

Abstract
H-Ryk is an atypical receptor tyrosine kinase which differs from other members of this family at a number of conserved residues in the activation and nucleotide binding domains. Using a chimeric receptor approach, we demonstrate that H-Ryk has impaired catalytic activity. Despite the receptor’s inability to undergo autophosphorylation or phosphorylate substrates, we demonstrate that ligand stimulation of the chimeric receptor results in activation of the mitogen-activated protein kinase pathway. The ability to transduce signals is abolished by mutation of the invariant lysine (K334A) in subdomain II of H-Ryk. Further, by in vitro mutagenesis, we show that the amino acid substitutions in the activation domain of H-Ryk account for the loss of catalytic activity. In addition to the essential aspartate residue, either phenylalanine or glycine is required in the activation domain to maintain proper conformation of the catalytic domain and thus ensure receptor autophosphorylation. Homology modelling of the catalytic domain of H-Ryk provides a rationale for these findings. Thus, the signalling properties of H-Ryk are divergent from those of other classical receptor tyrosine kinases.
The need for the coordinated regulation of cell growth and differentiation in multicellular organisms has given rise to a complex array of signalling pathways. Growth factors play pivotal roles in the coordination of these cellular programs, and their diverse biological effects are mediated primarily by a large family of cell surface receptors with intrinsic protein tyrosine kinase activity. Binding of a growth factor to the extracellular domain of its receptor induces receptor dimerization, resulting in autophosphorylation and conformational changes in the receptor that lead to the binding of downstream signalling proteins (53).
Although receptor protein tyrosine kinases (RPTK) exhibit variability in their regulation and intracellular signalling pathways, they share a highly conserved cytoplasmic catalytic domain that is responsible for kinase activity. Sequence alignments of protein kinases defined 11 distinct subdomains that are found throughout the broad family of protein kinases (22, 23). The highly conserved and invariant residues from these subdomains have been implicated in essential roles in ATP binding, substrate recognition, and phosphate transfer (29, 30, 39). In Ryk (also referred to as Nyk-r, Vik, Nbtk-1, Mrk, and Derailed [Drl]), a member of the RPTK family, some of the highly conserved protein kinase sequence motifs display variations (6, 7, 28, 36, 65, 71, 77). In H-Ryk, the human homologue, substitutions of glutamine (residue 307) for the first glycine of the GxGxxG (subdomain I) nucleotide binding motif and of asparagine and alanine (residues 454 and 455) for the highly conserved phenylalanine and glycine within the DFG activation loop motif represent the most notable changes (Fig. 1). In addition, the highly conserved alanine residue close to the essential lysine at the nucleotide cleft (subdomain II) and the invariant arginine residue in the catalytic loop (IHRDLAARN) are altered to phenylalanine and lysine, respectively. These sequence alterations suggest that the kinase activity of H-Ryk might be impaired (28, 36, 65, 71, 77).
FIG. 1.

ALSCRIPT (2) figure showing alignment of H-Ryk with other RTKs. The alignment of IRK and FGFR tyrosine kinase was performed by the STAMP structural alignment package (61), and the locations of alpha helices (blue cylinders) and beta strands (magenta arrows) for these two kinases, as assigned by DSSP (34) are shown at the bottom. Alignment of IRK with the others was performed using Clustal W (74) and merged with the STAMP alignment. Numbering above the alignment corresponds to H-Ryk. Boxed regions indicate approximate location of the 11 kinase subdomains described by Hanks et al. (22, 23), which are labelled below the aligned sequences. Residues are colored red if they show total conservation across the kinases in the alignment, yellow if they show conservation of hydrophobic character (73), green for polar character, and blue for small character. Residues in H-Ryk described in the text are indicated by circles above the H-Ryk sequence. The National Center for Biotechnology Information protein accession numbers for H-Ryk, H-Cck4, M-Mep1, H-Ror1, H-ErbB3, IR, and FGFR are 1710811, 2136061, 1911183, 346351, 119534, 124529, and 120046, respectively; the protein databank codes for IR and FGFR are 1irk and 1fgi, respectively (4).
In the absence of its ligand, the precise functional implications of the sequence variations on the catalytic activity and signalling of H-Ryk are unknown. A chimeric receptor approach in which the extracellular domain of the orphan receptor is replaced by the extracellular domain of another well-characterized receptor tyrosine kinase (RTK) whose ligand is available has been successfully used as a tool to study the signalling properties of orphan receptors (17, 49, 60). This type of approach permits analysis of the molecular events involved in the signal transduction pathway of the tyrosine kinase of interest, even when its ligands are unknown.
To address the effect of the sequence alterations on the catalytic function of H-Ryk, we constructed a TrkA:Ryk chimeric receptor composed of the extracellular domain of TrkA (human nerve growth factor [NGF] receptor) fused to the transmembrane and cytoplasmic domains of the H-Ryk receptor. We show that although the TrkA:Ryk chimera is catalytically impaired, ligand stimulation of the chimeric receptor results in activation of the mitogen-activated protein kinase (MAPK) pathway. These results imply that the receptor may mediate its biological activities by recruitment of a signalling-competent auxiliary protein through an as yet uncharacterized mechanism. We also demonstrate that the Phe and Gly residues in the DFG motif of the activation domain are essential for catalytic activity of RTKs.
MATERIALS AND METHODS
Growth factor, antibodies, and peptides.
The recombinant human NGF was purchased from R&D Systems. The N-terminal TrkA-specific monoclonal antibodies (MAbs) 220 and 5C3 were gifts from M. Barbacid and U. Saragovi, respectively. The H-Ryk polyclonal antibody was raised against the C-terminal region of H-Ryk (77). The antiphosphotyrosine (4G10) and Erk1/2 antibodies were purchased from TCS and Signal Transduction Laboratories, respectively. The p90 Rsk3, Src, Lck, Jak 1, 2, 3, and phospho-Erk antibodies were purchased from Santa Cruz Biotechnology. The horseradish peroxidase-conjugated anti-mouse/rabbit antibodies and the tyrosine kinase synthetic substrates R-R-L-I-E-D-A-E-Y-A-A-R-G and poly(Glu80Tyr20) were purchased from Sigma. The S6 peptide R-R-R-L-S-S-L-R-A was synthesized by the Imperial Cancer Research Fund peptide synthesis laboratory.
Construction of the chimeric receptors.
The extracellular ligand binding domain of TrkA was amplified by PCR (1 cycle of 95°C for 5 min and 72°C for 4 min; 35 cycles of 95°C for 1 min and 72°C for 4 min), using primers V-Famp C (5′-CGC CAG GGT TTT CCC AGT GAC GAG GTT GTA-3′), which anneals upstream of the 5′ untranslated region, and Trk-Ramp C (5′-CCC ACA GCC ACC GAG ACC CAA AAA CTA GTT TCG-3′), which anneals to the 3′ end of the extracellular domain of Trk A and also introduces a unique SpeI restriction site to the amplified PCR fragment. To amplify the Ryk chimeric cDNA, the gene-specific primer Ryk chimeric Famp (5′-ACT AGT ACG CGT GTC TTT TAT ATT-3′), which introduces a unique SpeI site into the amplified chimeric fragment, and the vector-specific primer M13 Rsp were used in the PCR (95°C for 1 min, 50°C for 1 min, 72°C for 1 min). The wild-type TrkA:Ryk chimera was constructed by fusing the amplified fragments at the unique SpeI site. A PCR-based primer extension overlap method (25) was used to introduce mutations into the H-Ryk catalytic domain. Four mutagenic primers were designed to create the activation domain mutants: (i) pRamp mut (5′-CTC CCG TAA CAG CAC CTA GAA TTG GAG GTC ACA-3′), (ii) pN457F Ryk (5′-ACA CTG CAG GTT AAG ATC ACA GAC TTT GCC CTC-3′), (iii) pA458G Ryk (5′-ACA CTG CAG GTT AAG ATC ACA GAC TTT GAC CTC-3′), and (iv) pN457F/A458G Ryk (5′-ACA CTG CAG GTT AAG ATC ACA GAC TTT GAC CTC-3′). PCR (95°C for 1 min, 60°C for 2 min, 72°C for 1 min) was carried with the proofreading DNA Pwo polymerase (Boehringer Mannheim). To introduce the relevant mutant Ryk fragments into the chimeric H-Ryk cDNA fragment, HindIII/XbaI restriction fragments were used to replace the wild-type HindIII/XbaI fragment in order to create the corresponding activation domain chimeras. Two primers, 5′-GTA CTC GGA GAA GGT ACT TTT GGG-3′ and 5′-CCC AAA AGT ACC TTC TCC GAG TAC-3′, were designed to introduce the glycine mutation in the nucleotide binding domain. The K334A mutation was introduced by amplifying the DNA template with primers 5′-CAA GCA TTT GTC GCA ACA GTT AAA CAA GCT-3′ and 5′-TCG AAC TAG AAA TTG ACA ACG CTG TTT ACG AAC-3′. Primers 5′-GCT TGA TCT TTA ACT GTT TTG ACA GCT GCT TGT TTT T-3′ and 5′-AAA AAC AAG CAG CTG TCA AAA CAG TTA AAG ATC AAG C-3′ were designed to introduce the F332A mutation. The K434R mutation was introduced by amplifying the H-Ryk cDNA with primers, 5′-GAC ACA GTT CCT GGC AGC CAG GTC TCT GTG GAT GAC-3′ and 5′-GTC ATC CAC AGA GAC CTG GCT GCC AGG AAC TGT GTC-3′. All chimeric cDNA constructs were cloned downstream of the cytomegalovirus promoter in the expression plasmid pcDNA3. This was accomplished by digesting the chimeric constructs with EcoRI/XbaI.
Transfection of chimeras into murine fibroblasts.
The high-efficiency calcium phosphate method of Chen and Okayama was used to transfect the cell lines (9, 10). The cells were maintained under selection (Dulbecco modified Eagle medium [DMEM], 10% fetal calf serum [FCS], 800 μg of neomycin per ml) for 2 weeks. The individual clones were then analyzed further to determine the expression levels of the chimeric receptor protein.
Dynal selection of the chimera-expressing cell lines.
