

Abstract

Phosphate linkages govern life as we know it. Their unique properties provide the foundation for many natural systems from cell biology and biosynthesis to the backbone of nucleic acids. Phosphates are ideal natural moieties; existing as ionized species in a stable P(V)-oxidation state, they are endowed with high stability but exhibit enzymatically unlockable potential. Despite intense interest in phosphorus catalysis and condensation chemistry, organic chemistry has not fully embraced the potential of P(V) reagents. To be sure, within the world of chemical oligonucleotide synthesis, modern approaches utilize P(III) reagent systems to create phosphate linkages and their analogs. In this Outlook, we present recent studies from our laboratories suggesting that numerous exciting opportunities for P(V) chemistry exist at the nexus of organic synthesis and biochemistry. Applications to the synthesis of stereopure antisense oligonucleotides, cyclic dinucleotides, methylphosphonates, and phosphines are reviewed as well as chemoselective modification to peptides, proteins, and nucleic acids. Finally, an outlook into what may be possible in the future with P(V) chemistry is previewed, suggesting these examples represent just the tip of the iceberg.
Short abstract
Phosphorus(V) is a key building block for life. This Outlook draws inspiration from Nature for the development of reagents that harness its unique reactivity and augments popular P(III)-based methods.
Introduction and Historical Perspective
The study of phosphate has led to a deep understanding of the chemistry that makes life on Earth possible. It is clear that phosphates and phosphate esters underpin almost all biological function including genetic information storage, energy storage, compartmentalization, lipid bilayers, and signaling. Multiple leaders in the field have published articles in which they rationalize this fact.1−4 For instance, in 1981 Todd stated, “Where there is life there is phosphorous”, in 1987 Westheimer penned the thought-provoking essay titled “Why Nature Chose Phosphates”, and more recently, in 2010 Blackburn posed and answered the question “Why did Nature select phosphorus for its dominant role in biology?”.5−7 Since this topic has been discussed by some of the leading minds in the field, this Outlook aims to take a more synthetic organic view at the dichotomy of phosphate utilization by Nature and by chemists. This difference is rather distinct and summarized through a series of case studies as outlined in Figure 1.
Figure 1.
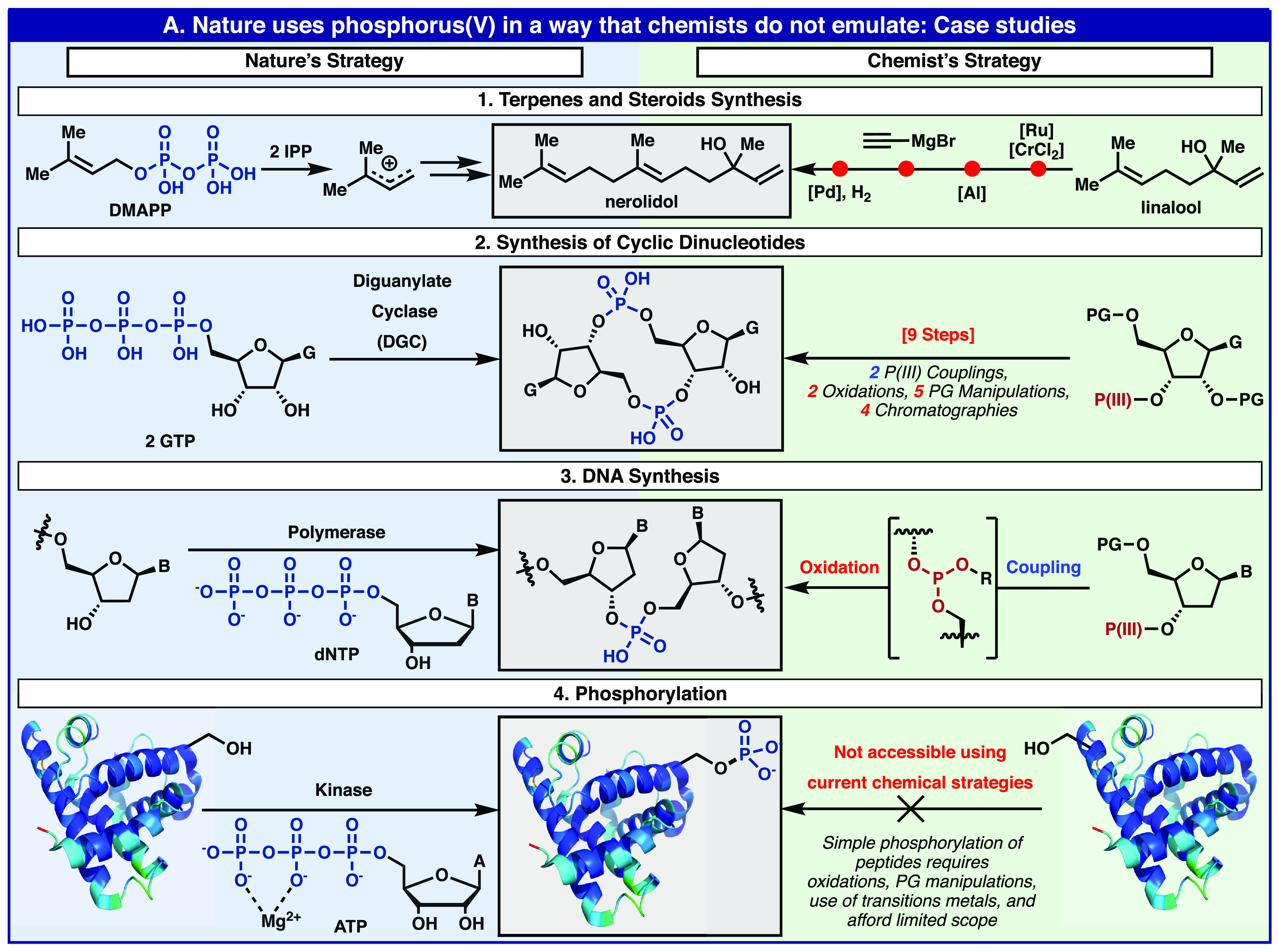
Unique uses of phosphorus in Nature as compared to synthesis.
The elementary enzymatic steps of terpene and steroid biosynthesis involve the assembly-line-like construction of polyunsaturated systems from a simple building block: dimethylallylpyrophosphate (DMAPP).8 The preorganized generation of carbocation intermediates, driven by pyrophosphate formation, allows for the controlled creation of C—C bonds that is currently outside the realm of chemical synthesis in the laboratory. In contrast, organic chemists generally use classical leaving groups and multistep sequences involving protecting group chemistry to accomplish the same overall construction.9 Cyclic dinucleotide synthesis is increasingly important in modern medicine. While Nature creates such structures by simple dimerization and macrocyclization of nucleoside triphosphate starting materials,10 the current chemical method to achieve the same goal is extremely complex in comparison (9 steps from a non-natural protected nucleoside).11 Finally, kinases have evolved to chemoselectively install phosphate groups, with some of the largest enzymatic rate enhancements yet identified,7 on natural products, peptides, and even proteins, at serine or tyrosine residues, with exquisite site selectivity.12 Currently, there is no chemical equivalent process for such reactivity. As mentioned by Westheimer, Todd, and Blackburn, there is a distinct reason that Nature chose phosphate: ionizability.5−7 To be sure, the ionizability of phosphates renders them hydrolytically stable (e.g., the half-life of the DNA phosphate backbone to background hydrolysis is ∼30 million years),13 unable to permeate a cell membrane (keeping them lodged inside a cell), and yet, under the right circumstances, still reactive. Westheimer and Todd astutely point out that no other conceivable functional group would exhibit such a perfect blend of characteristics. However, the absence of fast and controlled nonenzymatic means to activate and manipulate P(V) may hold a clue as to why chemists have long struggled to harness the utility of P(V).
The history of organophosphorus chemistry has been largely bifurcated within the synthetic organic community (Figure 2A): to those interested in synthesizing and studying molecules of life (oligonucleotides)2,14−18 and to those interested in basic organic transformations (Wittig, condensation chemistry, and catalysis).19−25 A brief timeline containing illustrative examples of this phenomenon is shown (Figure 2A). Another notable feature in the history of modern oligonucleotide synthesis is the abrupt shift from P(V)-based chemistry to the modern and ubiquitous P(III)-based methods, which have been summarized elsewhere.26 In 1955, the first oligonucleotide was synthesized by Todd through the use of P(V) phosphorochloridates.2 This groundbreaking work set the stage for Khorana’s seminal contributions, including “on/off” protecting groups which allowed for the synthesis of the first oligonucleotides of significant length and the synthesis of a 72mer tRNA.16,27−30 Letsinger later took the reins and introduced Solid Phase Oligonucleotide Synthesis (SPOS) as well as increasing coupling reactivity through the use of P(III) phosphite triesters, which eases access to the requisite trigonal bipyramidal transition state.14,31−35 The stage was then set for a revolution in SPOS. The development of the modern phosphoramidite method, by Caruthers, Beaucage, McBride, Marshall, Matteucci, and others, which exploited inorganic (nonswelling) supports and highly reactive P(III) reagents, coincided with the development of automated instrumentation and allowed for the birth of modern SPOS.17,36,37The impact of the phosphoramidite method on chemical, biochemical, and biological research cannot be overstated. The ability to synthesize long and short oligonucleotides in high purity and yield has enabled numerous technologies such as oligonucleotide therapeutics, high-throughput sequencing, and hybridization probes which have immensely impacted human health and basic science. As this history has been covered in detail elsewhere, this Outlook seeks to build on Westheimer, Todd, and Blackburn’s ideas and highlight some underexplored reactivity of P(V) which could augment existing P(III) methods in both synthetic organic and oligonucleotide chemistries.
Figure 2.
