

Abstract
Aryl hydrocarbon receptor (AHR) research has shifted from exploring dioxin toxicity to elucidation of various physiologic AHR functions. Exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is known to exert cellular stress-mediated sterile inflammatory responses in exposed human tissues but may be lethal in sensitive species. Inflammation can be thought of as the extreme end of a spectrum ranging from homeostasis to stress responses (sterile inflammation) and to defense against infection (infectious inflammation). Defense against bacterial infection by generation of reactive oxygen species has to be strictly controlled and may use up a considerable amount of energy. NAD+-mediated energy metabolism adapts to various inflammatory responses. As examples, the present commentary tries to integrate responses of AHR and NAD+-consuming enzymes (PARP7/TiPARP, CD38 and sirtuins) into infectious and stress-induced inflammatory responses, the latter exemplified by nonalcoholic fatty liver disease (NAFLD). TCDD toxicity models in sensitive species provide hints to molecular AHR targets of energy metabolism including gluconeogenesis and glycolysis. AHR research remains challenging and promising.
Keywords: AHR, CD38, PARP7, Sirtuins, Infectious and sterile inflammation
Introduction
Aryl hydrocarbon receptor (AHR) research has shifted from exploring dioxin toxicity to elucidation of various physiologic AHR functions. AHR has been characterized as ligand-activated, multifunctional transcription factor and environmental sensor (Gu et al. 2000; Avilla et al. 2020). Accumulating evidence suggests that AHR, mainly expressed in barrier organs such as skin, intestine and lung, is involved in multiple physiologic functions including development (Gasiewicz et al. 2014; Ko et al. 2016), chemical defense (Nebert et al. 2004), microbial defense (Lawrence and Vorderstrasse 2013), immunity and inflammation (Stockinger et al. 2014; Esser and Rannug 2015), reproduction (Baba et al. 2005), and energy metabolism (Diani-Moore et al. 2017).
Exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is known to exert sterile inflammatory responses in exposed tissues (Saurat et al. 2012). Classic causes of inflammation (infection and tissue injury) may be one end of a range of adverse conditions starting from tissue homeostasis to tissue stress-mediated sterile inflammation, and ending with infectious inflammation by pathogens (Medzhitov 2008; Wang et al. 2019). In infectious inflammation the innate immune system is activated by recognizing, e.g., pathogenic bacteria through pattern-recognition receptors (PRRs). PRRs such as TLRs (Toll-like receptors) are expressed by many cell types such as epithelial cells of barrier organs and associated immune cells including neutrophils, macrophages, and dendritic cells. PRRs recognize pathogen-associated molecular patterns (PAMPs). In the case of sterile inflammation, danger-associated molecular patterns (DAMPs) are similarly recognized by a variety of receptors and other factors triggered, e.g., by structures of dying cells.
Since inflammatory responses are energy-consuming processes they have to adapt to NAD+-dependent energy economy of the organism (Medzhitov 2008; Wang et al. 2019). NAD+ is an essential cofactor for redox enzymes and a substrate for signaling enzymes. Energy consumption by various inflammatory responses is regulated by NAD synthesis and consumption. NAD+-consumption is carried out by multiple enzymes such as CD38 (Verdin 2015; Yang and Sauve 2016), 17 PARPs including the AHR target gene PARP7/TiPARP (MacPherson et al. 2013; Gupte et al. 2018) and 7 NAD-dependent sirtuins (Chang and Guarente 2014; Singh et al. 2018).
Elucidation of AHR functions is challenged by dependence upon cell type, cellular context, and species differences, the latter suggested by TCDD toxicity in sensitive species leading to wasting syndrome and lethality (Poland and Knutson 1982). Therefore, the commentary is focused to inflammatory responses in intestine, liver and associated immune cells in humans. After an overview on AHR and NAD+-consuming enzymes (PARP7/TiPARP, CD38 and sirtuins) the commentary tries to integrate functions of AHR and NAD+-consuming enzymes in infectious and sterile inflammatory responses. TCDD toxicity models in sensitive species are discussed since they provide hints to molecular AHR targets in energy metabolism.
AHR ligands and signaling
A large number of AHR ligands has been identified and comprehensively reviewed (Denison and Nagy 2003; Nguyen and Bradfield 2008; Esser and Rannug 2015; Murray and Perdew 2017; Rothhammer and Quintana 2019). Prototypical AHR agonist TCDD is the most potent but poorly metabolized ligand leading to persistent AHR activation and dysregulation of AHR functions. 6-Formylindolo[3,2-b]carbazole (FICZ) is an equally potent AHR ligand that is rapidly metabolized by the AHR target CYP1A1 leading to an important negative feedback loop (Esser and Rannug 2015). Relevance of the AHR-CYP1-FICZ axis in vivo has been demonstrated by overexpression of CYP1A1 (Schiering et al. 2017). Notably, toxicity of TCDD and dioxin-like compounds leading to potent and persistent AHR activation has to be distinguished from that of polycyclic aromatic hydrocarbons (PAHs) such as benzo[a]pyrene that, e.g., do not lead to chloracne, the hallmark of dioxin toxicity (Avilla et al. 2020). Recently, vitamin B12 and folic acid have been identified as endogenous AHR antagonists (Kim et al. 2020).
There are diverse sources of AHR agonists and antagonists (Table 1). Dietary phytochemicals and microbial ligands represent natural AHR ligands since they have been characterized as physiological agents necessary for intestinal development (Kiss et al. 2011) and for homeostasis with commensal bacteria (Lamas et al. 2018). Many fruits and herbs contain AHR agonistic flavonoids such as quercetin and glucobrassicin, the latter generating the pro-ligand indole-3-carbinol that is further converted to the strong AHR ligand ICZ (indolo[3,2-b]carbazole). Dietary flavonoids such as quercetin belong to many indirect AHR agonists that inhibit CYP1A1 and thereby may increase the endogenous ligand FICZ. Microbiota such as Pseudomonas aeruginosa and mycobacterium tuberculosis have been demonstrated to generate AHR agonistic virulence factors such as pyocyanin, 1-hydroxyphenazine and phthiocol (Moura-Alves et al. 2014, 2019). Similar microbiota-generated virulence factors may be generated in the intestine. When recognized by AHR expressed in intestinal immune cells such as ILC3, neutrophils are recruited that generate the oxidative burst to kill pathogenic bacteria (Bock 2020a, for references).
Table 1.
