

Summary
Personalized in vitro models for dysplasia and carcinogenesis in the pancreas have been constrained by insufficient differentiation of human pluripotent stem cells (hPSCs) into the exocrine pancreatic lineage. Here, we differentiate hPSCs into pancreatic duct-like organoids (PDLOs) with morphological, transcriptional, proteomic, and functional characteristics of human pancreatic ducts, further maturing upon transplantation into mice. PDLOs are generated from hPSCs inducibly expressing oncogenic GNAS, KRAS, or KRAS with genetic covariance of lost CDKN2A, and from induced hPSCs derived from a McCune-Albright patient. Each oncogene causes a specific growth, structural, and molecular phenotype in vitro. While transplanted PDLOs with oncogenic KRAS alone form heterogenous dysplastic lesions or cancer, KRAS with CDKN2A-loss develop dedifferentiated pancreatic ductal adenocarcinomas. In contrast, transplanted PDLOs with mutant GNAS lead to intraductal papillary mucinous neoplasia-like structures. Conclusively, PDLOs enable in vitro and in vivo studies of pancreatic plasticity, dysplasia, and cancer formation from a genetically defined background.
Introduction
The ductal compartment of the pancreas is the origin of various diseases. These range from common diseases, such as pancreatic ductal adenocarcinoma (PDAC) (Ferreira et al., 2017, Lee et al., 2018, Kopp et al., 2018), to rare diseases, such as the Alagille syndrome, which is characterized by ductal malfunction (Golson et al., 2009, Gliwicz et al., 2016). In vitro disease models for PDAC are of major clinical relevance (Boj et al., 2015, Moreira et al., 2018), not least due to its dismal prognosis and recent predictions that PDAC will rank as the second most common cause of cancer-related deaths in the Western World by 2030 (Rahib et al., 2014).
Exploitation of self-renewing human pluripotent stem cells (hPSCs) and subsequent multi-lineage differentiation has led to the development of pre-clinical in vitro pancreatic disease models (Hohwieler et al., 2019, Rowe and Daley, 2019). Pancreatic exocrine and endocrine cells develop from a common precursor, the pancreatic progenitor cell (PP). Developmentally, tripotent PPs restrict their lineage potential to a tip domain, giving rise to the acinar lineage, and a bipotent trunk domain, forming the ductal lineage together with subsequently delaminating endocrine cell types (Zhou et al., 2007, Schaffer et al., 2010). While advances in PP differentiation enabled rapid success in generating endocrine pancreatic cells and respective disease models, approaches to guide hPSCs into the exocrine pancreas remained sparse up to the recently generated exocrine pancreatic organoids (exoPOs) (Hohwieler et al., 2017, Ito et al., 2019, Rodansky et al., 2015, Simsek et al., 2016). The resulting exoPOs are, however, heterogeneous, containing immature acinar, ductal, and non-pancreatic cell types. Although exocrine pancreatic development is neither in mice nor in men fully understood, the initial induction of a tip- or trunk-like domain might be of central importance for the successful in vitro derivation and the subsequent compartment-specific disease modelling of functional acinar or ductal cells from human PSCs.
PDACs can develop from both acini and ducts. While PDACs evolving from acinar cells frequently traverse pancreatic intraepithelial neoplasia (low-grade and high-grade PanIN), ductal cells appear more refractory to developing PanIN lesions, but instead rapidly progress to aggressive cancers (Ferreira et al., 2017, Lee et al., 2018). As an additional route of PDAC development, ductal cells can form cystic IPMN lesions (intraductal papillary mucinous neoplasia) which can also further progress to PDAC (Patra et al., 2017). Such differences in the pathophysiology of PDAC cannot be solely explained by the affected cell type. Instead, various combinations of oncogenes and tumor suppressors contribute in a probably cellular context-specific manner to define plasticity and cancer progression in the pancreas (Reichert et al., 2016). Oncogenic KRAS mutations regulate diverse phenotypes in sporadic and inherited PDAC, leading to different routes of tumorigenesis permitted by the loss of distinct tumor suppressor genes (e.g., TP53, CDKN2A, BRCA1/2). Indeed, a subset of approximately 10% of PDAC patients show an inheritable predisposition with mutations in the mentioned tumor suppressors also occurring in the germline (Roberts et al., 2016). Somatic GNAS mutations are more prevalent in IPMNs (Springer et al., 2015) and, vice versa, post-zygotic, mosaic mutations can cause McCune-Albright syndrome (MAS) associated with an increased risk of developing IMPNs (Gaujoux et al., 2014, Wood et al., 2017).
In order to establish in vitro-generated pancreatic ductal organoids as a pathophysiological model, the organoid cultures should be (i) of high purity, (ii) structurally and functionally comparable to in vivo ducts, (iii) provide access to disease progression intermediate cell types, (iv) reflect the mutation-triggered pathophysiology, (v) recapitulate germline features, and (vi) provide access to arising mutation dynamics. Such a disease model would allow studying initiation and progression of pancreatic cancer as well as the mimicry of genetic predisposition syndromes. The current study establishes such a hPSC-based pancreatic ductal differentiation platform to model dysplasia and cancer progression in a genetically and cellularly defined background.
Results
Engineering pancreatic duct-like organoids from human pluripotent stem cells
Our first goal was to develop a protocol giving rise to a homogeneous population of pancreatic duct-like organoids (PDLOs) from hPSCs by recapitulating pancreatic lineage commitment in vitro. We selected 30 compounds based on literature and previous results (Hohwieler et al., 2017, Rodansky et al., 2015). Compounds were screened in a 2-phased protocol over 17 days on PPs derived from human embryonic stem cells (hESC, HUES8) (Fig.1A). Within the first phase, we aimed to mimic the segregation of trunk cells, presumed bipotent progenitors, expressing SOX9 and PDX1. The simultaneous decrease of PTF1A, NKX6-1, INS, GCG, and ALB was assessed to ensure suppression of a pancreatic tip/acinar domain as well as an endocrine or hepatic fate. The second phase was designed to allow pancreatic trunk-like organoids (PTrLOs) to develop a duct-like expression profile indicated by upregulation of KRT19 (Fig.1B). Tubulogenesis, a morphological event important for maturation in vivo, is mediated by epithelial stratification, acquisition of cell polarity, and microlumen formation (Villasenor et al., 2010, Kesavan et al., 2009). Accordingly, we aimed for a homogenous culture of one-layered ring-like epithelial organoids (Fig.1B). While, in phase I, 7 out of 20 tested compounds improved ductal differentiation based on marker expression and organoid morphology on day 30, in phase II 4 out of 28 fulfilled this requirement (Suppl.Fig. 1A–D). Exemplarily, nicotinamide was a prerequisite for the formation of ring-like organoids (Fig.1C). FGF10 and EGF caused a strong upregulation of the ductal marker KRT19 (Fig.1D,E; Suppl.Fig. 1E), an observation previously reported in murine pancreatic explants (Rhodes et al., 2012). After EGF addition, we observed a tendency for higher PDLO formation capacity and proliferation rate, thereby significantly increasing PDLO size (Fig.1E,F). Zinc sulfate (ZnSO4), in phase I+II, and KGF in phase I, supported ductal specification by upregulating KRT19 and increasing culture homogeneity (Suppl.Fig. 1E–G). The putative NOTCH activator and WNT inhibitor MSC2530818 increased CFTR expression at a concentration of 0.05 μM (Czodrowski et al., 2016, Fryer et al., 2004). At higher MSC2530818 concentrations organoid morphology was disrupted (Fig.1G). RNA-seq analysis revealed a strongly dynamic expression pattern of NOTCH signaling genes during ductal differentiation indicative for a phase-dependent role of different NOTCH mediators. Addition of MSC2530818 during phase I augmented the expression of respective NOTCH-associated genes that were activated at the expected trunk like stage at day 20 (Fig.1H). In contrast, no clear inhibition of WNT target genes was found after MSC2530818 stimulation (Suppl.Fig. 1H), while, importantly, a CFTR-related gene set was enriched (Fig.1I). Thus, our final induction medium for PDLO generation consisted in phase I of KGF, MSC2530818, ROCK inhibitor, EGF, FGF10, nicotinamide, and ZnSO4, and in phase II of EGF, FGF10, nicotinamide, and ZnSO4 (Fig.1A).
Figure 1. Engineering pancreatic duct-like organoids (PDLOs) from human pluripotent stem cells.
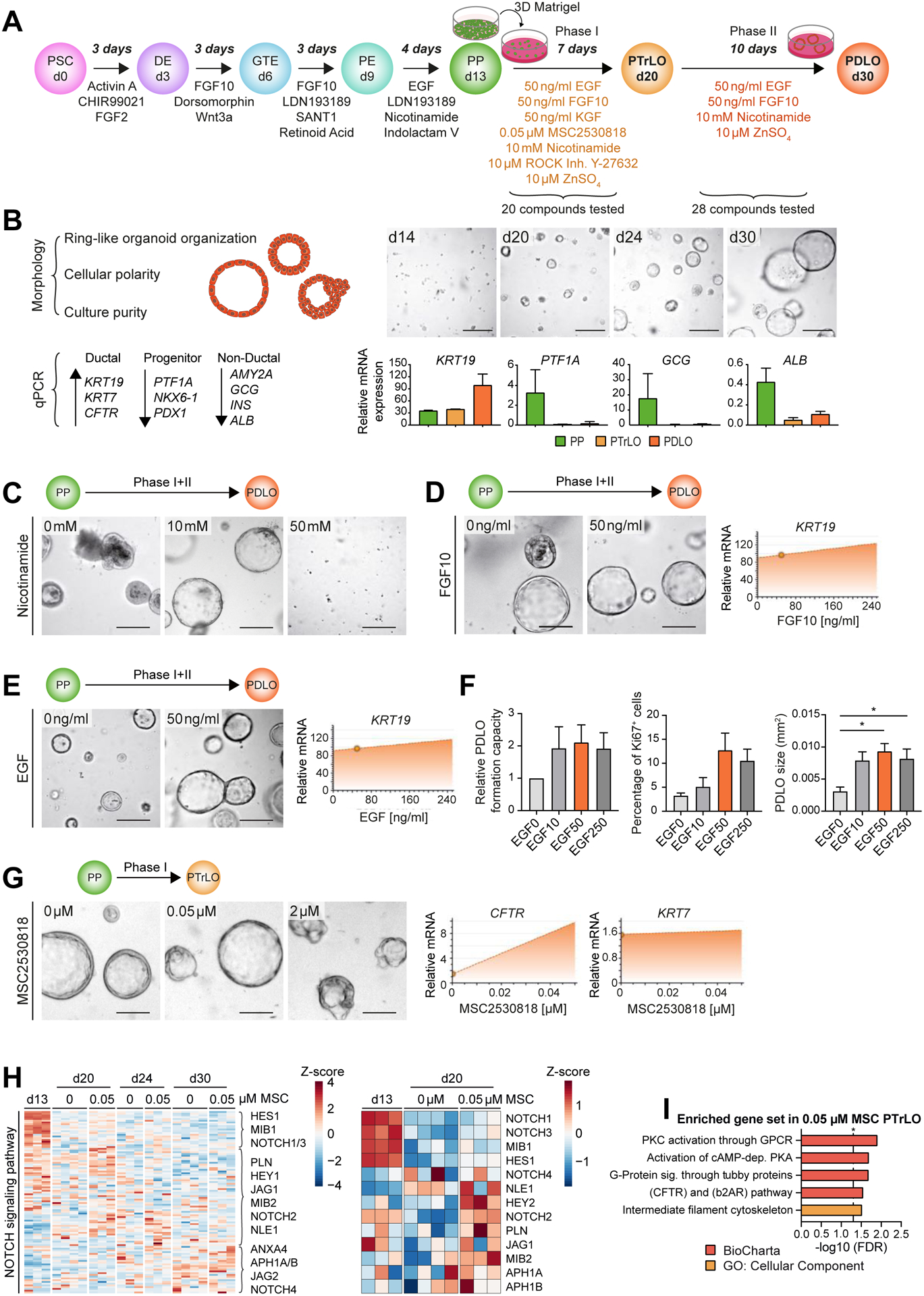
(A) Schematic overview of the 2-phase screening approach. Definitive endoderm (DE); gut tube endoderm (GTE); pancreatic endoderm (PE); pancreatic trunk-like organoid (PTrLO). (B) Left: Morphological criteria and marker for evaluation of duct formation. Right: Bright field (BF) images and marker profiles obtained from PP, PTrLOs, and PDLOs during differentiation; day (d). (C-E,G) BF images and dynamic marker profiles of PDLOs/PTrLOs. Compounds and screening phase as indicated. Dynamic marker profiles were interpolated from qPCR data using MODDE software and small circles indicate the applied concentration of the protocol version at the timepoint of testing. (C) Nicotinamide, (D) FGF10, (E) EGF, and (G) MSC2530818 titration. (F) Titration of EGF concentration in PDLO medium (0–250 ng/ml) and its effect on organoid growth characteristics (Mean±SEM; n=3; in duplicates, ordinary one-way Anova followed by Tukey’s multiple comparison test). (H,I) RNA-seq analysis of PPs, PTrLOs, and PDLOs with or without 0.05 μM MSC2530818 during phase I (0.00 μM: n=4, 0.05 μM: n=3). (H) Left: Plotting all identified genes from the GO term “NOTCH signaling pathway” (GO:0007219) over time. Right: Selection of genes at PP and PTrLO stage. (I) RNA-seq overrepresentation analysis of PTrLOs (d20) with 0.05 μM MSC2530818 treatment against PTrLOs (d20) without MSC. Scale bars: 100 μm. PDLO cultures were analyzed at day 30, if not stated elsewise. C,D,E, and G show data from one representative experiment in duplicates.
PDLOs recapitulate cell type-specific features
Next, we characterized the hESC-derived PDLOs for cell type-specific markers and conducted functional assays. PDLOs homogeneously expressed duct-specific proteins (e.g., KRT19, E-CAD, SOX9, HNF1B) with corresponding gene expression patterns (Fig.2A–C; Suppl.Fig. 2A). While progenitor markers (NKX6-1, PDX1) decreased during ductal differentiation, non-ductal markers (e.g., GCG, C-pep, AMY2A) were absent (Fig.2B–D; Suppl.Fig. 2B–D). Conclusively, immunofluorescence (IF) analysis confirmed the formation of a polarized ductal epithelium with highly organized expression of E-CAD, KRT19, KRT8 and of tight junction-associated proteins CLDN1, OCLN, and apical ZO-1 (Fig.2E,F). Complex structural organization was seen in both primary cilia staining (acTUB; Fig.2F) and transmission electron microscopy (microvilli margin, tight junctions; Fig.2G). Late ductal maturity markers (e.g. KRT7, CFTR) were only present in a subset of PDLOs (Fig.2E,F; Suppl.Fig. 2A).
Figure 2. PDLOs recapitulate cell type-specific features.
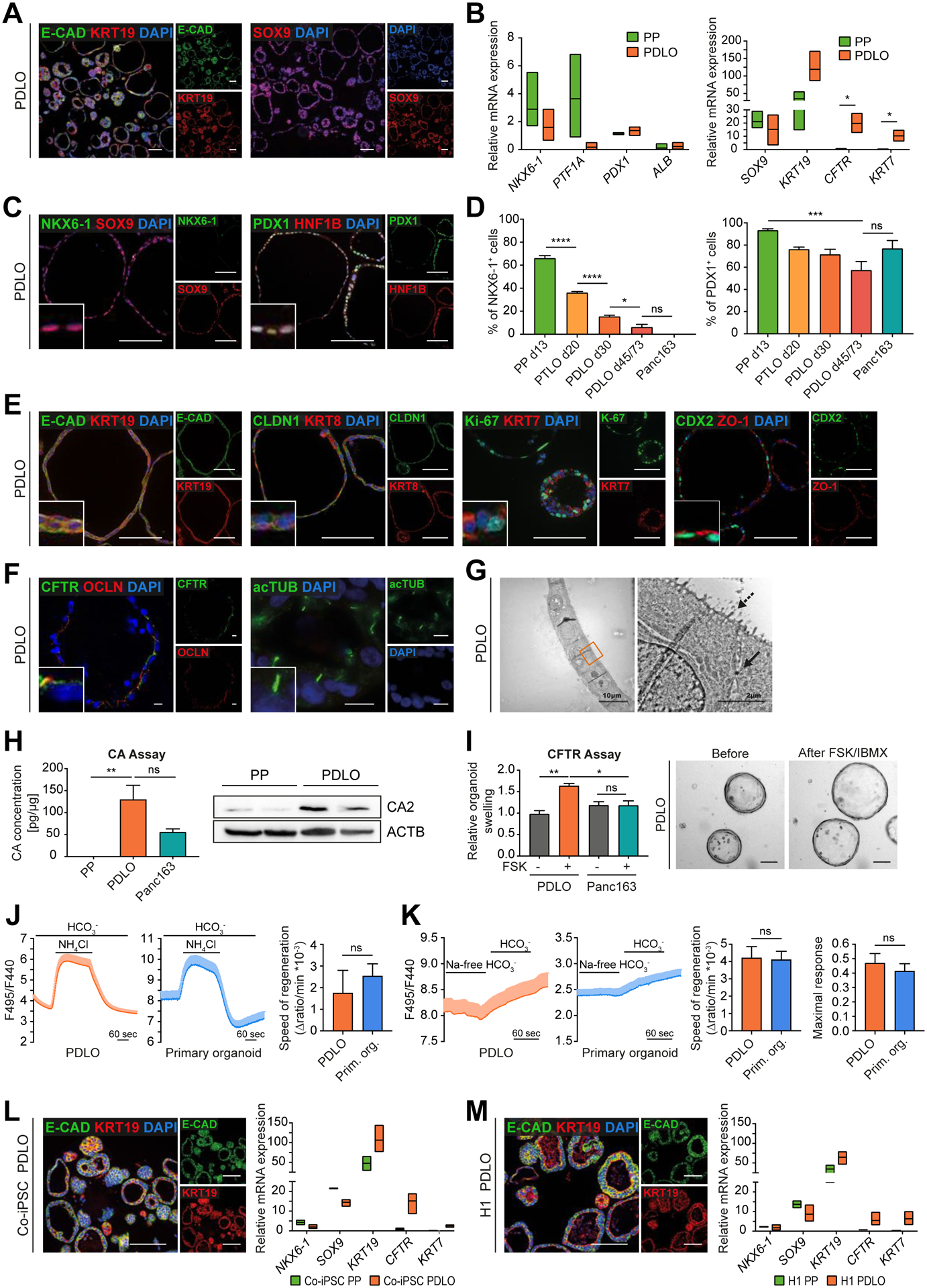
(A) Representative overview IF images of HUES8-derived PDLOs. (B) Downregulation of PP and upregulation of ductal markers in PDLOs in qPCR experiments (PTF1A, PDX1, KRT7, CFTR: n=3; NKX6-1, ALB, SOX9, KRT19: n=6; in duplicates). (C) Representative IF images of individual PDLOs. (D) Time-resolved downregulation of PP markers measured by flow cytometry (FC) in comparison with patient-derived human PDAC organoids (Panc163) (n=4; d45/73: n=3; Panc163: n=2; in duplicates). (E) IF images of PDLOs stained for ductal, epithelial, proliferation, and polarity markers. (F) IF staining for CFTR, tight junction protein Occludin (OCLN), and primary cilia (acTUB, acetylated Tubulin). Scale bar: 10 μm. (G) Transmission electron microscopy images of a PDLO. Arrow marks a desmosome, dashed arrow microvilli. (H) Carbonic anhydrase (CA) activity assay (n=3; in duplicates, 3 blinded measurements for each replicate). Right: Higher CA2 level in PDLOs than in PPs on Western blot (WB) (n=2; in duplicates). (I) PDLO swelling within the CFTR assay upon stimulation with 20 μM forskolin (FSK) and 100 μM IBMX for 18 h (n=3; in duplicates). Right: BF images of PDLOs (d44/45). (J,K) Confirmation of the functional similarity in ion secretion of PDLOs and adult primary tissue-derived ductal organoids by intracellular pH measurement. (J) Apical Cl−/HCO3– exchange activity (PDLOs: n=28; Primary organoids: n=12) (Maléth et al., 2015) and (K) basolateral Na+ dependent HCO3− uptake (PDLOs: n=15; Primary organoids: n=13; n=number of organoids) (Molnár et al., 2020) were estimated (Mean±SEM; analysis of variance/Mann-Whitney test). (L,M) IF images of KRT19 and E-CAD in PDLOs derived from Co-iPSCs or H1 together with progenitor and ductal mRNA marker expression (n=3; in duplicates). Scale bars: 100 μm, if not stated elsewise. Insets in the corners are 4x enlarged. PDLOs represent day 30 of the protocol. B,L,M: Floating bars spanning minimal and maximal values; multiple t-tests via the Holm-Sidak method; only significant comparisons are depicted. D,H,I: Mean±SEM; ordinary one-way Anova followed by Tukey’s multiple comparison test.
To test PDLO functionality, we compared the activity of carbonic anhydrase (CA), a key enzyme expressed in pancreatic ducts to catalyze bicarbonate (HCO3–) production to PPs and PDAC organoids (Panc163) (Wilschanski and Novak, 2013). Indeed, CA activity was significantly higher in PDLOs than in PPs, confirming upregulation of CA2 during ductal maturation (Fig.2H). CFTR mediates HCO3– secretion and subsequent osmosis-mediated water influx into the ductal lumen (Dekkers et al., 2013). Accordingly, stimulation with forskolin (FSK) led to a significant swelling of PDLOs indicative of CFTR ion channel activity (Fig.2I). Ki-67 was stained to exclude that the organoid size increases during the functional assay via proliferation (Suppl.Fig. 2I). Measurement of intracellular pH with the fluorescent indicator BCECF-AM demonstrated comparable apical Cl−/HCO3− exchange and basolateral Na2+ -dependent HCO3− uptake activities in PDLOs and adult human organ donor-derived pancreatic organoids (Fig.2J,K). To demonstrate broad applicability of our protocol, we differentiated a control hiPSC line (Co-iPSC) and the hESC line H1 into PDLOs. Both PDLO cultures showed comparable mRNA and protein marker expression to HUES8-derived PDLOs (Fig.2L,M; Suppl.Fig. 2E,F). The activity assays remained reproducible across multiple lines, which suggested the presence of functional CA enzyme and HCO3− secretion activity in all hPSC-derived PDLOs (Suppl.Fig. 2G–I). Still, the protocol performed best in HUES8 cells and further cell line tailored fine tuning will be necessary as described, e.g. for PP differentiation (Nostro et al., 2015).
Global transcriptomic and proteomic analyses confirm ductal identity
To validate the specificity, maturity, and developmental trajectories of PDLOs, we analyzed time-resolved transcriptomes in comparison to non-transformed human pancreatic ductal organoid controls derived from a resection specimen. Stage-specific clustering of differentially regulated genes revealed a closer proximity of PDLOs to primary organoids than to PPs (Fig.3A). Accordingly, progenitor genes were gradually lost during differentiation and duct-related genes (VI-VIII) including several claudins, annexins, and mucosal barrier-related genes (MUC13, TFF1/2), together with ion/water secretion-related genes like AQP3 were upregulated (Fig.3B,C, Suppl.Tab.1,2). Time-resolved dynamics of pancreatic ductal maturation and PP genes further validated the acquisition of a duct-like transcriptome in PDLOs, albeit the degree of maturation from primary adult ductal organoids could not be fully met (Fig.3C). Gene set enrichment analysis (GSEA) confirmed the loss of PP identity in PDLOs (Fig.3D) (Gerrard et al., 2016, Xie et al., 2013). To probe the transient acquisition of a trunk-like domain upon differentiation, trunk-specific gene sets derived from HES1+ NGN3− DBA+ E15.5 mouse pancreata (de Lichtenberg et al., 2018) and a scRNA-seq set from E15.5-E18.5 mice (Krentz et al., 2018) were employed. Indeed, transcriptomes of day 20/24 organoids were enriched for trunk-specific genes (Fig.3E), and trunk-specific markers such as ID2, ST3GAL6, or CXCL12 peaked at those time points (Cluster IV,V; Fig.3B, Suppl.Tab.1, Methods S5). Likewise, a ductal program was already initiated in PTrLOs as suggested by the upregulation of ductal markers such as SCTR and CFTR. At day 30, PDLOs were significantly enriched for ductal gene sets from two scRNA-seq studies (Baron et al., 2016, Enge et al., 2017) (Fig.3F). A recently developed cell population mapping algorithm for cell-type deconvolution (Frishberg et al., 2019) was additionally applied and assigned PDLOs among the different pancreatic cell types closest to ductal cells (Suppl.Fig. 3A). Furthermore, GSEA from the hallmark database and GO-term analysis indicated an acquisition of maturity in PDLOs, as proliferative and early developmental terms were depleted in exchange with enriched metabolic terms (Suppl.Fig. 3B–D).
Figure 3. Global transcriptomic and proteomic analyses confirm ductal identity.
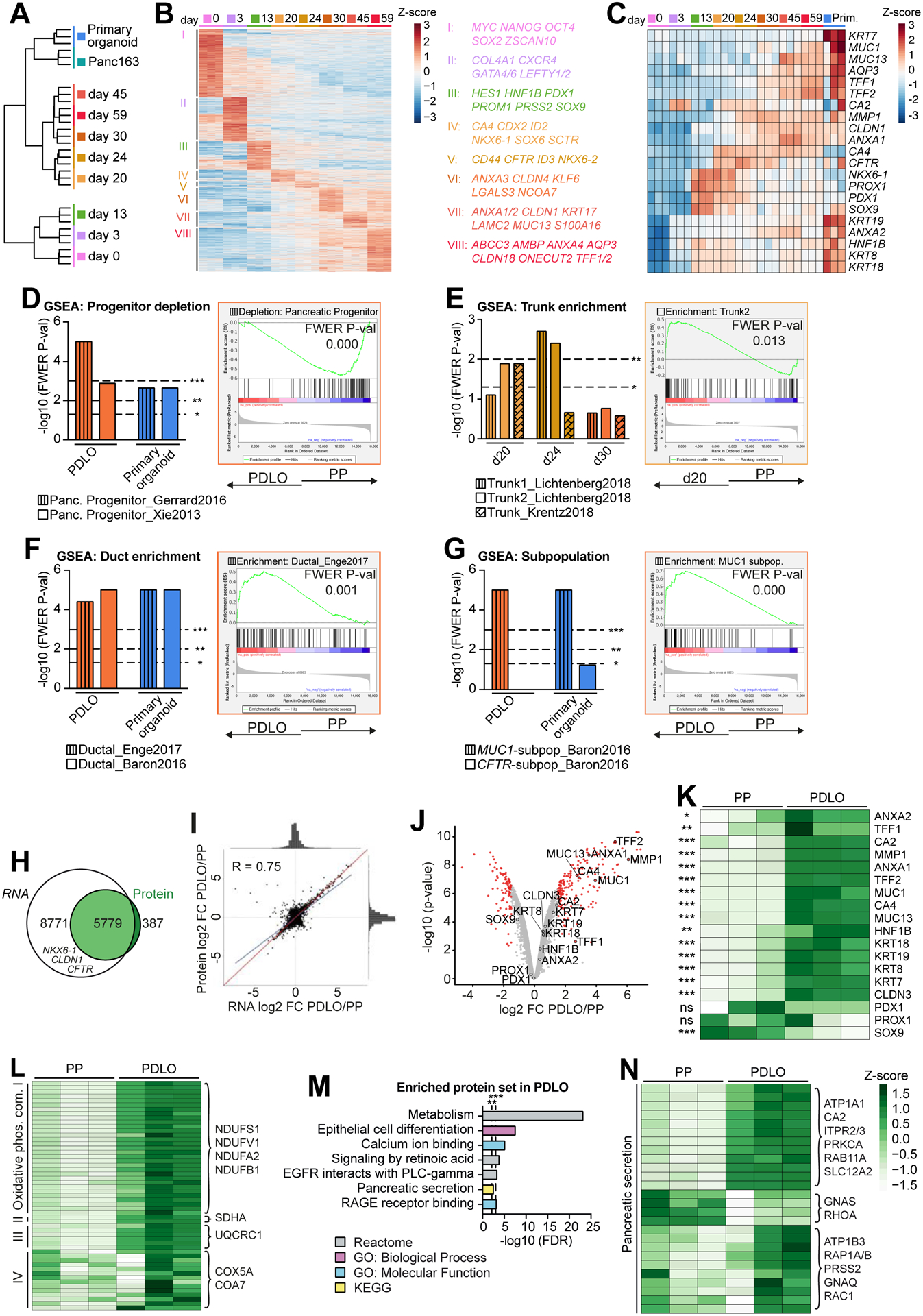
(A) Global RNA-seq data during PDLO differentiation and of patient-derived human ductal organoids (n=3). Ward clustering was performed with all processed genes. (B) Heatmap of stage-specific significant genes. (C) Temporally resolved heatmap of key progenitor and ductal genes. (D-G) Gene Set Enrichment Analysis (GSEA) of d20, d24, PDLOs (d30), and primary ductal organoids against PPs (d13) for distinct reference gene sets. Exemplary GSEA plots are highlighted in respective sample colors. (H) Venn diagram representing the overlap of transcripts measured by RNA-seq with proteins detected by mass spectrometry (n=3). (I) Pearson correlation of RNA-seq and proteome log2FC of PDLOs (d59) versus PPs (d13). The blue line indicates actual correlation, the red line ideal correlation of all 5779 shared genes/proteins. (J) Volcano plot of protein mass spectrometry data of PDLOs and PPs. Differentially regulated proteins in red (P-value ≤ 0.01 and FC ≥ |1.5|). (K) Heatmap of key progenitor and ductal proteins in PPs and PDLOs. (L,N) Heatmap illustration of proteins (L) from the four “Oxidative phosphorylation” complexes and (N) the KEGG term “Pancreatic Secretion”. (M) Enriched protein sets in PDLOs over PPs.