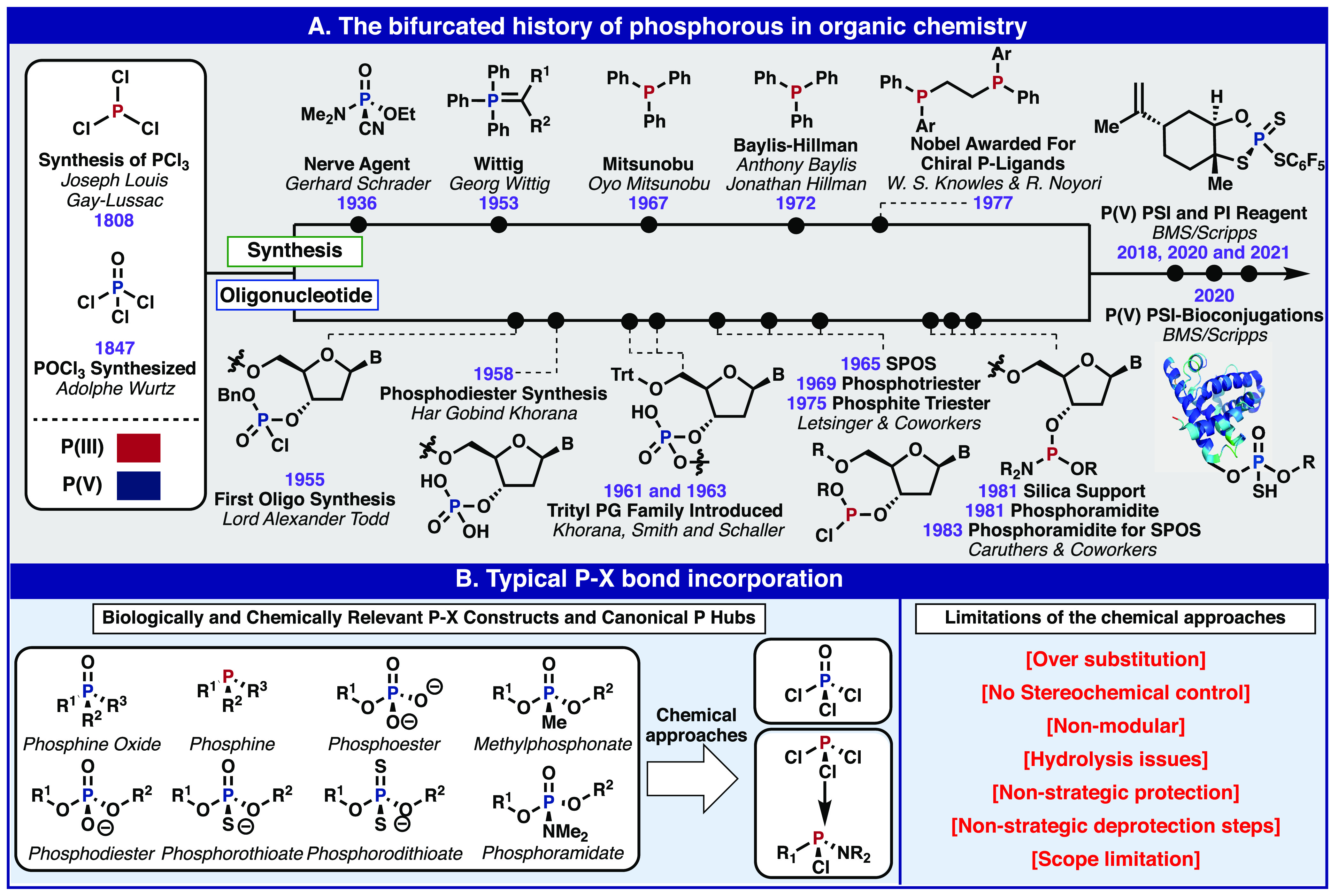
Brief illustrative history of phosphorus in organic chemistry (A) and reagents use for typical incorporation of P–X bonds (B).
Resurrecting P(V) for Oligonucleotide Synthesis
In both organic synthesis and biochemistry, two main feedstock building blocks (PCl3 and POCl3)19,20 have been employed to access the most versatile linkages and reagents based on phosphorus, eight of which are highlighted in Figure 2B.19,20 Approaches using these feedstock chemicals are challenged by the difficulty in controlling the stepwise substitution of the P–Cl bonds while simultaneously managing hydrolytic instability. As such, approaches that begin with POCl3 are not as straightforward as one would like. Indeed, the most common use of this reagent in synthesis is simply as a dehydrating agent.38 Historically, the P(V) reagents for oligonucleotide synthesis suffered from low reactivity and a high prevalence of side reactions. These shortcomings were tackled systematically, over years of research, through numerous tactical innovations which resulted in modern phosphonamidite chemistry.2 Today, the P(III)-based phosphoramidite platform represents the state of the art for oligonucleotide synthesis and is readily applicable for the formation of canonical phosphodiesters to phosphorothioates, which form the foundation of therapeutic oligonucleotides.39
Existing synthetic methods to control phosphorothioate stereochemistry (Figure 3A) commenced with the pioneering work of Stec and co-workers on P(V)-based oxathiaphospholane heterocycles.40−42 Although this method enabled studies elucidating the effects of phosphorothioate stereochemistry, the nonstereoselective synthesis of the monomers (which required resolution after synthesis from a P(III) starting material) and the subpar coupling efficiencies (92–94%) only allowed for the synthesis of stereopure oligonucleotides up to 15 bases.43,44 In a shift that mirrored phosphodiester synthesis, the field moved to P(III) to address this issue. Using chiral P(III) derivatives such as the N-acyl oxazaphospholidines,45 oxazaphosphorinanes,46−49 and oxazaphospholidines50,51 remedied the poor coupling efficiency problem, but suffered from a lack of stereoselectivity, either in synthesis of the monomer units or during SPOS (Figure 3A). A decade after their introduction, Wada and co-workers found that bicyclic oxazaphospholidine monomers could be stereospecifically coupled during SPOS when using a novel non-nucleophilic activator.52−56 This technology was improved upon and provided the foundation for Wave Life Sciences, a company that is currently investigating the impact of phosphorothioate stereochemistry in therapeutic ASOs.57,58
Figure 3.
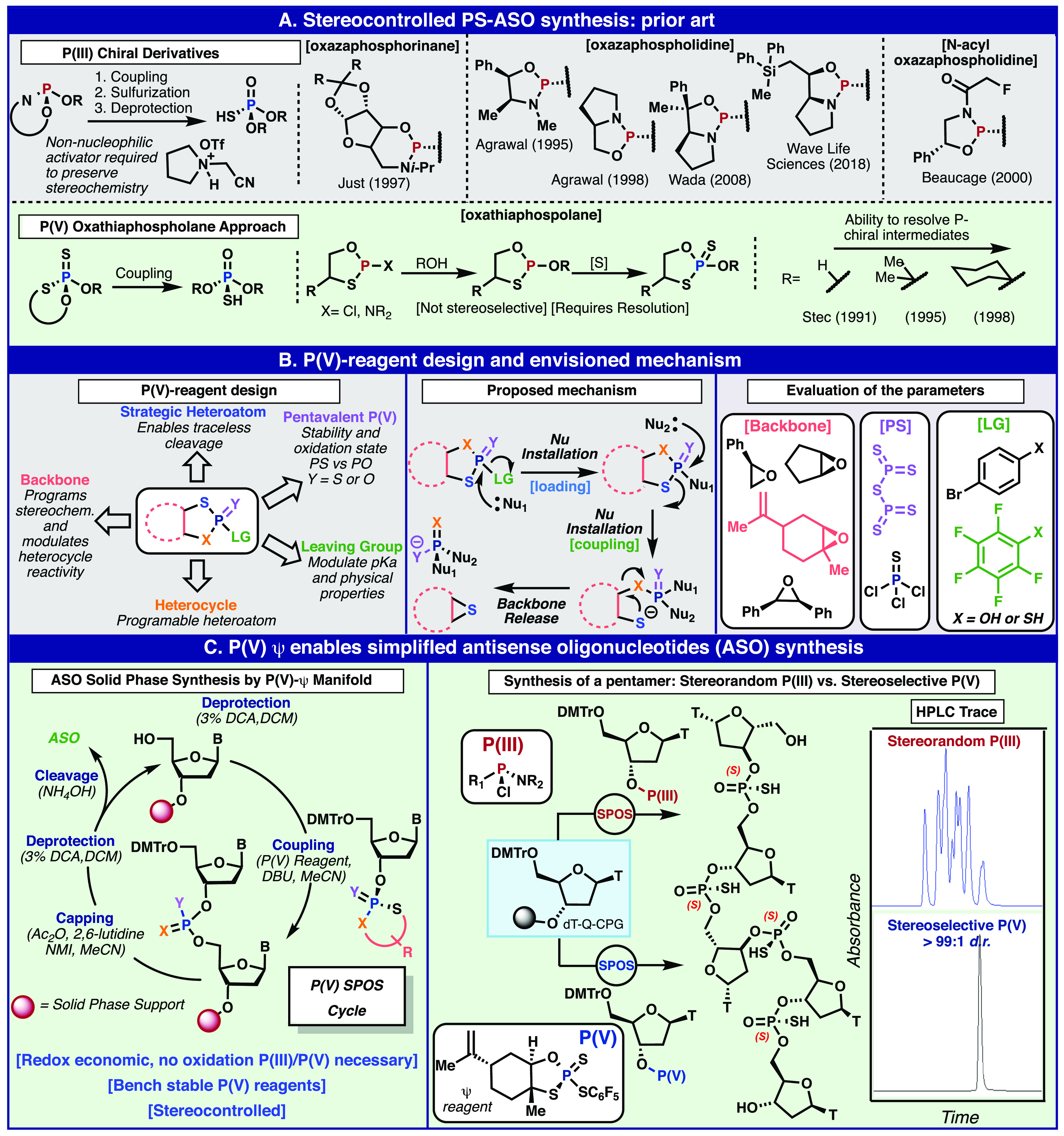
Stereocontrolled PS oligonucleotide synthesis, prior P(III) and P(V) strategies (A); design of a P(V) reagent platform to overcome current challenges (B); and the application of P(V)-based Ψ reagents for the synthesis of stereopure ASOs (C).
Despite the air-sensitive nature of P(III) reagents and the need for specific protecting groups, activators, and oxidation conditions that accompany its adoption, an entire industry has emerged (both reagents and equipment) to support P(III) chemistry in SPOS. In theory, a P(V) strategy could offer increased ideality59 and efficiency if the problem of sluggish reactivity could be resolved. Thus, a clean-slate approach to this challenge, in collaboration with scientists at Bristol Myers Squibb (BMS), was undertaken to answer the question of whether any simple P(V)-based solutions could be competitive or even offer advantages over P(III).
Inspired by Stec and co-workers’ research into oxathiaphospholane heterocycles,40−42,60 (Figure 3A) a modular reagent system was envisioned as outlined in Figure 3B. In their series of seminal publications, Stec demonstrated that such heterocycles, which were derived from PCl3, could be efficiently opened with a nucleophile triggering a self-immolation to deliver a thiirane byproduct and the desired P(V) linkage. We envisioned building on this important precedent to render the approach modular and only reliant on P(V). Thus, a suitable electrophilic heterocyclic phospholane might be subjected to an initial “loading” step to intercept Stec’s intermediate, followed by another displacement in a “coupling” step to deliver the desired product. To truly solve the problem at hand, the reagent would need to be inexpensive, amenable to multiple different P-X linkages, and stable to air and moisture, and offer P(III)-level reactivity. Such a construct would conceivably allow for the direct preparation of not only linkages of relevance to oligonucleotide research but also the separate realms of organic synthesis, medicinal chemistry, catalysis, and perhaps even bioconjugation. Thus, a systematic evaluation of key components such as the backbone, phosphorus source, and leaving group led to the identification of a reagent platform that could achieve these goals. Specifically, a reagent derived from limonene and P2S5 (a P(V) source) was disclosed in 2018 after a long series of exploration and optimization (vide infra).61
The most urgent application for the team was the stereocontrolled and redox neutral assembly of antisense oligonucleotides (ASOs, Figure 3C). As discussed above, only stereorandom or laborious multistep protocols based on P(III) were known at the time.42,45,50,52,57,62 Although these methods are indeed specialized, the most advanced of these methods was recently used to show that the precise control of phosphorothioate stereochemistry significantly increases the efficacy of ASOs.57 For this purpose, based on the logic described above, the phosphorus sulfur incorporation (or PSI, Ψ) reagent was born. The Ψ coupling cycle is simple, relying on direct P—O bond formation during loading and coupling steps, as well as the requisite deprotection/capping steps as the sequence grows. Critically, stereocontrol is entirely dictated by the reagent employed. Since both enantiomers of limonene are feedstock materials (derived from the citrus industry), they are relatively cheap and efficient. This stands in contrast to designer P(III) reagents (requiring a minimum of 7 steps)63 which rely on extraneous redox manipulations and protecting groups on phosphorus. As an early proof of concept, a simple pentamer was prepared (Figure 2C) with the Ψ approach providing only a single diastereomer product when compared with the stereorandom phosphoramidite method (see HPLC chromatogram inset, Figure 3C). ASO synthesis was only the beginning for this area of inquiry, as an equally pressing application awaited in both the medicinal and synthetic realms.