Selected agonists and antagonists of AHR
| Sources of ligands | Agonists and antagonistsb of AHR |
|---|---|
| Endogenous chemicals | Indole-3-acetaldehydea |
| FICZ | |
| Kynurenine | |
| Bilirubin | |
| Lipoxin A | |
| Prostaglandin G2 | |
| Vitamin B12b | |
| Folic acidb | |
| Phytochemicals | Indole-3-carbinola |
| Indole-3-carboxaldehydea | |
| ICZ | |
| Indigo | |
| Quercetinc | |
| Microbial products | Indole-3-acetaldehydea |
| 1-Hydroxyphenazine | |
| Pyocyanin | |
| Phthiocol | |
| Drugs | StemReginin1b |
| Omeprazolec | |
| Xenobiotic chemicals | TCDD |
| Benzo[a]pyrene | |
| 3-Methylcholanthrene | |
| ß-Naphthoflavone |
The list is by no means complete. Pro-ligands are converted to high-affinity ligands; for example, indole-3-carbinol to ICZ. Indirect agonists may inhibit CYP1A1 and thereby activate the endogenous ligand FICZ
aPro-ligands
bantagonists
cIndirect agonists
AHR is also operating by non-genomic signaling including protein kinases (Puga et al. 2009). For example, it operates as component of E3 ubiquitin ligases (Ohtake et al. 2009) and of a signalsome (Bunaciu et al. 2019). Furthermore, AHR often operates in cross-talk with other transcription factors including Nrf2, the key protector against oxidative stress (Kensler et al.2007) and NF-κB (Vogel et al. 2014). Interestingly, multiple mechanisms evolved to avoid sustained AHR activation including cross-talk with AHR repressor (Karchner et al. 2009), the discussed AHR-CYP1A1-FICZ axis (Esser and Rannug 2015), and nuclear-cytoplasmic shuttling (Ikuta et al. 2009).
Non-genomic AHR-mediated inflammatory signaling involving tissue-specific protein kinases has been substantiated in many cells/tissues from several species (Bock 2020b, for references). Presumably, AHR is activated in both genomic and non-genomic signaling by ligands or other cues including phosphorylation. Interestingly, non-genomic AHR signaling could be demonstrated in human platelets, an anucleated cell-mediating immunity, inflammation and thrombosis (Pombo et al. 2015). In this study, 1 nM TCDD and 10 nM omeprazole significantly increased AHR protein. AHR signaling was demonstrated by increased p38-MAPK and cPLA2 phosphorylation. Non-genomic pathways leading to TCDD-mediated tissue inflammation have been discussed before including membrane translocation and activation of c-Src. These transient responses are stabilized by genomic AHR signaling including IL-6 expression (Puga et al. 1997; Matsumura 2009).
NAD+ consuming enzymes (CD38, PARPs, and NAD+-dependent sirtuins)
As mentioned before, inflammatory responses may be energy-consuming processes and have to be metabolically adapted (Medzhitov 2008; Wang et al. 2019). Adaptation is achieved in part by NAD+ homeostasis. In addition to NAD synthesis, accumulating evidence suggests that NAD homeostasis is achieved by NAD-consuming enzymes including CD38, PARPs and sirtuins (Canto et al. 2015). Studies with CD38-deficient mice identified CD38 as NAD+-consuming enzyme involved in NAD+ homeostasis (Verdin 2015; Yang and Sauve 2017; Hogan et al. 2019). In addition, PARPs, particularly PARP1 and PARP7/TiPARP, are known NAD-consuming enzymes (Gupte et al. 2018; Cohen 2020; Fehr et al. 2021). Sirtuins have been identified as NAD-dependent protein deacetylases that consume a considerable amount of NAD+. The extent of NAD+ consumption by these enzymes under homeostatic or infectious stress conditions is presently unknown. It has been suggested that nicotinamide released by all NAD-consuming enzymes has to be almost completely reutilized to maintain NAD+ levels in nuclei, cytosol and mitochondria by the salvage pathway of NAD synthesis (Fig. 1). Estimates suggest that the entire NAD pool is replaced 2–4 times per day, and that only 0.1–0.2% nicotinamide is lost per turnover cycle (Yang and Sauve 2017). De novo NAD synthesis from tryptophan (TRP) is not sufficient to maintain NAD+ levels, particularly in stressed conditions such as inflammatory responses. Subcellular NAD+ pools are compartmentalized. For example, the mitochondrial NAD+ pool is distinct from the cytosolic pool. There has been much debate how mitochondrial NAD is generated. Recently, a mitochondrial NAD carrier has been identified (Luongo et al. 2020).
Fig. 1.

Simplified scheme of NAD+-consuming enzymes (CD38, PARPs and sirtuins), and reutilisation of generated nicotinamide (NAM) by the salvage pathway of NAD synthesis (dashed arrows). TRP, tryptophan; NA, nicotinic acid; NR, nicotinamide riboside; NRK, nicotinamide riboside kinase; NMN, nicotinamide mononucleotide; SLC, mitochondrial NAD+ transporter (Luongo et al. 2020). In the salvage pathway of NAD synthesis, NAM generated by all NAD-consuming enzymes is reutilized via the rate-limiting enzyme NAMPT (nicotinamide phosphoribosyltransferase) to synthesize NMN
The following brief overview of CD38, PARPs and sirtuins is focused on subsequently discussed interaction of AHR and NAD+-consuming enzymes in infectious diseases and energy metabolism.
CD38: subcellular locations and functions
CD38 hydrolyzes NAD+ at the energy-rich nicotinamide-ribose bond to ADP-ribose and nicotinamide. The enzyme catalyzes multiple reactions and is involved in many signaling pathways. In addition to generating extracellular adenosine (Horenstein et al. 2019), CD38 is a cyclic ADP-ribose synthase, mobilizing calcium for important signaling reactions from endoplasmic reticulum (ER) stores. Cyclic ADP-ribose-mediated calcium signaling is involved in many important processes, e.g., secretion of insulin and oxytocin (Jin et al. 2007; Lee and Zhao 2019). Loss of CD38 renders mice susceptible to bacterial infections due to loss of chemotaxis of neutrophils. This process has been shown to depend on calcium mobilization mediated by cyclic ADP-ribose (Partida-Sanchez et al. 2001). Interestingly, CD38 together with AHR is a constituent of a signalsome (Bunaciu et al. 2019), probably regulating microbial defense.
Early cell fractionation studies of rat liver suggested that, in addition to its role as plasma membrane-bound ecto-enzyme, NAD-glycohydrolase is also a constituent of the endoplasmic reticulum (Bock et al. 1971). NAD-glycohydrolase and CD38 have been demonstrated to be identical proteins (Bertellier et al. 1998; Cockayne et al. 1998). The enigma how the ecto-enzyme CD38 could use NAD to produce intracellular cyclic ADP-ribose has finally been solved by demonstrating that CD38 can be inserted into membranes in both type II and III orientation (Lee and Zhao 2019).
PARPs in NAD+ consumption
17 NAD+-consuming PARPs have been identified in the cell nucleus and cytoplasma (Gupte et al. 2018; Cohen 2020; Fehr et al. 2021). PARPs are NAD+-consuming enzymes, covalently linking a single ADP-ribose (ADPR) or a chain of ADPR units to proteins. PARP1 is the most abundant and best studied enzyme and belongs to the poly(ADP-ribosylating) proteins. It is mostly involved in DNA repair. With regard to cooperation with AHR, TCDD-induced PARP7/TiPARP (TCDD-inducible PARP) has been characterized as repressor of AHR expression (MacPherson et al. 2013) and mediator of NAD+-regulated dioxin toxicity (Diani-Moore et al. 2017), further discussed in the section on TCDD toxicity models in sensitive species. PARP7/TiPARP (together with PARP10, PARP12, PARP14) belongs to the mono(ADP-ribosylating) enzymes involved in sirtuin regulation (Cohen 2020).