Dynabeads M-450 (rat anti-mouse immunoglobulin G1 [IgG1] were washed three times with phosphate-buffered saline (PBS) to remove any traces of sodium azide prior to coating with TrkA-specific monoclonal antibody (MAb) 220 at a concentration of 5 μg of MAb per mg of beads. After a 2-h incubation at 4°C, the beads were collected by placement in a Dynal magnet. After removal of the supernatant, the beads were washed three times in a solution of PBS supplemented with 0.1% FCS. Adherent cells were lifted by a 15-min incubation in PBS–2 mM EDTA and then washed twice with PBSA and resuspended in PBS–6% sodium citrate before being incubated with the Dynabead-antibody complex. To reach the plateau in the binding kinetics after 10 min, 107 Dynabeads per ml were used. The beads were washed three times with PBS–0.1% FCS to remove unbound cells. The Dynabeads-antibody-cell complexes were cultured in DMEM supplemented with 800 μg of G418 per ml and 10% FCS. After the cells had grown to about 70% confluence, the Dynabead-antibody complex was dissociated from the cells by trypsinization. The clonal population of cells were maintained in DMEM–800 μg of G418 per ml–10% FCS.
Immunofluorescence analysis of the chimeric constructs.
Adherent cells were lifted by a 15-min incubation at 37°C in PBS–2 mM EDTA and washed with PBS–0.2% bovine serum albumin–2 mM sodium azide (FACS [fluorescence-activated cell sorting] wash buffer); 106 cells per sample were then incubated in the same sample buffer in 6-ml round-bottom polypropylene tubes (Falcon-2058) with the primary antibody for 60 min at 4°C. Samples were then washed once in excess FACS wash buffer and incubated with fluorescein isothiocyanate-conjugated goat anti-mouse serum for 60 min at 4°C. After a further wash in excess FACS wash buffer, stained cells were fixed in PBS with 0.5 ml 2% formaldehyde for 10 min at 4°C. At least 5,000 cells per sample were analyzed by flow cytometry on a Becton Dickinson FACSscan. Specific median fluorescence of a sample was calculated by subtracting the median fluorescence intensity of the negative control (usually cells labelled with secondary reagent only) from the median fluorescence intensity of the sample. The receptor density of the chimeras was calculated by extrapolating the median fluorescence intensity to that of the E 25-4-27 cell line (44).
Induction studies, immunoprecipitation, and immunoblotting.
For the induction studies, cells were grown to 80% confluence, washed twice with PBSA, and then incubated for 16 h in serum-free medium. The quiescent cells were stimulated with recombinant human NGF for different time intervals before being lysed on ice for 30 min in 1.00 ml of lysis buffer (1% Triton X-100, 0.5% Nonidet P-40, 150 mM NaCl, 50 mM Tris-HCl [pH 7.4], 1 mM EDTA, 0.5 mM EGTA) containing 2 mM sodium vanadate and freshly added protease inhibitors (10 μg each of aprotinin, leupeptin, pepstatin A, and trypsin inhibitor per ml, 100 μg of phenylmethylsulfonyl fluoride per ml). Insoluble material was removed by centrifugation for 30 min at 15,000 rpm at 4°C. The total protein concentration was measured by using the calorimetric bicinchoninic acid protein assay (Pierce); 800 μg of protein was incubated with 2 μg of mouse N-terminal TrkA MAb 5C3 preabsorbed onto 50 μl of protein G-agarose beads. After 12 h of incubation at 4°C, the immune complexes were washed three times in ice-cold cell lysis supplemented with protease inhibitors and twice in ice-cold lysis buffer supplemented with 1 M NaCl. The immunoprecipitates were eluted and denatured by boiling for 5 min in sodium dodecyl sulfate (SDS) sample buffer (50 mM Tris-HCl [pH 6.8], 2% SDS, 10% glycerol, 5% β-mercaptoethanol, 0.25% bromophenol blue). For immunoblotting, the immunoprecipitates were resolved by SDS-polyacrylamide gel electrophoresis (PAGE) according to standard protocols and then transferred onto nylon membranes. The membrane was blocked for 2 h in Tris-buffered saline (TBS) containing 5% nonfat milk and 0.1% Tween 20 and then incubated overnight at 4°C with 1 μg of antiphosphotyrosine antibody 4G10 (TCS) per ml. Afterwards the blot was washed with TBS containing 1% milk and 0.1% Tween 20. The blot was incubated for 1 h with peroxidase-conjugated anti-mouse immunoglobulin (diluted 1:5,000; Sigma). After a wash with TBS (1% milk, 0.1% Tween 20), antibody binding was localized by the enhanced chemiluminescence method (Amersham). All other antibodies were used according to manufacturer’s conditions.
In vitro kinase assays.
The NIH 3T3 TrkA:Ryk chimeric transfectants were incubated in DMEM supplemented with 1% FCS for 24 h and then in DMEM containing 0.1% FCS for another 24 h. After induction with NGF and lysis, the cell lysates were equalized for protein content prior to immunoprecipitation with either the C-terminal anti-p90 Rsk3 antibody (Santa Cruz) or N-terminal TrkA MAb 5C3. A total of 1.00 mg of cell lysate was used in the immunoprecipitation. The immune complex kinase assay was initiated by resuspending the final immune complexes in 25 μl of kinase assay buffer (1 mg of bovine serum albumin per ml, 30 mM Tris-Cl [pH 7.4], 20 mM MgCl2, 1 mM dithiothreitol, 200 mM ATP, 100 mM [γ-32P]ATP). The in vitro substrate kinase assays were initiated by resuspending the immune complexes in 25 μl of kinase assay buffer supplemented with either 250 mM S6 peptide or 10 mM tyrosine kinase substrate for the p90 Rsk3 and TrkA:Ryk substrate assays, respectively. The S6 and Src-related peptide reaction mixtures were incubated at 30°C for 10 min and then stopped by spotting onto pieces of P81 phosphocellulose (Whatman), followed by five washes in 150 mM phosphoric acid. The filter was dried, and the picomoles of [γ-32P]ATP incorporated into the substrates was determined by PhosphorImager analysis (Molecular Dynamics) after 4 h. The relative specific activity was calculated by determining the picomoles of [γ-32P]ATP incorporated into the synthetic peptide substrate. The following conversion factor was used: 1 μCi = 0.33 pmol = 2.2 × 106 cpm. By considering 1 PhosphorImager unit as equivalent to 1 cpm, the relative moles of [γ-32P]ATP incorporated into the synthetic peptide substrate can be determined. Mouse IgG was used as control in the experiment to estimate the background. The poly(Glu80Tyr20) substrate assay was initiated by incubating the reaction mixtures at 30°C for 10 min. The reactions were stopped by spotting the samples onto Whatman 3M paper and immersion into 10% trichloroacetic acid. The papers were extensively washed, and the radioactivity incorporated into the substrate was determined by counting the Whatman 3M paper dry in a scintillation counter. The specific activity of the [γ-32P]ATP in the kinase reaction was determined by spotting a small sample of the kinase reaction sample onto a paper and counting directly without washing to determine the specific activity. The moles of phosphate transferred in the reaction was determined by dividing the counts per minute obtained in the kinase reaction (minus blank) by the specific activity.
Comparative modelling of the H-Ryk receptor tyrosine kinase.
The sequence of H-Ryk was aligned to that of the insulin receptor (IR) tyrosine kinase (IRK; Brookhaven PDB code 1irk) by using the AMPS package (3), and the alignment was sent to the SWISSMODEL automated homology modelling server (54). Since the model was generated automatically, the conformation of most side chains in H-Ryk resembled the counterparts in IRK closely. Three nonconservative mutations within H-Ryk were thus modelled manually, using QUANTA (version 4.0; Molecular Simulations Inc.). Suitable side chain rotamers were chosen from the database (56) to model a putative interaction between (Gly 1003, IRK) Gln 307 (H-Ryk, 5th rotamer: χ1 = 71, χ2 = −166, χ3 = 27) and (Phe 1151, IRK) Asn 454 (4th rotamer: χ1 = 64, χ2 = −7). A suitable rotamer for Phe 332 (Ala 1028, IRK) was also chosen to accommodate the Ala-Phe change in H-Ryk.
RESULTS
Characterization of cells expressing the wild-type and mutant TrkA:Ryk chimeras.
A TrkA:Ryk chimeric construct consisting of the extracellular domain of the TrkA receptor (nucleotide residues 1 to 1444 according to accession no. M23102) and encompassing the H-Ryk transmembrane-to-carboxyl tail region (nucleotide residues 647 to 2300 according to accession no. X96588) was made as shown in Fig. 2A. To analyze the role of the individual amino acid alterations of the H-Ryk catalytic domain, we used a PCR-mediated primer extension overlap method to introduce point mutations into the open reading frame of the H-Ryk receptor. The resultant mutant fragments were fused to the extracellular domain of the TrkA receptor in order to create the various chimeric constructs (Fig. 2A). To allow functional analysis of the TrkA:Ryk chimeras, constructs encoding the wild-type and mutant receptors were transfected into NIH 3T3 cells, and stable G418-resistant clones were selected. Selection of the NIH 3T3 mouse fibroblast cell line was based on the fact that it does not express TrkA and therefore there is no interference due to endogenous receptor activity. To confirm the expression of the chimeras, lysates from the respective stable cell lines were immunoprecipitated with 15.2, a polyclonal antibody that recognizes the carboxyl-terminal region of H-Ryk (77). Immunoprecipitates were separated, transferred, and then immunoblotted with MAb 5C3, which recognizes the extracellular domain of human TrkA (41). MAb 5C3 recognizes the TrkA component of the chimeric receptor only under nonreducing conditions. A protein of approximately 125 kDa was detected in all of the NIH 3T3 cell lines transfected with wild-type and mutant chimeras but not in the parental NIH 3T3 cell line (Fig. 2B). The molecular weight of the chimeric receptor determined under nonreducing conditions is slightly larger than that of the 116-kDa protein that is detected under reducing conditions.
FIG. 2.