Bridging Both Worlds: Ψ and Π for Chemistry and Biology
In our 2018 disclosure, we showed how Ψ could dramatically simplify the synthesis of cyclic dinucleotides (CDNs) relative to P(III). Traditional P(III) approaches were mired in concession steps and multiple purifications while only delivering mixtures of diastereomers. The Ψ platform enabled medicinal chemists to create a large variety of CDN analogs in a fraction of the time previously required (Figure 4A)64,65 The Ψ platform could even be expanded to enable the facile introduction of stereopure thiophosphoramidate (P–N) linkages (Figure 4A). Initial attempts at stereocontrolled P–N bond formation by phosphoramidite chemistry or canonical Ψ reagents proved unsuccessful, but a competent backbone was quickly identified and deployed, increasing the scope and utility of Ψ-based reagents (Figure 4A).65 Methylphosphonates (MPOs), another linkage of importance to the field, were also explored. No stereocontrolled approach had been reported, and past work relied upon P(III)-based transformations that were wedded to tedious HPLC separation of isomers.66−72 In order to solve this problem, a new phosphorus incorporation reagent (PI, or Π) was invented.73 Using Π, a C—P bond could be forged in a stereocontrolled fashion followed by loading to a nucleoside, capping with MeI, and subsequent coupling to generate the MPO linked dinucleotide. For the first time, all 16 possible DNA-based MPO dinucleotides could be prepared as a single diastereomer. The utility of Π for forging C–P bonds led to exploration of the related synthetic challenge of modular access to enantiopure phosphines and phospine oxides. Thus, Π served as a surrogate for a chiral variant of POCl3 with controlled stepwise reactivity. For example, complex phosphines were accessible from Π through sequential additions of a Grignard reagent and two organolithium reagents to deliver bench-stable phosphine oxides that could be subsequently reduced to the desired phosphine (Figure 4C).
Figure 4.
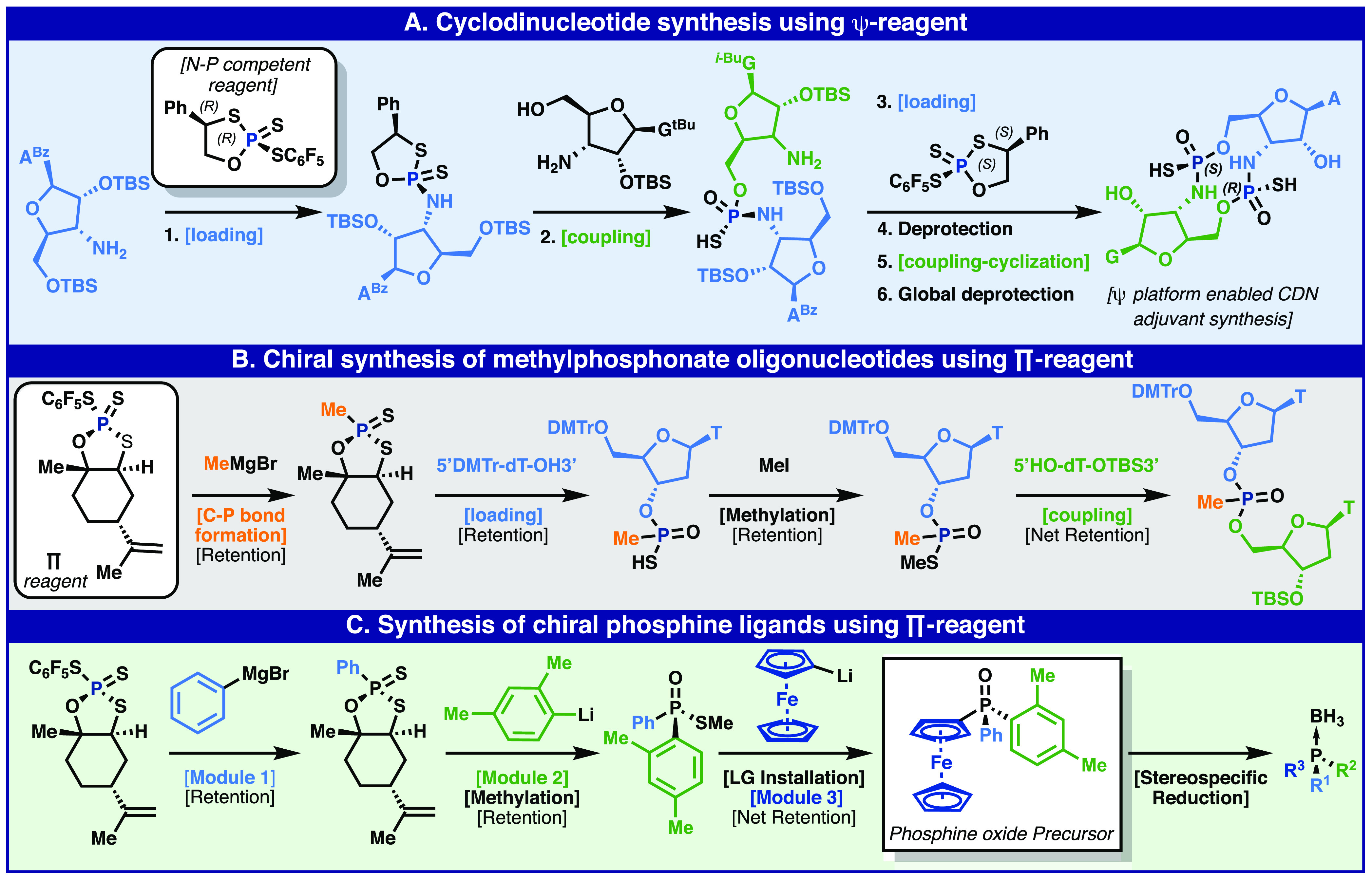
Beyond stereopure ASOs: Applications of the Ψ platform to the synthesis of CDNs (A), methylphosphonate-based oligonucleotides (B), and enantiopure phosphines and phosphine oxides (C).
Opportunities to exploit the enhanced reactivity of these P(V) reagents also arose in the natural product total synthesis. Specifically, the synthesis of the highly complex and polar natural product tagetitoxin, a decades-old unanswered puzzle for the field, was enabled by Ψ reagents.74 Some of the most difficult aspects of this synthesis were the incorporation of phosphorus at the right stage, the determination of the absolute configuration of the natural product, and the purification of noncrystalline intermediates (Figure 5B). The use of conventional P(III) and P(V) reagents for this purpose were only met with failure due to either a lack of chemoselectivity or reactivity. The use of Ψ solved all of the above problems, allowing for the phosphate to be installed (after SeO2-mediated desulfurization), rendering a key intermediate crystalline, and enabling a resolution of enantiomers. As a result, the synthesis could be completed and the absolute configuration could be assigned using a biochemical assay.74 The lessons learned in these strikingly different scenarios led to more daring applications of the P(V) platform.
Figure 5.
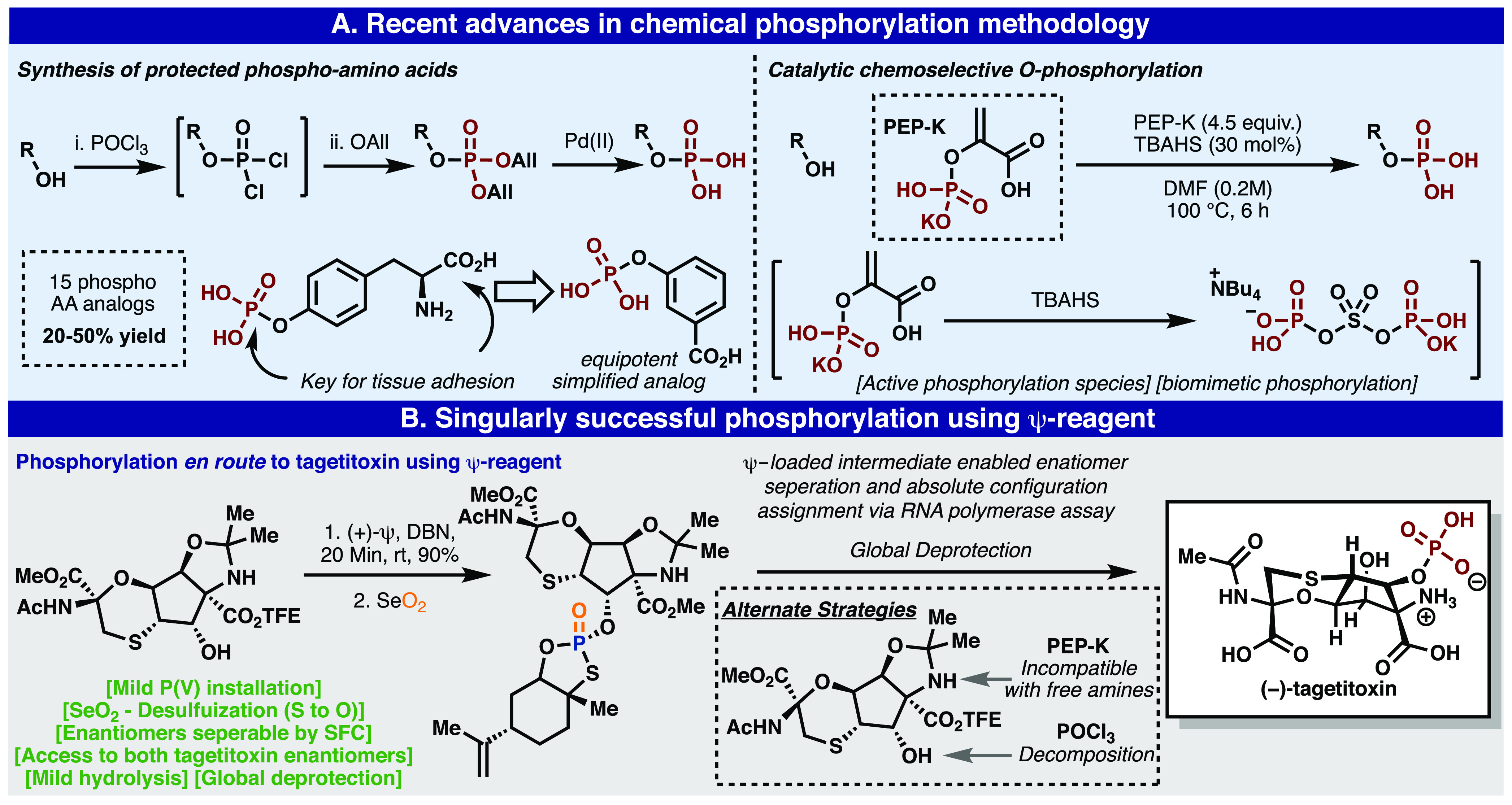
Emerging P(V)-based phosphorylation methods (A) and a singularly successful Ψ enable phosphorylation en route to the total synthesis of tagetitoxin (B).
Two recently emerging methods for phosphorylation, published in this Journal, suggest excitement surrounding new chemical phosphorylation methods (Figure 5A). The first, by Stevens and co-workers, involves the reaction of alcohols with POCl3 in the presence of a weak base followed by capping the intermediate phosphorochloridate with allyl alcohol. These isolable phosphotriesters can be cleanly deprotected with a Pd(II) source to furnish the desired phosphorylated product. This method was used to prepare protected phoso-amino acid analogs and to elucidate the roles they play as tissue cement adhesives.75 Alternatively, Kanai and co-workers demonstrated the alcohols could be phosphorylated using phosphoenolpyruvic acid monopotassium salt (PEP-K) catalyzed by tetrabutylammonium hydrogen sulfate.76 Unfortunately, neither method proved feasible for the synthesis of tagetitoxin, because POCl3 decomposed the starting material and PEP-K is not tolerant of free amines (Figure 5B).