Two studies indicate that in vivo ductal cells might be either predominantly important for water secretion to allow flow of zymogens (expression of CFTR) or are specialized to protect pancreatic tissue against digestive enzymes in the pancreatic juice (expression of MUC1) (Burghardt et al., 2003, Baron et al., 2016). PDLO transcriptomes were only enriched for a MUC1-positive signature, indicating the formation of ductal subpopulations in vitro (Fig.3G). Similarly, Qadir et al. (2020) identified five ductal subpopulations, from which four were enriched in PDLOs (Suppl.Fig. 3E). GO-terms for keratinocyte differentiation and RAGE receptor binding, known as receptors for advanced glycation end products and possibly linked to mucin production (Xiong et al., 2017), were enriched at later PDLO stages, supporting a MUC1 dominant subtype (Suppl.Fig. 3F).
To validate transcriptomic data, we measured the global proteomes of hPSC-derived PPs and PDLOs at the endpoint of differentiation, with more than 6000 proteins detected in both samples (Fig.3H). The analysis revealed an overall high correlation of protein and RNA levels (Fig.3I; Suppl.Fig. 3H), mirrored by an upregulation of ductal maturity proteins (e.g., MUC1/13, TFF2) in PDLOs (Fig.3J,K, Suppl.Tab.3). Increased protein expression of all detected members of the oxidative phosphorylation complex in PDLOs (Fig.3L) further suggests an increased metabolic turnover. In addition to similar GO-term enrichment/depletion patterns in transcriptome and proteome (Fig.3M; Suppl.Fig. 3B,I,J), terms such as retinoic acid signaling, EGFR signaling, and pancreatic secretion were enriched in PDLOs at protein level (Fig.3M,N). These terms were previously assigned to primary ductal organoids (Suppl.Fig. 3G) and further support the maturity of PDLOs.
Development of human duct-like tissue after xenotransplantation of PDLOs
Transplantation of in vitro differentiated cells into a suitable host site can support further maturation by mimicking a physiological niche (Rezania et al., 2012). To test this, we transplanted PDLOs or 3D-aggregated PPs into the anterior chamber of the mouse eye (ACE) (Fig.4A; Suppl.Fig. 4A) as previously described (Cohrs et al., 2020). Tripotent PPs mainly formed compact structures in vivo containing all pancreatic lineages, mainly CTRC-positive acinar, but also ductal and endocrine (GCG, C-pep) cells (Fig.4B–D). In contrast, PDLO grafts formed complex ring-like structures (Fig.4B,C) expressing the ductal markers KRT7, KRT8 and KRT19 (Fig.4D). PDLO in vivo imaging allowed to monitor engraftment on the iris, subsequent lumen formation, and the finally arising large duct-like tissue likely supported by vascularization (Suppl.Fig. 4B,C).
Figure 4. Development of human duct-like tissue after xenotransplantation of PDLOs.
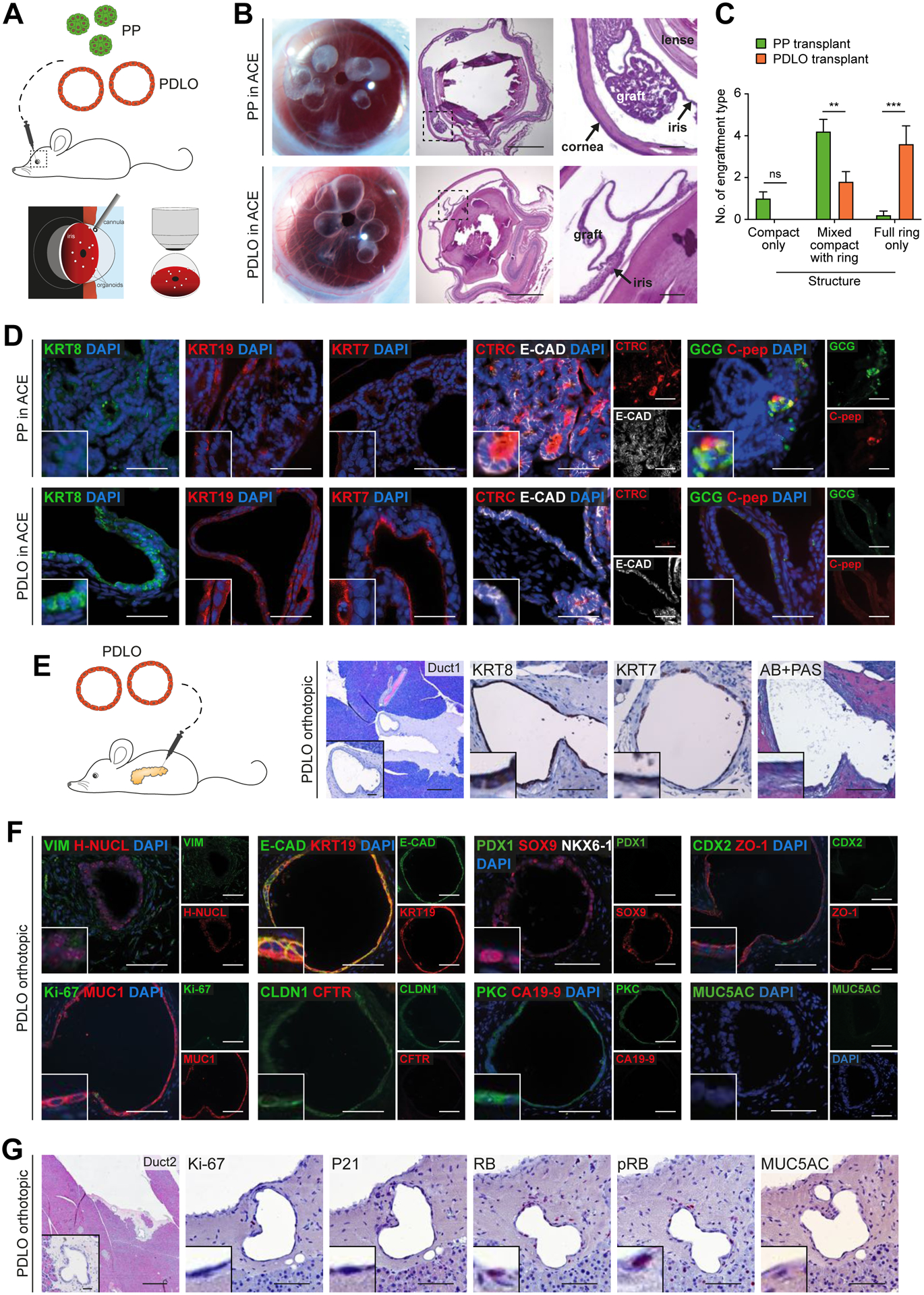
(A) Scheme of transplantation into the anterior chamber of the mouse eye (ACE). (B) Growth of grafted organoids on the iris 5 weeks after transplantation. Left panel: Image of eyes transplanted with PPs or PDLOs. Right panels: HE staining of sagittal section of explanted eyes with PDLO graft on the iris. (C) Quantification of observed engraftment types (Mean±SEM; n=5 mice per group; ordinary two-way Anova with Sidak’s multiple comparison test)(D) IF staining of PP-derived grafts revealed acinar, ductal, and endocrine cells in the ACE, while marker expression of PDLO-derived grafts was restricted to the ductal pancreatic lineage (CTRC, Chymotrypsin C; C-pep, C-peptide). (E) Orthotopic transplantation scheme and HE/IHC images demonstrating engraftment site 8 weeks after transplantation (n=5 mice). (F) PDLO transplants expressed ductal epithelium-specific proteins, MUC1, E-CAD, KRT19, and CLDN1, lost transcription factors, PDX1 and CDX2, but lacked CFTR and CA19-9 expression. (G) WT PDLO transplant stained for proliferation- and cell cycle-related proteins and the dysplastic marker MUC5AC (RB, retinoblastoma protein; pRB, phosphorylated RB). B,E-G: Scale bars: 100 μm. Insets in the corners are 4x enlarged. Except in overview staining: 500 μm, here insets: 50 μm. D: Scale bar: 50 μm, insets are 2x enlarged.
While kidney capsule transplantation is commonly used for endocrine engraftments (Hogrebe et al., 2020, Rezania et al., 2012), we and others have previously shown that the pancreatic niche is particularly suitable for exocrine engraftments (Hohwieler et al., 2017, Georgakopoulos et al., 2020). Orthotopic PDLO transplantation into immunocompromised mice led to the formation of tubularly organized structures, as demonstrated by consecutive sections (Fig.4E). The human tissue (identified by H-NUCL staining) within the transplantation site homogeneously expressed the ductal markers KRT8, KRT19, E-CAD, CLDN1 and SOX9 (Fig.4E,F). In agreement with the in vitro transcriptomic analysis, the majority of PDLOs in vivo developed duct-like tissue expressing MUC1 (Fig.4E,F). MUC1-negative duct-like tissue revealed in selected grafts CFTR expression, emphasizing the potential of our protocol to generate distinct ductal subpopulations of the pancreas (Suppl.Fig. 4D). Besides tubular organization of the graft and homogeneous MUC1 or CFTR expression, maturation upon transplantation was observed by downregulation of Ki-67, PDX1, and CDX2. Dysplasia indicating proteins were absent and respective checkpoints remained intact (Fig.4F,G). To validate marker expression, healthy human pancreatic tissue was stained with the same antibodies, further corroborating our results (Suppl.Fig. 4E).
KRASG12D expression induces lumen-filling and EMT in PDLOs
To establish PDLOs as a potential model for dysplasia and cancer, we investigated whether oncogenic KRAS or CDKN2A loss evoke specific phenotypes in vitro. For this we used piggyBac transposon-based vectors (Kim et al., 2016) to conditionally express mutant KRASG12D and CRISPR/Cas9 to knock out CDKN2A in the hESC line HUES8 (Fig.5A; Suppl.Fig. 5A,B). All engineered cell lines efficiently formed PPs and PDLOs, and expression of the HA-tagged KRASG12D construct was dose-dependently induced by doxycycline (Dox) (Suppl.Fig. 5C). Pulldown assays of active-KRAS and increased expression of phosphorylated ERK (pERK) levels validated functionality of the introduced KRASG12D (Suppl.Fig. 5D,E). Expression of KRASG12D in PDLOs of CDKN2A-deficient or -proficient background caused a lumen-filling and size reduction of PDLOs (Fig.5B; Suppl.Fig. 5F). KRASG12D induction significantly reduced proliferation depending on the Dox dosage as assessed by Ki-67 staining and cell cycle analysis (Fig.5C,D; Suppl.Fig. 5G,H).
Figure 5. KRASG12D expression induces lumen-filling and EMT in PDLOs.
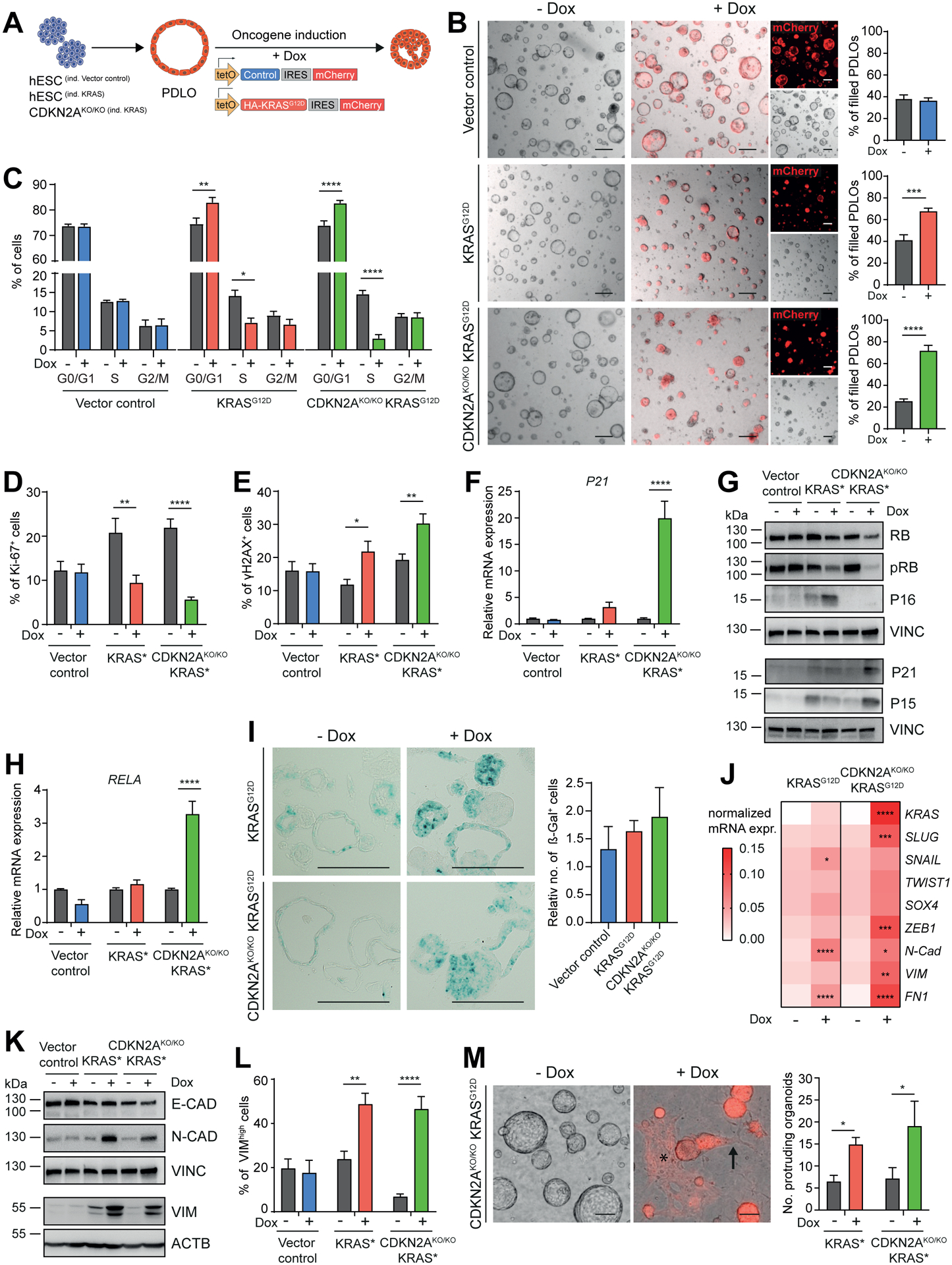
(A) Timed induction of a piggyBac KRASG12D transposon construct in engineered HUES8. (B) BF PDLO images after induction of the vector control in CDKN2AWT/WT cells, or the KRASG12D expression cassette in CDKN2AWT/WT and CDKN2AKO/KO cells. Formation of lumen-filled PDLOs was quantified (n=3; in triplicates). Scale bar: 200 μm. (C) Cell cycle analysis in PDLOs −/+ Dox (n=3; in duplicates). (D,E) FC quantification of proliferation marker Ki-67 and DNA-damage marker γH2AX (n=3; in duplicates). (F) qPCR analysis of P21 (n=3; in duplicates). (G) Genotype-dependent differential regulation of cell cycle regulators and checkpoint proteins. See Suppl.Fig. 5I,J for respective quantification of WB analysis. (H) qPCR analysis of senescence marker RELA (n=3; in duplicates). (I) Histological sections of PDLOs stained for senescence-associated β-galactosidase activity (dark cyan color) and respective quantification (vector control, CDKN2AKO/KO KRASG12D n=3; KRASG12D n=5). (J) Marker panel revealing increased EMT on mRNA level (n=3; in duplicates). (K) Regulation of EMT-related proteins after oncogene induction. See Suppl.Fig. 5O for respective quantification. (L) FC analysis of PDLO cells with high VIM expression (vector control, CDKN2AKO/KO KRASG12D n=4; KRASG12D n=3; in duplicates). (M) BF PDLO images reveal how cells adopt morphological features of EMT in response to KRAS activation. Arrow: single cells disseminating from a PDLO, asterisk: area of mesenchymal-like cells (red, mCherry). Right: Phenotype quantification (n=4; in duplicates). Refer to Suppl.Video S1,2 for respective live-cell imaging. All data were acquired in PDLOs at day 38, 9 days after Dox induction. Scale bar: 100 μm, if not stated elsewise. For all subfigures: Mean±SEM; only significant comparisons are depicted. B-F,H,J,L,M: Ordinary two-way Anova with Sidak’s multiple comparison test. I: Ordinary one-way Anova with Tukey’s multiple comparison test.
One explanation of the growth reduction could be oncogenic KRAS-provoked replication stress (Di Micco et al., 2006). Indeed, KRASG12D-induced cells upregulated the phosphorylated histone variant H2AX (γH2AX), indicative of DNA damage foci (Zeman and Cimprich, 2014) (Fig.5E). Checkpoints limiting cell growth in response to DNA damage and replicative stress can contribute to cancer protection (Bartek et al., 2007). Accordingly, we observed a KRASG12D-driven induction of P16 on the CDKN2A-proficient background, while P21 was particularly upregulated in Dox-treated CDKN2AKO/KO PDLOs (Fig.5F,G; Suppl.Fig. 5I). This suggests a functional P21 checkpoint to induce cell cycle arrest even in the absence P16 (Takeuchi et al., 2010). Upregulation of the NF-κB subunit RELA in CDKN2A-deficient PDLOs after KRASG12D induction supports this hypothesis (Fig.5H), as RELA can operate as a mediator of oncogene-induced senescence (OIS) (Lesina et al., 2016). As an additional putative mechanism of cell cycle inhibition in CDKN2AKO/KO cells, we detected increased levels of P15 (CDKN2B) after KRASG12D induction, which can replace P16 to inhibit CDK4/6, thus preventing inactivation of RB, a major G1/S checkpoint regulator (Tu et al., 2018, Kuilman et al., 2008) (Fig.5G; Suppl.Fig. 5I,J). Indeed, KRASG12D expression reduced phosphorylated RB (pRB) levels (Fig.5G; Suppl.Fig. 5J) while active (not hyperphosphorylated) RB prevents cells to enter the S-phase. We conclude that ablation of P16 alone was not sufficient to prevent KRAS-induced cell cycle arrest in PDLOs. These observations pointed toward OIS culminating in a tendency for increased senescence-associated β-galactosidase activity upon KRASG12D induction, independent of the genetic CDKN2A status (Fig.5I). Furthermore, the pro-apoptotic marker BAX was upregulated in CDKN2AKO/KO PDLOs, as was cleaved PARP after KRASG12D induction (Suppl.Fig. 5K,L). Intriguingly, senescence and apoptosis can operate as a tumorigenic roadblock in pre-neoplastic lesions (Lee and Schmitt, 2019). Epithelial-to-mesenchymal transition (EMT) has been described to potentially bypass such roadblocks during carcinogenesis (Ansieau et al., 2008, Song and Shi, 2018). Therefore, we analyzed EMT-related markers revealing differential and KRASG12D dose-dependent upregulation in the PDLO system on mRNA and protein level (Fig.5J–L; Suppl.Fig. 5M–P). Furthermore, cellular changes resembling the EMT-like character were detected by real-time imaging of CDKN2AKO/KO KRASG12D PDLOs, including outgrowth of single spindle-shaped cells from PDLOs, followed by cell flattening of migrating cells (Video S1,2; Fig.5M). Since a reduction of the E-CAD protein level could not be observed (Fig.5K; Suppl.Fig. 5N–P), we assume that the transcriptional and phenotypic pattern displays a partially initiated EMT program (Grigore et al., 2016, Aiello et al., 2018).
McCune-Albright syndrome-derived and GNASR201H-overexpressing PDLOs form large cysts
Somatic activating GNAS mutations, most frequently p.R201C or p.R201H, are major drivers of dysplastic growth in IPMNs (Tan et al., 2015, Wu et al., 2011). Postzygotic mosaic GNAS mutations cause fibrous dysplasia (FD), a rare disorder characterized by abnormal bone. McCune-Albright syndrome is associated with FD, café-au-lait macules, and endocrinopathies. Notably, several studies report pancreatic cysts in MAS patients accompanied by an increased PDAC risk (Robinson et al., 2018, Wood et al., 2017). In order to derive an in vitro MAS model to explore disease pathogenesis driven by GNAS mutations, a genetically mosaic culture of human bone marrow stromal cells was reprogrammed to generate clonal isogenic iPSC lines with either heterozygous p.R201C or WT GNAS genotype (Fig.6A,B; Suppl.Fig. 6A). MAS-iPSC lines were pluripotent and efficiently differentiated into PPs and PDLOs, irrespective of their GNAS status (Fig.6C–F; Suppl.Fig. 6B–D). While GNASWT/WT PDLOs resembled PDLOS derived from WT hESCs, GNASWT/R201C MAS-iPSCs formed large cystic PDLOs (Fig.6D,E; Suppl.Fig. 6D). Cystic growth can be explained by the increased proliferation of GNASWT/R201C cells in the PDLOs, indicated by Ki-67 expression and flow cytometry-based quantification of EdU incorporation (Fig.6F; Suppl.Fig. 6E). Next, we investigated downstream effectors of the GNAS-encoded protein Gαs, which mediates cAMP generation via the adenylyl cyclase. GNAS mutations lead to a constitutively active signaling axis upon stabilization of GTP-bound Gαs (Rosenbaum et al., 2009). Consistently, intracellular cAMP levels were elevated in both GNASWT/R201C iPSCs and PDLOs (Fig.6G). In line with this, GNASWT/R201C PDLOs displayed hyperactive protein-kinase-A (PKA) signaling shown by phosphorylation of target proteins including VASP. Inhibition of PKA reduced cyst size (Fig.6H,I). Of note, sustained PKA activation was reported to mediate Gαs signaling in the context of cystic pancreas tumorigenesis in mice (Patra et al., 2018).
Figure 6. McCune-Albright syndrome-derived and GNASR201H overexpressing PDLOs form large cysts.
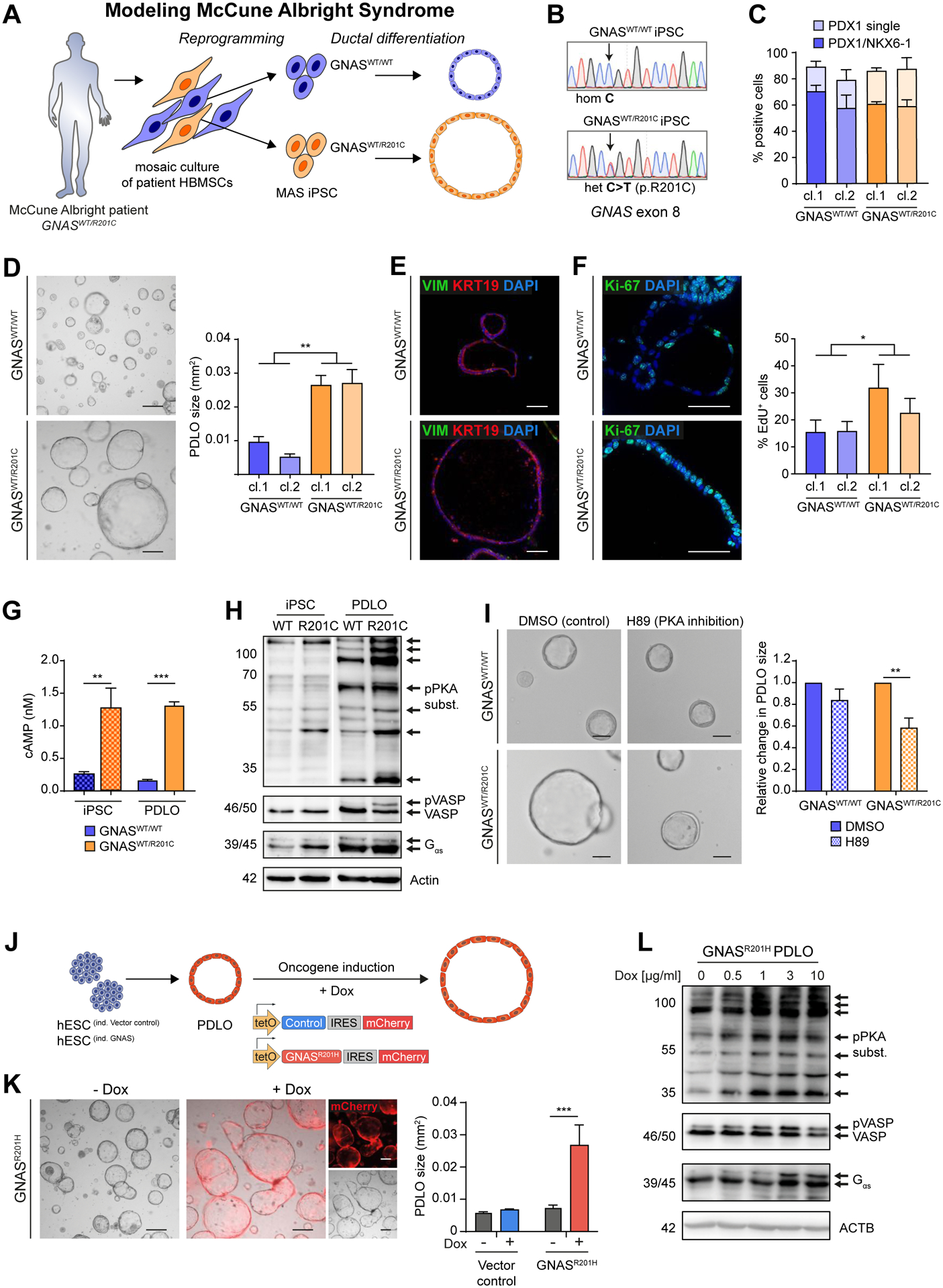
(A) Scheme of generating isogenic iPSC lines from a MAS patient carrying a mosaic GNASWT/R201C mutation followed by PDLO formation. (B) Sequencing results of selected iPSC clones. (C) FC-based PP quantification after differentiation of GNASWT/WT and GNASWT/R201C MAS-iPSCs (n=3; cl.: clonally derived iPSC line). (D) BF PDLO images from MAS-iPSCs. Right: Size comparison. (E) VIM and KRT19 IF staining of MAS-PDLOs. (F) Ki-67 IF staining (left images) and FC analysis after EdU-treatment (right) showed increased proliferation of GNASWT/R201C PDLOs. (G) Analysis of cAMP levels in MAS-iPSC and PDLO cells (n=1; in triplicates). (H) WB showing increased PKA signaling in GNASWT/R201C PDLOs. iPSC and PDLO samples shown separately were detected on the same blot, image was cropped due to additional loaded samples (n=1). (I) Representative BF images of MAS-iPSC-derived PDLOs treated with PKA inhibitor H89 or DMSO for 9 days. Right: Size quantification of PDLOs upon inhibition of PKA signaling (n=3). (J) Timed induction of a piggyBac GNASR201H transposon construct in engineered HUES8. (K) BF images of GNASR201H PDLO cultures after 7 days on Dox. (red: mCherry signal). Right: PDLO size quantification (n=3; in triplicates). (L) Dox concentration-dependent increase of PKA-signaling in PDLOs after Dox treatment for 3 days. Scale bar: 200 μm, if not stated elsewise. Mean±SEM; D,F: n=6 experiments per group (3 per individual clone), Mann-Whitney test. G: Ordinary one-way Anova with Tukey’s multiple comparison test. I,K: Ordinary two-way Anova with Sidak’s multiple comparison test; only significant comparisons are depicted.