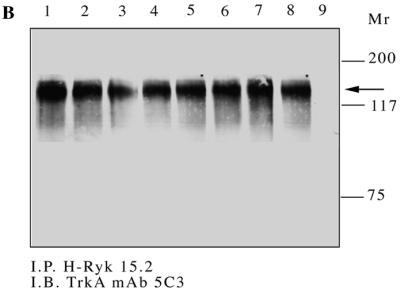
Characterization of the TrkA:Ryk chimera-expressing stable cell lines. (A) Schematic diagram of the chimeric fusion constructs consisting of the TrkA extracellular domain (nucleotides 1 to 1444) and the H-Ryk transmembrane (T.M.), juxtamembrane (J.M.), kinase (K.D.), and C-terminal (nucleotides 647 to 2300) domains. The TrkA:Ryk chimeric mutants are labelled according to amino acid change and location; thus, mutant Q307G corresponds to a glutamine-to-glycine change at amino acid 307. (B) Detection of chimeric receptors in NIH 3T3 transfectants. Equal amounts of cell extracts from the NIH 3T3 transfectants were immunoprecipitated (I.P.) with the anti-C-terminal H-Ryk antibody 15.2 and then resolved on an SDS–7.5% polyacrylamide gel under nonreducing conditions. The chimeric receptor proteins were detected by immunoblotting (I.B.) with anti-N-terminal TrkA MAb 5C3. The observed molecular mass (125 kDa) is slightly higher than that observed under reducing conditions. Lanes 1 to 9 represent TrkA:Ryk wild type, TrkA:Ryk Q307G, TrkA:Ryk F332A, TrkA:Ryk K4343R, TrkA:Ryk N454F, TrkA:Ryk A455G, TrkA:Ryk N454F/A455G, TrkA:Ryk Q307G/N454F/A455G chimeras, and the parental NIH 3T3 cell line respectively.
To ensure that the Trk-A:Ryk chimeric receptors were correctly synthesized and transported to the cell surface, the clonal cell lines were analyzed by immunostaining and FACS to determine receptor density. Immunostaining of the stable cell lines with MAb 5C3 indicated that the chimeric receptors were properly processed and transported to the cell surface (data not shown). The receptor density determinations were carried out at 4°C to prevent internalization of receptors. The number of receptors expressed was comparable to that for the NIH 3T3 TrkA-derived clone E25-4-27, which expresses approximately 3.1 × 104 receptors per cell (38, 44) (Table 1).
TABLE 1.
Receptor densities of TrkA:Ryk chimera-expressing stable cell linesa
| Chimera expressed by transfectant | No. of receptors (104/cell) |
|---|---|
| TrkA:Ryk wild type | 2.90 ± 9,600 |
| TrkA:Ryk N454F | 2.75 ± 13,000 |
| TrkA:Ryk A455G | 2.70 ± 10,500 |
| TrkA:Ryk N454F/A455G | 2.95 ± 7,500 |
| TrkA:Ryk Q307G | 2.80 ± 13,200 |
| TrkA:Ryk Q307G/N454F/A455G | 2.75 ± 12,500 |
| TrkA:Ryk F332A | 2.45 ± 12,700 |
| TrkA:Ryk K434R | 2.50 ± 9,700 |
| TrkA (E25-4-27) | 3.10 ± 11,500 |
Summary of FACSscan analysis of surface receptors expressing the TrkA ectodomain.
The TrkA:Ryk chimeric receptor is catalytically inactive.
To investigate the intrinsic tyrosine kinase activity of H-Ryk, the TrkA:Ryk immune complex from the TrkA:Ryk chimeric receptor-expressing stable cell line was subjected to an in vitro kinase assay. No phosphorylated band corresponding to the TrkA:Ryk receptor was detected in the immune complex kinase assay. In contrast, an in vitro kinase assay of the TrkA immune complex from the NIH 3T3 TrkA-expressing stable cell line (E25-4-27) revealed an autophosphorylated band of 140 kDa corresponding to the TrkA receptor (Fig. 3A to C). To investigate whether the TrkA:Ryk receptor had exogenous kinase activity, the TrkA:Ryk immune complexes before and after NGF induction were subjected to an in vitro kinase assay in the presence of either a synthetic Src tyrosine kinase peptide (R-R-L-I-E-D-A-E-Y-A-A-R-G) or poly(Glu80Tyr20) substrate. There was no evidence of TrkA:Ryk-mediated tyrosine kinase exogenous activity in the substrate assay (Fig. 3D and E). Addition of Mn2+ ions to the reaction had no effect on the exogenous kinase activity of the TrkA:Ryk chimeric receptor. In contrast, the TrkA immune complex was able to phosphorylate both substrates, as demonstrated by the incorporation of [γ-32P]ATP in each substrate assay (Fig. 3D and E).
FIG. 3.
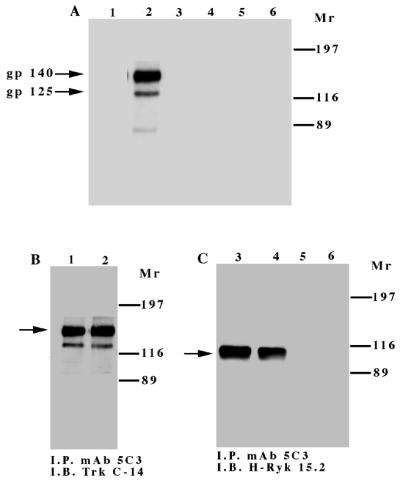

In vitro kinase assay of the TrkA and TrkA:Ryk chimeric receptors. Cells were incubated in DMEM supplemented with 0.1% FCS for 24 h prior to immunoprecipitation (I.P.) with MAb 5C3. Half of the sample was used in the in vitro kinase assay (A), while the other half was used to determine the relative amounts of the receptors by Western immunoblotting (I.B.) (B and C). Lanes 1 and 2 in panels A and B represent the TrkA receptor, lanes 3 and 4 in panels A and C represent the TrkA:Ryk chimeric receptor, and lanes 5 and 6 in panels A and C represent the immune complex from the NIH 3T3 parental cell line. Lanes 1, 3, and 5 and lanes 2, 4, and 6 represent before and after 30 min of stimulation with NGF, respectively. The exogenous tyrosine kinase assay was carried out in the presence of 10 mM synthetic Src peptide (D) and 0.2 mg of poly(Glu80Tyr20) substrate per ml (E). The assay was carried out in the presence of 10 mM MgCl2 ions (TrkA and TrkA:Ryk) or 10 mM MgCl2 and 2 mM Mn2+ in the case of the TrkA:Ryk* chimeric receptor. The relative specific activity was determined by calculating the nanomoles of ATP transferred per minute of the reaction by 1.00 mg of total protein.
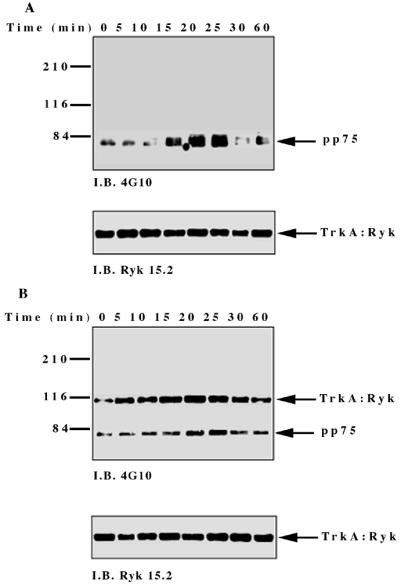
To investigate NGF-induced changes in tyrosine phosphorylation, total cell proteins from the TrkA:Ryk chimera-expressing cell line were immunoblotted with the antiphosphotyrosine antibody. Five major tyrosine-phosphorylated bands, pp115, pp75, pp60, pp42, and pp26, were observed (Fig. 4A). The tyrosine-phosphorylated bands corresponding to the pp42 was confirmed as Erk1/2 by immunoblotting (data not shown). The pp115 band (which corresponds to the approximate molecular weight of the TrkA:Ryk chimera) was not detected by either the H-Ryk polyclonal antibody 15.2 or TrkA MAb 5C3. This analysis indicated that the band did not represent the TrkA:Ryk chimera. The identities of the pp115, pp75, pp60, and pp26 bands are not known. The intrinsic kinase activity of the receptor was further evaluated by stimulating the stable cell line expressing the wild-type TrkA:Ryk chimera with NGF in a time-dependent manner. There was no detectable autophosphorylation of the TrkA:Ryk chimera after 60 min of stimulation of the chimeric receptor with NGF (Fig. 4B). RTKs usually become rapidly phosphorylated upon stimulation with their cognate ligand with the exception of the Eph and Discoidin domain family receptors, which require at least an hour for maximal receptor phosphorylation (18, 26, 64, 76). To eliminate the possibility that the kinetics of H-Ryk autophosphorylation are similar to the kinetics for the Eph and Discoidin domain family receptors, stimulation of cells expressing the chimeric receptor was assayed for up to 4 h. Autophosphorylation of the receptor was not detected after this time period (Fig. 4C). In contrast, upon stimulation of the E25-4-27 NIH 3T3 TrkA-expressing stable cell line with NGF, the gp140 TrkA receptor was stimulated within 5 min, with maximal stimulation being observed after 25 min (Fig. 4D).
FIG. 4.



(A) Analysis of total cell proteins for NGF-induced changes in tyrosine phosphorylation of cells expressing the wild-type TrkA:Ryk chimera. The TrkA:Ryk chimera-expressing cell line was cultured in DMEM supplemented with 0.1% for 16 h and then stimulated with NGF for the time periods indicated. The cells were lysed with 2× SDS sample buffer, and an aliquot of the lysate was resolved by SDS-PAGE. Tyrosine-phosphorylated proteins in the total cell lysate were visualized by immunoblotting (I.B.) with the antiphosphotyrosine antibody 4G10. The major phosphorylated bands are indicated by arrows. (B and C) Tyrosine phosphorylation mediated by the TrkA:Ryk chimeric receptor and the TrkA receptor (D). Cells were incubated for 16 h in DMEM containing 0.1% FCS prior to stimulation with NGF (100 ng/ml) for the time period indicated. After stimulation, the cell extracts were equalized for protein content, immunoprecipitated with TrkA MAb 5C3, and immunoblotted with antiphosphotyrosine antibody 4G10. The arrows indicate the coimmunoprecipitated phosphotyrosine-containing 75-kDa protein. The blots were stripped and blotted with either the H-Ryk anti-C terminal antibody (B and C, bottom panel) or the TrkA antibody (D, bottom panel). (E) Immunoprecipitation analysis of the wild-type TrkA:Ryk chimeric cell line under denaturing conditions. The chimera-expressing cells were made quiescent by incubation for 16 h in DMEM–0.1% FCS for 16 h. The cells were lysed in denaturing buffer (boiling SDS buffer) prior to immunoprecipitation (I.P.). Immunoblotting analysis with 4G10 (top panel) and H-Ryk 15.2 (bottom panel) shows absence of pp75 and presence of the TrkA:Ryk chimeric receptor, respectively.