Leveraging P(V) Reactivity for Exquisitely Selective Bioconjugation
The unique versatility, exquisite chemoselectivity, and high reactivity of these reagents led us to wonder if even more aspirational goals such as the selective modification of peptides, proteins, and nucleic acids could be achieved (Figure 6A). When one considers the vast field of chemical reactions specific to bioconjugation,77 click-chemistry,78 and biorthogonal chemistry,79 one notices a distinct lack of phosphate-based linkages. As shown in Figure 1, this stands in dramatic contrast to the functional group that Nature prefers, phosphate diester linkages, which was referred to by Westheimer as the basis of her “molecular glue”.5 Non-enzymatic means to form these linkages in this context are likely rare because of the heavy reliance on P(III) approaches which, due to the aforementioned hydrolytic instability and lack of chemoselectivity, makes them unsuitable for such applications. Prior work from the lab on DNA-encoded libraries80−83 gave us a framework from which to think about the direct modification of nucleic acids using Ψ and related reagents. In particular, the use of reversible adsorption to solid support (RASS) allowed native DNA constructs to be manipulated as if they were small organic compounds. Combining this technology with the exquisite chemoselectivity and high reactivity of Ψ toward O-nucleophiles enabled the direct synthetic elaboration of native DNA by RASS (SENDR, Figure 5B).84 DNA hybridization probes underpin much of modern biochemical, diagnostic, and genomic techniques; although ubiquitous, they are resource- and time-intensive to produce by total chemical synthesis when compared to biochemical processes.85 Numerous attempts to react the free alcohol residues of DNA (3′ or 5′) under conventional conditions (condensations, displacement, P(III)-based) were only met with failure or wildly unselective reactions. In contrast, Ψ coupling at the 3′ or 5′ alcohol proceeded smoothly producing DNA conjugates in high yields. The SENDR procedure allowed for the direct and expedient production of numerous useful constructs including electrophile-linked aptamers, DNA–protein conjugates, and diagnostic probes. SENDR should allow for novel constructs to be directly prepared from native or biosynthetically produced DNA.84 This late-stage functionalization approach to DNA conjugate synthesis may facilitate the development of novel constructs and experimental designs which may currently be inaccessible or cost-prohibitive.
Figure 6.
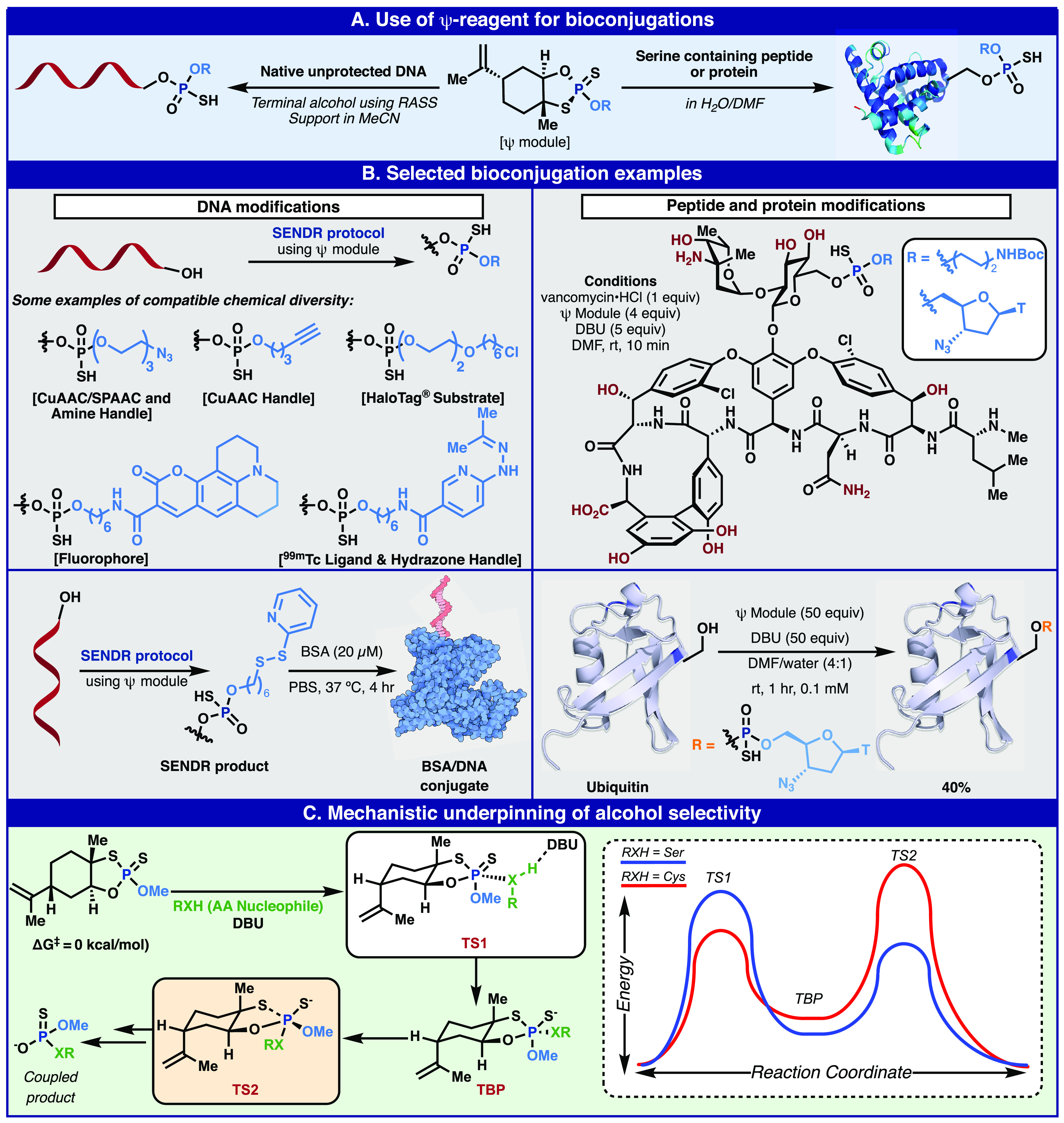
Bioconjugation of peptides, proteins, and nucleic acids using Ψ: proposal (A), execution (B), and mechanistic rationale (C).
Moving on to peptide and protein bioconjugation, the bar for acceptable chemoselectivity is even higher than of the 20 canonical amino acids; there are multiple nucleophilic side chains that can compete for an electrophile. This topic has been reviewed by us elsewhere, and it was clear that selective serine-modification, outside of the active site, is one of the large remaining challenges for the field.86 Ψ-Modules were demonstrated to have exquisite selectivity for serine residues even in the presence of lysine, histidine, cysteine, tyrosine, and threonine.87 It functioned reliably in the context of small peptides, natural products such as vancomycin, and even large proteins such as ubiquitin using an aqueous/organic solvent mixture (Figure 5B).
The remarkable selectivity of reactions between Ψ and O-nucleophiles was deconvoluted using a computational approach that revealed a substrate-dependent change in the rate-determining step. These studies of course benefitted from a multitude of early findings in P(V) nucleophilic substitution chemistry.88−92 In the first step, which is reversible, nucleophilic attack at the phosphorus center of a loaded Ψ-module leads to the formation of a trigonal bipyramidal intermediate (TBP, Figure 5C). Not surprisingly, the activation energy of this first step is lower for cysteine, which is much more nucleophilic than serine. The second step involves pseudorotation and the self-immolation of the TBP intermediate that leads to the formation of the P(V) adduct along with extrusion of the limonene backbone. For this second step, serine has a lower activation energy barrier than the other amino acids modeled. Thus, although the first step is rate-limiting for serine (as well as lys, tyr, and thr), the relatively fast pseudorotation and self-immolation explain the surprising selectivity for serine over the far more nucleophilic cysteine residue. These calculations help explain the observation that formation of the serine-P(V) adduct is the kinetic product.
In contrast with the transition state proposed during displacement at P(III), the TBP intermediate observed in the Ψ coupling mechanism mimics the TBP intermediate that is commonly invoked by kinases during enzymatic O-phosphorylation.93−95 Although no special precautions were taken during the work presented in this outlook, given the overlap in the O-selectivity that these reagents share with toxic nerve agents (tabun, sarin, VX, etc.), there exists a potential hazard that cannot be ruled out, and appropriate levels of caution should be exercised when working with such potentially reactive P-based reagents.
Putting It All Together: Chimeric Oligonucleotides Using a P(V)-Platform
Although progress has been made in resurrecting P(V) for the stereocontrolled synthesis of phosphorothioate oligonucleotides, much remains to be done in this area. For example, medicinal chemists often want access to the stereorandom variants,96 and thus, a racemic Ψ reagent that retains high reactivity and ensures a true 1:1 mixture of diastereomers would be useful (rac-Ψ). In contrast, P(III) stereorandom ASO synthesis often leads to scalemic mixtures due to substrate bias rather than reagent control.97 Similarly, since Ψ-based ASO synthesis does not use protecting groups on sulfur, it is not compatible with P(III)-based oxidative phosphodiester synthesis.98 A P(V) approach to phosphodiester P(O) synthesis (ΨO) would be a fitting tribute to the origin of this field that commenced with but later abandoned P(V) due to low reactivity (Figure 2A). Finally, phosphorodithioates P(S)2 are achiral linkages with reduced stereochemical complexity that maintain stability to nucleases are becoming increasingly common in this field.99 P(III) approaches are known; however, such reagents are unstable, and they are unavoidably plagued with varying degrees of desulfurization (furnishing phosphorothioates) upon removal of the phosphate protecting group after synthesis.100 The Ψ2 reagent solves these challenges.
Recently, after extensive optimization and nearly 4 years of work, these three new reagent systems were disclosed (rac-Ψ, ΨO, and Ψ2) that, when combined with the previously reported (+)-Ψ and (−)-Ψ, provide a unified P(V)-approach that entirely departs from the rubric of P(III)-based oligonucleotide synthesis and enables the at-will and controlled synthesis of specific chimeric oligonucleotides. Along with these new reagents are protocols for their unified application in commercial automated synthesizers using a single coupling protocol which spans all coupling types. This redox-neutral platform, based on the native P(V)-oxidation state, challenges past assumptions of sluggish reactivity and enables access to five relevant P-linkages across a range of sugar backbones (DNA and LNA) and bases (A, T, G, mC) in one oligonucleotide construct. Aside from enabling straightforward access to a wide range of chimeric oligonucleotides, the implementation of this new protocol benefits from a reduced reliance on protecting group chemistry (which eliminates the labile cyanoethyl group and therefore acrylonitrile production upon deprotection),101,102 bespoke additives,103 and redox manipulations. It is also of note that this new P(V) platform eliminates one full step in the standard SPOS protocol (namely, the phosphorus oxidation).
Figure 7 illustrates some of the most recent accomplishments in the development of P(V) reagents for chimeric ASO synthesis. A newly developed suite of reagents based off of the Ψ platform now allows for access to all common linkages found in current ASOs. Moreover, these reagents display kinetics on-par with phosphoramidite chemistry.104 The utility of the Ψ reagent suite is demonstrated by a few selected examples in which ASOs are synthesized in exquisite crude purity. The Ψ-enabled accessibility of stereopure ASOs could help address the currently controversial role of stereodefinition in ASO optimization.57,99,105
Figure 7.