To confirm the results from MAS-iPSCs, we engineered hESCs with the inducible GNAS variant (p.R201H) using the piggyBac system (Fig.6J). Overexpression of GNASR201H within hESC-derived PDLOs led to cystic growth and sustained cAMP-PKA-VASP signaling (Fig.6K,L; Suppl.Fig. 6F–H), which recapitulated MAS patient data. Thus, we present a patient-specific and an engineered in vitro model for activating GNAS mutations in human ductal organoids to study this oncogene as a driver of pancreatic cyst growth in humans.
Mutation dependent heterogeneity of KRASG12D driven PDAC-formation in PDLO grafts
To corroborate in vitro PDLO alterations in xenograft experiments, we orthotopically transplanted 51 mice with the previously described transgenic PDLOs. We induced KRASG12D alone or in genetic covariance of CDKN2A loss (CDKN2AKO/KO) as well as GNASR201H over 8 weeks in vivo (Fig.7A,G; Suppl.Fig. 7A). The average engraftment rate across all genotypes was around 75%, with the highest rates (100%) in CDKN2AKO/KO PDLOs. The reporter (HA-Tag, mCherry) signal of PDLO cells was robustly detected in the grafts (Suppl.Fig. 7A). Notably, the four PDLO grafts arising from KRASG12D PDLOs showed a substantial degree of dysplastic heterogeneity, resulting in one small glandular PDAC-like lesion forming a tumor mass with cells invading the murine host (PDAC 1). The second and third engraftment resembled high-grade preneoplastic lesions, smaller in size as PDAC 1, however, without invasion of the murine host (Fig.7B upper row) but with very strong MUC5AC and CA19-9 expression (Suppl.Fig. 7A, staining not shown). A fourth graft showed only moderate signs of dysplasia in line with less CA19-9 and MUC5AC expression (data not shown). In contrast to the heterogeneous dysplastic lesions in KRASG12D grafts, all CDKN2AKO/KO KRASG12D PDLO grafts with robust transgene expression formed large dedifferentiated, polymorphic PDAC-like lesions and invaded into the mouse host (Fig.7B lower row; Suppl.Fig. 7A). All KRASG12D grafts homogeneously expressed epithelial (e.g. KRT19) and partially mesenchymal markers (N-CAD, VIM) (Fig.7C upper row; Suppl.Fig. 7B upper row). Vice versa, only a few single tumor cells retained an epithelial phenotype in CDKN2AKO/KO KRASG12D tumors. Here, most tumor cells acquired a mesenchymal marker pattern when compared with CDKN2AKO/KO engraftments from the same organoid preparation lacking KRASG12D induction (Fig.7C lower row; Suppl.Fig. 7B lower row; compare to Suppl.Fig. 7C). To directly link KRASG12D dosage with EMT in the CDKN2AKO/KO genotype, we focused our analysis on areas with heterogeneous KRASG12D induction. Indeed, we observed spatial concordance of HA-tag expression and the occurrence of putative disseminating cells from the ductal epithelial layer expressing mesenchymal markers (Fig.7D). Notably, the CDKN2AKO/KO grafts formed well-differentiated pancreatic ducts with moderately increased proliferation compared to WT ducts (Suppl.Fig. 7C; compare to Fig.4G; Suppl.Fig. 7A). Likewise, we found signs of cellular atypia and dysplasia accompanied by the PDAC markers MUC5AC and/or CA19-9 in three out of six grafts without additional KRAS induction (Suppl.Fig. 7A,C).
Figure 7. Mutation-dependent PDAC- or IPMN-like tumor formation from PDLO grafts.
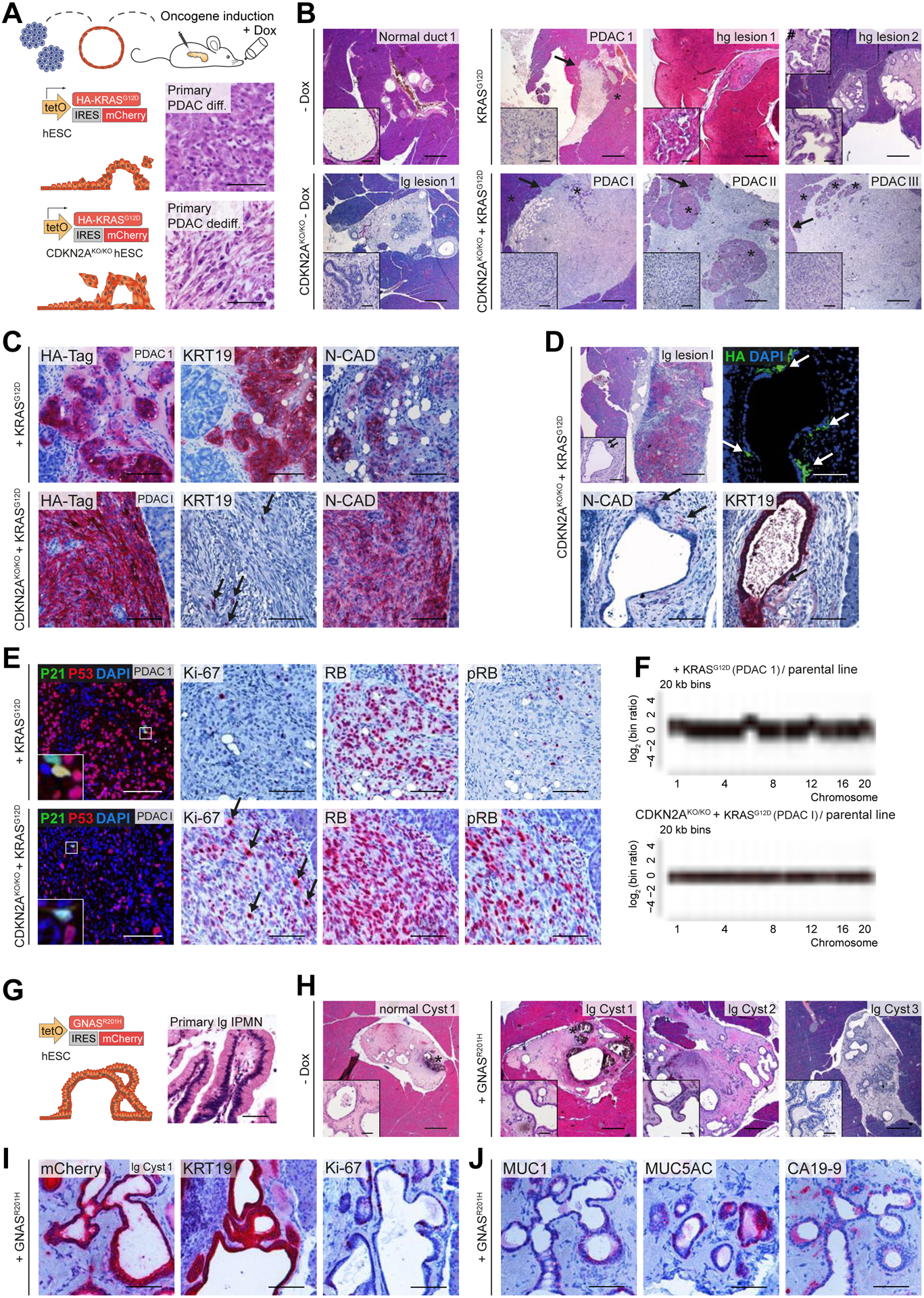
(A) Schema of orthotopic PDLO transplantations. Oncogene expression was induced in vivo for 8 weeks. Reference images of HE staining of primary PDAC patient tissue are depicted for illustration. (B) HE overview images of grafts that developed from KRASG12D and CDKN2AKO/KO KRASG12D PDLOs with and without oncogene induction (lg/hg lesion, low-grade/high-grade preneoplastic lesion). Asterisks label murine pancreas tissue, arrows indicate invasive tumor growth, hashtag marks a second inset from a different section of the same graft, demonstrating a higher grade of cellular atypia. See Suppl.Fig7A for number of transplanted mice. (C) IHC staining showing that KRASG12D induction (reflected by HA-Tag) alone led to differentiated PDAC, and CDKN2AKO/KO KRASG12D to dedifferentiated PDAC. Arrows highlight single epithelial cells in the CDKN2AKO/KO KRASG12D graft. (D) One specific CDKN2AKO/KO KRASG12D graft with heterogenous transgene induction. Sites of dissemination and EMT correlate with HA-tag expression (indicated by arrows). (E) Staining of cell cycle-associated proteins. Few P21 positive cells. RB/pRB staining indicated an intact checkpoint control in KRASG12D tumors, but nearly complete loss of active RB in CDKN2AKO/KO KRASG12D grafts resulting in increased proliferation (Ki-67) in CDKN2AKO/KO KRASG12D grafts. Arrows highlight several mitoses in the CDKN2AKO/KO KRASG12D graft. (F) lcWGS of PDLO-derived tumors. (G) Schema and reference HE image of primary patient IPMN tissue. (H) IPMN-like lesion formation observed after GNASR201H induction in vivo. HE overview of low-grade cystic GNASR201H grafts and control without Dox treatment. Asterisks label disrupted Matrigel, observed in few grafts. See Suppl.Fig7A for number of transplanted mice. (I,J) IHC staining indicating IPMN formation after GNASR201H induction (confirmed by mCherry expression). Scale bar: 100 μm, except for HE staining in B,D,H: 500 μm; in insets: 50 μm.
Oncogenic roadblocks are released in PDLO grafts
Next, we dissected the cell cycle roadblocks upon in vivo tumor formation in KRASG12D-driven tumorigenesis on a CDKN2A-proficient or -deficient background. Histopathological alterations and strongly increased proliferation in KRASG12D plus CDKN2AKO/KO were accompanied by attenuated P53 expression and an almost complete absence of P21 positive cells (Fig.7E lower row; Suppl.Fig. 7A). Similarly, the RB checkpoint was overcome either through increased phosphorylation of RB, indicative of its inactivation, or directly through reduced expression of RB (Fig.7E lower row; Suppl.Fig. 7D lower row). To interrelate checkpoint loss with structural aberrations, we performed low coverage whole genome sequencing (lcWGS) of two KRASG12D CDKN2AKO/KO tumors (PDAC I and III), but neither of them demonstrated substantial chromosomal rearrangements, indicating low strains in this genotype to overcome the threshold of cancerous growth (Fig.7F lower panel; Suppl.Tab.4). Accordingly, panel sequencing of PDAC II identified a P53S94P mutation with a variant allele frequency of 25% amongst other single-nucleotide variants (SNVs) (Suppl.Fig. 7E; Suppl.Tab.5).
In contrast, only KRASG12D-induced tumors with an intact CDKN2A/P16 checkpoint revealed a higher intra- and inter-tumoral heterogeneity than their CDKN2A-deficient counterparts, mirrored also in various degrees of proliferation (Fig.7E upper row; Suppl.Fig. 7D upper row). While P21 was only expressed in few cells of PDAC 1, the RB/pRB checkpoint appeared intact (Fig.7E upper row). Similarly, an intact RB and P53/P21 checkpoint correlated with the lack of proliferation in regions of high-grade lesion 1 where increased tissue dysplasia was observed (Suppl.Fig. 7D dashed arrows in upper row). lcWGS from KRASG12D-driven PDAC 1 graft revealed broader chromosomal rearrangements than in the KRASG12D CDKN2AKO/KO tumors (Fig.7F; Suppl.Tab.4). Interestingly, displayed CNVs in this tumor included previously reported regions of recurrent alterations such as arm-level gains of chromosomes 1, 6, 12, and 20 (Shain et al., 2012, Notta et al., 2016). To conclude, despite the possibility of tumor formation from KRASG12D induction only, CDKN2A operated as a roadblock to pancreatic ductal dysplasia and proliferation, and, in concert with KRASG12D, to EMT.
PDLOs expressing mutant GNAS can form IPMN-like lesions in vivo
Finally, we assessed the in vivo growth pattern of PDLOs exhibiting the inducible GNASR201H expression cassette (Fig.7G). GNASR201H PDLO engraftment occurred in 70% of the animals, independent of Dox-induction. In 8/9 GNASR201H PDLO engraftments, we detected the mCherry reporter (Suppl.Fig. 7A), from which 6 formed well-differentiated cystic ducts resembling human IPMNs with low-grade cellular atypia and tissue dysplasia (Fig.7G–J; Suppl.Fig. 7A,F). In line with our in vitro data, such GNASR201H grafts showed moderate proliferation (Fig.7I; Suppl.Fig. 7A,F), leading to branched structures with a variable degree of MUC1, MUC5AC, and CA19-9 expression (Fig.7J; Suppl.Fig. 7A,F). Expression of MUC1 and MUC5AC, without detection of MUC2 (data not shown), indicated a pancreatobiliary or gastric IPMN-like type (Furukawa et al., 2005, Klausen et al., 2019).
Discussion
We establish a scalable PDLO differentiation tool to model pancreatic dysplasia in vitro and pancreatic cancer development upon transplantation in vivo. In contrast to currently available cancer models derived from mice or fully developed PDAC tissue, PDLOs provide a defined and untransformed human genetic background with access to developmental intermediates. Complemented by gene-editing, PDLOs enable studying the impact of specific genetic mutations as the starting point of dysplasia and cancer formation from a ductal origin.
Homogeneous cell fate commitment from the PP stage to ductal cells was validated by comprehensive tests including ultrastructural and functional analyses. Temporally resolved RNA-seq complemented by mass spectrometry-based proteomics completed our in-depth characterization of PDLOs. Importantly, the PP signature was mainly lost in exchange with a ductal identity during differentiation, a hallmark segregating our PDLOs from previous progenitor or mixed exocrine organoids. In addition, PDLO transplantation into distinct in vivo niches like the ACE and the pancreas allowed the formation of more complex organized ductal structures. Transplantation studies employing the ACE as a niche for ductal pancreatic tissue could enable the longitudinal tracking of early human dysplasia and carcinogenesis in a living organism.
To establish PDLOs as a disease model, we asked whether common PDAC driver mutations induce an in vitro phenotype and distinguishable cancer types within an in vivo microenvironment. PDLOs generated via (i) GNASWT/R201C iPSCs derived from a McCune Albright syndrome patient or (ii) hESCs carrying an inducible GNASR201H formed large cystic structures compared to respective control counterparts. We confirmed that the Gαs-PKA-VASP signaling axis was important for the cystic growth of GNASWT/R201C PDLOs. GNASR201H-expressing PDLOs formed IPMN-like structures after orthotopic transplantation, thus underpinning the role of mutant GNAS as the main driver of cystic neoplastic growth in pancreatic ducts (Ideno et al., 2018, Patra et al., 2018).
In PDLOs overexpressing KRASG12D, we observed a specific morphological lumen-filling phenotype, a characteristic previously reported to indicate dysplastic growth from KRAS mutant organoids (Seino et al., 2018). On a molecular level, pro-apoptotic as well as features indicating OIS were upregulated upon KRASG12D induction. OIS has been established as a cancer progression roadblock, particularly in precursor lesions of various cancers including prostate and pancreas (Chen et al., 2005, Morton et al., 2010). As senescence-associated ß-galactosidase activity cannot be assessed on FFPE tissue (Caldwell et al., 2012), OIS effectors including P53, P21, and RB were investigated in PDLO grafts instead. Orthotopic PDLO engraftment was sufficient to partially trigger checkpoint evasion licensing various routes to cancerous growth and subsequent heterogeneity. Future studies need to clarify KRASG12D-driven cancer progression and metastatic traits by allowing graft development beyond the 8-week timepoint chosen in this study.
While apoptosis and OIS counteract tumorigenesis, EMT is considered to be a crucial driving process for cancer plasticity and invasion into the local tissue. Indeed, EMT-associated transcription factors can mediate a switch to bypass senescence and activate EMT by oncogenic stimuli such as high-dose KRAS (Ansieau et al., 2008, Ohashi et al., 2010). The combination of unleashed KRASG12D and lost CDKN2A again triggered EMT gene expression, accompanied by cells disseminating from the organoid body. Dissemination and EMT marker expression in PDLO grafts in vivo were spatially concordant with high levels of KRASG12D, an observation facilitated by the heterogeneous expression of the inducible transgene. This is intriguing, as PDAC appears to implement various routes to intrinsically amplify KRAS gene dosage (Mueller et al., 2018, Chan-Seng-Yue et al., 2020). Accordingly, large-scale sequencing of multiple established PDAC genomes identified CDKN2A as well as major allelic imbalance in mutant KRAS to mark a switch from a classical to a basal-like subtype, characterized by high EMT scores and frequently observed in stage IV disease (Moffitt et al., 2015, Chan-Seng-Yue et al., 2020, Puleo et al., 2018). However, neither of the afore mentioned studies started from a genetically defined untransformed pancreatic background. Thus, our PDAC modeling efforts that combine different oncogenic events in PDLOs are in line with the current picture of PDAC evolution, underpinning the value of the model.
Conclusion
Our PSC-derived PDLOs together with reprogramming technologies and advances in gene editing allow the customized design of a disease initiation and progression landscape. We, hereby, leverage opportunities for longitudinal studies alongside material access for distinct multi-dimensional analytics in vitro and in vivo. As an example, a head-to-head comparison of oncogene expression in ducts and in acini engineered from human pluripotent stem cells will help to answer a currently unresolved question during pancreatic carcinogenesis: How does the cell type of origin affect human PDAC biology, and why are ductal cells more refractory to developing PanIN lesions, but progress faster to PDACs (Lee et al., 2018, Ferreira et al., 2017)? As low-grade preneoplastic lesions were rare in our 8-week grafts and appeared to progress rapidly to cancer, transplanted PDLOs indeed have the capacity to provide answers for the duct-specific pathomechanisms of PDAC development. In summary, our robust PSC differentiation matrix opens a variety of different applications in pancreatic development and cancer research and fuels a versatile human research hub to gain access into the spatiotemporally resolved evolution of dysplasia and plasticity in pancreatic cancer.
Limitations of study
The aim of our study was to derive ductal cells from human PPs. Acinar cells generated from the same PP ancestors would be desirable to study oncogene and tumor suppressor gene function in a lineage-dependent manner. A long-term PDLO culture format has yet not been fully established, thus, requiring transplantation studies for further tumor progression studies. Also, the microenvironment in vivo is incomplete due to the lack of immune cells in the mice used.
STAR Methods
LEAD CONTACT
Further information and requests for resources and reagents should be directed to the Lead Contact Alexander Kleger (alexander.kleger@uni-ulm.de).
MATERIALS AVAILABILITY
Plasmids and cell lines generated in this study are available from the lead contact with some restrictions. piggyBac expression plasmids can only be provided with a completed Materials Transfer Agreement and the permission of Knut Woltjen, the provider of the original piggyBac plasmids. Transfer of hESCs can only be granted if permissions for the intended use are in place according to respective National Authorities and in compliance with the German “Stammzellgesetz”. Panc163 cells cannot be further distributed and requests have to be directly addressed to Bruno Sainz.
DATA AND CODE AVAILABILITY
The RNA-seq data for this study have been deposited in the European Nucleotide Archive (ENA) at EMBL-EBI under accession number PRJEB38015, and the lcWGS data under PRJEB42190. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD018785 (Reviewer Account: Username: reviewer_pxd018785@ebi.ac.uk, Password: hQt1By5o). The codes supporting the current study are available from the corresponding author on request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human bone marrow stromal cells (HBMSCs)
Human bone marrow stromal cells (HBMSCs, a gift from Natasha Cherman) were derived from a female patient with McCune-Albright syndrome. Cells were isolated elsewhere as described in Bianco et al. (1998), and were used to establish GNASWT/WT and GNASWT/R201C induced pluripotent stem cell (iPSC) lines. HBMSCs were cultured in growth medium consisting of α-Minimum Essential Medium (Thermo) supplemented with 20% non-heat inactivated, lot-selected fetal calf serum (FCS, Biochrom), 2 mM L-Glutamine, 1% Penicillin-Streptomycin (P/S), 10 nM Dexamethasone (Sigma) and 100 μM L-Ascorbic acid phosphate magnesium salt n-hydrate (AscP; Wako chemicals) at 37°C and 5% CO2. Cells were split using 0.05% Trypsin-EDTA (Sigma).
Embryonic and induced pluripotent stem cells
In this study, the human embryonic stem cell (hESC) lines HUES8 (Harvard University; RRID:CVCL_B207) and H1 (Wicell Research Institute) were used. Culture and differentiation of hESCs towards the pancreatic lineage were performed with permission from the Robert Koch Institute according to the “79. Genehmigung nach dem Stammzellgesetz, AZ 3.04.02/0084”. HUES8 cell authentication was confirmed with a DNA profile using nonaplex PCR of Short Tandem Repeats done by the Leibniz Institute DSMZ. The human control iPSC line Co-iPSC was established in-house from a healthy male donor.
Human ESCs and iPSCs were cultured on hESC Matrigel (Corning) coated plates (according to manufacturer’s recommendations) in mTesR1 medium (STEMCELL Technologies) at 5% CO2, 5% O2, and 37°C with daily media change. Splitting was done twice a week in a 1:4 – 1:6 ratio. Cells were washed with PBS, incubated with TrypLE (Thermo) for 3–5 min at 37°C for detachment and carefully collected in DMEM-F12+GlutaMAX (Gibco). After centrifugation at 200 × g for 5 min cells were resuspended in mTesR1 supplemented with 10 μM ROCK inhibitor (Y-27632; Abcam) and seeded again on hESC-qualified Matrigel.
Panc163
Panc163 cells have been previously established from a primary human PDAC xenograft model and were a generous gift from Bruno Sainz (Rubio-Viqueira et al., 2006). Cells were maintained as organoids in a Matrigel based culture, medium was changed twice a week, and organoids were split every 10 days using Collagenase/Dispase (Roche) and Accutase (Sigma) as described in more detail in the PDLO culture section. For cultivating PDAC organoids, the medium described by Tiriac et al. (2018) was used: DMEM/F12 medium was supplemented with 1x HEPES, 1x GlutaMAX, 1x P/S, 1x B27, 100 μg/ml Primocin (all Thermo), 1.25 mM N-acetyl-L-cysteine (Sigma), 50% Wnt3a-conditioned medium, 10% RSPO1-conditioned medium, 100 ng/ml recombinant Noggin (PeproTech), 50 ng/ml EGF (R&D), 10 nM Gastrin I (Sigma), 100 ng/ml FGF10 (R&D), 10 mM nicotinamide (Sigma), and 500 nM A83–01 (Tocris). Panc163 were used in qPCR, IF and FC analysis, CFTR and CA assay and RNA-seq experiments as control to PDLOs.
Human patient-derived organoids (PDO) from resection specimen
All patients were recruited, enrolled, and consented based on the institutional review board (IRB) project-number 207/15 and 1946/07 of the Technical University Munich. The isolation protocol is detailed in (Dantes et al., 2020) and was based on previously described protocols (Moreira et al., 2018, Boj et al., 2015, Biederstädt et al., 2020). To ensure a high take-rate of PDOs, the sample preparation was started within 15 min after receiving the biopsy. Samples were washed (splitting/washing media: Advanced DMEM/F12 with 1x GlutaMAX, 10 mM HEPES and 100 μg/ml Primocin (InvivoGen)) and centrifuged (5 min, 4°C, 1000 rpm). The supernatant was discarded, and the tissue sample was cut into small pieces followed by red blood cell lysis using ACK lysis buffer (Life Technologies) for 10–15 min at RT. The sample was then digested using 5 mg/ml collagenase type II (Life Technologies) for 1–2 h followed by enzymatic digestion (optional) with TrypLE (Life Technologies) for 5–10 min at 37°C. After one washing step, the pellet was mixed with Growth Factor Reduced (GFR)-Matrigel (Corning) and plated as 50 μl Matrigel domes in each well of a prewarmed 24-well plate. After incubation for 20 min at 37°C, 500 μl feeding media was added to each well. Normal feeding media (NFM) contained splitting media supplemented with 1x B27 (Life Technologies), 100 ng/ml recombinant human Wnt3a protein (R&D Systems) or 50% Wnt3a-conditioned medium, 10% R-Spondin 1-conditioned medium or 500 ng/ml recombinant human R-Spondin 1 protein (R&D), 1.25 mM N-acetyl-L-cysteine, 100 ng/ml mNoggin (PeproTech), 100 ng/ml FGF10, 10 nM Gastrin I, 50 ng/ml EGF (Life Technologies), 10 μM ROCK inhibitor (Sigma), 10 mM nicotinamide, 0.5 μM A83–01 (Tocris) and 1 μM prostaglandin E2 (Tocris).
To ensure a non-transformed state of these normal PDOs, whole exome sequencing was performed showing no copy number variants as well as no single nucleotide variants. Due to limited material only RNA could be isolated from three different individuals with a non-transformed state. Such material was used in RNA-seq experiments as control to PDLOs.
Establishment of human ductal organoid cultures derived from organ donors
Isolation and culturing of human ductal organoids was based on Boj et al. (2015): Pancreatic tissue samples from human cadaver donor were transferred in splitting media, composed of Advanced DMEM/F12 with 1x GlutaMAX, 10 mM HEPES and 1x Primocin. Minced tissue pieces were incubated in digestion media (splitting media supplemented with 1250 U/ml collagenase IV (Worthington), 0.5 U/ml dispase (Sigma), 2.5% v/v FBS, and 1 mg/ml trypsin inhibitor (Sigma)) at 37°C in a vertical shaker for approximately 30 min, depending on tissue density. Digestion of the tissue was verified by stereo microscopy every 5 min. Cells were collected by centrifugation in a 15-ml centrifuge tube (750 rpm, 10 min, 4°C). Collected cells were washed by wash media (splitting media with 2.5% v/v FBS, 1X Antibiotic-Antimycotic solution (Sigma), 1x kanamycin (Gibco) and 2 μg/ml voriconazole (Tocris) two times. The pellet was resuspended in washing media and Matrigel in a ratio of 1:5. Matrigel domes (10 μl) were placed in one well of a 24-well cell culture plate and after 10 min of solidification at 37°C, 500 μl feeding media were applied in each well. Feeding media was composed of splitting media supplemented with 50% L-WRN conditioned media, 500 nM A-83-01, 50 ng/ml EGF, 100 ng/ml FGF2 (Thermo), 0.01 μM Gastrin I, 1.25 mM N-acetyl-L-cysteine, 10 mM nicotinamide, 1x B27, 10.5 μM Y-27632, 1 μM prostaglandin E2, 1x Antibiotic-Antimycotic, 1x kanamycin and 2 μg/ml voriconazole. Media change was performed every second day. Domes were pooled and collected by centrifugation (750 rpm, 10 min, 4°C) for passaging during which Matrigel removal and cell separation were performed simultaneously by TrypLE at 37°C for 15 min in a vertical shaker followed by plating the cells in Matrigel as described above. Human pancreatic tissue samples were collected from transplantation donors (Ethical approval No.: 37/2017-SZTE). The herein described human primary organoids were used as controls for intracellular pH measurements as additional confirmation of PDLO functionality.
