

Abstract
Objective
In osteoarthritis (OA), a loss of healthy cartilage extracellular matrix (ECM) results in cartilage degeneration. Attracting chondrogenic progenitor cells (CPCs) to injury sites and stimulating them toward chondrogenic expression profiles is a regenerative approach in OA therapy. High mobility group box 1 protein (HMGB1) is associated with chemoattractant and proinflammatory effects in various pathological processes. Here, we investigate the migratory effects of HMGB1 in knee OA and CPCs for the first time.
Design
Immunohistochemistry, immunoblotting, and immunocytochemistry were performed to identify HMGB1 and its receptors, receptor for advanced glycation end products (RAGE) and toll-like receptor 4 (TLR4) in OA knee tissue, chondrocytes, and CPCs. In situ hybridization for HMGB1 mRNA was performed in CPCs ex vivo. The chemoattractant effects of HMGB1 on CPCs were analyzed in cell migration assays.
Results
HMGB1 expression in OA tissue and OA chondrocytes was higher than in healthy specimens and cells. HMGB1, RAGE, and TLR4 were expressed in CPCs and chondrocytes. In situ hybridization revealed HMGB1 mRNA in CPCs after migration into OA knee tissue, and immunohistochemistry confirmed HMGB1 expression at the protein level. Stimulation via HMGB1 significantly increased the migration of CPCs.
Conclusions
Our results show the chemoattractant role of HMGB1 in knee OA. HMGB1 is released by chondrocytes and has migratory effects on CPCs. These effects might be mediated via RAGE and TLR4. The in vitro and ex vivo results of this study need to be confirmed in vivo.
Keywords: HMGB1, RAGE, TLR4, chondrogenic progenitor cells, osteoarthritis, cell migration
Introduction
Osteoarthritis (OA) is a proliferative joint disease that severely restricts the quality of life and is becoming an increasing economic burden, as it is associated with aging and obesity.1,2 Chondrocytes are the dominant cells in the articular cartilage and facilitate cartilage homeostasis.3 During the pathogenesis of OA, mechanical stress by abnormal loading, cartilage aging processes or trauma can cause dysregulation of chondrocytes.4-6 As a result, extracellular matrix (ECM) stability is disturbed and leads to a loss of aggrecan and type II collagen.7,8 Although OA is often described as a degenerative disease, it also comprises inflammatory components, as it is driven by inflammatory mediators.9 Eventually, OA is characterized by articular cartilage degeneration, synovitis and changes in the periarticular and subchondral bone.10,11 As OA cartilage tissue shows little capacity for self-renewal, biological research aims to improve its regenerative potential. Recently, cells with a multipotent differentiation capacity, so-called chondrogenic progenitor cells (CPC), were isolated from repaired human OA knee tissue. CPCs exhibit stem cell characteristics such as clonogenicity, multipotency, and migratory activity. CPCs are controlled by the major transcription factors SOX9 and RUNX2. Therefore, they are able to produce a repair tissue in the diseased OA cartilage.12 Attracting CPCs to sites of OA injury and influencing them toward the expression of healthy cartilage ECM might be crucial for new OA treatment strategies. In cattle, one of the chemoattractant proteins responsible for the migratory effects of progenitor cells toward cartilage injury sites is high mobility group box 1 protein (HMGB1).13 HMGB1 is a highly conserved, ubiquitous nonhistone protein with a variety of functions inside and outside of the cell.14-16 For example, nuclear HMGB1 supports nucleosomal structure and stability, supports the binding of transcription factors to their associated DNA,17 enhances DNA transcription,18,19 and is associated with tumor growth.20,21 Although, HMGB1 is primarily located in the nucleus, it can translocate to the cytoplasm and can be secreted into the surrounding ECM during cell activation and cell death.22 Moreover, active release of HMGB1 by macrophages and monocytes is induced by oxidative stress, endotoxins, or proinflammatory cytokines.23-26 Necrotic or injured cells can passively release HMGB1.27 Extracellular HMGB1 is associated with chemoattractant and inflammatory signaling: in endochondral ossification, HMGB1 is released from differentiating chondrocytes; subsequently, HMGB1 regulates endochondral ossification by attracting osteoblasts, osteoclasts, and endothelial cells.28 In endotoxemia, hemorrhagic shock and arthritis, extranuclear expression of HMGB1 is considerably increased.22,23,29,30 HMGB1 signaling via toll-like receptor 4 (TLR4) affects the inflammatory reaction of autoimmune myositis and contributes to the secretion of interleukin-6 (IL-6) and IL-8 in upper airway inflammation.31,32 The binding of HMGB1 to receptor for advanced glycation end products (RAGE) on monocytes triggers their transendothelial migration, which ultimately contributes to inflammation.26 In heart disease, diabetic retinopathy, and skeletal muscle inflammation, RAGE-dependent HMGB1 binding to tissue-specific cells causes inflammation.33-36 Also, in synovitis and arthritis, several authors suggest a role for HMGB1 and RAGE.37-41 Yet their particular functions in inflammation and cell migration in OA remain unclear and need further elucidation.
The present study shows the expression of HMGB1 and its associated receptors RAGE and TLR4 in chondrocytes and CPCs and demonstrates the migratory effects of HMGB1 on CPCs in various migration assays. For the first time, HMGB1 expression is shown in CPCs in vitro and after migration into OA tissue ex vivo. Understanding the expression and the effects of HMGB1, RAGE, and TLR4 on CPCs might be helpful for developing novel regenerative therapeutic strategies in OA.
Methods
Tissue Sources and Preparation
Tissue samples were obtained from the lateral condyle of knee joints from patients who underwent total knee replacement. Tissue samples were collected from a region directly adjacent to the main defect with grade 4.0 to 4.5 (Pritzker et al.42) and exhibited deep surface fissures and chondrocyte clusters with a loss of the superficial zone. The patients (n = 15; 7 males and 8 females), with a mean age of 67.5 years (range 60-75 years), met the American College of Rheumatology classification43 and gave their written informed consent, consistent with the ethical regulations of the Medical Faculty of the University of Gottingen (application number 25/12/10). Healthier specimens from 3 accident victims displayed macroscopically and histologically intact hyaline cartilage with a smooth surface and a grade 0 to 1.42
Cell Isolation, Cell Culture, and Immortalization
Our work group has established a stock of more than 600 cell lines obtained by own production. They are named in continuous order according to the patient numbers; in this study, we used the numbers to distinguish between the different cell lines. Cell isolation was performed as described elsewhere12: CPCs (CPC241ht, CPC242ht, CPC531ht) were obtained using 8- to 15-mm3 tissue specimens taken from areas adjacent to the main defect. After 10 days, outgrown cells were harvested and 103 cells/cm2 transferred to a monolayer culture in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (GIBCO, lot number 41F2061K), supplemented with penicillin/streptomycin (50,000 U/50 mg) and l-glutamine (10 mM), and cultured under standard conditions. Differentiation and characterization of the used CPCs was described by Koelling et al.12
Chondrocytes from healthy (CH292ht) and OA (CH656-OA, CH657-OA) cartilage tissue samples were harvested after digestion for 6 hours at 37°C with the aid of collagenase I (152 U/mL; Invitrogen, Karlsruhe, Germany), collagenase II (280 U/mL; Biochrom, Berlin, Germany), and dispase (15 U/mL; Invitrogen). To eliminate cell matrix residues, the digested material was filtered through a 40-µm mesh sieve (BD Falcon, Heidelberg, Germany).
