

Key Points
Question
What are the prognostic implications of luminal and basal subtypes in metastatic castration-resistant prostate cancer?
Findings
In this cohort study including tissue samples from 634 patients with metastatic prostate cancer, distinct molecular patterns associated with luminal and basal subtypes were identified; in addition, enrichment of small cell/neuroendocrine prostate cancer samples were noted in the basal tumors. The subtypes were prognostic, and the patients with luminal subtypes preferentially benefited from androgen-signaling inhibitors.
Meaning
These findings suggest that patients with luminal tumors should be considered for androgen-signaling inhibitor therapy, and those with basal tumors could be considered for chemotherapeutic approaches given the similarity to small cell/neuroendocrine prostate cancer and the diminished benefit of androgen-signaling inhibitors; further investigation is warranted in prospective clinical trials.
Abstract
Importance
Luminal and basal subtypes of primary prostate cancer have been shown to be molecularly distinct and clinically important in predicting response to therapy. These subtypes have not been described in metastatic prostate cancer.
Objectives
To identify clinical and molecular correlates of luminal and basal subtypes in metastatic castration-resistant prostate cancer (mCRPC) and investigate differences in survival, particularly after treatment with androgen-signaling inhibitors (ASIs).
Design, Setting, and Participants
In this cohort study, a retrospective analysis was conducted of 4 cohorts with mCRPC (N = 634) across multiple academic centers. Treatment was at the physicians’ discretion. Details of the study cohorts have been published elsewhere between 2016 and 2019. Data were analyzed from March 2018 to February 2021.
Main Outcomes and Measures
The primary clinical end point was overall survival from the date of tissue biopsy/molecular profiling. Luminal and basal subtypes were also stratified by postbiopsy ASI treatment. The primary molecular analyses included associations with small cell/neuroendocrine prostate cancer (SCNC), molecular pathways, and DNA alterations.
Results
In the 634 patients, 288 (45%) had tumors classified as luminal, and 346 (55%) had tumors classified as basal. However, 53 of 59 (90%) SCNC tumors were basal (P < .001). Similar to primary prostate cancer, luminal tumors exhibited overexpression of AR pathway genes. In basal tumors, a significantly higher rate of RB1 loss (23% basal vs 4% luminal; P < .001), FOXA1 alterations (36% basal vs 27% luminal; P = .03) and MYC alterations (73% basal vs 56% luminal; P < .001) were identified. Patients with basal tumors had worse overall survival compared with those with luminal tumors only in patients treated with an ASI postbiopsy (East Coast Dream Team: hazard ratio [HR], 0.39; 95% CI, 0.20-0.74; P = .004; West Coast Dream Team: HR, 0.57; 95% CI, 0.33-0.97; P = .04). Among patients with luminal tumors, those treated with an ASI had significantly better survival (HR, 0.27; 95% CI, 0.14-0.53; P < .001), whereas patients with basal tumors did not (HR, 0.62; 95% CI, 0.36-1.04, P = .07). The interaction term between subtype and ASI treatment was statistically significant (HR, 0.42; 95% CI, 0.20-0.89; P = .02).
Conclusions and Relevance
These findings represent the largest integrated clinical, transcriptomic, and genomic analysis of mCRPC samples to date, and suggest that mCRPC can be classified as luminal and basal tumors. Analogous to primary prostate cancer, these data suggest that the benefit of ASI treatment is more pronounced in luminal tumors and support the use of ASIs in this population. In the basal tumors, a chemotherapeutic approach could be considered in some patients given the similarity to SCNC and the diminished benefit of ASI therapy. Further validation in prospective clinical trials is warranted.
This prognostic study examines the use of molecular profiling of luminal and basal tumors in estimation of survival in men with castration-resistant metastatic prostate cancer.
Introduction
Advances in genomic technologies and bioinformatics strategies have led to an unprecedented understanding of the molecular basis of prostate cancer. This work has focused on identifying common and recurrent genetic events, such as androgen receptor (AR) alterations and DNA-damage repair defects, with the goal of identifying drivers of oncogenesis and disease progression that may also be targets for pharmacologic intervention.1,2,3
Clinically relevant molecular subtypes of prostate cancer have been identified, analogous to breast cancer subtypes. Expression-based clustering has been extensively used to classify breast tumors into intrinsic molecular subtypes, including basal and luminal subtypes.4 This clustering has resulted in a commercial assay that provides prognostic risk assessment in patients with early-stage breast cancer and has been incorporated into international clinical practice guidelines.5 Similarly, basal and luminal subtypes have been identified across epithelial tumor types,6,7,8 including localized prostate cancers,9 suggesting that these intrinsic subtypes may share common features across cancers and have broad applicability.10 Classifying localized prostate cancers into luminal (A and B) and basal subtypes has been shown to be clinically prognostic for recurrence, metastasis, and survival.9 Furthermore, the luminal B subtype, which is characterized by increased AR signaling and clinical aggressiveness, has been shown to preferentially benefit from androgen deprivation therapy.9 These findings are being prospectively validated in a randomized multi-institutional National Clinical Trials Network clinical trial.11
To our knowledge, all studies evaluating luminal/basal subtypes within prostate cancer have been limited to primary prostate cancer tissue, in part due to the difficulties in obtaining biopsies from metastases, which are often confined to bone. Metastatic castration-resistant prostate cancer (mCRPC) is the lethal form of the disease. It is now well established that AR signaling persists in a substantial proportion of patients with mCRPC,1,3,12 and is the basis of the efficacy and US Food and Drug Administration approval of a number of androgen-signaling inhibitors (ASIs) in this patient population. We therefore hypothesized that, as in primary prostate cancer, luminal and basal subtypes exist in mCRPC and provide important molecular and clinical information that may be relevant to AR-targeted therapies.