Mouse model
NOD scid gamma (NSG) mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ strain (Charles River); RRID:BCBC_4142) were used for xenotransplantation of PDLOs into the pancreas with permission of the “Regierungspräsidium Tübingen” (TVA1406). For xenotransplantation of PDLOs into the ACE, male NOD scid mice (NOD.Cg-Prkdcscid/J strain (The Jackson Laboratory); RRID:IMSR_JAX:001303) were used with approval by the Committee on the Ethics of Animal Experiments of the State Directory of Saxony and the Ethics Committee of the Technische Universität Dresden (TVV57/2016). Husbandry was performed in standardized hygiene barrier rooms with reduced pathogen microorganism burden. Animals had an age between 6 to 12 weeks before experiments were started and male and female individuals were distributed equally to the different groups, although gender-specific effects were not expected. Housing was performed in groups of two to four mice per cage.
METHOD DETAILS
Generation of iPSCs by reprogramming
For the generation of MAS-iPSCs, a mixed population of mosaic GNASWT/WT/GNASWT/R201C HBMSCs were split using 0.05% Trypsin-EDTA to reach 50–60% confluency for viral infection after two days. Reprogramming with hOKSM-dTomato lentivirus (Warlich et al., 2011) was performed as previously described (Hohwieler et al., 2017): When 75% confluence was reached HBMSCs were infected once with 1 × 108 viral genome copies of hOKSM-dTomato lentivirus in growth medium supplemented with 8 μg/ml polybrene (Sigma). The next day, cells were detached using TrypLE and transferred to 6-wells covered with a feeder layer of 3.5 × 108 inactivated rat embryonic fibroblast (REF), which were gamma-irradiated with 30 Gy one day before (Linta et al., 2012). After that, cells were further cultured in hiPSC medium containing Knockout DMEM (Gibco), 20% knockout serum replacement (Gibco), 100 μM NEAA (Sigma), 2 mM L-Glutamine (Gibco), 1% Antibiotic-Antimycotic, 100 μM β-Mercaptoethanol (Merck Millipore), 50 μg/ml L-Ascorbic acid (Sigma), 10 ng/ml FGF2 (Novoprotein) and 10 μM ROCK inhibitor with daily media change at 5% CO2 and 5% O2. About 14 days later, iPSC colonies with appropriate size were mechanically picked on irradiated REFs to further expand cells. In the next step, colonies were again mechanically picked and plated onto Matrigel-coated dishes for feeder-free culture (Illing et al., 2013, Linta et al., 2012). Besides infection with the hOKSM-dTomato virus, in a different approach, HBMSCs were infected by using the CytoTune™-iPS 2.0 Sendai Reprogramming Kit (Thermo). CytoTune 2.0 KOS (hKlf4, hOct3/4, hSox2, MOI 1), hc-Myc (MOI 1) and CytoTune 2.0 hKlf4 (MOI 0.6) vectors were mixed with 10 μM ROCK inhibitor and 8 μg/ml polybrene in HBMSC medium and added to the cells. The subsequent reprogramming procedure was performed as described above. Generated MAS-iPSC clones were checked for p.R201C mutations by PCR amplification and sequencing (GNAS-exon8-fwd, CCAGACCTTTGCTTTAGATTGG (Salinas-Souza et al., 2015); GNAS-exon9-rev, CACAGCATCCTACCGTTGAAG) (Wood et al., 2017). Products were sent for Sanger sequencing (Eurofins Genomics).
Genome editing by CRISPR/Cas9 in hESCs
A large deletion in CDKN2A was created in the hESC line HUES8 by CRISPR/Cas9 gene editing by induction of two distinct double strand breaks (DSBs) flanking exon 2 and 3 of the CDKN2A gene. crRNAs were designed with an open-access online tool (http://crispor.tefor.net (Haeussler et al., 2016): crRNA-CDKN2A-exon2, GTAGGGGTAATTAGACACCT; crRNA-CDKN2A-exon3, GTCTCGAGTCTATCGATATG. Construct generation of the plasmid-based CRISPR/Cas9 approach was performed as described in Mali et al. (2013): For each target site, respective custom DNA oligonucleotides with complementary sequences were annealed to double stranded oligos using the Phusion Polymerase Reaction Kit (NEB). The gel purified fragment and the AflII-digested gRNA cloning vector (Addgene plasmid # 41824, a gift from George Church (Mali et al., 2013)) were combined by Gibson Assembly according to manufacturer’s instructions. Competent E. coli cells were transformed with the resulting construct, plasmid DNA was purified and checked by PCR and sequencing. The final gRNA expression plasmids for both gRNAs together with a Cas9 nuclease expression plasmid (Addgene plasmid #44719, a gift from Kiran Musunuru (Ding et al., 2013)) were used for transfection of HUES8 cells. Therefore, 200,000 cells were seeded on Matrigel-coated 6-wells and after 16 h the transfection mix consisting of 2 μg pCAS9_GFP vector, 2 μg gRNA plasmid1, 2 μg gRNA plasmid2 and 18 μl XtremeGene 9 DNA transfection reagent (Roche) (3:1 ratio) was added dropwise to the cells (see also manufacture’s protocol). 48 h post transfection, GFP-positive cells were sorted by FACS and plated at low density (500 cells/10 cm dish) for clonal expansion in media supplemented with 10 μM ROCK inhibitor and 0.5 μM Thiazovivin (Calbiochem). Multiple single cell-derived colonies were picked mechanically after 10–12 days for screening of gene edited clones.
Screening of edited clones: DNA isolation and PCR reaction
Clonal colonies were mechanically dissociated, and one half of the cells was further cultivated and expanded while the other half was used for genotyping. DNA was isolated using the Tissue Genomic DNA Purification Mini Prep Kit (Genaxxon) according to manufacturer’s instructions and amplified by RedMastermix (2x) Taq PCR Mastermix (Genaxxon). PCR screening was based on internal and external primer pairs detecting either the WT or mutated target region. An external PCR product from primers flanking the site of deletion was only obtained if the KO occurred (CDKN2AKO-external-fwd, GCGCTTGGATATACAGCAGTG; CDKN2AKO-external-rev, ACAGGAGCATCTCCCAACC). Internal primers are located within the deleted region and a product indicated the wildtype allele (CDKN2AKO-internal-fwd, GGCATTGTGAGCAACCACTG; CDKN2AKO-internal-rev, CCTGTAGGACCTTCGGTGAC). PCR products were sent for Sanger sequencing (Eurofins Genomics) for validation of clonal genotypes.
All-in-One piggyBac-system and Nucleofection
Dox-inducible KRASG12D, GNASR201H, and vector control overexpression lines were generated using a piggyBac (PB) transposon system. For the vector control a Luciferase (Luc2) construct was introduced. We modified an All-in-One-vector previously described by Kim et al. (2016) and introduced cDNA sequences of the target genes, that were amplified by PCR from the plasmids pBabe-KRAS G12D (Addgene plasmid #58902, a gift from Channing Der), pcDNA3.1+hsGNAS_EE(long) R201H and pGL4.10[luc2] (both kindly provided by Franz Oswald). A 2-step PCR approach was performed by first using the following gene-specific primers: for KRASG12D (attB1-SpeI-HindIII-(N-HA)KRAS_G12D-fwd; attB2-KRAS_G12D-rev), for GNASR201H (attB1-SpeI-HindIII-GNAS(EE)_R201H-fwd_new; attB2-GNAS_R201H-rev_new), for Luciferase (attB1-Luc2-for; attB2-Luc2-rev). In the second step the generated PCR products were further amplified with attB1/attB2 adapter primer (Gloeckner et al., 2009) to add the respective sequences necessary for the gateway cloning system. All corresponding sequences are listed in Methods S1. PCR-products were purified with the Wizard SV Gel and PCR Clean-Up System (Promega).
In the next step, PCR fragments were inserted into the pDONR201 vector (Thermo) and finally into the Destination vector PB-TAC-ERP2 (Addgene plasmid #80478, a gift from Knut Woltjen (Kim et al., 2016)) by gateway cloning (BP Clonase II/LR Clonase II enzyme mix, Life Technologies) as described by Gloeckner et al. (2009). Correct sequences of the generated plasmids PB-TAC-ERP2-(N-HA)KRAS_G12D, PB-TAC-ERP2-GNAS_R201H(EE) and PB-TAC-ERP2-Luc2 were confirmed by Sanger sequencing (PB-seq-fwd sequencing; PB-seq-rev sequencing; GNAS_AS189-fwd; Luc2_AS215-fwd; Methods S1).
For integration of the transposon elements into the genomic DNA, HUES8 cells were co-transfected with the transposase-expression vector (SBI Biosciences #PB200PA-1 (Rao et al., 2016)) and the respective targeting vector, either PB-KRAS, PB-GNAS, and PB-Luc2. Nucleofection of HUES8 cells was performed using the 4D Nucleofector (Lonza) and the P3 primary cell 4D Nucleofector X Kit S (Lonza) according to the manufacturer’s protocol. In brief, at 70–80% confluence, cells were harvested using TrypLE and 200,000 cells per reaction were centrifuged at 130 × g for 3 min. Cells were resuspended in P3-solution and plasmids were added in a 3.3:1 ratio of PB- to transposase plasmid with a total amount of 0.5–1 μg DNA. The reaction mix was transferred to the wells of a 16-well Nucleocuvette strip and pulsed with CA-137. mTesR1 with 10 μM ROCK inhibitor was added to the cells and after 3–5 min incubation at 37°C nucleofected cells were transferred to Matrigel-coated 96-wells. Cells with stable integration of the PB expression cassette were enriched by addition of 1 μg/ml puromycin (Sigma) to the media, starting after 24 h.
Pancreatic differentiation
Differentiation of hPSCs into PPs was performed based on previously published protocols (Nostro et al., 2015, Hohwieler et al., 2017). Cells were cultured in basal media (i) BE1: MCDB131 (Thermo) with 2 mM L-Glutamine, 1.174 g/l Sodium bicarbonate (Sigma), 0.8 g/l cell culture tested glucose (Sigma) and fatty acid free BSA (Proliant) in a concentration of 0.1% (BE1a) for d0-d2 or 0.5% (BE1b) for d3-5. (ii) BE3 (from d6): MCDB131 with 2 mM L-Glutamine, 1.754 g/l Sodium bicarbonate, 0.44 g/l cell culture tested glucose (Sigma), 0.5% ITS-X (Gibco), 44 mg/l L-Ascorbic acid and 2% fatty acid free BSA. 24-well cell culture plates were coated with GFR-Matrigel diluted 1:18 in DMEM-F12+GlutaMAX before 250,000–450,000 cells per well (depending on the cell line) were seeded in mTesR1 with 10 μM ROCK inhibitor. The day after seeding, differentiation was started with 80–90% cell confluence by washing with PBS (Gibco) and adding BE1a medium with 100 ng/ml Activin A (PeproTech) and 2 μM CHIR99021 (Axon MedChem) (day0-medium). Day0-Medium was replaced after 24 h for 2 days with BE1a containing 100 ng/ml Activin A and 5 ng/ml FGF2 (Novoprotein). At day 3 of differentiation cells reached DE stage and BE1b medium containing 50 ng/ml FGF10, 0.75 μM Dorsomorphin (Sigma) and 3 ng/ml Wnt3a (PeproTech) was added for three days. From day 6 (GTE stage) on BE3 media supplemented with 50 ng/ml FGF10, 200 nM LDN-193189 (Sigma), 0.25 μM SANT-1 (Sigma), 2 μM Retinoic acid (RA; Sigma), and 16 mM glucose was added for 3 days. The last four days of differentiation (PE stage, d9-d13) cells were cultured in BE3 with 100 ng/ml EGF (R&D), 200 nM LDN-193189, 10 mM nicotinamide, 330 nM Indolactam V (STEMCELL Technologies), and 16 mM glucose. Differentiation was performed at 37°C with 5% CO2 and medium was changed daily.
To standardize differentiation experiments we implemented purity thresholds of 95% CXCR4/cKIT double positive cells at DE stage and 60% of PDX1/NKX6-1 double positive cells at PP stage, measured by FC analysis. If the amount of PDX1/NKX6-1 double positive cells at PP stage was under 60%, Magnetic-Activated Cell Sorting (MACS) using glycoprotein 2 (GP2) as human pancreatic progenitor marker (Cogger et al., 2017) was done. In brief, cells were harvested and stained with anti-GP2-Antibody (MBL International, 1:5000) as outlined in the flow cytometry section for surface marker staining with the addition of 10 μM ROCK inhibitor to all buffers. After antibody incubation for 60 min at 4°C, cells were incubated another 15 min at 4 °C with anti-mouse IgG MicroBeads (Miltenyi) and sorted according to manufacturer’s instructions.
Compound screens for protocol development
In total, we performed more than ten rounds of compound screenings. Simplified, we applied morphological criteria (pure culture of ring-like organoids lined with one layered epithelium) together with mRNA expression criteria (increase of ductal markers by reduction of progenitor and non-pancreatic markers) as outlined in the results section. For experimental design and interpretation of qPCR data, we implemented a design of experiment approach to model, simulate, and analyze dependencies between the different biological compounds applied at various concentrations. The software tool MODDE was used to predict marker expression for different combinations of tested compounds, which allowed us to retrieve new recipe suggestions for subsequent compound screens. Promising compounds had been always tested in additional experiments and only compounds, which consistently improved morphology and marker expression, are applied in the current protocol. In this protocol 7 out of 20 compounds, tested in phase I, and 4 out of 28 compounds, tested in phase II, are implemented.
Most experiments were performed in duplicates (two wells per condition) and depending on the experiment intermediate stages (day 20, 24) and/or the endpoint of differentiation (day 30) was analyzed.
Following compounds were tested in phase I in at least one experiment in indicated concentrations: EGF (10–250 ng/ml), FGF10 (10–250 ng/ml), KGF (10–250 ng/ml), MSC2530818 (4 nM-1 μM), nicotinamide (2–50 mM), Y-27632 (10 μM), ZnSO4 (2–50 μM), and ALK5i-II (10 μM), Avagacestat/BMS-708163 (0.1–10 μM), BMP-4 (10 ng/ml), DAPT (0.1–50 μM), FGF1 (20–500 ng/ml), FGF2 (10 ng/ml), IL-V (33–660 nM), LDN-193189 (0.2 μM), Na2CO3 (1.5 mg/ml), RA (10 μM), R-Spondin (10% conditioned medium and 500 ng/ml), SANT-1 (0.25 μM), TGF-β (1 ng/ml).
Following compounds were tested in phase II in at least one experiment in indicated concentrations: EGF (5–250 ng/ml), FGF10 (5–250 ng/ml), KGF (5–250 ng/ml), nicotinamide (2–50 mM), Y-27632 (10 μM), ZnSO4 (0–50 μM), and Activin A (4–100 ng/ml), ALK5i-II (0.2–10 μM), Avagacestat (0.1–10 μM), Axitinib (0.1–10 μM), BMP-4 (0.2–100 ng/ml), BMP-7 (2–1000 ng/ml), CHIR99021 (0.4–10 μM), Dexamethasone (0.025–2.5 μM), FGF1 (12.5–500 ng/ml), FGF2 (10 ng/ml), Follistatin (20–5000 ng/ml), IL-V (66–1650 nM), IWP2 (0.4–10 μM), KGF (4–100 ng/ml), LDN (0.04–1 μM), MSC2530818 (0.05–10 μM), Na2CO3 (1.5 mg/ml), Neuregulin-4 (50–500 ng/ml), RA (0.1–100 μM), R-Spondin (10% conditioned medium and 500 ng/ml), Synthaxin/Epimorphin (20–1000 ng/ml), TGF-β (0.1–10 ng/ml), VEGF-164 (4–1000 ng/ml).
PDLO culture
Cell culture plates were coated with undiluted GFR-Matrigel using precooled tips and well plates, distributing the Matrigel in z-movements until the whole well was covered. Pancreatic progenitors on d13 of differentiation were harvested with TrypLE, washed in BE3, and resuspended in phase I differentiation media. 5% GFR was added additionally to the cold media directly before seeding 100,000 cells per 12-well. The above described Matrigel culture was based on Xiang and Muthuswamy (2006). The final PDLO culture medium consisted in phase I (d13-d19) of BE3 supplemented with 10 mM nicotinamide, 10 μM ZnSO4 (Sigma), 10 μM ROCK inhibitor, 50 ng/ml EGF, 50 ng/ml FGF10, 50 ng/ml KGF (PeproTech), and 50 nM MSC2530818 (Selleckchem). In phase II (from d20) BE3 medium contained 10 mM nicotinamide, 10 μM ZnSO4, 50 ng/ml EGF, 50 ng/ml FGF10. Media change was performed twice a week and media were supplemented with 5% GFR-Matrigel.
For splitting and harvesting organoids, cultures were washed with PBS and incubated with 1 mg/ml Collagenase/Dispase in DMEM-F12+GlutaMAX for 2–4 h at 37°C to degrade surrounding Matrigel. Enzymatic reaction was stopped by addition of equal amounts of neutralization solution (DMEM + 1% BSA + 1% P/S). After centrifugation (200 × g, 5 min) and PBS wash, recovered PDLOs were further processed to generate single cell suspensions or directly lysed for RNA or protein extraction. PDLOs were dissolved into single cells by treatment with Accutase (Sigma) for 30 min in a 37°C water bath and intermittent pipetting. Reaction was stopped again with neutralization solution followed by centrifugation. Finally, PDLO cells were resuspended in phase II medium with 10 μM ROCK inhibitor and seeded again as described above or used for FC analysis.
Analysis of phenotypic alterations upon oncogene induction including live-cell imaging
For experiments with inducible transgenic cell lines, PDLOs were split on day 27 and treated for 9 days with 5 μg/ml Dox starting from day 29. For titration experiments indicated Dox concentrations were applied. PDLOs were harvested on day 38 for respective applications. PDLO cultures were imaged 2, 5, 7, and 9 days after Dox treatment on a Keyence Biozero BZ-9000 microscope. Swelling was quantified using the ImageJ-based software Fiji (Schindelin et al., 2012). Therefore, an automated analysis was applied that calculates the area of organoids within one overview bright field image. In addition, the percentage of filled PDLOs was assessed semi-automatically by applying a second algorithm in Fiji. Analysis was performed in three experiments (independently started differentiations) with each experiment performed in triplicates (three wells per condition). To determine the number of protruding organoids, PDLOs showing outgrowth of single cells and areas of mesenchymal-like cells were manually counted per image of an individual well (comprising 60–80 organoids). Four independent experiments performed at least in duplicates were quantified.
Time series of organoid growth to track these cellular changes was recorded with an epifluorescence microscope (Zeiss Axio Observer 7) over a time period of 4 days starting 2 days after Dox addition. Longitudinal imaging was achieved by maintaining the PDLO culture plate on the microscope under 5% CO2, 85% humidity, and 37 °C within a small incubation chamber (PeCon). Fluorescence and bright field images were taken every 3 h with a 5x Zeiss Neofluar objective. Media was replenished after one day. Images were analyzed and videos were compiled using Zeiss ZEN blue imaging software.
PKA inhibition in PDLOs
PKA inhibitor treatment was initiated two days after cell seeding. PDLO cultures were incubated with 20 μM H89 2HCl (S1582, Selleckchem) or DMSO (1:1250) as negative control for 12 days in ductal medium. Pictures were captured using a Keyence Biozero BZ-9000 microscope and analyzed as detailed above. The mean organoid size was calculated in duplicates (two wells per condition) of three experiments and the PDLO size decrease was estimated relative to the controls.
Preparation of cell extracts and Western blot
For protein extraction, cell lysates were generated by incubating cell pellets in RIPA buffer (50 mM Tris pH 7.4 (AppliChem), 150 mM NaCl (Sigma), 1 mM EDTA (AppliChem), 1% NP40 (Fluka), 0.25% Sodium deoxycholate (Sigma), 0.1% SDS (Serva), supplemented with 1 mM PMSF (AppliChem), 1x phosphatase inhibitor and 1x EDTA-free protease inhibitor cocktail (cOmplete; both Roche) for 30 min on ice and vortexing every 10 min. After 8 min centrifugation at 10,600 × g, supernatant containing the protein fraction was collected. Protein concentration was determined using a Bradford reagent (Bio Rad) and equalized amounts of protein lysates were separated on a polyacrylamide gel (BioRad) in SDS-buffer followed by blotting to a methanol-activated Immobilon-P PVDF membrane (Millipore) by using transfer buffer (32 mM glycine, 44 mM Tris, and 20% methanol; Sigma) and the Transblot semidry transfer system (Bio-Rad). Effective protein transfer was confirmed by Ponceau staining (AppliChem) before membrane was blocked with 5% BSA (or 5% Milk) and 0.1% Tween20 (Sigma) in TBS for at least 1 h at RT. Membranes were incubated with primary antibodies diluted in blocking solution overnight (o/n) at 4°C. After washing three times with 0.1% Tween20 in TBS, incubation with secondary antibody anti-mouse-horseradish peroxidase (HRP) or anti-rabbit-HRP (ECL anti-rabbit or mouse IgG, GE Healthcare) was performed for 1 h at RT. For detection of HRP the SuperSignal West Dura Kit (Thermo) together with Chemiluminescence Imaging – Fusion SL system (VILBER) was applied. Quantification of Western blot bands was done with the ImageJ-based software Fiji (Schindelin et al., 2012). The following primary antibodies were used: anti-ACTB (Sigma Cat#A5316, 1:5000), anti-CA2 (Abcam Cat#ab124687, 1:1000), anti-E-CAD (Cell Signaling Cat#3195, 1:1000), anti-ERK (Cell Signaling Cat#9102, 1:1000), anti-Gαs/olf (G-10) (Santa Cruz Cat#sc-365855, 1:1000), anti-HA (Cell Signaling Cat#3724, 1:1000), anti-N-CAD (Cell Signaling Cat#13116, 1:1000), anti-P15 (Santa Cruz Cat#sc-271791, 1:500), anti-P16 (Cell Signaling Cat#80772, 1:1000), anti-P21 (Abcam Cat#ab109520, 1:1000), anti-PARP (Cell Signaling Cat#9542, 1:1000), anti-pERK (Cell Signaling Cat#4377, 1:1000), anti-pRB (Cell Signaling Cat#8516, 1:1000), anti-p-PKA substrates (RRXS*/T*) (Cell Signaling Cat#9624, 1:1000), anti-RAS (Thermo, Active Ras Pull-Down and Detection Kit, 1:200), anti-RB (Cell Signaling Cat#9309, 1:1000), anti-VASP (Cell Signaling Cat#3132, 1:1000), anti-Vimentin (VIM, Cell Signaling Cat#5741, 1:1000) and Vinculin (Sigma, V9264, 1:1000).
Pull-Down Assay
Active Ras Pull-Down and Detection Kit (Thermo) was used according to manufacturer’s instructions to verify functionality of the cloned KRASG12D and (N-HA)-KRASG12D constructs based on KRAS GTPase activity. Briefly, transgenic hESC lines KRASG12D HUES8 or KRASG12D(N-HA) HUES8 were cultured with or without Dox for 24 h, harvested and lysed with the provided lysis buffer supplemented with 1 mM PMSF, 1x phosphatase inhibitor and 1x EDTA-free protease inhibitor cocktail. As additional controls, equal amounts of the lysates (± Dox) were incubated with GTPγS (positive control activating Ras proteins) or GDP (negative control). Fusion protein of GST and Raf1-Ras-binding domain (RBD) linked to the glutathione agarose resin was prepared in spin cups. Then GTP- and GDP-pretreated as well as untreated lysates were added to such spin cups and incubated at 4°C for 1 h with gentle rocking. After washing three times with the included buffer, proteins were released from the resin by addition of 2x SDS sample buffer with β-Mercaptoethanol (Sigma). Samples were separated by SDS-PAGE and blotted on a PVDF membrane as described above. For KRAS protein detection the Anti-Ras antibody provided within the kit was used.
cAMP Assay
Cyclic AMP (cAMP) in PSCs or PDLOs was detected with the cAMP-Gs HiRange Kit (Cisbio) according to the manufacturer’s protocol. HUES8 as well as PDLOs inducibly expressing GNASR201H were pretreated with Dox (1 μg/ml for hESCs; 3 μg/ml for PDLOs) before performing the cAMP assay. GNASWT/WT and GNASWT/R201C iPSCs were used directly. 96-well low volume white microplates (Cisbio) were used and 20,000 cells were seeded per well in DMEM-F12+GlutaMAX. FRET-signal was measured in a homogeneous time resolved fluorescence (HTRF)-compatible plate reader (Infinite M1000 pro, Tecan). The “HTRF Europium cryptate donor/Red acceptor readout” setup recommendations were used. The assay was performed in technical triplicates (three wells per condition).
Carbonic anhydrase Assay
Carbonic anhydrase (CA) activity was measured by a colorimetric assay based on the imidazole-Tris method (Brion et al., 1988) including phenol red as a pH indicator. PDLOs at day 30 were harvested with Collagenase/Dispase, washed with PBS, and lysed at 4°C for 30 min in an SDS-free lysis buffer composed of 150 mM NaCl, 50 mM Tris-HCl (Sigma), pH 7.2, 2 mM EDTA (AppliChem), 1% NP40, and protease inhibitors. Analysis was performed with 60 μg protein per sample. A standard curve was established using serial dilutions of carbonic anhydrase II purified from bovine erythrocytes (Sigma). Incubation buffer containing 60 mM imidazole (Sigma), 30 mM Tris base, 1 mM phenol red (Fluka), and 0.1% BSA (pH 9.6) was added to the samples which were then gassed with CO2 at a constant flow rate. Time was measured until a colorimetric change was observed and CA concentrations were interpolated from the reference standard curve. The assay was performed blinded and each condition was analyzed in duplicates (two wells per condition) with repeated measurements (same lysate was measured twice or three times) in three independent experiments (independently started differentiations).
CFTR Assay
The implemented CFTR assay was based on FSK-induced organoid swelling as described previously (Dekkers et al., 2013). PDLOs were split on day 30 and 100,000 cells were seeded per 12-well as described in the PDLO culture section. At day 42–44 PDLOs were incubated with 20 μM forskolin (FSK) and 100 μM 3-isobutyl-1-methylxanthine (IBMX; both Sigma) for 18 h at 37°C. As negative control DMSO (Roth) was added in respective dilution. Directly before and after incubation, pictures of the PDLOs were captured on a Keyence Biozero BZ-9000 microscope with standardized settings. Swelling was quantified with an ImageJ-based automated analysis based on the SIOX segmentation tool. Here, the area/lumen of all organoids within one overview bright field image was measured. The area of the same organoids was calculated at time point 0 h and at time point 18 h to quantify the relative increase of PDLO size. Each condition was analyzed in duplicates (two wells per condition) and at least 400 organoids per condition were included in each of the three independent experiments.
pH measurements via fluorescence microscopy
PDLOs or human primary organoids from cadaveric organ donors were attached to a poly-l-lysine coated cover glass and were incubated in standard HEPES solution with 1.5 μM BCECF-AM (Thermo), a fluorescent pH indicator, for 30 min at 37°C. Cover glasses were transferred to a perfusion chamber mounted on an Olympus IX73 inverted microscope. Dye loaded samples were excited with an Olympus CoolLED PE-4000 illumination system. For BCECF the filter combination was as follows: 434/17 nm and 497/16 nm single-band bandpass filters for excitation (Semrock; P/N: FF01–434/17–25 and FF01–497/16–25, respectively), 511 nm edge single-edge standard epi-fluorescence dichroic beamsplitter (Semrock; P/N: FF511-Di01–25 × 36) and 537/26 nm single-band bandpass filters for emission (Semrock; P/N: FF01–537/26–25). The fluorescent signal was captured by a Hamamatsu ORCA-FLASH 4.0 CCD camera trough a 20x water immersion objective (Olympus; NA: 0.8) with a temporal resolution of 1 s. Ratiometric image analysis was performed by Olympus excellence software. For solution composition see Methods S2.
Flow cytometry
Staining of surface marker
At definitive endoderm (DE) stage differentiation efficiency was determined by c-Kit (CD117) and CXCR4 (CD184) marker staining. Therefore, cells were harvested with TrypLE, enzymatic reaction was stopped with FC buffer containing 2% FCS in PBS and washed once with FC buffer (200 × g, 5 min). Cells were blocked for at least 20 min on ice with blocking buffer consisting of 10% FCS in PBS and washed again with FC buffer. After resuspension of the cell pellets in 50 μl FC buffer incubation with PE-conjugated CXCR4 antibody (Life Technologies) was performed on ice for 30 min. In the next step, APC-conjugated c-Kit antibody (Thermo) was directly added and both antibodies were incubated for another 15 min. Samples were washed, resuspended in FC buffer, and filtered using a 50 μm polyamide mesh (Hartenstein). DAPI (Thermo) was added in a concentration of 150 ng/ml to distinguish viable and dead cells during analysis.