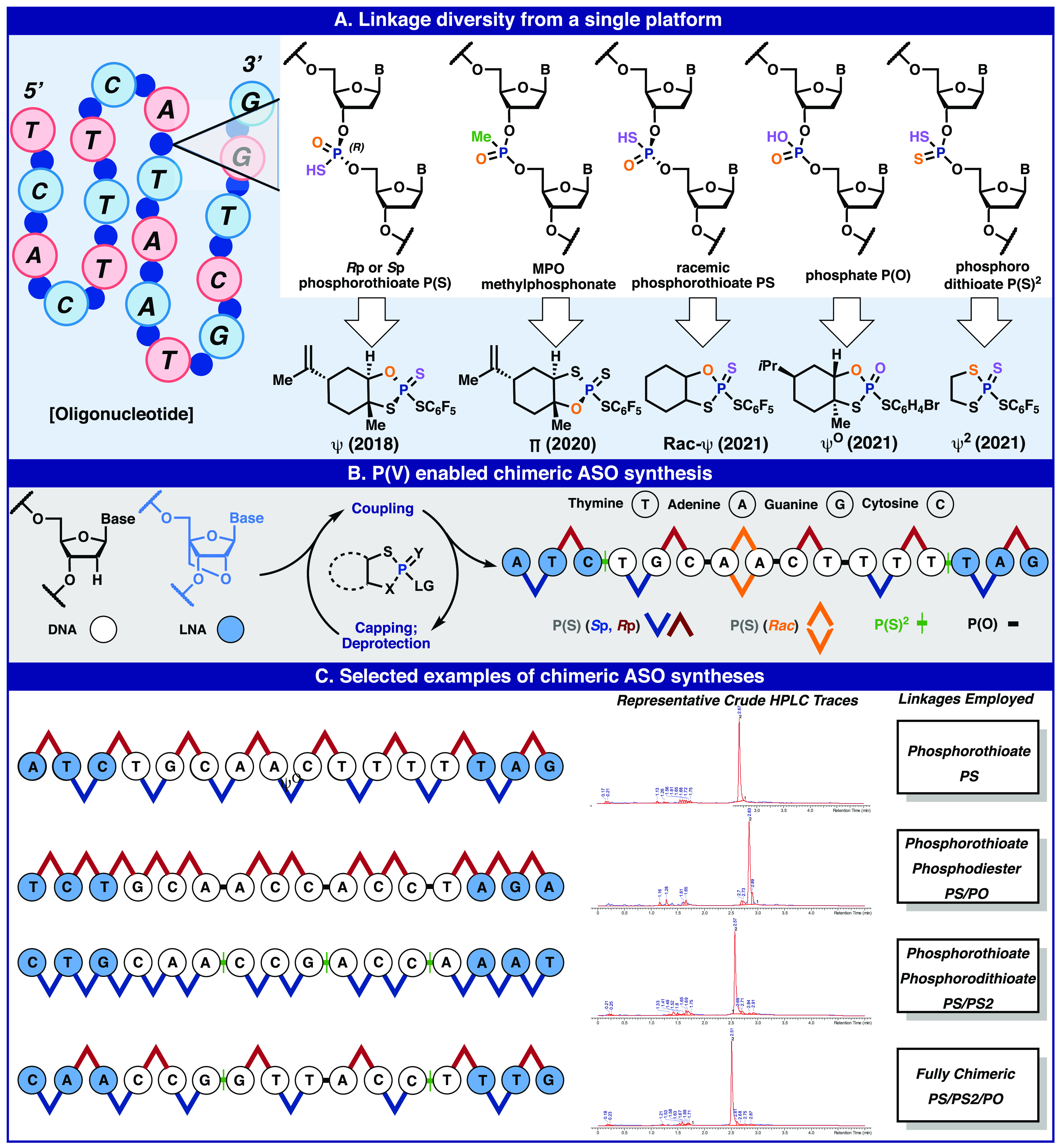
Future of ASO construction: P(V) reagents for the assembly of all P-based linkages (A), use of such a platform to access exotic chimeric oligonucleotide constructs (B), and selected examples of chimeric ASO synthesized (C).
Outlook: An Agnostic P(V)-Platform for the Future
Returning to one of the most basic of biochemical pathways, phosphorylation, it would be useful to have P(V)-based access to such a functionalization without the need for protecting groups, harsh conditions, or enzymes (Figure 8A). Recent studies in this area clearly demonstrate the need for such chemistry, but both methods enable access to a wide range of phosphorylated substrates but require either multiple steps or harsh conditions.75,76
Figure 8.
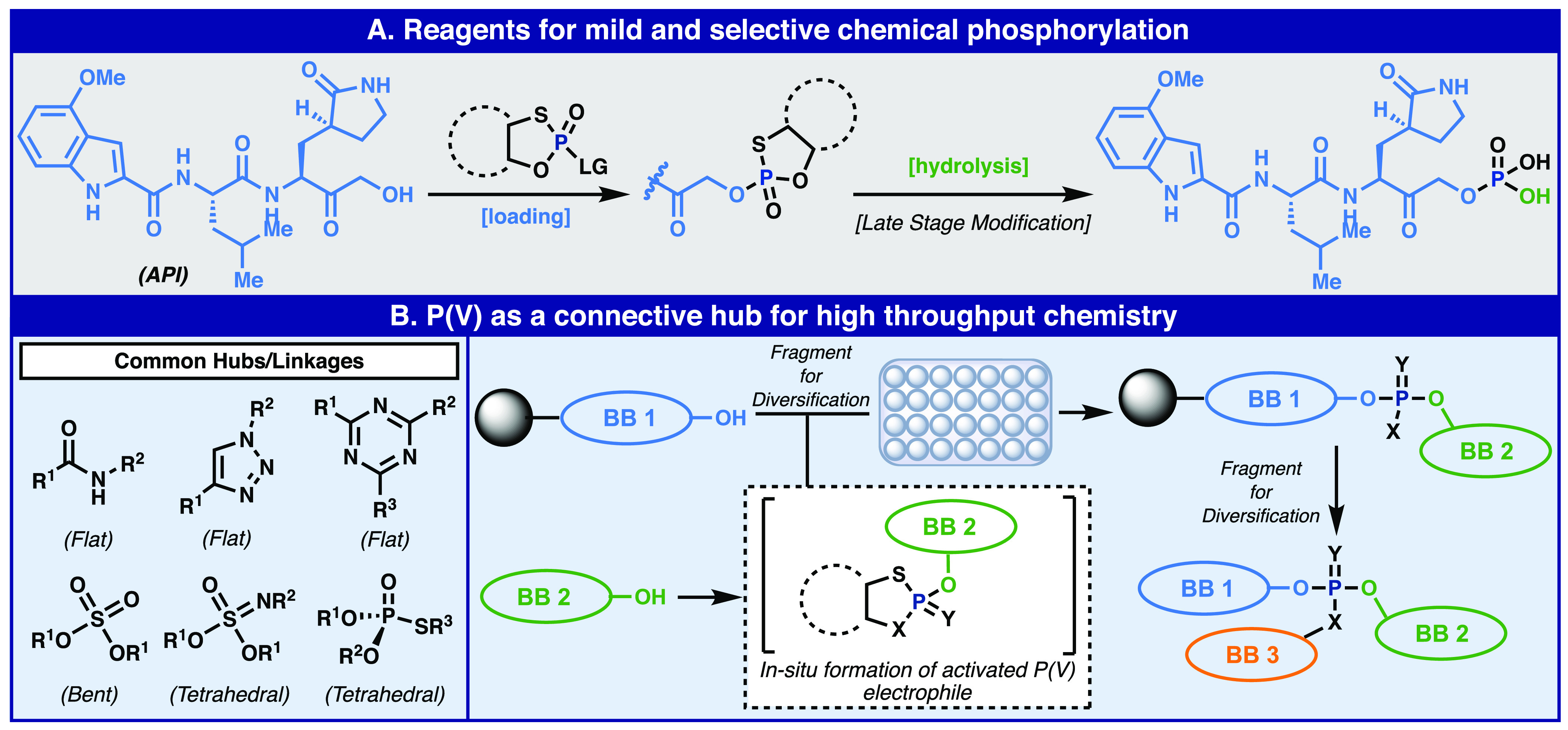
Outlook for P(V) beyond ASOs: a reagent for the facile late-stage introduction of phosphates, phosphorylation of valuable APIs as an example (A), and the development of P-based 3-dimensional connective hubs for high-throughput chemistry (B).
Finally, recent advances in SuFEx click chemistry suggest the need for “three-dimensional” connective hubs.106−108 In that chemistry, up to three different nucleophiles can be chemoselectively introduced onto an S-based electrophile to generate massive diversity with minimal effort. The advantage of such a strategy, unlike CuAAC, SPAAC, and other approaches that result in disubstituted 2-dimensional hubs, is that the resulting adducts are three-dimensional in nature. At this juncture, however, enantiocontrolled SuFEx methods are not known. In the same vein, a Ψ-based approach could lead to conceptually similar constructs with full stereocontrol allowing for unique positioning of each group in space (Figure 8B). While we do not believe the Ψ platform will supplant phosphoramidite-based SPOS, it may enable access to structures unavailable to P(III)-based methods. It might offer a more practical and sustainable approach to manufacturing oligonucleotides in the future. The exquisite chemoselectivity observed could make it a go-to strategy for bioconjugation efforts. Ultimately, the revitalization of P(V)-based methods gives chemists a new and orthogonal tool to install one of the most useful linkages that Nature has to offer.109
Acknowledgments
Financial support for this work was provided by Bristol Myers Squibb, the NIH (GM-118176), D.T.F. was supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant UL1 TR002551 and Linked Award TL1 TR002551. The authors would like to acknowledge Bin Zheng, Shenjie Qiu, Stephen Mercer, Richard Olson, Antonio Ramirez, Peter Park, Brian Fink, Rick Ewing, Matthew Winston, Yazhong Huang (BMS), Justine deGruyter, Javier Lopez, Shota Asai, Cian Kingston, Wei Hao, Dongmin Xu, Natalia Padial, Nazaret Rivas-Bascón, Rohan Narayan, Phillip Dawson, and Donna Blackmond (TSRI) for being an outstanding team of scientists and for making everything seen here possible.
Author Contributions
⊥ K.W.K., D.T.F., and J.C.V. contributed equally to this paper. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): Three of the authors are co-founders of Elsie Biotechnologies, a company that is in the process of licensing some of the technology patented in this outlook.