Immortalization of all cell lines was performed as follows.44
Virus Production
We seeded 5 × 105 293T-cells (ACC635; DSMZ) into a dish (diameter = 10 cm) and grew them to 80% confluence. At the next day, 10 μg lenti-plasmid-DNA of hTERT (customer order; amsbio) and 10 μg packing-plasmid-mix (LV053; abm) were mixed together with 1 ml DMEM. Furthermore, 80 μL lentifectin (G074; abm) with 1 ml DMEM were mixed. Both solutions rested for 5 minutes at room temperature, afterward they were mixed together to allow for the formation of the transfection complex. After 20 minutes, 4.5 mL DMEM was added to the transfection complex. The transfection complex mixture was pipetted to the cells and 0.65 mL heat inactivated fetal calf serum (FCS) was added after 6 hours. The next day, the medium was carefully removed from the cells and 10 mL DMEM or keratinocyte growth medium 2 + 10% heat-inactivated FCS + 1% bovine serum albumin (BSA) fraction V (BL63-0500; Equitech-Bio; Kerrville, TX, USA) was added. After 24 hours, the cells produced enough of the virus and the supernatant was harvested to get centrifuged and filtered (SLHA033SB; Merck Millipore, Burlington, MA, USA).
Transfection
A total of 1.8 × 105 freshly trypsinized cells were resuspended in 3 mL of the virus supernatant and 30 μL protaminsulfate (P3369; Sigma-Aldrich, St. Louis, MO, USA). Three wells of a 24-well plate were each filled with 1 mL of that solution. After 6 hours additionally, 1 mL of the medium was added to each well. The next day, the medium and the dead cells were removed, and the adhered cells received yet another treatment with 1 mL of virus supernatant together with 10 μL protaminsulfate per well over night.
Selection
The infected cells were transferred to a 75 cm2 flask and selected with up to 10 μg/mL of blasticidin.
For the performed experiments, cell lines were P7 to P10.
Green Fluorescent Protein Transfection
Green fluorescent protein (GFP) transfection of CPCs was performed using a human MSC Nucleofector Kit (VPE-1001, Lonza, Cologne, Germany). CPCs (5 × 105) were transfected with 2.5 μg of the vector pmaxGFP in 100 μL of Nucleofector solution using Nucleofector program U-23. Immediately afterward, the cells were transferred to DMEM supplemented with 20% FCS, incubated overnight, and cultured under standard conditions. The transfection efficiency reached approximately 75%. Fluorescence-activated cell sorting (FACS) enriched GFP-positive CPCs to greater than 99%. Before performing the ex vivo assay, sorted cells were cultured under standard conditions.
Migration Assays
Scratch Assay
Six-well plates were marked with 3 different reference lines that defined the survey area. The culture plates were coated with 1 mL of fibronectin (10 μg/mL) for 12 hours at 4°C; each well was blocked with 3 mL of BSA (2 mg/mL) at 37°C before 5 × 104 CPCs/well were seeded. The chemoattractant lipopolysaccharide (LPS)-free HMGB1 (10 or 50 ng/mL; HM-122, HMGBiotech, Milan, Italy) was added. CPCs without added HMGB1 served as controls. Scratches were executed perpendicularly to the defined survey area with the tip of a pipette following an established protocol45 to create standardized gaps within the cell monolayer. Confluence of the gap was determined visually under a light microscope after 0, 12, 48, and 72 hours.
Boyden Chamber Assay
Cell migration was performed in a Boyden chamber (CBA-100, Cell Biolabs, San Diego, CA, USA).46 Briefly, 5 × 104 CPCs were added to the upper transwell; the chamber was placed in serum-free medium alone or in medium with the chemoattractant LPS-free HMGB1. Three different concentrations of HMGB1 (10, 50, and 100 ng/mL) were tested. Micro-membranes with a defined pore size (8 μm) ensured an active migration from the upper transwell to the medium. Plates were incubated for 24 hours prior to processing at 37°C. Cells were detached with trypsin EDTA (ethylenediaminetetraacetic acid) and transferred to another 24-well plate containing serum-free medium with 8 μmol/L calcein-AM (calcein-acetoxymethyl ester) and incubated for 45 minutes. Fluorescence analysis was performed using a multimode microplate reader at a wavelength of 560 nm (SpectraMax M2, Molecular Devices, Sunnyvale, CA, USA). Absolute fluorescence values correlate linearly to the amount of living migrated cells.
Ex Vivo Assay
Samples of diseased tibia from 9 patients (69-79 years old) with OA who underwent total knee replacement were obtained using commercial arc punches; no bone tissue was included. Stainless steel barrels with a diameter of 10 mm and a height of 15 mm provided a vessel for cartilage specimens and GFP-labeled CPCs for an aligned migration within the reservoir. Barrels containing the specimens were transferred to 6-well plates containing DMEM with 10% FCS supplemented with gentamycin (50 μg/mL), and cells were placed on top using a sterile micropipette. Migration was carried out for 5 days under standard conditions. Specimens were released from the steel barrels and washed with phosphate buffered saline (PBS) before being transferred to flasks containing 4% paraformaldehyde. Fixation was carried out for 6 hours at 4°C. Specimens were dehydrated for 8 hours in 70% ethanol. Visualization was carried out via fluorescence microscopy.
Antibodies
A rabbit polyclonal anti-HMGB1 antibody (ab18256, Abcam, Cambridge, UK), a rabbit polyclonal anti-RAGE antibody (R12-2327; Assay Biotechnology, Fremont, CA, USA), a mouse monoclonal anti-TLR4 antibody (14-9917-80; Thermo Fisher Scientific, Waltham, MA, USA), a mouse monoclonal anti-GFP antibody (sc-9996; Santa Cruz Biotechnology, Dallas, TX, USA), a monoclonal mouse anti-α-tubulin antibody (T6199, Sigma-Aldrich, Steinheim, Germany) and a goat anti-DIG antibody (CU-3210-0488-IgPG, Biogenesis, Poole, UK) were used as primary antibodies. All antibodies have been demonstrated to be specific for each protein.47-53
Immunoblotting
A total of 1.5 × 105 cells were dissolved in 30 µL 3 × sodium dodecyl sulfate (SDS) with 10% β-mercaptoethanol and heated at 95°C for 5 minutes. SDS–polyacrylamide gel electrophoresis (PAGE) was performed with 5% acrylamide in the stacking gel and 15 % in the separation gel. After SDS-PAGE, the separated proteins were blotted on an Immobilon-P Transfer Membrane (PVH07850; Merck Millipore). General detection of the proteins was performed with Coomassie blue staining. After destaining, the membrane was blocked with 5% milk in TRIS-buffered saline with TWEEN (TBS-T) for 1 hour, followed by 5 washing steps with TBS-T. Then, the primary antibodies were dissolved in 5% milk in TBS-T, according to the dilution instructions provided by the manufacturer, and incubated for 12 hours at 4°C. Again, 5 washing steps were performed. Then, secondary antibodies (anti-rabbit-IgG-PO, A0545, Sigma-Aldrich; anti-mouse-IgG-PO, A9917, Sigma-Aldrich) were incubated for 1 hour at room temperature, followed by 5 washing steps. Proteins were visualized by applying WesternBright Sirius HRP substrate (cat. R03027-D10; Advansta, Menlo Park, CA, USA). To control the immunoreactions of the Western blotting, membranes were incubated with the secondary antibody only.
