

Abstract
Estrogens modulate different physiological functions, including reproduction, inflammation, bone formation, energy expenditure, and food intake. In this review, we highlight the effect of estrogens on food intake regulation and the latest literature on intracellular estrogen signaling. In addition, gut satiety hormones, such as cholecystokinin, glucagon-like peptide 1 and leptin are essential to regulate ingestive behaviors in the postprandial period. These peripheral signals are sensed by vagal afferent terminals in the gut wall and transmitted to the hindbrain axis. Here we 1. review the role of the vagus-hindbrain axis in response to gut satiety signals and 2. consider the potential synergistic effects of estrogens on gut satiety signals at the level of vagal afferent neurons and nuclei located in the hindbrain. Understanding the action of estrogens in gut-brain axis provides a potential strategy to develop estrogen-based therapies for metabolic diseases and emphasizes the importance of sex difference in the treatment of obesity.
Introduction
Overnutrition and changes in lifestyle have increased the incidence of obesity. The global prevalence of overweight and obese individuals is higher in women than men [1]. There are several factors including cultural factors, social behaviors, pregnancy and menopause that might contribute to this sex difference in the prevalence of obesity. Food intake and energy expenditure vary across the menstrual cycle and ovulation is followed by high energy expenditure and low food intake [2-5]. These observations suggest that ovarian hormones affect energy balance in women. Estradiol (E2), a major circulating estrogen, is mainly produced and secreted from the ovary in premenopausal women. Of note, other organs, such as brain, bone, muscle, and adipose tissue, synthesize small amounts of E2 that can act locally through paracrine pathways [6]. The termination of ovulation in menopause leads to a decrease in production of E2 and a dramatic drop of circulating E2. Postmenopausal women and ovariectomized (OVX) rodents have increased weight gain, with increased food intake and reduced energy expenditure [7-9]. It is interesting to note, however, that genetic deletion of aromatase, the key enzyme that synthesizes estrogens, increases body weight and adiposity in both male and female mice, suggesting estrogens have a sex-independent metabolic effect, but not androgens. [10]. Intact female rodents ingesting a high-fat diet (HFD) have less weight gain, adiposity and obese phenotype than males, suggesting ovulating females are generally protected from diet-induced obesity, possibly through estrogen signaling [11-16]. Thus, ovarian hormones, estrogens, have a negative effect on energy balance and seem to provide some metabolic protection.
Energy balance is controlled by multiple complex mechanisms that influence both food intake and energy expenditure. One mechanism that regulates food intake is satiety signals arising from the gastrointestinal tract during eating. There are two pathways that conduct satiety signals from the periphery to the central nervous system (CNS): 1) blood-borne mediated hormonal pathway and 2) the vagal afferent neuron mediated paracrine pathway. The interaction between estrogens and satiety signals in the CNS have been reviewed in other articles [17-19]. In this review, we highlight recent studies on how estrogens may modulate gut satiety signals at the level of vagus-hindbrain axis.
Estrogen, Estrogen Receptors, and the Effects on Energy Balance
In the classic mode of action, estrogens bind to the nuclear receptors, estrogen receptor alpha (ERα) and estrogen receptor beta (ERβ), acting as transcription factors and providing a slow-onset, long-lasting effect (Figure 1). Male and female mice with a global knockout of ERα have an increase in body weight and adiposity, with reduced energy expenditure [20]. It has also been shown that female ERα knockout mice have increased food intake and reduced responsiveness to the gut satiety hormone, cholecystokinin (CCK) [21]. These data suggest that ERα-mediated signaling has a negative effect on energy balance through reduced food intake and increased energy expenditure. In contrast, standard laboratory chow-diet-fed male and female mice with a global knockout of ERβ have no significant increase in body weight [22,23]. However, female mice with deletion of ERβ are prone to HFD-induced weight gain and increased adiposity [24], and consistent with this observation, administration of ERβ ligands are sufficient to eliminate HFD-induced weight gain and increased fat mass [25]. This ERβ-mediated anti-obesity effect is due to indirect suppression of peroxisome proliferation-activated receptor gamma (PPARγ) in adipocytes [24]. Taken together, these data suggest that the metabolically-protective effect of estrogens is mediated by both ERα and ERβ.