However, despite the lack of TrkA:Ryk receptor autophosphorylation, a coimmunoprecipitating 75-kDa phosphoprotein (pp75) was observed after stimulation of the TrkA:Ryk cell line. pp75 was observed at the basal level after 16 h of serum starvation, suggesting that this association may be ligand independent. The p75 phosphoprotein is unlikely to be an artifact, as two independent TrkA antibodies (5C3 and 220) (41, 44) repeatedly coimmunoprecipitated pp75 from the TrkA:Ryk chimera-expressing cell line. In addition, pp75 was not observed after immunoprecipitation of the parental NIH 3T3 cell line or the TrkA-expressing cell line (E25-4-27) with the TrkA-specific MAb 5C3 (Fig. 4C). It is well established that NGF binding can be mediated by both the high-affinity gp140 TrkA receptor and the low-affinity p75 NGF receptor (1, 31). Therefore, to rule out the possibility that pp75 observed upon stimulation of the TrkA:Ryk chimera is the low-affinity p75 NGF receptor, the TrkA:Ryk chimeric immunoprecipitates were immunoblotted with the p75 low-affinity receptor antibody. No detectable expression of the p75 low-affinity receptor was observed (data not shown). These observations suggest that pp75 associates with the Ryk intracellular part of the TrkA:Ryk chimera. This inference is further supported by the inability to immunoprecipitate pp75 from the TrkA:Ryk chimeric receptor-expressing cell line after lysis in denaturing buffer (Fig. 4E).
The catalytically impaired H-Ryk receptor signals through the MAPK pathway.
The observation that overexpression of the H-Ryk receptor leads to in vitro transformation of NIH 3T3 cells and tumor formation in vivo (35) raised the possibility that H-Ryk activates downstream signalling pathways. The conserved MAPK signal transduction pathway plays a central role in cell behavior since depending on cellular context it can induce either cell proliferation or differentiation (15, 16, 43, 55). Erk kinase is activated by phosphorylation on both Tyr and Thr residues (43), which can be used to assess its activation. Upon ligand stimulation of the TrkA:Ryk chimera, phosphorylation of Erk was first detected after 10 min, with maximal stimulation after 25 min (Fig. 5A). Consistent with the lack of TrkA receptor expression, no Erk kinase activation was observed in the parental NIH 3T3 cell line after induction with NGF (data not shown). To verify the activation of the extracellular regulated kinases, we assayed the specific activity of p90 Rsk3, one of the ribosomal S6 kinases which lies downstream of the Erk kinases (82). p90 Rsk3 specific activity was stimulated threefold after 20 min of induction with NGF (Fig. 5B). This is consistent with the expected two- to fourfold induction of p90 Rsk3 observed after stimulation with other growth factors such as epidermal growth factor (EGF) as previously published (82, 83).
FIG. 5.




Activation of the catalytically inactive TrkA:Ryk chimera stimulates the MAPK pathway. The TrkA:Ryk chimera-expressing cell line was made quiescent by incubation in DMEM supplemented with 1% FCS for 24 h followed by another 24 h of incubation in DMEM containing 0.1% FCS prior to stimulation with 100 ng of NGF per ml. The lysates were immunoprecipitated (I.P.) with the Erk1- or Rsk3-specific antibodies and subjected to either immunoblotting (I.B.) with the antiphosphotyrosine antibody (A) or the S6 peptide kinase assay (B). (C) Tyrosine phosphorylation mediated by the TrkA:Ryk K334A chimeric receptor. The TrkA:Ryk K334A chimera-expressing cell line was serum starved for 16 h prior to stimulation with NGF for the time periods indicated. Immunoblotting with 4G10 indicates significantly lower levels of coimmunoprecipitating pp75 compared to the wild-type TrkA:Ryk chimera (prolonged exposure, 30 min). The bottom panel indicates the relative levels of TrkA:Ryk K334A chimera immunoprecipitated. The dominant negative K334A mutant abolishes Erk (D) and p90 Rsk3 (G) activation. Total cellular proteins were resolved by SDS-PAGE and immunoblotted with phospho-Erk (D) and Erk (E). This analysis revealed the absence of Erk activation. (F) Total cellular lysates were equalized for protein content prior to immunoprecipitation with the p90 Rsk3 antibody. Half of the immune complex was used for immunoblot analysis, while the other half was used in the S6 peptide kinase assay (G). The S6 peptide kinase assay demonstrates that the TrkA:Ryk K334A chimera abolishes p90 Rsk3 activation.
To conclusively establish whether residual low H-Ryk catalytic activity is required for MAPK activation, we mutated the invariant lysine in subdomain II to alanine and constructed the TrkA:Ryk K334A chimera. There was no autophosphorylation of the chimeric receptor upon stimulation with NGF, similar to findings for the wild-type TrkA:Ryk receptor. In addition, significantly lower levels of pp75 were coimmunoprecipitated from the TrkA-Ryk K334A chimera-expressing cell line (Fig. 5C). Further, NGF stimulation of the TrkA:Ryk K334A chimera failed to induce phosphorylation of both Erk1/2 and p90 Rsk3 (Fig. 5D to G).
The activation domain residues phenylalanine and glycine are essential for receptor autophosphorylation.
We next wanted to determine the relevance of each of the amino acid changes in the activation loop of H-Ryk. To examine whether H-Ryk catalytic activity could be restored by alteration of the asparagine residue to phenylalanine in the DNA motif, the mutant TrkA:Ryk N454F chimera was constructed by in vitro mutagenesis. Autophosphorylation of the receptor was detected after 5 min and peaked after 20 min (Fig. 6A). Surprisingly, mutation of the alanine to glycine (A455G) also restored the kinase activity of the Ryk receptor (Fig. 6B). A double-mutant TrkA:Ryk N454F/A455G chimera was, as expected, kinase active (Fig. 6C). To determine if activation domain mutants had exogenous catalytic activity, the immune complexes from the mutants were subjected to a kinase assay using the substrate poly(Glu80Tyr20). All of the mutants were able to phosphorylate the exogenous substrate, albeit to different extents (Fig. 6D). However, no significant differences in specific activities of the various mutants were observed. In addition, the activation domain mutants induced Erk MAPK in response to NGF stimulation with stronger kinetics of activation than the wild-type TrkA:Ryk chimeric receptor, possibly as a consequence of the reactivated autophosphorylation activity of the activation domain mutants (Fig. 6E to G).
FIG. 6.



Analysis of the kinetics of tyrosine phosphorylation of the TrkA:Ryk activation domain chimeric mutants in response to NGF. TrkA:Ryk N454F (A), TrkA:Ryk A455G (B), and TrkA:Ryk N454F/A455G (C) chimera-expressing cell lines were made quiescent by incubation in DMEM medium supplemented with 0.1% FCS for 16 h and then stimulated with 100 ng of NGF per ml for the time periods indicated. The tyrosine-phosphorylated chimeric and associated proteins are indicated with arrows (top panel). The blots were stripped and immunoblotted (I.B.) with H-Ryk polyclonal antibody 15.2 (bottom panel). (D) Phosphorylation of exogenous substrate by the activation domain mutants. The synthetic substrate polyGlu80Tyr20 was phosphorylated with labelled ATP. Incorporation of ATP into the substrate was determined by a filter paper assay. The graph represents the mean of two independent experiments. (E to G) Activation of the Erk MAPK pathway by the TrkA:Ryk N454F (E), A455G (F), and N454F/A455G (G) chimeric receptors, respectively.
Alteration of the glutamine residue to glycine in the nucleotide binding loop does not restore catalytic activity.
The glycine-rich loop, GxGxxG, in the ATP binding site is one of the most conserved sequence motifs in protein kinases. To assess the effect of the glutamine substitution in H-Ryk, the autophosphorylation activity of the Q307G mutant TrkA:Ryk chimera was determined by induction with NGF. Alteration of the glutamine residue to glycine had no detectable effect on the autophosphorylation activity of the receptor (Fig. 7A). Although the Q307G mutation is a drastic change in that it replaces a large side chain with hydrogen, this alteration on its own presumably cannot restore catalytic activity. A cooperative effect with the DFG tripeptide motif may be required to restore activity. This possibility is supported by the kinase activity of the triple-mutant TrkA:Ryk Q307G/N454F/A455G chimera (Fig. 7B), wherein receptor autophosphorylation is restored. In vitro tyrosine kinase activity of the chimeric receptors as measured by phosphorylation of the exogenous poly(Glu80Tyr20) demonstrated kinase activity of the TrkA:Ryk Q307/N454F/A455G mutant. In contrast, no tyrosine kinase activity was observed for the TrkA:Ryk Q307G chimera (Fig. 7C). However, NGF stimulation of both chimeras resulted in Erk activation, again reaffirming that H-Ryk autophosphorylation activity does not appear to be required for activation of the MAPK pathway (Fig. 7D and E).
FIG. 7.


Stimulation of the TrkA:Ryk Q307G (A) and TrkA:Ryk Q307G/N454F/A455G (B) chimera-expressing cell lines in response to NGF. Serum-starved cells (DMEM, 0.1% FCS, 16 h) were stimulated with NGF (100 ng/ml). After cell lysis, the cell lysates immunoprecipitated with TrkA MAb 5C3. Immunoprecipitates were analyzed by Western immunoblotting (I.B.) with the antiphosphotyrosine antibody (top panel) and blotted with the H-Ryk C-terminal antibody (bottom panel). (C) Phosphorylation of the synthetic poly(Glu80Tyr20) substrate by the TrkA:Ryk Q307 and Q307G/N454F/A455G chimeric mutants. The graph represents the mean of two independent experiments. (D and E) Activation of the Erk MAPK pathway by the TrkA:Ryk Q307 (D) and Q307G/N454F/A455G (E) chimeric mutants in response to NGF stimulation for the time period indicated.
The invariant arginine residue in the catalytic loop and the conserved phenylalanine in subdomain II are not essential for catalysis.