Immunohistochemistry
Immunoperoxidase staining was performed on paraffin-embedded tissue sections as follows. Tissue samples were deparaffinized, rehydrated, and rinsed for 10 minutes in PBS. Endogenous peroxidase was blocked for 45 minutes with 3% H2O2/methanol in the dark. Each reaction was followed by rinsing for 10 minutes in PBS. The sections were pretreated for 5 minutes with 10 μg/mL protease XXIV (P8038, Sigma-Aldrich, St. Louis, MO, USA). The anti-HMGB1 antibody was applied at a dilution of 1:200 in PBS and the anti-GFP antibody at 1:100 for 12 hours at room temperature. For detection, a HiDef Detection Alk Phos Polymer System (962D, Cell Marque, Rocklin, CA, USA) or a HiDef Detection HRP System (954D, Cell Marque) was used. Intracellular detection of HMGB1 was accomplished via antigen retrieval with 1 mM citrate buffer (pH 6.0) for 10 minutes at 90°C. Negative controls were processed by treating the sections with swine serum instead of the rabbit polyclonal antibodies and with isotype-matched IgGs instead of the mouse monoclonal antibodies. Counterstaining was performed using Fast Green or Hemalaun.
Immunocytochemistry
Immunocytochemistry (ICC) was conducted as described elsewhere.12 Briefly, P1 cells were grown on coverslips, fixed, and incubated with 100 μL of primary antibody (1:50 dilution in PBS) for 1 hour at room temperature. As secondary antibodies, Alexa Fluor 555 donkey anti-rabbit IgG (ab150062, Abcam) was used to detect HMGB1 and RAGE, and Alexa Fluor 555 goat anti-mouse IgG (ab150114, Abcam) was used to detect TLR4. Dilution of the secondary antibodies was 1:500 in PBS, and staining was carried out for 20 minutes at room temperature. Cell nuclei were stained with DAPI in PBS (1:500) for 20 minutes at 37°C. Negative controls were treated with secondary antibody only.
In Situ Hybridization
In situ hybridization (ISH) was performed as described elsewhere.53 Briefly, probes were generated from DNA plasmids for the genes of interest using T7 (EP0111, Thermo Fisher Scientific) and Sp6 RNA polymerases (EP0131, Thermo Fisher Scientific). Cloning was carried out using the pGEM-T Easy vector kit (TM042, Promega, Fitchburg, WI, USA) and the restriction enzymes Spe1 (FD1254, Thermo Fisher Scientific) and Nco1 (FD0574, Thermo Fisher Scientific).
The following cDNA was used to generate antisense probes: HMGB1 forward primer 5′-AAGCACCCAGATGCTTCAGT and reverse primer 5′-GCAACATCACCAATGGACAG.
The hybridization solution contained 50% deionized formamide, tRNA (0.25 mg/mL), 1× Denhardt’s Solution (750018, Thermo Fisher Scientific), 10% dextran sulfate and 4× saline-sodium citrate (SSC). Cartilage tissue slides were incubated with digoxigenin (DIG)-labeled antisense probes (50-100 ng/μL), and hybridization was performed at 39°C for 16 hours. Posthybridization treatment consisted of washing with 5× SSC followed by 2× SSC and additional washing with PBS for 5 minutes. Detection was carried out with NBT/BCIP (11681451001; Sigma-Aldrich) as a substrate for anti-DIG-alkaline phosphatase (11093274910, Roche, Basel, Switzerland), and the hybridized probes were visualized using light microscopy. For confocal microscopy, the goat anti-DIG antibody (dilution 1:500 in PBS) and Alexa Fluor 555 donkey anti-goat IgG antibody (ab150130, Abcam) (dilution 1:500 in PBS) were used to detect HMGB1 mRNA.
Statistical Analysis
We reported representative data from at least 3 independent experiments and statistically tested our results using separate specimens. For immunoblotting, we performed 1-way analysis of variance (ANOVA) after testing for normal distribution; then we performed Tukey’s multiple comparison testing. For the scratch assay, cells were counted within defined survey areas using digital photographs of light microscopic magnifications and ImageJ 1.51 software. After testing the data for normal distribution, we performed 1-way ANOVA and, if significant differences were encountered, Tukey’s multiple comparison testing. For the Boyden chamber assay, we acquired representative data from 3 CPC cell lines and a total of 19 independent experiments. Since the data were not normally distributed, we determined the medians and performed Kruskal-Wallis testing. If significant differences were encountered, Mann-Whitney U testing was performed for multiple comparisons.
The analyses were executed using SPSS software (SPSS, IBM Corp, Armonk, NY, USA) and GraphPad Prism 7 (GraphPad Software, La Jolla, CA, USA). The results are reported as the mean values and standard deviations; P values <0.05 were considered statistically significant.
Results
IHC was performed to detect HMGB1 in human OA articular cartilage. OA specimens showed typical surface fissures and breaks in the tidemarks (Fig. 1a and c), whereas healthy specimens had smooth articular surfaces and intact tidemarks (Fig. 1b and d). HMGB1 was found in the nucleus, cytoplasm and the pericellular matrix of chondrocytes (Fig. 1a), as well as in the ECM (Fig. 1c). In healthy cartilage, HMGB1 was detectable in the nuclei of some chondrocytes, but most chondrocytes showed no HMGB1 expression (Fig. 1b and d).
Figure 1.

Light microscopic detection of HMGB1 via IHC (a-d) and fluorescence microscopic detection of HMGB1, RAGE, and TLR4 via ICC (e). (a and c) Chondrocytes in OA cartilage express HMGB1. Insets: Detail of chondrocyte clusters with HMGB1 expression in nucleus, cytoplasm, and pericellular regions; the arrow indicates a break in the tidemark. (b and d) HMGB1 expression in healthy cartilage is restricted to few chondrocytes. Insets: details of chondrocytes without HMGB1 expression. Scale bars shown in figure a are transferable to figures b, c, and d. (e) CPCs and chondrocytes express HMGB1, RAGE, and TLR4. Note that HMGB1 expression in CPCs is restricted to nuclear regions, whereas in chondrocytes it is expressed in cytoplasmic areas as well. The scale bar is transferable to all figures in e. OA = osteoarthritis; CPCs = chondrogenic progenitor cells; HMGB1 = high mobility group box 1 protein; RAGE = receptor for advanced glycation end products; TLR4 = toll-like receptor 4; IHC = immunohistochemistry; ICC = immunocytochemistry.