References
- Baddiley J.; Michelson A. M.; Todd A. R. Synthesis of adenosine triphosphate. Nature 1948, 161 (4098), 761. 10.1038/161761a0. [DOI] [PubMed] [Google Scholar]
- Michelson A. M.; Todd A. R. Nucleotides part XXXII. Synthesis of a dithymidine dinucleotide containing a 3′: 5′-internucleotidic linkage. J. Chem. Soc. 1955, 0 (0), 2632–2638. 10.1039/JR9550002632. [DOI] [Google Scholar]
- Khorana H. G.; Todd A. R. 465. Studies on phosphorylation. Part XI. The reaction between carbodi-imides and acid esters of phosphoric acid. A new method for the preparation of pyrophosphates. J. Chem. Soc. 1953, 0, 2257–2260. 10.1039/jr9530002257. [DOI] [Google Scholar]
- Sturtevan J. M.; Gerlt J. A.; Westheimer F. H. Enthalpy of hydrolysis of simple phosphate diesters. J. Am. Chem. Soc. 1973, 95 (24), 8168–8169. 10.1021/ja00805a036. [DOI] [Google Scholar]
- Westheimer F. H. Why nature chose phosphates. Science 1987, 235 (4793), 1173–8. 10.1126/science.2434996. [DOI] [PubMed] [Google Scholar]
- Todd L. Where there’s life there’s phosphorus. Science and Scientists 1981, 275. 10.1007/978-94-009-7755-6_35. [DOI] [Google Scholar]
- Bowler M. W.; Cliff M. J.; Waltho J. P.; Blackburn G. M. Why did Nature select phosphate for its dominant roles in biology?. New J. Chem. 2010, 34 (5), 784–794. 10.1039/b9nj00718k. [DOI] [Google Scholar]
- Johnson L. A.; Dunbabin A.; Benton J. C. R.; Mart R. J.; Allemann R. K. Modular Chemoenzymatic Synthesis of Terpenes and their Analogues. Angew. Chem., Int. Ed. 2020, 59 (22), 8486–8490. 10.1002/anie.202001744. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Sorensen E. J.. Classics in Total Synthesis; VCH: Weinheim, 1996. [Google Scholar]
- Schirmer T. C-di-GMP Synthesis: Structural Aspects of Evolution, Catalysis and Regulation. J. Mol. Biol. 2016, 428 (19), 3683–701. 10.1016/j.jmb.2016.07.023. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Watt S.; Wang J.; Nakayama S.; Sayre D. A.; Lam Y. F.; Lee V. T.; Sintim H. O. Potent suppression of c-di-GMP synthesis via I-site allosteric inhibition of diguanylate cyclases with 2′-F-c-di-GMP. Bioorg. Med. Chem. 2013, 21 (14), 4396–404. 10.1016/j.bmc.2013.04.050. [DOI] [PubMed] [Google Scholar]
- Roskoski R. Jr. A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol. Res. 2015, 100, 1–23. 10.1016/j.phrs.2015.07.010. [DOI] [PubMed] [Google Scholar]
- Mikkola S.; Lonnberg T.; Lonnberg H. Phosphodiester models for cleavage of nucleic acids. Beilstein J. Org. Chem. 2018, 14, 803–837. 10.3762/bjoc.14.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letsinger R. L.; Ogilvie K. K. Nucleotide chemistry. XIII. Synthesis of oligothymidylates via phosphotriester intermediates. J. Am. Chem. Soc. 1969, 91 (12), 3350–3355. 10.1021/ja01040a042. [DOI] [Google Scholar]
- Reese C. B. The chemical synthesis of oligo- and poly-nucleotides by the phosphotriester approach. Tetrahedron 1978, 34 (21), 3143–3179. 10.1016/0040-4020(78)87013-6. [DOI] [Google Scholar]
- Gilham P. T.; Khorana H. G. Studies on Polynucleotides. I. A New and General Method for the Chemical Synthesis of the C5″-C3″ Internucleotidic Linkage. Syntheses of Deoxyribo-dinucleotides1. J. Am. Chem. Soc. 1958, 80 (23), 6212–6222. 10.1021/ja01556a016. [DOI] [Google Scholar]
- Beaucage S. L.; Caruthers M. H. Deoxynucleoside phosphoramidites—A new class of key intermediates for deoxypolynucleotide synthesis. Tetrahedron Lett. 1981, 22 (20), 1859–1862. 10.1016/S0040-4039(01)90461-7. [DOI] [Google Scholar]
- Sinha N. D.; Biernat J.; McManus J.; Köster H. Polymer support oligonucleotide synthesis XVIII: use of beta-cyanoethyl-N,N-dialkylamino-/N-morpholino phosphoramidite of deoxynucleosides for the synthesis of DNA fragments simplifying deprotection and isolation of the final product. Nucleic Acids Res. 1984, 12 (11), 4539–4557. 10.1093/nar/12.11.4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thénard G.-L. Extrait de plusieurs notes sur les métaux de la potasse et de la soude, lues à l’Institut depuis le 12 janvier jusqu’au 16 mai. Gazette Nationale, Ou le Maniteur Universel 1808, 40 (148), 581–582. [Google Scholar]
- Wurtz A. Sur l’acide sulfophosphorique et le chloroxyde de phosphore. Annals de Chimie et de Physique, 3rd Series 1847, 20, 472–481. [Google Scholar]
- Tucker J.War of Nerves: Chemical Warfare From World War I to Al-Qaeda. Pantheon Books: 2006. [Google Scholar]
- Wittig G.; Schöllkopf U. Über Triphenyl-phosphin-methylene als olefinbildende Reagenzien (I. Mitteil. Chem. Ber. 1954, 87 (9), 1318–1330. 10.1002/cber.19540870919. [DOI] [Google Scholar]
- Mitsunobu O.; Yamada M. Preparation of Esters of Carboxylic and Phosphoric AcidviaQuaternary Phosphonium Salts. Bull. Chem. Soc. Jpn. 1967, 40 (10), 2380–2382. 10.1246/bcsj.40.2380. [DOI] [Google Scholar]
- Baylis A. B.; Hillman M. E. D. German Patent 2155113, 1972.
- Vineyard B. D.; Knowles W. S.; Sabacky M. J.; Bachman G. L.; Weinkauff D. J. Asymmetric hydrogenation. Rhodium chiral bisphosphine catalyst. J. Am. Chem. Soc. 1977, 99 (18), 5946–5952. 10.1021/ja00460a018. [DOI] [Google Scholar]
- Brown D. M.A Brief History of Oligonucleotide Synthesis. In Protocols for Oligonucleotides and Analogs: Synthesis and Properties, Agrawal S., Ed.; Humana Press: Totowa, NJ, 1993; pp 1–17. [Google Scholar]
- Smith M.; Rammler D. H.; Goldberg I. H.; Khorana H. G. Studies on Polynucleotides. XIV.1 Specific Synthesis of the C3″-C5″ Interribonucleotide Linkage. Syntheses of Uridylyl-(3″5″)-Uridine and Uridylyl-(3″5″)-Adenosine2. J. Am. Chem. Soc. 1962, 84 (3), 430–440. 10.1021/ja00862a023. [DOI] [Google Scholar]
- Sekiya T.; Takeya T.; Brown E. L.; Belagaje R.; Contreras R.; Fritz H. J.; Gait M. J.; Lees R. G.; Ryan M. J.; Khorana H. G.; Norris K. E. Total synthesis of a tyrosine suppressor transfer RNA gene. XVI. Enzymatic joinings to form the total 207-base pair-long DNA. J. Biol. Chem. 1979, 254 (13), 5787–801. 10.1016/S0021-9258(18)50482-8. [DOI] [PubMed] [Google Scholar]
- Sekiya T.; Brown E. L.; Belagaje R.; Fritz H. J.; Gait M. J.; Lees R. G.; Ryan M. J.; Khorana H. G.; Norris K. E. Total synthesis of a tyrosine suppressor tRNA gene. XV. Synthesis of the promoter region. J. Biol. Chem. 1979, 254 (13), 5781–6. 10.1016/S0021-9258(18)50481-6. [DOI] [PubMed] [Google Scholar]
- Belagaje R.; Brown E. L.; Fritz H. J.; Lees R. G.; Khorana H. G. Total synthesis of a tyrosine suppressor transfer RNA gene. XIV. Chemical synthesis of oligonucleotide segments corresponding to the terminal regions. J. Biol. Chem. 1979, 254 (13), 5765–80. 10.1016/S0021-9258(18)50480-4. [DOI] [PubMed] [Google Scholar]
- Letsinger R. L.; Kornet M. J.; Mahadevan V.; Jerina D. M. Reactions on Polymer Supports. J. Am. Chem. Soc. 1964, 86 (23), 5163–5165. 10.1021/ja01077a024. [DOI] [Google Scholar]
- Letsinger R. L.; Mahadevan V. Oligonucleotide Synthesis on a Polymer Support1,2. J. Am. Chem. Soc. 1965, 87 (15), 3526–3527. 10.1021/ja01093a058. [DOI] [PubMed] [Google Scholar]
- Letsinger R. L.; Ogilvie K. K.; Miller P. S. Nucleotide chemistry. XV. Developments in syntheses of oligodeoxyribonucleotides and their organic derivatives. J. Am. Chem. Soc. 1969, 91 (12), 3360–3365. 10.1021/ja01040a044. [DOI] [Google Scholar]
- Letsinger R. L.; Finnan J. L.; Heavner G. A.; Lunsford W. B. Nucleotide chemistry. XX. Phosphite coupling procedure for generating internucleotide links. J. Am. Chem. Soc. 1975, 97 (11), 3278–3279. 10.1021/ja00844a090. [DOI] [PubMed] [Google Scholar]
- Letsinger R. L.; Lunsford W. B. Synthesis of thymidine oligonucleotides by phosphite triester intermediates. J. Am. Chem. Soc. 1976, 98 (12), 3655–3661. 10.1021/ja00428a045. [DOI] [PubMed] [Google Scholar]
- Matteucci M. D.; Caruthers M. H. Synthesis of deoxyoligonucleotides on a polymer support. J. Am. Chem. Soc. 1981, 103 (11), 3185–3191. 10.1021/ja00401a041. [DOI] [PubMed] [Google Scholar]
- McBride L. J.; Caruthers M. H. An investigation of several deoxynucleoside phosphoramidites useful for synthesizing deoxyoligonucleotides. Tetrahedron Lett. 1983, 24 (3), 245–248. 10.1016/S0040-4039(00)81376-3. [DOI] [Google Scholar]
- Meier M. S.; Ruder S. M.; Malona J. A.; Frontier A. J. Phosphorus Oxychloride. Encyclopedia of Reagents for Organic Synthesis 2008, 1. 10.1002/047084289X.rp162.pub2. [DOI] [Google Scholar]
- Roy S.; Caruthers M. Synthesis of DNA/RNA and Their Analogs via Phosphoramidite and H-Phosphonate Chemistries. Molecules 2013, 18 (11), 14268–14284. 10.3390/molecules181114268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stec W. J.; Grajkowski A.; Koziolkiewicz M.; Uznanski B. Novel route to oligo(deoxyribonucleoside phosphorothioates). Stereocontrolled synthesis of P-chiral oligo(deoxyribonucleoside phosphorothioates). Nucleic Acids Res. 1991, 19 (21), 5883–5888. 10.1093/nar/19.21.5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stec W. J.; Grajkowski A.; Kobylanska A.; Karwowski B.; Koziolkiewicz M.; Misiura K.; Okruszek A.; Wilk A.; Guga P.; Boczkowska M. Diastereomers of Nucleoside 3′-O-(2-Thio-1,3,2-oxathia(Selena)phospholanes): Building Blocks for Stereocontrolled Synthesis of Oligo(nucleoside phosphorothioate)s. J. Am. Chem. Soc. 1995, 117 (49), 12019–12029. 10.1021/ja00154a001. [DOI] [Google Scholar]
