

SUMMARY
eIF4F plays diverse roles in human cancers, which complicate development of an overarching understanding of its functional and regulatory impacts across tumor types. Typically, eIF4F drives initiation from the mRNA 5’ end (cap), and is composed of eIF4G1, eIF4A1, and cap-binding eIF4E. Cap-independent initiation is possible without eIF4E, from internal ribosomal entry sites (IRESs). By analyzing large public datasets, we found that cancers selectively overexpress EIF4G1 more than EIF4E. That expression imbalance supports EIF4G1 as a prognostic indicator in patients with cancer. It also attenuates “house-keeping” pathways that are usually regulated in a tissue-specific manner via cap-dependent initiation in healthy tissues, and reinforces regulation of cancer-preferred pathways in cap-independent contexts. Cap-independent initiation is mechanistically attributable to eIF4G1 hyperphosphorylation that promotes binding to eIF4A1, and reduced eIF4E availability. Collectively, these findings reveal a novel model of dysregulated eIF4F function, and highlight the clinical relevance of cap-(in)dependent initiation in cancer.
eTOC blurb
Wu and Wagner leverage mRNA/proteomic correlations to investigate the translation initiation complex eIF4F in human cancers using large public datasets. They find selective expression imbalance of EIF4G1 and EIF4E in cancer, associated with attenuated cap-dependent initiation for “house-keeping” genes, and reinforced cap-independent initiation for cancer-preferred genes.
Graphical Abstract
INTRODUCTION
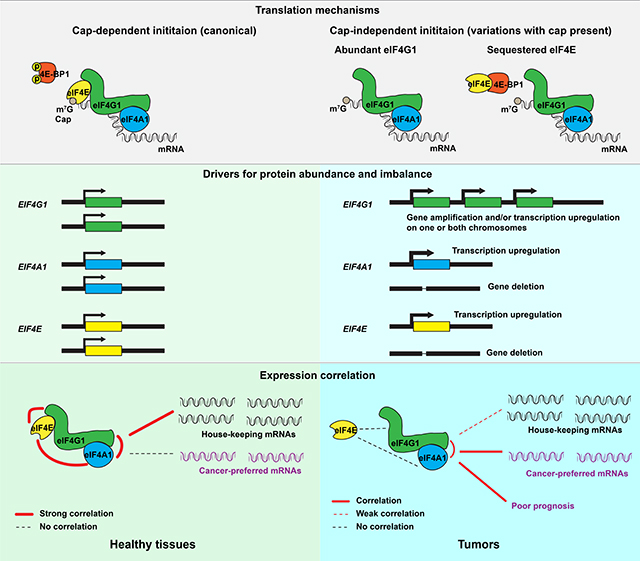
Translation is often stimulated as an onco-sustenance strategy (Wu and Naar, 2019) that permits malignant cells to survive, proliferate, and metastasize under adverse (e.g. anaerobic and nutrient-deficient) conditions that generally suppress protein synthesis in normal cells (Spriggs et al., 2010). The first step of translation initiation is a rate-limiting process in which ribosomes are recruited to an activated mRNA (Aitken and Lorsch, 2012). Its regulatory nexus is eIF4F, a protein complex typically consisting of eIF4G1, eIF4E1 (often, and hereinafter, referred to as eIF4E), and eIF4A1 (Merrick, 2015). eIF4G1 is a scaffold protein that directly binds the 5’ untranslated region (5’UTR) of mRNA and anchors eIF4E, eIF4A1, the eIF3 complex, PABP1, and Mnk1/2 (Jackson et al., 2010). eIF4E binds to N7-methylated GTP at the cap, where it modulates eIF4G1’s ability to stimulate eIF4A1, which in turn (together with eIF4B/eIF4H) unwinds mRNA secondary structure and thus permits ribosome attachment (Feoktistova et al., 2013; Merrick, 2015). This cap-dependent translation initiation mechanism typically requires both eIF4E and its cap binding for subsequent eIF4F activity on most eukaryotic mRNAs. There are also cap-independent mechanisms, for which the cap need not bind to eIF4E even when both are present, and neither the cap nor eIF4E is strictly required (Pelletier and Sonenberg, 2019). In most cases (e.g. poliovirus, encephalomyocarditis, hepatitis A virus, and some cellular mRNAs), cap-independent initiation relies on eIF4G1, or eIF4G2, to recognize the IRES at the 5’UTR and facilitate ribosome recruitment (Walsh and Mohr, 2011).
Prevailing thought holds that tumors frequently activate eIF4E to enhance cap-dependent initiation in support of transformation, tumorigenesis and metastasis (Graff et al., 2008). In this model, cellular proliferation and metabolism require continuous stimulation of cap-dependent initiation by environmental nutrients, growth factors, and hormones, via the mTOR pathway (Saxton and Sabatini, 2017). When active, mTORC1 phosphorylates eIF4E Binding Protein 1 (4E-BP1), thus rendering eIF4E free from 4E-BP1 inhibition, and thereby promotes cap-dependent initiation (Gingras et al., 1999; Gruner et al., 2016; Igreja et al., 2014). The postulated dependence on eIF4E or cap-dependent initiation to sustain malignancy has guided the development of cancer therapies (Bhat et al., 2015). However, physiological stresses such as hypoxia, nutrient deprivation, and chemo and radiation treatments tend to deactivate mTORC1; and hypophosphorylated 4E-BP can then block eIF4E●eIF4G1 interaction and inhibit cap-dependent initiation (Hara et al., 1998; Liu et al., 2006). Under stress, cap-dependent initiation is possible only with overabundant eIF4E, consistent with a previous finding from our lab that hypoxia upregulates EIF4E transcription (Yi et al., 2013). However, enablement of translation initiation under physiological stresses is requisite for viability in malignant tumors whether or not eIF4E is overabundant. Several cancer-related mRNAs (VEGFA, FGF2, and HIF1A) reportedly have recourse to both cap-dependent and -independent mechanisms (Badura et al., 2012). eIF4G1 and eIF4A1 are necessary for both cap-dependent and -independent mechanisms (with rare exceptions), but eIF4E is dispensable in cap-independent initiation. Such mechanistic diversity threatens to confound development of therapies related to eIF4F (or eIF4E specifically).
The prevalence and clinical relevance of cap-dependent and -independent mechanisms are ill-understood, yet important for advancement of cancer treatment. Although direct observation of eIF4F protein activity would be ideal, precise protein quantification of eIF4F subunits, particularly the 250-kDa eIF4G1, remains challenging with modern proteomic techniques (Timp and Timp, 2020). However, positive correlations between RNA-Seq and proteomics data for eIF4F have been reported in cancer cell lines (Nusinow et al., 2020), breast invasive carcinoma (Mertins et al., 2016), and lung adenocarcinoma (LUAD) (Gillette et al., 2020), due to their highly stable mRNAs and proteins (Schwanhausser et al., 2011). With EIF4F mRNA as a valid proxy for protein abundance, mechanistic dissection of eIF4F under physiological conditions is possible. A quantitative understanding of subunit abundance and stoichiometry, and consequent influence upon malignant phenotypes, would clarify which initiation mechanisms are employed. RNA-Seq data from large-scale biopsy studies offer statistical power and clinical relevance that traditional wet-lab biochemistry studies seldom achieve (Hutter and Zenklusen, 2018).
We analyzed copy number variation (CNV) and RNA-Seq data from The Cancer Genome Atlas (TCGA) to gain insights into initiation mechanisms in more than 10,000 tumors, and the Genotype Tissue Expression (GTEx) database to investigate normal tissues. We found that most tumors gain gene copies of EIF4G1. Additionally, among initiation factors, elevated EIF4G1 expression most closely correlates with poor survival. In healthy tissues, EIF4F subunit expressions collectively distinguish between healthy tissues, and strong correlation of EIF4G1, EIF4A1 and EIF4E with house-keeping genes suggests cap-dependent initiation. In contrast, loss of functional alignment of EIF4G1 and EIF4E is evident for tumors. EIF4G1 and EIF4A1 achieve correlation with cancer-preferred genes that are uncorrelated with EIF4E. From Clinical Proteomic Tumor Analysis Consortium (CPTAC) phospho-proteomics data, we found evidence of altered biochemical interactions among eIF4F subunits that are mechanistically linked to cap-independent initiation. Hence, tumors, among initiation mechanisms they employ, rely for survival upon heightened adoption of cap-independent initiation.
RESULTS
Among Initiation Factor Genes, EIF4G1 Is Frequently Amplified and Overexpressed in Tumors
EIF4E, EIF4EBP1, and EIF4G1 gene amplifications have each been attributed to mRNA and protein overexpression in many cancers (Jaiswal et al., 2018; Rutkovsky et al., 2019; Sorrells et al., 1998; Sorrells et al., 1999). It remains unclear whether eIF4F subunits are equally prone to gene amplification, and if so, whether commensurate overexpression of the entire complex is implied. We analyzed EIF4F subunit CNVs in all 10,845 TCGA tumors combined (Figures 1A–1B). CNVs were categorized into five statuses: amplification, duplication, diploid, heterozygous deletion, and homozygous deletion, according to array-based DNA copy number data (Mermel et al., 2011). We found contrasting CNV statuses. EIF4G1 is frequently duplicated (26.79%) and amplified (7.74%). EIF4G1 and EIF4A2 CNV statuses display strong statistical association, consistent with chromosomal proximity at 3q27. Frequent duplication of EIF4H (36.83%) is attributable to its microduplication-prone location at 7q11.23 (Abbas et al., 2016). Heterozygous deletions of EIF4A1 (40%) and TP53 (39.76%) show strong association, consistent with their proximity on 17q13.1. Heterozygous deletions of EIF4E are also frequent (30.21%), possibly due to chromosomal fragility at 4q23 (Morgan et al., 1988).
Figure 1. Among Initiation Factor Genes, EIF4G1 Is Frequently Amplified and Overexpressed in Tumors.

(A) The stacked bar plot shows overall CNV statuses for translation initiation genes in all tumors combined from 33 TCGA cancer types. We used TCGA groupings of CNV value estimates that were derived from whole genome microarray data by the GISTIC2 method. The estimated gene-level CNV values were grouped with thresholds 3+, 3, 2, 1, 0, to represent high-level copy number gain (amplification), low-level copy number gain (duplication), diploid, shallow (possibly heterozygous) deletion, or deep (possibly homozygous) deletion, respectively. Percentage contributions of each group are labeled on the bars.
(B) The matrix plot shows Pearson correlation coefficients for translation initiation gene CNVs. For each gene, a list of estimated CNV values for 10,845 tumors (all TCGA study groups) is correlated with the corresponding list for another gene. Each cell is colored based on the magnitude of the resulting coefficient, and cells with ‘X’ indicate statistical insignificance (p > 0.05, p values not shown). Aside from identity relationships (topmost diagonal cells), strong co-occurrence (dark blue) is evident for TP53/EIF4A1 and EIF4G1/EIF4A2 – gene pairs with neighboring chromosomal locations.