ICC showed HMGB1 expression in both CPCs and chondrocytes. Unlike in cartilage tissue sections, in monolayer culture, the CPCs and chondrocytes were fibroblast-like in shape. HMGB1 expression was detectable in chondrocytes in both the nucleus and cytoplasm, but in CPCs it was restricted to the nucleus. RAGE and TLR4, 2 receptors for HMGB1, were observed in CPCs via ICC and showed an equal distribution in chondrocytes. Staining of cell nuclei was achieved via DAPI (Fig. 1e).
Immunoblotting was performed in 7 different cell lines: 2 OA chondrocyte cell lines (CH656-OA, CH657-OA), 1 healthy chondrocyte cell line (CH292ht) and 3 CPC cell lines (CPC531ht, CPC241ht, CPC242ht); mesenchymal stem cells (MSC577) served as positive control. CPCs from different passages (P20 to P50) were chosen in order to show if long term cell culture altered HMGB1 expression patterns of the cells. Immunoblotting results confirmed HMGB1 expression in all tested cells (Fig. 2a). OA chondrocytes showed significantly higher expression levels of HMGB1 (P < 0.05) than healthy chondrocytes. CPCs from different passages did not differ significantly in terms of HMGB1 expression (Fig. 2b).
Figure 2.
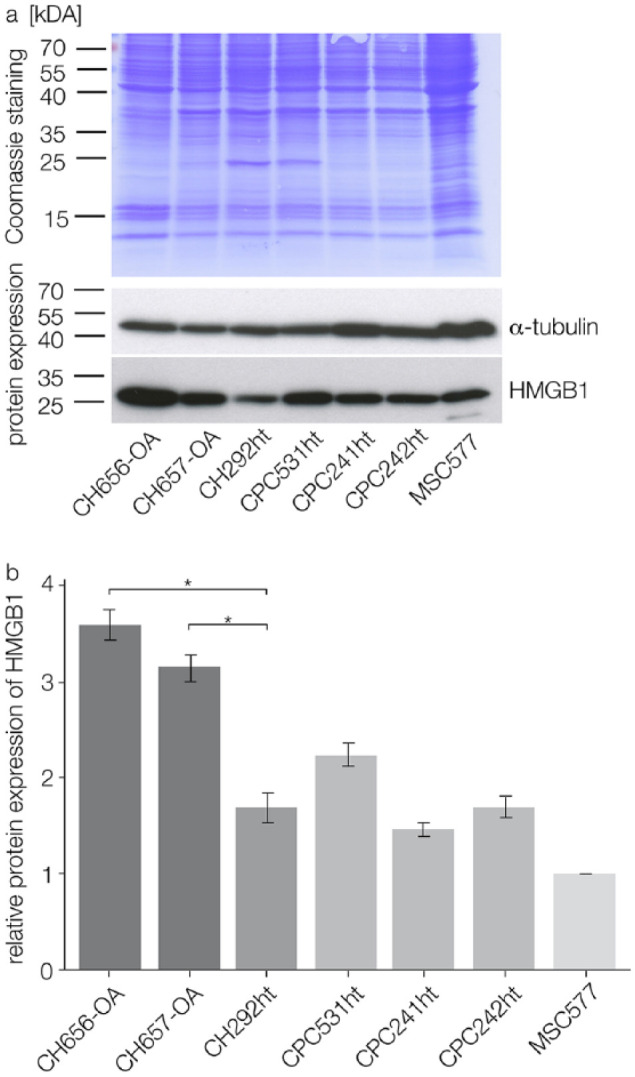
immunoblotting results from 2 OA chondrocyte cell lines (CH656-OA, CH657-OA), 1 healthy chondrocyte cell line (CH292ht), and 3 CPC cell lines from different cell culture passages (CPC531ht, CPC241ht, CPC242ht); MSCs (MSC577) served as positive control. (a) Coomassie staining of the transfer membrane stained proteins after separation; immunoblotting showed HMGB1 and α-tubulin expression in the tested cells (merged figure). (b) Relative HMGB1 expression after normalization with α-tubulin: OA chondrocytes express significantly more HMGB1 than healthy chondrocytes; CPCs from different cell culture passages do not differ significantly in their HMGB1 expression; statistical significance (P < 0.05) is indicated by an asterisk. OA = osteoarthritis; CPCs = chondrogenic progenitor cells; HMGB1 = high mobility group box 1 protein; MSCs = mesenchymal stem cells.
In vitro migration assays were performed to investigate the migratory effects of HMGB1 on CPCs. In a scratch assay, after 12 and 48 hours, the gaps between CPCs were smaller in specimens with 10 and 50 ng/mL HMGB1 than in control specimens (Fig. 3a, second and third column from left). After 72 hours, the gaps were confluent after stimulation with 10 and 50 ng/mL HMGB1. In the control group, the created gaps were still visible after 72 hours (Fig. 3a, fourth column from left). Quantification of the results showed that after 0 hours, migration was not significantly different between the tested groups. After 12 hours, HMGB1 stimulation with 10 and 50 ng/mL increased migration of CPCs significantly (P < 0.05) compared with the control group. After 48 hours, migration of CPCs was significantly higher (P < 0.05) after stimulation with 10 ng/mL HMGB1 than after stimulation with 0 ng/mL (control) or 50 ng/mL HMGB1. After 72 hours, both test groups exhibited significantly higher CPC migration (P < 0.05) than the control group (Fig. 3b).
Figure 3.

Scratch assay results performed on CPCs after 0, 12, 48, and 72 hours of stimulation with 0 (control), 10, or 50 ng/mL HMGB1. (a) Light microscopy, the gap between the white lines indicates the initial scratch marks: after 0 and 12 hours, the gap between CPCs is present in all groups; after 48 hours, confluence of CPCs stimulated by 10 and 50 ng/mL HMGB1 is increasing, while in the control group the initial gap is still present; after 72 hours, the CPC monolayer is confluent in specimens with stimulation via HMGB1; in the control group, a small gap is still visible. Scale bars are transferable to all figures in (a). (b) Quantification of the scratch assay results (n = 3): The number of cells between the white lines was counted and analyzed; statistical significance (P < 0.05) between results is indicated by an asterisk. CPCs = chondrogenic progenitor cells; HMGB1 = high mobility group box 1 protein.
Using a Boyden chamber assay, the migration of CPCs stimulated by different concentrations of HMGB1 was observed via fluorescence analysis after 6, 12, and 24 hours. At all points in time, cell migration was significantly higher in specimens with any of the tested HMGB1 concentrations than in unstimulated controls (P < 0.05). After 6 hours, higher concentrations of HMGB1 led to significantly higher migration rates (P < 0.05) than lower concentrations. After 12 hours, cell migration was significantly higher for 100 ng/mL HMGB1 than for 10 and 50 ng/mL HMGB1 (P < 0.05). After 24 hours, cell migration was significantly higher for 50 and 100 ng/mL HMGB1 than for 10 ng/mL HMGB1 (P < 0.05) (Fig. 4).
Figure 4.

Boyden chamber results (n = 19) performed on CPCs after 6, 12, and 24 hours of stimulation with 0 (control), 10, 50, or 100 ng/mL HMGB1; absolute fluorescence was measured and analyzed; statistical significance (P < 0.05) between results is indicated by an asterisk. CPCs = chondrogenic progenitor cells; HMGB1 = high mobility group box 1 protein.