The alanine residue in subdomain II (xVAV/LK, where x represents any amino acid) of the protein kinase domain represents a semiconserved residue. To determine the significance of this substitution, the TrkA:Ryk F332A chimera was stimulated with NGF. No detectable autophosphorylation of the receptor was observed after 60 min of induction with NGF (Fig. 8A). However, introduction of the F332A mutation in conjunction with the N454F mutation restored receptor autophosphorylation, again emphasizing the role of the phenylalanine residue in the activation domain in catalysis (data not shown).
FIG. 8.


The TrkA:Ryk F332A (A) and TrkA:Ryk K434R (B) chimeric receptors are catalytically inactive. NIH 3T3 cell lines expressing the various chimeras were made quiescent by incubating them in DMEM supplemented with 0.1% FCS for 16 h. Immunoprecipitates were analyzed for tyrosine phosphorylation by immunoblotting (I.B.) with the antiphosphotyrosine antibody (top panel). The coimmunoprecipitated p75 protein is indicated with an arrow. The blot was stripped and probed with the H-Ryk C-terminal antibody (bottom panel). (C) Substrate phosphorylation assay of the TrkA:Ryk F332A and TrkA:Ryk K434R chimeric receptors. The graph represents the mean of two independent experiments.
No receptor autophosphorylation was detected after stimulation of the TrkA:Ryk K434R chimera (Fig. 8B). The arginine residue (IHRDLAARN) represents one of the invariant residues in the tyrosine kinase family. In the tertiary structure of the serine threonine kinase, cyclic AMP (cAMP), this residue is important in the stabilization of the phosphorylated threonine residue (39). However, in the fibroblast growth factor receptor (FGFR) and IRK structures, arginine does not have a stabilization role in the phosphorylated forms of the receptors, suggesting that the residue plays no role in catalysis in the RTK family (29, 30, 46). This supports our observation, as restoration of this residue had no effect on the kinase activity of the mutant chimeric receptor. Analysis of the in vitro protein tyrosine kinase activity of each chimeric receptor demonstrated lack of tyrosine kinase activity, consistent with lack of intrinsic tyrosine kinase autophosphorylation activity (Fig. 8C).
Homology modelling of the H-Ryk receptor.
Recently, the tertiary structures of the IR and FGFR kinase domains were reported (29, 30, 46). Sequence similarity suggests that gross structural details of the IRK catalytic domain are likely to hold for H-Ryk. In the absence of an experimentally determined three-dimensional structure, an approximate model of the tertiary structure of H-Ryk can be made via homology modelling. The coordinates of IRK and an alignment of H-Ryk and IRK were used to build a model (see Materials and Methods). Regions around the active site were scrutinized to ensure that automatically assigned residue replacements had reasonable side chain rotamers and that nonconservative substitutions (e.g., hydrophobic for polar) were suitably accommodated in the model (Fig. 9).
FIG. 9.

Homology modelling of the H-Ryk catalytic domain. Alignment of sequences of H-Ryk and IRK and manual modelling of three nonconservative mutations within H-Ryk were done as described in Materials and Methods. Corresponding residues of IRK (A) and H-Ryk (B) are labelled. The model is magnified to display the cleft between the two lobes.
Two unusual features in H-Ryk involve the change of a highly conserved glycine. The glycine-to-glutamine change in the glycine-rich loop of H-Ryk is drastic in that it replaces a hydrogen with a large side chain. Glycine, due to its lack of a side chain, is able to adopt a wider range of backbone conformations. The glycine residue 1003 in IRK has backbone ψ/φ values of −178 and 149°, respectively, which occurs in a disallowed region of ψ/φ space for nonglycine residues (5). Thus, the substitution of glycine for glutamine in H-Ryk would likely be accompanied by changes in the backbone (ψ/φ) conformation (Fig. 9). Gly 1152 in IRK has backbone ψ/φ values of 50.0 and 112.7° and is also in a region of ψ/φ space forbidden to nonglycine residues (5). The glycine-alanine mutation in the highly conserved DFG tripeptide is conservative in that glycine and alanine have similar physicochemical properties. However, the change to alanine is likely to be accompanied by some changes in backbone (ψ/φ) conformation.
With the exception of the lysine-for-arginine substitution in the catalytic loop, all of the amino acid changes in H-Ryk cluster around the active site of the enzyme. The change of the mostly conserved Ala (Ala 1028, IRK) in subdomain II to Phe puts a large side chain into a space normally occupied by Phe from the DFG loop. The Phe-Asn change seen in H-Ryk replaces a hydrophobic residue by a polar one, and we thus predict that this side chain, which would be partly displaced by the Ala-Phe substitution, would be forced into a more solvent exposed position (Fig. 9). This would put it closer to the Gln from the glycine rich loop and may even form an electrostatic interaction. The interaction between the glycine-rich loop and the activation domain is mediated by the conserved Gly 1003 and residues Phe 1151 and Gly 1152 in the IRK structure (29, 30). In the homology model of H-Ryk, this interaction appears to be mediated by Gln 307 (Gly 1003, IRK) and Asn 454 (Phe 1151, IRK). The net effect of all these changes would be to add approximately nine noncarbon atoms to the region around the kinase active site. Such changes, together with the alterations of the highly conserved glycines, would likely disrupt the precise association of the two kinase lobes, which is essential for proper catalytic activity.
DISCUSSION
Recently a number of RPTKs with alterations to conserved and invariant amino acids in the catalytic domain have been cloned. These include Cck-4, Mep (Eph-B6), Ror 1, and Ror 2 (21, 45, 48, 80). Similar to H-Ryk, these RPTKs all have sequence alterations in either the nucleotide binding, catalytic, or activation domains (Fig. 1). This subfamily of atypical RPTKs also share a common feature: they are all non-RD kinases, a category which essentially encompasses those kinases in which the aspartate in the catalytic loop (IHRDLAARN) is not preceded by the arginine residue (33). The distinguishing feature of these kinases is that they do not appear to be regulated by phosphorylation in the activation segment, in direct contrast to the RD kinases (i.e., MAPK, IRK, and FGFR kinase) (33). The intrinsic protein tyrosine kinase activity of this subfamily and the way in which these putative receptors mediate their biological functions remain undetermined. It has been demonstrated previously by an in vitro kinase assay that a FLAG:Ryk fusion protein expressed in bacteria does not have intrinsic kinase activity (28). This observation is consistent with the in vitro experimental findings of this study. In the absence of a ligand, a chimeric receptor approach using the NGF receptor TrkA was used to analyze the function of H-Ryk. The inability to identify any detectable autophosphorylation of the chimeric receptor after 4 h of stimulation with NGF suggests that H-Ryk is catalytically inactive under these experimental conditions. This possibility is supported by the inability of H-Ryk to phosphorylate exogenous substrate.
The Ryk RTK has been implicated in a number of developmental processes, and its conservation across species, as evidenced by its widespread expression (28, 32, 36, 40, 42, 51, 52, 57, 63, 65, 67, 70, 77, 78, 81), suggests that it plays a fundamental role in development. H-Ryk expression has been demonstrated to be regulated during hematopoietic development by lineage commitment and stage maturation (65). In the ovary, H-Ryk is overexpressed in malignant epithelial ovarian tumors and NIH 3T3 H-Ryk-overexpressing stable cell lines are transforming in vivo and in vitro (35, 77). The Drosophila homologue Drl has been implicated in both neuronal pathway and muscle attachment site selection (6, 7). To carry out these functions, H-Ryk must be able to transduce a signalling cascade upon stimulation which leads to either proliferation or differentiation. We demonstrate in this report the activation of Erk1/2 and the downstream p90 Rsk3 kinase upon stimulation of the TrkA:Ryk chimera with NGF, which is consistent with the ability of Ryk to carry out these biological functions. However, the intriguing question is how the kinase-impaired H-Ryk receptor is able to transduce its signals to the MAPK pathway. One possibility may be that H-Ryk uses members of the Src and Jak nonreceptor tyrosine kinases to propagate its intracellular signals, analogous to the way cytokine and T-cell receptors mediate their biological functions. Unlike receptor tyrosine kinases, cytokine and T-cell receptors do not have intrinsic tyrosine kinase activity but still retain the ability to activate downstream signalling pathways by coupling to the Jak family of cytoplasmic kinases and nonreceptor tyrosine kinases, respectively (13, 59, 79). However, analysis of the TrkA:Ryk immune complexes revealed no presence of coimmunoprecipitating Jak 1, 2, or 3 or the nonreceptor tyrosine kinases Src and Lck after NGF induction of the chimeric receptor-expressing cell line (data not shown). Further, there were no temporal changes in phosphoproteins identified in the chimeric TrkA:Ryk cell lysates upon ligand stimulation (Fig. 4A) compared to basal levels.
A highly conserved feature of both serine-threonine and tyrosine kinases is the invariant lysine residue, which is K334 in H-Ryk and is located carboxyl terminal to the Gly-rich loop. This residue interacts with the α and β phosphates of ATP and is critical for the proper alignment of the triphosphate chain in the active site to promote the in-line phosphotransfer reaction. Mutation of the invariant lysine abolishes catalytic activity of kinases while only moderately reducing the ability to bind ATP (19, 72). In our study, we demonstrate that the K334A mutant chimeric receptor does not induce the MAPK pathway. The simplest explanation of this result is that H-Ryk retains very low autophosphorylation activity that is not detectable under the present experimental conditions, which enables it to transduce signals through the MAPK pathway. H-Ryk might phosphorylate a specific substrate(s) which acts as an adapter or auxiliary protein in the activation of downstream signalling pathways. It is tempting to speculate that pp75 acts as a conduit between H-Ryk and the downstream signalling components. This proposition is supported by the inability of the TrkA:Ryk K334A chimera to coimmunoprecipitate pp75 and the concomitant loss of MAPK activation. However, until pp75 is identified and the nature of its association with H-Ryk is characterized, this remains mere speculation. Alternatively, H-Ryk may transmit downstream signals independent of receptor autophosphorylation. This possibility is supported by the inability of the TrkA:Ryk chimeric receptor to bind the nonhydrolyzable ATP analogue 5′-p-fluorosulfonylbenzoyl adenosine (FSBA) (data not shown). However, to our knowledge, no RTK which is capable of inducing downstream signalling in the absence of receptor autophosphorylation or catalytic activity has been identified. Recently, the cyclin-dependent kinase-activating kinase in budding yeast, Cak1p, has been shown to be functionally active in vitro and in vivo despite mutation of the invariant lysine residue (11, 14, 75). This finding suggests that the invariant lysine residue in Cak1p is not required for catalysis. It is possible that the effect of the K334A mutation in the TrkA:Ryk chimera is to alter the compensated structure of the catalytic domain of H-Ryk, inhibiting any interaction with putative substrates. Further, for some functions such as the muscle attachment site selection in the Drosophila homologue Drl, the catalytic domain is not required (4a).