Figure 1. Estrogen receptor-mediated signaling in neurons.

Estrogen acting on classic estrogen nuclear receptor modulates the transcription of downstream genes. Acting on membrane-bound estrogen receptors, including GPER and mER, induces signaling cascade with a fast response in neurons. GPER is coupled with Gs subunit that conducts cAMP/PKA signaling. mER conjugates with RTK and mGluR and possibly modulates the signaling cascade of these two types of receptors, including JAK/STAT, MAPK, PI3K/Akt, cAMP/KPA, and DAG/IP3 signaling. GPER, G-protein-coupled estrogen receptor; RTK, receptor tyrosine kinase; ER, estrogen receptor; mGluR, metabotropic glutamate receptor; mER, membrane-associated estrogen receptor; PKA, protein kinase A; DAG, diacylglycerol; IP3, inositol trisphosphate.
In addition to acting at nuclear receptors, estrogens can induce a rapid signaling cascade by acting on the membrane-associated estrogen receptor (mER) alpha and beta in various cell types, including neurons [26]. Estrogens acting at mER interacts with two types of membrane receptors to initiate a signaling cascade: 1) Metabotropic glutamate receptors, coupled to either Gi/o or Gq, that either negatively or positively modulate the protein kinase cascade, including protein kinase A (PKA) and protein kinase C (PKC), as well as inducing inositol trisphosphate (IP3)-mediated Ca2+ influx [27]; 2) Receptor tyrosine kinase that signals via the PI3K/Akt and JAK/STAT pathways [28]. These signaling cascades may crosstalk with other intracellular signaling pathways and modulate neuronal excitability. In addition to the interaction with membrane receptors, in the hypothalamus protein phosphatase 2A is critical to mediate mER signaling in regulating energy expenditure [29].
In addition to mER-mediated rapid signaling, G-protein coupled estrogen receptor (GPER, formerly known as GPR30) is a membrane-bound receptor that mediates estrogen-induced activation of intracellular signaling cascades [30]. The evidence for a role of GPER in energy balance and regulation of body weight is contradictory. Female mice with global knockout of GPER show no difference in body weight during feeding of a standard laboratory chow diet [31-34]. Unexpectedly, female GPER knockout mice are resistant to high fat diet-induced obesity, due to an increase in energy expenditure [35]. These studies GPER-mediated estrogen signaling is necessary for weight gain at least during feeding of HFD. In contrast, other studies demonstrate that global knockout of GPER leads to an increase in body weight and fat mass during normal diet feeding, with no difference in food intake or physical activity [36-38]. The increased body weight in GPER knockout mice is primarily because of reduced energy expenditure from brown adipose tissue thermogenesis [38]. A recent study using the GPER agonist in OVX mice shows that activation of GPER is sufficient to significantly reduce OVX-induced weight gain and adiposity by increasing energy expenditure [39]. Taken together, these results suggest that estrogens acting on GPER might be involved in energy balance via upregulation of energy expenditure, but has no effect on food intake regulation.