(C to F) For each cancer study group, a stacked bar plot shows CNV status for a translation initiation gene (marked at the bottom of each plot), and a box and whisker plot shows corresponding mRNA expression of the same gene in tumor samples and Normal Adjacent Tissues (NATs). The X axes of box plots represent normalized gene level expression (transcripts per million) in log2 scale. TCGA uses the same bioinformatics pipeline to process and normalize RNA-Seq data from different cancer study groups, to minimize batch effects of sequencing data processing
To further characterize tumors in terms of CNV statuses, we considered normal adjacent tissues (NATs) and analyzed average tumor:NAT CNV ratios (Figures S1A–S1B). In most cancers, the mean CNV ratios of EIF4G1, EIF4A2, EIF4B, and EIF4H are higher than 1, indicating duplication/amplification tendencies in tumors. Mean ratios for EIF4E, EIF4A1, EIF4G2, EIF4G3, EIF4E2, EIF4E3, and EIF3D are lower than 1, indicating deletion tendencies in tumors. The average ratios for EIF4EBP1 are close to 1, due to the similar frequencies of duplication and heterozygous deletion in Figure 1A.
CNVs affect large regions that contain many genes of diverse functions. Transcription regulation of individual gene expression permits fine-grained exploitation or counteraction of CNV impacts. We analyzed the differential mRNA expression for EIF4F subunits in tumors vs NATs. We found frequent EIF4G1 duplication and amplification from most TCGA cancer types and markedly elevated EIF4G1 expression in tumors vs. NATs (Figure 1C). Lung squamous cell carcinomas (LUSCs) have the highest average EIF4G1 expression among all cancers but no apparent EIF4G1 upregulation in NATs, suggesting that EIF4G1 overexpression in lung tumors is not lung tissue-specific. We verified that EIF4G1 expression is significantly elevated in metastatic and primary tumors from all TCGA cancer studies combined (Figure S3E). EIF4H exhibits frequent duplication and elevated expression vs NATs, among cancer types (Figure S3B). EIF4G2, EIF4E2, and EIF4E3 exhibit frequent heterozygous deletions, and low expression vs NATs, among cancer types (Figure S2A, S2D and S2E). Low expression is also evident for combined primary and combined metastatic tumors (Figure S3C).
CNV and differential expression can exhibit loose correlation that fluctuates by cancer type, as with EIF4EBP1 (Figure 1F), or even exhibit clear independence. Despite frequent heterozygous deletions, overexpression of EIF4E, EIF4A1, EIF4G3, and EIF3D occurs among cancer types (Figures 1D, 1E, S2B and S2F). EIF4E is mildly elevated for combined primary tumors. EIF4A1 and EIF3D are significantly elevated in combined primary and combined metastatic tumors (Figure S3C). Conversely, despite frequent duplication and amplification, neither EIF4A2 nor EIF4B is elevated in most individual cancer types, (Figures S2C and S3A), nor in all tumor types collectively (Figure S3C). c-Myc regulatory (E-box) motifs are present in promoter regions for EIF4G1, EIF4A1, and EIF4E, but not for EIF4A2 (Jones et al., 1996; Lin et al., 2008). Transcription regulation by c-Myc may contribute to mRNA overexpression of EIF4A1 and EIF4E despite their gene deletions. Overexpression of canonical eIF4F subunits (EIF4G1, EIF4A1, and EIF4E) suggests enhanced initiation activity in multiple cancer types.
EIF4G1 Expression Is a Strong Predictor for Survival in Patients with Cancer
To assess how cap-(in)dependent initiation is related to cancer progression, we performed Kaplan-Meier (KM) analysis to associate survival probabilities with EIF4F gene expression. In all TCGA cancer types combined, the survival probabilities of patients with high expression (top 20% or 30%) are significantly worse than those with low expression (bottom 20% or 30%), of EIF4G1 (Figures 2A and S4G), EIF4A1 (Figures 2C and S4I), EIF4EBP1 (Figures 2D and S4J), EIF4G2 (Figures 2E and S4K), EIF4E2 (Figures 2G and S4M), MKNK1 (Figures 2K and S4U), EIF4H (Figures S4D and S4T), PABPC1 (Figures S4E and S4W), and MYC (Figures S4F and S4X). eIF4G2 participates only in cap-independent initiation (Hundsdoerfer et al., 2005), which, judging by correlation with poor prognosis (Figures 2E and S4K), may benefit tumors. eIF4E2 participates in cap-dependent initiation by interacting with eIF4G3 under hypoxia (Ho et al., 2016), and its mRNA expression correlates with poor prognosis (Figures 2G and S4M), hinting at functional relevance of cap-dependent initiation with eIF4E2 to cancer progression. In contrast, the survival probabilities are significantly greater for the patients with high expressions of EIF4E3 (Figures 2H and S4N), EIF4A2 (Figures 2I and S4O), MKNK2 (Figure 2L and S4V), EIF4EBP2 (Figures S4A and S4P), and EIF4B (Figures S4C and S4S). We observed no significant difference regarding EIF4E (Figures 2B and S4H), EIF4G3 (Figures 2F and S4L), EIF3D (Figures 2J and S4Q), or EIF3E (Figures S4B and S4R). Although mRNA expressions of canonical eIF4F subunits are upregulated in tumors, EIF4G1, EIF4A1 but not EIF4E correlate with poor prognosis (Figures 2A–2C, S4G–S4I). These results hint at functional relevance of cap-independent initiation with eIF4G1 and eIF4A1 to cancer progression.
Figure 2. EIF4G1 Expression Is a Strong Predictor for Survival in Patients with Cancer.

(A to L) Each Kaplan-Meier plot shows survival probabilities of TCGA patients with cancer according to mRNA expressions of a specific translation initiation gene (marked inside each box, at top) in their tumors. Survival probability (Y axis) is the probability of individual survival from the time origin (e.g. initial cancer diagnosis) to a specified time (X axis). We ranked all 10,295 TCGA patients with cancer based on the indicated individual gene expressions from their tumor biopsies, and selected two groups of patients with the top or bottom 20% of gene expression. Differences in survival probabilities between the two selected groups were assessed with the log-rank test. The shaded areas around each curve depict a 95% confidence region for that curve.
(M and N) Univariable (M) and multivariable (N) Cox proportional-hazards regression models for expression of translation initiation genes in all 10,235 patients with cancer from TCGA. P value indicates the statistical significance of association between gene expression and survival (i.e. a significant fit in the Cox-PH model). “P value for interaction” is calculated using the Schoenfeld residual method. P value for interaction < 0.05 indicates statistically significant interaction between gene expression and time, a violation of the Cox proportional-hazards model. Values without violation are marked with a blue asterisk. “CI” means “confidence interval”.
We then used the Cox proportional-hazards (PH) regression to quantitatively relate patient survival and gene expression in tumors (hazard ratio and P value). PH models assume that hazard rates are constant over time, i.e. gene expression has no time dependence. We tested for violation of this assumption by assessing the likelihood of violation (P value for interaction). We performed univariable Cox-PH analyses using a single gene expression as the dependent variable (Figure 2M). As EIF4G1 or MYC expression increases, the chance of death significantly increases (36% in the case of EIF4G1 and 16% MYC), with no PH assumption violation. In contrast, as MKNK2 expression increases, the chance of death significantly decreases by 20%, also without violation. Although MKNK1, EIF4A1, EIF4E2, EIF4EBP1, EIF4G2, and EIF4H expressions are each associated with poor prognosis while EIF4B, EIF4E3, and EIF4A2 each associated with good prognosis, they exhibit significant PH assumption violations and thus cannot be related conclusively to survival expectation. We performed multivariable Cox-PH analyses to model patient survival and expressions of all initiation factors together (Figure 2N). EIF4G1 expression significantly correlates with poor prognosis and MKNK2 with good prognosis, without violation. Some genes become statistically significant in multivariable analysis, despite lacking significant prognostic value in univariable analysis (Figure 2M). This may be attributable to confounding influences (e.g. of EIF4E2 upon EIF4G3, or EIF4EBP1 upon MTOR). In sum, EIF4G1 is the only initiation factor whose expression significantly correlates with poor prognosis in the KM, univariable and multivariable Cox-PH analyses, suggesting a critical role in disease progression.
To validate our findings in a specific cancer type, we performed KM analysis on 517 patients with LUAD. The survival probabilities of patients with high expression of EIF4G1 (Figure S5A), EIF4E (Figure S5B), EIF4A1 (Figure S5C) and EIF3D (Figure S5J) are significantly worse than those with low expression. In contrast, the survival probability of the patients with high EIF4E3 expression (Figure S5H) is significantly greater. However, we observed no significant difference related to EIF4EBP1 (Figure S5D), EIF4G2 (Figure S5E), EIF4G3 (Figure S5F), EIF4E2 (Figure S5G), EIF4A2 (Figure S5I), MKNK1 (Figure S5K), and MKNK2 (Figure S5L) expression. Univariable Cox-PH analyses indicate that EIF4G1 and EIF4A1 expressions each have negative survival association, without violation (Figure S5M). Multivariable analysis (Figure S5N) indicates that EIF4G1 and EIF3E expressions together predict poor prognosis, without violation. Thus, among KM, univariable and multivariable Cox PH analyses, multiple initiation factors exhibit correlation with poor prognosis in LUADs; but EIF4G1 alone exhibits correlation in all three.
Tumors Have Altered EIF4G1:EIF4E and Characteristic EIF4G1:(EIF4E + EIF4EBP1) Ratios
To investigate whether tumor-specific overexpression perturbs the stoichiometry of initiation subunits, we analyzed TCGA RNA-Seq data, which has normalized sequencing counts of each gene relative to gene length and total RNA counts. We first compared the abundances of all EIF4F subunits in tumors from 33 TCGA cancer types. EIF4A1, EIF4A2, EIF4G1, and EIF4G2 are much more abundant than EIF4G3, EIF4E, EIF4E2, EIF4E3, and EIF4EBP1 in all cancer types (Figure 3A). Higher expressions of EIF4G1 and EIF4A1 than EIF4E may be due to participation of eIF4G1 and eIF4A1 in cap-independent initiation. Consistently, eIF4E was reported less abundant than other subunits in eIF4F protein complexes purified by various biochemical methods (Duncan et al., 1987; Galicia-Vazquez et al., 2012). We then calculated the ratios of EIF4F subunits within each tumor. The optimal binding of eIF4G and eIF4E on the cap occurs at a 1:1 ratio (Haghighat and Sonenberg, 1997), and indeed our observation is close to 1:1 for EIF4G3:EIF4E2 specifically, in most tumors and NATs (Figures 3B, S6A and S6B). However, although EIF4G1, EIF4A1 and EIF4E expressions are all elevated in tumors, the elevation is greater for EIF4G1 and EIF4A1 than for EIF4E. EIF4G1:EIF4E and EIF4A1:EIF4E ratios are significantly elevated in most cancer types, with the highest ratios in metastatic tumors (Figures 3B and S6B), indicating EIF4F expression imbalance in cancer.
Figure 3. Tumors Have Altered EIF4G1:EIF4E and Characteristic EIF4G1:(EIF4E + EIF4EBP1) Ratios.

(A) Comparison of EIF4F gene expressions across different tumor types. The box plot represents, for each indicated cancer type (labelled at bottom) expression (transcripts per million) for 9 genes in log2 scale. The Y axis indicates normalized RNA-Seq counts. The graph overall depicts 9,806 malignant tumors of 33 cancer groups in TCGA.
(B and C) Gene expression ratios for tumors and NATs in different cancer types. Each box plot depicts ratios of RNA counts (X-axis, linear scale) between two genes (marked at top), computed from each sample of a particular cancer type (left), for tumors and NATs considered separately. In the last two panels of C, the summed expression of EIF4E and EIF4EBP1 (E+EBP1) was calculated within each sample, then used to compute the ratio between EIF4G1 or EIF4A1 (marked at top) and E+EBP1 for that sample. The dashed line indicates where the 4:1 ratio falls on the X axis.