In addition to the difficulties associated with accessing metastases, the identification of subtypes in metastatic prostate cancer can be challenging because not all subtypes are equally represented. Luminal A tumors have the lowest propensity for metastasis,9 and thus would be expected to be under-represented in metastatic tumors. Thus, in the CHAARTED clinical trial undertaken in men with metastatic (hormone-naive) prostate cancer, examination of the original prostate tissue from biopsies showed almost no luminal A tumors.13 Because of these differences, we developed an approach specific to the metastatic prostate cancer disease state to identify luminal (B) and basal subtypes in 634 mCRPC samples from 4 independent clinical cohorts. We then explored the molecular and clinical implications of these subtypes.
Methods
We used data from 4 mCRPC cohorts to assess luminal and basal subtypes. Clinical, processed RNA sequencing, variant call, and copy number data were downloaded from cBioPortal14 for 3 cohorts: an autopsy cohort from the Fred Hutchinson Cancer Research Center (FHCRC) (n = 157),15 a neuroendocrine prostate cancer–enriched cohort from Weill Cornell Medicine (WCM) (n = 49),16 and an mCRPC cohort from the Stand Up 2 Cancer/Prostate Cancer Foundation (SU2C/PCF) East Coast Dream Team (ECDT) (n = 266 using the poly-A–enriched data set).1,17 We also obtained clinical, processed RNA sequencing, variant call, and copy number data from an expansion of the SU2C/PCF West Coast Dream Team (WCDT) (n = 162) cohort.3,12,18 Details of the study cohorts have been published elsewhere between 2016 and 2019.15,16,17,18 Data were analyzed from March 2018 to February 2021. In our primary analysis of the RNA sequencing data, we included genes that had expression data in all samples. We next sought to adjust for the variability between cohorts due to batch effects, different processing techniques, and experimental conditions. We first converted gene expression levels into a per-sample gene rank to standardize genes across cohorts. However, a large batch effect persisted (eFigure 1A in the Supplement) and so we applied an empirical bayes framework for batch correction19 that was able to successfully eliminate the batch effects (eFigure 1B in the Supplement). The present study was approved by the UCSF institutional review board, and patients provided written informed consent. Data for other participants were publicly available.
Because of the lack of the luminal A subtype in metastatic prostate cancer,13 the standard centroid-based approach used previously to define primary prostate cancer subtypes cannot be applied directly to mCRPC samples.20,21 Therefore, we used unsupervised hierarchical clustering of all samples as an unbiased approach in identifying metastatic prostate cancer subtypes using gene expression of the canonical 50 genes that make up the PAM50 signature, which has been examined across breast, prostate, and other cancers.7,9,22 DNA alterations and clinical data were not used in the clustering. Hierarchical clustering used the nonsquared Euclidean distance measure and the ward.D2 clustering method, which implements the Ward clustering criterion.23 No other parameters were changed from the defaults in the R hclust function. The 2 clusters identified were identified as luminal and basal based on increased and decreased expression of luminal and basal markers, and these classes were used for subsequent analysis. To visualize how the luminal-ness or basal-ness of a sample varied between patients and by location within individual patients, we developed a continuous luminal-basal score using an elastic-net regression linear model24 and the gene expression data from the PAM50 genes. This score was only used for plotting purposes, and not for sample classification, which was obtained from the unbiased clustering.
Pathways enriched in luminal vs basal samples were evaluated using gene set enrichment analysis (GSEA).25 The batch-corrected values were then scaled to 0 to 1 to remove negative values and standardize all genes and the log2 fold-change between luminal and basal samples was then calculated for each gene. The log2 fold-change was then processed by the GSEA preranked algorithm to identify luminal- and basal-enriched pathways. For the GSEA, we evaluated gene sets from the Molecular Signatures Database hallmark gene set collection26 excluding pathways unrelated to prostate cancer.27 Significance of enrichment was evaluated using the Benjamini-Hochberg procedure to determine a multiple-testing adjusted P value, which controls for the false discovery rate.28
Statistical Analysis
Overall survival data were available only for the ECDT and WCDT cohorts. In patients who underwent multiple biopsies, only the first biopsy was used for the survival analysis. Time to event was defined as the time from biopsy to death or last contact, as this was the only end point available for both cohorts. Survival was compared using the Kaplan-Meier method. The proportional hazards assumption was checked using the Schoenfeld test,29 with no violation of the proportional hazards assumption identified. The Cox proportional hazards model P value was used to test for a significant difference in survival between samples. When comparisons of the continuous luminal-basal score based on tissue site of biopsy were made, the Kruskal-Wallis 1-way analysis of variance test was used (a nonparametric alternative to the analysis of variance test), and a Wilcoxon signed-rank test was used to compare groups against each other. As with the GSEA, the Benjamini-Hochberg method was used for multiple testing correction when appropriate. All statistical testing was 2-sided with a significance threshold of 0.05. All analysis was completed using R, version 4.0.2 (R Foundation for Statistical Computing).