Estrogen and the Neural Gut-brain Axis
The vagus nerve is a the neural pathway linking visceral organs to the CNS. The vagal pathway is bidirectional; vagal afferents conduct signals from visceral organs toward the CNS and vagal efferents relay commands from the CNS to the periphery (Figure 2). Vagal afferent neurons (VAN) are pseudobipolar neurons and cell bodies are located in the nodose and jugular ganglia. Recent studies using single-cell RNA sequencing reveal the heterogeneous population of VAN in response to different signals, such as nutrients, gastrointestinal distention, and pulmonary volume [40-43]. These studies match the understating of vagal afferent function in sensing and regulating respiratory and digestive system obtained from many years of electrophysiological and physiological studies [44-46]. In the gastrointestinal tract, there are three types of vagal afferent that are characterized by their terminal endings; intraganglionic laminar endings (IGLE), intramuscular arrays (IMA), and mucosal afferent endings. IGLE and IMA sense the stretch and distension of gastrointestinal tract [47]. The mucosal afferent endings are located in close proximity to the basolateral membrane of epithelium cells and enteroendocrine cells, and respond to mucosal stroking as well as luminal nutrient stimuli and epithelial factors, including hormones and many other neuromodulatory factors [47]. Anorexigenic signals secreted from gastrointestinal tract, such as CCK, glucagon-like peptide 1 (GLP1), peptide YY (PYY) and leptin, increase neuronal excitability and induce depolarization of VAN [48-51]. The depolarization of VAN induces the release of neuronal transmitters, including glutamate and cocaine and amphetamine regulated transcript (CART), from central terminals of vagal afferents in the hindbrain, which in turn activates the second order neurons in the nucleus of solitary tract (NTS) [52,53]. NTS neurons ultimately project the signals to different nuclei located in hindbrain and other brain regions, which influence overall feeding behavior [54].
Figure 2. Graphic illustration of vagus-hindbrain axis and ovarian hormones on sensing gastrointestinal satiety signals.
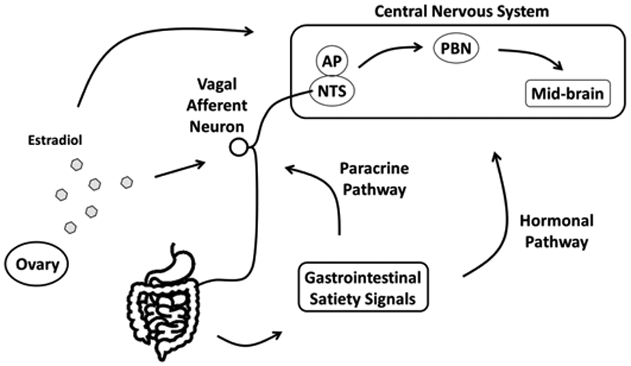
Meal-related gastrointestinal signals are paracrinally sensed by vagal afferent neurons (VAN), and the signals are projected secondary neurons located in the hindbrain, NTS and AP. The signals are integrated and conducted to high-order neurons, including PBN or nuclei in the mid-brain region, which regulates feeding behavior. Other than the paracrine pathway, gastrointestinal satiety signals are mediated by hormonal pathway and acting in the central nervous system (CNS). Circulating estradiol that secreted from ovary acting on both VAN and the nuclei located in the CNS modulates the responsiveness to gastrointestinal satiety signals. NTS, nucleus of the solitary tract; AP, area postrema; PBN, parabrachial nuclei.
The major form of estrogen receptor subtypes expressed in VAN is ERα, with much lower expression of ERβ and GPER [55]. The expression of ERα in VAN is positively regulated by plasma estradiol and the expression fluctuates through estrous cycle with highest levels in the estrus phase and lowest in the diestrus phase [56]. The density of axonal projections of vagal afferents in the hindbrain is also positively regulated by administration of E2 in OVX rats [57]. Replacement of E2 in OVX rats increases the excitability of myelinated vagal afferent neurons (A fiber) and this is mediated by GPER [58]. The mechanostimulation-induced excitability of gastric vagal afferents is potentiated by administration of E2 [55]. These results suggest that E2 modulates the primary functions of VAN, including neuronal projections and excitability. On the other hand, NTS neurons also express ERα, with much lower expression of ERβ [59]. In OVX rats replacement of E2 into the fourth ventricle have reduces food intake and this effect is blunted by co-administration of the ERα antagonist [60,61]. Taken together, these data suggest that estrogen signaling in both vagal afferent neurons and NTS neurons is also involved in the control of food intake. However, direct effects of ERα-mediated estrogen signaling on NTS neurons to change neuronal excitability has yet to be demonstrated.
