

Abstract
Hapalindoles, fischerindoles, ambiguines and welwitindolinones are all members of a class of indole alkaloid natural products that have been isolated from the Stigonematales order of cyanobacteria. These compounds possess a polycyclic ring system, unique functional groups and various stereo- and regiochemical isomers. Since their initial isolation in 1984, they have been explored as potential therapeutics due to their wide variety of biological activities. Although numerous groups have pursued total syntheses of these densely functionalized structures, hapalindole biosynthesis has only recently been unveiled. Several groups have uncovered a wide range of novel enzymes that catalyze formation and tailoring of the hapalindole-type metabolites. In this article, we provide an overview of these natural products, their biological activities, highlight general synthetic routes, and provide an extensive review on the surprising biosynthetic processes leading to these structurally diverse metabolites.
Keywords: hapalindole, fischerindole, ambiguine, welwitindolinone, Stig cyclase
Graphical Abstract
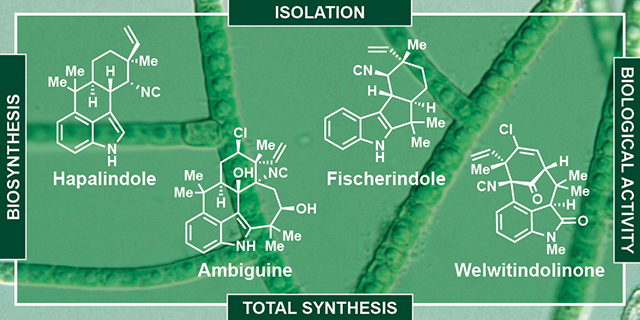
This review covers isolation, biological activity, an overview of total synthesis efforts and recent biosynthetic discoveries related to hapalindole-type indole alkaloids.
1.1. Introduction
Terrestrial, marine and aquatic cyanobacteria thrive in many different habitats worldwide. Due to their resilience in even the most extreme environments, they have been explored extensively for specialized metabolites that may aid in their survival.1 This has led to the discovery of biologically active metabolites such as cryptophycin-1, dolastatin, noscomin and pahayokolide A.2 A derivative of dolastatin (monomethyl auristatin E) serves as a cytotoxic payload in several FDA-approved antibody drug conjugates.3
In 1984, Richard Moore and colleagues isolated the first hapalindoles from the cyanobacterial strain Hapalosiphon fontinalis. The authors were searching for a compound responsible for the antimicrobial effects against Anabaena oscillarioides. Their work uncovered hapalindoles A 12 and B 13.4 Following initial isolation, hapalindoles C-Q (5,7,8,10,14,16–18,20,24–25,28,30–31) and T-V (4,15,22) (Figure 1) were isolated from the same strain.5 Investigation of other cyanobacterial strains have uncovered a variety of tri-, tetra- and pentacyclic hapalindole–type alkaloids. These compounds have been shown to have a wide scope of biological activities, which include antibacterial,1,6–9 antimycotic,1,4,10 insecticidal,11,12 antitumoral,13–15 and immunomodulatory.16 To date, over 80 hapalindole-type metabolites have been isolated from 18 distinct cyanobacterial strains, all arising from the Stigonematales order.1
Figure 1:

Currently reported hapalindole metabolites grouped into tetracyclic and tricyclic categories.
The polycyclic compounds are divided into four subgroups, the hapalindoles (tri-/tetracyclic), fischerindoles (tetracyclic), ambiguines (tetra-/pentacyclic) and welwitindolinones. The core fused ring system arises from a cis-indole isonitrile moiety and a geranyl monoterpene unit, and contains similar structural features such as a functionalized D-ring, which include late-stage C-H oxidation and halogenation (Figure 2).
Figure 2:

Common hapalindole-type indole alkaloid cores with letter labels for each ring. Key carbon stereocenters and fused ring points are numbered.
In 2009, Gault and Marler published a handbook on cyanobacteria in which Moffitt and Burns contributed a chapter with the first comprehensive review on the hapalindoles.17 Bhat and co-workers subsequently published an extensive review on the hapalindole-type metabolites.1 This review highlighted each class within the family of molecules, their biological properties, the proposed biosynthesis, and significant advances in their total syntheses. Since that time, no major update on the hapalindole-type metabolites has been published, spanning a period where remarkable progress has been made on their biosynthetic assembly. Significant advances in total syntheses of the pentacyclic ambiguines have also been reported. In this review, we will provide a brief overview of the hapalindole-type metabolites and their biological properties. General advancements in the landscape of total syntheses will be highlighted before reviewing the fascinating results uncovered from recent studies on enzymes involved in hapalindole/fischerindole biosynthesis.
1.2. Structural Diversity
1.2.1. Hapalindoles
The hapalindoles are the most prevalent group of compounds comprising this class of indole alkaloids, and account for at least 31 molecules. The majority of the hapalindoles possess a polycyclic system containing four fused rings with the key connection between the C-4 and C-16 positions (Figure 2). Stereocenters at the C-10, C-11, C-12 and C-15 positions endow these structures with a spectrum of stereoisomers. Adding further to this diversity are a group of tricyclic hapalindoles that lack the C-C bond between the C-4 and C-16 positions. Lastly, the presence of a halogen or hydroxyl group in select compounds brings more variety to an already distinct family of indole alkaloids (Figure 1). Most recently, two new compounds, hapalindole A formamide 34 and hapalindole J formamide 35 were isolated from the branching cyanobacterium Hapalosiphon sp. CBT1235.16
1.2.2. Fischerindoles
First isolated from Fischerella muscicola in 1992 by Moore and colleagues, the fischerindoles represented a structurally distinct, but related group of compounds.18 These metabolites contain many of the same characteristics that defined the hapalindoles; a polycyclic system containing four rings, stereocenters at the C-10, C-11, C-12 and C-15 positions and the presence of a halogen or oxygenation in some of the compounds. The main difference is the key connection is now between the C-2 and C-16 positions as opposed to the C-4 and C-16 positions. Isolated in 2012, the Orjala group discovered fischerindoles that contain a nitrile moiety instead of the hallmark isonitrile (Figure 3, 41, 43, 44). A nitrile group had previously been observed only in the ambiguines (Figure 4, 51, 61).19
Figure 3:

Current group of characterized fischerindole metabolites.
Figure 4:

Current group of characterized ambiguine and fischambiguine metabolites.
1.2.3. Ambiguines
Further work by the Moore group in 1992 uncovered ambiguines A-F (45–50) from Fischerella ambigua, revealing pentacyclic hapalindole-type indole alkaloids for the first time (Figure 4).10 The seven membered E-ring encompasses a connection between the C-2 and C-11 positions. The search for additional ambiguines expanded when the Orjala group identified metabolites that contain a 6-membered E-ring (Fischambiguines A 62 and B 63).20 Because ambiguines A 45, B 46, C 47, and H 52 are comprised of a reverse prenyl group at the C-2 position, the E-ring is proposed to arise from further 7-endo-trig (ambiguine) or 6-exo-trig (fischambiguine) cyclization via a radical mechanism catalyzed by a putative Rieske-type protein (Figure 5). Further halogenation and oxidation of both the E-ring and core hapalindole scaffold give rise to further structural diversity observed in this family of molecules.
Figure 5:

Proposed radical cyclization mechanism for the formation of the E-ring in the ambiguines and fischambiguines.
1.2.4. Welwitindolinones
Moore’s group reported that the lipophilic extract of Hapalosiphon welwitschii reversed P-glycoprotein-mediated multi-drug resistance in human ovarian adenocarcinoma cells.11 Upon isolation, the compound responsible for this action was identified as N-methylwelwitindolinone C 69, a similar, but structurally unique compound related to the hapalindoles. From this strain, six structurally related congeners were also discovered (64, 65, 66, 67, 68, 70).11 The welwitindolinones represent a unique structural class in this family of indole alkaloids. Their defining feature is the bicyclo[4.3.1]decane ring system except for welwitindolinone A isonitrile 64, which contains a spirooxindole moiety. Every compound is also oxidized at the C-2 position, which raises additional questions about the biogenesis of these highly functionalized molecules (Figure 6).
Figure 6:

Currently identified welwitindolinone metabolites.
1.2.5. Other related hapalindole-type indole alkaloids
There have been several hapalindole-type indole alkaloids isolated from various extracts that do not fit directly within the families discussed above (Figure 7). Isolated from another Fischerella strain, the hapalindolinones (75–76) contain an oxidized C-2 position (oxo-indole) and a highly strained spiro-cyclopropane moiety.21 Moore and colleagues also reported the isolation of several other oxidized hapalindoles from Hapalosiphon fontinalis (77–82, 84–86).22,23 More recently, the Orjala group isolated 13-hydroxydechlorofontonamide 83 from Fischerella muscicola, which alongside the fontonamides and the hapalonamides, lacks the indole B-ring.24
Figure 7:

Hapalindole-type metabolites with atypical structural modifications.
1.3. Biological activity
1.3.1. Antibacterial
Many isolated hapalindole-type molecules have displayed antibiotic activity against a wide variety of both Gram-positive and Gram-negative bacteria. Hapalindoles A-H (5,10,12–13,24–25,30–31) were reported to have MIC values ranging from 0.06 to 64 μg/mL against Gram-positive pathogens such as Staphlyococcus aureus and Streptococcus pneumoniae.1 Hapalindoles J-Q (7–8,14,16–18,21,28) and T-V (4,15,22) were also shown to be active against numerous multi-drug resistant (MDR) bacteria.7 Hapalindole T 22, isolated from a Fischerella strain growing on the bark of the Neem tree, showed very low concentration values for inhibition of MDR bacteria including 0.25 μg/mL against S. aureus and 2.0 μg/mL against Pseudomonas aeruginosa.6
In 2007, ambiguines H 52 and I 53 were shown to have MIC values of 0.625 μg/mL and 0.078 μg/mL, respectively against Staphlyococcus albus and 1.25 μg/mL and 0.312 μg/mL against Bacillus subtilis.7 These values were similar, if not better than the clinical antibiotic streptomycin. Orjala and co-authors reported numerous ambiguines with low micromolar MIC values against Mycobacterium tuberculosis, Bacillus anthracis and S. aureus.8 These values were similar to approved antibiotics such as rifampin, ciprofloxacin and gentamicin.
It is believed that the presence of halogens, hydroxyl groups, and epoxides give this family of indole alkaloids their biological activity against numerous pathogens.7 However, the target of these molecules remained unknown until 2001 when Smith and colleagues showed inhibition of the bacterial RNA polymerase with 12-epi-hapalindole E isonitrile 32 from a Fischerella strain. The authors calculated an IC50 value of 500 μM for the RNA polymerase and the compound was shown to prevent elongation of the growing nucleic acid chain. However, the authors note that initial assays only required an IC50 of 3 μM to inhibit growth of B. subtilis suggesting that there is likely more than one biological target for the hapalindole-type alkaloids.9 As such, the result reported should be regarded as preliminary and requires further rigorous studies.
1.3.2. Antimycotic
Initial discovery of the hapalindoles was driven by the antimicrobial activity observed against Anabaena oscillarioides.4 Since then, numerous hapalindole-type alkaloids have also shown antimycotic activity against a wide variety of fungi including Aspergillus, Penicillium, Fusarium, and Candida strains.7 Ambiguines A-F (45–50) exhibited low μg/mL values against C. albicans and Trichophyton mentagrophytes.10 While these results are encouraging, further medicinal chemistry efforts will be necessary to improve potency compared to already approved fungicides.
1.3.3. Insecticidal
While not all hapalindole-type alkaloids have been screened for insecticidal activity, some have shown promise against specific agricultural pests. Becher and colleagues noted that at 26 μM 12-epi-hapalindole J isonitrile 9 killed 100% of growing Chironomus riparius larvae over 48 hours. Similar activity was observed with 12-epi-hapalindole C 26, hapalindole L 14 and 32.12 While the Moore group noted insecticidal activity for N-methylwelwitindolinone C isothiocyanate 69, more recent attention has focused on the compound’s antitumoral activities and P-glycoprotein transporter inhibition.13,15
1.3.4. Antitumoral and other activity
Another promising area for hapalindole-type metabolites is in the realm of antitumoral therapeutics. Hapalindoles A 12, C 24, H 5, I 20 and J 7 along with fischerindole L 39 showed low micromolar inhibition of many human carcinoma cell lines including colon, breast, large cell lung and glioblastoma.14 As noted earlier, Moore’s group uncovered the P-glycoprotein inhibition of 69 (IC50=3.03 μM), N-methylwelwitindolinone C isonitrile 70 (IC50=0.12 μM) and welwitindolinone C isothiocyanate 68 (IC50=0.13 μM) in breast cancer cell lines.13 P-glycoprotein mutations are a common resistance mechanism observed in cancer cells and its inhibition allows for more effective chemotherapy treatments. Roughly one year later, Zhang and Smith revealed another mechanism of action for 68 against ovarian cancer cell lines. The authors showed that the welwitindolinone analog caused disorganization of the microtubules at the reported IC50 value of 0.13 μM and by 3.3 μM showed complete loss of the microtubules.15
In other therapeutic areas, 12 showed inhibition of T-cell proliferation. As reported recently by Grundemann and colleagues, the IC50 value of 12 (1.56 μM) shows promise as a new drug lead to treat auto-immune disorders. Interestingly, the authors also uncovered hapalindoles D 25, M 8 and hapalindoles A and J formamide (34, 35) from Hapalosiphon sp. CBT1235. None of these compound showed the same potency compared to 12, which indicated that a chlorine at the C-13 and isonitrile functionality at C-11 position were critical for activity (Figure 2).16 Another potential biological target was uncovered in 2014 by Botana et al. Using isolated 7, 14, 26, and 32; these investigators reported the hapalindoles to be sodium channel modulators. Neuroblastoma cell lines were treated with veratridine and a bis-oxonol fluorescent dye to activate sodium channels and monitor the membrane potential changes induced by hapalindole compounds. Addition of the hapalindoles reduced the intensity of the dye, showing a decrease in membrane potential. Using linear regression, the IC50 values for almost all the compounds were determined (4.8 μM for 26, 6.7 μM for 32 and 10.6 μM for 14) suggesting that the hapalindoles may be a starting point for development of more potent neuroactive agents.25
1.4. Review of total synthesis efforts
1.4.1. Introduction
To date, 25 hapalindole-type metabolites have been synthesized, which include efforts to develop novel routes or improve earlier ones.26–60 The overview below highlights the general routes used in the total syntheses of each class of compounds, which sets the stage for contrasting the widely variant strategies employed between total synthesis and biosynthesis (discussed in the next section) of these molecules. Rawal’s excellent 2014 review contains in-depth explanations of the total synthesis efforts, which we will not cover here.1
1.4.2. General routes to tri- and tetracyclic hapalindoles, ambiguines, fischerindoles and pentacyclic ambiguines
Routes toward the synthesis of tri- and tetracyclic hapalindoles and ambiguines along with fischerindoles rely on functionalized indole derivatives coupled with various cyclohexanone derivatives to produce a tricyclic-like intermediate. Final tailoring reactions complete the construction of tricyclic hapalindoles, while Friedel-Crafts type reactions are used to complete the tetracyclic framework of hapalindoles and fischerindoles (Figure 8A).27,29,30,34–36,38,49–54,57–59 Currently, ambiguine H remains the only synthesized tetracyclic ambiguine with prenyl-9-BBN and light used to induce a rearrangement to install the reverse prenyl group at the C-2 position (Figure 8A).30,34,39,58 Two groups have synthesized pentacyclic ambiguine P 60, and both routes start by cross-coupling indole and either (S) or (R)-carvone. Different strategies are then employed to install the ambiguine E-ring followed by further transformations to complete the molecule (Figure 8B).55,56
Figure 8:


(A): General synthesis route used to produce tri- and tetracyclic hapalindoles along with fischerindoles. A functionalized indole derivative is coupled with a cyclohexanone derivative to produce a tricyclic-like intermediate. Final tailoring reactions complete the routes to tricyclic hapalindoles, and tetracyclic hapalindoles and fischerindoles, (B): General synthesis route used to produce pentacyclic ambiguine P 60.
1.4.3. General routes to welwitindolinones
Various routes have been explored to construct the unique bicyclo[4.3.1]decane ring system or the spirooxindole functionality of the welwitindolinones. Baran et al. uncovered an acid catalyzed rearrangement reaction from 12-epi-fischerindole I isonitrile 40, which gives rise to welwitindolinone A 64 (Figure 9A).57,58 Reisman and Wood explored another route to 64 and the spirooxindole moiety. Using a synthesized hydroxyl-enone derivative and a diazo Grignard reagent, the spirooxindole can be effectively produced leading to the total synthesis of 64 (Figure 9B).37,60 For the bicyclo[4.3.1]decane ring system, Rawal and Garg developed efficient routes starting from functionalized indole and cyclohexanone derivatives. Rawal utilized a Friedel-Crafts type reaction to complete the ring system, while Garg has relied upon a unique benzyne intermediate to produce the framework. For both groups, further chemistry led to the total synthesis of various welwitindolinone compounds (Figure 9C).26,40–48
Figure 9:

(A) Acid-catalyzed rearrangement route employed by Baran et al. to produce welwitindolinone A.57,58 (64). (B) The route used by Reisman et al. in the total synthesis of 64. A hydroxyl-enone derivative is reacted with a diazo Grignard reagent to aid in producing the spirooxindole moiety.37,60 (C) General synthesis routes used by Rawal and Garg to produce various welwitindolinone compounds. Both routes use functionalized cyclohexanone and indole derivatives with key intermediates highlighted.26, 40–48
1.4.4. Summary
The general synthetic approaches described above highlight the creative strategies employed by a number of groups to access the hapalindole-type natural products. Alternative routes have also been utilized to produce the compounds described above. The isolation of numerous natural products and total synthesis efforts helped motivate initiation of biosynthetic studies to determine how the compounds are produced by the Stigonematales order of cyanobacteria. Following the identification of a series of hapalindole-type biosynthetic gene clusters through genome sequencing,61–70 a period of intense exploration has led to surprising and fascinating conclusions, described below.
1.5. Hapalindole biosynthesis: past and present
1.5.1. Early biosynthetic proposals
Concurrent with their discovery, Moore and colleagues developed a hypothesis regarding hapalindole biogenesis. By examining each compound, they surmised an indole isonitrile core 1 and geranyl monoterpene subunit, and reasoned that every hapalindole-type compound was derived from an initial tricyclic hapalindole intermediate. A chloronium ion or proton-catalyzed enzymatic reaction was postulated to produce the tricyclic intermediate. Further enzyme catalyzed acidic condensation reactions were proposed to complete the tetracyclic hapalindole and fischerindole core. For the welwitindolinones, Moore and colleagues also proposed that an early stage oxidation of the indole ring played a role in an acid-catalyzed cyclization reaction to produce the unusual core.11 Isolation of ambiguine H 52 led to the suggestions that the pentacyclic ambiguines are produced by a reverse prenylation with dimethylallyl pyrophosphate (DMAPP) followed by an enzyme catalyzed cyclization reaction.
In 2006, Raveh and Carmeli proposed a new biosynthetic mechanism for assembly of the hapalindole core. This was motivated by their inability to isolate any tricyclic hapalindole compound from a Fischerella strain, and they hypothesized a condensed chloronium ion or proton catalyzed cyclization reaction to produce the hapalindole system.7 In 2008, Baran revised the hypothesis for welwitindolinone formation suggesting that welwitindolinone A 64 arises from an oxidative ring contraction from 12-epi-fischerindole I 40 followed by epoxide formation and rearrangement to produce welwitindolinone B isonitrile (Figure 10).38
Figure 10:

Summary of previously proposed biosynthetic mechanisms for hapalindole, fischerindole, ambiguine and welwitindolinone formation. The cis-indole isonitrile core (1) is highlighted in blue and the geranyl monoterpene unit (GPP) is highlighted in red. Magenta arrows highlight the mechanism hypothesized by Moore et. al.11 Green arrows highlight the mechanism hypothesized by Carmeli et. al.7 Orange arrows highlight the mechanism hypothesized by Baran et. al.38
Since Baran’s proposal, no further mechanistic hypothesis has been proposed for welwitindolinone formation. Given the recent work into the biosynthesis of the hapalindole-type metabolites (see below), we propose a new mechanism for welwitindolinone formation that starts from the tricyclic hapalindole species and includes an epoxidation at the indole C-2/C-3 positions. Similar to Moore’s mechanism, this step triggers a cyclization cascade to open the epoxide and, through further oxidation, leads to the formation of the spirooxindole intermediate observed in welwitindolinone A 64. As in Baran’s mechanism, this intermediate is subject to epoxidation and subsequent formation of a charged intermediate following rearrangement. Through a Friedel-Crafts type mechanism, the cyclodecane ring system is formed and the general welwitindolinone core is completed (Figure 11).
Figure 11:

New mechanism for welwitindolinone core formation building from Moore and Baran’s previous proposals. Formation of an epoxide triggers the cyclization cascade to generate the spirooxindole intermediate. A second epoxidation reaction is proposed to initiate the rearrangement and Friedel-Crafts type mechanism to form the core cyclodecane ring system.
1.5.2. Biosynthetic gene cluster analysis and revised proposal for hapalindole biogenesis
Recent advances in bioinformatics have enabled more in depth analysis of the biosynthetic gene clusters (BGCs) of the hapalindole-type producing cyanobacterial strains. In 2014, the Liu group published the first annotated BGC from Fischerella ambigua UTEX 1903 (amb).61 In their work with the amb gene cluster, they were able to use heterologous expression to characterize several proteins from the BGC. AmbI1–3 were shown to be homologues of the isonitrile synthases IsnA and IsnB. These proteins have been previously shown to produce the trans-indole isonitrile derivative.71,72 However, AmbI1–3 were shown to produce the cis-indole isonitrile 1, a proposed subunit for hapalindole metabolites. This unique isonitrile functionality is believed to be derived from a β-keto imine formation between L-tryptophan and ribulose-5-phosphate followed by two retro-aldol type condensations.71–73 They also annotated three genes that encode prenyltransferases, AmbP1–3. AmbP2 was uncovered to catalyze formation of geranyl pyrophosphate (GPP) from isopentenyl pyrophosphate (IPP) and DMAPP. AmbP3 was found to prenylate hapalindole G 10 with DMAPP to afford ambiguine A 45, thus confirming Moore’s original hypothesis that the tetracyclic ambiguines are the result of a reverse prenylation with DMAPP. A small substrate screen was performed with various other indole derivatives, but none of them were found to be prenylated by AmbP3 suggesting that the terpenoid component in the cyclization of the hapalindole core was needed for further prenylation. The authors did not examine AmbP1, but hypothesized that it plays a role in geranylating 1 with GPP to give an intermediate before the cyclized core is completed. The pathway was found to contain five non-heme iron dependent oxygenases, including four Rieske-type proteins (AmbO1–5). These proteins were hypothesized to mediate halogenation and further oxidative modifications to the hapalindole and ambiguine cores (Figure 12).61 Later, AmbO5 was characterized to halogenate various hapalindole, fischerindole and ambiguine metabolites.74
Figure 12:

Summary of Hillwig et al.61,69 discoveries from annotation of the amb BGC from Fisherella ambigua UTEX 1903. Solid arrows represent enzymes whose function was confirmed. Dotted arrows represent hypothesized enzyme functions.
Soon thereafter, Liu and co-workers published the BGC of a welwitindolinone producing strain, Hapalosiphon welwitschii UTEX B1830 (wel).62 They discovered many similarities to the amb gene cluster, suggesting horizontal gene transfer across the Stigonematales order of cyanobacteria. In order to confirm that the annotated homologues performed the same function, heterologous expression was again employed to access the proteins. WelP2 (homolog of AmbP2) was found to function as a geranyl diphosphate synthase. WelI1–3 were responsible for biosynthesis of 1, consistent with the function of AmbI1–3. However, there were eight proteins annotated that did not correlate with any protein from the amb gene cluster. One of the proteins, WelM, was determined to be a SAM-dependent methyltransferase responsible for the N-methylation of the welwitindolinone core. Enzyme kinetic studies revealed that welwitindolinone C isothiocyanate 68 is likely the native substrate for WelM. Substrate scope studies of WelM revealed that the enzyme is highly selective for an oxindole core and likely is the final biosynthetic enzyme in the formation of N-methylwelwitindolinone C isothiocyanate 69. Five non-heme iron dependent oxygenases (WelO1–5) shared moderate (61–79%) identity sequences with their amb counterparts; nevertheless, they were all hypothesized to play a role in late-stage tailoring (Hillwig and Liu later characterized WelO5 as the halogenase responsible for chlorinating 36 and 2669).62
Building from this initial gene cluster analysis, Micallef et al. performed an extensive bioinformatic analysis from the genome sequences of numerous Stigonematales cyanobacterial strains.63 Seven strains were analyzed, including Fischerella and Hapalosiphon strains, in addition to Westiella intricata UH strain HT-29–1. The goal of the study was to compare numerous hapalindole/fischerindole BGCs and examine similarities and differences. The majority of the annotated gene clusters showed overall similarities in architecture, with notable differences. Welwitindolinone gene clusters were all highly conserved but were distinct from hapalindole and ambiguine BGCs in the sequence identity of their late-stage tailoring enzymes (i.e. N-methyltransferases, oxygenases). In all strains, the enzymes that catalyze formation of 1, GPP, and prenyltransferases were highly conserved. Interestingly, the isonitrile synthases from Westiella intricata UH strain HT-29–1 were found to produce both 1 and its trans isomer uncovering the first isonitrile synthases from a hapalindole-type producing strain to generate the trans isomer. Based on phylogenetic analysis, the authors hypothesized that gene cluster transmission is vertical (rather than horizontal as suggested by Liu) because their analysis identified a monophyletic clade between all the strains examined in the study.63 This extensive study set the ground work for further elucidation of the biosynthetic pathway responsible for hapalindole-type metabolite formation.
In 2013, the Sherman lab initiated a study to examine the prenyltransferases and proteins with “unknown” annotations responsible for hapalindole core formation. Li et al. examined the BGC of Fischerella ambigua UTEX 1903 (which they elected to rename fam).64 Their efforts revealed four novel cyclase-type enzymes (FamC1-C4) alongside previously annotated proteins. Upon more in depth exploration, they discovered that FamD2 (identical to AmbP1) catalyzes indole C-3 geranylation in a pH dependent manner. At relatively acidic pH, a 1–2 shift of the geranyl group at C-3 to the indole C-2 position results in formation of a terminal shunt metabolite. This result was later confirmed by Liu and co-workers upon examination of the magnesium dependence of AmbP1.70 Li et al. also explored the function of FamD1 (identical to AmbP3). They showed that while FamD1 can catalyze prenylation of 1 with both GPP and DMAPP; its higher selectivity for DMAPP leads to formation of ambiguine H 52 from hapalindole U 4. Analysis of a cell free extract from F. ambigua 1903 in the presence of 1 and GPP resulted in formation of a new metabolite 12-epi-hapalindole U 3. Proteomic analysis of the soluble protein fraction revealed the identity of the thermostable cyclase FamC1. Cloning and expression of famC1 devoid of the leader peptide provided ready access to soluble, functional recombinant protein, which in the presence of FamD2, 1 and GPP, catalyzed formation of 3. These results led to a new hypothesis for the biosynthesis of hapalindole-type natural products. The process begins when 1 is geranylated at the C-3 position to give 2, and in the presence of a Stig cyclase, undergoes a remarkable three-step reaction including 1) [3,3]-sigmatropic (Cope) rearrangement, 2) 6-exo-trig cyclization and 3) terminal electrophilic aromatic substitution (EAS) reaction to generate either the hapalindole or fischerindole core (Figure 13 & 14). Further late stage prenylation, cyclization and oxidation reactions are presumed to be responsible for the formation of the ambiguines and welwitindolinones.64 This work revealed one of the first biosynthetic Cope rearrangements,75 which represents the initial reaction in the three-step cascade catalyzed by FamC1 and related Stig cyclases.
Figure 13:

Biosynthetic mechanism for assembly of the hapalindole and fischerindole cores based on independent studies of Sherman64 and Liu.61 Cis-indole isonitrile core (1) is highlighted in blue. GPP is highlighted in red.
Figure 14:

Detailed cyclization cascade starting from 2. Top row highlights mechanism to product formation with epi stereochemistry. Bottom row highlights mechanism to products with opposing (regular) stereochemistry.
1.5.3. Further exploration of Stig cyclases
Upon the discovery of FamC1 as the central hapalindole core forming biocatalyst, the Sherman and Liu groups independently continued to explore the newly named Stig cyclases (named after the Stigonematales cyanobacterial strains they have been isolated from) further. In early 2017, Zhu and Liu reported the characterization of all WelU cyclases from Hapalosiphon welwitschii UTEX B1830 and IC-52–3.65 They discovered that WelU1 assembled 12-epi-fischerindole U 36 and WelU3 assembled the tricyclic 12-epi-hapalindole C 26. They also noted that the cyclases were more efficient at lower pHs, but made the reaction more challenging as acidic pHs induce the 1,2-shift of 2.65
Concurrently and independently, the Sherman lab examined the famC gene cluster and discovered that a combination of different cyclases leads to variant stereochemical outcomes at the molecule’s four chiral centers (C-10, C-11, C-12, C-15). A reaction mixture containing FamC2-FamC3 showed production of hapalindole H 5.66 Li suggested that these varying product profiles resulted from different interactions in these heterodimeric combinations, which was shown by protein pull down experiments supporting a direct association between FamC2 and FamC3.66 Examination of close homologs HpiC1 (from Fischerella sp. ATCC43239) and FilC1 (from Fischerella sp. IL-199–3-1) showed production of 3, as expected. Fischerindole production was explored in cyclases from Fischerella muscicola UTEX1829 (FimC5) and Fischerella sp. SAG 46.79 (FisC). Both enzymes produced 36 even though they were about 90% identical in sequence to FamC1 (although FimC1-FimC4, from the same BGC as FimC5, was found to produce 4).66 With HpiC1 and FimC5, the tricyclic hapalindole 26 was observed as a minor product. This metabolite was not converted to the tetracyclic form when provided exogenously to the cyclases, supporting the hypothesis that the C-3 geranylated intermediate is the substrate for the cyclases and the tricyclic hapalindoles are shunt products, not intermediates, in the pathway as originally hypothesized by Moore. 66
Later in 2017, Zhu and Liu published another follow up study examining the cyclases AmbU1–4.67 Already knowing that AmbU4 (identical to FamC1) produces 3, they elected to look at AmbU1–3. Through heterologous expression, all four proteins were incubated together, which lead to the production of 3, 4, and 6, a result which was consistent with previous studies.66 However, Zhu and Liu made an important observation by adjusting the stoichiometric ratios of select Stig cyclases, and were able to shift product profiles of AmbU2-AmbU3 (FamC2/C3) and AmbU1-AmbU4 (FamC1/C4). Upon addition of supplementary Ca2+ to AmbU1-AmbU4, they were able to shift the product profile exclusively to 4. Likewise, the addition of Ca2+ to AmbU2-AmbU3 shifted the product profile to assemble 5 over 4. They also noted that AmbU2 (FamC2) by itself could assemble 5, but less efficiently compared to the combination. To confirm the Ca2+ dependence, the authors dialyzed the proteins (including WelU1) and showed that EDTA abolished activity, which was restored upon addition of Ca2+.67
After this initial series of publications that revealed a fundamentally new type of biosynthetic cascade for indole alkaloid assembly, attention was drawn to crystallographic and mechanistic insights for the Stig cyclases (see below). However, in early 2020, the Sherman lab published a new study on this unique class of enzymes. Li et al. decided to further explore cyclases from the fil gene cluster concurrently with a newly annotated gene cluster from Westiellopsis prolifica SAG 16.93 (wep).68 FilC1-FilC3 was shown to catalyze formation of 3, with 4 as a minor product. FilC1-FilC4 heterodimer led to production of 4, and FilC2-FilC3 catalyzed assembly of a new metabolite, 12-epi-hapalindole H 6. FilC2 could also generate 6 as a homodimer, with lower efficiency than the heterodimeric combination. FilC2-FilC3 was also reported to assemble 26 and 12-epi-hapalindole Q 29 as minor products. This broader product profile from FilC2-FilC3 was shown to be cofactor dependent as the addition of supplementary Ca2+ shifted the product profile to produce 6 as the major product. As described in Liu’s study67, all Stig cyclase enzymatic activity was abolished by dialyzing the proteins with EDTA. Further analysis of the Wep pathway derived cyclases showed similar results. WepC1 produced 3 and heterodimeric combination WepC1-WepC2 produced three metabolites including, 4 and hapalindole C 24 as major products alongside 5 as a minor product. Li et al. were able to shift product profiles by incubating the Stig cyclase homologues from different gene clusters together. Thus, FamC2-FilC3 showed production of 6 unlike FamC2-FamC3, which produces 5. Mutational analysis of active site amino acids along the presumed dimeric interface (identified by Newmister et al.76) (Figure 15) led to alterations in the product profiles of specific hetero-oligomeric cyclase combinations. These results suggest that key residues in a composite active site play a role in affecting the stereochemical outcome of the cyclization cascade.68 Further studies on heterodimeric and oligomeric combinations of Stig cyclases are expected from studies on the growing list of annotated Stigonematales cyanobacterial genome derived BGCs (Figure 16). Currently characterized Stig cyclases and their respective products are shown in Figure 17.
Figure 15:

Crystal structure of dimeric interface of HpiC1 reported in Newmister et al.76 and supported by functional data in Li et al.68 Dimeric interface is highlighted by the blue cavity. Red brackets encompass the combined active site while green brackets encompass the active site from the individual Stig cyclase monomers. Blue cavity calculated using CAVER software.90
Figure 16:

Currently annotated hapalindole/fischerindole encoding BGCs from Stigonematales cyanobacteria.
Figure 17:

Currently characterized Stig cyclases and the compounds derived from their three-, or two-step biocatalytic reactions.64–68
1.5.4. Stig cyclase and prenyltransferase protein structure and mechanistic insights
Following the initial functional characterization of various Stig cyclases in 2017, there was a shift towards X-ray crystal structure studies to identify the active site and elucidate the mechanistic basis of the three-step cyclization cascade. In 2018, Newmister et al. reported the first high resolution structure of a Stig cyclase,76 and subsequent mutational analysis of HpiC1 revealed some remarkable insights. The HpiC1 hapalindole-forming cyclase exists in a homodimeric state with four Ca2+ coordination sites, which confirmed and provide molecular details behind the metal cofactor dependence noted in previous studies.66,67 Analysis of a likely active site region identified by sequence comparisons uncovered numerous aromatic residues, and the presence of a conserved aspartic acid residue suggested that the cyclization cascade may be acid catalyzed (Figure 18). Mutating the active site aspartic acid residue to an alanine abrogated Stig cyclase (HpiC1) activity, and confirmed its importance in the cyclization cascade. Comparing the proposed active site of HpiC1 to FimC5 (catalyzes fischerindole formation) facilitated the identification of key amino acid residues responsible for selectivity of the indole C-4 versus C-2 position in the terminal EAS step. Thus, Y101 and F138 were mutated to their corresponding residues in FimC5 (F101 and S138) to interrogate their impact on hapalindole vs fischerindole production. The Y101F mutant did not show a shift in product profile; however the F138S mutant catalyzed production of both 36 and 3 (1:2 ratio), suggesting that these active site amino acids play a more important role in selectivity of the terminal cyclization step. This profile was shifted even further with the double mutant (Y101F/F138S) form of HpiC1, which produced 36 as the major product in (2:1 ratio) over 3.76 Molecular dynamics (MD) and density-functional theory (DFT) calculations were performed to understand further the basis for hapalindole vs fischerindole formation. MD simulations showed that the aspartate residue retains its conformation toward the inner cavity of the active site while F138 acts as a wall on the side of the pocket. However in the F138S mutant, there is additional space and the active site becomes more flexible offering an explanation for the change in selectivity of the C-2 vs C-4 EAS. DFT calculations showed that in the proposed chair transition state for the Cope rearrangement, the aspartate residue can lower the energy barrier from 20.6 kcal/mol to 18.4 kcal/mol (a 40-fold rate enhancement) suggesting that the aspartate residue catalyzes a proton transfer with the indolenine nitrogen, inducing the acid catalyzed Cope rearrangement. This was further confirmed from computations showing that the 6-exo-trig cyclization can only occur when the indole nitrogen is protonated, while suppressing reaction at the C-3 and C-16 positions to form a cyclobutane ring.76 Lastly, the intrinsic energy difference that favors the terminal EAS occurring at the C-2 position versus the C-4 position is about 8 kcal/mol (fischerindole production). This suggests that HpiC1 and likely all hapalindole producing cyclases overcome the intrinsic energy barrier by controlling the terminal EAS at C-4 instead of the intrinsic lower energy at C-2.76 A crystal structure was also obtained with HpiC1 homodimers bearing the dimeric interface comprising the individual active sites. This interface was functionally confirmed by Li et al. through mutagenesis studies that confirmed the dependence of both Asp residues at the interface of a dimeric Stig cyclase (Figure 15).68 These remarkable findings gave the first in-picture view of how the Stig cyclase class of enzymes catalyze a complex cyclization cascade.
Figure 18:

Left: Crystal structure of HpiC1 obtained by Newmister et al.76 Cyclase exists in dimeric state with each individual subunit colored. Ca2+ ions highlighted in magenta. Right: Structure of active site with important residues highlighted in yellow and labeled. Ca2+ ion highlighted in magenta.
Chen et al. followed this study by publishing X-ray crystal structures of FamC1, FilC1 and FisC.77 The majority of their work confirmed insights gleaned from HpiC1, while also revealing that the cyclases exhibit significant conformational changes around the active site. They also showed that hydrophobic residues in FamC1 and FilC1 push the terminal EAS towards the C-4 position while the polar residues found in FisC and FimC5 allow for the energetically favored cyclization at the C-2 position.77
Other groups elected to explore the prenyltransferases and reported structural studies of both FamD1 (AmbP3) and FamD2 (AmbP1). Wang et al. disclosed the first X-ray crystal structure of FamD1,78 which is similar to all other ABBA prenyltransferases, in that the protein contains a central β-barrel of ten anti-parallel strands surrounded by ten α-helices. Obtaining a crystal structure with DMAPP bound to the enzyme, the authors observed the pyrophosphate group interacting with a group of basic residues on the top of the active site in the center of the β-barrel. They also uncovered two tyrosine residues that may help facilitate pyrophosphate release. Furthermore, a crystal structure with both 1 and DMAPP bound was obtained. They observed 1 interacting with a group of hydrophobic residues in the lower half of the active site, opposite of DMAPP. From this complex structure, the authors proposed a mechanism wherein DMAPP is dephosphorylated and reacts via an SN1 type reaction with 1. Following proton abstraction, the formation of the C-3 and C-2 prenylated products originally identified by Li et al. are produced.64
Soon after publication of the Wang study, the Liu and Abe groups collaborated to determine the first crystal structure of FamD2 (AmbP1),79 which was shown to contain the same ABBA prenyltransferase structure as FamD1 (ten anti-parallel β-barrel strands surrounded by α-helices). They uncovered a unique Mg2+ binding site that was not observed in FamD1. However, this proved to not be the Mg2+ responsible for FamD2’s selectivity as another Mg2+ binding site was uncovered in the ternary complex of the protein, 1, and GSPP (a non-hydrolyzable GPP analog) at pH 8. As in the case of FamD1, the top half of the active site is rich in polar residues while the bottom half is replete in hydrophobic residues, which helped orient GSPP. Interestingly, in structures without Mg2+ bound, 1 turned by nearly 100 degrees showing the importance of Mg2+ for catalysis. The presence of Mg2+ actually shifts the C-2 of 1 2Å further away from C-1 of GPP helping to favor geranylation at the C-3 position in the predicted65,67,76 (R) configuration.79 This was the first structural work on a prenyltransferase where a metal co-factor was shown to play a role in enzymatic selectivity.
These two groups also produced a structure of FamD1 (AmbP3) bound to both 4 and hapalindole A 12.80 The authors observed the expected reverse prenylation at the C-2 position with 4 but observed normal prenylation at the C-2 position with 12. Upon obtaining separate crystal structures with 4 and 12 in complex with dimethylallyl S-thiolodiphosphate (DMSPP), they observed DMSPP bound to the polar region, near the top of the β-barrel cavity. Compounds 4 and 12 were bound to the more hydrophobic region of the pocket and 4 formed a hydrogen bond with Glu207; with 12 this key hydrogen bond was not observed. Both 4 and 12 were found to have completely altered orientations suggesting that the terpenoid component of the molecule is more important for substrate recognition by the prenyltransferase. For compound 4, the distance from indole C-2 to the C-3 of DMSPP was shorter than with compound 12, supporting the original observation of reverse versus normal prenylation (Figures 19 & 20). 80Along with the other studies discussed in this section, we now have an in-depth picture regarding how the Stig cyclases and allied prenyltransferases catalyze formation of hapalindole-type metabolites.
Figure 19:

Left: Crystal structure of FamD1 with 4 obtained by Wong et al.80 Looking down the β-barrel 4 and DMSPP are visible. Right: Structure of active site with Glu207 highlighted forming key hydrogen bond with 4. This hydrogen bond places 4 into a configuration favoring reverse prenylation over normal prenylation at indole C-2. Distances between carbons are also displayed.
Figure 20:

Left: Crystal structure of FamD1 with 12 obtained by Wong et al.80 Looking down the β-barrel 12 and DMSPP are visible. Right: Structure of active site with Glu207 highlighted without forming a key hydrogen bond with 12. The loss of this hydrogen bond places 12 into a configuration favoring normal prenylation over reverse prenylation at indole C-2. Distances between carbons are also displayed.
1.5.5. Further analysis of WelO5 halogenase and homologs
As noted previously, Hillwig and Liu were able to characterize WelO5 as a Fe(II)/α-ketoglutarate dependent halogenase responsible for chlorination of 36 and 26.69 Because halogens are important bioisosteres and late-stage halogenation continues to be a challenge for medicinal chemists, halogenase enzymes have attracted much interest to solve these problems.81,82 Due to the higher selectivity of WelO5 compared to its counterpart AmbO5, Liu et al. continued their work by showing that WelO5 could stereoselectively halogenate 36 with both chlorine and bromine, although the enzyme showed a 10:1 preference for Cl− over Br−.83
With these results in hand, Liu (in collaboration with Boal) obtained a crystal structure of WelO5.84 Like most Fe(II)/α-ketoglutarate dependent proteins, it contained an eight-stranded β-sandwich topology; however, it contained a glycine where normally a glutamate or aspartate would be found, thus removing a carboxylate moiety that enables direct coordination between the substrate and chloride ion. The active site is largely hydrophobic but key hydrogen bonds are made between the indole N-H and the isonitrile functionality. This leaves C-13 of 36 a short distance from the Fe(II) center but farther from the chloride ion, thus indicating that hydroxylation would occur instead of chlorination (Figure 21). From this crystal structure, site directed mutagenesis was performed to convert the noted glycine residue into an aspartate. This led to the formation of C-13 hydroxylated 36. With these results in hand, the investigators hypothesized that 36 would have to shift positions to allow for selective chlorination. Closer observation of the active site revealed a key serine residue (Ser189) that could form a hydrogen bond with the iron-oxo intermediates, thus allowing 36 to shift positions and suppress hydroxylation. Mutagenesis of Ser189 to an alanine led to 1:1 production of chlorinated and hydroxylated metabolites.84 Building off this work, Zhang and co-workers used quantum mechanical (QM) and molecular mechanical (MM) calculations to confirm both the mechanism for chlorination and the importance of the serine residue.85 This crystallographic study and further follow up analysis revealed the unique features of WelO5 as a halogenating biocatalyst and how various residues work together to abrogate hydroxylation.
Figure 21:

Left: Crystal structure of WelO5 with 36 obtained by Mitchell et al.84 Right: Structure of active site with key residues highlighted. His164 and His259 coordinate to Fe(II) (orange sphere). Ala82 and Met225 hydrogen bond to 36 for proper configuration. Ser189 coordinates the chloride ion (green sphere).
Continuing their work, Liu and Zhu characterized the homologous halogenase from Hapalosiphon welwitschii IC-52–3 (also known as the haw BGC), which they referred to as WelO5*.86 WelO5* was found to be more selective for 26 while still showing some promiscuity for 36. A comparative analysis of the two halogenases found an 11 amino acid residue difference at the C-terminus. This C-terminus difference is believed to account for selectivity as a similar sequence was observed in AmbO5.86
Recently, two groups used the results learned from the previous studies to perform directed evolution on WelO5 homologs.87,88 Hayashi et al. examined directed evolution of WelO5* to halogenate the core of martinelline natural products, which have been shown to be potent bradykinin receptor agonists and have antitumoral activity.89 Their work uncovered two residue differences (V81R & I161S) that improved halogenation of the non-native core 400 fold from the wild-type enzyme. Variant V81L_I161M shifted the reactivity towards hydroxylation of the unnatural core as well (Figure 22A). 87 Studies by Duewel et al. showcased directed evolution of the WelO5 homolog from Westiella intricata HT-29–1 which the authors named Wi-WelO15.88 Because the previous crystallographic work84 showed that a key hydrogen bond with the isonitrile functionality was important for substrate reactivity, the authors pursued a directed evolution campaign to halogenate substrates without the isonitrile moiety. Upon replacing the isonitrile with a carbonyl, all activity in the wild-type enzyme was abolished, thus demonstrating the importance of this hydrogen bond for activity. Select mutants were produced after three and four generations of mutagenesis that was able to stereoselectively chlorinate a suite of unnatural tricyclic hapalindole-type species in fairly moderate yields (7–48%) on milligram scale (Figure 22B).88 These studies with WelO5 and close homologs showcase the power and versatility of cyanobacterial derived biocatalysts for difficult late-stage halogenation reactions.
Figure 22:

(A) Isolated yields of selective halogenation and hydroxylation of the martinelline core by WelO5* variants as reported by Hayashi et al.87 (B) Isolated yields of selective chlorination of unnatural hapalindole-type core compounds as reported by Duewel et al.88
1.6. Conclusion
Cyanobacteria generate an amazing array of structurally complex natural products, which include the hapalindole-type metabolites. With their unique polycyclic ring system, functional groups and stereochemical differences, these indole alkaloids continue to attract attention. The diverse biological activities of these metabolites enhance their importance as we continue to search for novel small molecules to combat infectious agents, cancer and other human diseases. While there have been numerous total syntheses, we continue to search for new approaches to explore and diversify these natural products for on-going drug development efforts. Because nature has developed elegant ways to synthesize natural products, numerous groups have taken the challenge to dissect the biosynthetic construction and tailoring of these compounds. This work has uncovered a collection of fascinating enzymes that catalyze distinctive reactions. Isonitrile synthases, aromatic prenyltransferases, the newly discovered Stig cyclases, carrier protein independent halogenases and various Rieske oxygenases all work in unison to produce these remarkable indole alkaloids. Currently, we have only scratched the surface of potential versatility of these unique biocatalysts and their ability to create structural diversity in metabolite assembly. As bioinformatics, synthetic biology and heterologous expression tools improve, we expect to characterize additional hapalindole-type metabolite pathways and expand our ability to harness them to aid in drug discovery and development efforts.
Acknowledgements
We are grateful to the National Science Foundation under the CCI Center for Selective C−H Functionalization (CHE-1700982), the National Institutes of Health, the Hans W. Vahlteich Professorship (to D.H.S.) and the ACS MEDI Pre-Doctoral Fellowship (to R.M.H) for financial support. We thank Dr. Shasha Li for her inspiration and design for Figure 10 and Dr. Sean Newmister for generating Figure 14.
Footnotes
The authors declare no competing financial interest.
References
- (1).Bhat V; Dave A; MacKay JA; Rawal VH Chapter Two - The Chemistry of Hapalindoles, Fischerindoles, Ambiguines, and Welwitindolinones. In The Alkaloids: Chemistry and Biology; Knölker H-J, Ed.; Chemistry and Biology; Academic Press, 2014; Vol. 73, pp 65–160. 10.1016/B978-0-12-411565-1.00002-0. [DOI] [PubMed] [Google Scholar]
- (2).Singh RK; Tiwari SP; Rai AK; Mohapatra TM Cyanobacteria: An Emerging Source for Drug Discovery. The Journal of Antibiotics 2011, 64 (6), 401–412. 10.1038/ja.2011.21. [DOI] [PubMed] [Google Scholar]
- (3).Polakis P Antibody Drug Conjugates for Cancer Therapy. Pharmacol Rev 2016, 68 (1), 3–19. 10.1124/pr.114.009373. [DOI] [PubMed] [Google Scholar]
- (4).Moore RE; Cheuk C; Patterson GML Hapalindoles: New Alkaloids from the Blue-Green Alga Hapalosiphon Fontinalis. J. Am. Chem. Soc 1984, 106 (21), 6456–6457. 10.1021/ja00333a079. [DOI] [Google Scholar]
- (5).Moore RE; Cheuk C; Yang XQG; Patterson GML; Bonjouklian R; Smitka TA; Mynderse JS; Foster RS; Jones ND; Swartzendruber JK; Deeter JB Hapalindoles, Antibacterial and Antimycotic Alkaloids from the Cyanophyte Hapalosiphon Fontinalis. J. Org. Chem 1987, 52 (6), 1036–1043. 10.1021/jo00382a012. [DOI] [Google Scholar]
- (6).Asthana RK; Srivastava A; Singh AP; Deepali; Singh SP; Nath G; Srivastava R; Srivastava BS Identification of an Antimicrobial Entity from the Cyanobacterium Fischerella Sp. Isolated from Bark of Azadirachta Indica (Neem) Tree. J. Appl. Phycol 2006, 18 (1), 33–39. 10.1007/s10811-005-9011-9. [DOI] [Google Scholar]
- (7).Raveh A; Carmeli S Antimicrobial Ambiguines from the Cyanobacterium Fischerella Sp. Collected in Israel. J. Nat. Prod 2007, 70 (2), 196–201. 10.1021/np060495r. [DOI] [PubMed] [Google Scholar]
- (8).Mo S; Krunic A; Chlipala G; Orjala J Antimicrobial Ambiguine Isonitriles from the Cyanobacterium Fischerella Ambigua. J. Nat. Prod 2009, 72 (5), 894–899. 10.1021/np800751j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Doan NT; Stewart PR; Smith GD Inhibition of Bacterial RNA Polymerase by the Cyanobacterial Metabolites 12- Epi -Hapalindole E Isonitrile and Calothrixin A. FEMS Microbiol. Lett 2001, 196 (2), 135–139. 10.1111/j.1574-6968.2001.tb10554.x. [DOI] [PubMed] [Google Scholar]
- (10).Smitka TA; Bonjouklian R; Doolin L; Jones ND; Deeter JB; Yoshida WY; Prinsep MR; Moore RE; Patterson GML Ambiguine Isonitriles, Fungicidal Hapalindole-Type Alkaloids from Three Genera of Blue-Green Algae Belonging to the Stigonemataceae. J. Org. Chem 1992, 57 (3), 857–861. 10.1021/jo00029a014. [DOI] [Google Scholar]
- (11).Stratmann K; Moore RE; Bonjouklian R; Deeter JB; Patterson GML; Shaffer S; Smith CD; Smitka TA Welwitindolinones, Unusual Alkaloids from the Blue-Green Algae Hapalosiphon Welwitschii and Westiella Intricata. Relationship to Fischerindoles and Hapalinodoles. J. Am. Chem. Soc 1994, 116 (22), 9935–9942. 10.1021/ja00101a015. [DOI] [Google Scholar]
- (12).Becher PG; Keller S; Jung G; Süssmuth RD; Jüttner F Insecticidal Activity of 12-Epi-Hapalindole J Isonitrile. Phytochemistry 2007, 68 (19), 2493–2497. 10.1016/j.phytochem.2007.06.024. [DOI] [PubMed] [Google Scholar]
- (13).Smith CD; Zilfou JT; Stratmann K; Patterson GM; Moore RE Welwitindolinone Analogues That Reverse P-Glycoprotein-Mediated Multiple Drug Resistance. Mol. Pharmacol 1995, 47 (2), 241–247. [PubMed] [Google Scholar]
- (14).Kim H; Lantvit D; Hwang CH; Kroll DJ; Swanson SM; Franzblau SG; Orjala J Indole Alkaloids from Two Cultured Cyanobacteria, Westiellopsis Sp. and Fischerella Muscicola. Bioorg. Med. Chem 2012, 20 (17), 5290–5295. 10.1016/j.bmc.2012.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zhang X; Smith CD Microtubule Effects of Welwistatin, a Cyanobacterial Indolinone That Circumvents Multiple Drug Resistance. Mol. Pharmacol 1996, 49 (2), 288–294. [PubMed] [Google Scholar]
- (16).Chilczuk T; Steinborn C; Breinlinger S; Zimmermann-Klemd AM; Huber R; Enke H; Enke D; Niedermeyer THJ; Gründemann C Hapalindoles from the Cyanobacterium Hapalosiphon Sp. Inhibit T Cell Proliferation. Planta Med. 2019. 10.1055/a-1045-5178. [DOI] [PubMed] [Google Scholar]
- (17).Handbook on Cyanobacteria: Biochemistry, Biotechnology and Applications; Gault PM, Marler HJ, Eds.; Bacteriology research developments series; Nova Science Publishers: New York, 2009. [Google Scholar]
- (18).Park A; Moore RE; Patterson GML Fischerindole L, a New Isonitrile from the Terrestrial Blue-Green Alga Fischerella Muscicola. Tetrahedron Lett. 1992, 33 (23), 3257–3260. 10.1016/S0040-4039(00)92061-6. [DOI] [Google Scholar]
- (19).Kim H; Krunic A; Lantvit D; Shen Q; Kroll DJ; Swanson SM; Orjala J Nitrile-Containing Fischerindoles from the Cultured Cyanobacterium Fischerella Sp. Tetrahedron 2012, 68 (15), 3205–3209. 10.1016/j.tet.2012.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Mo S; Krunic A; Santarsiero BD; Franzblau SG; Orjala J Hapalindole-Related Alkaloids from the Cultured Cyanobacterium Fischerella Ambigua. Phytochemistry 2010, 71 (17–18), 2116–2123. 10.1016/j.phytochem.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Schwartz RE; Hirsch CF; Springer JP; Pettibone DJ; Zink DL Unusual Cyclopropane-Containing Hapalindolinones from a Cultured Cyanobacterium. J. Org. Chem 1987, 52 (16), 3704–3706. 10.1021/jo00392a045. [DOI] [Google Scholar]
- (22).Moore RE; Yang XQG; Patterson GML Fontonamide and Anhydrohapaloxindole A, Two New Alkaloids from the Blue-Green Alga Hapalosiphon Fontinalis. J. Org. Chem 1987, 52 (17), 3773–3777. 10.1021/jo00226a009. [DOI] [Google Scholar]
- (23).Moore RE; Yang XG; Patterson GML; Bonjouklian R; Smitka TA Hapalonamides and Other Oxidized Hapalindoles from Hapalosiphon Fontinalis. Phytochemistry 1989, 28 (5), 1565–1567. 10.1016/S0031-9422(00)97798-7. [DOI] [Google Scholar]
- (24).Cagide E; Becher PG; Louzao MC; Espiña B; Vieytes MR; Jüttner F; Botana LM Hapalindoles from the Cyanobacterium Fischerella: Potential Sodium Channel Modulators. Chem. Res. Toxicol 2014, 27 (10), 1696–1706. 10.1021/tx500188a. [DOI] [PubMed] [Google Scholar]