Results
Luminal and Basal Subtypes
To define luminal and basal subtypes in mCRPC, RNA sequencing data from a total of 634 mCRPC samples across 4 cohorts were collected and analyzed. Expression of the PAM50 genes across all samples are depicted in Figure 1A. Hierarchical clustering of the samples identified 2 distinct clusters: a basal group with 346 samples (55%) that demonstrates higher expression of basal markers (PTTG1, CDC20, ORC6L, KIF2C, UBE2C, MELK, BIRC5, NUF2, CEP55, EXO1, CENPF, NDC80, TYMS, UBE2T, ANLN, CCNB1, RRM2, and MKI67)30 and a luminal group with 288 samples (45%) that demonstrated lower expression of the basal genes, with higher to mixed expression of luminal markers (ESR1, PGR, BCL2, FOXA1, CDC6, CXXC5, MLPH, MAPT, NAT1, MDM2, MMP11, and BLVRA).30 Gene set enrichment analysis with luminal and basal prostate cancer genes from the literature31 further confirmed that luminal genes were enriched in the luminal subtype (GSEA, P < .001) and basal genes were enriched in the basal subtype (GSEA, P = .001). Our results showing only 2 distinct clusters in metastatic prostate cancer are consistent with the low propensity of luminal A tumors to metastasize,13 as opposed to the 3 clusters found in primary prostate cancer.9
Figure 1. Luminal and Basal Subtypes of Metastatic Castration-Resistant Prostate Cancer (mCRPC).
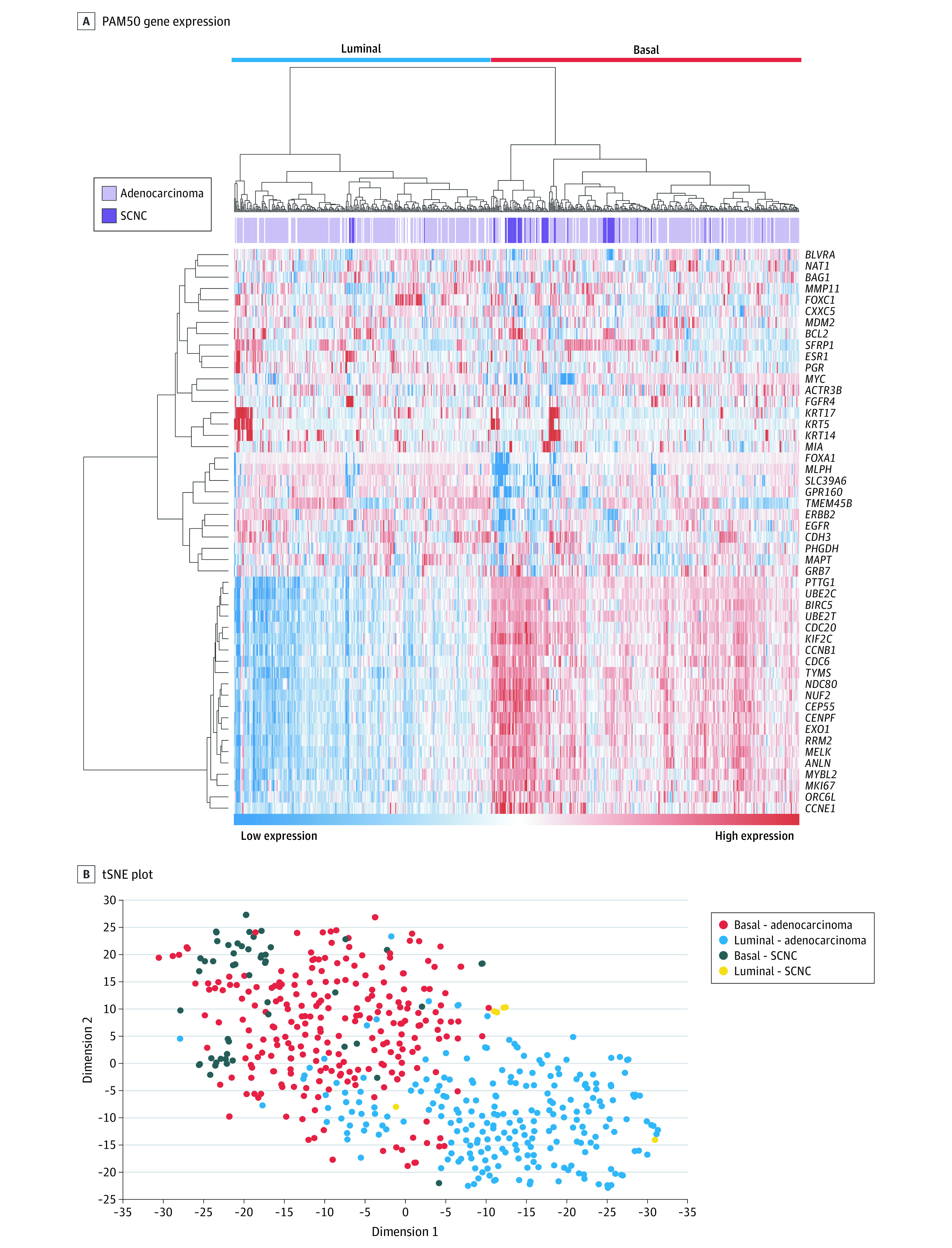
A, PAM50 expression (rows) for each sample (columns). B, t-distributed stochastic neighbor-embedding (tSNE) plot.