With regard to the current COVID-19 pandemia, it should be mentioned that preliminary studies with murine hepatitis virus and human SARS-CoV-2 infection suggest increases of PARP7 and PARP10 expression and dysregulation of the NAD metabolome by decreasing de novo NAD synthesis and increasing the salvage pathway (Heer et al. 2020). However, implications of these findings are still unclear.
NAD+-consuming sirtuins
Mammalian sirtuins are seven members belonging to a group of histone and protein deacetylases with different subcellular localization and function (Chang and Guarente 2014; Canto et al. 2015; Singh et al. 2018). Nuclear SIRT1 and mitochondrial SIRT3 are major NAD+-dependent deacetylases involved in energy, lipid and glucose metabolism. They are emerging as key regulators of energy metabolism in normal physiology and a variety of oxidative stress-mediated pathological situations. Evidence has been obtained that sirtuins regulate key rate-limiting metabolic enzymes and cofactors including AMPK (AMP-activated protein kinase) (Hou et al. 2008) and PGC1α (Rodgers et al. 2005). It is conceivable that in this way SIRT1 and mitochondrial SIRT3 are involved in adapting energy metabolism.
AHR and NAD+-consuming enzymes in infectious inflammation of the intestine
Homeostasis
As discussed in a previous commentary (Bock 2020a), AHR is involved in intestinal homeostasis via microbial indoles and in bacterial defense via microbial virulence factors such as pyocyanin and 1-hydroxyphenazine (Moura-Alves et al. 2014, 2019). AHR-expressing ILC3 cells are generating anti-inflammatory Il-22. They are involved in commensal-host homeostasis, and maintenance of intestinal epithelial integrity via stabilizing the tight-junction barrier (Scott et al. 2020). Evidence for an essential role for ILC3 cells has been obtained in maintaining tissue homeostasis and limiting chronic infection in the intestine of humans and mice (Qiu et al. 2013; Blander et al. 2017; Sonnenberg and Artis 2015).
Microbial defense
AHR-deficient mice are more susceptible to viral and bacterial diseases (Lawrence and Vorderstrasse 2013). AHR sensing of bacterial pigments alerts the host of invading pathogens. Microbial virulence factors have been identified as AHR agonists (Moura-Alves et al. 2014, 2019). AHR is involved in quorum sensing, i.e., a communication system to modify bacterial behavior through signaling molecules. In this way, intestinal epithelial cells and associated immune cells recognize these virulence factors as PAMPs, and orchestrate microbial defense recruiting neutrophils and Th17 cells that generate reactive oxygen species (ROS) to kill bacteria in the so-called respiratory burst (Esser and Rannug 2015; Medzhitov 2008; Wang et al. 2019). In addition, AHR and CD38 are constituents of a signalsome involved in neutrophil differentiation and activation (Fig. 2) (Bunaciu et al. 2019). The respiratory burst in colitis consumes a lot of energy (Wang et al. 2019). AHR has been demonstrated to regulate a subunit of the NADPH oxidase complex (NOX) (Wada et al. 2013) and to promote macrophage survival (Kimura et al. 2013). CD38 may be an integrating factor, mediating neutrophil trafficking (Partida-Sanchez et al. 2001) and possibly maintaining energy economy. It is conceivable that CD38 in the discussed signalsome may be involved in adapting energy metabolism to demands of the organism. However, underlying mechanisms are unknown.
Fig. 2.

Illustration of proposed roles of AHR in inflammatory responses of bacterial colitis. Pathogenic bacteria (black rectangles) generate AHR agonistic virulence factors (1, black dots) leading to recruitment of ROS-generating Th17 cells and neutrophils, the latter expressing (i) AHR target gene p40phox subunit of the NADPH oxidase complex (NOX) (Wada et al. 2013) and (ii) plasma membrane-bound signalsome (2) containing AHR (crossed circle) and CD38 (striped circle) (Bunaciu et al. 2019)
Resolution phase of inflammation
To prevent collateral tissue damage, ROS production has to be strictly controlled (Roy et al. 2017). In the resolution phase of inflammation, Th17 cells are reprogrammed to Treg cells, and macrophages switch to secrete anti-inflammatory cytokines (Ip et al. 2017). In addition, AHR-associated Src kinase promotes phosphorylation of indolamine-2,3-dioxygenase (IDO-1) resulting in a tolerant state (Bessede et al. 2014). Roles of NAD+-dependent SIRT1 as well as epigenetic and metabolic programming in endotoxin tolerance have been discussed (Liu et al. 2011; Vachharajani and McCall 2019).
AHR and CD38 in sterile inflammation, obesity-mediated nonalcoholic fatty liver disease (NAFLD) as example
Obesity is a worldwide complex health problem. It is mainly produced by an imbalance between food intake and energy expenditure, which leads to an excessive accumulation of adipose tissue leading to dyslipidemia affecting many organs including the liver as the major organ responsible for lipid homeostasis (Jung and Choi 2014). Here, the discussion is focused on roles of AHR and CD38 in NAFLD and NASH (nonalcoholic steatohepatitis).
Roles of AHR
As discussed previously, AHR has been demonstrated to be involved in opposing processes (Bock 2020a). Sustained AHR activation by TCDD or in constitutively active AHR-expressing transgenic mice sensitizes mice to NASH (He et al. 2013). However, transient AHR activation has also been demonstrated to be involved in beneficial effects: Murine AHR activation by ß-naphthoflavone is involved in decreased lipogenesis (Alexander et al. 1998; Tanos et al. 2012). AHR activation by multiple agents and factors including quercetin and indigo has been demonstrated to prevent NASH (Yang et al. 2019; Lin et al. 2019). Activation of the Notch signaling pathway indirectly activates AHR (Alam et al. 2010; Lee et al. 2012) leading to enhanced anti-inflammatory IL-22 secretion in ILC3 and Th22 cells (Fig. 3). Effects of IL-22 have been comprehensively reviewed (Gulhane et al. 2016; Mizoguchi et al. 2018).
Fig. 3.
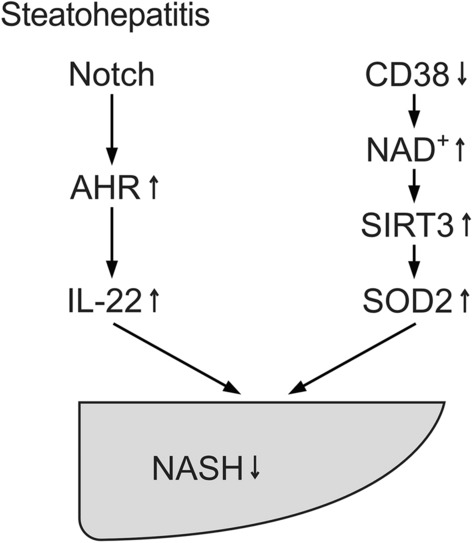
Discussed AHR- and CD38-mediated pathways attenuating steatohepatitis. AHR activation may lead to generation of anti-inflammatory IL-22. Inhibition of CD38 increases NAD+, leading to increased SIRT3 and SOD2 activity. SOD2 detoxifies reactive oxygen species (ROS) and thereby decreases nonalcoholic steatohepatitis (NASH). SOD superoxide dismutase
Notably, although beneficial effects of AHR may be achieved in isolated experimental models, sustained TCDD-activated AHR always bears the risk of facilitating chronic inflammatory responses. Interestingly, there is also a connection of fatty liver with vitamin B12 and folic acid as AHR antagonists: Mice pretreated with these vitamins prevented fatty liver produced by long-term treatment with TCDD (Kim et al. 2020).