Furthermore, we compared abundance among homologs. Average EIF4G1:EIF4G3 ratios are significantly higher in most tumors than in NATs, while EIF4G2:EIF4G1 ratios are lower. These results suggest that EIF4G1 is preferred over its homologs in tumors. Similarly, average EIF4A1:EIF4A2 ratios are significantly higher in most tumors, suggesting a preference for EIF4A1 (Figures 3C, S6A and S6B). EIF4E2:EIF4E ratios remain close, between tumors and NATs. In sum, these results hint a preference of EIF4G1 and EIF4A1 over other homologs by tumors, so the altered EIF4G1:EIF4E and EIF4A1:EIF4E ratios likely have the strongest impact on initiation.
Average EIF4E1:EIF4EBP1 ratios are over 1 in both tumors and NATs from all cancer types, but dramatically decrease in tumors, most of all in metastatic tumors (Figures 3C, S6A and S6B). We calculated the summed expression of EIF4E and EIF4EBP1 (E+EBP) within each sample. The average EIF4G1:(E+EBP) ratios remain relatively constant around 4:1 in most cancer types but vary greatly across NATs (Figures 3C). The 4:1 ratio of EIF4G1:(E+EBP) is consistent across metastatic and primary tumors (Figure S6B). EIF4E and EIF4EBP1 promoters contain hypoxia-responsive elements (HREs) that can be directly regulated by HIF1α (Azar et al., 2013; Yi et al., 2013). Since the HRE and the E-box motif overlap on the EIF4E promoter, HIF1 α and c-Myc compete to transcriptionally regulate EIF4E (Gordan et al., 2007), which may perturb c-Myc regulation of EIF4G1 and EIF4E. The EIF4G1:(E+EBP1) ratio in tumors probably reflects the interplay among c-Myc, HIF1α and other factors on transcription regulation under hypoxia. Detailed mechanistic explanation of the 4:1 ratio will require additional biochemical investigation.
EIF4F Expressions Collectively Better Distinguish Healthy Tissues than Tumor Types
The mRNA overexpression and altered ratios of canonical EIF4F subunits in tumors suggest enhanced but altered initiation activities. We used principal component analysis (PCA) to assess the collective activities of EIF4F subunits across biopsies. We also included PABPC1, MNK1, and MNK2, because the proteins coded by those genes biochemically interact with eIF4G1 and are deemed peripheral factors of the eIF4F complex. We performed three individual PCAs.
First, in a PCA of RNA-Seq data from the 30 healthy tissue types in GTEx (Figures 4A), samples separate widely along Figure 4A-PC1 and partially along Figure 4A-PC2, as multiple distinct blobs representing brain (cyan), pancreas (light green), muscle (grey), and blood samples (yellow). PC1 and PC2 both play a part in sample separation. All seven genes contribute to PC1. Only EIF4G1, EIF4E and MKNK1 are meaningful contributors to PC2 (Figure S7A). We reason that the gene expressions are distinctly controlled and bounded for individual tissue types.
Figure 4. EIF4F Expressions Collectively Better Distinguish Healthy Tissues than Tumor Types.

(A) Principal Component Analysis (PCA) of RNA-Seq-derived counts of EIF4G1, EIF4A1, EIF4E, EIF4EBP1, PABPC1, MKNK1, and MKNK2 from 7,388 tissue samples of various healthy tissue types in GTEx. Tissue types were observations – not used to construct PCs but colored for visualization. The PCA biplot shows the PCs with most significant sample variation. Axis titles show the percentage of variances (squared loadings) explained by PC1 or PC2. Axis values show the PCA scores of individual samples. Arrows show the influence of each gene variable on the PCs.
(B) PCA of normalized RNA-Seq-derived counts of indicated genes from 9,162 primary tumors of 33 cancer types in TCGA. Cancer types were colored after analysis, for visualization.
(C) PCA of normalized RNA-Seq-derived counts of indicated genes from 392 metastatic tumors of 33 cancer types in TCGA. Cancer types were colored afterwards. Note: 366 metastatic tumors are from TCGA skin cutaneous melanoma group.
(D) PCA of normalized RNA-Seq-derived counts of indicated genes from 9,162 primary and 392 metastatic tumors from TCGA, and 7,388 healthy tissue samples from GTEx. RNA-Seq data from TCGA and GTEx have been processed and normalized with the same bioinformatics pipeline to minimize batch effects of sequencing experiments. Sample types were colored after analysis, for visualization.
(E) The matrix plot shows the cos2 value for the contribution of each gene to each PC, from the PCA of TCGA tumor samples and GTEx normal tissue samples in Figure 4D. The sum of values in a given row across all PCs is equal to one (+/− epsilon introduced by rounding each value to two significant figures).
(F) PCA of standardized RNA-Seq-derived counts of indicated genes from 1011 primary lung tumors (517 LUADs and 494 LUSCs) and 109 NATs from TCGA LUAD and LUSC study groups, and 287 healthy lung tissues from GTEx. Sample types were colored after analysis, for visualization.
Second, in a PCA of primary tumor data from 32 TCGA cancer types (Figure 4B), most cancer types form an overlapping cluster at the center of biplot. A few cancer types, e.g. head & neck squamous cell carcinoma (blue on the right) and pheochromocytoma (pink on the left), separate horizontally along Figure 4B-PC1. Most cancer types have distributions above and below the Figure 4B-PC1 axis, suggesting PC2 does not play a significant role in separating cancer types. Overall, data separation is less distinct than in Figure 4A. Tumor gene contributions are distributed across all 7 PCs (Figure S7B). Contrastingly, the PCA for healthy tissues (Figure S7A) made little use of PCs beyond PC3 (almost zero for PC6 or PC7). We reason that general EIF4F overexpression in cancers blurs tissue-specific expression boundaries. Moreover, no PC for the primary tumors has significant contribution from both EIF4E and EIF4G1 (Figure S7B), whereas PC1 and PC2 for the healthy tissues both have contributions from both genes (Figure S7A). This result is consistent with our earlier observation of EIF4F expression imbalance in primary tumors, and furthermore hints at a functional separation between EIF4E and other EIF4F subunits.
Third, in a PCA of metastatic tumor data from 11 TCGA cancer types (Figures 4C and S7C), Figure 4C-PC1 and Figure 4C-PC2 fail to separate the samples, which is expected because skin cutaneous melanoma is predominant. In sum, three PCA results showed that EIF4F is well able to distinguish healthy tissue identities but is much less able for tumors. These results support the notion that eIF4F activities are distinctly-regulated in individual healthy tissue types to carry out tissue-specific functions, and such regimes are lost across cancer types (Ruggero, 2013).
To further compare the collective activities of EIF4F subunits between tumors and healthy tissues, we applied PCA to the normalized RNA-Seq data from both TCGA tumors and GTEx healthy samples (Figures 4D and 4E). Tumor samples separate from healthy ones (Figure 4D). The nature of the PCs in Figure 4D is partly revealed by Figures S8A–S8F, in which the same PCs are preserved and the isolated subsets are color-coded by tissue or tumor types. PC1 strongly distinguishes between healthy brain tissues (magenta in Figure S8D) and metastatic skin cutaneous melanomas (brown in Figure S8F). PC2 strongly distinguishes between healthy brain tissues (magenta in Figure S8D), and healthy blood samples (yellow in Figure S8D). Most primary tumors from various cancers cluster with significant overlap (Figures S8B and S8E). PC1 is composed primarily of EIF4G1, EIF4A1, EIF4EBP1 and PABPC1; PC2 is of EIF4E, MKNK1 and MKNK2. These results suggest that EIF4G1 and EIF4E expressions each contribute to separate malignant from healthy tissues.
To verify functional alignment of genes from the same PC, we repeated the PCA on combined tumor and healthy samples with four additional controls (Figure S7D): MYC – a gene relying on eIF4F for translation initiation (Nanbru et al., 1997); JUN – a gene relying on eIF3D but not eIF4F for translation (Lee et al., 2016); EIF4B; and EIF4H. MYC, EIF4G1, EIF4A1, EIF4EBP1, and PABPC1 primarily contribute to PC1. Figure S7E-PC1 contains all elements of Figure 4E-PC1, plus additional genes including MYC. The EIF4F genes (except for EIF4E) are aligned with MYC. This result is expected given the cap-independent mechanism for MYC translation during mitosis, apoptosis or cell stress (Kim et al., 2003; Stoneley et al., 2000; Subkhankulova et al., 2001), and the established use of MYC expression as a malignancy biomarker (Trop-Steinberg and Azar, 2018). In contrast, JUN dominates PC4 and does not meaningfully coincide with EIF4F subunits in other PCs (Figure S7E), consistent with its eIF4F-independent translation mechanism (Lee et al., 2016). Finally, EIF4B and EIF4H both contribute to PC1 (Figure S7E), consistent with their auxiliary roles for eIF4F during initiation (Rogers et al., 2001). EIF4B and EIF4H also contribute to other PCs, likely due to eIF4F-independent functions (Methot et al., 1996). These results confirm functional alignment of genes in the same PCs. We reason that the contribution of EIF4G1 and EIF4E to separate PCs (Figures S7B and 4E) is consistent with both expression imbalance and functional separation of eIF4F subunits, and furthermore indicates functional dysregulation of eIF4F overall.
Finally, we performed PCA on individual cancer types with matched healthy tissues, using RNA-Seq and proteomics data. PCA of normalized RNA-Seq data from TCGA (LUAD and LUSC studies) and GTEx (healthy lung tissues) shows wide separation between lung tumors and healthy tissues along the PC1 axis (Figure 4F). PCA of proteomics data from CPTAC shows similar separation between LUADs and NATs (Figures S7F). Thus, both mRNA and protein expressions of EIF4F distinguished malignant from healthy lung samples. We further performed PCA with RNA-Seq data from the following tumor/tissue pairs: lowergrade glioma and glioblastoma multiforme vs. healthy brain tissue (Figure S9A), breast invasive carcinoma vs. healthy mammary tissue (Figure S9B), colon adenocarcinoma vs. healthy colon tissue (Figure S9C), pancreatic adenocarcinoma vs. healthy pancreas tissue (Figure S9D), prostate adenocarcinoma vs. healthy prostate tissue (Figure S9E), skin cutaneous melanoma vs. healthy skin tissue (Figure S9F). Wide separations between tumors and healthy tissues are present in the PCA plots of all cases. Overall, these results confirm that EIF4F genes collectively serve as biomarkers to distinguish tumors from healthy tissues.
EIF4F Subunits Have More and Stronger Expression Correlations in Healthy Tissues than in Tumors
We sought an underlying biological basis for our findings that EIF4F subunits have imbalanced expression in tumors, and are predictive of cancer progression. Because positive RNA/protein correlations of EIF4G1, EIF4A1 and EIF4E (Figure 5A) were observed in 375 cell lines from various cancer types (Nusinow et al., 2020), we took their RNA expressions as proxies for protein levels in tumors. We used RNA-Seq data to identify genes dependent upon each eIF4F subunit in tumors or healthy tissues. We calculated Pearson’s correlation coefficients between each gene (EIF4E, EIF4G1, EIF4A1, or EIF4EBP1) and all detected genes across 10,323 TCGA tumor samples, to identify genes with positive correlations (posCORs) or negative correlations (negCORs). We similarly identified posCORs and negCORs for each EIF4F gene across 7,414 GTEx healthy tissues.