We next examined the samples categorized as small cell/neuroendocrine prostate cancer (SCNC) (96 samples were missing these data). Of 59 SCNC tumors, 53 (90%) were classified as basal (Fisher exact test, P < .001). We then used a t-distributed stochastic neighbor-embedding (tSNE) plot, which reduces the dimensionality of the RNA sequencing data so that samples with similar RNA expression profiles are near each other in a 2-dimensional plot. The stochastic neighbor embedding plot in Figure 1B identifies 2 primary clusters of samples—basal and luminal—confirming that our hierarchical clustering results are similar regardless of the unsupervised approach used; however, there appeared to be a more continuous distribution along a luminal-basal axis rather than 2 clearly different subpopulations. The SCNC tumors represent the most basal tumors along this axis. Most of the SCNC tumors that were classified as luminal appear near the border between luminal and basal. These results suggest the existence of luminal and basal subtypes of mCRPC.
Molecular Characteristics of Luminal and Basal Subtypes
We next evaluated the molecular differences between these 2 groups based on enriched pathways from RNA sequencing data along with mutation and copy number changes in key prostate cancer oncogenes and tumor suppressor genes. Gene set enrichment analysis (Figure 2A and 2B) identified the androgen-response pathway as the most luminal-enriched pathway, consistent with previously published data on primary prostate cancer7,9 and the enrichment of SCNC in basal tumors. The most basal-enriched pathways were associated with the cell cycle and cell division, consistent with the increased expression of proliferation genes, such as MKI67. We then examined somatic mutations and copy number calls for the 3 most frequently mutated or copy number altered3 tumor suppressor genes (PTEN, RB1, and TP53) and oncogenes (AR, FOXA1, and MYC) in prostate cancer (Figure 3). Owing to potential technical differences in processing between cohorts, we examined each cohort individually as well as together (reported P values are for the pooled comparisons). We identified a significantly higher rate of 2 RB1 alterations (presumed biallelic inactivation from copy number loss and/or mutation) in basal samples (23% basal vs 4% luminal; Fisher exact test, P < .001), with no significant difference in PTEN loss (29% basal vs 32% luminal; Fisher exact test, P = .45) or TP53 loss (33% basal vs 28% luminal; Fisher exact test, P = .19). When examining key prostate cancer oncogenes, we identified a significantly higher alteration rate in basal tumors for FOXA1 (36% basal vs 27% luminal; Fisher exact test, P = .03) and MYC (73% basal vs 56% luminal; Fisher exact test, P < .001), with no significant difference in AR (74% basal vs 79% luminal; Fisher exact test, P = .22). These findings are consistent across cohorts, except for MYC alterations in WCM, a cohort with a smaller sample size and higher SCNC enrichment, which may account for this discrepancy.
Figure 2. Gene Set Enrichment Analysis.
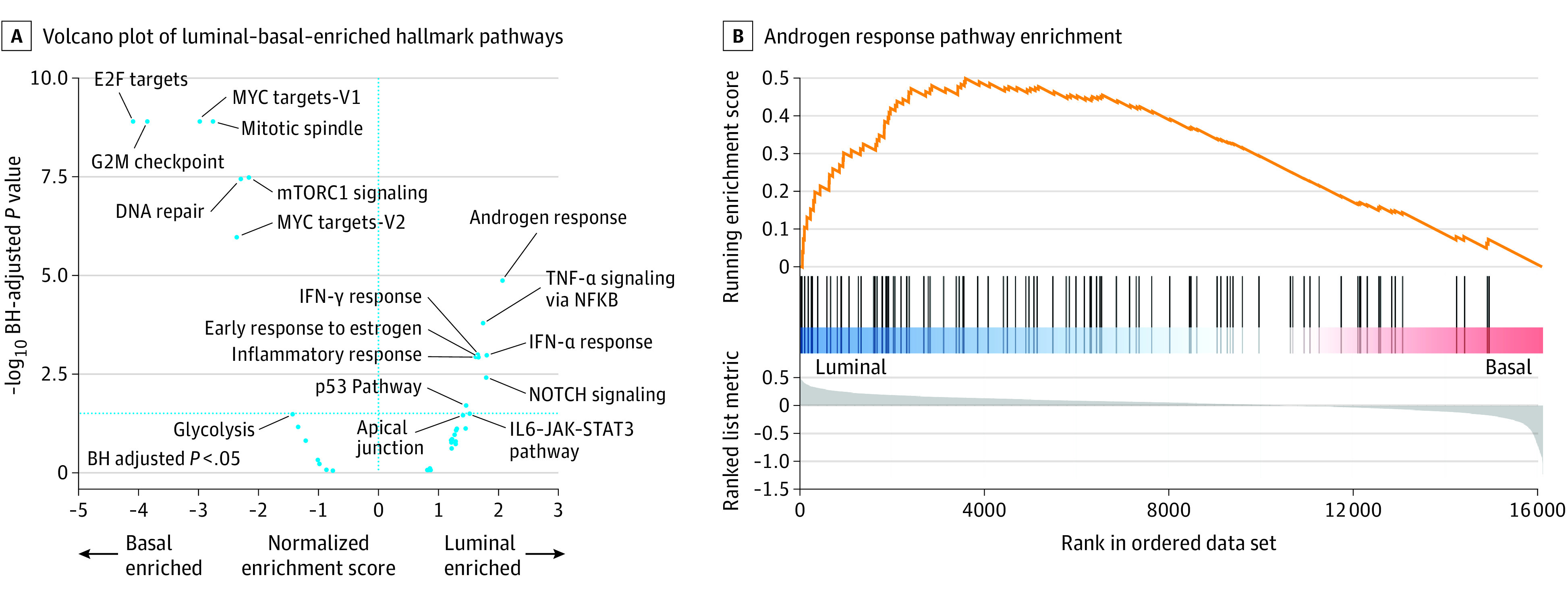
A, Gene set enrichment analysis (GSEA) was completed on the Molecular Signatures Database hallmark gene set collection, focusing on hallmark pathways involved in prostate cancer. A volcano plot of the GSEA results depicts log2 geometric mean fold change in gene expression across each gene set between basal vs luminal samples plotted against the − log10 Benjamini-Hochberg (BH)–corrected P values, to control for the false discovery rate with multiple testing. IFN-γ indicates interferon-γ; Il-6, interleukin 6; and TNF-α, tumor necrosis factor-α. The vertical dotted blue line indicates P < .05. B, GSEA enrichment plot of the androgen response hallmark pathway, showing that the androgen response pathway is positively enriched in basal samples (P < .001).
