: e1006560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugo E, and Erben ED (2020) Identifying trypanosome protein-RNA interactions using RIP-Seq. Methods Mol Biol 2116: 285–294. [DOI] [PubMed] [Google Scholar]
- Müller-McNicoll M, Botti V, de Jesus Domingues AM, Brandl H, Schwich OD, Steiner MC, Curk T, Poser I, Zarnack K, and Neugebauer KM (2016) SR proteins are NXF1 adaptors that link alternative RNA processing to mRNA export. Genes Dev 30: 553–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obado SO, Brillantes M, Uryu K, Zhang W, Ketaren NE, Chait BT, Field MC, and Rout MP (2016) Interactome mapping reveals the evolutionary history of the nuclear pore complex. PLOS Biology 14: e1002365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehwinkel J, Herold A, Gari K, Köcher T, Rode M, Ciccarelli FL, Wilm M, and Izaurralde E (2004) Genome-wide analysis of mRNAs regulated by the THO complex in Drosophila melanogaster. Nat Struct Mol Biol 11: 558–566. [DOI] [PubMed] [Google Scholar]
- Rico E, Ivens A, Glover L, Horn D, and Matthews KR (2017) Genome-wide RNAi selection identifies a regulator of transmission stage-enriched gene families and cell-type differentiation in Trypanosoma brucei. PLoS Pathog 13: e1006279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rink C, and Williams N (2019) Unique interactions of the nuclear export receptors TbMex67 and TbMtr2 with components of the 5S ribonuclear particle in Trypanosoma brucei. mSphere 4: e00471–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwede A, Manful T, Jha BA, Helbig C, Bercovich N, Stewart M, and Clayton C (2009) The role of deadenylation in the degradation of unstable mRNAs in trypanosomes. Nucl Acids Res 37: 5511–5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DD, Aguilar LC, Kramar M, and Oeffinger M (2019) It’s not the destination, it’s the journey: Heterogeneity in mRNA export mechanisms. Adv Exp Med Biol 1203: 33–81. [DOI] [PubMed] [Google Scholar]
- Serpeloni M, Moraes CB, Muniz JR, Motta MC, Ramos AS, Kessler RL, Inoue AH, daRocha WD, Yamada-Ogatta SF, Fragoso SP, Goldenberg S, Freitas-Junior LH, and Avila AR (2011a) An essential nuclear protein in trypanosomes is a component of mRNA transcription/export pathway. PLoS One 6: e20730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpeloni M, Vidal NM, Goldenberg S, Avila AR, and Hoffmann FG (2011b) Comparative genomics of proteins involved in RNA nucleocytoplasmic export. BMC Evol Biol 11: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strambio-De-Castillia C, Niepel M, and Rout MP (2010) The nuclear pore complex: bridging nuclear transport and gene regulation. Nat Rev Mol Cell Biol 11: 490–501. [DOI] [PubMed] [Google Scholar]
- Tetenbaum-Novatt J, and Rout MP (2010) The mechanism of nucleocytoplasmic transport through the nuclear pore complex. Cold Spring Harb Symp Quant Biol 75: 567–584. [DOI] [PubMed] [Google Scholar]
- Tudek A, Schmid M, and Jensen TH (2019) Escaping nuclear decay: the significance of mRNA export for gene expression. Curr Genet 65: 473–476. [DOI] [PubMed] [Google Scholar]
- Tudek A, Schmid M, Makaras M, Barrass JD, Beggs JD, and Jensen TH (2018) A nuclear export block triggers the decay of newly synthesized polyadenylated RNA. Cell Rep 24: 2457–2467.e2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tutucci E, and Stutz F (2011) Keeping mRNPs in check during assembly and nuclear export. Nature Rev Mol Cell Biol 12: 377–384. [DOI] [PubMed] [Google Scholar]
- Verner Z, Basu S, Benz C, Dixit S, Dobáková E, Faktorová D, Hashimi H, Horáková E, Huang Z, Paris Z, Peña-Diaz P, Ridlon L, Týč J, Wildridge D, Zíková A, and Lukeš J (2015) Malleable mitochondrion of Trypanosoma brucei. Int Rev Cell Mol Biol 315: 73–151. [DOI] [PubMed] [Google Scholar]
- Wickramasinghe VO, Andrews R, Ellis P, Langford C, Gurdon JB, Stewart M, Venkitaraman AR, and Laskey RA (2014) Selective nuclear export of specific classes of mRNA from mammalian nuclei is promoted by GANP. Nucl Acids Res 42: 5059–5071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickramasinghe VO, and Laskey RA (2015) Control of mammalian gene expression by selective mRNA export. Nat Rev Mol Cell Biol 16: 431–442. [DOI] [PubMed] [Google Scholar]
- Zimmer SL, Simpson RM, and Read LK (2018) High throughput sequencing revolution reveals conserved fundamentals of U-indel editing. Wiley Interdiscip Rev RNA: e1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.