Figure 5. EIF4F Subunits Have More and Stronger Expression Correlations in Healthy Tissues than in Tumors.

(A) The scatter plots show the positive correlation between protein and mRNA expression levels of EIF4G1, EIF4A1 and EIF4E (indicated at top) in tumors. RNA expression levels (log2 transformed and upper quartile normalized) were from Cancer Cell Line Encyclopedia (CCLE) RNA-Seq data (Barretina et al., 2012). Protein expression levels (quantified as the relative abundance of detected peptides to reference, and log2 transformed) were from CCLE proteomics data (Nusinow et al., 2020). The plots include all 375 CCLE cell lines for which both RNA-seq and proteomic data are available.
(B and C) Pearson’s correlation coefficients between EIF4F (EIF4E, EIF4G1, EIF4A1, or EIF4EBP1) and each of 58,582 other genes were calculated separately across 10,323 TCGA tumor samples from different cancer types, or across 7,414 GTEx healthy samples from different tissue types, using the Toil recomputed RNA-Seq datasets. Genes with significant positive (R > 0.3) or negative (R < −0.3) correlations were selected for analysis. Venn diagrams show overlapping posCOR counts for EIF4F genes in healthy tissue samples (B) or in tumors (C).
(D and E) The heatmap (D) shows correlation strengths of posCORs and negCORs in healthy tissues and in tumors. Each row indicates correlation of a gene with each EIF4F gene in healthy tissues (grouped in left-side columns) and tumors (right-side columns). The color and intensity of each heatmap cell represents a Pearson’s correlation coefficient. posCORs are red, negCORs are blue, non-correlations are white. The dendrogram on the top indicates similarity as a hierarchical relationship between the columns: more-similar columns are linked lower in the dendrogram. Rows were ordered by K-means clustering to partition the dataset into three non-overlapping subgroups. Dot plots (E) show the enriched pathways for the three heatmap (D) row clusters, according to REACTOME pathway analysis. The 8 most significantly enriched pathways of each cluster were plotted.
(F) The scatter plots show the positive correlation between protein and mRNA expression levels of EIF4G1, EIF4A1 and EIF4E across 109 LUADs from CPTAC. RNA expression levels (log2 transformed and upper quartile normalized) were from the CPTAC RNA-Seq dataset. Protein expression levels (two-component normalized) were from CPTAC proteomics dataset (Gillette et al., 2020). The plots include all CPTAC LUADs for which both RNA-seq and proteomic data are available.
(G and H) Pearson’s correlation coefficients between EIF4F (EIF4E, EIF4G1, EIF4A1, or EIF4EBP1) and 58,582 other genes were calculated separately across 1,122 lung tumors from LUSC and LUAD TCGA study groups, or across 287 healthy lung tissues from GTEx, using the Toil recomputed RNA-Seq datasets. Genes with significant positive (R > 0.3) or negative (R < −0.3) correlations were selected for analysis. The Venn diagrams show overlapping posCOR counts for EIF4F genes in healthy lungs (G) or lung tumors (H).
(I and J) Analysis is similar to (D and E), but specific to lung samples. The heatmap (I) shows the correlation strengths of posCORs and negCORs for EIF4E, EIF4G1, EIF4A1 and EIF4EBP1 in healthy lungs and in lung tumors. The dot plots (J) show the enriched pathways in three clusters (K-means) of the heatmap (I) yielded by REACTOME pathway analysis.
In general, EIF4F subunits have more posCORs in healthy tissues than in tumors (Figure S10A). Healthy tissues and tumors alike contain more posCORs than negCORs (Figures S10B–S10D). In healthy tissues, EIF4A1 and EIF4G1 share most posCORs; and EIF4E shares more than half of its posCORs with EIF4G1 and EIF4A1 (Figure 5B). In tumors, EIF4A1 shares a third of its posCORs with EIF4G1; and EIF4E, EIF4G1 and EIF4A1 share only 50 posCORs (Figure 5C). These results suggest that healthy tissues – but not tumors – have largely overlapping posCORs for EIF4F genes. Hierarchical clustering confirmed the similarity of the EIF4F posCORs in healthy tissues (Figure 5D), and showed that posCORs from healthy tissues have stronger correlations than those from tumors. These results suggest that many mRNAs across healthy samples are subject to strong regulation by EIF4E, EIF4G1 and EIF4A1.
We used partitioning to classify correlating genes into three clusters (Figure 5D) and performed pathway enrichment analysis (Figure 5E). The “cluster one” genes are posCORs for EIF4E in healthy tissues but not in tumors. They are involved in the neuronal function-related pathways. The “cluster two” genes are moderate posCORs for EIF4E, EIF4G1 and EIF4A1 in healthy tissues, but their correlation strengths became weaker – particularly for EIF4E – in tumors. They are involved in extracellular matrix organization and interleukin signaling pathways. The “cluster three” genes are strong posCORs for EIF4E, EIF4G1 and EIF4A1 in healthy tissues, but – as cluster two – their correlation strengths become weaker in tumors. They are involved in house-keeping pathways including translation, pre-mRNA and ribosomal RNA processing, which are reportedly linked to translation initiation (Moore and Proudfoot, 2009). For a given posCOR of all EIF4F genes in tumors, correlation strength with each EIF4F gene varies widely, which we regard as the likely biological basis for the varying prognostic effects among EIF4F expressions (Figures 2A–2D and 2M).
To validate our findings in a specific cancer type, we performed correlation analyses in lung tumors (LUSC and LUAD), and in healthy lung tissues. We confirmed the positive RNA/protein correlations for EIF4G1, EIF4A1 and EIF4E in LUADs from CPTAC (Figure 5F) (Gillette et al., 2020). Healthy lung tissues contain more posCORs for EIF4F than lung tumors (Figure S10E). There are fewer negCORs than posCORs in healthy lung tissues and in lung tumors (Figures S10F–S10H). In healthy tissues, EIF4E, EIF4A1 and EIF4G1 shared most of their posCORs (Figure 5G) suggesting overlapping functions. In lung tumors, EIF4E, EIF4G1 and EIF4A1 share only 176 posCORs (Figure 5H). The healthy posCORs have much higher correlation coefficients than tumor posCORs (Figure 5I), which indicates similar and strong regulation of EIF4E, EIF4A1 and EIF4G1 over posCORs in healthy lungs.
We again classified correlating genes in three clusters (Figure 5I) and performed pathway enrichment analysis (Figure 5J). The “cluster one” genes are weak posCORs for EIF4E, EIF4G1, EIF4A1, and EIF4EBP1 in healthy lungs. In lung tumors, their correlations with EIF4G1 and EIF4A1 are stronger by comparison, and their correlations with EIF4E are weaker. They are involved in keratinization (formation of cornified envelope) – a pathological process associated with LUSC progression (Park et al., 2017) – and cell cycle, which suggest that EIF4G1 and EIF4A1 reinforce their regulation of pathways that benefit malignancy. The “cluster two” genes are moderate posCORs for all four genes in healthy lungs, but are negCORs for EIF4G1, EIF4A1 and EIF4EBP1 in lung tumors. They are involved in extracellular matrix organization and cell communication. The “cluster three” genes are strong posCORs for all four genes in healthy lungs, but weak posCORs in lung tumors. They are involved in house-keeping pathways such as translation and RNA processing. Altogether, lung tumors exhibit dysregulation of EIF4F function, characterized by weakened regulation of housekeeping pathways, and strengthened regulation specifically by EIF4G1 and EIF4A1 of certain pathological pathways.
Dysregulated eIF4F Function Favors Cap-Independent Initiation in LUADs
To verify the effects of dysregulated eIF4F function, we analyzed correlation among eIF4G1, eIF4A1 and eIF4E protein levels in LUADs. We observed a strong positive correlation (hence, potentially overlapping function) between eIF4A1 and eIF4G1, but no correlation between eIF4A1 and eIF4E (Figure 6A). We verified strong positive correlation of eIF4G1 and eIF4A1, but not eIF4E, with 40S ribosomal protein S2 (Rps2) (Figure 6B) and other initiation factors, such as eIF3B, eIF3G, eIF2S2 and eIF2S3 (Figure 6C). Among mRNAs, EIF4G1 and EIF4A1 correlate more strongly than EIF4E with “cluster one” genes in lung tumors (Figures 5I–5J). EIF4F mRNAs can furthermore be related to eIF4F proteins (Figure 5F). We further found positive protein correlations of eIF4A1 and eIF4G1 – but not eIF4E – with cell-cycle-related proteins (a subset of “cluster one”), e.g. ERCC6L, CKAP2, CCNA2 and MCM7 (Figure 6D). We reason that eIF4G1 and eIF4A1 are more influential than eIF4E over cell cycle gene translation in LUADs. By implication, the biochemical basis of eIF4F dysregulation may be the adoption of cap-independent initiation for certain genes.
Figure 6. Dysregulated eIF4F Function Favors Cap-Independent Initiation in LUADs.

(A) The scatter plots show correlation of eIF4G1, eIF4A1 and eIF4E protein expression levels with each other across 109 LUADs from CPTAC. The expression level is shown as log2 ratio of protein abundance in sample to a reference by the two-component normalization method (Gillette et al., 2020).
(B) The scatter plots show correlation of protein expression levels between Rps2 and each of three eIF4F subunits across 109 LUADs.
(C) The scatter plots show protein expression correlation of indicated translation initiation factors (rows) and each of three eIF4F subunits (columns) across 109 LUADs.
(D) The scatter plots show protein expression correlation of indicated protein factors involved in cell division (rows) and each of three eIF4F subunits (columns) across 109 LUADs.
(E and F) The box plots show, for eIF4F subunits, ratios to paired NATs (Y axis) of mean peptide abundance (either whole protein or phospho-peptide with phosphorylation at indicated serine or threonine residues). Depicted for NATs and LUADs at each tumor stage are: eIF4G1 (whole protein or phosphorylation at serine 1029, 1093, or 1098), eIF4A1 (whole protein or phosphorylation at serine 78), eIF4E (whole protein or phosphorylation at threonine 68), 4E-BP1 (whole protein, or phosphorylation at threonines 36 and 46, or at serine 44 and threonine 46, or at serine 65). Mean expression from NATs was normalized at 1. Phospho-peptides, containing one or more fully localized phosphorylation modifications, are mapped to reference protein sites for eIF4G1(NP_886553.3), eIF4A1(NP_001407.1), eIF4E(NP_001959.1), and 4E-BP1 (NP_004086.1). Tumor stages (for each column) and sample counts (n, for each plot) are marked on X axes. The dashed red line marks average abundance in all tumor stages combined, relative to NATs. Elevation is pronounced for eIF4G1 phosphorylations (note variation in Y axis ranges). The two-tailed Student’s t tests were performed. ns, not significant; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001
(G) The box plots compare abundances of Mnk1/2 and Akt1, and their phosphorylations (Akt1 at serine 477, Mnk1 at serines 209 and 214, Mnk2 at serine 76). Phospho-peptides, containing one or more fully localized phosphorylation modifications, are mapped to reference protein sites for Mnk1(NP_003675.3), Mnk2(NP_951009.1), and Akt1(NP_001014431.1).