The impaired TrkA:Ryk catalytic activity suggests that in certain cellular contexts, Ryk may use nonphosphorylation-dependent mechanisms in vivo to couple Ryk activation to downstream pathways. The recently characterized PDZ protein recognition module represents such a mechanism. PDZ proteins are modular proteins that act as adapters by selectively attaching through their PDZ domains to the C termini of membrane receptors while also binding to signalling proteins via PDZ or other protein modules (12). The C-terminal consensus sequence X-Ser/Thr-X-Val-COOH, where Val-COOH represents the C-terminal valine residue, was initially proposed for PDZ domains (37). However, there appear to be additional determinants for binding, as different PDZ domains can distinguish between binding partners containing identical sequence motifs, and additional C-terminal motifs such as V-K-I and Y-Y-V have also been shown to bind PDZ domains (27, 66). The consensus sequence motif G-A-Y-V-COOH, which is conserved in mouse and human Ryk, provides a potential PDZ binding domain. The recent identification of a potential PDZ domain that binds to the C terminus of the Drosophila Ryk homologue Drl (62) in the consensus sequence T-R-Y-V-COOH suggests that a similar mechanism may exist in mammals. This would either enable the coupling of the receptor to downstream signalling pathways or cluster the Ryk receptor in specific subcellular positions which contain its substrates or are critical for its proper functioning.
Most RTKs are able to directly recruit the Grb2-Sos complex which is central to the activation of the Ras/MAPK pathway upon stimulation. However, the recruitment of the Grb2-Sos complex can also occur indirectly via tyrosine phosphorylation of Shc as is the case for the EGF receptor (50). Insulin stimulation, on the other hand, leads to tyrosine phosphorylation of Shc and IRS1, and it is both of these proteins that recruit the Grb2-Sos complex (58, 68). The type I family of receptors, in contrast, extend their signalling diversity by transphosphorylation of the kinase-impaired c-ErbB3, which leads to the recruitment of phosphatidylinositol 3-kinase (8). The kinase-impaired c-ErbB3 receptor therefore modulates signals of the type I family of receptors by heterodimerization with c-ErbB2 and c-ErbB4, which are kinase active. The mapping of Ryk to two distinct loci, Ryk-1 and Ryk-2, on human chromosomes 3q11-22 and 17p13.3, respectively, raises the possibility that there is a kinase-active partner for the Ryk-1 locus cDNA analyzed in this study (20, 69). If the Ryk-2 locus does harbor a kinase-active partner, one would speculate that heterodimerization of the two receptors would be sufficient to activate the MAPK pathway. Whether the coimmunoprecipitating pp75 represents the Ryk-2 locus receptor remains to be determined, as the cDNA for Ryk-2 has not been isolated. However, the inability of the pp75 to cross-react with the Ryk polyclonal antibody 15.2 raises doubts as to whether this protein is closely related to Ryk (data not shown).
Irrespective of the variation in specificity, the activation segment is important in aligning the catalytic residues in the protein kinase family. It has been previously demonstrated that the aspartate residue of the tripeptide DFG motif in the activation segment is critical for receptor autophosphorylation. Even relatively conservative mutations of the aspartate residue to glutamate or asparagine in v-Fps yielded an inactive kinase (47). In this study we have addressed the role of both the Phe and Gly residues in receptor autophosphorylation. The interaction between the Phe and Gly residues of the DFG tripeptide and the second glycine of the nucleotide binding loop are particularly important for correct orientation of the C- and N-terminal lobes and hence the high degree of conservation of these residues in the protein kinase family (29, 30, 46). In H-Ryk, the Asn residue in the tripeptide motif DNA would probably be accommodated by outward movement of the polar Asn side chain. This may result in the Asn interacting with the glutamine from the nucleotide binding motif (QxGxxG). The net effect of the side chain replacements is thus likely to be a congestion of the active-site cleft (Fig. 9). This together with the Gly backbone changes is likely to produce a disruption of the interdomain orientation. Restoration of either the Phe or the Gly residue is sufficient to induce the catalytic activity of the H-Ryk receptor. Presumably, restoration of these residues allows for proper orientation of the lobes, thereby enabling the receptor to autophosphorylate. In particular, the N454F mutant apparently results in the nonpolar residue Phe being buried in the active-site cleft, which would correct the disruption of the interdomain orientation brought about by the Asn residue. We speculate that the N454F mutant restores the interaction of the Phe residue of the activation domain with Gly residue of the nucleotide binding motif and subsequently the proper orientation of this lobe. The Ala-to-Gly amino acid change is often viewed as a conservative substitution owing to the similarity in physicochemical properties. However, Gly residues, particularly those that are highly conserved, play a structurally very important role in proteins owing to their ability to adopt main chain ψ/φ conformations forbidden to other amino acids, including alanine. The A455G mutation would result in the adoption of a wider range of backbone confirmations. Presumably the TrkA:Ryk A455G chimera adopts a confirmation that results in the proper alignment of the two lobes, thereby overcoming catalytic constraints brought about by the alterations in this domain. Therefore, in addition to the critical Asp, either the Phe or the Gly residue is required to maintain the proper orientation of the two lobes, as demonstrated by the N454F and A455G mutants in this study.
We have demonstrated that restoration of the Gly residue in the nucleotide binding loop of H-Ryk does not restore the catalytic activity of the receptor. Although this mutation on its own is sufficient to affect the catalytic activity of the receptor, it is not dominant enough to completely abolish its activity. This view is supported by the study carried out by Hemmer et al., in which they demonstrated that substitution of the first glycine in protein kinase A caused a five- to eightfold reduction in ATP binding and a three- to fivefold drop in catalytic activity (24). This is broadly supported by sequence analysis and mutational studies which predicts a gradation of functional importance of the three glycines (Gly 2 ≫ Gly 1 > Gly 3). Therefore, although the glutamine substitution is expected to have an adverse effect on ATP binding and catalysis, this amino acid alteration on its own does not account for the loss of intrinsic kinase activity of the H-Ryk RTK. It is possible therefore that although the Q307G mutant may affect ATP binding, it is unlikely to have a significant effect on the overall catalytic activity due to the mutations in the activation segment of the H-Ryk catalytic domain.
The arginine residue in the catalytic loop (IHRDLAARN) represents one of the most invariant amino acid residues in the tyrosine kinase family. In the cAMP kinase structure, the residue has been implicated in the stabilization of the phosphorylated threonine (39). However in the FGFR structure, stabilization of the phosphorylated tyrosine is mediated by the conserved Pro residue 663 (Pro 475, H-Ryk) (46). Mutation of this residue to glutamine in the IRK structure results in impaired kinase activity. In the crystal structures of both the FGF and IRK receptors, the arginine residue does not appear to play a role in catalysis. The results of this study appear to support that finding as the K434R mutant does not have any effect on the kinase activity of the H-Ryk receptor. The same applies to the substitution of alanine with phenylalanine in subdomain II of H-Ryk. In H-Ryk, the Ala-to-Phe change in subdomain II (VFV/LK) of H-Ryk is likely accommodated by placement of the Phe side chain in the same location as the Phe from DFG in other kinases. The presence of the Phe residue in subdomain II appeared to contribute to the bulking up of the active-site cleft in the comparative model of the H-Ryk receptor (Fig. 9). However, restoration of the alanine residue does not have an effect on the kinase activity of the receptor. This residue does not compensate for the absent Phe residue in the DFG mutant, as demonstrated by the lack of phosphorylation of the wild-type TrkA:Ryk chimera.
In conclusion, this first detailed functional analysis of an atypical receptor tyrosine kinase with impaired catalytic function suggests an unusual mechanism by which signals can be transduced independent of receptor autophosphorylation. Further, the results extend the observation of the requirement of the DFG motif in substrate catalysis. The focus of future investigations will be on characterizing the mechanism by which Ryk transduces signals to downstream signalling proteins and determining the tertiary structure of H-Ryk.
ACKNOWLEDGMENTS
We thank M. Barbacid for generously providing the TrkA MAb, TrkA cDNA, and TrkA-expressing stable cell line, E25-4-27. We are grateful to Uri Saragovi for providing the N-terminal TrkA MAb and to Mary Gregoriou and Mike Sternberg for helpful discussions on the crystal model. We also acknowledge Chris Norbury, Julian Downward, and Elisabeth Trivier for critical reading of the manuscript.
This work was supported by the Imperial Cancer Research Fund. R.M.K. is a recipient of the Valerie Rosina Howell Fellowship.
REFERENCES
- 1.Barker P A, Murphy R A. The nerve growth-factor receptor a multi-component system that mediates the actions of the neurotrophin family of proteins. Mol Cell Biol. 1992;110:1–15. doi: 10.1007/BF02385000. [DOI] [PubMed] [Google Scholar]
- 2.Barton G J. “ALSCRIPT”—a tool to format multiple sequence alignments. Protein Eng. 1993;6:47–50. doi: 10.1093/protein/6.1.37. [DOI] [PubMed] [Google Scholar]
- 3.Barton G J. Protein multiple sequence alignment and flexible pattern matching. Methods Enzymol. 1990;183:403–428. doi: 10.1016/0076-6879(90)83027-7. [DOI] [PubMed] [Google Scholar]
- 4.Bernstein F C, Koetzle T F, Williams G J, Meyer E E, Brice M D, Rodgers J R, Kennard O, Shimanouchi T, Tasumi M. The Protein Data Bank: a computer-based archival file for macromolecular structures. J Mol Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 4a.Bonkowsky, J. L., and J. B. Thomas. Personal communication.