Figure 3. DNA Alterations and Subtypes.
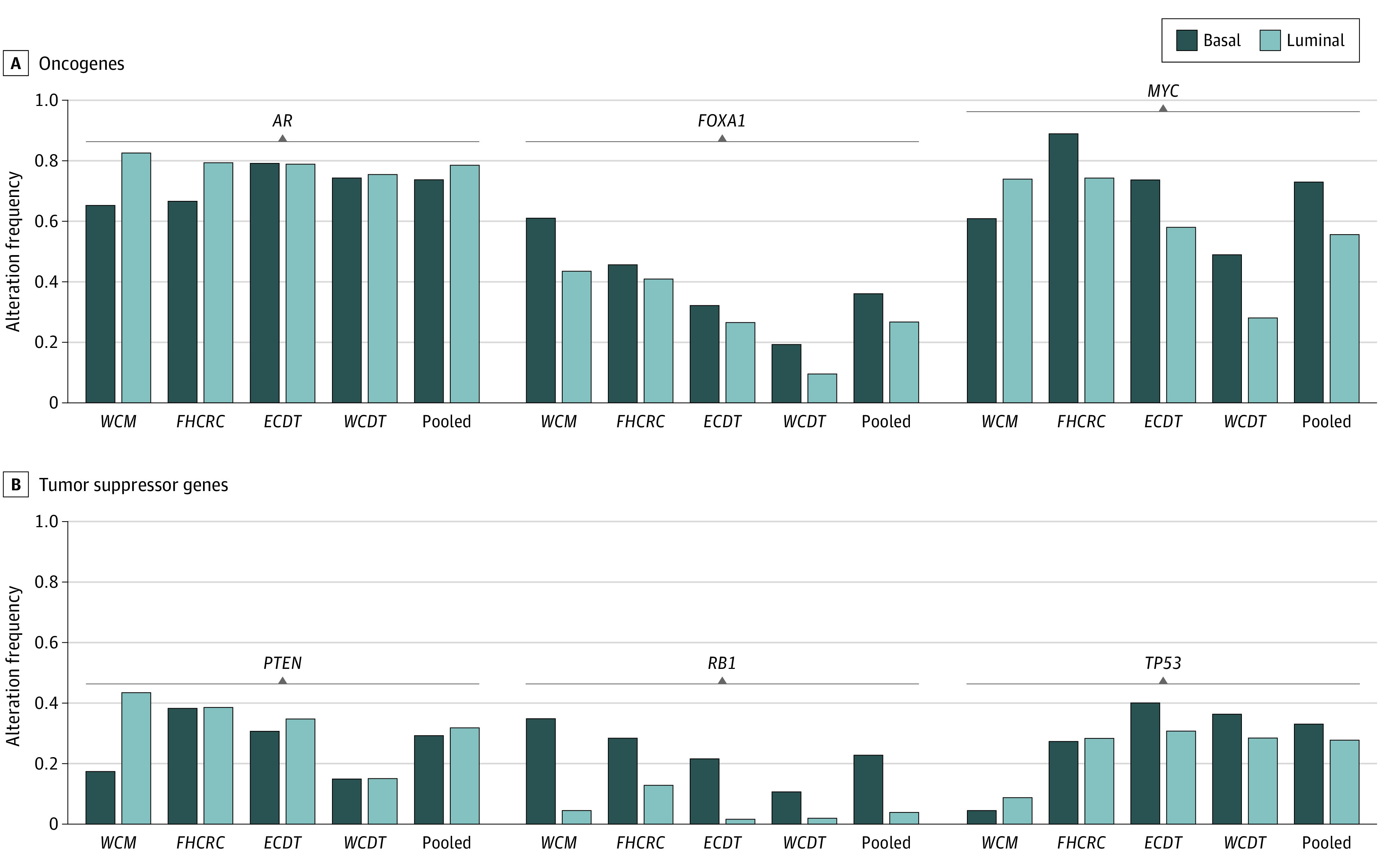
Proportion of samples with a biallelic loss of function mutation/copy loss of a tumor suppressor gene and gain of function mutations in oncogenes in each individual cohort as well as pooled.