- Stec W. J.; Karwowski B.; Boczkowska M.; Guga P.; Koziołkiewicz M.; Sochacki M.; Wieczorek M. W.; Błaszczyk J. Deoxyribonucleoside 3′-O-(2-Thio- and 2-Oxo-“spiro”-4,4-pentamethylene-1,3,2-oxathiaphospholane)s: Monomers for Stereocontrolled Synthesis of Oligo(deoxyribonucleoside phosphorothioate)s and Chimeric PS/PO Oligonucleotides. J. Am. Chem. Soc. 1998, 120 (29), 7156–7167. 10.1021/ja973801j. [DOI] [Google Scholar]
- Koziolkiewicz M.; Krakowiak A.; Kwinkowski M.; Boczkowska M.; Stec W. J. Stereodifferentiation--the effect of P chirality of oligo(nucleoside phosphorothioates) on the activity of bacterial RNase H. Nucleic Acids Res. 1995, 23 (24), 5000–5005. 10.1093/nar/23.24.5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guga P.; Stec W. J. Synthesis of phosphorothioate oligonucleotides with stereodefined phosphorothioate linkages. Curr. Protoc Nucleic Acid Chem. 2003, 4.17. 10.1002/0471142700.nc0417s14. [DOI] [PubMed] [Google Scholar]
- Wilk A.; Grajkowski A.; Phillips L. R.; Beaucage S. L. Deoxyribonucleoside Cyclic N-Acylphosphoramidites as a New Class of Monomers for the Stereocontrolled Synthesis of Oligothymidylyl- and Oligodeoxycytidylyl- Phosphorothioates. J. Am. Chem. Soc. 2000, 122 (10), 2149–2156. 10.1021/ja991773u. [DOI] [Google Scholar]
- Jin Y.; Biancotto G.; Just G. A stereoselective synthesis of dinucleotide phosphorothioates, using chiral phosphoramidites as intermediates. Tetrahedron Lett. 1996, 37 (7), 973–976. 10.1016/0040-4039(95)02355-0. [DOI] [Google Scholar]
- Wang J.-C.; Just G. A stereoselective synthesis of dinucleotide phosphorothioates, using chiral indol-oxazaphosphorine intermediates. Tetrahedron Lett. 1997, 38 (22), 3797–3800. 10.1016/S0040-4039(97)00749-1. [DOI] [Google Scholar]
- Wang J.-C.; Just G. A stereoselective synthesis of dinucleotide phosphorothioate triesters through a chiral indol-oxazaphosphorine intermediate. Tetrahedron Lett. 1997, 38 (5), 705–708. 10.1016/S0040-4039(96)02420-3. [DOI] [Google Scholar]
- Jin Y.; Just G. Stereoselective Synthesis of Dithymidine Phosphorothioates Using Xylose Derivatives as Chiral Auxiliaries. J. Org. Chem. 1998, 63 (11), 3647–3654. 10.1021/jo972318o. [DOI] [Google Scholar]
- Iyer R. P.; Yu D.; Ho N.-H.; Tan W.; Agrawal S. A novel nucleoside phosphoramidite synthon derived from 1R, 2S-ephedrine. Tetrahedron: Asymmetry 1995, 6 (5), 1051–1054. 10.1016/0957-4166(95)00122-6. [DOI] [Google Scholar]
- Guo M.; Yu D.; Iyer R. P.; Agrawal S. Solid-phase stereoselective synthesis of 2′-O-methyl-oligoribonucleoside phosphorothioates using nucleoside bicyclic oxazaphospholidines. Bioorg. Med. Chem. Lett. 1998, 8 (18), 2539–2544. 10.1016/S0960-894X(98)00450-8. [DOI] [PubMed] [Google Scholar]
- Oka N.; Yamamoto M.; Sato T.; Wada T. Solid-Phase Synthesis of Stereoregular Oligodeoxyribonucleoside Phosphorothioates Using Bicyclic Oxazaphospholidine Derivatives as Monomer Units. J. Am. Chem. Soc. 2008, 130 (47), 16031–16037. 10.1021/ja805780u. [DOI] [PubMed] [Google Scholar]
- Oka N.; Wada T.; Saigo K. Diastereocontrolled Synthesis of Dinucleoside Phosphorothioates Using a Novel Class of Activators, Dialkyl(cyanomethyl)ammonium Tetrafluoroborates. J. Am. Chem. Soc. 2002, 124 (18), 4962–4963. 10.1021/ja017275e. [DOI] [PubMed] [Google Scholar]
- Oka N.; Wada T.; Saigo K. An Oxazaphospholidine Approach for the Stereocontrolled Synthesis of Oligonucleoside Phosphorothioates. J. Am. Chem. Soc. 2003, 125 (27), 8307–8317. 10.1021/ja034502z. [DOI] [PubMed] [Google Scholar]
- Oka N.; Kondo T.; Fujiwara S.; Maizuru Y.; Wada T. Stereocontrolled Synthesis of Oligoribonucleoside Phosphorothioates by an Oxazaphospholidine Approach. Org. Lett. 2009, 11 (4), 967–970. 10.1021/ol802910k. [DOI] [PubMed] [Google Scholar]
- Nukaga Y.; Yamada K.; Ogata T.; Oka N.; Wada T. Stereocontrolled Solid-Phase Synthesis of Phosphorothioate Oligoribonucleotides Using 2′-O-(2-Cyanoethoxymethyl)-nucleoside 3′-O-Oxazaphospholidine Monomers. J. Org. Chem. 2012, 77 (18), 7913–7922. 10.1021/jo301052v. [DOI] [PubMed] [Google Scholar]
- Iwamoto N.; Butler D. C. D.; Svrzikapa N.; Mohapatra S.; Zlatev I.; Sah D. W. Y.; Meena; Standley S. M.; Lu G.; Apponi L. H.; Frank-Kamenetsky M.; Zhang J. J.; Vargeese C.; Verdine G. L. Control of phosphorothioate stereochemistry substantially increases the efficacy of antisense oligonucleotides. Nat. Biotechnol. 2017, 35 (9), 845–851. 10.1038/nbt.3948. [DOI] [PubMed] [Google Scholar]
- Bowman K. A.; Vargeese C.; Butler D. C. D.; Kandasamy P.; Alam M. R.; Shimizu M.; Standley S. M.; Aduda V.; Bommineni G. R.; Tripathi S., Korboukh I.. Technologies For Oligonucleotide Preparation. International Patent WO 2019/055951 A1, March 21, 2019.
- Gaich T.; Baran P. S. Aiming for the Ideal Synthesis. J. Org. Chem. 2010, 75 (14), 4657–4673. 10.1021/jo1006812. [DOI] [PubMed] [Google Scholar]
- Guga P.; Stec W. J. Synthesis of Phosphorothioate Oligonucleotides with Stereodefined Phosphorothioate Linkages. Current Protocols in Nucleic Acid Chemistry 2003, 14 (1), 4.17.1–4.17.28. 10.1002/0471142700.nc0417s14. [DOI] [PubMed] [Google Scholar]
- Knouse K. W.; deGruyter J. N.; Schmidt M. A.; Zheng B.; Vantourout J. C.; Kingston C.; Mercer S. E.; McDonald I. M.; Olson R. E.; Zhu Y.; Hang C.; Zhu J.; Yuan C.; Wang Q.; Park P.; Eastgate M. D.; Baran P. S. Unlocking P(V): Reagents for chiral phosphorothioate synthesis. Science 2018, 361 (6408), 1234–1238. 10.1126/science.aau3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.; Lightfoot H. L.; Halloy F.; Malinowska A. L.; Berk C.; Behera A.; Schümperli D.; Hall J. Synthesis and cellular activity of stereochemically-pure 2′-O-(2-methoxyethyl)-phosphorothioate oligonucleotides. Chem. Commun. 2017, 53 (3), 541–544. 10.1039/C6CC08473G. [DOI] [PubMed] [Google Scholar]
- Shimizu M.; Wada T.. Asymmetric Auxiliary Group. US Patent 9,598,458 B2, March 31, 2017.
- Fink B. E.; Zhao Y.; Chen L.; Huang A.. Cyclic dinucleotides as anticancer agents. International Patent WO/2019/079261, April 25, 2019.
- Schmidt M. A.; Wang Q.; Rogers A.; Cohen B.; Eastgate M.; Park H.; Ayers S.; Tai H.; Fu W.; He B.; Shackman J.; Paulson J.; Hritzko B.; Zhu G.; Guo S.; Freitag A.; Zhu Y.; Yuan C.; Yu M.; Treitler D.; Sezen-Edmonds M.; Purdum G.; Zhu J.; Hang C.; Zheng B. P(III) vs. P(V): A P(V) Reagent for Thiophosphoramidate Linkages and Application to An Asymmetric Synthesis of a Cyclic Dinucleotide STING Agonist. ChemRxiv 2021, 1. 10.26434/chemrxiv.14207360.v1. [DOI] [PubMed] [Google Scholar]
- Engels J.; Jäger A. Eine neue Synthese von Nukleosidmethylphosphonaten. Angew. Chem., Int. Ed. Engl. 1982, 21 (S12), 2010–2015. 10.1002/anie.198220100. [DOI] [Google Scholar]
- Helinski J.; Dabkowski W.; Michalski J. N,N-diisopropyl-O-P-nitrophenyl-P-methylphosphonoamidite: novel difunctional PIII reagent in oligonucleoside methylphosphonate synthesis containing 4-nitrophenoxy group. Tetrahedron Lett. 1991, 32 (37), 4981–4984. 10.1016/S0040-4039(00)93513-5. [DOI] [Google Scholar]
- Lebedev A. V.; Wenzinger G. R.; Wickstrom E. A new DMAP-catalyzed phosphonamidite coupling reaction for synthesis of oligonucleotide methylphosphonate derivatives. Tetrahedron Lett. 1990, 31 (6), 851–854. 10.1016/S0040-4039(00)94645-8. [DOI] [Google Scholar]
- Miller P. S.; Yano J.; Yano E.; Carroll C.; Jayaraman K.; Ts’o P. O. P. Nonionic nucleic acid analogs. Synthesis and characterization of dideoxyribonucleoside methylphosphonates. Biochemistry 1979, 18 (23), 5134–5143. 10.1021/bi00590a017. [DOI] [PubMed] [Google Scholar]
- Reynolds M. A.; Hogrefe R. I.; Jaeger J. A.; Schwartz D. A.; Riley T. A.; Marvin W. B.; Daily W. J.; Vaghefi M. M.; Beck T. A.; Knowles S. K.; Klem R. E.; Arnold L. J. Jr Synthesis and Thermodynamics of Oligonucleotides Containing Chirally Pure R P Methylphosphonate Linkages. Nucleic Acids Res. 1996, 24 (22), 4584–4591. 10.1093/nar/24.22.4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosmanitz P.; Eisenhardt S.; Bats J. W.; Engels J. W. New proline derived chiral building blocks for nucleoside methylphosphonate synthesis. Tetrahedron 1994, 50 (19), 5719–5734. 10.1016/S0040-4020(01)85640-4. [DOI] [Google Scholar]
- Schell P.; Engels J. W. Rp-Diastereoselective synthesis of dinucleoside methylphosphonates by the phosphoramidite approach. Tetrahedron Lett. 1998, 39 (47), 8629–8632. 10.1016/S0040-4039(98)01972-8. [DOI] [Google Scholar]
- Xu D.; Rivas-Bascón N.; Padial N. M.; Knouse K. W.; Zheng B.; Vantourout J. C.; Schmidt M. A.; Eastgate M. D.; Baran P. S. Enantiodivergent Formation of C-P Bonds: Synthesis of P-Chiral Phosphines and Methylphosphonate Oligonucleotides. J. Am. Chem. Soc. 2020, 142 (12), 5785–5792. 10.1021/jacs.9b13898. [DOI] [PubMed] [Google Scholar]
- He C.; Chu H.; Stratton T. P.; Kossler D.; Eberle K. J.; Flood D. T.; Baran P. S. Total Synthesis of Tagetitoxin. J. Am. Chem. Soc. 2020, 142 (32), 13683–13688. 10.1021/jacs.0c06641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spicer C. D.; Pujari-Palmer M.; Autefage H.; Insley G.; Procter P.; Engqvist H.; Stevens M. M. Synthesis of Phospho-Amino Acid Analogues as Tissue Adhesive Cement Additives. ACS Cent. Sci. 2020, 6 (2), 226–231. 10.1021/acscentsci.9b01149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domon K.; Puripat M.; Fujiyoshi K.; Hatanaka M.; Kawashima S. A.; Yamatsugu K.; Kanai M. Catalytic Chemoselective O-Phosphorylation of Alcohols. ACS Cent. Sci. 2020, 6 (2), 283–292. 10.1021/acscentsci.9b01272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt E. A.; Cal P. M. S. D.; Oliveira B. L.; Bernardes G. J. L. Contemporary approaches to site-selective protein modification. Nature Reviews Chemistry 2019, 3 (3), 147–171. 10.1038/s41570-019-0079-1. [DOI] [Google Scholar]