- 5.Branden C, Tooze J. Introduction to protein structure. New York, N.Y: Garland Publishing Inc.; 1991. [Google Scholar]
- 6.Callahan C A, Bonkovsky J L, Scully A L, Thomas B J. derailed is required for muscle attachment site selection in Drosophila. Development. 1996;127:2761–2767. doi: 10.1242/dev.122.9.2761. [DOI] [PubMed] [Google Scholar]
- 7.Callahan C A, Muralidhar M G, Lundgren S E, Scully A L, Thomas J B. Control of neuronal pathway selection by a drosophila receptor protein-tyrosine kinase family member. Nature. 1995;376:171–174. doi: 10.1038/376171a0. [DOI] [PubMed] [Google Scholar]
- 8.Carraway K L, Soltoff S P, Diamonti A J, Cantley L C. Heregulin stimulates mitogenesis and phosphatidylinositol 3-kinase in mouse fibroblasts transfected with erbb2/neu and erbb3. J Biol Chem. 1995;270:7111–7116. doi: 10.1074/jbc.270.13.7111. [DOI] [PubMed] [Google Scholar]
- 9.Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen C, Okayama H. A high efficient system for stably transforming cells with plasmid DNA. BioTechniques. 1988;6:632–638. [PubMed] [Google Scholar]
- 11.Chun K T, Goebl M G. Mutational analysis of Cak1p, an essential protein kinase that regulates cell cycle progression. Mol Gen Genet. 1997;256:365–375. doi: 10.1007/s004380050580. [DOI] [PubMed] [Google Scholar]
- 12.Craven S, Bredt D. PDZ proteins organize synaptic signaling pathways. Cell. 1998;93:495–498. doi: 10.1016/s0092-8674(00)81179-4. [DOI] [PubMed] [Google Scholar]
- 13.Duplay P, Thome M, Herve F, Acuto O. p56lck interacts via its src homology 2 domain with the ZAP-70 kinase. J Exp Med. 1994;179:1163–1172. doi: 10.1084/jem.179.4.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Enke D A, Kaldis P, Holmes J K, Solomon M J. The CDK-activating kinase (Cak1p) from budding yeast has an unusual ATP-binding pocket. J Biol Chem. 1999;274:1949–1956. doi: 10.1074/jbc.274.4.1949. [DOI] [PubMed] [Google Scholar]
- 15.Fabian J R, Morrison D K, Daar I O. Required for Raf and MAP kinase function during the meiotic maturation of Xenopus oocytes. J Cell Biol. 1993;122:645–652. doi: 10.1083/jcb.122.3.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fabian J R, Vojtek A B, Cooper J A, Morrison D K. A single amino acid change in Raf-1 inhibits Ras binding and alters Raf-1 function. Proc Natl Acad Sci USA. 1994;91:5982–5986. doi: 10.1073/pnas.91.13.5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fedi P, Pierce J H, di Fiore P P, Kraus M H. Efficient coupling with phosphatidylinositol 3-kinase, but not phospholipase C gamma or GTPase-activating protein, distinguishes ErbB-3 signaling from that of other ErbB/EGFR family members. Mol Cell Biol. 1994;14:492–500. doi: 10.1128/mcb.14.1.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gale N W, Holland S J, Valenzuela D M, Flenniken A, Pan L, Ryan T E, Henkemeyer M, Strebhardt M, Hirai K, Wilkinson D G. Eph receptors and ligands comprise two major specificity subclasses and are reciprocally compartimentalized during embryogenesis. Neuron. 1996;17:8–19. doi: 10.1016/s0896-6273(00)80276-7. [DOI] [PubMed] [Google Scholar]
- 19.Gibbs C S, Zoller M J. Identification of electrostatic interactions that determine the phosphorylation site specificity of the cAMP-dependent protein kinase. Biochemistry. 1991;30:5329–5334. doi: 10.1021/bi00236a001. [DOI] [PubMed] [Google Scholar]
- 20.Gough N M, Rakar S, Hovens C M, Wilks A. Localization of two mouse genes encoding the protein tyrosine kinase receptor-related protein RYK. Mamm Genome. 1995;6:255–256. doi: 10.1007/BF00352411. [DOI] [PubMed] [Google Scholar]
- 21.Gurniak C B, Berg L J. A new member of the Eph family of receptors that lacks protein tyrosine kinase activity. Oncogene. 1996;13:777–786. [PubMed] [Google Scholar]
- 22.Hanks S K, Hunter T. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 1995;9:576–596. [PubMed] [Google Scholar]
- 23.Hanks S K, Quinn A M, Hunter T. Conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241:42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- 24.Hemmer W, McGlone M, Tsigelny I, Taylor S S. Role of the glycine triad in the ATP-binding site of cAMP-dependent protein kinase. J Biol Chem. 1997;272:16946–16954. doi: 10.1074/jbc.272.27.16946. [DOI] [PubMed] [Google Scholar]
- 25.Ho S N, Hunt H D, Horton R M, Pullen J K, Pease L R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 26.Holland S J, Gale N W, Gish G D, Roth R A, Songyang Z, Cantley L C, Henkemeyer M, Yancopoulos G D, Pawson T. Juxtamembrane tyrosine residues couple the Eph family receptor AphB2/Nuk to specific SH2 domain proteins in neuronal cells. EMBO J. 1997;16:3877–3888. doi: 10.1093/emboj/16.13.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoskins R, Hajnal A, Harp S, Kim S. The C. elegans vulval induction gene lin-2 encodes a member of the MAGUK family of cell junction proteins. Development. 1996;122:97–111. doi: 10.1242/dev.122.1.97. [DOI] [PubMed] [Google Scholar]
- 28.Hovens C M, Stacker S A, Andres A C, Harpur A G, Ziemiecki A, Wilks A F. RYK, a receptor tyrosine kinase-related molecule with unusual kinase domain motifs. Proc Natl Acad Sci USA. 1992;89:11818–11822. doi: 10.1073/pnas.89.24.11818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hubbard S R. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 1997;16:5572–5581. doi: 10.1093/emboj/16.18.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hubbard S R, Wei L, Ellis L, Hendrickson W A. Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature. 1994;372:746–754. doi: 10.1038/372746a0. [DOI] [PubMed] [Google Scholar]
- 31.Huber L J, Chao M V. A potential interaction of p75 and Trka NGF receptors revealed by affinity cross-linking and immunoprecipitation. J Neurosci Res. 1995;40:557–563. doi: 10.1002/jnr.490400415. [DOI] [PubMed] [Google Scholar]
- 32.Iwama A, Okano K, Sudo T, Matsuda Y, Suda T. Molecular cloning of a novel receptor tyrosine kinase gene, STK, derived from enriched hematopoietic stem cells. Blood. 1994;83:3160–3169. [PubMed] [Google Scholar]
- 33.Johnson L N, Noble M E, Owen D J. Active and inactive protein kinases: structural basis for regulation. Cell. 1996;85:149–158. doi: 10.1016/s0092-8674(00)81092-2. [DOI] [PubMed] [Google Scholar]
- 34.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 35.Katso R, Manek S, Biddolph S, Whittaker R, Charnock F, Wells M, Ganesan T. Overexpression of H-Ryk in mouse fibroblasts confers transforming ability in vitro and in vivo: correlation with up-regulation in epithelial ovarian cancer. Cancer Res. 1999;59:2265–2270. [PubMed] [Google Scholar]
- 36.Kelman Z, Simon-Chazottes D, Guénet J L, Yarden Y. The murine vik gene (chromosome 9) encodes a putative receptor with unique protein kinase motifs. Oncogene. 1993;8:37–44. [PubMed] [Google Scholar]
- 37.Kim E, Niethammer M, Rothchild A, Jan Y, Sheng M. Clustering of the Shaker-type K+ channels by interaction with a family of membrane associated guanylate kinases. Nature. 1995;378:85–88. doi: 10.1038/378085a0. [DOI] [PubMed] [Google Scholar]
- 38.Klein R, Jing S, Nanduri V, O’Rourke E, Barbacid M. The trk proto-oncogene encodes a receptor for nerve growth factor. Cell. 1991;65:189–197. doi: 10.1016/0092-8674(91)90419-y. [DOI] [PubMed] [Google Scholar]
- 39.Knighton D R, Zheng J, Ten E L F, Ashford V A, Xuong N H, Taylor S S, Sowadski J M. Crystal structure of the catalytic subunit of cyclic AMP dependent protein kinase. Science. 1991;253:407–414. doi: 10.1126/science.1862342. [DOI] [PubMed] [Google Scholar]
- 40.Larsson-Blomberg L, Dzierzak E. Isolation of tyrosine kinase related genes expressed in the early hematopoietic system. FEBS Lett. 1994;348:119–125. doi: 10.1016/0014-5793(94)00577-x. [DOI] [PubMed] [Google Scholar]
- 41.LeSauteur L, Maliartchouk S, Le Jeune H, Quirion R, Saragovi H U. Potent human p140-TrkA agonists derived from an anti-receptor monoclonal antibody. J Neurosci. 1996;16:1308–1316. doi: 10.1523/JNEUROSCI.16-04-01308.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maminta M L, Williams K L, Nakagawara A, Enger K T, Guo C, Brodeur G M, Deuel T F. Identification of a novel tyrosine kinase receptor-like molecule in neuroblastomas. Biochem Biophys Res Commun. 1992;189:1077–1083. doi: 10.1016/0006-291x(92)92314-n. [DOI] [PubMed] [Google Scholar]
- 43.Marshall C J. MAP kinase kinase kinase, MAP kinase kinase and MAP kinase. Curr Opin Genet Dev. 1994;4:82–89. doi: 10.1016/0959-437x(94)90095-7. [DOI] [PubMed] [Google Scholar]
- 44.Martin-Zanca D, Oskam R, Mitra G, Copeland T, Barbacid M. Molecular and biochemical characterization of the human trk proto-oncogene. Mol Cell Biol. 1989;9:24–33. doi: 10.1128/mcb.9.1.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Masiakowski P, Carroll D. A novel family of cell surface receptors with tyrosine kinase-like domain. J Biol Chem. 1992;267:26181–26190. [PubMed] [Google Scholar]