- (25).Bhat V; Allan KM; Rawal VH Total Synthesis of N -Methylwelwitindolinone D Isonitrile. J. Am. Chem. Soc 2011, 133 (15), 5798–5801. 10.1021/ja201834u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Chandra A; Johnston JN Total Synthesis of the Chlorine-Containing Hapalindoles K, A, and G. Angew. Chem. Int. Ed 2011, 50 (33), 7641–7644. 10.1002/anie.201100957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Connon R; Guiry PJ Recent Advances in the Development of One-Pot/Multistep Syntheses of 3,4-Annulated Indoles. Tetrahedron Lett. 2020, 151696. 10.1016/j.tetlet.2020.151696. [DOI] [Google Scholar]
- (28).Dethe DH; Das S; Kumar VB; Mir NA Enantiospecific Total Syntheses of (+)-Hapalindole H and (−)-12-Epi-Hapalindole U. Eur. J. Chem 2018, 24 (36), 8980–8984. 10.1002/chem.201800970. [DOI] [PubMed] [Google Scholar]
- (29).Gademann K; Bonazzi S Total Synthesis of Complex Cyanobacterial Alkaloids without Using Protecting Groups. Angew. Chem. Int. Ed 2007, 46 (30), 5656–5658. 10.1002/anie.200701881. [DOI] [PubMed] [Google Scholar]
- (30).Kinsman AC; Kerr MA Total Synthesis of (±)-Hapalindole Q. Org. Lett 2001, 3 (20), 3189–3191. 10.1021/ol0165138. [DOI] [PubMed] [Google Scholar]
- (31).Kinsman AC; Kerr MA The Total Synthesis of (+)-Hapalindole Q by an Organomediated Diels−Alder Reaction. J. Am. Chem. Soc 2003, 125 (46), 14120–14125. 10.1021/ja036191y. [DOI] [PubMed] [Google Scholar]
- (32).Lu Z; Yang M; Chen P; Xiong X; Li A Total Synthesis of Hapalindole-Type Natural Products. Angew. Chem. Int. Ed 2014, 53 (50), 13840–13844. 10.1002/anie.201406626. [DOI] [PubMed] [Google Scholar]
- (33).Maimone TJ; Ishihara Y; Baran PS Scalable Total Syntheses of (−)-Hapalindole U and (+)-Ambiguine H. Tetrahedron 2015, 71 (22), 3652–3665. 10.1016/j.tet.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Rafferty RJ; Williams RM Total Synthesis of Hapalindoles J and U. J. Org. Chem 2012, 77 (1), 519–524. 10.1021/jo202139k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Rafferty RJ; Williams RM Formal Synthesis of Hapalindole O and Synthetic Efforts towards Hapalindole K and Ambiguine A. Heterocycles 2012, 86 (1), 219–231. 10.3987/COM-12-S(N)3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Reisman SE; Ready JM; Hasuoka A; Smith CJ; Wood JL Total Synthesis of (±)-Welwitindolinone A Isonitrile. J. Am. Chem. Soc 2006, 128 (5), 1448–1449. 10.1021/ja057640s. [DOI] [PubMed] [Google Scholar]
- (37).Richter JM; Ishihara Y; Masuda T; Whitefield BW; Llamas T; Pohjakallio A; Baran PS Enantiospecific Total Synthesis of the Hapalindoles, Fischerindoles, and Welwitindolinones via a Redox Economic Approach. J. Am. Chem. Soc 2008, 130 (52), 17938–17954. 10.1021/ja806981k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Sahu S; Das B; Maji MS Stereodivergent Total Synthesis of Hapalindoles, Fischerindoles, Hapalonamide H, and Ambiguine H Alkaloids by Developing a Biomimetic, Redox-Neutral, Cascade Prins-Type Cyclization. Org. Lett 2018, 20 (20), 6485–6489. 10.1021/acs.orglett.8b02804. [DOI] [PubMed] [Google Scholar]
- (39).Weires NA; Styduhar ED; Baker EL; Garg NK Total Synthesis of (−)-N-Methylwelwitindolinone B Isothiocyanate via a Chlorinative Oxabicycle Ring-Opening Strategy. J. Am. Chem. Soc 2014, 136 (42), 14710–14713. 10.1021/ja5087672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Styduhar ED; Huters AD; Weires NA; Garg NK Enantiospecific Total Synthesis of N-Methylwelwitindolinone D Isonitrile. Angew. Chem. Int. Ed 2013, 52 (47), 12422–12425. 10.1002/anie.201307464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Quasdorf KW; Huters AD; Lodewyk MW; Tantillo DJ; Garg NK Total Synthesis of Oxidized Welwitindolinones and (−)-N-Methylwelwitindolinone C Isonitrile. J. Am. Chem. Soc 2012, 134 (3), 1396–1399. 10.1021/ja210837b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Huters AD; Styduhar ED; Garg NK Total Syntheses of the Elusive Welwitindolinones with Bicyclo[4.3.1] Cores. Angew. Chem. Int. Ed 2012, 51 (16), 3758–3765. 10.1002/anie.201107567. [DOI] [PubMed] [Google Scholar]
- (43).MacKay JA; Bishop RL; Rawal VH Rapid Synthesis of the N-Methylwelwitindolinone Skeleton. Org. Lett 2005, 7 (16), 3421–3424. 10.1021/ol051043t. [DOI] [PubMed] [Google Scholar]
- (44).Bhat V; Rawal VH Stereocontrolled Synthesis of 20,21-Dihydro N-Methylwelwitindolinone B Isothiocyanate. Chem. Commun 2011, 47 (34), 9705–9707. 10.1039/C1CC13498A. [DOI] [PubMed] [Google Scholar]
- (45).Bhat V; MacKay JA; Rawal VH Lessons Learned While Traversing the Welwitindolinone Alkaloids Obstacle Course. Tetrahedron 2011, 67 (52), 10097–10104. 10.1016/j.tet.2011.09.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Bhat V; MacKay JA; Rawal VH Directed Oxidative Cyclizations to C2- or C4-Positions of Indole: Efficient Construction of the Bicyclo[4.3.1]Decane Core of Welwitindolinones. Org. Lett. 2011, 13 (12), 3214–3217. 10.1021/ol201122f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Allan KM; Kobayashi K; Rawal VH A Unified Route to the Welwitindolinone Alkaloids: Total Syntheses of (−)-N-Methylwelwitindolinone C Isothiocyanate, (−)-N-Methylwelwitindolinone C Isonitrile, and (−)-3-Hydroxy-N-Methylwelwitindolinone C Isothiocyanate. J. Am. Chem. Soc 2012, 134 (3), 1392–1395. 10.1021/ja210793x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Muratake H; Natsume M Total Synthesis of Marine Alkaloids (±)-Hapalindoles J and M. Tetrahedron Lett. 1989, 30 (14), 1815–1818. 10.1016/S0040-4039(00)99587-X. [DOI] [Google Scholar]
- (49).Muratake H; Natsume M Synthetic Studies of Marine Alkaloids Hapalindoles. Part 2. Lithium Aluminum Hydride Reduction of the Electron-Rich Carbon-Carbon Double Bond Conjugated with the Indole Nucleus. Tetrahedron 1990, 46 (18), 6343–6350. 10.1016/S0040-4020(01)96006-5. [DOI] [Google Scholar]
- (50).Muratake H; Kumagami H; Natsume M Synthetic Studies of Marine Alkaloids Hapalindoles. Part 3 Total Synthesis of (±)-Hapalindoles H and U. Tetrahedron 1990, 46 (18), 6351–6360. 10.1016/S0040-4020(01)96007-7. [DOI] [Google Scholar]
- (51).Vaillancourt V; Albizati KF Synthesis and Absolute Configuration of (+)-Hapalindole Q. J. Am. Chem. Soc 1993, 115 (9), 3499–3502. 10.1021/ja00062a013. [DOI] [Google Scholar]
- (52).Fukuyama T; Chen X Stereocontrolled Synthesis of (−)-Hapalindole G. J. Am. Chem. Soc 1994, 116 (7), 3125–3126. 10.1021/ja00086a053. [DOI] [Google Scholar]
- (53).Baran PS; Richter JM Direct Coupling of Indoles with Carbonyl Compounds: Short, Enantioselective, Gram-Scale Synthetic Entry into the Hapalindole and Fischerindole Alkaloid Families. J. Am. Chem. Soc 2004, 126 (24), 7450–7451. [DOI] [PubMed] [Google Scholar]
- (54).Johnson RE; Ree H; Hartmann M; Lang L; Sawano S; Sarpong R Total Synthesis of Pentacyclic (−)-Ambiguine P Using Sequential Indole Functionalizations. J. Am. Chem. Soc 2019, 141 (6), 2233–2237. 10.1021/jacs.8b13388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Xu J; Rawal VH Total Synthesis of (−)-Ambiguine P. J. Am. Chem. Soc 2019, 141 (12), 4820–4823. 10.1021/jacs.9b01739. [DOI] [PubMed] [Google Scholar]
- (56).Baran PS; Richter JM Enantioselective Total Syntheses of Welwitindolinone A and Fischerindoles I and G. J. Am. Chem. Soc 2005, 127 (44), 15394–15396. 10.1021/ja056171r. [DOI] [PubMed] [Google Scholar]
- (57).Baran PS; Maimone TJ; Richter JM Total Synthesis of Marine Natural Products without Using Protecting Groups. Nature 2007, 446 (7134), 404–408. 10.1038/nature05569. [DOI] [PubMed] [Google Scholar]
- (58).Liu Y; Cheng L-J; Yue H-T; Che W; Xie J-H; Zhou Q-L Divergent Enantioselective Synthesis of Hapalindole-Type Alkaloids Using Catalytic Asymmetric Hydrogenation of a Ketone to Construct the Chiral Core Structure. Chem. Sci 2016, 7 (7), 4725–4729. 10.1039/C6SC00686H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Ready JM; Reisman SE; Hirata M; Weiss MM; Tamaki K; Ovaska TV; Wood JL A Mild and Efficient Synthesis of Oxindoles: Progress Towards the Synthesis of Welwitindolinone A Isonitrile. Angew. Chem. Int. Ed 2004, 43 (10), 1270–1272. 10.1002/anie.200353282. [DOI] [PubMed] [Google Scholar]
- (60).Hillwig ML; Zhu Q; Liu X Biosynthesis of Ambiguine Indole Alkaloids in Cyanobacterium Fischerella Ambigua. ACS Chem. Biol 2014, 9 (2), 372–377. 10.1021/cb400681n. [DOI] [PubMed] [Google Scholar]