Roles of CD38
CD38 is necessary for high-fat diet-induced NAFLD (Barbosa et al. 2007). Hence, inhibition of CD38 may prevent NASH: Inhibition of CD38 leads to increased NAD+ levels (Escande et al. 2013). SIRT3 and SOD2 activity are enhanced, and oxidative stress and NASH may be attenuated (Fig. 3) (Qiu et al. 2018; Camacho-Pereira et al. 2016). Furthermore, the CD38 component of the discussed signalsome (Bunaciu et al. 2019) may contribute to the signalsome’s proposed integrating function of energy economy during inflammatory responses.
Modulation of AHR signaling and NAD+ homeostasis may provide therapeutic options. Transient AHR activation by multiple agents (including quercetin and indigo) may stimulate anti-inflammatory responses and prevent NASH (Yang et al. 2019; Lin et al. 2019). Similarly, dietary indol-3-carbinol-generating phytochemicals (Diaz-Diaz et al. 2016) and indole-3-acetate-generated microbial products have been demonstrated to stimulate anti-inflammatory responses (Krishnan et al. 2018; Scott et al. 2020). Recently, phytochemicals and bacterial indole derivatives including indole-3-carboxaldehyde have been suggested to improve intestinal integrity via the AHR and Il-10 by stimulating antimicrobial peptide generation and goblet cell development (Powell et al. 2020). These indole derivatives may also limit age-associated systemic inflammation. Nevertheless, as comprehensively discussed (Smirnova et al. 2016), there is no consensus about direct and indirect AHR ligands operating within particular cells and in particular contexts. In addition to AHR modulation, search for NAD-boosting molecules including nicotinamide riboside (NR) may synergistically attenuate inflammatory responses (Rajman et al. 2018). NR enhances oxidative metabolism and protects against high-fat diet-induced obesity via enhancing SIRT1 and SIRT3 activation (Canto et al. 2012). Safety and metabolism of NR has been tested in a clinical trial (Conze et al. 2019). In addition, NR depressed levels of circulating inflammatory cytokines in aged adults (Dollerup et al. 2018).
AHR is involved in IDO-kynurenine-mediated tolerance and immunosuppression (Bessede et al. 2014). In this way, AHR is involved in cancer cell-mediated immunosuppression, PD-1 up-regulation in CD8+ T cells, and resistance to immune checkpoint inhibitors in different IDO-1 overexpressing cancer types. In these conditions it has been demonstrated that blockade of AHR would overcome this limitation (Campesato et al. 2020). Hence, anti-inflammatory AHR activation may not be justified in cancer patients. A similar note of caution is warranted in chronic inflammatory disease leading to autoimmunity.
TCDD toxicity models in sensitive species providing hints to molecular AHR targets of energy metabolism
TCDD toxicity is known to be species dependent leading in sensitive species to wasting syndrome and lethality (Poland and Knutson 1982). Early studies suggested that TCDD reduced gluconeogenic enzymes in liver including PEPCK (phosphoenolpyruvate carboxykinase) (Weber et al. 1991). TCDD toxicity sensitive and resistant rat strains were also identified (Viluksela et al. 1999). Targets of TCDD toxicity have been further identified in the chick embryo model: AHR-regulated PARP7/TiPARP was found to decrease NAD+ levels and suppress gluconeogenesis by decreasing the expression of PEPCK and its coactivator PGC1α due to decreased SIRT1-mediated ADP-ribosylation of these genes. Addition of nicotinamide, the major substrate of the NAD+ salvage pathway, corrected these effects (Diani-Moore et al. 2010, 2013). NAD+ loss was found to be attributable to increased PARP activity in thymus and liver, leading to thymus atrophy and hepatosteatosis (Diani-Moore et al. 2017). Effects of TCDD were found to be organ specific leading to decreased glycolytic enzymes and glucose transporters in thymus and increased glycolytic enzymes and glucose transporters in liver. In TCDD-treated primary cultures of human hepatocytes glycolysis and GLUT1 were also increased (Diani-Moore et al. 2020). These findings provide hints to molecular AHR targets of energy metabolism. However, many questions remain.
Conclusions
Accumulating evidence suggests that AHR is involved in infectious and sterile inflammatory responses. These processes may use up a considerable amount of energy. Therefore, energy metabolism has to be metabolically adapted (Medzhitov 2008; Wang et al. 2019). An excess of bactericidal ROS may lead to collateral host tissue injury and has to be strictly controlled. Metabolic adaptation appears to be achieved by NAD homeostasis (Verdin 2015; Yang and Sauve 2017; Hogan et al. 2019). In addition to NAD synthesis, NAD+ homeostasis appears to be regulated by NAD+-consuming enzymes including CD38, PARPs such as PARP1, PARP7/TiPARP and sirtuins such as SIRT1 and SIRT3. Roles of AHR and NAD-consuming enzymes have been discussed using infectious inflammation (bacterial infection) and sterile inflammation (nonalcoholic fatty liver disease) as examples.
Multiple sources of endogenous, phytochemical and microbial AHR ligands have been identified allowing intense interaction of the receptor and sensor between barrier organs and the environment. Connections between AHR and NAD+-consuming enzymes (PARP7/TiPARP, CD38 and sirtuins) have been demonstrated. In addition, both AHR and CD38 are constituents of a signalsome of neutrophils (Bunaciu et al. 2019) that is probably involved in metabolic adaptation between inflammation and associated energy demands.
Roles of AHR and NAD+-consuming enzymes in tissue-dependent inflammatory diseases have been discussed using two examples: infectious colitis and obesity-mediated NAFLD and NASH. In intestinal inflammation, beneficial effects have been discussed in homeostasis, microbial defense and the resolution phase of inflammation, provided that AHR is transiently activated. In NASH, both AHR and CD38 may be involved in anti-inflammatory processes. Therefore, both AHR-activating and CD38-inhibiting phytochemicals and microbial products as well as NAD+ boosting compounds may provide therapeutic options. However, anti-inflammatory tolerance mechanisms bear the risk to facilitate autoimmune diseases and carcinogenesis. Therefore, clinical studies have to evaluate risks and benefits in particular cases. The discussed perceptions may be applicable to inflammatory diseases of other organs including skin and lung.
TCDD toxicity is known to be species dependent leading to wasting syndrome and lethality in sensitive species (Poland and Knutson 1982). TCDD toxicity models in sensitive species, in particular in the chick embryo model, provide valuable hints to molecular AHR targets of energy metabolism. The results point to NAD+-dependent nuclear SIRT1 and mitochondrial SIRT3 as key regulators of gluconeogenesis and glycolytic enzymes. Hence, AHR research remains a challenging and promising field.