Survival Differences of Luminal and Basal Subtypes
We next examined overall survival, subtype, and the first postbiopsy treatment for 80 patients from the ECDT and 123 patients from the WCDT cohorts. All patients examined in the ECDT cohort were taxane-naive and had received first-line ASI therapy postbiopsy.1,17 On Cox proportional hazards analysis, patients from the ECDT with luminal tumors had better overall survival compared with those who had basal tumors, with a median overall survival of 33.1 months, compared with 18.7 months (hazard ratio [HR], 0.39; 95% CI, 0.20-0.74; P = .004) (Figure 4A). In the WCDT cohort, there were patients who received ASI therapy postbiopsy as well as non-ASI therapy. All patients initially had prostate adenocarcinoma—de novo SCNC was excluded. Only 19 patients (15.4%) in the cohort received chemotherapy for mCRPC before enrollment, because this was an exclusion criterion after August 2014. As in the ECDT, luminal tumors treated with ASI therapy postbiopsy had better overall survival compared with basal tumors, with a median overall survival of 21.7 months, compared with 32.0 months (HR, 0.57; 95% CI, 0.33-0.97; P = .04), but there was no significant difference in patients not treated with ASI therapy postbiopsy (Figure 4B). Among patients with luminal tumors, we found that those treated with an ASI had significantly better survival compared with those who were not treated with an ASI (median survival, 32.0 vs 8.7 months; HR, 0.27; 95% CI, 0.14-0.53; P < .001). Among patients with basal tumors, this effect size was diminished and was no longer significant (HR, 0.62; 95% CI, 0.36-1.04; P = .07), suggesting that the benefit of ASI treatment may be higher in luminal tumors compared with basal tumors. The median overall survival was similar between the ECDT patients (all of whom received postbiopsy ASI treatment) and the WCDT patients who received postbiopsy ASI treatment (28.1 vs 25.7 months). Therefore, we pooled the 2 cohorts for Cox proportional hazards interaction analysis, also adjusting for prebiopsy exposure to ASI therapy. We found that the interaction term between subtype and postbiopsy ASI therapy was statistically significant (HR, 0.42; 95% CI, 0.20-0.89; P = .02), consistent with a bona fide predictive biomarker. When we performed subgroup analysis dichotomizing our cohort by earlier ASI treatment, in patients who had never before received ASI therapy, patients with luminal tumors had better overall survival than those with basal tumors (HR, 0.39; 95% CI, 0.20-0.74; P = .005), matching the results from the ECDT. The interaction term continued to be significant in this ASI-naive subset (HR, 0.07; 95% CI, 0.01-0.48; P = .007). However, when we examined patients who received prior ASI therapy, the difference between luminal and basal tumors disappeared (HR, 1.03; 95% CI, 0.61-1.72; P = .92).
Figure 4. Clinical Outcomes and Subtypes.

A, Survival compared between luminal and basal subtypes within the East Coast Dream Team, where all patients received postbiopsy androgen-signaling inhibitor (ASI) treatment. Hazard ratio (HR), 0.39; P = .004. B, Survival compared between luminal and basal subtypes within the West Coast Dream Team further stratified by whether a patient received postbiopsy ASI treatment. Basal with vs without ASI: HR, 0.62; P = .07. Luminal with vs without ASI: HR, 0.27; P < .001. Hazard ratios determined using Cox proportional hazards model.
To confirm that random genes do not give the same result, we re-ran our analysis using 50 random genes, repeated 100 times. We found that none of these attempts were able to identify significant P values (with false discovery rate multiple testing correction) for the survival analyses and interaction terms. Furthermore, to ensure that the clustering performed in the ECDT or WCDT cohort was not biasing our survival analyses via information leakage, we identified the luminal and basal clusters in the FHCRC or WCM cohort and then assigned samples in the ECDT or WCDT cohort based on the nearest median centroid (using the nonsquared Euclidean distance). The results of this analysis were similar, with all significant P values remaining significant.
Variation in Subtype
Information regarding the location of the profiled metastatic site was available in 633 patients from all 4 cohorts. Given the more continuous appearance of the luminal-basal spectrum (Figure 1B), we developed a similarity score that could be used to better visualize this variability across tumor sites. A significant difference in the luminal-basal score across the site of biopsy was observed (Kruskal-Wallis test, χ2 = 13.01; P = .001) (eFigure 2 in the Supplement). On further pairwise comparisons, a significant increase in the luminal-basal score, indicating a more basal-like sample, was identified in lymph node samples compared with primary site samples (primary site median, 0.45; interquartile range,0.29-0.64; lymph node median, 0.60; interquartile range, 0.43-0.72; distant metastasis median, 0.63; interquartile range, 0.47-0.75) (Benjamini-Hochberg–adjusted Wilcoxon signed-rank test, P = .01) and distant metastasis samples compared with primary site samples (Benjamini-Hochberg–adjusted Wilcoxon signed-rank test, P = .002). This result is consistent with our finding that basal subtypes have a more aggressive phenotype.
The FHCRC cohort was a rapid autopsy cohort that profiled multiple tumor sites from each patient. This unique data set allowed for an analysis of the intraindividual variation of subtype. Figure 5 depicts the variability in the luminal-basal score across and within patients from the FHCRC cohort. Only 7 of 42 patients (17%) had both luminal and basal tumors identified, suggesting that the subtypes appear to be relatively stable within individual patients.
Figure 5. Intrapatient Subtype Variability.