To understand the molecular basis for cap-independent initiation in LUADs, we analyzed eIF4F proteins, and phosphorylations that reportedly regulate subunit interactions and initiation mechanisms. Among eIF4F subunits, eIF4G1 has the strongest elevation in LUADs from all stages: 3.546 times the level in NATs (Figure 6E). eIF4G1 phosphorylation is elevated in tumors at Ser1029 (4.001 times NATs) and Ser1093 (4.256 times NATs). A higher multiple for phosphorylation than protein suggests a higher proportion of eIF4G1 is phosphorylated. Ser1093 phosphorylation confirms elevated initiation activity (Dobrikov et al., 2018). Most eIF4G1 phosphorylation is clustered at the interdomain linker region (IDLR) that separates the HEAT-1 and HEAT-2 domains (Figures 6E and S11A). Intense IDLR phosphorylation has been reported in mitotic cancer cells to enhance eIF4A1 binding to the HEAT-2 domain (Dobrikov et al., 2014). The eIF4A1 protein level is higher in LUADs than in NATs (Figure 6E). Only two phosphorylation sites are detectable (Figures 6E and S11A). Ser78 phosphorylation, which likely interferes with ATP binding (Schutz et al., 2010), is curtailed in LUADs to 20.6% of its level in NATs. We reason that tumor-specific Ser78 dephosphorylation enables ATP binding, which may subsequently promote eIF4A1 binding to the HEAT-1 domain (Marintchev et al., 2009). Furthermore, protein levels of eIF4B and eIF4H, auxiliary factors for eIF4G1/eIF4A1, are elevated in tumors (Figures S11C and S11D). Ser406 and Ser422 phosphorylations, essential for eIF4B’s activity (Shahbazian et al., 2006; van Gorp et al., 2009), are also elevated in tumors. In sum, these results suggest strong elevation of eIF4G1 activities and eIF4A1●eIF4G1 interaction in tumors, a possible mechanism for selective adoption of cap-independent initiation.
We explored other possible mechanisms for the adoption of cap-independent initiation. First, sequestration of eIF4E by hypophosphorylated 4E-BP1 in tumors may inhibit cap-dependent and increase cap-independent initiation (Braunstein et al., 2007). The eIF4E protein level is higher in LUADs than in NATs (Figure 6F). Thr68 and Thr11 phosphorylation mildly increases in LUADs (Figures 6F and S11A), but their function in eIF4E is unclear. The 4E-BP1 level is lower in LUADs than in NAT. However, we found no difference in Thr36, Thr46 or Ser65 phosphorylation between tumors and NATs to account for blocked eIF4E binding (Sekiyama et al., 2015). Despite significant elevation of Thr70 and Ser83 phosphorylations in tumors, 4E-BP1 with only those phosphorylations reportedly binds to eIF4E in mitotic cancer cells (Sun et al., 2019). These results suggest that 4E-BP1 may be capable of sequestering even elevated eIF4E, as one mechanism for cap-independent initiation.
Second, Akt/mTORC1 signaling causes phosphorylation of 4E-BP1 at Thr36/Thr46/Ser65 thus inhibits eIF4E sequestration (Gingras et al., 1999). Akt/mTORC1 signaling suppression therefore frees 4E-BP1 to inhibit cap-dependent initiation. We observe significant decreases of Akt1 level and phosphorylation at Ser477 and Thr450, and of mTORC1 phosphorylation at Ser2450 and Ser2481 in LUADs (Figures 6G and S11B). Consistent with that suppression of Akt1 and mTORC1 activity, we observe a lack of 4E-BP1 phosphorylation at Thr36, Thr46 or Ser65 in Figure 6F, which hints at hypoxic conditions (Wangpaichitr et al., 2008).
Finally, Mnk1 and Mnk2 phosphorylate eIF4E and regulate translation initiation (Knauf et al., 2001; Pyronnet et al., 2001). Mnk1 levels are on average similar in tumors and NATs (Figure 6G), but phosphorylations at Ser209 and/or Ser214 are significantly elevated in tumors (Figures 6G and S11B). These phosphorylation sites are within the Mnk1 activation segment (Jauch et al., 2006), indicating kinase activity (Waskiewicz et al., 1999). The functional implication of this is unclear, because Mnk1 supports cap-dependent initiation only while interacting with eIF4G1 (Pyronnet et al., 2001). Mnk2 negatively regulates translation overall by inhibiting eIF4G1 phosphorylation (Hu et al., 2012). Consistently, in tumors, the Mnk2 level is only 40.5% that in NATs (Figure 6G), and eIF4G1 is more phosphorylated (Figure 6E). Mnk2 Ser76 phosphorylation is significantly lower in LUADs (Figures 6G and S11B), but its function is unknown. These results suggest that Mnk1 (but not Mnk2) maintains kinase activity in LUADs. However, further investigation is needed to fully explain the influence of Mnk1 and Mnk2 upon cap-dependent and -independent initiation.
DISCUSSION
Our work leverages previously-established mRNA-protein correlations to uncover a novel type of translation dysregulation in cancers: cancers selectively overexpress EIF4G1, to a greater extent than EIF4E, diverging from mRNA ratios typical of the trimetric eIF4F complex in healthy cells. The resulting mRNA expression imbalance supports use of EIF4G1 as a prognostic indicator in patients with cancer, and causes dysregulation of eIF4F function that distinguishes healthy tissues by type. Tumors attenuate biological “house-keeping” pathways that are usually regulated by cap-dependent initiation in healthy tissue, and reinforce regulation of cancer-preferred pathways in cap-independent contexts. eIF4F dysregulation may be mechanistically attributable to hyperphosphorylation of eIF4G1 that promotes binding to eIF4A1, and/or to 4E-BP1 sequestration of eIF4E and consequently diminished cap-dependent initiation.
Positive Selection of EIF4G1 Favors Cap-Independent Initiation in Cancers
Gene amplification, transcription upregulation, and correlation with poor prognosis across various cancer types indicate strong positive selection by tumors towards increased EIF4G1 expression for its tumor- beneficial function. In contrast, the cap-dependent initiation factor EIF4E demonstrates no prognostic utility (Figure 2B), as expected given that its overexpression induces cellular senescence overridden only by concurrent oncogene expression (i.e. MYC), to promote tumorigenesis (Ruggero et al., 2004). EIF4E2 expression correlates with poor prognosis in KM analysis (Figure 2G), but not in Cox-PH analyses (Figures 2M and 2N). Because eIF4E2 interacts only with eIF4G3 (not eIF4G1) for cap-dependent initiation (Ho et al., 2016), it may provide an alternate means of cap-dependent initiation during hypoxia without canonical eIF4F. EIF4E3 expression correlates with good prognosis (Figure 2H), hinting at a tumor-suppressive function (Osborne et al., 2013). Furthermore, EIF4G1 overexpression reportedly drives malignant transformation even when EIF4E expression is unchanged (Fukuchi-Shimogori et al., 1997). Thus, widespread expression imbalance of EIF4G1 and EIF4E in tumors (Figures 3B and S6B) suggests that tumors can and do better exploit cap-independent initiation than healthy cells. This reasoning is in line with findings that human breast cancer cells under radiation treatment rely on eIF4G1 overexpression and cap-independent initiation to translate genes for cell survival and DNA repair (Badura et al., 2012). EIF4G1 mRNA even contains an IRES selected for its own translation by cap-independent initiation under stress conditions (Gan et al., 1998; Johannes and Sarnow, 1998), a positive feedback mechanism.
Cap-Independent Initiation Is One Manifestation of Dysregulated Translation Initiation in Cancers
Elevated initiation activity is regarded as the major characteristic of translation dysregulation in malignancy (Ruggero, 2013), largely because distinct regulation of protein synthesis among somatic tissues is crucial for tumor suppression, and for establishment and maintenance of differences in tissue identity, function and homeostasis (Buszczak et al., 2014). Our work confirms that EIF4F subunit expressions are well- and distinctly-controlled in various healthy tissues, whereas cancers overexpress canonical initiation factors and (Figures 1C–1F) abandon such tissue-specific regulation (Figures 4A–4B). eIF4E overexpression is found in many cancers and has been thought to account for altered eIF4F activity (Graff et al., 2009; Hsieh and Ruggero, 2010; Rosenwald et al., 1999). However, it has never been conclusively resolved that the eIF4E expression level is the only determinant for eIF4F activity. Our finding that loss of functional alignment for EIF4G1 and EIF4E expression distinguishes malignant from healthy tissues (Figure 4E) portrays a novel dysregulation mechanism. Correlations of EIF4G1, EIF4A1 and EIF4E with house-keeping genes in healthy tissues (Figures 5D–5E) are in line with biochemical findings that most mRNAs rely on cap-dependent initiation for translation (Merrick, 2004). Weakened correlation of posCORs for EIF4F genes in tumors provides strong evidence for eIF4F dysregulation. Finally, in lung cancers, we observed that EIF4G1 and EIF4A1 mRNAs (Figures 5I–5J) and their proteins (Figure 6D) correlate with cell cycle factors, but EIF4E and its protein do not. This suggests reliance by lung cancers upon cap-independent initiation. Thus, diversification of initiation mechanisms, rather than diminished relevance of eIF4E, may be the salient consequence of expression imbalance.
Altered Protein Interaction Serves as the Molecular Basis of Cap-Independent Initiation
Cap-dependent initiation, most common in mammals (Merrick, 2004), ordinarily requires eIF4E●eIF4G1 interaction that enhances cap binding with eIF4E (Haghighat and Sonenberg, 1997). Cap-independent initiation, in contrast, has no reliance upon eIF4E, interacts with RNA at the IRES rather than the cap (Kolupaeva et al., 2003), and typically requires enhanced eIF4A1●eIF4G1 interaction at HEAT1 and HEAT2 (Imataka and Sonenberg, 1997; Lomakin et al., 2000). Both mechanisms require eIF4A1●HEAT1 interaction (Lomakin et al., 2000; Morino et al., 2000). Therefore, the adoption of cap-independent initiation depends upon eIF4A1●HEAT2 interaction and eIF4E availability (Svitkin et al., 2005). As we discuss above, lung cancers employ cap-independent initiation. We specifically observed eIF4G1 hyperphosphorylation (conducive to HEAT2 binding) (Dobrikov et al., 2014) and eIFA1 dephosphorylation (conducive to HEAT1 binding) (Marintchev et al., 2009) in LUADs (Figure 6E). We also observed evidence of reduced eIF4E availability: low phosphorylation of Akt1, mTORC1 and 4E-BP1 at Thr37/Thr46 (Figure 6F and 6G) suggests that physiological conditions intrinsically inhibit mTORC1 and enable 4E-BP1 to sequester eIF4E. Finally, considering eIF4G1 hyperphosphorylation jointly with absent eIF4E phosphorylation at Ser209, we infer that eIF4G1●eIF4E interaction is altered to facilitate cap-independent initiation as previously reported in vitro for mitotic cancer cells (Pyronnet et al., 2001). Avenues for further investigation remain. There may be additional regulatory factors that influence translation initiation. Additionally, we contemplate an eIF4F complex having eIF4E●eIF4G1 interaction as well as eIF4A1●eIF4G1 interactions at both HEAT1 and HEAT2. It is unclear which initiation mechanism such a complex might employ in a physiological context.