Acknowledgements
Valuable help of Thomas Staiger in preparing the figures is greatly appreciated.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Alam MS, Maekawa Y, Kitamura A, Tanigaki K, Yoshimoto T, Kishihara YK. Notch signaling drives IL-22 secretion in CD4+ T cells by stimulating the aryl hydrocarbon receptor. Proc Natl Acad Sci USA. 2010;107:5943–5948. doi: 10.1073/pnas.0911755107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander DL, Ganem LG, Fernandez-Salguero P, Gonzalez F. Aryl-hydrocarbon receptor is an inhibitory regulator of lipid and of commitment to adipogenesis. J Cell Sci. 1998;111:3311–3322. doi: 10.1242/jcs.111.22.3311. [DOI] [PubMed] [Google Scholar]
- Avilla MN, Malecki KMC, Hahn ME, Wilson RM, Bradfield CA. The Ah receptor: adaptive metabolism, ligand diversity, and the xenokine model. Chem Res Toxicol. 2020;33:860–896. doi: 10.1021/acs.chemrestox.9b00476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Mimura J, Nakamura N, Harada N, Yamamoto M, Morohashi K, Fujii-Kuriyama Y. Intrinsic function of the aryl hydrocarbon (dioxin) receptor as a key factor in female reproduction. Mol Cell Biol. 2005;25:10040–10051. doi: 10.1128/MCB.25.22.10040-10051.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa MTP, Soares SM, Novak CM, Sinclair D, Levine JA, Aksoy P, Chini EN. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB J. 2007;21:3629–3639. doi: 10.1096/fj.07-8290com. [DOI] [PubMed] [Google Scholar]
- Bertellier V, Tixier JM, Muller-Steffner H, Schuber F, Deterre P. Human CD38 is an authentic NAD(P) glycohydrolase. Biochem J. 1998;330:1383–1390. doi: 10.1042/bj3301383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessede A, Gargaro M, Pallotta MT, Matino D, Servillo G, Brunacci C, Bicciato S, Mazza EMC, Macchiarulo A, Vacca C, Ianniti R, Tissi L, Volpi C, Belladonna ML, Orabona C, Bianchi R, Lanz TV, Platten M, Della Facia MA, Piobbico D, Zelante T, Funakoshi H, Nakamura T, Gilot D, Denison MS, Guillemin GJ, DuHadaway JB, Prendergast GC, Metz R, Geffard M, Boon L, Pirro M, Iorio A, Veyret B, Romani L, Grohmann U, Fallino F, Puccetti P. Aryl hydrocarbon receptor control of a disease tolerance defense pathway. Nature. 2014;511:184–190. doi: 10.1038/nature13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blander JM, Longman RS, Iliev ID, Sonnenberg GF, Artis D. Regulation of inflammation by microbiota interactions with the host. Nat Immunol. 2017;18:851–860. doi: 10.1038/ni.3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock KW. Aryl hydrocarbon receptor (AHR) functions: balancing opposing processes including inflammatory reactions. Biochem Pharmacol. 2020;178:114093. doi: 10.1016/j.bcp.2020.114093. [DOI] [PubMed] [Google Scholar]
- Bock KW. Aryl hydrocarbon receptor (AHR)-mediated inflammation and resolution: non-genomic and genomic signalling. Biochem Pharmacol. 2020;182:114220. doi: 10.1016/j.bcp.2020.114220. [DOI] [PubMed] [Google Scholar]
- Bock KW, Siekevitz P, Palade GE. Localization and turnover studies of membrane nicotinamide adenine dinucleotide glycohydrolase in rat liver. J Biol Chem. 1971;246:188–195. doi: 10.1016/S0021-9258(18)62548-7. [DOI] [PubMed] [Google Scholar]
- Bunaciu RP, MacDonald RJ, Jensen HA, Gao F, Wang X, Johnson L, Varner JD, Yen A. Retinoic acid and 6-formylindolo[3,2-b]carbazole (FICZ) combination therapy reveals putative targets for enhancing response in non-APL AML. Leuk Lymphoma. 2019;60:1697–1708. doi: 10.1080/10428194.2018.1543880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho-Pereira J, Tarrogo MG, Chini CCS, Nin V, Escanda C, Warner GM, Puranik AS, Schoon RA, Reid JM, Galina A, Chini EN. CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab. 2016;23:1127–1139. doi: 10.1016/j.cmet.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campesato LF, Budhu S, Tchaicha J, Weng CH, Gigoux M, Cohen IJ, Redmond D, Mangarin L, Pourpe S, Liu C, Zappasodi R, Zamarin D, Cavanough J, Castro C, Manfredi MG, McGovern K, Merghoub T, Wolchok JD. Blockade of the AHR restricts a Treg-macrophage suppressive axis induced by L-kynurenine. Nat Commun. 2020;11:4011. doi: 10.1038/s41467-020-17750-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P, Gademann K, Rinsch C, Schoonjans K, Sauve AA, Auwerx J. The NAD+ precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012;15:838–847. doi: 10.1016/j.cmet.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Menzies KJ, Auwerx J. NAD+ metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 2015;22:31–53. doi: 10.1016/j.cmet.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HC, Guarente L. Sirt 1 and other sirtuins in metabolisms. Trends Endocinol Metab. 2014;25:138–145. doi: 10.1016/j.tem.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockayne DA, Muchamuel T, Grimaldi JC, Muller-Steffner H, Randall TD, Lund FE, Murray R, Schuber F, Howard MC. Mice deficient for the ecto-nicotinamide adenine dinucleotide glycohydrolase CD38 exhibit altered humoral immune responses. Blood. 1998;92:1324–1333. doi: 10.1182/blood.V92.4.1324. [DOI] [PubMed] [Google Scholar]
- Cohen MS. Interplay between compartmentalized NAD+ synthesis and consumption: a focus on the PARP family. Genes Dev. 2020;34:254–262. doi: 10.1101/gad.335109.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conze D, Brenner C, Kruger CL. Safety and metabolism of long-term administration of NIAGEN (nicotinamide riboside chloride) in a randomized, double-blind, placebo-controlled clinical trial of healthy overweight adults. Sci Rep. 2019;9:9772. doi: 10.1038/s41598-019-46120-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol. 2003;43:309–334. doi: 10.1146/annurev.pharmtox.43.100901.135828. [DOI] [PubMed] [Google Scholar]