Staining of intracellular marker
Differentiation efficiency was analyzed at the pancreatic endoderm (PE) and pancreatic progenitor (PP) stage by FC-based analysis of PDX1 and NKX6-1 expression. Besides, a panel of other intracellular maker proteins were used to characterize and investigate PDLOs. Cells were harvested as illustrated before (see section “PDLO culture“), washed with PBS (200 × g, 5 min) and fixed on ice for 25 min in 4% PFA in PBS with 100 mM sucrose (both Sigma). Samples were washed twice with PBS and blocked with 5% normal donkey serum (DS, Jackson ImmunoResearch) in 0.1% Triton X-100/PBS for 30 min on ice. After centrifugation (1000 × g, 5 min), cells were resuspended in blocking solution with primary antibodies. Incubation was either performed o/n at 4°C or 90 min on ice if antibodies were directly coupled to a fluorescence protein. Next, the samples were washed three times with 2% DS, 0.1% Triton X-100 in PBS (wash solution) and, if primary antibodies were not directly coupled to a fluorochrome, incubated with Alexa Fluor secondary antibodies (Thermo) for 90 min on ice. Again, cells were washed twice, resuspended in washing solution, and filtered to obtain single cells before measurement.
Following antibodies were used within this study: anti-PDX1 (R&D Cat#AF2419, 1:500), anti-PDX1-PE (BD Cat#562161, 1:35), anti-NKX6-1 (DSHB Cat#F55A12, 1:150), anti-NKX6-1-APC (BD Cat#563338, 1:35), anti-Ki-67 (Thermo Cat#MA5–14520, 1:1000), anti-Ki-67 (Dako Cat# M7240, 1:1000), anti-H2AX (pS139)-APC (BD Cat#560447, 2.5 μl per sample), anti-HA (Cell Signaling Cat#3724, 1:1600), anti-C-peptide (Cell Signaling Cat#4593, 1:100), anti-GCG (Sigma Cat#G2654, 1:500) and anti-Vimentin (Cell Signaling Cat#5741, 1:100).
Cell cycle analysis (EdU staining)
Cell cycle was analyzed by FC using the Click-iT EdU Alexa Fluor 647 Assay Kit (Life Technologies). EdU at a concentration of 10 μM was added to the cell cultures 4 h before samples were harvested. Staining was performed following to manufacturer’s recommendations, but the volume of antibody/reaction mix was reduced to 200 μl per sample and the next steps were also adapted to 1 ml for washing and 350 μl final resuspension volume. Staining of intracellular markers prior to EdU staining procedure was possible. DNA was stained with 3 μM DAPI to define the different cell cycle phases.
FC measurement was performed on the LSR II flow cytometer (BD).
ICC staining
Immunocytochemistry (ICC/IF) staining were performed of cells cultivated and differentiated on Matrigel-coated ibidi-precoated glass-bottom 24-well μ-plates (IBIDI). Cells were washed with PBS, fixed in 4% PFA+100 mM sucrose solution at RT for 20 min, and washed with PBS three times. Quenching was done with 50 mM NH4Cl (Sigma) for 10 min and wells were washed three times with PBS before permeabilization with 0.1% Triton X-100/PBS was performed. Cells were blocked with 5% normal goat (Jackson ImmunoResearch) or DS in 0.1% Triton X-100/PBS for 45 min and incubated with the primary antibody solution at 4°C o/n. On the next day, cells were washed three times with PBS and incubated with secondary antibodies diluted in blocking solution at RT for 1 h in the dark. After PBS wash, 500 ng/ml DAPI in PBS was added to the cells for 10 min before well-plates were stored in PBS at 4°C prior to imaging. Used antibodies were: OCT3/4 (Santa Cruz Cat#sc-5279, 1:200), NANOG (Cell Signaling Cat#3580, 1:500), SSEA4 (stained without permeabilization step, Cell Signaling Cat#4755, 1:500) and Alexa-conjugated secondary antibodies. Cells on IBIDI-plates were imaged on a Keyence Biozero BZ-9000 microscope.
Transmission electron microscopy
PDLOs were harvested at day 30 by degrading surrounding Matrigel either with Collagenase/Dispase or Cell recovery solution (Corning). After collection and several washing steps with PBS to remove residual Matrigel, PDLOs were fixed in 0.1 M phosphate buffer pH 7.3, containing 2.5% glutaraldehyde, 1% sucrose and osmicated for 1 h in 2% OsO4. Afterwards they were dehydrated in graded series of ethanol, contrasted in 2% uranyl acetate and embedded in epoxy resin (Sigma) at 60°C. Thin sections of 70–80 nm were cut with a diamond knife on a Reichert ultramicrotome and collected on 300 mesh grids. The sections were contrasted with 0.3% lead citrate for 1 min and analyzed on the transmission electron microscope EM 10 (Zeiss) at 80 kV. Sample embedding and processing were carried out by the Central Electron Microscopy Unit at Ulm University.
RNA isolation, reverse transcription and qPCR
RNA extraction was performed with the GeneJET RNA Purification Kit (Thermo) according to manufacturer’s instructions. Reverse transcription of 50–1000 ng of total RNA was done with the iScript cDNA Synthesis Kit (Bio-Rad) and cDNA was utilized for quantitative real-time PCR (qPCR) with SensiMix SYBR No-ROX Kit (Bioline) on the QuantStudio 3 Real-Time PCR System (Thermo). Self-designed or commercially available QuantiTect (Qiagen) qPCR primers of target genes are listed in Methods S3. Hydroxymethylbilane synthase (HMBS) was used as endogenous reference gene for normalization with the 2−ΔΔCt method. RNA values were normalized to the control treated samples of each experiment. For heatmap generation the relative RNA expression of EMT-associated genes was additionally scaled by the sum of each row.
RNA-seq experiments
RNA was isolated from different days of differentiation (day 0, 3, 13, 20, 24, 30, 45, and 59) and from Panc163 cells as described above. For each condition, RNA samples from three different wells were sequenced. For non-transformed human patient-derived ductal organoids from resection specimens, RNA was isolated from three independent lines using the AllPrep DNA/RNA/miRNA Universal Kit (Qiagen) according to the manufacturer’s instructions. Library preparation for bulk 3′-sequencing of poly(A)-RNA was done as described previously (Parekh et al., 2016). Briefly, barcoded cDNA of each sample was generated with a Maxima RT polymerase (Thermo) using oligo-dT primer containing barcodes, unique molecular identifiers (UMIs) and an adapter. 5′-ends of the cDNA were extended by a template switch oligo (TSO); after pooling of all samples, full-length cDNA was amplified with primers binding to the TSO-site and the adapter. cDNA was tagmented with the Nextera XT Kit (Illumina) and 3′-end-fragments finally amplified using primers with Illumina P5 and P7 overhangs. In comparison to Parekh et al. (2016), the P5 and P7 sites were exchanged to allow sequencing of the cDNA in read1 and barcodes and UMIs in read2 to achieve a better cluster recognition. The library was sequenced on a NextSeq 500 (Illumina) with 75 cycles for the cDNA in read1 and 16 cycles for the barcodes and UMIs in read2.
Full proteome measurement
In-solution digest
Protein was isolated from PPs (day 13) and PDLOs (day 59) from three different wells per condition (n=3). After harvesting and washing twice with PBS, cells were lysed in urea lysis buffer (8 M urea, 40 mM Tris (pH 7.6), 1x EDTA-free protease inhibitor and 1x phosphatase inhibitor (in-house, composition resembling phosphatase inhibitor cocktail 1, 2 and 3; Sigma)) for 10 min. After lysing, protein concentration was determined from the cell-free supernatant (20 min, 20,000 × g, 4°C) via Bradford protein assay (Pierce™ Coomassie Plus (Bradford) Assay Kit; Thermo). 100 μg protein of each sample was further digested. Here, samples were first reduced with 10 mM DTT for 45 min at 37°C at 700 rpm on a Thermoshaker and then alkylated with 55 mM chloroacetamide (CAA) for 30 min at RT in the dark. After diluting the samples <1.6 M urea with 40 mM Tris (pH 7.6) and 2 mM CaCl2, trypsin (Trypsin Sequencing Grade; Roche) was added 1:100 (enzyme:protein) and incubated for 3 h at 37°C and 700 rpm. After the pre-incubation, trypsin was added again 1:100 (enzyme:protein) and incubated o/n at 37°C and 700 rpm.
SepPak desalting
On the next day, samples were acidified with formic acid (FA) to pH 2–3 and further desalted using 50 mg Sep-Pak columns (Waters Corp.). The columns were first wetted with 100% acetonitrile (ACN), followed by 0.1% FA in 50% ACN and further equilibrated with three washes of 0.1% FA. The samples were slowly loaded onto the column and the flow-through was reloaded onto the column to increase peptide binding. After washing off unspecific binders with three times 0.1% FA, peptides were eluted using 2×150 μl of 0.1% FA in 50% ACN. Samples were frozen at −80°C and dried using a Speed-Vac.
TMT labeling
TMT10-plex labeling was performed as previously described (Zecha et al., 2019). Briefly, 45 μg of peptides (measured on Nanodrop) were reconstituted in 20 μl of 50 mM HEPES (pH 8.5) and 5 μl of 11.6 mM TMT 10-plex (Thermo) in 100% ACN was added to each sample. After 1 h incubation at 25°C and 400 rpm, the reaction was stopped using 2 μl of 5% hydroxylamine. All TMT labeled samples were pooled. Remnants in sample vessels were rinsed with 20 μl of 10% FA in 10% ACN for 5 min and 400 rpm and added to pooled samples. The samples were frozen at −80°C and dried using a Speed-Vac.
Sep-Pak desalting
Pooled samples were again desalted using 50 mg Sep-Pak columns (Waters Corp.). The columns were first wetted with 100% ACN, followed by 0.1% FA in 50% ACN and further equilibrated with three washes of 0.1% FA. The dried pooled samples were reconstituted in 1 ml of 0.1% FA and loaded twice onto the column. After washing columns three times with 0.1% FA, peptides were eluted using 200 μl of 0.1% FA in 50% ACN. Samples were frozen at −80°C and dried using a Speed-Vac.
HpH reversed phase fractionation
For high pH reversed phase fractionation, dried samples were reconstituted in MS-grade water with 10% fractionation buffer A (25 mM ammonium bicarbonate (pH 8)) and centrifuged for 5 min at 20.000 × g and 4°C. The supernatant was then loaded on a C18 column (XBridge BEH130, 3.5 μm, 2.1×150 mm, Waters Corp), which was connected to a Dionex Ultimate 3000 HPLC system (Thermo). After injecting 100 μg peptides at a flow rate of 200 μg/min, the system was equilibrated for 5 min with 85% fractionation buffer B (MS-grade water), 10% fractionation buffer A and 5% fractionation buffer C (ACN). Peptides were eluted in a three-step linear gradient from 5% to 7% buffer C in 1 min with a constant amount of 10% buffer A. Then, a linear gradient from 7% to 42% buffer C in 44 min and from 42% to 80% buffer C in 6 min (with buffer A being constant at 10%) was used.
Starting from minute 3, 48 fractions (1 fraction/min) were collected in a 96-well plate and pooled to 24 fractions. For that, column 4 was pooled to column 1, column 5 was pooled to column 2 and column 6 was pooled to column 3. All fractions were frozen at −80°C and dried using a Speed-Vac.
LC-MS/MS data acquisition
Fractionated samples were measured in data-dependent acquisition mode using a nanoflow LC-MS/MS by coupling a Dionex Ultimate 3000 UHPLC+ system to a Fusion Lumos Tribrid mass spectrometer (Thermo). Dried samples were reconstituted in 0.1% FA and approximately 200 ng peptides were inserted. The sample was loaded to a trap column (75 μm × 2 cm, packed in-house with 5 μm C18 resin; Reprosil PUR AQ, Dr. Maisch, Ammerbruch-Etringen, Germany) with a flow rate of 5 μl/min and washed for 10 min with 0.1% FA. Subsequently, peptides were separated on an analytical column (75 μm × 40 cm, packed in-house with 3 μm C18 resin; Reprosil PUR AQ, Dr. Maisch, Ammerbruch-Etringen, Germany) with a flow rate of 300 nl/min and a linear 50 min gradient from 8% to 34% LC buffer B (0.1% FA, 5% DMSO in ACN) in LC buffer A (0.1% FA, 5% DMSO in MS-grade water). The eluate was sprayed via a stainless-steel emitter into the mass spectrometer, which was run in positive ion mode. Fullscan MS1 spectra were accorded in the Orbitrap with 60,000 resolution and a scan range from 360–1300 m/z (automatic gain control target of 4e5 charges, maximum injection time of 50 ms). A cycle time of 2 sec and a dynamic exclusion of 90 sec was used. MS2 spectra were recorded in the Ion Trap in rapid mode via sequential isolation of up to 10 precursors and the following settings: an automatic gain control target of 2e4, maximum injection time of 60 ms, isolation window of 0.7 m/z, and fragmentation via CID (NCE of 35%). For the following MS3 scan the ten most intense precursors were further fragmented via HCD (NCE of 55%) and acquired in the Orbitrap with 50,000 resolution, scan range of 100–1,000 m/z, automatic gain control target of 1.2e5 charges, maximum injection time of 120 sec and a charge-dependent isolation window from 1.3 (2+) to 0.7 (5–6+).
Transplantation into the ACE
Suspension cultures of PPs and PDLOs were generated in preparation for the ACE injection. To generate PP-spheroids, cells at the PE-stage (day 9 of the differentiation) were detached with TrypLE for 5–6 min resulting in clumps of 3 to 10 cells. After centrifugation at 400 rpm for 5 min the pellet was washed in BE3 medium and resuspended in day 9 medium (detailed above) supplemented with 10 μM ROCK inhibitor. Cells were replated into suspension on an ultra-low attachment (ULA) plate and further cultivated in differentiation medium until day 13. PDLOs in Matrigel were scraped off from the culture plate at day 23, directly resuspended and washed in cold BE3 medium, and transferred in a ULA plate in PDLO medium with 10 μM Y-27632 for shipping. Transplantation of organoid clusters to the anterior chamber of the eye (ACE) and in vivo imaging was performed as previously described (Chmelova et al., 2015). Briefly, mice were anesthetized by inhalation of 2% isoflurane in 100% oxygen via a face mask. A 25-gauge needle was used to make a small incision in the cornea, close to the corneal limbus, and 10–15 organoids in PBS were slowly injected into the ACE, using a custom-made beveled glass cannula (outer diameter, 0.4 mm; inner diameter, 0.32 mm; Hilgenberg). For in vivo imaging, mice were intubated and anesthetized with 2% isoflurane in 100% oxygen. A drop of 0.4% pilocarpine (Pilomann; Bausch & Lomb) in saline was placed on the cornea shortly before imaging to limit pupil dilation and iris movement. Animals were fixated and kept on a heating pad during the imaging procedure. Repetitive in vivo imaging was performed at indicated time points on an upright laser scanning microscope (LSM780 NLO; Zeiss) with a water dipping objective (W Plan-Apochromat 20×/1.0 DIC M27 75 mm; Zeiss) using vidisic eye gel (Bausch & Lomb) as immersion. Z-stacks of entire organoids were acquired at 1.5 μm intervals by detection of 633 nm laser backscatter. Additionally, FITC-Dextran (0.2 mg/ml in 100 μl PBS, Thermo) was injected into the tail vein to visualize blood vessels. Dextran was excited at 488 nm and detected at 468–607 nm. Z-stacks were processed using Imaris 8.1 software (Bitplane AG) and Fiji. Mice were sacrificed and eyes were collected 5 weeks after PDLO engraftment. Tissue specimens were fixed in 4% phosphate-buffered paraformaldehyde for 30 min at RT and then incubated in 25% sucrose solution o/n at 4°C. Eyes were embedded in Tissue-Tek OCT (Sakura Finetek) compound and snap-frozen in liquid nitrogen.
Orthotopic transplantation of PDLOs
For xenotransplantation of PDLOs into the pancreas, NSG mice were used. PDLOs between day 27 and day 31 were harvested and singularized as described above. After washing in BE3 medium, PDLO cells were resuspended in PDLO medium phase II supplemented with 20 μM Y-27632 and GFR-Matrigel in a 1:1 ratio. Aliquots with cell/Matrigel mixture were kept on ice until applied for transplantation.
Mice were pain mediated starting three days before transplantation by addition of 1 mg/ml Tramadol (Grünenthal) to the drinking water. After anesthesia with isoflurane transplantation side was disinfected with ethanol-containing skin antiseptic. A small 0.8 cm long cutaneous midline incision was made and subsequently a small subcutaneous pocket was prepared. After a 0.8 cm small incision into the peritoneum, the pancreas was mobilized and exposed. A volume of 40–50 μl with 0.5–1 ×106 PDLO cells was injected per mouse directly into the pancreatic tail. Carefully, pancreas and spleen were repositioned in the abdomen before the peritoneum was closed by medical sewing using 5–0 polyglactin coated vicryl suture (Ethicon). Surgical staples were used for closing the skin and removed one week after transplantation, when also Tramadol treatment was stopped. For oncogene induction, Dox was added in a final concentration of 400 μg/ml to the drinking water supplemented with 5% sucrose starting the day of transplantation. Mice were sacrificed after eight weeks and pancreata were collected and processed for histological analysis. Similarly, as described in the paraffin embedding of PDLOs section, tissue samples were fixated, dehydrated, embedded, sectioned, and mounted.
All animal experiments were performed in compliance with the institutional guidelines, under ethical and animal protection regulations of Ulm University.
Paraffin embedding of PDLOs
PDLO cultures were washed with PBS and 4% PFA with 100 mM sucrose was directly added to the wells for fixation. After incubation at 4°C o/n, PFA was removed carefully and PDLOs were washed twice with PBS. By scratching with a pipet tip, all organoids were removed from the well, transferred to a tube and centrifuged at 1000 × g for 3 min. Samples were pre-embedded in 2% agarose (Sigma) and further processed according to standard histology procedures. After serial dehydration, PDLOs as well as pancreas tissue samples were embedded in paraffin and sectioned at 4 μm. Tissue slices were mounted on SuperFrost Ultra Plus microscope slides (Thermo).
Histological standard techniques
Histological staining including Hematoxylin and Eosin (H&E) staining as well as Alcian Blue (AB) and Alcian Blue+Periodic Acid-Schiff reaction (AB+PAS) were performed according to standard protocols. In brief, after deparaffinization and hydration AB staining was done by incubating 3 min in 1% acetic acid solution (Sigma), 5 min in Alcian Blue (Roth) and rinsed shortly first with 1% acetic acid solution and then with tap water. Nuclei were counterstained with 0.1% Nuclear Fast Red solution (Merck) for 10 min and washed with dH2O. After dehydration slides were embedded in Entellan (Merck).
For AB+PAS staining procedure was identical but after washing with 1% acetic acid solution and tap water another short washing step with dH2O was necessary before incubation with periodic acid solution (Sigma) was performed for 5 min. Slides were rinsed three times in dH2O, incubated in Schiff’s reagent (Merck) for 15 min and washed with running tap water for 5 min. Finally, 20% Haematoxylin was applied for 90 sec, slides were washed with running tap water for 5 min, dehydrated and embedded in Entellan.
IF and IHC staining on paraffin tissue sections
Paraffin sections of PDLOs or pancreatic tissue were rehydrated in ethanol series followed by either heat-mediated or enzymatic antigen retrieval, depending on the antibody (Methods S4). Commercial Tris buffer (pH 9) or Citrate buffer (pH 6, both Vector Laboratories) were used for heat mediated antigen retrieval in the microwave or steamer, while a self-made Citrate buffer (pH 6, 1.9 g/l citric acid; Sigma) was used in the pressure cooker. For enzymatic antigen retrieval, sections were treated with 0.05 mg/ml pronase (Sigma) for 30 min at 37°C.
To continue with immunofluorescence (IF) staining, tissue permeabilization was performed with 0.5% Triton X-100/PBS (PBS-T) for 30 min at RT. After washing twice, primary antibodies diluted in Antibody Diluent (Zytomed) were added to the slides, which were then incubated o/n at 4°C in a wet chamber. After washing three times with PBS-T for 5 min, slides were stained with Alexa Fluor secondary antibodies (Thermo) and 500 ng/ml DAPI diluted in Antibody Diluent for 90 min at RT in the dark. Slides where washed three times with PBS-T and finally with dH2O before sections were mounted with Fluoromount-G (SouthernBiotech).
For immunohistochemistry (IHC) staining, antigen retrieval was directly followed by incubation with primary antibody solution for 30 min at RT in a wet chamber. Slides were washed and antibody detection was performed applying the Dako Detection Kit (ABC, Dako); different secondary antibodies were used to detect goat or rat IgGs (Vector Laboratories). In brief, slides were incubated first with a biotinylated secondary antibody, washed and next incubated with Streptavidin both for 30 min at RT. After another washing step, slides were incubated with Red Detection Dye for 3–16 min depending on antibody recommendation, counterstained with 20% Hematoxylin solution (Merck) for 30 sec, washed with tap water for 5 min and finally mounted with Aquatex (Merck).
IF staining on cryo sections
PDLOs were frozen in Shandon Cryomatrix (Thermo) and stored at −20°C as described previously (Molnár et al., 2020). Cyrosections of 7 μm thickness were cut, fixed in 4% PFA-PBS and washed in 1x TBS. For staining of CFTR (Alomone) and Occludin (Thermo) (Methods S4), antigen retrieval was performed in Sodium Citrate/Tween20 buffer (0.001 M Sodium Citrate Buffer, pH 6.0 and 0.05% Tween20) at 94° C for 30 min. During antigen retrieval, the slide was placed in a glass flask and the temperature was controlled on a heating block with a thermometer. Sections were blocked with 0.1% goat serum and 10% BSA-TBS for 1 h. Incubation with primary antibodies was performed o/n at 4°C. Incubation with Alexa Fluor secondary antibodies was performed at RT for 2 h. Sections were sealed with ProLong Gold Antifade Mounting Medium with DAPI (Thermo) then left to dry. Images were captured with a Zeiss LSM880 confocal microscope using a 40x oil immersion objective (Zeiss, NA: 1.4).
IF analysis of the eyes was performed on serial cryo sections of 8 μm. Frozen tissue slices were rehydrated for 10 min with PBS and quenched for 15 min using 100 mM Glycine (Sigma). Permeabilization and blocking were performed in 5% DS and 2% BSA in PBS-T (0.05% Tween20 in PBS) for 1 h. Subsequently, tissue sections were incubated with the primary antibodies diluted in 5% BSA/0.2% Triton X-100/PBS o/n at 4°C. Slides were washed three times for 5 min each with PBS-T followed by incubation with the secondary antibody solution supplemented with, 500 ng/ml DAPI for 1 h. Again, slides were washed, twice with 0.2% Triton X-100/PBS and once with PBS for 10 min each, and sections were mounted with Fluoromount-G. Fluorescence images were acquired on a Zeiss Axioscope2 microscope with ApoTome and Axiovision software was used for analysis. Imaging of IHC staining was performed with Olympus CKX41 microscope. Specific staining conditions for each antibody are listed in Methods S4.
B-Galactosidase staining
Organoids were washed with PBS and fixed in 4% PFA with 100 mM sucrose for 30 min at RT. Organoids were removed from the well by scratching with a pipet tip and transferred to a tube. After centrifugation at 1000 × g for 3 min supernatant was removed and organoids were incubated in 25% sucrose solution o/n at 4°C with rotation. After embedding in Tissue-Tek OCT compound, samples were frozen and stored at −80 °C until serial cryo sections of 8 μm thickness were cut. Staining for β-Galactosidase was done with the Senescence β-Galactosidase Staining Kit (Cell Signaling) according to manufacturer’s instructions. In brief, staining solution was freshly prepared, adjusted to pH between 5.9 and 6.1 and added onto the slides. After incubation o/n (14–16 h), slides were carefully washed with PBS, mounted with Aquatex and finally imaged with an Olympus CKX41 microscope.
Processing of images
Most BF, HE, IHC and IF images were cropped and brightness and contrast was modified using Photoshop or ImageJ for improved illustration. For IF staining, the single channels were modified, and compositions were created afterwards. All modifications were applied to the whole image and modified images clearly reflect the original images.
DNA isolation of FFPE tumor tissue
Tumor tissue (KRASG12D PDAC 1 and CDKN2AKO/KO KRASG12D PDAC I, III) was isolated from formalin-fixed paraffin embedded (FFPE) tissue by microdissection using fine needles. DNA was isolated from that tissue with an adapted version of the Maxwell RSC Blood Kit (Promega). Briefly, FFPE tissue was incubated in incubation buffer (Promega) for 10 min at 80°C under agitation. After cool-down on ice, ProteinaseK (20mg/ml, Sigma) was added and samples were incubated ON at 65°C with gentle agitation. Subsequently, lysis buffer (Promega) was added and samples were incubated for 30 min at 65°C, cooled down on ice and further processed on a Maxwell RSC instrument according to manufacturer’s instructions. Eluted DNA was quantified using Qubit dsDNA HS reagents (Thermo Fisher).
Low Coverage Whole Genome Sequencing (lcWGS) of hESC lines and FFPE tissue
DNA isolated from FFPE tumor tissue was sequenced together with DNA from their respective parental cell line (HUES8 KRASG12D and HUES8 CDKN2AKO/KO KRASG12D). In addition, HUES8 cells prior to piggyBAC gene editing were sequenced. Library preparation was performed with 50–200 ng DNA per sample using the NEBNext Ultra II FS DNA Library Prep Kit for Illumina following the manufacturer’s instructions. An adapted library preparation protocol was used for low-input (<50ng) or FFPE-isolated DNA. In this case, AMPure XP bead purification was omitted after adapter ligation and all DNA bound to AMPure XP beads (reaction volume:bead ratio: 0.8) directly eluted into PCR Mastermix. Further, denaturation, annealing and extension cycle number was varied according to DNA input (50–200 ng DNA: 4 cycles, <50 ng DNA/FFPE: 6 cycles). Samples were sequenced on NextSeq 500 (Illumina), resulting in ~20 Mio. single-end, 75 bp long, reads per tumor sample and ~10 Mio. reads per parental cell lines. Resulting reads were trimmed using “Trimmomatic” [version 0.39] (Bolger et al., 2014) and mapped to the human reference genome GRCh38.p12 using “bwa mem” [version 0.7.17] (Heng, 2013). “Sambamba” [version 0.7.0] (Tarasov et al., 2015) was used to identify read duplicates and the “GATK” toolkit [version 4.1.4.1] (Poplin et al., 2018) was used for base recalibration. Data from the parental hESC lines served as control for calling of somatic copy number alterations using the “CopywriteR” [version 2.16.0] package in R (Kuilman, 2020) (window size: 20kb, otherwise default settings). Raw sequencing data are available under the accession number PRJEB42190. Copy number alterations with a cut-off of log2(PDAC/parental cell line) ≥ I0.75I can be found in Suppl.Tab.4.
Cancer-panel sequencing
DNA from CDKN2AKO/KO KRASG12D PDAC II was isolated as described above with the difference that laser microdissection was performed instead of fine-needle microdissection. DNA was subsequently sequenced with a targeted sequencing approach using QIA-seq V3 chemistry (Qiagen) and the Human Comprehensive Cancer Panel (Qiagen). Target enrichment, amplicon processing, and library generation were performed according to the manufacturer’s instructions. For target enrichment, we included 40 ng genomic DNA. Successful target enrichment and library generation was controlled using the High Sensitivity DNA kit on a bioanalyzer device (Agilent). Libraries were diluted to 10 pM solutions and sequencing was performed on a MiSeq platform (Illumina) using a V3 FlowCell. The resulting FASTQ files were further analyzed to identify somatic mutations using a common workflow in the CLC Genomic Workbench [version 20.0.3] (Qiagen). The mean read depth on target regions was about 3000-fold, and 95% of bases reached a UMI depth of >100x. All identified mutations were manually reanalyzed using the Integrated Genome Viewer Software (Broad Institute) (Robinson et al., 2011, Cho et al., 2012). The parental cell line was Sanger-sequenced (Eurofins Genomics) for the identified P53S94P mutation and no mutation could be detected in the parental cell line.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
In general, data summarize three independent experiments (independently started differentiations) with each analysis performed in duplicates (two wells per condition), unless otherwise stated.