In an ex vivo setting, GFP-tagged CPCs were placed on top of OA tissue explants to observe their aligned migration toward OA injury sites. After 5 days of migration, GFP-tagged CPCs were detected within the tissue using confocal microscopy (Fig. 5a). In the migrated CPCs, HMGB1 was identified at the mRNA and protein levels. HMGB1 mRNA in the migrated CPCs was detected by ISH and visualized by light microscopy (Fig. 5b and c) and confocal microscopy (Fig. 5d). At the protein level, HMGB1 and GFP expression in the migrated CPCs was confirmed by IHC (Fig. 5e and f).
Figure 5.

Migration of CPCs into OA tissue and their HMGB1 expression ex vivo. (a) After 5 days of migration, GFP-tagged CPCs are detected in OA cartilage tissue by confocal microscopy. (b) detection of HMGB1 mRNA in migrated CPCs via ISH. (c) Detail of a HMGB1 mRNA-positive migrated CPC. (d) Confocal reflection microscopy confirms HMGB1 mRNA (red) in migrated GFP-tagged CPCs (green). (e) IHC results show that migrated CPCs are GFP-positive. (f) IHC double-staining shows that migrated GFP-positive CPCs (brown) express HMGB1 (red). CPCs = chondrogenic progenitor cells; HMGB1 = high mobility group box 1 protein; OA, osteoarthritis; GFP = green fluorescent protein; ISH = in situ hybridization; IHC = immunohistochemistry
Discussion
In the present study, the distribution of HMGB1 in human late stage OA knee cartilage was shown. The IHC results of late-stage OA specimens showed nuclear, cytosolic, and pericellular expression of HMGB1, whereas in healthy specimens, HMGB1 expression was restricted to the nuclei of few chondrocytes, and most cells in the healthy tissue showed no HMGB1 expression. The observation that nuclear expression in healthy chondrocytes was higher than cytosolic expression is in line with previous findings showing that the ratio of nuclear to cytoplasmic HMGB1 is approximately 30:1.54 Perhaps in resting chondrocytes, tight nucleosomal HMGB1 binding causes steric hindrance of immunostaining. OA-mediated chondrocyte activation leads to nuclear staining, and the proinflammatory cytokines IL-1β and tumor necrosis factor α (TNFα) cause the translocation of HMGB1 from the nucleus to the cytosol.41,55 The ratio of positive cytosolic expression of HMGB1 within chondrocytes tends to increase with the OA histological grade, while the nuclear-positive cell ratio remains unaffected.55 In the present study, the increase in cytosolic HMGB1 expression in OA tissue was confirmed. Interestingly, the ICC results differed from the IHC results: Chondrocytes taken from healthy specimens showed strong nuclear but weak cytosolic HMGB1 expression in monolayer culture. This might be explainable by the biological and biochemical differences between in vitro and in vivo conditions because monolayer culture does not mimic chondrocyte functions that occur in native cartilage ECM or 3-dimensional culture.56 For the first time, the present study demonstrated HMGB1 expression in human CPCs by ICC; CPCs express nuclear HMGB1 but no cytosolic HMGB1. Further research is needed to clarify if stimulation by proinflammatory cytokines causes the cytosolic translocation of HMGB1 not only in chondrocytes but in CPCs as well.
Moreover, ICC showed RAGE and TLR4 expression in chondrocytes and CPCs. RAGE and TLR4 are receptors for HMGB1 involved in proinflammatory responses.57-59 In articular chondrocytes, RAGE expression has already been demonstrated via IHC, immunoblotting, and reverse transcription1–polymerase chain reaction37; our results supplement these findings via ICC. HMGB1 and RAGE overexpression in OA knee tissue indicates a role in the disease pathogenesis.38 The presence of TLR4 in chondrocytes and CPCs suggests that it is another receptor for HMGB1 in OA proinflammatory signaling besides RAGE.
In line with findings by Seol et al.,13 the current results of the migration assays showed that stimulation by HMGB1 significantly increased the migration of CPCs compared with control groups. Generally, the higher the concentration of the chemoattractant HMGB1, the higher was the migration rate of CPCs in the Boyden chamber assay. On the other hand, in the scratch assay, this linear correlation was not observed at all times: after 48 hours, migration was significantly lower in specimens with the highest tested HMGB1 concentration than in specimens with a lower HMGB1 concentration. Perhaps CPCs were damaged when the scratches were executed and, subsequently, released cytokines falsified the migratory effects of HMGB1, to some extent. The results from 2 different migration assay methods clearly showed that HMGB1 enhanced migration of CPCs.
Although there is various evidence about the migratory effects of HMGB1 on different cell types,60-63 there are contrary conclusions about its RAGE-dependency in progenitor and stem cells. Meng et al.64 showed that bone marrow mesenchymal stem cell migration is RAGE-independent, whereas Seol et al.13 suppose that CPC migration is mediated in part by RAGE. The present ICC results showed that chondrocytes and CPCs express both HMGB1 and RAGE, but future research needs to clarify if they interact as partners in cell migration.
In an ex vivo migration assay, CPCs were shown to migrate into human OA knee tissue. To our knowledge, this has previously been described for bovine tissue only.13 Although the physiological function of CPCs remains unknown, the present results strongly support the assumption that CPCs migrate to sites of tissue injury in order to participate in repair and regeneration processes. We suggest that migration is stimulated via HMGB1 released by chondrocytes at the injury site and mediated by binding to RAGE; both proteins were expressed by chondrocytes in the present study. Moreover, migrated CPCs expressed HMGB1 as shown by ISH. For methodical reasons, in the ex vivo assay, we were not able to examine HMGB1 expression in living CPCs on top of the explants. It would be of interest to know if HMGB1 expression in CPCs changes or remains unaffected during migratory activity. In human bronchial epithelial cells, HMGB1 promotes ECM synthesis and wound repair mediated by RAGE and TLR4.65 Possibly, HMGB1 in CPCs stimulates ECM restoration and healing processes at sites of tissue injury. Future research needs to clarify the expression profile of HMGB1 during migration of CPCs and its role in tissue regeneration.
The present study investigated the expression and chemotaxis of HMGB1 both in vitro and ex vivo. The results we obtained using CPC, MPC and chondrocyte cell lines need to be confirmed in primary cells to make them transferable to clinical situations.
In conclusion, the results of the present study give insight into the migratory role of HMGB1 in knee OA beyond its ubiquitous nuclear role. Proinflammatory cytokines IL-1β and TNFα are known to upregulate cytoplasmic HMGB1 expression in chondrocytes.55 Inflammasomes regulate HMGB1 release by immune and damaged cells.66-69 Extracellular HMGB1 participates in migratory, inflammatory, and repair responses by binding to RAGE and TLR4 on effector cells.23,30,39,57,70-75 In the present study, we demonstrated that extracellular HMGB1 enhances migration of CPCs and we suggest the mediation of this effect via RAGE and TLR4 binding. Our ex vivo results showed that CPCs that migrate into OA tissue express HMGB1. Future investigation is required to define the role of CPCs at OA injury sites and the possible involvement of HMGB1in further inflammation and repair processes.
Footnotes
Acknowledgments and Funding: We would like to thank the UMG central light microscopy service unit “Klinische Optische Mikroskopie” for their support and access to the microscopes. The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work was supported by the German Research Foundation (Mi 573/10-2).