- Diani-Moore S, Ram P, Li X, Mondal P, Youn DY, Sauve AA, Rifkind AB. Identification of aryl hydrocarbon receptor target gene TiPARP as mediator of suppression of hepatic gluconeogenesis by 2,3,7,8-tetrachlorodibenzo-p-dioxin and nicotinamide as a corrective agent for this effect. J Biol Chem. 2010;285:38801–38810. doi: 10.1074/jbc.M110.131573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diani-Moore S, Zhang S, Ram P, Rifkind AB. Aryl hydrocarbon receptor activation by dioxin targets phosphoenolpyruvate carboxykinase (PEPCK) for ADP-ribosylation via 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-inducible poly(ADP-ribose) polymerase (TiPARP) J Biol Chem. 2013;288:21514–21525. doi: 10.1074/jbc.M113.458067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diani-Moore S, Shoots J, Singh R, Zuk JB, Rifkind AB. NAD+ loss, a new player in AhR biology: prevention of thymus atrophy and hepatosteatosis by NAD+ repletion. Sci Rep. 2017;7:2268. doi: 10.1038/s41598-017-02332-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diani-Moore S, Marques Pedro T, Rifkind AB. Organ-specific effects on glycolysis by the dioxin-activated aryl hydrocarbon receptor. PLoS One. 2020;15:e0243842. doi: 10.1371/journal.pone.0243842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Diaz CJ, Ronnekleiv-Kelly SM, Nukaya M, Geiger PG, Hecht SS, Balbo S, Megna BW, Carney PR, Bradfield CA, Kennedy GD. The aryl hydrocarbon receptor mediates the chemopreventive effect of indole-3-carbinol in an inflammation-associated colorectal tumorigenesis model. Ann Surg. 2016;264:429–436. doi: 10.1097/SLA.0000000000001874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dollerup OL, Christensen B, Svart M, Schmidt MS, Sulek K, Ringgaard S, Stodkilde-Jorgensen H, Moller N, Brenner C, Treeback JT, Jessen N. A randomized placebo-controlled clinical trial of nicotinamide riboside in obese men: safety, insulin-sensitivity, and lipid-mobilizing effects. Amer J Clin Nutrition. 2018;108:343–353. doi: 10.1093/ajcn/nqy132. [DOI] [PubMed] [Google Scholar]
- Escande C, Nin V, Price NL, Capellini V, Gomes AP, Barbosa MT, O'Neil L, White TA, Sinclaire DA, Chini EN. Flavonoid apigenin is an inhibitor of the NAD+ase CD38. Diabetes. 2013;62:1084–1093. doi: 10.2337/db12-1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser C, Rannug A. The aryl hydrocarbon receptor in barrier organ physiology, immunology and toxicology. Physiol Rev. 2015;67:259–279. doi: 10.1124/pr.114.009001. [DOI] [PubMed] [Google Scholar]
- Fehr AR, Singh SA, Kerr CM, Mukai S, Higashi H. The impact of PARPs and ADP-ribosylation on inflammation and host-pathogen interactions. Genes Dev. 2021;34:341–359. doi: 10.1101/gad.334425.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasiewicz TA, Singh KP, Bennett JA. The Ah receptor in stem cell cycling, regulation, and quiescence. Ann NY Acad Sci USA. 2014;1310:1–7. doi: 10.1111/nyas.12361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu YZ, Hogenesch JB, Bradfield CA. The PAS superfamily: sensors of environmental and developmental signals. Annu Rev Pharmacol Toxicol. 2000;40:519–561. doi: 10.1146/annurev.pharmtox.40.1.519. [DOI] [PubMed] [Google Scholar]
- Gulhane M, Murray L, Lourie R, Tong H, Sheng YH, Wang R, Kang A, Schreiber V, Wong KY, Magor G, Denman S, Begun J, Florin TH, Perkins A, Cuiv PO, McGuckin MA, Hasnain SZ. High fat diets induce colonic epithelial stress and inflammation that is reversed by IL-22. Sci Rep. 2016;6:28990. doi: 10.1038/srep28990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupte R, Liu Z, Kraus WL. PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev. 2018;31:101–126. doi: 10.1101/gad.291518.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Hu B, Shi X, Weidert ER, Lu P, Xu M, Huang M, Kelley EE, Xie W. Activation of the aryl hydrocarbon receptor sensitizes mice to nonalcoholic steatohepatitis by deactivating mitochondrial sirtuin deacetylase Sirt3. Mol Cell Biol. 2013;33:2047–2055. doi: 10.1128/MCB.01658-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heer CD, Sanderson DJ, Voth LS, Alhammad YMO, Schmidt MS, Trammell SAJ, Perlman S, Cohen MS, Fehr AR, Brenner C. Coronavirus infection and PARP expression dysregulates the NAD metabolome: an actional component of innate immunity. J Biol Chem. 2020;295:17986–17996. doi: 10.1074/jbc.RA120.015138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan KA, Chini CCS, Chini EN. The multi-faceted ecto-enzyme CD38: roles in immunomodulation, cancer, aging and metabolic diseases. Frontiers Immunol. 2019;10:1187. doi: 10.3389/fimmu.2019.01187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horenstein AL, Bracci C, Morandi F, Malavasi F. CD38 in adenosinergic pathways and metabolic re-programming in human multiple myeloma cells: in-tandem insights from basic science to therapy. Front Immunol. 2019;10:760. doi: 10.3389/fimmu.2019.00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou X, Xu S, Maitland-Toolan KA, Sato K, Jiang B, Ido Y, Lan F, Walsh K, Wiezbicki M, Verbeuren TJ, Cohen RA, Zang M. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J Biol Chem. 2008;283:20015–20026. doi: 10.1074/jbc.M802187200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikuta T, Namiki T, Fujii-Kuriyama Y, Kawajiri K. AhR protein trafficking and function in the skin. Biochem Pharmacol. 2009;77:588–596. doi: 10.1016/j.bcp.2008.10.003. [DOI] [PubMed] [Google Scholar]
- Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science. 2017;356:513–519. doi: 10.1126/science.aal3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin D, Liu HX, Hirai H, Torashima T, Nagai T, Lopatina O, Shnayder NA, Yamada K, Noda M, Seike T, Fujita K, Takasawa S, Yokoyama S, Koizumi K, Shiraishi Y, Tanaka S, Hashii M, Yoshihara T, Higashida K, Islam MS, Yamada N, Hayashi K, Noguchi N, Kato I, Okamoto H, Matsishima A, Salmina A, Munesue T, Shimizi N, Mochda S, Asano M, Higashida H. CD38 is critical for social behaviour by regulating oxytocin secretion. Nature. 2007;446:41–45. doi: 10.1038/nature05526. [DOI] [PubMed] [Google Scholar]
- Jung UJ, Choi MS. Obesity and its metabolic complications: the role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int J Mol Sci. 2014;15:6184–6223. doi: 10.3390/ijms15046184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karchner SI, Jenny MJ, Tarrant AM, Evans BR, Kang HJ, Bae I, Sherr DH, Hahn ME. The active form of human aryl hydrocarbon receptor (AHR) repressor lacks exon 8, and its Pro185 and Ala185 variants repress both AHR and hypoxia-inducible factor. Mol Cell Biol. 2009;29:3465–3477. doi: 10.1128/MCB.00206-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- Kim DJ, Venkataraman A, Jain PC, Wiesler EP, DeBlasio M, Klein J, Tu SS, Lee S, Medzhitov R. Vitamin B12 and folic acid alleviate symptoms of nutritional deficiency by antagonizing aryl hydrocarbon receptor. Proc Natl Acad Sci USA. 2020;117:15837–15845. doi: 10.1073/pnas.2006949117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura A, Abe H, Tsuruta S, Chuba S, Fujii-Kuriyama Y, Sekiya T, Morita R, Yoshimura A. Aryl hydrocarbon receptor protects against bacterial infection by promoting macrophage survival and reactive oxygen production. Int J Immunol. 2013;26:209–220. doi: 10.1093/intimm/dxt067. [DOI] [PubMed] [Google Scholar]