Statistical analysis was performed using the GraphPad Prism 8 software, if not stated elsewise, and detailed information regarding the different applied tests are indicated in the figure legends. Statistical significance was defined as follows: * P-value < 0.05, ** P-value < 0.01, *** P-value < 0.001, **** P-value < 0.0001.
RNA-seq data analysis
Processing and Generation of DEG lists
Raw sequencing data are available under the accession number PRJEB38015. Gene annotations of the human reference genome GRCh38 were derived from the Gencode homepage (EMBL-EBI). “Dropseq tool” [version 1.12] (Macosko et al., 2015) was used for mapping raw sequencing data to the reference genome. The resulting UMI filtered count matrix was imported into R [version 3.4.4]. Prior differential expression analysis with “DESeq2” [version 1.18.1] (Love et al., 2014), dispersion of the data was estimated with a parametric fit using the day of differentiation (d0, d3, d13, d20, d24, d30, d45, d59, and ductal controls) as covariate in the model matrix.
The Wald test was used for determining differentially regulated genes between all pairwise group comparisons and shrunken log2 fold changes were calculated afterward, with setting the type argument of the “lfcShrink” function to “apeglm” (Pairwise comparisons are listed in Suppl.Tab.2). A gene was determined to be differentially regulated if the absolute log2 fold change was at least 2 and the adjusted P-value was below 0.01.
Deviating from this setting, the absolute log2 fold change threshold was set to 1 while keeping the same alpha level for comparisons between d20 vs d13 and d59 vs d30, and the absolute log2 fold change threshold was set to 0 for the comparison between 0.00 μM and 0.05 μM MSC2530818 d20. Overrepresentation analyses of the differentially regulated genes from depicted pairwise comparisons were conducted using “EnrichR” (Kuleshov et al., 2016) within the KEGG, GO-term, Reactome, and BioCharta common database. Rlog transformation of the data was performed for visualization and further downstream analysis.
Reanalysis of publicly available data
Within this study 15 reference gene lists from seven different studies were implemented (listed in Methods S5). Ten gene lists were directly retrieved from the literature, one gene set for pancreatic progenitors (Xie et al., 2013), three gene sets for trunk cells (de Lichtenberg et al., 2018, Krentz et al., 2018), and six gene sets for putative ductal subpopulations (Qadir et al., 2020). Gene sets from (de Lichtenberg et al., 2018) were compiled by filtering the published pairwise comparisons of a putative trunk domain against either tip (trunk1) or early-endocrine (trunk2) cells by a P-value lower than 0.05 and sorting according to the 100 genes with the highest FC.
Five additional gene sets were compiled from raw data. A second pancreatic progenitor gene set was generated from FASTQ files (friendly provided by Neil Hanley) (Gerrard et al., 2016). Data was mapped to the human reference genome GRCh38 and the Gencode gene annotation [v29] with “Star aligner” version 2.6.1.c] (Dobin et al., 2013). Prior mapping sequencing reads were trimmed with “Trimmomatic” [version 0.36] (Bolger et al., 2014). Bases at the start and end of each read, for which the “phread” score was below 25 were removed. Furthermore, reads were clipped if the average quality within a sliding window of 10 fell below a “phread” score of 25. Conclusively, reads smaller than 50 bases were removed. “FeatureCounts” from the “Subread” package [v1.6.3] (Liao et al., 2019) was used to get sample wise gene counts in the stranded mode having the parameter “ignoreDup” set to “False”. Resulting gene count lists were imported into R and quantile normalized with “Limma” (Ritchie et al., 2015) using the organ description as covariate during model fitting. Organ specific gene lists have been generated by pairwise comparisons between all investigated organs. All genes found significant in at least on pairwise comparison at an FDR level of 0.05 were subsequently sorted according to their peak expression to assign organ specific gene lists comprising of 100 genes.
Duct-specific gene lists have been generated using scRNA-seq data. For the first data set (Enge et al., 2017), raw transcript count matrix and annotation data including donor age and cell type clustering information were downloaded from the GEO databank under accession number GSE81547. The dataset contained 2527 cells and 23359 genes and for downstream analysis python package “scanpy” (Wolf et al., 2018) was used. For preprocessing, cells with less than 300 counts, cells with less than 300 expressed genes, and genes expressed in less than 10 cells were filtered out. Normalization of the preprocessed raw data was performed using “scran” (Lun et al., 2016) and batch correction for donors was performed using “ComBat” (Johnson et al., 2007). Cluster were assigned as in the original publication (acinar, ductal, mesenchymal, alpha, beta, delta, and unsure). The top 4000 genes with the highest variance were selected to generate UMAPs (McInnes et al., 2018). Marker genes were identified using “sc.tl.rank_genes_groups” function (Wolf et al., 2020) for each cell type with default parameters except for “groupby” set to “Celltype”. To generate reference input lists a P-value lower than 0.01 was used as threshold and the top 100 genes with highest FC were subsequently used for GSEA.
For the second scRNA-seq data set (Baron et al., 2016), the raw transcript count matrix and annotation data was downloaded from the GEO databank under accession number GSE84133. The dataset contained 8569 cells and 20125 genes. Preprocessing including filtering, normalization, and batch correction was done exactly as described above. Clustering of cells was redone in this study: Cells were clustered using the Louvain algorithm (Traag, 2015, Blondel et al., 2008, Levine et al., 2015) implemented with “scanpy.tl.louvain” function with resolution=1.0 and further parameters set as default. The clusters obtained from Louvain algorithm were merged into new custom clusters. As part of reclustering, we only focused on cells of interest (acinar, ductal, alpha, beta, combined gamma-epsilon, and delta cells), while omitting non-parenchymal cells (stellate, endothelial, immune, and unknown cells). Subsequent steps including identification of marker genes were identical as in the first scRNA-seq data set.
From the second scRNA-seq data set, we additionally generated ductal subpopulation-specific gene lists by assigning CFTRhigh MUC1low and CFTRlow MUC1high cell clusters based on the original publication. For that, cells were ranked once according to their expression of CFTR and a second time according to their expression of MUC1. The difference in ranks was calculated and all cells with a rank score difference (rank_CFTR_minus_MUC1) of at least 100 were assigned to a “ductal_CFTR” cluster and vice versa. Remaining cells were assigned to a third “ductal_other” cluster, which was not further investigated. The additional ranked marker gene list was calculated using the “sc.tl.rank_genes_groups” function (Wolf et al., 2020) for each cell type with “groupby” set to “subClusters” and “groups” set to [“ductal_MUC1”, “ductal_CFTR”, “ductal_other”] in order to compare “ductal_CFTR” versus “ductal_MUC1”. To generate the respective reference input lists a P-value lower than 0.01 was used as threshold and all genes, fulfilling the P-value criteria (less than 100), were subsequently used for GSEA.
Gene set enrichment analysis (GSEA, Broad Institute)
Gene lists for pairwise comparisons were ranked using log2(“FC_Shrink-apegIm”) and ranked lists were analyzed with the “GSEAPreranked” tool of GSEA [version 4.0.3] (Subramanian et al., 2005) using except for the Collapse parameters (“No_collaps”) default settings including 1000 permutations with a weighted statistical analysis. For comparison with reference gene sets, gene lists have been either directly compiled from literature or raw data has been reanalyzed as indicated above. A complete list of applied gene sets can be found in Methods S5.
Cell Type Deconvolution using cell population mapping (CPM)
A recently published cell population mapping algorithm for cell type deconvolution (Frishberg et al., 2019) was implemented with the help of the R package “scBio”. Our Rlog transformed bulk RNA data matrix was used in conjunction with the processed scRNA-seq dataset to determine similarity scores of PDLOs (day 30) and human primary ductal organoids with the distinct cell types of a human adult pancreas (Enge et al., 2017). Prior running the CPM algorithm, the bulk RNA dataset was reduced to the number of genes that have been used to generate the UMAP representations for the scRNA-seq data (see above).
Heatmap of most significant genes
A heatmap of stage-specific significant genes was generated based on a data matrix including only samples of the time course of differentiation (d0, d3, d13, d20, d24, d30, d45, and d59). Read counts were processed as described above. All genes with a P-value ≤ 0.01 and a log2FC ≥ 2 in at least one pairwise comparison between different time points were ordered according to their peak expression value (Stage-specific Peak expression is listed in Suppl.Tab.1).
Heatmap of target genes/proteins and ward clustering
Heatmaps to illustrate target gene or protein expression were conducted using the packages “pheatmap” for plotting and the package “biomaRt” for retrieval of gene sets from common databases. The function “pheatmap” was used with parameter “scale” set to “row” and “clustering_method” set to “ward.D” and with further parameters set as default. To cluster all samples of the RNA-seq experiment the same function was applied on the entire data matrix containing all processed (15630) genes.
Proteome data analysis
Acquired raw files were mapped with “Maxquant” [version 1.5.7.4] (Cox and Mann, 2008) against the UniProtKB human reference list (downloaded 22.07.2013). For the search settings, up to 2 missed cleavages were allowed, carbamidomethylation was defined as a fixed modification and oxidation of methionine as well as N-terminal protein acetylation were set as variable modifications. Reporter ion MS3 was set as quantification type and TMT10plex as isobaric labels. The first search peptide tolerance was set to 20 ppm and the main search peptide tolerance was set to 4.5 ppm. Results were filtered by setting the protein and peptide false discovery rate to 1% using a classical target-decoy approach. All the following data analysis was performed using R [version 6.1.7601]. From the “Maxquant” output, all reversed and “only identified by site” protein entries were filtered out and the log2 reporter ion intensities of the three replicates were further “ComBat” (Johnson et al., 2007) adjusted with the “sva” package [version 3.30.1] (Leek et al., 2012) to correct for batch effects across the measured samples. P-values were calculated with the “limma” package [version 3.38.3] and corrected for multiple testing using the Benjamini-Hochberg method (Benjamini and Hochberg, 1995) (Comparison of protein expressionis shown in Suppl.Tab.3).
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD018785 (Reviewer Account: Username: <reviewer76493@ebi.ac.uk, Password: 9Wzfw38m).
RNA-Proteome data comparison
To adjust for different modes of measurement in RNA-seq and full proteome (read counts versus protein intensities), the normalized counts per million (CPM) values and the batch effect corrected reporter ion intensities were normalized with the modified “MComBat” procedure (Frejno et al., 2017) based on the protein data and the correlation was further visualized by histogram plots (package “ggExtra”, [version 0.8]).
Supplementary Material
Live-cell imaging of CDKN2AKO/KO KRASG12D PDLOs w/o Dox stimulation
Related to Fig.5.M
* Time-lapse video of PDLO culture monitored every 3 h over 4 days. The frame rate was 5 frames per second (fps).
Live-cell imaging of CDKN2AKO/KO KRASG12D PDLOs + Dox stimulation
Related to Fig.5.M
Time-lapse video of PDLO culture monitored every 3 h over 4 days. The frame rate was 5 frames per second (fps) and the red mCherry fluorescence signal indicates KRASG12D expression.
Supplemental Table 3. Differentially expressed proteins in PDLOs vs PPs
Related to Fig.3H–N and Suppl.Fig3H–J (Results)
Limma protein expression comparison of d59 vs d13.
Supplemental Table 4. Copy number variations identified in PDAC 1, PDAC I, and III using lcWGS
CNV with a log2ratio ≥ I0.75I are depicted.
Supplemental Table 5. Point mutations identified in PDAC II using a targeted sequencing approach
Related to Fig.7 and Suppl.Fig. 7E (Results)
Point mutations identified in a comprehensive cancer panel (Qiagen). Variants marked in red are not reliable, as clarified in the “tag-defs” spreadsheet.
Reference gene sets used within this study
Related to “Gene set enrichment analysis” (STAR Methods)
Supplemental Table 1. Stage-specific RNA-seq peak expression
All genes with a P-value ≤ 0.01 and a log2FC ≥ 2 in at least one pairwise comparison were ordered according to their peak expression value.
Supplemental Table 2. Differentially expressed genes in RNA-seq analysis
Related to Fig.3A–G and Suppl.Fig3A–G (Results)
Pairwise DESeq2 expression comparisons.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Secondary antibody anti-mouse-HRP | GE Healthcare | Cat# NA931; RRID:AB_772210 |
| Secondary antibody anti-rabbit-HRP | GE Healthcare | Cat# NA934; RRID:AB_772206 |
| anti-mouse IgG MicroBeads | Miltenyi | Cat# 130-048-402; RRID:AB_244361 |
| Rabbit monoclonal AcTUB | Abcam | Cat# ab179484 |
| Rabbit polyclonal AMY2A | SIGMA | Cat# A8273; RRID:AB_258380 |
| Mouse monoclonal anti-ACTB | Sigma | Cat# A5316; RRID:AB_476743 |
| Rabbit polyclonal C-peptide | Cell Signaling | Cat# 4593; RRID:AB_10691857 |
| Mouse monoclonal CA19-9 | Thermo | Cat# CA1003-200UL; RRID:AB_212741 |
| Rabbit monoclonal anti-CA2 | Abcam | Cat# ab124687; RRID:AB_10972000 |
| Rabbit monoclonal CDX2 | Cell Marque - RabMab | Cat# MU392-UC; RRID:AB_2335627 |
| Mouse monoclonal CFTR | R&D | Cat# MAB1660; RRID:AB_2260674 |
| Rabbit polyclonal CLDN1 | Abcam | Cat# ab15098; RRID:AB_301644 |
| Mouse monoclonal PE-conjugated CXCR4 antibody | Life Technologies | Cat# MHCXCR404; RRID:AB_10373097 |
| Mouse monoclonal APC-conjugated c-Kit antibody | Thermo | Cat# CD11705; RRID:AB_2536476 |
| Mouse monoclonal E-CAD | Dako/Agilent | Cat# M3612; RRID:AB_2341210 |
| Mouse monoclonal E-CAD | BD Bioscience | Cat# 610182; RRID:AB_397581 |
| Rabbit monoclonal E-CAD | Cell Signaling | Cat# 3195; RRID:AB_2291471 |
| Mouse monoclonal Gαs/olf (G-10) | Santa Cruz | Cat# sc-365855; RRID:AB_10842167 |
| Mouse monoclonal GCG | SIGMA | Cat# G2654; RRID:AB_259852 |
| Rabbit polyclonal GFP | Thermo | Cat# A-6455 RRID: AB_221570 |
| Mouse monoclonal GP2 | MBL International | Cat# D277-3, RRID:AB_10598500 |
| Rabbit polyclonal C-pep | Cell Signaling | Cat# 4593; RRID:AB_10691857 |
| Rat monoclonal CD31 | BD Bioscience | Cat# 557355; RRID:AB_396660 |
| Mouse monoclonal CTRC | Millipore | Cat# MAB1476; RRID:AB_2261190 |
| Rabbit polyclonal ERK | Cell Signaling | Cat# 9102; RRID:AB_330744 |
| Mouse monoclonal H-NUCL | Abcam | Cat# ab190710 |
| Rabbit monoclonal HA | Cell Signaling | Cat# 3724; RRID:AB_1549585 |
| Mouse monoclonal HNF1B | Abcam | Cat# ab236759 |
| Rabbit monoclonal Ki67 | Thermo | Cat# MA5-14520; RRID:AB_10979488 |
| Mouse monoclonal Ki67 | Dako/Agilent | Cat# M7240; RRID:AB_2142367 |
| Mouse monoclonal KRT7 | Dako/Agilent | Cat# M7018; RRID:AB_2134589 |
| Mouse monoclonal KRT8 | BD Bioscience | Cat# 345779; RRID:AB_2800363 |
| Mouse monoclonal KRT19 | Dako/Agilent | Cat# M0888; RRID:AB_2234418 |
| Rabbit polyclonal mCherry | Abcam | Cat# ab167453; RRID:AB_2571870 |
| Mouse monoclonal MUC1 | Santa Cruz | Cat# sc-7313; RRID:AB_626983 |
| Mouse monoclonal MUC5AC | Santa Cruz | Cat# sc-33667; RRID:AB_627973 |
| Rabbit monoclonal N-CAD | Cell Signaling | Cat# 13116; RRID:AB_2687616 |
| Rabbit polyclonal NANOG | Cell Signaling | Cat# 3580; RRID:AB_2150399 |
| Mouse monoclonal NKX6-1 | DSHB Hybridoma | Cat# F55A12; RRID:AB_532379 |
| Mouse monoclonal NKX6-1-APC | BD | Cat# 563338; RRID:AB_2738144 |
| Mouse monoclonal OCT4 | Santa Cruz | Cat# sc-5279; RRID:AB_628051 |
| Mouse monoclonal P15 | Santa Cruz | Cat# sc-271791; RRID:AB_10709436 |
| Rabbit monoclonal P16 | Cell Signaling | Cat# 80772; RRID:AB_2799960 |
| Rabbit monoclonal P21 | Abcam | Cat# ab109520; RRID:AB_10860537 |
| Mouse monoclonal P53 | Santa Cruz | Cat# sc-47698; RRID:AB_628083 |
| Rabbit monoclonal pERK | Cell Signaling | Cat# 4377; RRID:AB_331775 |
| Rabbit polyclonal PARP | Cell Signaling | Cat# 9542; RRID:AB_2160739 |
| Goat polyclonal PDX1 | R&D | Cat# AF2419; RRID:AB_355257 |
| Mouse monoclonal PDX1-PE | BD | Cat# 562161; RRID:AB_10893589 |
| Rabbit polyclonal PKC | Abcam | Cat# ab59364; RRID:AB_944858 |
| Rabbit monoclonal p-PKA substrates (RRXS*/T*) | Cell Signaling | Cat# 9624; RRID:AB_331817 |
| Rabbit monoclonal pRB | Cell Signaling | Cat# 8516; RRID:AB_11178658 |
| Mouse monoclonal RB | Cell Signaling | Cat# 9309; RRID:AB_823629 |
| Rabbit polyclonal SOX9 | Millipore | Cat# AB5535; RRID:AB_2239761 |
| Mouse monoclonal SSEA4 | Cell Signaling | Cat# 4755; RRID:AB_1264259 |
| Rabbit polyclonal Turbo GFP | Thermo | Cat# PA5-22688 RRID: AB_2540616 |
| Rabbit monoclonal VASP | Cell Signaling | Cat# 3132; RRID:AB_2213393 |
| Rabbit monoclonal VIM | Cell Signaling | Cat# 5741; RRID:AB_10695459 |
| Mouse monoclonal VINC | Sigma | Cat# V9264; RRID:AB_10603627 |
| Mouse monoclonal H2AX (pS139) Clone N1-431-APC | BD | Cat# 560447; RRID:AB_1645414 |
| Rabbit polyclonal Zeb1 | Santa Cruz | Cat# sc-25388; RRID:AB_2217979 |
| Mouse monoclonal ZO1 | Thermo | Cat# 33-9100; RRID:AB_2533147 |
| Antibody conditions for IHC/IF are listed in Suppl.Table6 | This paper | N/A |
| Bacterial and Virus Strains | ||
| Lentivirus hOKSM-dTomato | (Warlich et al., 2011) | N/A |
| Non-integrating Sendai virus (CytoTuneTM-iPS 2.0 Sendai Reprogramming Kit) | Thermo | Cat# A16517 |
| One Shot™ ccdB Survival™ 2 T1R Competent Cells | Thermo | Cat# A10460 |
| Subcloning Efficiency DH5α Competent Cells | Thermo | Cat# 18265017 |
| Biological Samples | ||
| Human pancreatic ductal organoids from cadaveric organ donors | Tamara Madácsy, József Maléth (based on Boj et al. (2015)) | N/A |
| FPC patient keratinocytes | This paper | N/A |
| Human bone marrow stromal cells (HBMSCs) | Natasha Cherman (Pamela G. Robey) | N/A |
| Transformed human patient-derived organoids (Panc163) | Bruno Sainz, Patrick Hermann (Rubio-Viqueira et al., 2006) | N/A |
| Untransformed human patient-derived organoids from resection specimen | Maximilian Reichert, Zahra Dantes (Dantes et al., 2020) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 3,3,5-Triiodo-L-thyronine | Sigma | Cat# T0281; CAS: 5817-39-0 |
| A-83-01 | Tocris | Cat# 2939; CAS: 909910-43-6 |
| Activin A | PeproTech | Cat# 120-14; SDS: 25-120-14 |
| Carbonic anhydrase II from bovine erythrocytes | Sigma | Cat# C2273; CAS: 9001-03-0 |
| CHIR99021 | Axon MedChem | Cat# 1386; CAS: 252917-06-9 |
| Choleratoxin | Sigma | Cat# C9903; CAS: 131096-89-4 |
| Collagenase/Dispase | Roche | Cat# 11097113001 |
| Collagenase II | Life Technologies | Cat# 17101015 |
| Collagenase IV | Worthington | Cat# LS0004186; CAS: 9007-34-5 |
| Dexamethasone | Sigma | Cat# D1756; CAS: 50-02-2 |
| Dextran, Fluorescein 500,000 MW | Thermo | Cat# D7136 |
| Dispase | Sigma | Cat# D4693; CAS: 42613-33-2 |
| Dorsomorphin | Sigma | Cat# P5499; CAS: 866405-64-3 |
| Doxycycline hyclate (Dox) | Sigma | Cat# D9891; CAS: 24390-14-5 |
| EGF human | R&D | Cat# 236-EG-200 |
| Fatty acid free BSA | Proliant | Cat# 608068; CAS: 9048-46-8 |
| FGF2 | Novoprotein | Cat# C046 |
| FGF10 | R&D | Cat# 345-FG-250 |
| FGF-Basic | Thermo | Cat# PHG0360 |
| Forskolin (FSK) | Sigma | Cat# F 3917; CAS: 66575-29-9 |
| Gastrin I | Sigma | Cat# G9020; CAS: 10047-33-3 |
| KGF | PeproTech | Cat# 100-19; SDS: 25-100-19 |
| LDN-193189 (=DM3189) | Sigma | Cat# SML0559; CAS: 1062368-24-4 |
| MSC2530818 | Selleckchem | Cat# S8387; CAS: 1883423-59-3 |
| Matrigel Basement Membrane Matrix | Corning | Cat# 354234 |
| Matrigel Basement Membrane Matrix Growth Factor Reduced (GFR) | Corning | Cat# 354230 |
| Matrigel hESC-qualified Matrix | Corning | Cat# 354277 |
| N-acetyl-L-cysteine | Sigma | Cat# A7250; CAS: 616-91-1 |
| Nicotinamide (NA) | Sigma | Cat# N0636; CAS: 98-92-0 |
| Noggin murine | PeproTech | Cat# 250-38; SDS: 25-250-38 |
| Nu-Serum IV | Corning | Cat# 355104; |
| PKA inhibitor H 89 2HCl | Selleckchem | Cat# S1582; CAS: 130964-39-5 |
| Prostaglandin E2 | Tocris | Cat# 2296; CAS: 363-24-6 |
| R-Spondin 1 protein | R&D | Cat# 4645-RS |
| Retinoic acid (RA) | Sigma | Cat# R2625; CAS: 302-79-4 |
| ROCK inhibitor (Y-27632) | Abcam | ab120129; CAS: 129830-38-2 |
| SANT1 | Sigma | Cat# S4572; CAS: 304909-07-7 |
| TMT 10-plex | Thermo | Cat# A37725 |
| Wnt3a mouse | PeproTech | Cat# 315-20; SDS: 25-315-20 |
| XtremeGene 9 DNA transfection reagent | Roche | Cat# 6365787001 |
| ZnSO4 | Sigma | Cat# Z0251; CAS: 7446-20-0 |
| (-)-Indolactam V | STEMCELL Technologies | Cat# 72312; CAS: 90365-57-4 |
| 3-isobutyl-1-methylxanthine (IBMX) | Sigma | Cat# I5879; CAS: 28822-58-4 |
| Critical Commercial Assays | ||
| Active Ras Pull-Down and Detection Kit | Thermo | Cat# 16117 |
| AllPrep DNA/RNA/miRNA Universial Kit | Qiagen | Cat# 80224 |
| BCECF, AM | Thermo | Cat# B1170 |
| BP Clonase II enzyme mix | Life Technologies | Cat# 11789020 |
| cAMP-Gs HiRange Kit | Cisbio | Cat# 62AM6PEB |
| Click-iT EdU Alexa Fluor 647 Assay Kit | Life Technologies | Cat# C10635 |
| Gibson Assembly® Master Mix | New England BioLabs | Cat# E2611 |
| Human Comprehensive Cancer Panel | Qiagen | Cat# DHS-3501Z |
| LR Clonase II enzyme mix | Life Technologies | Cat# 11791100 |
| Maxwell RSC Blood Kit | Promega | Cat# AS1400 |
| Nextera XT Kit | Illumina | Cat# FC-131-1096 |
| NEBNext Ultra II FS DNA Library Prep Kit | NEB | Cat# E6177 |
| P3 primary cell 4D Nucleofector X Kit S | Lonza | Cat# V4XP-3032 |
| Senescence β-Galactosidase Staining Kit | Cell Signaling | Cat#9860S |
| SuperSignal West Dura Kit | Thermo | Cat# 34076 |
| Tissue Genomic DNA Purification Mini Prep Kit | Genaxxon | Cat# S5378.0050 |
| Qubit dsDNA HS reagents | Thermo | Cat# Q32851 |
| Deposited Data | ||
| Human reference proteome | UniProt | UP000005640 (downloaded 22.07.2013); https://www.uniprot.org/proteomes/UP000005640 |
| Human reference genome GRCh38.p13 | EMBL-EBI | GCF_000001405.39; https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.39/ |
| Raw and analyzed RNA data | This paper (ENA at EMBL-EBI) | ENA: PRJEB38015 |
| Raw and analyzed protein mass spectrometry data | This Paper (PRIDE) | PRIDE: PXD018785 |
| Pancreatic progenitor first gene set | (Xie et al., 2013) | DOI: 10.1016/j.stem.2012.11.023 |
| Pancreatic progenitor second gene set | (Gerrard et al., 2016) | E-MTAB-3928 |
| scRNA-seq data (first set) | (Enge et al., 2017) | GEO: GSE81547 |
| scRNA-seq data (second set) | (Baron et al., 2016) | GEO: GSE84133 |
| scRNA-seq data | (Qadir et al., 2020) | DOI: 10.1073/pnas.1918314117/-/DCSupplemental. |
| Trunk domain gene sets Trunk1/Trunk2 | (de Lichtenberg et al., 2018) | DOI: doi.org/10.1101/336305 |
| Trunk third gene set | (Krentz et al., 2018) | DOI: 10.1016/j.stemcr.2018.11.008 |
| Compiled reference gene sets applied in GSEA are listed in Suppl.Table7 | Indicated above | Indicated above |
| Experimental Models: Cell Lines | ||
| Human: HUES8 hESC line (NIH approval number NIHhESC-10-0021) | HSCI iPS Core, Harvard University, Cambridge, MA, USA | hES Cell Line: HUES-8; RRID:CVCL_B207 |
| HUES8-CDKN2Ako/ko (+/− KRASG12D) | This paper | N/A |
| HUES8-KRASG12D line | This paper | N/A |
| HUES8-GNASR201H line | This paper | N/A |
| HUES8-Luciferase line (Vector control) | This paper | N/A |
| Human: H1 ES cells | Wicell Research Institute, Madison, WI, USA | https://www.wicell.org/ |
| CoiPSC (Control iPSC) | This paper | N/A |
| MAS-GNASWT/R201C iPSCs | This paper | N/A |
| MAS-GNASWT/WT iPSCs | This paper | N/A |
| Rat embryonic fibroblast (REF) | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| NSG mice (strain: NOD.Cg-Prkdc<scid<tm1Wjl>/SzJ GVO) | Charles River | RRID:BCBC_4142 |
| Nod scid mice (strain: NOD.Cg-Prkdcscid/J) | The Jackson Laboratory | RRID:IMSR_JAX:001303 |
| Oligonucleotides | ||
| crRNA-CDKN2A exon2: GTAGGGGTAATTAGACACCT | This paper | N/A |
| crRNA-CDKN2A exon3: GTCTCGAGTCTATCGATATG | This paper | N/A |
| CDKN2AKO-external-fwd: GCGCTTGGATATACAGCAGTG | This paper | N/A |
| CDKN2AKO-external-rev: ACAGGAGCATCTCCCAACC | This paper | N/A |
| CDKN2AKO-internal-fwd: GGCATTGTGAGCAACCACTG | This paper | N/A |
| CDKN2AKO-internal-rev: CCTGTAGGACCTTCGGTGAC | This paper | N/A |
| Cloning primer for the “All-in-One piggyBac”-system are listed in Suppl.Table3 | This paper | N/A |
| CDKN2AiPSC-exon2-fwd: CCGCAGAAGTTCGGAGGATA | This paper | N/A |
| CDKN2AiPSC-exon2-rev: CTTTGGAAGCTCTCAGGGTACA | This paper | N/A |
| GNAS-exon8-fwd: CCAGACCTTTGCTTTAGATTGG | (Salinas-Souza et al., 2015) | N/A |
| GNAS-exon9-rev: CACAGCATCCTACCGTTGAAG | (Wood et al., 2017) | N/A |
| Primers for qPCR are listed in Suppl.Table5 | This paper | N/A or Qiagen Cat# |
| Recombinant DNA | ||
| pCAS9_GFP plasmid | Kiran Musunuru; (Ding et al., 2013) | Addgene plasmid #44719 |
| gRNA cloning vector | George Church; (Mali et al., 2013) | Addgene plasmid #41824 |
| pBabe-KRASG12D | Channing Der | Addgene plasmid # 58902 |
| pcDNA3.1+hsGNAS_EE(long)R201H | Franz. Oswald | N/A |
| pGL4.10[luc2] | Franz Oswald | N/A |
| pDONR201 | Thermo | Cat# 11798-014 |
| Destination vector PB-TAC-ERP2 | (Kim et al., 2016) | Addgene plasmid #80478 |
| PB-TAC-ERP2-(N-HA)KRAS_G12D | This paper | N/A |
| PB-TAC-ERP2-KRAS_G12D (w/o HA) | This paper | N/A |
| PB-TAC-ERP2-GNAS_R201H(EE) | This paper | N/A |
| PB-TAC-ERP2-Luc2 | This paper | N/A |
| Transposase-encoding vector | SBI Biosciences (Rao et al., 2016) | #PB200PA-1 |
| Software and Algorithms | ||
| Adobe Illustrator | Adobe | Creative Cloud |
| Adobe Photoshop | Adobe | Creative Cloud |
| AxioVision software | ZEISS | https://www.microshop.zeiss.com/de/de/system/software+axiovision-axiovision+basissoftware-axiovision+software/10221/ |
| “bwa mem” | (Heng, 2013) | Version 0.7.17 |
| CLC Genomic Workbench | Qiagen | Version 20.0.3 https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-clc-genomics-workbench/ |
| CRISPOR sgRNA prediction and designing platform | (Haeussler et al., 2016) | http://crispor.tefor.net/ |
| EnrichR webtool | Ma’ayan lab (Kuleshov et al., 2016) | https://amp.pharm.mssm.edu/Enrichr/ |
| Fiji | (Schindelin et al., 2012) | https://imagej.net/Fiji |
| “GATK” toolkit | (Poplin et al., 2018) | Version 4.1.4.1 |
| GraphPad Prism 8 | GraphPad Software, San Diego, California USA | www.graphpad.com |
| GSEA software | Broad Institute (Subramanian et al., 2005) | Version 4.0.3; https://www.gsea-msigdb.org/gsea/index.jsp |
| Imaris 8.1 software | Bitplane AG | https://imaris.oxinst.com/downloads |
| Interactive Genome Viewer (IGV) | Broad Institute (Robinson et al., 2011) | https://software.broadinstitute.org/software/igv/ |
| Maxquant software | (Cox and Mann, 2008) | Version v.1.5.7.4; https://www.maxquant.org/ |
| Modde | Umetrics, Sartorius | https://umetrics.com/product/modde-go |
| Olympus excellence software | Olympus | N/A |
| Phyton | Python Software Foundation | https://www.python.org/psf/ |
| Python package “scanpy” | (Wolf et al., 2018) | N/A |
| Python package “scran” | (Lun et al., 2016) | N/A |
| R | The R Project | Version 6.1.7601 https://cran.r-project.org/mirrors.html |
| R package “CopywriteR” | (Kuilman, 2020) | Version 2.16.0 |
| R package “DESeq2” | (Love et al., 2014) | Version 1.18.1 |
| R package “Limma” | (Ritchie et al., 2015) | https://www.bioconductor.org/packages/release/bioc/html/limma.html |
| R package “scBio” for Cell population mapping | (Frishberg et al., 2019) | https://github.com/amitfrish/scBio |
| R package “sva” | (Johnson et al., 2007; Leek et al., 2012) | Version 3.34.0 |
| “Sambamba” | (Tarasov et al., 2015) | Version 0.7.0 |
| Star aligner | (Dobin et al., 2013) | Version 2.6.1.c https://github.com/alexdobin/STAR |
| Trimmomatic | (Bolger et al., 2014) | Version 0.36 / 0.39 http://www.usadellab.org/cms/index.php?page=trimmomatic |
| Other | ||
| Biozero BZ-9000 microscope | Keyence | N/A |
| Dionex Ultimate 3000 HPLC system | Thermo | Cat# 6130-7-Sys-004 |
| Zeiss Axioscope2 | Zeiss | N/A |
| Ibidi-ibiTreat-precoated glass-bottom 24-well μ-plates | IBIDI | Cat# 82406 |
| Ibidi-ibiTreat-precoated 15-well polymer coverslip bottom μ slides | IBIDI | Cat# 81506 |
| Infinite M1000 pro | Tecan | N/A |
| Fusion SL system | VILBER | N/A |
| LSM780 NLO confocal microscope | Zeiss | N/A |
| LSM880 confocal microscope | Zeiss | N/A |
| LSR II flow cytometer | BD | N/A |
| NextSeq 500 Sequencing System | Illumina | Cat# 770-2013-053-F |
| Olympus CKX41 | Olympus | N/A |
| Olympus Cool LED PE-4000 illumination system | Olympus | N/A |
| Olympus IX73 | Olympus | N/A |
| Orbitrap Fusion Lumos Tribrid mass spectrometer | Thermo | Cat# IQLAAEGAAPFADBMBHQ |
| SteREO Discovery.V12 | Zeiss | N/A |
| 4D Nucleofector Core Unit | Lonza | Cat# AAF-1002B |
| 4D Nucleofector X Unit | Lonza | Cat# AAF-1002X |
Acknowledgements
The authors thank Katrin Köhn, Aref Saed, Ulrike Mayr-Beyrle, Juliane Nell, Rashmi Bijegatte, Claudia Ruhland, and Ralf Köhntop for excellent technical assistance, and Franz Oswald, Barbara Möpps, Johann Gout, Elodie Roger, Frank Arnold, Tim Eiseler, Florian Weeber, Karolin Walter, and Kanishka Tiwary for helpful discussions. They acknowledge Rupert Öllinger for performing RNA-seq experiments and Olga Baranov and Sebastian Lange for assisting with lcWGS analysis. We also want to thank Ivan Costa and Julia Gehrmann for providing input on RNA-seq data evaluation and Ralf Marienfeld for performing panel sequencing. They also thank Paul Walther and the Electron Microscopy Group, and Inken Beck and the team of the “Tierforschungszentrum” at Ulm University. The authors acknowledge O.D. Madsen and the Developmental Studies Hybridoma Bank (DSHB) for providing the NKX6-1 antibody.