Conclusion
Our work indicates that a prevailing role of eIF4G1 in cap-independent initiation influences disease progression in most cancers. eIF4G1’s adaptable biochemical interactions with eIF4F subunits can employ cap-dependent or -independent initiation mechanisms to sustain cancer functions. These findings reveal clinical relevance of interactions among eIF4F subunits, and highlight the value of computational analysis to guide biochemical research efforts in the hunt for better cancer treatments.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Gerhard Wagner (gerhard wagner@hms.harvard.edu).
Materials Availability
This study did not generate any new reagents.
Data and Code Availability
This paper analyzes existing, publicly available data. These links for the datasets are listed in the key resources table.
KEY RESOURCES TABLE.
Original code for the data analysis and reproducing figures in the manuscript can be found in the GitHub repository: https://github.com/a3609640/EIF-analysis.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
METHOD DETAILS
Copy Number Variation Analysis
Gene-level copy number data for 33 TCGA cancer types were obtained from the UCSC Xena data hub (Goldman et al., 2020) (https://tcga.xenahubs.net and https://pancanatlas.xenahubs.net). Genomic copy number variants from biopsies were measured by Affymetrix Genome-Wide Human SNP 6.0 arrays at the TCGA genome characterization center. The array data were normalized and estimated for raw copy number of genomic segments through the copy number variation (CNV) pipeline at NCI’s Genomic Data Commons (GDC). Gene-level CNVs were estimated from the copy numbers of genomic segments using the Genomic Identification of Significant Targets in Cancer 2.0 (GISTIC2) method (Mermel et al., 2011). The estimated gene-level CNV values were grouped with thresholds 3+, 3, 2, 1, 0, to represent high-level copy number gain (amplification), low-level copy number gain (duplication), diploid, shallow (possibly heterozygous) deletion, or deep (possibly homozygous) deletion. We downloaded the CNV and annotation data from the UCSC Xena data hub.
To construct percentage stacked bar plots for CNV statuses of EIF4F genes across TCGA cancer types, we used the TCGA pan-cancer gene-level CNV threshold dataset that combined GISTIC2-thresholded data from all TCGA cohorts, with the Xena dataset ID: TCGA.PANCAN.sampleMap/Gistic2_CopyNumber_Gistic2_all_thresholded.by_genes.
For the correlation matrix of CNVs among EIF4F genes, we used the TCGA pan-cancer estimated gene-level CNV value data that combined GISTIC2 analyzed data from all TCGA cohorts, with the Xena dataset ID: TCGA.PANCAN.sampleMap/Gistic2_CopyNumber_Gistic2_all_data_by_genes. We used rcorr() function from the R package “Hmisc” to compute correlation coefficients by Pearson correlation method and p-values.
To analyze CNV ratios between tumor and normal tissues across TCGA cancer types, we used the copy ratio data between tumor and normal tissues that were generated from the Affymetrix SNP6.0 array data by Tangent Normalization method (Tabak et al., 2019), with the Xena dataset ID: broad.mit.edu_PANCAN_Genome_Wide_SNP_6_whitelisted.gene.xena. CNV ratios were calculated by dividing the estimated gene-level CNV values in malignant tumors to the average CNV value in normal adjacent tissues (NATs) of the same cancer type.
To assess the differential gene expression, we used the batch normalized RNA-Seq data published by from The Cancer Genome Atlas (TCGA) Pan-Cancer analysis project (Cancer Genome Atlas Research et al., 2013), with Xena dataset ID: EB++AdjustPANCAN_IlluminaHiSeq_RNASeqV2.geneExp.xena.
We acquired the clinically relevant phenotype information for each TCGA sample, including sample type and primary disease annotation datasets from all individual TCGA cohorts, with the Xena dataset ID: TCGA_phenotype_denseDataOnlyDownload.tsv.
Survival Analysis
Tumors from 11,160 patients of 33 different cancer types were collected at TCGA. Those tumors were originally diagnosed from 1978 to 2013 and mostly primary tumors (except skin cutaneous melanoma). For primary tumors, the biopsies were removed from the patient at or close to the time of diagnosis. Overall Survival (OS) data were selected as the clinical endpoints for pan-cancer survival analysis. OS was defined as the period from the date of diagnosis until the date of death from any cause, with the date of diagnosis chosen as time zero.
The curated clinical data were from the TCGA Pan-Cancer Clinical Data Resource (Liu et al., 2018), with the Xena dataset ID: Survival_SupplementalTable_S1_20171025_xena_sp. We used the batch normalized RNA-Seq data from TCGA Pan-Cancer analysis project, with the Xena dataset ID: EB++AdjustPANCAN_IlluminaHiSeq_RNASeqV2.geneExp.xena for the survival analyses. The unit of gene expression was log2(norm_value+1) in the expression dataset. The sample type annotation data with the Xena dataset ID: TCGA_phenotype_denseDataOnlyDownload.tsv were used, to exclude the “solid tissue normal” samples from survival analysis. We examined the data distribution of EIF4F relevant genes (log2(norm_value+1)) by density plots. Although RNA-Seq data do not form normal distribution, all analyzed genes displayed bell-shaped curves without obviously extreme values across 10,295 tumor samples.
For Kaplan-Meier analysis on gene expression, two subgroups (top and bottom 20% groups) were selected out of patients with cancer, according to the ranking of EIF4F expression level within their tumors. We created the survival plots for two subgroups using survfit() function and compared the difference of survival curves by the log-rank test using survdiff() function from the R package “survival”.
The Cox proportional hazards (PH) regression model was used to develop a predictive model of overall survival, based on the gene expression values and clinical outcomes from TCGA patients with cancer. We used the coxph() function to compute Hazard Ratio (HR), 95% confidence interval, and statistical significance for gene expression in relation to overall survival (P value). The proportional hazards assumption was assessed by testing the dependence of Schoenfeld residual on time, using the cox.zph() function from the “survival” package. For each gene, the statistically significant correlation between Schoenfeld residual and time is shown as the P value for interaction. A significant P value for interaction (< 0.05) indicates that the PH assumption is violated.
RNA-Seq Data for Gene Expression Analysis
The original underlying mRNA sequencing of tumor samples were obtained from TCGA, and healthy samples from GTEx (Mele et al., 2015). All RNA-Seq experiments used the poly(A) enrichment method for RNA preparation. To treat data from TCGA and GTEx in a consistent way and minimize computational batch effects on read alignment and quantification, we used the reprocessed RNA-Seq read count data for both sources, available from the UC Santa Cruz computational genomics Lab, computed with the Toil-based RNA-Seq bioinformatic pipeline (Vivian et al., 2017). In the Toil-based pipeline, paired-end reads of RNA-seq samples from the TCGA and GTEx projects were processed by STAR to align sequence reads to the GRCh38/hg38 human reference genome and generate read coverage, and by RSEM to quantify number of RNA-Seq reads that aligned to a transcript. Transcript-level expression was estimated as transcripts per million (TPM), which divided the mapped RNA-Seq reads by the length of transcript to give transcript-level expression independent of transcript length. Gene-level expression data were calculated by summing up all transcript-level TPM for each gene. Gene-level expression data were quantile normalized to facilitate comparison across samples and experiments, and then log2(x+1) transformed to remove extreme values. Gene-level expression datasets were presented in terms of the counting unit: log2(normalized_TPM+1). We used the sample annotation dataset for clinically relevant phenotype information of each sample, including tumor primary site, tumor subtypes, and primary tissues. We obtained the gene expression and sample annotation datasets from UCSC Xena data hub (https://toil.xenahubs.net), with the Xena dataset IDs: TcgaTargetGtex_RSEM_Hugo_norm_count, andTcgaTargetGTEX_phenotype.txt. To compute the sum expression or expression ratios, we back-transformed counting units to obtain the normalized TPM values for each gene.
Principal Component Analysis
PCA analysis on RNA-Seq was performed using combined gene expression data from TCGA and GTEx, upon variances in gene expressions (log2(normalized_TPM+1)). The gene expression data were scaled to even out the variances between principal components (PCs), prior to the PCA transformation. We used the function PCA() implemented in the R package “FactoMineR” to draw PCA biplots. We used the function get_pca_var() to extract cos2 of gene variables, and corrplot() (from “corrplot” R package) to visualize representation of the gene variable on each PC.
Because among 33 TCGA cancer types, eighteen study groups contain fewer than 10 matched NATs from patients with cancer, we combined TCGA tumor samples (primary and metastatic tumors) and GTEX normal tissue samples from healthy individuals for PCA analysis. When we performed PCA analysis on individual cancer types, we included tumor samples and two types of normal samples: TCGA’s solid normal adjacent tissues from patients with cancer, and matched GTEx’s normal tissues from healthy individuals.
PCA analysis on proteomics was performed using the proteomics data from CPTAC LUAD, upon variances in relative protein abundance (log2(ratio)). The proteomics data were scaled to even out the variances between PCs prior to performing the PCA transformation. Because the CPTAC proteomics dataset missed expression values for some proteins, we used the regularized iterative PCA algorithm to impute the missing expression data. We used the function impute.PCA() from the R package “missMDA” (two dimensions were chosen) to impute the proteomics dataset, and then performed PCA on output dataset using the PCA() function from “FactoMineR” package.
Correlation analysis and Venn diagram
The linear dependence between each of EIF4F genes and all other genes identified by RNA-Seq (58,582 genes in total) were measured by Pearson’s correlation method in 10,323 TCGA tumor samples, or in 7,414 GTEx healthy tissues. For consistency, we used the reprocessed RNA-Seq data for the TCGA and GTEx datasets with the Toil-based pipeline. Gene expression data for correlation analysis were in the count unit log2(normalized_TPM+1). We used the cor.test() function in R to measure the association between paired genes, with the Pearson’s correlation coefficient (r) and statistical significance of the correlation (p-value). We selected the genes with r values greater than 0.3 (p-value ≤ 0.05) as the positive correlation genes (posCORs), and the genes with r values less than −0.3 (p-value ≤ 0.05) as the negative correlation genes (negCORs). Venn diagram was used to illustrates levels of overlap between correlating genes. We used the VennCounts() function from the R package “limma” to count the overlapping genes between gene groups, and then used the counts to draw the proportional Venn diagrams with euler() function from the R package “eulerr”.
Heatmap, Clustering and Pathway Enrichment Analysis
The heatmap was created to visualize the Pearson’s correlation coefficients of posCORs and negCORs for EIF4E, EIF4G1, EIF4A1 and EIF4EBP1 from tumors or normal samples. Using Heatmap() function of the R package “ComplexHeatmap”, we drew heatmaps using rows to represent posCORs and negCORs. The heatmap rows were ordered and partitioned into three non-overlapping subgroups by K-means clustering method (k = 3). The heatmap columns that represent EIF4F genes and sample types were ordered by the hierarchical clustering method. The gene list within each K-means cluster from heatmap was retrieved for pathway enrichment analysis.
The pathway-based analysis was performed on the clustered gene lists with the enrichPathway() function from the R package “ReactomePA”, which used Reactome as a source of pathway data. Statistical analysis and visualization of enriched biological pathways were performed with the compareCluster() function from R package “clusterProfiler”, by over-representation analysis (ORA) method. The statistical significance (p-value) of the overlap between genes from a given pathway and the clustered gene list was determined by the hypergeometric distribution test. The p-values were adjusted for multiple comparison by the Hochberg’s and Hommel’s method, using p.adjust() function (from the R package “stats”) with method = “BH”. The enriched pathways were ranked according to their adjusted p-values. The ratios between the number of genes associated with a given pathway and the total number of genes in the clustered list were calculated as gene ratios.