- Kiss EA, Vornarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C, Diefenbach A. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science. 2011;334:1561–1565. doi: 10.1126/science.1214914. [DOI] [PubMed] [Google Scholar]
- Ko CI, Fan Y, De Gannes M, Wang Q, Xia Y, Puga A. Repression of aryl hydrocarbon receptor is required to maintain mitotic progression and prevent loss of pluripotency of embryonic stem cells. Stem Cells. 2016;34:2825–2839. doi: 10.1002/stem.2456. [DOI] [PubMed] [Google Scholar]
- Krishnan S, Ding Y, Saedi N, Choi M, Sridharan GV, Sherr DH, Yarmush M, Alaniz RC, Jayaraman A, Lee K. Gut microbiota-derived tryptophan metabolites modulate inflammatory response in hepatocytes and macrophages. Cell Rep. 2018;23:1099–1111. doi: 10.1016/j.celrep.2018.03.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamas B, Natividad JM, Sokol H. Aryl hydrocarbon receptor and intestinal immunity. Mucosal Immunol. 2018;11:1024–1038. doi: 10.1038/s41385-018-0019-2. [DOI] [PubMed] [Google Scholar]
- Lawrence BP, Vorderstrasse BA. New insights into the aryl hydrocarbon receptor as a modulator of host responses to infection. Semin Immunopathol. 2013;35:615–625. doi: 10.1007/s00281-013-0395-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HC, Zhao YJ. Resolving the topological enigma in Ca2+-signaling by cyclic ADP-ribose and NAADP. J Biol Chem. 2019;294:19831–19843. doi: 10.1074/jbc.REV119.009635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Cella M, McDonald K, Garlanda C, Kennedy GD, Nukaya M, Mantovani A, Kopan R, Bradfield CA, Newberry RD, Colonna M. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol. 2012;13:144–151. doi: 10.1038/ni.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YH, Luck H, Khan S, Schneeberger PHH, Tsai S, Clemente-Casares X, Lei H, Leu YL, Chan YT, Chen HY, Yang SH, Coburn B, Winer S, Winer DA. Aryl hydrocarbon receptor agonist indigo protects against obesity-related insulin resistance through modulation of intestinal and metabolic tissue immunity. Int J Obes. 2019;43:2407–2421. doi: 10.1038/s41366-019-0340-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TF, Yoza BK, El Gazzar M, Vachharajani VT, McCall CE. NAD+-dependent SIRT1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance. J Biol Chem. 2011;286:9856–9864. doi: 10.1074/jbc.M110.196790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luongo TS, Eller JM, Lu MJ, Niere M, Raith F, Perry C, Bornstein MR, Oliphint P, Wang L, McReynolds MR, Migaud ME, Rabinowitz JD, Johnson FB, Johnsson K, Ziegler M, Cambronne XA, Baur JA. SLC25A51 is a mammalian mitochondrial NAD+ transporter. Nature. 2020;588:174–179. doi: 10.1038/s41586-020-2741-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson L, Tamblyn L, Rajendra S, Bralha F, McPherson JP, Matthews J. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-inducible poly(ADP-ribose) polymerase (TiPARP, ARTD14) is a mono-ADP-ribosyltransferase and repressor of aryl hydrocarbon receptor transactivation. Nucleic Acid Res. 2013;41:1604–1621. doi: 10.1093/nar/gks1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura F. The significance of the nongenomic pathway in mediating signaling of the dioxin-activated Ah receptor to cause toxic effects. Biochem Pharmacol. 2009;77:608–626. doi: 10.1016/j.bcp.2008.10.013. [DOI] [PubMed] [Google Scholar]
- Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- Mizoguchi A, Yano A, Himuro H, Ezaki Y, Sadanaga T, Mizoguchi E. Clinical importance of IL-22 cascade in IBD. J Gastroenterol. 2018;53:465–474. doi: 10.1007/s00535-017-1401-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moura-Alves P, Fae K, Houthouys E, Dorhoi A, Kreuschwig A, Furkert J, Barison N, Diehl A, Munder A, Constant P, Skrahina T, Guhlich-Bornhof U, Klemm M, Koehler AB, Bandermann S, Goosmann C, Mollenkopf HJ, Hurwitz R, Brinkmann V, Fillatreu S, Daffe M, Tümmler B, Kolbe M, Oschkinat H, Krause G, Kaufmann SHE. AhR sensing of bacterial pigments regulate antibacterial defence. Nature. 2014;512:387–392. doi: 10.1038/nature13684. [DOI] [PubMed] [Google Scholar]
- Moura-Alves P, Puyskens A, Stinn A, Klemm M, Guhlich-Bornhof U, Dorhoi A, Furkert J, Kreuschwig A, Protze J, Lozza L, Pei G, Saikali P, Perdomo C, Mollenkopf HJ, Hurwitz R, Kirschhoefer F, Brenner-Weiss G, Weiner J, Oschkinat H, Kolbe M, Krause G, Kaufmann SHE. Host monitoring of quorum sensing during Pseudomonas aeroginosa infection. Science. 2019;366:1472. doi: 10.1126/science.aaw1629. [DOI] [PubMed] [Google Scholar]
- Murray IA, Perdew GH. Ligand activation of the Ah receptor contributes to gastrointestinal homeostasis. Curr Opin Toxicol. 2017;2:15–23. doi: 10.1016/j.cotox.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebert DW, Dalton TP, Okey AB, Gonzalez FJ. Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J Biol Chem. 2004;279:23847–23850. doi: 10.1074/jbc.R400004200. [DOI] [PubMed] [Google Scholar]
- Nguyen LP, Bradfield CA. The search for endogenous activators of the aryl hydrocarbon receptor. Chem Res Toxicol. 2008;21:102–116. doi: 10.1021/tx7001965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtake F, Fujii-Kuriyama Y, Kato S. AhR acts as an E3 ubiquitin ligase to modulate steroid receptor functions. Biochem Pharmacol. 2009;77:474–484. doi: 10.1016/j.bcp.2008.08.034. [DOI] [PubMed] [Google Scholar]
- Partida-Sanchez S, Cockayne DA, Monard S, Jacobson EL, Oppenheimer N, Garvy B, Kusser K, Goodrich S, Howard M, Harmsen A, Randall TD, Lund FE. Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat Med. 2001;7:1209–1216. doi: 10.1038/nm1101-1209. [DOI] [PubMed] [Google Scholar]
- Poland A, Knutson JC. 2,3,7,8-Tetrachlorodibenzo-p-dioxin and related hagenated aromatic hydrocarbons: examination of the mechanisms of toxicity. Annu Rev Pharmacol Toxicol. 1982;22:517–554. doi: 10.1146/annurev.pa.22.040182.002505. [DOI] [PubMed] [Google Scholar]
- Pombo M, Lame MW, Walker NJ, Huynh DH, Tablin F. TCDD and omeprazole prime platelets through the aryl hydrocarbon receptor (AhR) non-genomic pathway. Toxicol Lett. 2015;235:28–36. doi: 10.1016/j.toxlet.2015.03.005. [DOI] [PubMed] [Google Scholar]