Estrogens and Gut Hormones on Food Intake
Cholecystokinin
The major site of CCK synthesis and secretion is the small intestine, particularly the proximal duodenum although it is also found in neurons in several nuclei in the CNS, including the NTS. CCK has been extensively studied in the postprandial regulation of digestion and food intake, including secretion of bile acids and pancreatic enzymes, inhibition of gastric acid secretion and delay of gastric emptying [62]. Dietary lipid and amino acids stimulate the release of CCK from duodenal enteroendocrine cells. That CCK could act as a satiety signal was reported around the mid-1970s, when James Gibbs and his colleagues were the first to show that CCK inhibited food intake [63]. Abdominal vagotomy blunts CCK-induced satiety in rats; however, lesion of the ventromedial hypothalamus has no effect [64]. Peripheral terminals of vagal afferent neurons are located in the duodenal mucosa near enteroendocrine cells, suggesting that VAN might sense these gut signals locally [65]. This is further supported by evidence to show that there is no correlation between plasma CCK and suppression on food intake during intestinal infusion of different nutrients [66]. These data suggest that CCK-induced satiety is acting through a paracrine pathway and mainly mediated by vagal afferents, instead of via the blood-borne endocrine route.
There are two types of CCK receptor, type A (CCK1R) and type B (CCK2R); the CCK1R has higher affinity to sulfated CCK, and nonsulfated CCK mainly binds to CCK2R. Vagal afferents that innervate the duodenum expresses CCK1R [67,68]. The CCK1R antagonist, devazepide, blocks CCK-induced suppression on food intake, but the CCK2R antagonist has no effect [69,70]. These data suggest that CCK-induced satiety is mediated by CCK1R. CCK acting on CCK1R, a G-protein coupled receptor (GPCR) coupled to Gq induces IP3 signaling that initiates Ca2+ influx and depolarization of VAN [62]. In addition to the IP3-induced Ca2+ influx, other ion channels, including L-type calcium channel, transient receptor potential channel, A-type potassium channel, and calcium-activated chloride channel, are all involved in CCK-induced depolarization in VAN [71-74].
Exogenous CCK-induced satiety fluctuates with the estrous cycle; in rats CCK suppression of food intake is most potent during the estrus phase when plasma E2 is at its peak [75]. In OVX rats, subcutaneous replacement of E2 potentiates suppression of total food intake induced by CCK [76-78]. Other studies using the CCK1R antagonist, devazepide, have shown that estrogen potentiates endogenous CCK-induced suppression of food intake using both E2 replacement in OVX rats and determination of estrus phase in intact female rats [79-81]. These observations strongly support the hypothesis that E2 synergizes with CCK to suppress food intake. Female mice with global knockout of ERα have reduced CCK-induced activation of NTS neurons, which suggests that the synergism between estrogens and CCK is mediated by ERα at the level of vagal afferent neurons, rather than NTS neurons [21]. Although there is robust evidence to support the synergistic effect of estrogen, the intracellular mechanism remains unclear. Further studies are needed to determine whether there is crosstalk between mER-induced protein kinase cascade and CCK1R-induced signaling transduction or modulation of ion channels involved in CCK-induced neuronal depolarization (Figure 3). In addition to VAN, a significant portion of NTS neurons activated by CCK express ERα, suggesting that estrogen signaling in NTS neurons might also modulate the postsynaptic function in response to the input from vagal afferent-mediated anorexigenic signals [81].
Figure 3. Potential interaction between cholecystokinin (CCK) and estrogen signaling in neurons.
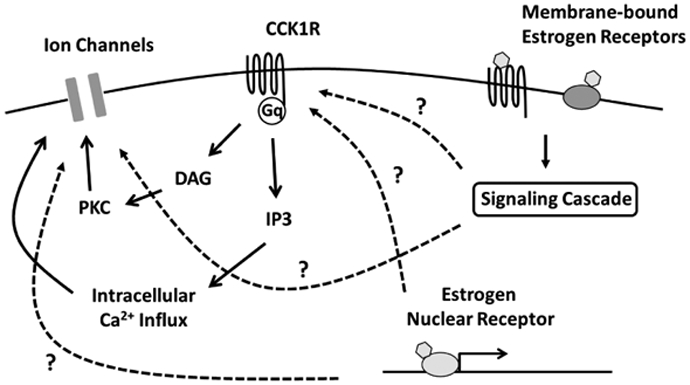
CCK acting on CCK1R induces intracellular Ca2+ influx and modulates activity of ion channels that involves in neuronal depolarization through DAG/IP3 signaling. Estrogens acting on membrane-bound estrogen receptors and estrogen nuclear receptor possibly enhances orexigenic action of CCK through modulation of CCK1R or the ion channels. CCK1R, cholecystokinin receptor type A; DAG, diacylglycerol; IP3, inositol trisphosphate; PKC, protein kinase C.