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval: This study conformed to the ethical regulations of the Medical Faculty of the University of Gottingen (application number 25/12/10).
Informed Consent: All patients gave their written informed consent.
Trial Registration: Not applicable.
References
- 1.Felson DT.Clinical practice. Osteoarthritis of the knee. N Engl J Med. 2006;354(8):841-8. doi: 10.1056/NEJMcp051726 [DOI] [PubMed] [Google Scholar]
- 2.Xie F, Kovic B, Jin X, He X, Wang M, Silvestre C.Economic and humanistic burden of osteoarthritis: a systematic review of large sample studies. Pharmacoeconomics. 2016;34(11):1087-100. doi: 10.1007/s40273-016-0424-x [DOI] [PubMed] [Google Scholar]
- 3.Goldring MB.Chondrogenesis, chondrocyte differentiation, and articular cartilage metabolism in health and osteoarthritis. Ther Adv Musculoskelet Dis. 2012;4(4):269-85. doi: 10.1177/1759720x12448454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldring MB, Marcu KB.Cartilage homeostasis in health and rheumatic diseases. Arthritis Res Ther. 2009;11(3):224. doi: 10.1186/ar2592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buckwalter JA, Mankin HJ.Articular cartilage: degeneration and osteoarthritis, repair, regeneration, and transplantation. Instr Course Lect. 1998;47:487-504. [PubMed] [Google Scholar]
- 6.Martin JA, Buckwalter JA.Roles of articular cartilage aging and chondrocyte senescence in the pathogenesis of osteoarthritis. Iowa Orthop J. 2001;21:1-7. [PMC free article] [PubMed] [Google Scholar]
- 7.Miosge N, Waletzko K, Bode C, Quondamatteo F, Schultz W, Herken R.Light and electron microscopic in-situ hybridization of collagen type I and type II mRNA in the fibrocartilaginous tissue of late-stage osteoarthritis. Osteoarthritis Cartilage. 1998;6(4):278-85. doi: 10.1053/joca.1998.0121 [DOI] [PubMed] [Google Scholar]
- 8.Huang K, Wu LD.Aggrecanase and aggrecan degradation in osteoarthritis: a review. J Int Med Res. 2008;36(6):1149-60. [DOI] [PubMed] [Google Scholar]
- 9.Loeser RF, Goldring SR, Scanzello CR, Goldring MB.Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 2012;64(6):1697-707. doi: 10.1002/art.34453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldring MB, Goldring SR.Osteoarthritis. J Cell Physiol. 2007;213(3):626-34. doi: 10.1002/jcp.21258 [DOI] [PubMed] [Google Scholar]
- 11.Brandt KD, Radin EL, Dieppe PA, van de, Putte L.Yet more evidence that osteoarthritis is not a cartilage disease. Ann Rheum Dis. 2006;65(10):1261-4. doi: 10.1136/ard.2006.058347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koelling S, Kruegel J, Irmer M, Path JR, Sadowski B, Miro X, et al. Migratory chondrogenic progenitor cells from repair tissue during the later stages of human osteoarthritis. Cell Stem Cell. 2009;4(4):324-35. doi: 10.1016/j.stem.2009.01.015 [DOI] [PubMed] [Google Scholar]
- 13.Seol D, McCabe DJ, Choe H, Zheng H, Yu Y, Jang K, et al. Chondrogenic progenitor cells respond to cartilage injury. Arthritis Rheum. 2012;64(11):3626-37. doi: 10.1002/art.34613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paonessa G, Frank R, Cortese R.Nucleotide sequence of rat liver HMG1 cDNA. Nucleic Acids Res. 1987;15(21):9077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferrari S, Ronfani L, Calogero S, Bianchi ME.The mouse gene coding for high mobility group 1 protein (HMG1). J Biol Chem. 1994;269:28803-8. [PubMed] [Google Scholar]
- 16.Wen L, Huang JK, Johnson BH, Reeck GR.A human placental cDNA clone that encodes nonhistone chromosomal protein HMG-1. Nucleic Acids Res. 1989;17(3):1197-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bustin M.Regulation of DNA-dependent activities by the functional motifs of the high-mobility-group chromosomal proteins. Mol Cell Biol. 1999;19(8):5237-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verrier CS, Roodi N, Yee CJ, Bailey LR, Jensen RA, Bustin M, et al. High-mobility group (HMG) protein HMG-1 and TATA-binding protein-associated factor TAF(II)30 affect estrogen receptor-mediated transcriptional activation. Mol Endocrinol. 1997;11(8):1009-19. doi: 10.1210/mend.11.8.9962 [DOI] [PubMed] [Google Scholar]
- 19.Boonyaratanakornkit V, Melvin V, Prendergast P, Altmann M, Ronfani L, Bianchi ME, et al. High-mobility group chromatin proteins 1 and 2 functionally interact with steroid hormone receptors to enhance their DNA binding in vitro and transcriptional activity in mammalian cells. Mol Cell Biol. 1998;18(8):4471-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taguchi A, Blood DC, del Toro G, Canet A, Lee DC, Qu W, et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature. 2000;405(6784):354-60. doi: 10.1038/35012626 [DOI] [PubMed] [Google Scholar]
- 21.Zhu J, Luo J, Li Y, Jia M, Wang Y, Huang Y, et al. HMGB1 induces human non-small cell lung cancer cell motility by activating integrin αvβ3/FAK through TLR4/NF-κB signaling pathway. Biochem Biophys Res Commun. 2016;480(4):522-7. doi: 10.1016/j.bbrc.2016.10.052 [DOI] [PubMed] [Google Scholar]
- 22.Harris HE, Andersson U, Pisetsky DS.HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat Rev Rheumatol. 2012;8(4):195-202. doi: 10.1038/nrrheum.2011.222 [DOI] [PubMed] [Google Scholar]
- 23.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285(5425):248-51. [DOI] [PubMed] [Google Scholar]
- 24.Rendon-Mitchell B, Ochani M, Li J, Han J, Wang H, Yang H, et al. IFN-γ induces high mobility group box 1 protein release partly through a TNF-dependent mechanism. J Immunol. 2003;170(7):3890-7. [DOI] [PubMed] [Google Scholar]
- 25.Tang D, Shi Y, Kang R, Li T, Xiao W, Wang H, et al. Hydrogen peroxide stimulates macrophages and monocytes to actively release HMGB1. J Leukoc Biol. 2007;81(3):741-7. doi: 10.1189/jlb.0806540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rouhiainen A, Kuja-Panula J, Wilkman E, Pakkanen J, Stenfors J, Tuominen RK, et al. Regulation of monocyte migration by amphoterin (HMGB1). Blood. 2004;104(4):1174-82. doi: 10.1182/blood-2003-10-3536 [DOI] [PubMed] [Google Scholar]
- 27.Scaffidi P, Misteli T, Bianchi ME.Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418(6894):191-5. doi: 10.1038/nature00858 [DOI] [PubMed] [Google Scholar]