Main funding is provided by the Deutsche Forschungsgemeinschaft (DFG) “Sachbeihilfe” KL 2544/7-1 and “Heisenberg-Programm” KL 2544/6-1 as well as the Baden-Württemberg-Foundation ExPoChip. A.K. and T.S. are PIs in the HEIST RTG funded by the DFG GRK 2254/1. Additional funding comes from the German Cancer Aid grant to A.K. (111879), the DFG (K.L. 2544/1-1 and 1-2 and 5-1), the INDIMED-Verbund PancChip, and the Else-Kröner-Fresenius Excellence funding to A.K., L.P., M.H., S.H. received supportive funds by the Bausteinprogramm of Ulm University. P.G.R. was supported by the DIR of the NIDCR, a part of the IRP, NIH, DHHS. M.R. was supported by the German Cancer Aid (Max Eder Program, Deutsche Krebshilfe 111273, MR) and the German Research Foundation (DFG, SFB1321 Modeling and Targeting Pancreatic Cancer INST 95/1470-1 and INST 95/1461-1 as well as RE 3723/4-1). C.M.C. and S.S. received supportive funds by de German Research Foundation (DFG, SFB TRR 127 Biology of Xenogeneic Cell and Organ Transplantation) and the Paul Langerhans Institute Dresden (PLID) of Helmholtz Zentrum München at the University Clinic Carl Gustav Carus of Technische Universität Dresden.
Footnotes
Declaration of Interests: The authors declare no competing interests.
Literature:
- AIELLO NM, MADDIPATI R, NORGARD RJ, BALLI D, LI J, YUAN S, YAMAZOE T, BLACK T, SAHMOUD A, FURTH EE, BAR-SAGI D & STANGER BZ 2018. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev Cell, 45, 681–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ANSIEAU S, BASTID J, DOREAU A, MOREL AP, BOUCHET BP, THOMAS C, FAUVET F, PUISIEUX I, DOGLIONI C, PICCININ S, MAESTRO R, VOELTZEL T, SELMI A, VALSESIA-WITTMANN S, CARON DE FROMENTEL C & PUISIEUX A 2008. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell, 14, 79–89. [DOI] [PubMed] [Google Scholar]
- BARON M, VERES A, WOLOCK SL, FAUST AL, GAUJOUX R, VETERE A, RYU JH, WAGNER BK, SHEN-ORR SS, KLEIN AM, MELTON DA & YANAI I 2016. A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-cell Population Structure. Cell Syst, 3, 346–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARTEK J, BARTKOVA J & LUKAS J 2007. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene, 26, 7773–9. [DOI] [PubMed] [Google Scholar]
- BENJAMINI Y & HOCHBERG Y 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal statistical society: series B (Methodological), 57, 289–300. [Google Scholar]
- BIANCO P, KUZNETSOV SA, RIMINUCCI M, FISHER LW, SPIEGEL AM & ROBEY PG 1998. Reproduction of human fibrous dysplasia of bone in immunocompromised mice by transplanted mosaics of normal and Gsalpha-mutated skeletal progenitor cells. The Journal of clinical investigation, 101, 1737–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BIEDERSTÄDT A, HASSAN Z, SCHNEEWEIS C, SCHICK M, SCHNEIDER L, MUCKENHUBER A, HONG Y, SIEGERS G, NILSSON L, WIRTH M, DANTES Z, STEIGER K, SCHUNCK K, LANGSTON S, LENHOF HP, COLUCCIO A, ORBEN F, SLAWSKA J, SCHERGER A, SAUR D, MÜLLER S, RAD R, WEICHERT W, NILSSON J, REICHERT M, SCHNEIDER G & KELLER U 2020. SUMO pathway inhibition targets an aggressive pancreatic cancer subtype. Gut, 69, 1472–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BLONDEL VD, GUILLAUME J-L, LAMBIOTTE R & LEFEBVRE E 2008. Fast unfolding of communities in large networks. Journal of statistical mechanics: theory and experiment, 2008, P10008. [Google Scholar]
- BOJ SF, HWANG CI, BAKER LA, CHIO II, ENGLE DD, CORBO V, JAGER M, PONZ-SARVISE M, TIRIAC H, SPECTOR MS, GRACANIN A, ONI T, YU KH, VAN BOXTEL R, HUCH M, RIVERA KD, WILSON JP, FEIGIN ME, OHLUND D, HANDLY-SANTANA A, ARDITO-ABRAHAM CM, LUDWIG M, ELYADA E, ALAGESAN B, BIFFI G, YORDANOV GN, DELCUZE B, CREIGHTON B, WRIGHT K, PARK Y, MORSINK FH, MOLENAAR IQ, BOREL RINKES IH, CUPPEN E, HAO Y, JIN Y, NIJMAN IJ, IACOBUZIO-DONAHUE C, LEACH SD, PAPPIN DJ, HAMMELL M, KLIMSTRA DS, BASTURK O, HRUBAN RH, OFFERHAUS GJ, VRIES RG, CLEVERS H & TUVESON DA 2015. Organoid models of human and mouse ductal pancreatic cancer. Cell, 160, 324–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOLGER AM, LOHSE M & USADEL B 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRION LP, SCHWARTZ JH, ZAVILOWITZ BJ & SCHWARTZ GJ 1988. Micro-method for the measurement of carbonic anhydrase activity in cellular homogenates. Analytical biochemistry, 175, 289–297. [DOI] [PubMed] [Google Scholar]
- BURGHARDT B, ELKAER ML, KWON TH, RACZ GZ, VARGA G, STEWARD MC & NIELSEN S 2003. Distribution of aquaporin water channels AQP1 and AQP5 in the ductal system of the human pancreas. Gut, 52, 1008–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CALDWELL ME, DENICOLA GM, MARTINS CP, JACOBETZ MA, MAITRA A, HRUBAN RH & TUVESON DA 2012. Cellular features of senescence during the evolution of human and murine ductal pancreatic cancer. Oncogene, 31, 1599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAN-SENG-YUE M, KIM JC, WILSON GW, NG K, FIGUEROA EF, O’KANE GM, CONNOR AA, DENROCHE RE, GRANT RC, MCLEOD J, WILSON JM, JANG GH, ZHANG A, DODD A, LIANG SB, BORGIDA A, CHADWICK D, KALIMUTHU S, LUNGU I, BARTLETT JMS, KRZYZANOWSKI PM, SANDHU V, TIRIAC H, FROELING FEM, KARASINSKA JM, TOPHAM JT, RENOUF DJ, SCHAEFFER DF, JONES SJM, MARRA MA, LASKIN J, CHETTY R, STEIN LD, ZOGOPOULOS G, HAIBE-KAINS B, CAMPBELL PJ, TUVESON DA, KNOX JJ, FISCHER SE, GALLINGER S & NOTTA F 2020. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat Genet, 52, 231–240. [DOI] [PubMed] [Google Scholar]
- CHEN Z, TROTMAN LC, SHAFFER D, LIN HK, DOTAN ZA, NIKI M, KOUTCHER JA, SCHER HI, LUDWIG T, GERALD W, CORDON-CARDO C & PANDOLFI PP 2005. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature, 436, 725–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHMELOVA H, COHRS CM, CHOUINARD JA, PETZOLD C, KUHN M, CHEN C, ROEDER I, KRETSCHMER K & SPEIER S 2015. Distinct roles of β-cell mass and function during type 1 diabetes onset and remission. Diabetes, 64, 2148–2160. [DOI] [PubMed] [Google Scholar]
- CHO CH, HANNAN NR, DOCHERTY FM, DOCHERTY HM, JOAO LIMA M, TROTTER MW, DOCHERTY K & VALLIER L 2012. Inhibition of activin/nodal signalling is necessary for pancreatic differentiation of human pluripotent stem cells. Diabetologia, 55, 3284–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COGGER KF, SINHA A, SARANGI F, MCGAUGH EC, SAUNDERS D, DORRELL C, MEJIA-GUERRERO S, AGHAZADEH Y, ROURKE JL & SCREATON RA 2017. Glycoprotein 2 is a specific cell surface marker of human pancreatic progenitors. Nature Communications, 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COHRS CM, CHEN C & SPEIER S 2020. Transplantation of Islets of Langerhans into the Anterior Chamber of the Eye for Longitudinal In Vivo Imaging. Methods Mol Biol, 2128, 149–157. [DOI] [PubMed] [Google Scholar]
- COX J & MANN M 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol, 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- CZODROWSKI P, MALLINGER A, WIENKE D, ESDAR C, PÖSCHKE O, BUSCH M, ROHDICH F, ECCLES SA, ORTIZ-RUIZ MJ, SCHNEIDER R, RAYNAUD FI, CLARKE PA, MUSIL D, SCHWARZ D, DALE T, URBAHNS K, BLAGG J & SCHIEMANN K 2016. Structure-Based Optimization of Potent, Selective, and Orally Bioavailable CDK8 Inhibitors Discovered by High-Throughput Screening. J Med Chem, 59, 9337–9349. [DOI] [PubMed] [Google Scholar]
- DANTES Z, YEN HY, PFARR N, WINTER C, STEIGER K, MUCKENHUBER A, HENNIG A, LANGE S, ENGLEITNER T, ÖLLINGER R, MARESCH R, ORBEN F, HEID I, KAISSIS G, SHI K, TOPPING G, STÖGBAUER F, WIRTH M, PESCHKE K, PAPARGYRIOU A, REZAEE-OGHAZI M, FELDMANN K, SCHÄFER AP, RANJAN R, LUBESEDER-MARTELLATO C, STANGE DE, WELSCH T, MARTIGNONI M, CEYHAN GO, FRIESS H, HERNER A, LIOTTA L, TREIBER M, VON FIGURA G, ABDELHAFEZ M, KLARE P, SCHLAG C, ALGÜL H, SIVEKE J, BRAREN R, WEIRICH G, WEICHERT W, SAUR D, RAD R, SCHMID RM, SCHNEIDER G & REICHERT M 2020. Implementing cell-free DNA of pancreatic cancer patient-derived organoids for personalized oncology. JCI Insight, 5, e137809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE LICHTENBERG KH, SEYMOUR PA, JØRGENSEN MC, KIM Y-H, GRAPIN-BOTTON A, MAGNUSON MA, NAKIC N, FERRER J & SERUP P 2018. Notch Controls Multiple Pancreatic Cell Fate Regulators Through Direct Hes1-mediated Repression. Available: https://www.biorxiv.org/content/10.1101/336305v1.full [Accessed 01/22/2021].
- DEKKERS JF, WIEGERINCK CL, DE JONGE HR, BRONSVELD I, JANSSENS HM, DE WINTER-DE GROOT KM, BRANDSMA AM, DE JONG NW, BIJVELDS MJ, SCHOLTE BJ, NIEUWENHUIS EE, VAN DEN BRINK S, CLEVERS H, VAN DER ENT CK, MIDDENDORP S & BEEKMAN JM 2013. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nature Medicine, 19, 939–45. [DOI] [PubMed] [Google Scholar]
- DI MICCO R, FUMAGALLI M, CICALESE A, PICCININ S, GASPARINI P, LUISE C, SCHURRA C, GARRE M, NUCIFORO PG, BENSIMON A, MAESTRO R, PELICCI PG & D’ADDA DI FAGAGNA F 2006. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature, 444, 638–42. [DOI] [PubMed] [Google Scholar]
- DING Q, REGAN SN, XIA Y, OOSTROM LA, COWAN CA & MUSUNURU K 2013. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell stem cell, 12, 393–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOBIN A, DAVIS CA, SCHLESINGER F, DRENKOW J, ZALESKI C, JHA S, BATUT P, CHAISSON M & GINGERAS TR 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENGE M, ARDA HE, MIGNARDI M, BEAUSANG J, BOTTINO R, KIM SK & QUAKE SR 2017. Single-Cell Analysis of Human Pancreas Reveals Transcriptional Signatures of Aging and Somatic Mutation Patterns. Cell, 171, 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FERREIRA RMM, SANCHO R, MESSAL HA, NYE E, SPENCER-DENE B, STONE RK, STAMP G, ROSEWELL I, QUAGLIA A & BEHRENS A 2017. Duct- and Acinar-Derived Pancreatic Ductal Adenocarcinomas Show Distinct Tumor Progression and Marker Expression. Cell Rep, 21, 966–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FREJNO M, ZENEZINI CHIOZZI R, WILHELM M, KOCH H, ZHENG R, KLAEGER S, RUPRECHT B, MENG C, KRAMER K, JARZAB A, HEINZLMEIR S, JOHNSTONE E, DOMINGO E, KERR D, JESINGHAUS M, SLOTTA-HUSPENINA J, WEICHERT W, KNAPP S, FELLER SM & KUSTER B 2017. Pharmacoproteomic characterisation of human colon and rectal cancer. Mol Syst Biol, 13, 951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRISHBERG A, PESHES-YALOZ N, COHN O, ROSENTUL D, STEUERMAN Y, VALADARSKY L, YANKOVITZ G, MANDELBOIM M, IRAQI FA, AMIT I, MAYO L, BACHARACH E & GAT-VIKS I 2019. Cell composition analysis of bulk genomics using single-cell data. Nat Methods, 16, 327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRYER CJ, WHITE JB & JONES KA 2004. Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol Cell, 16, 509–20. [DOI] [PubMed] [Google Scholar]
- FURUKAWA T, KLOPPEL G, VOLKAN ADSAY N, ALBORES-SAAVEDRA J, FUKUSHIMA N, HORII A, HRUBAN RH, KATO Y, KLIMSTRA DS, LONGNECKER DS, LUTTGES J, OFFERHAUS GJ, SHIMIZU M, SUNAMURA M, SURIAWINATA A, TAKAORI K & YONEZAWA S 2005. Classification of types of intraductal papillary-mucinous neoplasm of the pancreas: a consensus study. Virchows Arch, 447, 794–9. [DOI] [PubMed] [Google Scholar]
- GAUJOUX S, SALENAVE S, RONOT M, RANGHEARD AS, CROS J, BELGHITI J, SAUVANET A, RUSZNIEWSKI P & CHANSON P 2014. Hepatobiliary and Pancreatic neoplasms in patients with McCune-Albright syndrome. J Clin Endocrinol Metab, 99, E97–101. [DOI] [PubMed] [Google Scholar]
- GEORGAKOPOULOS N, PRIOR N, ANGRES B, MASTROGIOVANNI G, CAGAN A, HARRISON D, HINDLEY CJ, ARNES-BENITO R, LIAU SS, CURD A, IVORY N, SIMONS BD, MARTINCORENA I, WURST H, SAEB-PARSY K & HUCH M 2020. Long-term expansion, genomic stability and in vivo safety of adult human pancreas organoids. BMC Dev Biol, 20, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GERRARD DT, BERRY AA, JENNINGS RE, PIPER HANLEY K, BOBOLA N & HANLEY NA 2016. An integrative transcriptomic atlas of organogenesis in human embryos. Elife, 5, e15657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GLIWICZ D, JANKOWSKA I, WIERZBICKA A, MISKIEWICZ-CHOTNICKA A, LISOWSKA A & WALKOWIAK J 2016. Exocrine pancreatic function in children with Alagille syndrome. Sci Rep, 6, 35229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GLOECKNER CJ, BOLDT K, SCHUMACHER A & UEFFING M 2009. Tandem affinity purification of protein complexes from mammalian cells by the Strep/FLAG (SF)-TAP tag. Methods Mol Biol, 564, 359–72. [DOI] [PubMed] [Google Scholar]
- GOLSON ML, LOOMES KM, OAKEY R & KAESTNER KH 2009. Ductal malformation and pancreatitis in mice caused by conditional Jag1 deletion. Gastroenterology, 136, 1761–1771. [DOI] [PubMed] [Google Scholar]
- GRIGORE AD, JOLLY MK, JIA D, FARACH-CARSON MC & LEVINE H 2016. Tumor Budding: The Name is EMT. Partial EMT. J Clin Med, 5, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAEUSSLER M, SCHONIG K, ECKERT H, ESCHSTRUTH A, MIANNE J, RENAUD JB, SCHNEIDER-MAUNOURY S, SHKUMATAVA A, TEBOUL L, KENT J, JOLY JS & CONCORDET JP 2016. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol, 17, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HENG L 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM [Online]. arXiv: 1303.3997. Available: https://arxiv.org/abs/1303.3997 [Accessed 01/22/2021].