In addition to actions of peripheral CCK, a group of NTS neurons expressing CCK have axonal projections toward parabrachial nucleus (PBN) and paraventricular nucleus of the hypothalamus (PVH) [82]. Optogenetic stimulation of this group of NTS neurons leads to the suppression on food intake, with activation of neurons located in the PBN and PVH [83,84]. CCK acts directly on melanocortin-4 receptor neurons in the PVH that express CCK1R to induce neuronal depolarization, suggesting that central CCK serves as a neurotransmitter and might be involved in food intake regulation [83]. Interestingly, an earlier study shows that intracerebroventricular injection of E2 into the PVH increases the potency of CCK-induced satiety [85]. This suggests that the synergism between estrogens and CCK on food intake regulation is not only at the level of vagus-hindbrain axis but also at other regions in the CNS where CCK serves as a neurotransmitter.
Leptin
Leptin is an anorexigenic hormone secreted by adipose tissues, and the plasma level of leptin is positive correlated with fat mass. Of note in the context of discussion of the gut-brain axis, the stomach also synthesizes and secretes leptin in response to meal ingestion [86]. Leptin plays a critical role in long-term energy homeostasis by suppressing food intake and upregulating energy expenditure. Mice lacking leptin or the leptin receptor (LepR), ob/ob and db/db mice, respectively, have an increase in weight gain, adiposity, and food intake [87]. Leptin acting on the long-form of the LepR, a receptor tyrosine kinase, initiates a classic JAK-STAT signal transduction pathway and possibly activates the MAPK pathway [87]. Neurons in the midbrain (hypothalamus) and hindbrain expressing the LepR mediate the major portion of the anorexigenic action of leptin [88,89]. In addition to the hypothalamus as a site of leptin action, there is good evidence for an important role of leptin in the hindbrain. Administration of leptin directly into the NTS reduces food intake and body weight gain [90,91]. Moreover, knockdown of LepR by adeno-associated virus short hairpin RNA-interference in NTS neurons leads to increased food intake, which is associated with reduced responsiveness of gut satiety signal, CCK [92]. In addition to the CNS, leptin receptor is expressed on VAN that innervate the gastrointestinal tract, and it has been shown that leptin induces depolarization of VAN and synergizes with CCK-evoked Ca2+ influx [93,94]. Mice with a conditional deletion of the LepR in VAN are hyperphagic with decreased responsiveness to CCK-induced satiety [56,95]. These data suggest that both parts of vagus-hindbrain axis, vagal afferent and NTS neurons, respond to leptin and contribute to leptin-induced anorexia; furthermore, leptin modulates the potency of gut satiety signals in both sets of neurons.
Chronic HFD feeding leads to an increases in plasma leptin and bacterial lipopolysaccharide, and induces chronic immune activation and inflammation, which reduces leptin sensitivity, known as leptin resistance [89]. These factors are mediated by several intracellular signals, such as suppressor of cytokine signaling (SOCS3), protein tyrosine phosphatase 1B (PTP1B), and T-cell protein tyrosine phosphatase (TCPTP) and desensitizes the LepR in both peripheral and central neurons [96-99]. Leptin resistance leads to hyperphagia and an increase in body weight and fat mass [100,101].