- 28.Taniguchi N, Yoshida K, Ito T, Tsuda M, Mishima Y, Furumatsu T, et al. Stage-specific secretion of HMGB1 in cartilage regulates endochondral ossification. Mol Cell Biol. 2007;27(16):5650-63. doi: 10.1128/mcb.00130-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ombrellino M, Wang H, Ajemian MS, Talhouk A, Scher LA, Friedman SG, et al. Increased serum concentrations of high-mobility-group protein 1 in haemorrhagic shock. Lancet. 1999;354(9188):1446-7. doi: 10.1016/s0140-6736(99)02658-6 [DOI] [PubMed] [Google Scholar]
- 30.Taniguchi N, Kawahara K, Yone K, Hashiguchi T, Yamakuchi M, Goto M, et al. High mobility group box chromosomal protein 1 plays a role in the pathogenesis of rheumatoid arthritis as a novel cytokine. Arthritis Rheum. 2003;48(4):971-81. doi: 10.1002/art.10859 [DOI] [PubMed] [Google Scholar]
- 31.Shimizu S, Kouzaki H, Kato T, Tojima I, Shimizu T.HMGB1-TLR4 signaling contributes to the secretion of interleukin 6 and interleukin 8 by nasal epithelial cells. Am J Rhinol Allergy. 2016;30(3):167-72. doi: 10.2500/ajra.2016.30.4300 [DOI] [PubMed] [Google Scholar]
- 32.Wan Z, Zhang X, Peng A, He M, Lei Z, Wang Y.TLR4-HMGB1 signaling pathway affects the inflammatory reaction of autoimmune myositis by regulating MHC-I. Int Immunopharmacol. 2016;41:74-81. doi: 10.1016/j.intimp.2016.10.009 [DOI] [PubMed] [Google Scholar]
- 33.Bangert A, Andrassy M, Müller AM, Bockstahler M, Fischer A, Volz CH, et al. Critical role of RAGE and HMGB1 in inflammatory heart disease. Proc Natl Acad Sci U S A. 2016;113(2):E155-E164. doi: 10.1073/pnas.1522288113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu Y, Yang L, Lv J, Huang X, Yi J, Pei C, et al. The role of high mobility group box 1 (HMGB-1) in the diabetic retinopathy inflammation and apoptosis. Int J Clin Exp Pathol. 2015;8(6):6807-13. [PMC free article] [PubMed] [Google Scholar]
- 35.Muth IE, Zschuntzsch J, Kleinschnitz K, Wrede A, Gerhardt E, Balcarek P, et al. HMGB1 and RAGE in skeletal muscle inflammation: implications for protein accumulation in inclusion body myositis. Exp Neurol. 2015;271:189-97. doi: 10.1016/j.expneurol.2015.05.023 [DOI] [PubMed] [Google Scholar]
- 36.Liang Y, Hou C, Kong J, Wen H, Zheng X, Wu L, et al. HMGB1 binding to receptor for advanced glycation end products enhances inflammatory responses of human bronchial epithelial cells by activating p38 MAPK and ERK1/2. Mol Cell Biochem. 2015;405(1-2):63-71. doi: 10.1007/s11010-015-2396-0 [DOI] [PubMed] [Google Scholar]
- 37.Loeser RF, Yammani RR, Carlson CS, Chen H, Cole A, Im HJ, et al. Articular chondrocytes express the receptor for advanced glycation end products: potential role in osteoarthritis. Arthritis Rheum. 2005;52(8):2376-85. doi: 10.1002/art.21199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun XH, Liu Y, Han Y, Wang J.Expression and significance of high-mobility group protein B1 (HMGB1) and the receptor for advanced glycation end-product (RAGE) in knee osteoarthritis. Med Sci Monit. 2016;22:2105-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kokkola R, Sundberg E, Ulfgren AK, Palmblad K, Li J, Wang H, et al. High mobility group box chromosomal protein 1: a novel proinflammatory mediator in synovitis. Arthritis Rheum. 2002;46(10):2598-603. doi: 10.1002/art.10540 [DOI] [PubMed] [Google Scholar]
- 40.Pullerits R, Jonsson IM, Verdrengh M, Bokarewa M, Andersson U, Erlandsson-Harris H, et al. High mobility group box chromosomal protein 1, a DNA binding cytokine, induces arthritis. Arthritis Rheum. 2003;48(6):1693-700. doi: 10.1002/art.11028 [DOI] [PubMed] [Google Scholar]
- 41.Heinola T, Kouri VP, Clarijs P, Ciferska H, Sukura A, Salo J, et al. High mobility group box-1 (HMGB-1) in osteoarthritic cartilage. Clin Exp Rheumatol. 2010;28(4):511-8. [PubMed] [Google Scholar]
- 42.Pritzker KP, Gay S, Jimenez SA, Ostergaard K, Pelletier JP, Revell PA, et al. Osteoarthritis cartilage histopathology: grading and staging. Osteoarthritis Cartilage. 2006;14(1):13-29. doi: 10.1016/j.joca.2005.07.014 [DOI] [PubMed] [Google Scholar]
- 43.Altman R, Asch E, Bloch D, Bole G, Borenstein D, Brandt K, et al. Development of criteria for the classification and reporting of osteoarthritis. Classification of osteoarthritis of the knee. Diagnostic and Therapeutic Criteria Committee of the American Rheumatism Association. Arthritis Rheum. 1986;29(8):1039-49. [DOI] [PubMed] [Google Scholar]
- 44.Schminke B, Vom Orde F, Gruber R, Schliephake H, Burgers R, Miosge N.The pathology of bone tissue during peri-implantitis. J Dent Res. 2015;94(2):354-61. doi: 10.1177/0022034514559128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liang CC, Park AY, Guan JL.In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2(2):329-33. doi: 10.1038/nprot.2007.30 [DOI] [PubMed] [Google Scholar]
- 46.Boyden S.The chemotactic effect of mixtures of antibody and antigen on polymorphonuclear leucocytes. J Exp Med. 1962;115:453-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aslani F, Schuppe HC, Guazzone VA, Bhushan S, Wahle E, Lochnit G, et al. Targeting high mobility group box protein 1 ameliorates testicular inflammation in experimental autoimmune orchitis. Hum Reprod. 2015;30(2):417-31. doi: 10.1093/humrep/deu320 [DOI] [PubMed] [Google Scholar]
- 48.Tabeta K, Yamazaki K, Akashi S, Miyake K, Kumada H, Umemoto T, et al. Toll-like receptors confer responsiveness to lipopolysaccharide from Porphyromonas gingivalis in human gingival fibroblasts. Infect Immun. 2000;68(6):3731-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Holmdahl R, Rubin K, Klareskog L, Larsson E, Wigzell H.Characterization of the antibody response in mice with type II collagen-induced arthritis, using monoclonal anti-type II collagen antibodies. Arthritis Rheum. 1986;29(3):400-10. [DOI] [PubMed] [Google Scholar]