- Kolb H. C.; Finn M. G.; Sharpless K. B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem., Int. Ed. 2001, 40 (11), 2004–2021. . [DOI] [PubMed] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew. Chem., Int. Ed. 2009, 48 (38), 6974–6998. 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood D. T.; Asai S.; Zhang X.; Wang J.; Yoon L.; Adams Z. C.; Dillingham B. C.; Sanchez B. B.; Vantourout J. C.; Flanagan M. E.; Piotrowski D. W.; Richardson P.; Green S. A.; Shenvi R. A.; Chen J. S.; Baran P. S.; Dawson P. E. Expanding Reactivity in DNA-Encoded Library Synthesis via Reversible Binding of DNA to an Inert Quaternary Ammonium Support. J. Am. Chem. Soc. 2019, 141 (25), 9998–10006. 10.1021/jacs.9b03774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Lundberg H.; Asai S.; Martin-Acosta P.; Chen J. S.; Brown S.; Farrell W.; Dushin R. G.; O’Donnell C. J.; Ratnayake A. S.; Richardson P.; Liu Z.; Qin T.; Blackmond D. G.; Baran P. S. Kinetically guided radical-based synthesis of C(sp(3))-C(sp(3)) linkages on DNA. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (28), E6404–E6410. 10.1073/pnas.1806900115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood D. T.; Kingston C.; Vantourout J. C.; Dawson P. E.; Baran P. S. DNA Encoded Libraries: A Visitor’s Guide. Isr. J. Chem. 2020, 60 (3–4), 268–280. 10.1002/ijch.201900133. [DOI] [Google Scholar]
- Flood D. T.; Zhang X.; Fu X.; Zhao Z.; Asai S.; Sanchez B. B.; Sturgell E. J.; Vantourout J. C.; Richardson P.; Flanagan M. E.; Piotrowski D. W.; Kölmel D. K.; Wan J.; Tsai M.-H.; Chen J. S.; Baran P. S.; Dawson P. E. RASS-Enabled S/P–C and S–N Bond Formation for DEL Synthesis. Angew. Chem., Int. Ed. 2020, 59 (19), 7377–7383. 10.1002/anie.201915493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood D. T.; Knouse K. W.; Vantourout J. C.; Kitamura S.; Sanchez B. B.; Sturgell E. J.; Chen J. S.; Wolan D. W.; Baran P. S.; Dawson P. E. Synthetic Elaboration of Native DNA by RASS (SENDR). ACS Cent. Sci. 2020, 6 (10), 1789–1799. 10.1021/acscentsci.0c00680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolpashchikov D. M. Evolution of Hybridization Probes to DNA Machines and Robots. Acc. Chem. Res. 2019, 52 (7), 1949–1956. 10.1021/acs.accounts.9b00098. [DOI] [PubMed] [Google Scholar]
- deGruyter J. N.; Malins L. R.; Baran P. S. Residue-Specific Peptide Modification: A Chemist’s Guide. Biochemistry 2017, 56 (30), 3863–3873. 10.1021/acs.biochem.7b00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vantourout J. C.; Adusumalli S. R.; Knouse K. W.; Flood D. T.; Ramirez A.; Padial N. M.; Istrate A.; Maziarz K.; deGruyter J. N.; Merchant R. R.; Qiao J. X.; Schmidt M. A.; Deery M. J.; Eastgate M. D.; Dawson P. E.; Bernardes G. J. L.; Baran P. S. Serine-Selective Bioconjugation. J. Am. Chem. Soc. 2020, 142 (41), 17236–17242. 10.1021/jacs.0c05595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd D. B. Mechanism of hydrolysis of cyclic phosphate esters. J. Am. Chem. Soc. 1969, 91 (5), 1200–1205. 10.1021/ja01033a028. [DOI] [Google Scholar]
- Uchimaru T.; Stec W. J.; Taira K. Mechanism of the Chemoselective and Stereoselective Ring Opening of Oxathiaphospholanes: An Ab Initio Study. J. Org. Chem. 1997, 62 (17), 5793–5800. 10.1021/jo962220u. [DOI] [Google Scholar]
- Uchimaru T.; Stec W. J.; Tsuzuki S.; Hirose T.; Tanabe K.; Taira K. Ab initio investigation on nucleophilic ring opening of 1,3,2-oxathiaphospholane: nucleophilic substitution at phosphorus coupled with pseudorotation. Chem. Phys. Lett. 1996, 263 (5), 691–696. 10.1016/S0009-2614(96)01262-6. [DOI] [Google Scholar]
- Arantes G. M.; Chaimovich H. Thiolysis and Alcoholysis of Phosphate Tri- and Monoesters with Alkyl and Aryl Leaving Groups. An ab Initio Study in the Gas Phase. J. Phys. Chem. A 2005, 109 (25), 5625–5635. 10.1021/jp0449556. [DOI] [PubMed] [Google Scholar]
- Westheimer F. H. Pseudo-rotation in the hydrolysis of phosphate esters. Acc. Chem. Res. 1968, 1 (3), 70–78. 10.1021/ar50003a002. [DOI] [Google Scholar]
- Xu Y.-W.; Moréra S.; Janin J.; Cherfils J. AlF3 mimics the transition state of protein phosphorylation in the crystal structure of nucleoside diphosphate kinase and MgADP. Proc. Natl. Acad. Sci. U. S. A. 1997, 94 (8), 3579–3583. 10.1073/pnas.94.8.3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles A. F.; Kaplan N. O. Oxidative phosphorylation and ATPase activities of human tumor mitochondria. Biochim. Biophys. Acta, Bioenerg. 1980, 590 (2), 170–181. 10.1016/0005-2728(80)90022-5. [DOI] [PubMed] [Google Scholar]
- Grueninger D.; Schulz G. E. Structure and reaction mechanism of L-rhamnulose kinase from Escherichia coli. J. Mol. Biol. 2006, 359 (3), 787–97. 10.1016/j.jmb.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Østergaard M. E.; De Hoyos C. L.; Wan W. B.; Shen W.; Low A.; Berdeja A.; Vasquez G.; Murray S.; Migawa M. T.; Liang X.-h.; Swayze E. E.; Crooke S. T.; Seth P. P. Understanding the effect of controlling phosphorothioate chirality in the DNA gap on the potency and safety of gapmer antisense oligonucleotides. Nucleic Acids Res. 2020, 48 (4), 1691–1700. 10.1093/nar/gkaa031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahns H.; Roos M.; Imig J.; Baumann F.; Wang Y.; Gilmour R.; Hall J. Stereochemical bias introduced during RNA synthesis modulates the activity of phosphorothioate siRNAs. Nat. Commun. 2015, 6 (1), 6317. 10.1038/ncomms7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radzikowska E.; Baraniak J. Synthesis of PS/PO-chimeric oligonucleotides using mixed oxathiaphospholane and phosphoramidite chemistry. Org. Biomol. Chem. 2015, 13 (1), 269–276. 10.1039/C4OB01837K. [DOI] [PubMed] [Google Scholar]
- Wan W. B.; Seth P. P. The Medicinal Chemistry of Therapeutic Oligonucleotides. J. Med. Chem. 2016, 59 (21), 9645–9667. 10.1021/acs.jmedchem.6b00551. [DOI] [PubMed] [Google Scholar]
- Wiesler W. T.; Caruthers M. H. Synthesis of Phosphorodithioate DNA via Sulfur-Linked, Base-Labile Protecting Groups1. J. Org. Chem. 1996, 61 (13), 4272–4281. 10.1021/jo960274y. [DOI] [PubMed] [Google Scholar]
- Guo X.; Stolee J. A.; Fillon Y. A.; Zou L. F. Trace-Level Determination of Acrylonitrile Generated in the Manufacturing Process of Oligonucleotides by Static Headspace Gas Chromatography with an Electron Impact(+) Mass Detector. Org. Process Res. Dev. 2021, 25 (2), 318–326. 10.1021/acs.oprd.0c00527. [DOI] [Google Scholar]
- Capaldi D. C.; Gans H.; Krotz A. H.; Arnold J.; Carty R. L.; Moore M. N.; Scozzari A. N.; Lowery K.; Cole D. L.; Ravikumar V. T. Synthesis of high-quality antisense drugs. Addition of acrylonitrile to phosphorothioate oligonucleotides: Adduct characterization and avoidance. Org. Process Res. Dev. 2003, 7 (6), 832–838. 10.1021/op020090n. [DOI] [Google Scholar]
- Oka N.; Wada T.; Saigo K. An oxazaphospholidine approach for the stereocontrolled synthesis of oligonucleoside phosphorothioates. J. Am. Chem. Soc. 2003, 125 (27), 8307–17. 10.1021/ja034502z. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Knouse K. W.; Qiu S.; Hao W.; Padial N. M.; Vantourout J. C.; Zheng B.; Mercer S. E.; Lopez J. O.; Narayan R.; Olson R. E.; Blackmond D. G.; Eastgate M. D.; Schmidt M. A.; McDonald I. M.; Baran P. S. A P(V)-Platform for Oligonucleotide Synthesis. Science 2021, 10.1126/science.abi9727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G.; Fu W.; Zhang Z.; He Y.; Yu H.; Wang Y.; Wang X.; Zhao Y. L.; Deng Z.; Wu G.; He X. Structural basis for the recognition of sulfur in phosphorothioated DNA. Nat. Commun. 2018, 9 (1), 4689. 10.1038/s41467-018-07093-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J.; Sharpless K. B.; Kwisnek L.; Oakdale J. S.; Fokin V. V. SuFEx-Based Synthesis of Polysulfates. Angew. Chem., Int. Ed. 2014, 53 (36), 9466–9470. 10.1002/anie.201403758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J.; Krasnova L.; Finn M. G.; Sharpless K. B. Sulfur(VI) Fluoride Exchange (SuFEx): Another Good Reaction for Click Chemistry. Angew. Chem., Int. Ed. 2014, 53 (36), 9430–9448. 10.1002/anie.201309399. [DOI] [PubMed] [Google Scholar]
- Li S.; Wu P.; Moses J. E.; Sharpless K. B. Multidimensional SuFEx Click Chemistry: Sequential Sulfur(VI) Fluoride Exchange Connections of Diverse Modules Launched From An SOF4 Hub. Angew. Chem., Int. Ed. 2017, 56 (11), 2903–2908. 10.1002/anie.201611048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recently we have demonstrated how the PSI platform can be used to realize direct phosphorylation of alcohols:Ociepa M.; Knouse K. W.; He D.; Vantourout J. C.; Flood D. T.; Padial N. M.; Chen J. S.; Sanchez B. B.; Sturgell E. J.; Zheng B.; Qiu S.; Schmidt M. A.; Eastgate M. D.; Baran P. S.. Mild and Chemoselective Phosphorylation of Alcohols Using a PSI-Reagent. ChemRxiv 2021, 10.33774/chemrxiv-2021-4ksng. [DOI] [PMC free article] [PubMed]