- Powell DN, Swimm A, Sonowal R, Bretin A, Gewirtz AT, Jones RM, Kalman D. Indoles from the commensal microbiota act via the AHR and IL-10 to tune the cellular composition of the colonic epithelium during aging. Proc Natl Acad Sci USA. 2020;117:21519–21526. doi: 10.1073/pnas.2003004117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puga A, Hoffer S, Zhou S, Bohm JM, Leikauf GD, Shertzer HG. Sustained increase in intracellular free calcium and activation of cyclooxygenase-2 expression in mouse hepatoma cells treated with dioxin. Biochem Pharmacol. 1997;54:1287–1296. doi: 10.1016/S0006-2952(97)00417-6. [DOI] [PubMed] [Google Scholar]
- Puga A, Ma C, Marlowe JL. The aryl hydrocarbon receptor cross-talks with multiple signal transduction pathways. Biochem Pharmacol. 2009;77:713–722. doi: 10.1016/j.bcp.2008.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Guo X, Chen ZE, He L, Sonnenberg GF, Artis D, Fu YX, Zhou L. Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity. 2013;39:386–399. doi: 10.1016/j.immuni.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Caloric restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2018;12:662–667. doi: 10.1016/j.cmet.2010.11.015. [DOI] [PubMed] [Google Scholar]
- Rajman L, Chwalek K, Sinclair DA. Therapeutic potential of NAD-boosting molecules: the in vivo evidence. Cell Metab. 2018;27:529–547. doi: 10.1016/j.cmet.2018.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1a and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- Rothhammer V, Quintana FJ. The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nature Rev Immunol. 2019;19:184–196. doi: 10.1038/s41577-019-0125-8. [DOI] [PubMed] [Google Scholar]
- Roy J, Galano JM, Durand T, Le Guennec JY, Lee JCY. Physiological role of reactive oxygen species as promoters of natural defenses. FASEB J. 2017;31:3729–3745. doi: 10.1096/fj.201700170R. [DOI] [PubMed] [Google Scholar]
- Saurat JH, Kaya G, Saxer-Sekulic N, Pardo B, Becker M, Fantao L, Mottu F, Caraux P, Pham XC, Parde C, Fontao F, Zennec M, Schmid P, Schaad O, Descombes P, Sorg O. The cutaneous lesions of dioxin exposure: lessons from the poisoning of Victor Yushchenko. Toxicol Sci. 2012;125:310–317. doi: 10.1093/toxsci/kfr223. [DOI] [PubMed] [Google Scholar]
- Schiering C, Wincent E, Mediji A, Iseppon A, Li Y, Potcnik AJ, Omenetti S, Henderson CJ, Wolf CR, Nebert DW, Stockinger B. Feedback control of AHR signalling regulates intestinal immunity. Nature. 2017;542:242–245. doi: 10.1038/nature21080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott SA, Fu J, Chang PV. Microbial tryptophan metabolites regulate gut barrier function via the aryl hydrocarbon receptor. Proc Natl Acad Sci USA. 2020;117:19376–19387. doi: 10.1073/pnas.2000047117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh CK, Chhabra G, Ndiaye MA, Garcia-Peterson LM, Mack NJ, Ahmad N. The role of sirtuins in antioxidant and redox signaling. Antioxid Redox Signal. 2018;28:643–661. doi: 10.1089/ars.2017.7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova A, Wincent E, Vikström Bergander L, Alsberg T, Bergman J, Rannug A, Rannug U. Evidence for new light-independent pathways for generation of the endogenous aryl hydrocarbon receptor agonist FICZ. Chem Res Toxicol. 2016;29:75–86. doi: 10.1021/acs.chemrestox.5b00416. [DOI] [PubMed] [Google Scholar]
- Sonnenberg GF, Artis D. Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat Medicine. 2015;21:698–708. doi: 10.1038/nm.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockinger B, Di Meglio P, Gialitakis M, Duarte JH. The aryl hydrocarbon receptor: multitasking in the immune system. Annu Rev Immunol. 2014;32:403–432. doi: 10.1146/annurev-immunol-032713-120245. [DOI] [PubMed] [Google Scholar]
- Tanos R, Murray IA, Smith PB, Patterson A, Perdew GH. Role of the Ah receptor in homeostatic control of fatty acid synthesis. Toxicol Sci. 2012;129:372–379. doi: 10.1093/toxsci/kfs204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vachharajani V, McCall CE. Epigenetic and metabolic programming of innate immunity in sepsis. Innate Immun. 2019;25:267–279. doi: 10.1177/1753425919842320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin E. NAD+ in aging, metabolism, and neurodegeneration. Science. 2015;340:1208–1213. doi: 10.1126/science.aac4854. [DOI] [PubMed] [Google Scholar]
- Viluksela M, Unkila M, Pohjanvirta R, Tuomisto JT, Stahl BU, Rozman KK, Tuomisto J. Effects of 2,3,7.8-tetrachlorodibenzo-p-dioxin (TCDD) on liver phosphoenolpyruvate carboxykinase (PEPCK) activity, glucose homeostasis and plasma amino acid concentrations in the most TCDD-susceptible and the most TCDD-resistant rat strains. Arch Toxicol. 1999;73:323–336. doi: 10.1007/s002040050626. [DOI] [PubMed] [Google Scholar]
- Vogel CFA, Khan EM, Leung PSC, Me G, Chang WLW, Wu D, Haarmann-Stemmann T, Hoffmann A, Denison MS. Cross-talk between aryl hydrocarbon receptor and the inflammatory response, a role for nuclear factor-kB. J Biol Chem. 2014;289:1866–1875. doi: 10.1074/jbc.M113.505578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada T, Sunuga H, Ohkawara R, Shimba S. Aryl hydrocarbon receptor modulates NADPH oxidase activity via direct transcriptional regulation of p40phox expression. Mol Pharmacol. 2013;83:1133–1140. doi: 10.1124/mol.112.083303. [DOI] [PubMed] [Google Scholar]
- Wang A, Luan HH, Medzhitov R. An evolutionary perspective on immunometabolism. Science. 2019;363:140. doi: 10.1126/science.aar3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber LW, Lebofsky M, Greim H, Rozman K. Key enzymes of gluconeogenesis are dose-dependently reduced in 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-treated rats. Arch Toxicol. 1991;65:119–123. doi: 10.1007/BF02034937. [DOI] [PubMed] [Google Scholar]
- Yang Y, Sauve AA. NAD+ metabolism: bioenergetics, signaling and manipulation for therapy. Biochim Biophys Acta. 2016;1864:1787–1800. doi: 10.1016/j.bbapap.2016.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Yang T, Heng C, Zhou Y, Jiang Z, Qian X, Du L, Mao S, Yin X, Lu Q. Quercetin improves nonalcoholic fatty liver by ameliorating inflammation, oxidative stress, and lipid metabolism in db/db mice. Phytotherapy Res. 2019;8:1–13. doi: 10.1002/ptr.6486. [DOI] [PubMed] [Google Scholar]