- 50.Danshina PV, Geyer CB, Dai Q, Goulding EH, Willis WD, Kitto GB, et al. Phosphoglycerate kinase 2 (PGK2) is essential for sperm function and male fertility in mice. Biol Reprod. 2010;82(1):136-45. doi: 10.1095/biolreprod.109.079699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liao Y, Lv G, Wang B, Kuang L, Wang X.Imatinib promotes apoptosis of giant cell tumor cells by targeting microRNA-30a-mediated runt-related transcription factor 2. Mol Med Rep. 2016;13(2):1739-45. doi: 10.3892/mmr.2015.4722 [DOI] [PubMed] [Google Scholar]
- 52.Cidlinsky N, Dogliotti G, Pukrop T, Jung R, Weber F, Krahn MP.Inactivation of the LKB1-AMPK signaling pathway does not contribute to salivary gland tumor development—a short report. Cell Oncol (Dordr). 2016;39(4):389-96. doi: 10.1007/s13402-016-0290-8 [DOI] [PubMed] [Google Scholar]
- 53.Kruegel J, Sadowski B, Miosge N.Nidogen-1 and nidogen-2 in healthy human cartilage and in late-stage osteoarthritis cartilage. Arthritis Rheum. 2008;58(5):1422-32. doi: 10.1002/art.23480 [DOI] [PubMed] [Google Scholar]
- 54.Kuehl L, Salmond B, Tran L.Concentrations of high-mobility-group proteins in the nucleus and cytoplasm of several rat tissues. J Cell Biol. 1984;99(2):648-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Terada C, Yoshida A, Nasu Y, Mori S, Tomono Y, Tanaka M, et al. Gene expression and localization of high-mobility group box chromosomal protein-1 (HMGB-1) in human osteoarthritic cartilage. Acta Med Okayama. 2011;65(6):369-77. [DOI] [PubMed] [Google Scholar]
- 56.Caron MM, Emans PJ, Coolsen MM, Voss L, Surtel DA, Cremers A, et al. Redifferentiation of dedifferentiated human articular chondrocytes: comparison of 2D and 3D cultures. Osteoarthritis Cartilage. 2012;20(10):1170-8. doi: 10.1016/j.joca.2012.06.016 [DOI] [PubMed] [Google Scholar]
- 57.Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, et al. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279(9):7370-7. doi: 10.1074/jbc.M306793200 [DOI] [PubMed] [Google Scholar]
- 58.Hudson BI, Lippman ME.Targeting RAGE signaling in inflammatory disease. Annu Rev Med. 2017;69:349-64. doi: 10.1146/annurev-med-041316-085215 [DOI] [PubMed] [Google Scholar]
- 59.Kang R, Zhang Q, Zeh HJ, 3rd, Lotze MT, Tang D.HMGB1 in cancer: good, bad, or both? Clin Cancer Res. 2013;19(15):4046-57. doi: 10.1158/1078-0432.ccr-13-0495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Di Maggio S, Milano G, De Marchis F, D’Ambrosio A, Bertolotti M, Palacios BS, et al. Non-oxidizable HMGB1 induces cardiac fibroblasts migration via CXCR4 in a CXCL12-independent manner and worsens tissue remodeling after myocardial infarction. Biochim Biophys Acta. 2017;1863(11):2693-704. doi: 10.1016/j.bbadis.2017.07.012 [DOI] [PubMed] [Google Scholar]
- 61.Fahmy-Garcia S, van Driel M, Witte-Buoma J, Walles H, van Leeuwen JP, van Osch G, et al. Nell-1, HMGB1 and CCN2 enhance migration and vasculogenesis, but not osteogenic differentiation compared to BMP2. Tissue Eng Part A. 2017;24(3-4):207-18. doi: 10.1089/ten.TEA.2016.0537 [DOI] [PubMed] [Google Scholar]
- 62.Pang X, Zhang Y, Zhang S.High-mobility group box 1 is overexpressed in cervical carcinoma and promotes cell invasion and migration in vitro. Oncol Rep. 2017;37(2):831-40. doi: 10.3892/or.2016.5317 [DOI] [PubMed] [Google Scholar]
- 63.Lin F, Xue D, Xie T, Pan Z.HMGB1 promotes cellular chemokine synthesis and potentiates mesenchymal stromal cell migration via Rap1 activation. Mol Med Rep. 2016;14(2):1283-9. doi: 10.3892/mmr.2016.5398 [DOI] [PubMed] [Google Scholar]
- 64.Meng E, Guo Z, Wang H, Jin J, Wang J, Wang H, et al. High mobility group box 1 protein inhibits the proliferation of human mesenchymal stem cells and promotes their migration and differentiation along osteoblastic pathway. Stem Cells Dev. 2008;17(4):805-13. doi: 10.1089/scd.2008.0276 [DOI] [PubMed] [Google Scholar]
- 65.Ojo OO, Ryu MH, Jha A, Unruh H, Halayko AJ.High-mobility group box 1 promotes extracellular matrix synthesis and wound repair in human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2015;309(11):L1354-66. doi: 10.1152/ajplung.00054.2015 [DOI] [PubMed] [Google Scholar]
- 66.Lu B, Wang H, Andersson U, Tracey KJ.Regulation of HMGB1 release by inflammasomes. Protein Cell. 2013;4(3):163-7. doi: 10.1007/s13238-012-2118-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Walle LV, Kanneganti TD, Lamkanfi M.HMGB1 release by inflammasomes. Virulence. 2011;2(2):162-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Willingham SB, Allen IC, Bergstralh DT, Brickey WJ, Huang MT, Taxman DJ, et al. NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J Immunol. 2009;183(3):2008-15. doi: 10.4049/jimmunol.0900138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lamkanfi M, Sarkar A, Walle LV, Vitari AC, Amer AO, Wewers MD, et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol. 2010;185(7):4385-92. doi: 10.4049/jimmunol.1000803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Levy RM, Mollen KP, Prince JM, Kaczorowski DJ, Vallabhaneni R, Liu S, et al. Systemic inflammation and remote organ injury following trauma require HMGB1. Am J Physiol Regul Integr Comp Physiol. 2007;293(4):R1538-44. doi: 10.1152/ajpregu.00272.2007 [DOI] [PubMed] [Google Scholar]
- 71.Abraham E, Arcaroli J, Carmody A, Wang H, Tracey KJ.HMG-1 as a mediator of acute lung inflammation. J Immunol. 2000;165(6):2950-4. [DOI] [PubMed] [Google Scholar]
- 72.Dumitriu IE, Baruah P, Valentinis B, Voll RE, Herrmann M, Nawroth PP, et al. Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J Immunol. 2005;174(12):7506-15. [DOI] [PubMed] [Google Scholar]
- 73.van Zoelen MA, Yang H, Florquin S, Meijers JC, Akira S, Arnold B, et al. Role of toll-like receptors 2 and 4, and the receptor for advanced glycation end products in high-mobility group box 1-induced inflammation in vivo. Shock. 2009;31(3):280-4. doi: 10.1097/SHK.0b013e318186262d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tancharoen S, Gando S, Binita S, Nagasato T, Kikuchi K, Nawa Y, et al. HMGB1 promotes intraoral palatal wound healing through RAGE-dependent mechanisms. Int J Mol Sci. 2016;17(11):E1961. doi: 10.3390/ijms17111961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fages C, Nolo R, Huttunen HJ, Eskelinen E, Rauvala H.Regulation of cell migration by amphoterin. J Cell Sci. 2000;113(Pt 4):611-20. [DOI] [PubMed] [Google Scholar]