- HOGREBE NJ, AUGSORNWORAWAT P, MAXWELL KG, VELAZCO-CRUZ L & MILLMAN JR 2020. Targeting the cytoskeleton to direct pancreatic differentiation of human pluripotent stem cells. Nat Biotechnol, 38, 460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOHWIELER M, ILLING A, HERMANN PC, MAYER T, STOCKMANN M, PERKHOFER L, EISELER T, ANTONY JS, MULLER M, RENZ S, KUO CC, LIN Q, SENDLER M, BREUNIG M, KLEIDERMAN SM, LECHEL A, ZENKER M, LEICHSENRING M, ROSENDAHL J, ZENKE M, SAINZ B JR., MAYERLE J, COSTA IG, SEUFFERLEIN T, KORMANN M, WAGNER M, LIEBAU S & KLEGER A 2017. Human pluripotent stem cell-derived acinar/ductal organoids generate human pancreas upon orthotopic transplantation and allow disease modelling. Gut, 66, 473–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOHWIELER M, MULLER M, FRAPPART PO & HELLER S 2019. Pancreatic Progenitors and Organoids as a Prerequisite to Model Pancreatic Diseases and Cancer. Stem Cells Int, 2019, 9301382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IDENO N, YAMAGUCHI H, GHOSH B, GUPTA S, OKUMURA T, STEFFEN DJ, FISHER CG, WOOD LD, SINGHI AD, NAKAMURA M, GUTKIND JS & MAITRA A 2018. GNAS(R201C) Induces Pancreatic Cystic Neoplasms in Mice That Express Activated KRAS by Inhibiting YAP1 Signaling. Gastroenterology, 155, 1593–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ILLING A, STOCKMANN M, SWAMY TELUGU N, LINTA L, RUSSELL R, MULLER M, SEUFFERLEIN T, LIEBAU S & KLEGER A 2013. Definitive Endoderm Formation from Plucked Human Hair-Derived Induced Pluripotent Stem Cells and SK Channel Regulation. Stem Cells International, 2013, 360573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ITO K, MATSUURA K, MIHARA Y, SAKAMOTO Y, HASEGAWA K, KOKUDO N & SHIMIZU T 2019. Delivery of pancreatic digestive enzymes into the gastrointestinal tract by pancreatic exocrine tissue transplant. Sci Rep, 9, 5922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOHNSON WE, LI C & RABINOVIC A 2007. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics, 8, 118–127. [DOI] [PubMed] [Google Scholar]
- KESAVAN G, SAND FW, GREINER TU, JOHANSSON JK, KOBBERUP S, WU X, BRAKEBUSCH C & SEMB H 2009. Cdc42-mediated tubulogenesis controls cell specification. Cell, 139, 791–801. [DOI] [PubMed] [Google Scholar]
- KIM SI, OCEGUERA-YANEZ F, SAKURAI C, NAKAGAWA M, YAMANAKA S & WOLTJEN K 2016. Inducible Transgene Expression in Human iPS Cells Using Versatile All-in-One piggyBac Transposons. Methods Mol Biol, 1357, 111–31. [DOI] [PubMed] [Google Scholar]
- KLAUSEN P, KOVACEVIC B, TOXVAERD A, KALAITZAKIS E, KARSTENSEN JG, RIFT CV, HANSEN CP, STORKHOLM J, VILMANN P & HASSELBY JP 2019. Subtyping of intraductal papillary mucinous neoplasms - pitfalls of MUC1 immunohistochemistry. Apmis, 127, 27–32. [DOI] [PubMed] [Google Scholar]
- KOPP JL, DUBOIS CL, SCHAEFFER DF, SAMANI A, TAGHIZADEH F, COWAN RW, RHIM AD, STILES BL, VALASEK M & SANDER M 2018. Loss of Pten and Activation of Kras Synergistically Induce Formation of Intraductal Papillary Mucinous Neoplasia From Pancreatic Ductal Cells in Mice. Gastroenterology, 154, 1509–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRENTZ NAJ, LEE MYY, XU EE, SPROUL SLJ, MASLOVA A, SASAKI S & LYNN FC 2018. Single-Cell Transcriptome Profiling of Mouse and hESC-Derived Pancreatic Progenitors. Stem Cell Reports, 11, 1551–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUILMAN T 2020. CopywriteR: Copy number information from targeted sequencing using off-target reads [Online]. GitHub. Available: https://github.com/PeeperLab/CopywriteR [Accessed 01/22/2021]. [Google Scholar]
- KUILMAN T, MICHALOGLOU C, VREDEVELD LC, DOUMA S, VAN DOORN R, DESMET CJ, AARDEN LA, MOOI WJ & PEEPER DS 2008. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell, 133, 1019–31. [DOI] [PubMed] [Google Scholar]
- KULESHOV MV, JONES MR, ROUILLARD AD, FERNANDEZ NF, DUAN Q, WANG Z, KOPLEV S, JENKINS SL, JAGODNIK KM & LACHMANN A 2016. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic acids research, 44, W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE AYL, DUBOIS CL, SARAI K, ZAREI S, SCHAEFFER DF, SANDER M & KOPP JL 2018. Cell of origin affects tumour development and phenotype in pancreatic ductal adenocarcinoma. Gut, 68, 487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE S & SCHMITT CA 2019. The dynamic nature of senescence in cancer. Nat Cell Biol, 21, 94–101. [DOI] [PubMed] [Google Scholar]
- LEEK JT, JOHNSON WE, PARKER HS, JAFFE AE & STOREY JD 2012. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics, 28, 882–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LESINA M, WORMANN SM, MORTON J, DIAKOPOULOS KN, KORNEEVA O, WIMMER M, EINWACHTER H, SPERVESLAGE J, DEMIR IE, KEHL T, SAUR D, SIPOS B, HEIKENWALDER M, STEINER JM, WANG TC, SANSOM OJ, SCHMID RM & ALGUL H 2016. RelA regulates CXCL1/CXCR2-dependent oncogene-induced senescence in murine Kras-driven pancreatic carcinogenesis. J Clin Invest, 126, 2919–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEVINE JH, SIMONDS EF, BENDALL SC, DAVIS KL, AMIR EL AD, TADMOR MD, LITVIN O, FIENBERG HG, JAGER A, ZUNDER ER, FINCK R, GEDMAN AL, RADTKE I, DOWNING JR, PE’ER D & NOLAN GP 2015. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell, 162, 184–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIAO Y, SMYTH GK & SHI W 2019. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Research, 47, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LINTA L, STOCKMANN M, KLEINHANS KN, BOCKERS A, STORCH A, ZAEHRES H, LIN Q, BARBI G, BOCKERS TM, KLEGER A & LIEBAU S 2012. Rat embryonic fibroblasts improve reprogramming of human keratinocytes into induced pluripotent stem cells. Stem Cells Dev, 21, 965–76. [DOI] [PubMed] [Google Scholar]
- LOVE MI, HUBER W & ANDERS S 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology, 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUN AT, MCCARTHY DJ & MARIONI JC 2016. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Res, 5, 2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MACOSKO EZ, BASU A, SATIJA R, NEMESH J, SHEKHAR K, GOLDMAN M, TIROSH I, BIALAS AR, KAMITAKI N & MARTERSTECK EM 2015. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell, 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MALÉTH J, BALÁZS A, PALLAGI P, BALLA Z, KUI B, KATONA M, JUDÁK L, NÉMETH I, KEMÉNY LV, RAKONCZAY Z JR., VENGLOVECZ V, FÖLDESI I, PETŐ Z, SOMORÁCZ Á, BORKA K, PERDOMO D, LUKACS GL, GRAY MA, MONTERISI S, ZACCOLO M, SENDLER M, MAYERLE J, KÜHN JP, LERCH MM, SAHIN-TÓTH M & HEGYI P 2015. Alcohol disrupts levels and function of the cystic fibrosis transmembrane conductance regulator to promote development of pancreatitis. Gastroenterology, 148, 427–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MALI P, YANG L, ESVELT KM, AACH J, GUELL M, DICARLO JE, NORVILLE JE & CHURCH GM 2013. RNA-guided human genome engineering via Cas9. Science, 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCINNES L, HEALY J & MELVILLE J 2018. Umap: Uniform manifold approximation and projection for dimension reduction [Online]. arXiv:1802.03426. Available: https://arxiv.org/abs/1802.03426 [Accessed 01/22/2021].
- MOFFITT RA, MARAYATI R, FLATE EL, VOLMAR KE, LOEZA SG, HOADLEY KA, RASHID NU, WILLIAMS LA, EATON SC, CHUNG AH, SMYLA JK, ANDERSON JM, KIM HJ, BENTREM DJ, TALAMONTI MS, IACOBUZIO-DONAHUE CA, HOLLINGSWORTH MA & YEH JJ 2015. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet, 47, 1168–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOLNÁR R, MADÁCSY T, VARGA Á, NÉMETH M, KATONA X, GÖRÖG M, MOLNÁR B, FANCZAL J, RAKONCZAY Z & HEGYI P 2020. Mouse pancreatic ductal organoid culture as a relevant model to study exocrine pancreatic ion secretion. Laboratory Investigation, 100, 84–97. [DOI] [PubMed] [Google Scholar]
- MOREIRA L, BAKIR B, CHATTERJI P, DANTES Z, REICHERT M & RUSTGI AK 2018. Pancreas 3D Organoids: Current and Future Aspects as a Research Platform for Personalized Medicine in Pancreatic Cancer. Cell Mol Gastroenterol Hepatol, 5, 289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORTON JP, TIMPSON P, KARIM SA, RIDGWAY RA, ATHINEOS D, DOYLE B, JAMIESON NB, OIEN KA, LOWY AM, BRUNTON VG, FRAME MC, EVANS TR & SANSOM OJ 2010. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci U S A, 107, 246–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MUELLER S, ENGLEITNER T, MARESCH R, ZUKOWSKA M, LANGE S, KALTENBACHER T, KONUKIEWITZ B, OLLINGER R, ZWIEBEL M, STRONG A, YEN HY, BANERJEE R, LOUZADA S, FU B, SEIDLER B, GOTZFRIED J, SCHUCK K, HASSAN Z, ARBEITER A, SCHONHUBER N, KLEIN S, VELTKAMP C, FRIEDRICH M, RAD L, BARENBOIM M, ZIEGENHAIN C, HESS J, DOVEY OM, ESER S, PAREKH S, CONSTANTINO-CASAS F, DE LA ROSA J, SIERRA MI, FRAGA M, MAYERLE J, KLOPPEL G, CADINANOS J, LIU P, VASSILIOU G, WEICHERT W, STEIGER K, ENARD W, SCHMID RM, YANG F, UNGER K, SCHNEIDER G, VARELA I, BRADLEY A, SAUR D & RAD R 2018. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature, 554, 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NOSTRO MC, SARANGI F, YANG C, HOLLAND A, ELEFANTY AG, STANLEY EG, GREINER DL & KELLER G 2015. Efficient generation of NKX6-1+ pancreatic progenitors from multiple human pluripotent stem cell lines. Stem Cell Reports, 4, 591–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NOTTA F, CHAN-SENG-YUE M, LEMIRE M, LI Y, WILSON GW, CONNOR AA, DENROCHE RE, LIANG SB, BROWN AM, KIM JC, WANG T, SIMPSON JT, BECK T, BORGIDA A, BUCHNER N, CHADWICK D, HAFEZI-BAKHTIARI S, DICK JE, HEISLER L, HOLLINGSWORTH MA, IBRAHIMOV E, JANG GH, JOHNS J, JORGENSEN LG, LAW C, LUDKOVSKI O, LUNGU I, NG K, PASTERNACK D, PETERSEN GM, SHLUSH LI, TIMMS L, TSAO MS, WILSON JM, YUNG CK, ZOGOPOULOS G, BARTLETT JM, ALEXANDROV LB, REAL FX, CLEARY SP, ROEHRL MH, MCPHERSON JD, STEIN LD, HUDSON TJ, CAMPBELL PJ & GALLINGER S 2016. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature, 538, 378–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OHASHI S, NATSUIZAKA M, WONG GS, MICHAYLIRA CZ, GRUGAN KD, STAIRS DB, KALABIS J, VEGA ME, KALMAN RA, NAKAGAWA M, KLEIN-SZANTO AJ, HERLYN M, DIEHL JA, RUSTGI AK & NAKAGAWA H 2010. Epidermal growth factor receptor and mutant p53 expand an esophageal cellular subpopulation capable of epithelial-to-mesenchymal transition through ZEB transcription factors. Cancer Res, 70, 4174–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAREKH S, ZIEGENHAIN C, VIETH B, ENARD W & HELLMANN I 2016. The impact of amplification on differential expression analyses by RNA-seq. Scientific reports, 6, 25533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PATRA KC, BARDEESY N & MIZUKAMI Y 2017. Diversity of Precursor Lesions For Pancreatic Cancer: The Genetics and Biology of Intraductal Papillary Mucinous Neoplasm. Clin Transl Gastroenterol, 8, e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PATRA KC, KATO Y, MIZUKAMI Y, WIDHOLZ S, BOUKHALI M, REVENCO I, GROSSMAN EA, JI F, SADREYEV RI, LISS AS, SCREATON RA, SAKAMOTO K, RYAN DP, MINO-KENUDSON M, CASTILLO CF, NOMURA DK, HAAS W & BARDEESY N 2018. Mutant GNAS drives pancreatic tumourigenesis by inducing PKA-mediated SIK suppression and reprogramming lipid metabolism. Nat Cell Biol, 20, 811–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PEREZ-RIVEROL Y, CSORDAS A, BAI J, BERNAL-LLINARES M, HEWAPATHIRANA S, KUNDU DJ, INUGANTI A, GRISS J, MAYER G, EISENACHER M, PEREZ E, USZKOREIT J, PFEUFFER J, SACHSENBERG T, YILMAZ S, TIWARY S, COX J, AUDAIN E, WALZER M, JARNUCZAK AF, TERNENT T, BRAZMA A & VIZCAINO JA 2019. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res, 47, D442–d450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POPLIN R, RUANO-RUBIO V, DEPRISTO MA, FENNELL TJ, CARNEIRO MO, VAN DER AUWERA GA, KLING DE, GAUTHIER LD, LEVY-MOONSHINE A, ROAZEN D, SHAKIR K, THIBAULT J, CHANDRAN S, WHELAN C, LEK M, GABRIEL S, DALY MJ, NEALE B, MACARTHUR DG & BANKS E 2018. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv [Online]. Available: https://www.biorxiv.org/content/biorxiv/early/2018/07/24/201178.full.pdf [Accessed 01/22/2021]. [Google Scholar]
- PULEO F, NICOLLE R, BLUM Y, CROS J, MARISA L, DEMETTER P, QUERTINMONT E, SVRCEK M, ELAROUCI N, IOVANNA J, FRANCHIMONT D, VERSET L, GALDON MG, DEVIÈRE J, DE REYNIÈS A, LAURENT-PUIG P, VAN LAETHEM JL, BACHET JB & MARÉCHAL R 2018. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology, 155, 1999–2013. [DOI] [PubMed] [Google Scholar]
- QADIR MMF, ÁLVAREZ-CUBELA S, KLEIN D, VAN DIJK J, MUÑIZ-ANQUELA R, MORENO-HERNÁNDEZ YB, LANZONI G, SADIQ S, NAVARRO-RUBIO B, GARCÍA MT, DÍAZ Á, JOHNSON K, SANT D, RICORDI C, GRISWOLD A, PASTORI RL & DOMÍNGUEZ-BENDALA J 2020. Single-cell resolution analysis of the human pancreatic ductal progenitor cell niche. Proc Natl Acad Sci U S A, 117, 10876–10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAHIB L, SMITH BD, AIZENBERG R, ROSENZWEIG AB, FLESHMAN JM & MATRISIAN LM 2014. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res, 74, 2913–21. [DOI] [PubMed] [Google Scholar]
- RAO J, PFEIFFER MJ, FRANK S, ADACHI K, PICCINI I, QUARANTA R, ARAÚZO-BRAVO M, SCHWARZ J, SCHADE D & LEIDEL S 2016. Stepwise clearance of repressive roadblocks drives cardiac induction in human ESCs. Cell stem cell, 18, 341–353. [DOI] [PubMed] [Google Scholar]
- REICHERT M, BLUME K, KLEGER A, HARTMANN D & VON FIGURA G 2016. Developmental Pathways Direct Pancreatic Cancer Initiation from Its Cellular Origin. Stem Cells Int, 2016, 9298535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REZANIA A, BRUIN JE, RIEDEL MJ, MOJIBIAN M, ASADI A, XU J, GAUVIN R, NARAYAN K, KARANU F, O’NEIL JJ, AO Z, WARNOCK GL & KIEFFER TJ 2012. Maturation of human embryonic stem cell-derived pancreatic progenitors into functional islets capable of treating pre-existing diabetes in mice. Diabetes, 61, 2016–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RHODES JA, CRISCIMANNA A & ESNI F 2012. Induction of mouse pancreatic ductal differentiation, an in vitro assay. In Vitro Cell Dev Biol Anim, 48, 641–9. [DOI] [PubMed] [Google Scholar]
- RITCHIE ME, PHIPSON B, WU D, HU Y, LAW CW, SHI W & SMYTH GK 2015. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res, 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROBERTS NJ, NORRIS AL, PETERSEN GM, BONDY ML, BRAND R, GALLINGER S, KURTZ RC, OLSON SH, RUSTGI AK, SCHWARTZ AG, STOFFEL E, SYNGAL S, ZOGOPOULOS G, ALI SZ, AXILBUND J, CHAFFEE KG, CHEN YC, COTE ML, CHILDS EJ, DOUVILLE C, GOES FS, HERMAN JM, IACOBUZIO-DONAHUE C, KRAMER M, MAKOHON-MOORE A, MCCOMBIE RW, MCMAHON KW, NIKNAFS N, PARLA J, PIROOZNIA M, POTASH JB, RHIM AD, SMITH AL, WANG Y, WOLFGANG CL, WOOD LD, ZANDI PP, GOGGINS M, KARCHIN R, ESHLEMAN JR, PAPADOPOULOS N, KINZLER KW, VOGELSTEIN B, HRUBAN RH & KLEIN AP 2016. Whole Genome Sequencing Defines the Genetic Heterogeneity of Familial Pancreatic Cancer. Cancer Discov, 6, 166–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROBINSON C, ESTRADA A, ZAHEER A, SINGH VK, WOLFGANG CL, GOGGINS MG, HRUBAN RH, WOOD LD, NOE M, MONTGOMERY EA, GUTHRIE LC, LENNON AM, BOYCE AM & COLLINS MT 2018. Clinical and Radiographic Gastrointestinal Abnormalities in McCune-Albright Syndrome. J Clin Endocrinol Metab, 103, 4293–4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROBINSON JT, THORVALDSDÓTTIR H, WINCKLER W, GUTTMAN M, LANDER ES, GETZ G & MESIROV JP 2011. Integrative genomics viewer. Nat Biotechnol, 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RODANSKY ES, JOHNSON LA, HUANG S, SPENCE JR & HIGGINS PD 2015. Intestinal organoids: a model of intestinal fibrosis for evaluating anti-fibrotic drugs. Exp Mol Pathol, 98, 346–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROSENBAUM DM, RASMUSSEN SG & KOBILKA BK 2009. The structure and function of G-protein-coupled receptors. Nature, 459, 356–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROWE RG & DALEY GQ 2019. Induced pluripotent stem cells in disease modelling and drug discovery. Nat Rev Genet, 20, 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RUBIO-VIQUEIRA B, JIMENO A, CUSATIS G, ZHANG X, IACOBUZIO-DONAHUE C, KARIKARI C, SHI C, DANENBERG K, DANENBERG PV, KURAMOCHI H, TANAKA K, SINGH S, SALIMI-MOOSAVI H, BOURAOUD N, AMADOR ML, ALTIOK S, KULESZA P, YEO C, MESSERSMITH W, ESHLEMAN J, HRUBAN RH, MAITRA A & HIDALGO M 2006. An in vivo platform for translational drug development in pancreatic cancer. Clin Cancer Res, 12, 4652–61. [DOI] [PubMed] [Google Scholar]
- SALINAS-SOUZA C, DE ANDREA C, BIHL M, KOVAC M, PILLAY N, FORSHEW T, GUTTERIDGE A, YE H, AMARY MF, TIRABOSCO R, TOLEDO SR, BAUMHOER D & FLANAGAN AM 2015. GNAS mutations are not detected in parosteal and low-grade central osteosarcomas. Mod Pathol, 28, 1336–42. [DOI] [PubMed] [Google Scholar]
- SCHAFFER AE, FREUDE KK, NELSON SB & SANDER M 2010. Nkx6 transcription factors and Ptf1a function as antagonistic lineage determinants in multipotent pancreatic progenitors. Developmental Cell, 18, 1022–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHINDELIN J, ARGANDA-CARRERAS I, FRISE E, KAYNIG V, LONGAIR M, PIETZSCH T, PREIBISCH S, RUEDEN C, SAALFELD S, SCHMID B, TINEVEZ JY, WHITE DJ, HARTENSTEIN V, ELICEIRI K, TOMANCAK P & CARDONA A 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods, 9, 676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEINO T, KAWASAKI S, SHIMOKAWA M, TAMAGAWA H, TOSHIMITSU K, FUJII M, OHTA Y, MATANO M, NANKI K, KAWASAKI K, TAKAHASHI S, SUGIMOTO S, IWASAKI E, TAKAGI J, ITOI T, KITAGO M, KITAGAWA Y, KANAI T & SATO T 2018. Human Pancreatic Tumor Organoids Reveal Loss of Stem Cell Niche Factor Dependence during Disease Progression. Cell Stem Cell, 22, 454–467. [DOI] [PubMed] [Google Scholar]
- SHAIN AH, GIACOMINI CP, MATSUKUMA K, KARIKARI CA, BASHYAM MD, HIDALGO M, MAITRA A & POLLACK JR 2012. Convergent structural alterations define SWItch/Sucrose NonFermentable (SWI/SNF) chromatin remodeler as a central tumor suppressive complex in pancreatic cancer. Proc Natl Acad Sci U S A, 109, E252–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIMSEK S, ZHOU T, ROBINSON CL, TSAI SY, CRESPO M, AMIN S, LIN X, HON J, EVANS T & CHEN S 2016. Modeling Cystic Fibrosis Using Pluripotent Stem Cell-Derived Human Pancreatic Ductal Epithelial Cells. Stem Cells Transl Med, 5, 572–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SONG J & SHI W 2018. The concomitant apoptosis and EMT underlie the fundamental functions of TGF-β. Acta Biochim Biophys Sin (Shanghai), 50, 91–97. [DOI] [PubMed] [Google Scholar]
- SPRINGER S, WANG Y, DAL MOLIN M, MASICA DL, JIAO Y, KINDE I, BLACKFORD A, RAMAN SP, WOLFGANG CL, TOMITA T, NIKNAFS N, DOUVILLE C, PTAK J, DOBBYN L, ALLEN PJ, KLIMSTRA DS, SCHATTNER MA, SCHMIDT CM, YIP-SCHNEIDER M, CUMMINGS OW, BRAND RE, ZEH HJ, SINGHI AD, SCARPA A, SALVIA R, MALLEO G, ZAMBONI G, FALCONI M, JANG JY, KIM SW, KWON W, HONG SM, SONG KB, KIM SC, SWAN N, MURPHY J, GEOGHEGAN J, BRUGGE W, FERNANDEZ-DEL CASTILLO C, MINO-KENUDSON M, SCHULICK R, EDIL BH, ADSAY V, PAULINO J, VAN HOOFT J, YACHIDA S, NARA S, HIRAOKA N, YAMAO K, HIJIOKA S, VAN DER MERWE S, GOGGINS M, CANTO MI, AHUJA N, HIROSE K, MAKARY M, WEISS MJ, CAMERON J, PITTMAN M, ESHLEMAN JR, DIAZ LA JR., PAPADOPOULOS N, KINZLER KW, KARCHIN R, HRUBAN RH, VOGELSTEIN B & LENNON AM 2015. A combination of molecular markers and clinical features improve the classification of pancreatic cysts. Gastroenterology, 149, 1501–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUBRAMANIAN A, TAMAYO P, MOOTHA VK, MUKHERJEE S, EBERT BL, GILLETTE MA, PAULOVICH A, POMEROY SL, GOLUB TR & LANDER ES 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences, 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAKEUCHI S, TAKAHASHI A, MOTOI N, YOSHIMOTO S, TAJIMA T, YAMAKOSHI K, HIRAO A, YANAGI S, FUKAMI K, ISHIKAWA Y, SONE S, HARA E & OHTANI N 2010. Intrinsic cooperation between p16INK4a and p21Waf1/Cip1 in the onset of cellular senescence and tumor suppression in vivo. Cancer Res, 70, 9381–90. [DOI] [PubMed] [Google Scholar]
- TAN MC, BASTURK O, BRANNON AR, BHANOT U, SCOTT SN, BOUVIER N, LAFEMINA J, JARNAGIN WR, BERGER MF, KLIMSTRA D & ALLEN PJ 2015. GNAS and KRAS Mutations Define Separate Progression Pathways in Intraductal Papillary Mucinous Neoplasm-Associated Carcinoma. J Am Coll Surg, 220, 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TARASOV A, VILELLA AJ, CUPPEN E, NIJMAN IJ & PRINS P 2015. Sambamba: fast processing of NGS alignment formats. Bioinformatics, 31, 2032–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TIRIAC H, BUCOBO JC, TZIMAS D, GREWEL S, LACOMB JF, ROWEHL LM, NAGULA S, WU M, KIM J, SASSON A, VIGNESH S, MARTELLO L, MUNOZ-SAGASTIBELZA M, SOMMA J, TUVESON DA, LI E & BUSCAGLIA JM 2018. Successful creation of pancreatic cancer organoids by means of EUS-guided fine-needle biopsy sampling for personalized cancer treatment. Gastrointest Endosc, 87, 1474–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRAAG V 2015. louvain-igraph: v0.5.3 [Online]. Zenodo. Available: https://zenodo.org/record/35117#.YAoEuy337BI [Accessed 01/22/2021]. [Google Scholar]
- TU Q, HAO J, ZHOU X, YAN L, DAI H, SUN B, YANG D, AN S, LV L, JIAO B, CHEN C, LAI R, SHI P & ZHAO X 2018. CDKN2B deletion is essential for pancreatic cancer development instead of unmeaningful co-deletion due to juxtaposition to CDKN2A. Oncogene, 37, 128–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VILLASENOR A, CHONG DC, HENKEMEYER M & CLEAVER O 2010. Epithelial dynamics of pancreatic branching morphogenesis. Development, 137, 4295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WARLICH E, KUEHLE J, CANTZ T, BRUGMAN MH, MAETZIG T, GALLA M, FILIPCZYK AA, HALLE S, KLUMP H & SCHÖLER HR 2011. Lentiviral vector design and imaging approaches to visualize the early stages of cellular reprogramming. Molecular Therapy, 19, 782–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILSCHANSKI M & NOVAK I 2013. The cystic fibrosis of exocrine pancreas. Cold Spring Harbor perspectives in medicine, 3, a009746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WOLF FA, ANGERER P, RAMIREZ F, VIRSHUP I, RYBAKOV S, ERASLAN G, WHITE T, LUECKEN M, CITTARO D, CALLIES T, LANGE M & MUNOZ-ROJAS AR 2020. scanpy.tl.rank_genes_groups function [Online]. GitHub. Available: https://github.com/theislab/scanpy/blob/099be48b860388cf6b8b0aa6006311f6f39de712/scanpy/tools/_rank_genes_groups.py#L416-L628 [Accessed 01/22/2021]. [Google Scholar]
- WOLF FA, ANGERER P & THEIS FJ 2018. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol, 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WOOD LD, NOË M, HACKENG W, BROSENS LA, BHAIJEE F, DEBELJAK M, YU J, SUENAGA M, SINGHI AD & ZAHEER A 2017. Patients with McCune-Albright syndrome have a broad spectrum of abnormalities in the gastrointestinal tract and pancreas. Virchows Archiv, 470, 391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WU J, MATTHAEI H, MAITRA A, DAL MOLIN M, WOOD LD, ESHLEMAN JR, GOGGINS M, CANTO MI, SCHULICK RD, EDIL BH, WOLFGANG CL, KLEIN AP, DIAZ LA JR., ALLEN PJ, SCHMIDT CM, KINZLER KW, PAPADOPOULOS N, HRUBAN RH & VOGELSTEIN B 2011. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci Transl Med, 3, 92ra66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XIANG B & MUTHUSWAMY SK 2006. Using Three-Dimensional Acinar Structures for Molecular and Cell Biological Assays. Methods in enzymology, 406, 692–701. [DOI] [PubMed] [Google Scholar]
- XIE R, EVERETT LJ, LIM HW, PATEL NA, SCHUG J, KROON E, KELLY OG, WANG A, D’AMOUR KA, ROBINS AJ, WON KJ, KAESTNER KH & SANDER M 2013. Dynamic chromatin remodeling mediated by polycomb proteins orchestrates pancreatic differentiation of human embryonic stem cells. Cell Stem Cell, 12, 224–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XIONG J, ZHAO WQ, HUANG GH, YAO LH, DONG HM, YU CH, ZHAO HJ & CAI SX 2017. [Receptor for advanced glycation end products upregulates MUC5AC expression and promotes mucus overproduction in mice with toluene diisocyanate-induced asthma]. Nan Fang Yi Ke Da Xue Xue Bao, 37, 1301–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZECHA J, SATPATHY S, KANASHOVA T, AVANESSIAN SC, KANE MH, CLAUSER KR, MERTINS P, CARR SA & KUSTER B 2019. TMT Labeling for the Masses: A Robust and Cost-efficient, In-solution Labeling Approach. Mol Cell Proteomics, 18, 1468–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZEMAN MK & CIMPRICH KA 2014. Causes and consequences of replication stress. Nat Cell Biol, 16, 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHOU Q, LAW AC, RAJAGOPAL J, ANDERSON WJ, GRAY PA & MELTON DA 2007. A multipotent progenitor domain guides pancreatic organogenesis. Dev Cell, 13, 103–14. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Live-cell imaging of CDKN2AKO/KO KRASG12D PDLOs w/o Dox stimulation
Related to Fig.5.M
* Time-lapse video of PDLO culture monitored every 3 h over 4 days. The frame rate was 5 frames per second (fps).
Live-cell imaging of CDKN2AKO/KO KRASG12D PDLOs + Dox stimulation
Related to Fig.5.M
Time-lapse video of PDLO culture monitored every 3 h over 4 days. The frame rate was 5 frames per second (fps) and the red mCherry fluorescence signal indicates KRASG12D expression.
Supplemental Table 3. Differentially expressed proteins in PDLOs vs PPs
Related to Fig.3H–N and Suppl.Fig3H–J (Results)
Limma protein expression comparison of d59 vs d13.
Supplemental Table 4. Copy number variations identified in PDAC 1, PDAC I, and III using lcWGS
CNV with a log2ratio ≥ I0.75I are depicted.
Supplemental Table 5. Point mutations identified in PDAC II using a targeted sequencing approach
Related to Fig.7 and Suppl.Fig. 7E (Results)
Point mutations identified in a comprehensive cancer panel (Qiagen). Variants marked in red are not reliable, as clarified in the “tag-defs” spreadsheet.
Reference gene sets used within this study
Related to “Gene set enrichment analysis” (STAR Methods)
Supplemental Table 1. Stage-specific RNA-seq peak expression
All genes with a P-value ≤ 0.01 and a log2FC ≥ 2 in at least one pairwise comparison were ordered according to their peak expression value.
Supplemental Table 2. Differentially expressed genes in RNA-seq analysis
Related to Fig.3A–G and Suppl.Fig3A–G (Results)
Pairwise DESeq2 expression comparisons.
Data Availability Statement
The RNA-seq data for this study have been deposited in the European Nucleotide Archive (ENA) at EMBL-EBI under accession number PRJEB38015, and the lcWGS data under PRJEB42190. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD018785 (Reviewer Account: Username: reviewer_pxd018785@ebi.ac.uk, Password: hQt1By5o). The codes supporting the current study are available from the corresponding author on request.