Biopsy samples from the Fred Hutchinson Cancer Research Center autopsy cohort, depicting intrapatient variability in both the luminal-basal score and subtype class. Only patients with more than 1 biopsy were included. Each patient is plotted along a single line from the y-axis, with each symbol representing a separate biopsy. Biopsies are color coded based on the luminal-basal cluster group and variability of the linear luminal-basal score is plotted along the x-axis.
Discussion
In the first, to our knowledge, evaluation of the molecular and clinical characteristics of luminal and basal subtypes in mCRPC, we examined RNA and DNA sequencing data from 634 mCRPC tissue samples reported from 4 different cohorts. We demonstrated the existence of 2 distinct subtypes (luminal and basal) and that the molecular characteristics of these subtypes are consistent with data from primary prostate cancer. These clusters are clinically significant, as the luminal or basal designation serves as both a prognostic and predictive biomarker. We have also noted smaller intraindividual variability compared with interindividual variability, consistent with other molecular analyses of this autopsy series, supporting the feasibility of classifying patients on basis of a single biopsy of a metastatic tumor.
As in primary prostate cancer,9 luminal mCRPC tumors demonstrate increased expression of androgen-signaling genes. In addition, analogous to findings in primary prostate cancer,9 our data suggest that patients with luminal tumors obtain preferential benefit for ASI therapy compared with those with basal tumors. These results are also consistent with the enrichment of AR-independent SCNC in the basal tumor group. The ASI treatment to subtype interaction term was statistically significant, suggesting that luminal and basal subtypes could serve as predictive biomarkers for ASI therapy in mCRPC, primarily in the first-line setting. Multiple Food and Drug Administration–approved therapeutic options exist for men with mCRPC. Molecular subtypes that predict which patients respond best to ASI therapy represent a potential tool that could be used to select the optimal treatment strategy for each patient.
We also observed that SCNC was enriched within the basal subtype tumors. Small cell/neuroendocrine prostate cancer is thought to be androgen independent, and thus one would expect it to be more similar to basal tumors compared with luminal tumors. De novo SCNC has also been shown to originate from basal cells in mouse models,32 which is consistent with the results reported herein. Furthermore, the data presented in this report suggest that the luminal-ness and basal-ness of metastatic prostate tumors lies on a spectrum, and that SCNC tumors largely represent the most basal-like in nature along this continuum. Treatment-associated SCNC is thought to arise as the tumor evolves in response to multiple lines of AR-targeted therapy via trans-differentiation,33 which would be consistent with our observations of a spectrum in which only the most pronounced basal-ness was identified as SCNC. The basal subtype includes but is not exclusive to SCNC, which raises the potential that non-SCNC basal tumors (the majority) may share enough similarity with their SCNC counterparts that similar chemotherapy-based treatment strategies may be effective, especially given the diminished benefit of ASI therapy. However, there is still a spectrum even within the basal tumors, and this heterogeneity may be why there is a trend toward benefit of ASIs in the basal tumors.
Limitations
This study has limitations. Although molecular subtype information was not available at the time of clinical decision-making, an important limitation of these data are that treatment information is available only on a subset of our total cohort, and treatment was not protocol-specified, so treatment selection bias was possible. However, our results are consistent with a small enzalutamide phase 2 trial in men with mCRPC demonstrating that luminal gene signatures were enriched in responders and basal gene signatures were enriched in nonresponders.34 Together, these data suggest that ASI therapy should be considered for patients with luminal tumors.
Conclusions
To our knowledge, these findings represent the largest integrated clinical, transcriptomic, and genomic analysis of mCRPC samples to date, and an important step toward better personalizing therapy for men with mCRPC based on the molecular information of their tumors. The results warrant further validation in larger prospective cohorts and clinical trials.
eFigure 1. Clustering Before and After Batch Correction
eFigure 2. Tumor Location
References
- 1.Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215-1228. doi: 10.1016/j.cell.2015.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pritchard CC, Mateo J, Walsh MF, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375(5):443-453. doi: 10.1056/NEJMoa1603144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Quigley DA, Dang HX, Zhao SG, et al. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell. 2018;174(3):758-769.e9. doi: 10.1016/j.cell.2018.06.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parker JS, Mullins M, Cheang MC, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27(8):1160-1167. doi: 10.1200/JCO.2008.18.1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harris LN, Ismaila N, McShane LM, et al. ; American Society of Clinical Oncology . Use of biomarkers to guide decisions on adjuvant systemic therapy for women with early-stage invasive breast cancer: American Society of Clinical Oncology Clinical Practice guideline. J Clin Oncol. 2016;34(10):1134-1150. doi: 10.1200/JCO.2015.65.2289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siegfried JM, Lin Y, Diergaarde B, et al. Expression of PAM50 genes in lung cancer: evidence that interactions between hormone receptors and HER2/HER3 contribute to poor outcome. Neoplasia. 2015;17(11):817-825. doi: 10.1016/j.neo.2015.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao SG, Chen WS, Das R, et al. Clinical and genomic implications of luminal and basal subtypes across carcinomas. Clin Cancer Res. 2019;25(8):2450-2457. doi: 10.1158/1078-0432.CCR-18-3121 [DOI] [PubMed] [Google Scholar]