- (61).Hillwig ML; Fuhrman HA; Ittiamornkul K; Sevco TJ; Kwak DH; Liu X Identification and Characterization of a Welwitindolinone Alkaloid Biosynthetic Gene Cluster in the Stigonematalean Cyanobacterium Hapalosiphon Welwitschii. ChemBioChem 2014, 15 (5), 665–669. 10.1002/cbic.201300794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Micallef ML; Sharma D; Bunn BM; Gerwick L; Viswanathan R; Moffitt MC Comparative Analysis of Hapalindole, Ambiguine and Welwitindolinone Gene Clusters and Reconstitution of Indole-Isonitrile Biosynthesis from Cyanobacteria. BMC Microbiol. 2014, 14 (1), 213. 10.1186/s12866-014-0213-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Li S; Lowell AN; Yu F; Raveh A; Newmister SA; Bair N; Schaub JM; Williams RM; Sherman DH Hapalindole/Ambiguine Biogenesis Is Mediated by a Cope Rearrangement, C–C Bond-Forming Cascade. J. Am. Chem. Soc 2015, 137 (49), 15366–15369. 10.1021/jacs.5b10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Zhu Q; Liu X Molecular and Genetic Basis for Early Stage Structural Diversifications in Hapalindole-Type Alkaloid Biogenesis. Chem. Commun 2017, 53 (19), 2826–2829. 10.1039/C7CC00782E. [DOI] [PubMed] [Google Scholar]
- (65).Li S; Lowell AN; Newmister SA; Yu F; Williams RM; Sherman DH Decoding Cyclase-Dependent Assembly of Hapalindole and Fischerindole Alkaloids. Nat. Chem. Biol 2017, 13 (5), 467–469. 10.1038/nchembio.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Zhu Q; Liu X Discovery of a Calcium-Dependent Enzymatic Cascade for the Selective Assembly of Hapalindole-Type Alkaloids: On the Biosynthetic Origin of Hapalindole U. Angew. Chem. Int. Ed 2017, 56 (31), 9062–9066. 10.1002/anie.201703932. [DOI] [PubMed] [Google Scholar]
- (67).Li S; Newmister SA; Lowell AN; Zi J; Chappell CR; Yu F; Hohlman RM; Orjala J; Williams RM; Sherman DH Control of Stereoselectivity in Diverse Hapalindole Metabolites Is Mediated by Cofactor-Induced Combinatorial Pairing of Stig Cyclases. Angew. Chem. Int. Ed 2020, 59 (21), 8166–8172. 10.1002/anie.201913686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Hillwig ML; Liu X A New Family of Iron-Dependent Halogenases Acts on Freestanding Substrates. Nat. Chem. Biol 2014, 10 (11), 921–923. 10.1038/nchembio.1625. [DOI] [PubMed] [Google Scholar]
- (69).Liu X; Hillwig ML; Koharudin LMI; Gronenborn AM Unified Biogenesis of Ambiguine, Fischerindole, Hapalindole and Welwitindolinone: Identification of a Monogeranylated Indolenine as a Cryptic Common Biosynthetic Intermediate by an Unusual Magnesium-Dependent Aromatic Prenyltransferase. Chem. Commun 2016, 52 (8), 1737–1740. 10.1039/C5CC10060G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Brady SF; Clardy J Systematic Investigation of the Escherichia Coli Metabolome for the Biosynthetic Origin of an Isocyanide Carbon Atom. Angew. Chem. Int. Ed 2005, 44 (43), 7045–7048. 10.1002/anie.200501942. [DOI] [PubMed] [Google Scholar]
- (71).Brady SF; Clardy J Cloning and Heterologous Expression of Isocyanide Biosynthetic Genes from Environmental DNA. Angew. Chem. Int. Ed 2005, 44 (43), 7063–7065. 10.1002/anie.200501941. [DOI] [PubMed] [Google Scholar]
- (72).Chang W; Sanyal D; Huang J-L; Ittiamornkul K; Zhu Q; Liu X In Vitro Stepwise Reconstitution of Amino Acid Derived Vinyl Isocyanide Biosynthesis: Detection of an Elusive Intermediate. Org. Lett 2017, 19 (5), 1208–1211. 10.1021/acs.orglett.7b00258. [DOI] [PubMed] [Google Scholar]
- (73).Hillwig ML; Zhu Q; Ittiamornkul K; Liu X Discovery of a Promiscuous Non-Heme Iron Halogenase in Ambiguine Alkaloid Biogenesis: Implication for an Evolvable Enzyme Family for Late-Stage Halogenation of Aliphatic Carbons in Small Molecules. Angew. Chem. Int. Ed 2016, 55 (19), 5780–5784. 10.1002/anie.201601447. [DOI] [PubMed] [Google Scholar]
- (74).Mahmoodi N; Qian Q; Luk L; Tanner M Rearrangements in the Mechanisms of the Indole Alkaloid Prenyltransferases. Pure Appl. Chem 2013, 85 (10), 1935–1948. 10.1351/pac-con-13-02-02. [DOI] [Google Scholar]
- (75).Newmister SA; Li S; Garcia-Borràs M; Sanders JN; Yang S; Lowell AN; Yu F; Smith JL; Williams RM; Houk KN; Sherman DH Structural Basis of the Cope Rearrangement and Cyclization in Hapalindole Biogenesis. Nat. Chem. Biol 2018, 14 (4), 345–351. 10.1038/s41589-018-0003-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Chen C-C; Hu X; Tang X; Yang Y; Ko T-P; Gao J; Zheng Y; Huang J-W; Yu Z; Li L; Han S; Cai N; Zhang Y; Liu W; Guo R-T The Crystal Structure of a Class of Cyclases That Catalyze the Cope Rearrangement. Angew. Chem. Int. Ed 2018, 57 (46), 15060–15064. 10.1002/anie.201808231. [DOI] [PubMed] [Google Scholar]
- (77).Wang J; Chen C-C; Yang Y; Liu W; Ko T-P; Shang N; Hu X; Xie Y; Huang J-W; Zhang Y; Guo R-T Structural Insight into a Novel Indole Prenyltransferase in Hapalindole-Type Alkaloid Biosynthesis. Biochem. Biophys. Res. Commun 2018, 495 (2), 1782–1788. 10.1016/j.bbrc.2017.12.039. [DOI] [PubMed] [Google Scholar]
- (78).Awakawa T; Mori T; Nakashima Y; Zhai R; Wong CP; Hillwig ML; Liu X; Abe I Molecular Insight into the Mg2+-Dependent Allosteric Control of Indole Prenylation by Aromatic Prenyltransferase AmbP1. Angew. Chem. Int. Ed 2018, 57 (23), 6810–6813. 10.1002/anie.201800855. [DOI] [PubMed] [Google Scholar]
- (79).Wong CP; Awakawa T; Nakashima Y; Mori T; Zhu Q; Liu X; Abe I Two Distinct Substrate Binding Modes for the Normal and Reverse Prenylation of Hapalindoles by the Prenyltransferase AmbP3. Angew. Chem. Int. Ed 2018, 57 (2), 560–563. 10.1002/anie.201710682. [DOI] [PubMed] [Google Scholar]
- (80).Hernandes MZ; Cavalcanti SMT; Moreira DRM; de Azevedo Junior WF; Leite ACL Halogen Atoms in the Modern Medicinal Chemistry: Hints for the Drug Design. Curr. Drug Targets 2010, 11 (3), 303–314. 10.2174/138945010790711996. [DOI] [PubMed] [Google Scholar]
- (81).Fraley AE; Sherman DH Halogenase Engineering and Its Utility in Medicinal Chemistry. Bioorg. Med. Chem. Lett 2018, 28 (11), 1992–1999. 10.1016/j.bmcl.2018.04.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Zhu Q; Hillwig ML; Doi Y; Liu X Aliphatic Halogenase Enables Late-Stage C−H Functionalization: Selective Synthesis of a Brominated Fischerindole Alkaloid with Enhanced Antibacterial Activity. ChemBioChem 2016, 17 (6), 466–470. 10.1002/cbic.201500674. [DOI] [PubMed] [Google Scholar]
- (83).Mitchell AJ; Zhu Q; Maggiolo AO; Ananth NR; Hillwig ML; Liu X; Boal AK Structural Basis for Halogenation by Iron- and 2-Oxo-Glutarate-Dependent Enzyme WelO5. Nat. Chem. Biol 2016, 12 (8), 636–640. 10.1038/nchembio.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Zhang X; Wang Z; Gao J; Liu W Chlorination versus Hydroxylation Selectivity Mediated by the Non-Heme Iron Halogenase WelO5. Phys. Chem. Chem. Phys 2020, 22 (16), 8699–8712. 10.1039/D0CP00791A. [DOI] [PubMed] [Google Scholar]
- (85).Zhu Q; Liu X Characterization of Non-Heme Iron Aliphatic Halogenase WelO5* from Hapalosiphon Welwitschii IC-52–3: Identification of a Minimal Protein Sequence Motif That Confers Enzymatic Chlorination Specificity in the Biosynthesis of Welwitindolelinones. Beilstein J. Org. Chem 2017, 13 (1), 1168–1173. 10.3762/bjoc.13.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Hayashi T; Ligibel M; Sager E; Voss M; Hunziker J; Schroer K; Snajdrova R; Buller R Evolved Aliphatic Halogenases Enable Regiocomplementary C−H Functionalization of a Pharmaceutically Relevant Compound. Angew. Chem. Int. Ed 2019, 58 (51), 18535–18539. 10.1002/anie.201907245. [DOI] [PubMed] [Google Scholar]
- (87).Duewel S; Schmermund L; Faber T; Harms K; Srinivasan V; Meggers E; Hoebenreich S Directed Evolution of an FeII-Dependent Halogenase for Asymmetric C(Sp3)–H Chlorination. ACS Catal. 2020, 10 (2), 1272–1277. 10.1021/acscatal.9b04691. [DOI] [Google Scholar]
- (88).Chung P-Y; Tang JC-O; Cheng C-H; Bian Z-X; Wong W-Y; Lam K-H; Chui C-H Synthesis of Hexahydrofuro[3,2-c]Quinoline, a Martinelline Type Analogue and Investigation of Its Biological Activity. SpringerPlus 2016, 5 (1), 271. 10.1186/s40064-016-1890-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Chovancová E, Pavelka A, Beneš P, Strnad O, Brezovský J, Kozlíková B, Gora A, Šustr V, Klvaňa M, Medek P, Biedermannová L, Sochor J, Damborský J CAVER 3.0: A Tool for the Analysis of Transport Pathways in Dynamic Protein Structures. PLoS Computational Biology 2012, 8 (10), e1002708. 10.1371/journal.pcbi.1002708. [DOI] [PMC free article] [PubMed] [Google Scholar]