Proteomics and Phosphor-Proteomics Data for Protein Abundance Correlation Analysis
Proteomics and phosphor-proteomics data of 109 LUADs and 102 paired normal adjacent tissue samples were generated by CPTAC. An isobaric peptide labeling approach (iTRAQ) was employed to quantify protein and phosphor-peptides across samples. For proteomics, protein extractions from biopsies were labeled with 10-plex tandem mass tags (TMT) reagents and analyzed by liquid chromatography with tandem mass spectrometry LC-MS/MS (Mertins et al., 2016). For phosphor-proteomics, phosphor-peptides were enriched by immobilized metal-ion affinity chromatography and then analyzed by LC-MS/MS. The phosphorylated amino acids within peptides were mapped to the corresponding protein sites. To facilitate quantitative comparison between all samples across experiments, a pooled reference sample from all samples was included in each 10-plex experiment. All data processed steps including peptide sequence alignment to proteins and peptide quantitation were performed with the common data analysis pipeline (CDAP) at CPTAC. RNA-Seq, proteomics and phosphor-proteomics data were obtained from (Gillette et al., 2020). The sample annotation data were obtained from CPTAC_LUAD_metadata.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics were performed on R (version 3.6). The p-value for Pearson’s correlation was calculated using a t-distribution with n – 2 degrees of freedom. Unpaired Student’s t tests or one-way analysis of variance (ANOVA) were used to compare the expression data. The p-value for Kaplan-Meier analysis was determined by the log-rank test. The p-value for Cox regression analysis was determined by the likelihood-ratio test. The p-value for the pathway enrichment analysis was determined by the hypergeometric distribution test and further adjusted for multiple comparison by the Hochberg and Hommel method. p-values < 0.05 were considered statistically significant.
Supplementary Material
Highlights.
RNA-Seq data that has proteomic correlation offers insight into translation initiation
Tumors often have elevated EIF4G1 expression, which correlates with poor survival
EIF4F subunit expression imbalance disrupts tissue distinctiveness, gene correlations
Such imbalance, common in tumors, facilitates cap-independent translation initiation
ACKNOWLEDGMENTS
The results published here are based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga and the National Cancer Institute Clinical Proteomic Tumor Analysis Consortium (CPTAC). This work was supported by the National Cancer Institute [ 5R01CA200913-05 and 5R01AI037581-25, Gerhard Wagner].
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abbas E, Cox DM, Smith T, and Butler MG (2016). The 7q11.23 Microduplication Syndrome: A Clinical Report with Review of Literature. J Pediatr Genet 5, 129–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitken CE, and Lorsch JR (2012). A mechanistic overview of translation initiation in eukaryotes. Nat Struct Mol Biol 19, 568–576. [DOI] [PubMed] [Google Scholar]
- Azar R, Lasfargues C, Bousquet C, and Pyronnet S (2013). Contribution of HIF-1alpha in 4E-BP1 gene expression. Molecular cancer research : MCR 11, 54–61. [DOI] [PubMed] [Google Scholar]
- Badura M, Braunstein S, Zavadil J, and Schneider RJ (2012). DNA damage and eIF4G1 in breast cancer cells reprogram translation for survival and DNA repair mRNAs. Proceedings of the National Academy of Sciences of the United States of America 109, 18767–18772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat M, Robichaud N, Hulea L, Sonenberg N, Pelletier J, and Topisirovic I (2015). Targeting the translation machinery in cancer. Nat Rev Drug Discov 14, 261–278. [DOI] [PubMed] [Google Scholar]
- Braunstein S, Karpisheva K, Pola C, Goldberg J, Hochman T, Yee H, Cangiarella J, Arju R, Formenti SC, and Schneider RJ (2007). A hypoxia-controlled cap-dependent to cap-independent translation switch in breast cancer. Molecular cell 28, 501–512. [DOI] [PubMed] [Google Scholar]
- Buszczak M, Signer RA, and Morrison SJ (2014). Cellular differences in protein synthesis regulate tissue homeostasis. Cell 159, 242–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N., Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, and Stuart JM (2013). The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 45, 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrikov MI, Dobrikova EY, and Gromeier M (2018). Ribosomal RACK1:Protein Kinase C betaII Phosphorylates Eukaryotic Initiation Factor 4G1 at S1093 To Modulate Cap-Dependent and -Independent Translation Initiation. Molecular and cellular biology 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrikov MI, Shveygert M, Brown MC, and Gromeier M (2014). Mitotic phosphorylation of eukaryotic initiation factor 4G1 (eIF4G1) at Ser1232 by Cdk1:cyclin B inhibits eIF4A helicase complex binding with RNA. Molecular and cellular biology 34, 439–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan R, Milburn SC, and Hershey JW (1987). Regulated phosphorylation and low abundance of HeLa cell initiation factor eIF-4F suggest a role in translational control. Heat shock effects on eIF-4F. The Journal of biological chemistry 262, 380–388. [PubMed] [Google Scholar]
- Feoktistova K, Tuvshintogs E, Do A, and Fraser CS (2013). Human eIF4E promotes mRNA restructuring by stimulating eIF4A helicase activity. Proceedings of the National Academy of Sciences of the United States of America 110, 13339–13344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuchi-Shimogori T, Ishii I, Kashiwagi K, Mashiba H, Ekimoto H, and Igarashi K (1997). Malignant transformation by overproduction of translation initiation factor eIF4G. Cancer research 57, 5041–5044. [PubMed] [Google Scholar]
- Galicia-Vazquez G, Cencic R, Robert F, Agenor AQ, and Pelletier J (2012). A cellular response linking eIF4AI activity to eIF4AII transcription. RNA 18, 1373–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan W, LaCelle M, and Rhoads RE (1998). Functional characterization of the internal ribosome entry site of eIF4G mRNA. The Journal of biological chemistry 273, 5006–5012. [DOI] [PubMed] [Google Scholar]
- Gillette MA, Satpathy S, Cao S, Dhanasekaran SM, Vasaikar SV, Krug K, Petralia F, Li Y, Liang WW, Reva B, et al. (2020). Proteogenomic Characterization Reveals Therapeutic Vulnerabilities in Lung Adenocarcinoma. Cell 182, 200–225 e235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, Aebersold R, and Sonenberg N (1999). Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev 13, 1422–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman MJ, Craft B, Hastie M, Repecka K, McDade F, Kamath A, Banerjee A, Luo Y, Rogers D, Brooks AN, et al. (2020). Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol 38, 675–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordan JD, Thompson CB, and Simon MC (2007). HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 12, 108–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff JR, Konicek BW, Carter JH, and Marcusson EG (2008). Targeting the eukaryotic translation initiation factor 4E for cancer therapy. Cancer research 68, 631–634. [DOI] [PubMed] [Google Scholar]
- Graff JR, Konicek BW, Lynch RL, Dumstorf CA, Dowless MS, McNulty AM, Parsons SH, Brail LH, Colligan BM, Koop JW, et al. (2009). eIF4E activation is commonly elevated in advanced human prostate cancers and significantly related to reduced patient survival. Cancer research 69, 3866–3873. [DOI] [PubMed] [Google Scholar]
- Gruner S, Peter D, Weber R, Wohlbold L, Chung MY, Weichenrieder O, Valkov E, Igreja C, and Izaurralde E (2016). The Structures of eIF4E-eIF4G Complexes Reveal an Extended Interface to Regulate Translation Initiation. Molecular cell 64, 467–479. [DOI] [PubMed] [Google Scholar]
- Haghighat A, and Sonenberg N (1997). eIF4G dramatically enhances the binding of eIF4E to the mRNA 5’-cap structure. The Journal of biological chemistry 272, 21677–21680. [DOI] [PubMed] [Google Scholar]
- Hara K, Yonezawa K, Weng QP, Kozlowski MT, Belham C, and Avruch J (1998). Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. The Journal of biological chemistry 273, 14484–14494. [DOI] [PubMed] [Google Scholar]
- Ho JJD, Wang M, Audas TE, Kwon D, Carlsson SK, Timpano S, Evagelou SL, Brothers S, Gonzalgo ML, Krieger JR, et al. (2016). Systemic Reprogramming of Translation Efficiencies on Oxygen Stimulus. Cell Rep 14, 1293–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh AC, and Ruggero D (2010). Targeting eukaryotic translation initiation factor 4E (eIF4E) in cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 16, 4914–4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu SI, Katz M, Chin S, Qi X, Cruz J, Ibebunjo C, Zhao S, Chen A, and Glass DJ (2012). MNK2 inhibits eIF4G activation through a pathway involving serine-arginine-rich protein kinase in skeletal muscle. Sci Signal 5, ra14. [DOI] [PubMed] [Google Scholar]
- Hundsdoerfer P, Thoma C, and Hentze MW (2005). Eukaryotic translation initiation factor 4GI and p97 promote cellular internal ribosome entry sequence-driven translation. Proceedings of the National Academy of Sciences of the United States of America 102, 13421–13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter C, and Zenklusen JC (2018). The Cancer Genome Atlas: Creating Lasting Value beyond Its Data. Cell 173, 283–285. [DOI] [PubMed] [Google Scholar]
- Igreja C, Peter D, Weiler C, and Izaurralde E (2014). 4E-BPs require non-canonical 4E-binding motifs and a lateral surface of eIF4E to repress translation. Nat Commun 5, 4790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imataka H, and Sonenberg N (1997). Human eukaryotic translation initiation factor 4G (eIF4G) possesses two separate and independent binding sites for eIF4A. Molecular and cellular biology 17, 6940–6947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RJ, Hellen CU, and Pestova TV (2010). The mechanism of eukaryotic translation initiation and principles of its regulation. Nature reviews Molecular cell biology 11, 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal PK, Koul S, Shanmugam PST, and Koul HK (2018). Eukaryotic Translation Initiation Factor 4 Gamma 1 (eIF4G1) is upregulated during Prostate cancer progression and modulates cell growth and metastasis. Sci Rep 8, 7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jauch R, Cho MK, Jakel S, Netter C, Schreiter K, Aicher B, Zweckstetter M, Jackle H, and Wahl MC (2006). Mitogen-activated protein kinases interacting kinases are autoinhibited by a reprogrammed activation segment. EMBO J 25, 4020–4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannes G, and Sarnow P (1998). Cap-independent polysomal association of natural mRNAs encoding c-myc, BiP, and eIF4G conferred by internal ribosome entry sites. RNA 4, 1500–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RM, Branda J, Johnston KA, Polymenis M, Gadd M, Rustgi A, Callanan L, and Schmidt EV (1996). An essential E box in the promoter of the gene encoding the mRNA cap-binding protein (eukaryotic initiation factor 4E) is a target for activation by c-myc. Molecular and cellular biology 16, 4754–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Paek KY, Choi K, Kim TD, Hahm B, Kim KT, and Jang SK (2003). Heterogeneous nuclear ribonucleoprotein C modulates translation of c-myc mRNA in a cell cycle phase-dependent manner. Molecular and cellular biology 23, 708–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knauf U, Tschopp C, and Gram H (2001). Negative regulation of protein translation by mitogen- activated protein kinase-interacting kinases 1 and 2. Molecular and cellular biology 21, 5500–5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolupaeva VG, Lomakin IB, Pestova TV, and Hellen CU (2003). Eukaryotic initiation factors 4G and 4A mediate conformational changes downstream of the initiation codon of the encephalomyocarditis virus internal ribosomal entry site. Molecular and cellular biology 23, 687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AS, Kranzusch PJ, Doudna JA, and Cate JH (2016). eIF3d is an mRNA cap-binding protein that is required for specialized translation initiation. Nature 536, 96–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CJ, Cencic R, Mills JR, Robert F, and Pelletier J (2008). c-Myc and eIF4F are components of a feedforward loop that links transcription and translation. Cancer research 68, 5326–5334. [DOI] [PubMed] [Google Scholar]
- Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, et al. (2018). An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 173, 400–416 e411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Cash TP, Jones RG, Keith B, Thompson CB, and Simon MC (2006). Hypoxia-induced energy stress regulates mRNA translation and cell growth. Molecular cell 21, 521–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomakin IB, Hellen CU, and Pestova TV (2000). Physical association of eukaryotic initiation factor 4G (eIF4G) with eIF4A strongly enhances binding of eIF4G to the internal ribosomal entry site of encephalomyocarditis virus and is required for internal initiation of translation. Molecular and cellular biology 20, 6019–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marintchev A, Edmonds KA, Marintcheva B, Hendrickson E, Oberer M, Suzuki C, Herdy B, Sonenberg N, and Wagner G (2009). Topology and regulation of the human eIF4A/4G/4H helicase complex in translation initiation. Cell 136, 447–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mele M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, Young TR, Goldmann JM, Pervouchine DD, Sullivan TJ, et al. (2015). Human genomics. The human transcriptome across tissues and individuals. Science 348, 660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, and Getz G (2011). GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 12, R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrick WC (2004). Cap-dependent and cap-independent translation in eukaryotic systems. Gene 332, 1–11. [DOI] [PubMed] [Google Scholar]
- Merrick WC (2015). eIF4F: a retrospective. The Journal of biological chemistry 290, 24091–24099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, Wang X, Qiao JW, Cao S, Petralia F, et al. (2016). Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methot N, Song MS, and Sonenberg N (1996). A region rich in aspartic acid, arginine, tyrosine, and glycine (DRYG) mediates eukaryotic initiation factor 4B (eIF4B) self-association and interaction with eIF3. Molecular and cellular biology 16, 5328–5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, and Proudfoot NJ (2009). Pre-mRNA processing reaches back to transcription and ahead to translation. Cell 136, 688–700. [DOI] [PubMed] [Google Scholar]
- Morgan R, Morgan SS, Hecht BK, and Hecht F (1988). Fragile sites at 4q23 and 7q11.23 unique to bone marrow cells. Cancer Genet Cytogenet 31, 47–53. [DOI] [PubMed] [Google Scholar]
- Morino S, Imataka H, Svitkin YV, Pestova TV, and Sonenberg N (2000). Eukaryotic translation initiation factor 4E (eIF4E) binding site and the middle one-third of eIF4GI constitute the core domain for cap-dependent translation, and the C-terminal one-third functions as a modulatory region. Molecular and cellular biology 20, 468–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanbru C, Lafon I, Audigier S, Gensac MC, Vagner S, Huez G, and Prats AC (1997). Alternative translation of the proto-oncogene c-myc by an internal ribosome entry site. The Journal of biological chemistry 272, 32061–32066. [DOI] [PubMed] [Google Scholar]
- Nusinow DP, Szpyt J, Ghandi M, Rose CM, McDonald ER 3rd, Kalocsay M, Jane-Valbuena J, Gelfand E, Schweppe DK, Jedrychowski M, et al. (2020). Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell 180, 387–402 e316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne MJ, Volpon L, Kornblatt JA, Culjkovic-Kraljacic B, Baguet A, and Borden KL (2013). eIF4E3 acts as a tumor suppressor by utilizing an atypical mode of methyl-7-guanosine cap recognition. Proceedings of the National Academy of Sciences of the United States of America 110, 3877–3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HJ, Cha YJ, Kim SH, Kim A, Kim EY, and Chang YS (2017). Keratinization of Lung Squamous Cell Carcinoma Is Associated with Poor Clinical Outcome. Tuberc Respir Dis (Seoul) 80, 179–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier J, and Sonenberg N (2019). The Organizing Principles of Eukaryotic Ribosome Recruitment. Annual review of biochemistry 88, 307–335. [DOI] [PubMed] [Google Scholar]
- Pyronnet S, Dostie J, and Sonenberg N (2001). Suppression of cap-dependent translation in mitosis. Genes Dev 15, 2083–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers GW Jr., Richter NJ, Lima WF, and Merrick WC (2001). Modulation of the helicase activity of eIF4A by eIF4B, eIF4H, and eIF4F. The Journal of biological chemistry 276, 30914–30922. [DOI] [PubMed] [Google Scholar]
- Rosenwald IB, Chen JJ, Wang S, Savas L, London IM, and Pullman J (1999). Upregulation of protein synthesis initiation factor eIF-4E is an early event during colon carcinogenesis. Oncogene 18, 2507–2517. [DOI] [PubMed] [Google Scholar]
- Ruggero D (2013). Translational control in cancer etiology. Cold Spring Harb Perspect Biol 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggero D, Montanaro L, Ma L, Xu W, Londei P, Cordon-Cardo C, and Pandolfi PP (2004). The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nature medicine 10, 484–486. [DOI] [PubMed] [Google Scholar]
- Rutkovsky AC, Yeh ES, Guest ST, Findlay VJ, Muise-Helmericks RC, Armeson K, and Ethier SP (2019). Eukaryotic initiation factor 4E-binding protein as an oncogene in breast cancer. BMC Cancer 19, 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA, and Sabatini DM (2017). mTOR Signaling in Growth, Metabolism, and Disease. Cell 168, 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutz P, Karlberg T, van den Berg S, Collins R, Lehtio L, Hogbom M, Holmberg-Schiavone L, Tempel W, Park HW, Hammarstrom M, et al. (2010). Comparative structural analysis of human DEAD-box RNA helicases. PloS one 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, and Selbach M (2011). Global quantification of mammalian gene expression control. Nature 473, 337–342. [DOI] [PubMed] [Google Scholar]
- Sekiyama N, Arthanari H, Papadopoulos E, Rodriguez-Mias RA, Wagner G, and Leger-Abraham M (2015). Molecular mechanism of the dual activity of 4EGI-1: Dissociating eIF4G from eIF4E but stabilizing the binding of unphosphorylated 4E-BP1. Proceedings of the National Academy of Sciences of the United States of America 112, E4036–4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazian D, Roux PP, Mieulet V, Cohen MS, Raught B, Taunton J, Hershey JW, Blenis J, Pende M, and Sonenberg N (2006). The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J 25, 2781–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrells DL, Black DR, Meschonat C, Rhoads R, De Benedetti A, Gao M, Williams BJ, and Li BD (1998). Detection of eIF4E gene amplification in breast cancer by competitive PCR. Ann Surg Oncol 5, 232–237. [DOI] [PubMed] [Google Scholar]
- Sorrells DL, Ghali GE, Meschonat C, DeFatta RJ, Black D, Liu L, De Benedetti A, Nathan CO, and Li BD (1999). Competitive PCR to detect eIF4E gene amplification in head and neck cancer. Head Neck 21, 60–65. [DOI] [PubMed] [Google Scholar]
- Spriggs KA, Bushell M, and Willis AE (2010). Translational regulation of gene expression during conditions of cell stress. Molecular cell 40, 228–237. [DOI] [PubMed] [Google Scholar]
- Stoneley M, Chappell SA, Jopling CL, Dickens M, MacFarlane M, and Willis AE (2000). c-Myc protein synthesis is initiated from the internal ribosome entry segment during apoptosis. Molecular and cellular biology 20, 1162–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subkhankulova T, Mitchell SA, and Willis AE (2001). Internal ribosome entry segment-mediated initiation of c-Myc protein synthesis following genotoxic stress. The Biochemical journal 359, 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun R, Cheng E, Velasquez C, Chang Y, and Moore PS (2019). Mitosis-related phosphorylation of the eukaryotic translation suppressor 4E-BP1 and its interaction with eukaryotic translation initiation factor 4E (eIF4E). The Journal of biological chemistry 294, 11840–11852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkin YV, Herdy B, Costa-Mattioli M, Gingras AC, Raught B, and Sonenberg N (2005). Eukaryotic translation initiation factor 4E availability controls the switch between cap-dependent and internal ribosomal entry site-mediated translation. Molecular and cellular biology 25, 10556–10565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabak B, Saksena G, Oh C, Gao GF, Meyers BH, Reich M, Schumacher SE, Westlake LC, Berger AC, Carter S.L.b., et al. (2019). The Tangent copy-number inference pipeline for cancer genome analyses. bioRxiv, 566505. [Google Scholar]
- Timp W, and Timp G (2020). Beyond mass spectrometry, the next step in proteomics. Sci Adv 6, eaax8978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trop-Steinberg S, and Azar Y (2018). Is Myc an Important Biomarker? Myc Expression in Immune Disorders and Cancer. Am J Med Sci 355, 67–75. [DOI] [PubMed] [Google Scholar]
- van Gorp AG, van der Vos KE, Brenkman AB, Bremer A, van den Broek N, Zwartkruis F, Hershey JW, Burgering BM, Calkhoven CF, and Coffer PJ (2009). AGC kinases regulate phosphorylation and activation of eukaryotic translation initiation factor 4B. Oncogene 28, 95–106. [DOI] [PubMed] [Google Scholar]
- Vivian J, Rao AA, Nothaft FA, Ketchum C, Armstrong J, Novak A, Pfeil J, Narkizian J, Deran AD, Musselman-Brown A, et al. (2017). Toil enables reproducible, open source, big biomedical data analyses. Nat Biotechnol 35, 314–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D, and Mohr I (2011). Viral subversion of the host protein synthesis machinery. Nat Rev Microbiol 9, 860–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangpaichitr M, Savaraj N, Maher J, Kurtoglu M, and Lampidis TJ (2008). Intrinsically lower AKT, mammalian target of rapamycin, and hypoxia-inducible factor activity correlates with increased sensitivity to 2-deoxy-D-glucose under hypoxia in lung cancer cell lines. Molecular cancer therapeutics 7, 1506–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waskiewicz AJ, Johnson JC, Penn B, Mahalingam M, Kimball SR, and Cooper JA (1999). Phosphorylation of the cap-binding protein eukaryotic translation initiation factor 4E by protein kinase Mnk1 in vivo. Molecular and cellular biology 19, 1871–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, and Naar AM (2019). SREBP1-dependent de novo fatty acid synthesis gene expression is elevated in malignant melanoma and represents a cellular survival trait. Sci Rep 9, 10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi T, Papadopoulos E, Hagner PR, and Wagner G (2013). Hypoxia-inducible factor-1alpha (HIF-1alpha) promotes cap-dependent translation of selective mRNAs through up-regulating initiation factor eIF4E1 in breast cancer cells under hypoxia conditions. The Journal of biological chemistry 288, 18732–18742 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper analyzes existing, publicly available data. These links for the datasets are listed in the key resources table.
KEY RESOURCES TABLE.
Original code for the data analysis and reproducing figures in the manuscript can be found in the GitHub repository: https://github.com/a3609640/EIF-analysis.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.