- 8.Choi W, Porten S, Kim S, et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell. 2014;25(2):152-165. doi: 10.1016/j.ccr.2014.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao SG, Chang SL, Erho N, et al. Associations of luminal and basal subtyping of prostate cancer with prognosis and response to androgen deprivation therapy. JAMA Oncol. 2017;3(12):1663-1672. doi: 10.1001/jamaoncol.2017.0751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao SG, Chen WS, Das R, et al. Clinical and genomic implications of luminal and basal subtypes across carcinomas. Clin Cancer Res. 2019;25(8):2450-2457. doi: 10.1158/1078-0432.CCR-18-3121 [DOI] [PubMed] [Google Scholar]
- 11.Biomarker trial of apalutamide and radiation for recurrent prostate cancer. ClinicalTrials.gov identifier NCT03371719. Updated March 2, 2021. Accessed August 17, 2021. https://clinicaltrials.gov/ct2/show/NCT03371719
- 12.Zhao SG, Chen WS, Li H, et al. The DNA methylation landscape of advanced prostate cancer. Nat Genet. 2020;52(8):778-789. doi: 10.1038/s41588-020-0648-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamid A, V. WX, Chen YH, et al. Luminal B subtype as a predictive biomarker of docetaxel benefit for newly diagnosed metastatic hormone sensitive prostate cancer (mHSPC): a correlative study of E3805 CHAARTED. Presented at: ASCO Annual Meeting; 2019; Chicago, IL. [Google Scholar]
- 14.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401-404. doi: 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar A, Coleman I, Morrissey C, et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med. 2016;22(4):369-378. doi: 10.1038/nm.4053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beltran H, Prandi D, Mosquera JM, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016;22(3):298-305. doi: 10.1038/nm.4045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abida W, Cyrta J, Heller G, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci U S A. 2019;116(23):11428-11436. doi: 10.1073/pnas.1902651116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen WS, Aggarwal R, Zhang L, et al. ; West Coast Prostate Cancer Dream Team . Genomic drivers of poor prognosis and enzalutamide resistance in metastatic castration-resistant prostate cancer. Eur Urol. 2019;76(5):562-571. doi: 10.1016/j.eururo.2019.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical bayes methods. Biostatistics. 2007;8(1):118-127. doi: 10.1093/biostatistics/kxj037 [DOI] [PubMed] [Google Scholar]
- 20.Lusa L, McShane LM, Reid JF, et al. Challenges in projecting clustering results across gene expression-profiling datasets. J Natl Cancer Inst. 2007;99(22):1715-1723. doi: 10.1093/jnci/djm216 [DOI] [PubMed] [Google Scholar]
- 21.Zhao X, Rødland EA, Tibshirani R, Plevritis S. Molecular subtyping for clinically defined breast cancer subgroups. Breast Cancer Res. 2015;17:29. doi: 10.1186/s13058-015-0520-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perou CM, Sørlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747-752. doi: 10.1038/35021093 [DOI] [PubMed] [Google Scholar]
- 23.Murtagh F, Legendre P. Ward’s hierarchical agglomerative clustering method: which algorithms implement Ward’s criterion? J Classif. 2014;31(3):274-295. doi: 10.1007/s00357-014-9161-z [DOI] [Google Scholar]
- 24.Zou H, Hastie T.. Regularization and variable selection via the elastic net. J Royal Statistical Soc Series B. 2005;67(2):301-320. doi: 10.1111/j.1467-9868.2005.00503.x [DOI] [Google Scholar]
- 25.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545-15550. doi: 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1(6):417-425. doi: 10.1016/j.cels.2015.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao SG, Lehrer J, Chang SL, et al. The immune landscape of prostate cancer and nomination of PD-L2 as a potential therapeutic target. J Natl Cancer Inst. 2019;111(3):301-310. doi: 10.1093/jnci/djy141 [DOI] [PubMed] [Google Scholar]
- 28.Benjamini Y, Hochberg Y.. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Statistical Soc Series B. 1995;57(1):289-300. doi: 10.1111/j.2517-6161.1995.tb02031.x [DOI] [Google Scholar]
- 29.Grambsch PM, Therneau TM. Proportional hazards tests and diagnostics based on weighted residuals. Biometrika. 1994;81(3):515-526. doi: 10.1093/biomet/81.3.515 [DOI] [Google Scholar]
- 30.Mathews JC, Nadeem S, Levine AJ, Pouryahya M, Deasy JO, Tannenbaum A. Robust and interpretable PAM50 reclassification exhibits survival advantage for myoepithelial and immune phenotypes. NPJ Breast Cancer. 2019;5:30. doi: 10.1038/s41523-019-0124-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang D, Park D, Zhong Y, et al. Stem cell and neurogenic gene-expression profiles link prostate basal cells to aggressive prostate cancer. Nat Commun. 2016;7:10798. doi: 10.1038/ncomms10798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee DK, Liu Y, Liao L, Li W, Danielpour D, Xu J. Neuroendocrine prostate carcinoma cells originate from the p63-expressing basal cells but not the pre-existing adenocarcinoma cells in mice. Cell Res. 2019;29(5):420-422. doi: 10.1038/s41422-019-0149-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beltran H, Hruszkewycz A, Scher HI, et al. The role of lineage plasticity in prostate cancer therapy resistance. Clin Cancer Res. 2019;25(23):6916-6924. doi: 10.1158/1078-0432.CCR-19-1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alumkal JJ, Sun D, Lu E, et al. Transcriptional profiling identifies an androgen receptor activity-low, stemness program associated with enzalutamide resistance. Proc Natl Acad Sci U S A. 2020;117(22):12315-12323. doi: 10.1073/pnas.1922207117 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eFigure 1. Clustering Before and After Batch Correction
eFigure 2. Tumor Location