- 46.Mohammadi M, Schlessinger J, Hubbard S R. Structure of the FGF receptor tyrosine kinase domain reveals a novel autoinhibitory mechanism. Cell. 1996;86:577–587. doi: 10.1016/s0092-8674(00)80131-2. [DOI] [PubMed] [Google Scholar]
- 47.Moran M F, Koch C A, Sadowski I, Pawson T. Mutational analysis of a phosphotransfer motif essential for v-fps tyrosine kinase activity. Oncogene. 1988;3:665–672. [PubMed] [Google Scholar]
- 48.Mossie K, Jallal B, Alves F, Sures I, Plowman G D, Ullrich A. Colon carcinoma kinase-4 defines a new subclass of the receptor tyrosine kinase family. Oncogene. 1995;11:2179–2184. [PubMed] [Google Scholar]
- 49.Mothe I, Tartare S, Kowalskichauvel A, Kaliman P, Vanobberghen E, Ballotti R. Tyrosine kinase-activity of a chimeric insulin-like-growth-factor-1 receptor-containing the insulin-receptor C-terminal domain—comparison with the tyrosine kinase-activities of the insulin and insulin-like-growth-factor-1 receptors using a cell-free system. Eur J Biochem. 1995;228:842–848. doi: 10.1111/j.1432-1033.1995.0842m.x. [DOI] [PubMed] [Google Scholar]
- 50.Okabayashi Y, Kido Y, Okutani T, Sugimoto Y, Sakaguchi K, Kasuga M. Tyrosines 1148 and 1173 of activated human epidermal growth factor receptors are binding sites of Shc in intact cells. J Biol Chem. 1994;269:18674–18678. [PubMed] [Google Scholar]
- 51.Partanen J, Makela T P, Alitalo R, Lehvaslaiho H, Alitalo K. Putative tyrosine kinases expressed in K-562 human leukemia cells. Proc Natl Acad Sci USA. 1990;87:8913–8917. doi: 10.1073/pnas.87.22.8913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paul S R, Merberg D, Finnerty H, Morris G E, Morris J C, Jones S S, Kriz R, Turner K J, Wood C R. Molecular cloning of the cDNA encoding a receptor tyrosine kinase-related molecule with a catalytic region homologous to c-met. Int J Cell Cloning. 1992;10:309–314. doi: 10.1002/stem.5530100509. [DOI] [PubMed] [Google Scholar]
- 53.Pawson T, Gish G D. SH2 and SH3 domains—from structure to function. Cell. 1992;71:359–362. doi: 10.1016/0092-8674(92)90504-6. [DOI] [PubMed] [Google Scholar]
- 54.Peitsch M C. Protein modelling by E-mail. BioTechnology. 1995;13:658–660. [Google Scholar]
- 55.Perrimon N. Signalling pathways initiated by receptor protein tyrosine kinases in Drosophila. Curr Opin Cell Biol. 1994;6:260–266. doi: 10.1016/0955-0674(94)90145-7. [DOI] [PubMed] [Google Scholar]
- 56.Ponder J W, Richards F M. Tertiary templates for proteins. Use of packing criteria in the enumeration of allowed sequences for different structural classes. J Mol Biol. 1987;193:775–791. doi: 10.1016/0022-2836(87)90358-5. [DOI] [PubMed] [Google Scholar]
- 57.Potworowski E F, Beauchemin C. Expression of protein tyrosine kinases in the murine thymus stroma. Immunol Lett. 1996;50:65–69. doi: 10.1016/0165-2478(96)02520-5. [DOI] [PubMed] [Google Scholar]
- 58.Pronk G J, Mcglades J, Pelicci G, Pawson T, Bos J L. Insulin-induced phosphorylation of the 46- and 52-kDa Shc proteins. J Biol Chem. 1993;268:5748–5753. [PubMed] [Google Scholar]
- 59.Ravichandran K S, Burakoff S J. The adapter protein Shc interacts with the interleukin-2 (IL-2) receptor upon IL-2 stimulation. J Biol Chem. 1994;269:1599–1602. [PubMed] [Google Scholar]
- 60.Reichman F M, Dickson B, Hafen E, Shilo B Z. Elucidation of the role of breathless, a Drosophila FGF receptor homolog, in tracheal cell migration. Genes Dev. 1994;8:428–439. doi: 10.1101/gad.8.4.428. [DOI] [PubMed] [Google Scholar]
- 61.Russell R B, Barton G J. Multiple protein sequence alignment from tertiary structure comparison: assignment of global and residue confidence levels. Proteins Struct Funct Genet. 1992;14:309–323. doi: 10.1002/prot.340140216. [DOI] [PubMed] [Google Scholar]
- 62.Schneider S, Buchert M, Georgiev O, Catimel B, Halford M, Stacker S, Baechi T, Moelling K, Hovens C. Mutagenesis and selection of PDZ domains that bind new protein targets. Nat Biotechnol. 1999;17:170–175. doi: 10.1038/6172. [DOI] [PubMed] [Google Scholar]
- 63.Serfas M S, Tyner A L. Ryk is expressed in a differentiation-specific manner in epithelial tissues and is strongly induced in decidualizing uterine stroma. Oncogene. 1998;17:3435–3444. doi: 10.1038/sj.onc.1202266. [DOI] [PubMed] [Google Scholar]
- 64.Shrivastava A, Radziejewski C, Campbell E, Kovac L, McGglynn M, Ryan T, Davis S, Goldfarb M, Glass D, Lemke G, Yancopoulos G. An orphan receptor tyrosine kinase family whose members serve as nonintegrin collagen receptors. Mol Cell. 1997;1:25–34. doi: 10.1016/s1097-2765(00)80004-0. [DOI] [PubMed] [Google Scholar]
- 65.Simoneaux D K, Fletcher F A, Jurecic R, Shilling H G, Van N T, Patel P, Belmont J W. The receptor tyrosine kinase-related gene (ryk) demonstrates lineage and stage-specific expression in hematopoietic cells. J Immunol. 1995;154:1157–1166. [PubMed] [Google Scholar]
- 66.Simske J, Kaech S, Harp S, Kim S. Let-23 receptor localisation by the cell junction protein LIN-7 during C. elegans vulval induction. Cell. 1996;85:195–204. doi: 10.1016/s0092-8674(00)81096-x. [DOI] [PubMed] [Google Scholar]
- 67.Siyanova E Y, Serfas M S, Mazo I A, Tyner A L. Tyrosine kinase gene expression in the mouse small intestine. Oncogene. 1994;9:2053–2057. [PubMed] [Google Scholar]
- 68.Skolnik E Y, Lee C H, Batzer A, Vicentini L M, Zhou M, Daly R, Myers M J, Backer J M, Ullrich A, White M F. The sh2 sh3 domain-containing protein grb2 interacts with tyrosine-phosphorylated irs1 and shc—implications for insulin control of ras signaling. EMBO J. 1993;12:1929–1936. doi: 10.1002/j.1460-2075.1993.tb05842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stacker S A, Hovens C M, Vitali A, Pritchard M A, Baker E, Sutherland G R, Wilks A F. Molecular cloning and chromosomal localisation of the human homologue of a receptor related to tyrosine kinases (RYK) Oncogene. 1993;8:1347–1356. [PubMed] [Google Scholar]
- 70.Stark K L, McMahon J A, McMahon A P. FGFR-4, a new member of the fibroblast growth factor receptor family, expressed in the definitive endoderm and skeletal muscle lineages of the mouse. Development. 1991;113:641–651. doi: 10.1242/dev.113.2.641. [DOI] [PubMed] [Google Scholar]
- 71.Tamagnone L, Partanen J, Armstrong E, Lasota J, Ohgami K, Tazunoki T, LaForgia S, Huebner K, Alitalo K. The human ryk cDNA sequence predicts a protein containing two putative transmembrane segments and a tyrosine kinase catalytic domain. Oncogene. 1993;8:2009–2014. [PubMed] [Google Scholar]
- 72.Taylor S S, Knighton D R, Zheng J, Sowadski J M, Gibbs C S, Zoller M J. A template for the protein kinase family. Trends Biochem Sci. 1993;18:84–89. doi: 10.1016/0968-0004(93)80001-r. [DOI] [PubMed] [Google Scholar]
- 73.Taylor W R. The classification of amino acid conservation. J Theor Biol. 1986;119:205–208. doi: 10.1016/s0022-5193(86)80075-3. [DOI] [PubMed] [Google Scholar]
- 74.Thompson J D, Higgins D G, Gibson T J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thuret J Y, Valay J G, Faye G, Mann C. Civ1 (CAK in vivo), a novel Cdk-activating kinase. Cell. 1996;86:565–576. doi: 10.1016/s0092-8674(00)80130-0. [DOI] [PubMed] [Google Scholar]
- 76.Vogel W, Gish G D, Alves F, Pawson T. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol Cell. 1997;1:13–23. doi: 10.1016/s1097-2765(00)80003-9. [DOI] [PubMed] [Google Scholar]
- 77.Wang X C, Katso R, Butler R, Hanby A M, Poulsom R, Jones T, Sheer D, Ganesan T S. H-RYK, an unusual receptor kinase: isolation and analysis of expression in ovarian cancer. Mol Med. 1996;2:189–203. [PMC free article] [PubMed] [Google Scholar]
- 78.Weiner H L, Rothman M, Miller D C, Ziff E B. Pediatric brain tumors express multiple receptor tyrosine kinases including novel cell adhesion kinases. Pediatr Neurosurg. 1996;25:64–71. doi: 10.1159/000121099. [DOI] [PubMed] [Google Scholar]
- 79.Wilks A, Harpur A. Cytokine signal transduction and the Jak family of protein tyrosine kinases. Bioessays. 1994;16:313–319. doi: 10.1002/bies.950160505. [DOI] [PubMed] [Google Scholar]
- 80.Wilson C, Goberdhan C I, Steller H. Dror, apotential neurotrophic receptor gene, encodes a Drosophila homolog of the vertebrate Ror family of Trk-related receptor tyrosine kinases. Proc Natl Acad Sci USA. 1993;90:7190–7113. doi: 10.1073/pnas.90.15.7109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yee K, Bishop T R, Mather C, Zon L I. Isolation of a novel receptor tyrosine kinase cDNA expressed by developing erythroid progenitors. Blood. 1993;82:1335–1343. [PubMed] [Google Scholar]
- 82.Zhao Y, Bjorbaek C, Moller D. Regulation and interaction of pp90rsk isoforms with mitogen activated protein kinases. J Biol Chem. 1996;271:29773–29779. doi: 10.1074/jbc.271.47.29773. [DOI] [PubMed] [Google Scholar]
- 83.Zhao Y, Bjorbaek C, Weremowicz S, Morton C, Moller D. RSK3 encodes a novel pp90rsk isoform with a unique N-terminal sequence: growth factor-stimulated kinase function and nuclear translocation. Mol Cell Biol. 1995;15:4353–4363. doi: 10.1128/mcb.15.8.4353. [DOI] [PMC free article] [PubMed] [Google Scholar]