It is controversial whether estrogens interact with leptin to induce its anorexigenic action. In intact female rats, the potency of chronic leptin-induced suppression on food intake does not fluctuate with estrus cycle [102]. In addition, replacement of E2 in OVX rats does not change the effects of chronic leptin on energy balance [103,104]. These data suggest there is no synergistic effect between estrogens and leptin. Recent studies show that E2-induced anorexigenic action is leptin signaling-independent in the hypothalamus, and replacement of E2 does not increase leptin-induced JAK-STAT signaling in the hypothalamus of OVX mice [105,106] (Figure 4). Another study demonstrates that E2 decreases about 50 % of food intake in ob/ob and db/db mice, and this effect is possibly mediated by mERα and JAK-STAT signaling transduction in hypothalamus [107,108]. In addition to mERα, activation of GPER also induces JAK-STAT signaling in the hypothalamus, which results in a decrease in food intake and weight gain [109]. These results suggest that E2 acting on membrane-bound estrogen receptors in the hypothalamus might mimic the anorexigenic action of leptin.
Figure 4. Interaction between leptin and estrogen signaling in neurons.
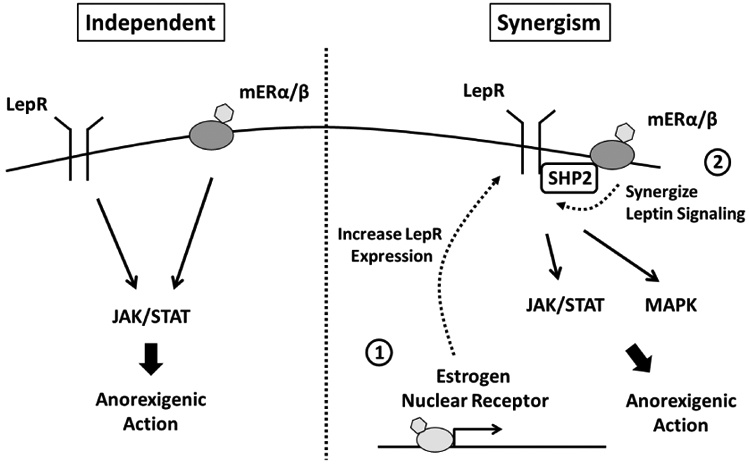
Estrogen acting on mER conducts the leptin-independent JAK/STAT signaling, suggesting that the estrogen and leptin signaling are parallel in mediation of anorexigenic action. In contrast, estrogens synergizes leptin signaling, including JAK/STAT and MAPK, through increases expression of leptin receptor ① and conjugation between mER and tyrosine phosphatase Shp2 ②. LepR, leptin receptor; mER, membrane-associated estrogen receptor.
On the other hand, E2 replacement in OVX rats does potentiate the anorexigenic action induced by administration of leptin into the third ventricle [110-112]. Systemic administration of E2 restores leptin sensitivity that is impaired by chronic consumption of HFD in the hypothalamus and prevents diet-induced weight gain, an effect that is accompanied by reduced food intake [113]. These studies show that E2 not only potentiates the anorexigenic action of leptin but also eliminates the hyperphagia caused by leptin resistance. There are two mechanisms that explain how estrogens modulate leptin potency in the hypothalamus; 1) Estrogens increase the expression of the long-form of the LepR, the form conducting JAK-STAT signaling [114,115] and 2) The signal transduction mechanism from mER crosstalk with leptin signaling through the linker, tyrosine phosphatase Shp2 [116] (Figure 4). Although LepR and ERα are both expressed in either NTS neurons or VAN, whether estrogens interact with leptin signaling at the level of vagus-hindbrain axis remains unclear.
Glucagon-like peptide 1 (GLP-1)
GLP-1 is a peptide produced in both peripheral tissues, primarily by the small intestine and proximal colon, and in NTS neurons in the central nervous system [117]. There is evidence to suggest that peripheral GLP-1 and stable GLP-1 analogs can cross the blood brain barrier and reach neurons located in CNS [118,119]. However, under physiological conditions, endogenous GLP-1 is rapidly degraded by dipeptidyl peptidase-4 [117]; therefore, current concepts suggest that the ability of peripheral GLP-1 to regulate food intake is mainly mediated in the periphery and by the vagal afferent pathway [120,121]. The main metabolic actions of GLP-1 includes improvement of glycemic control by inducing insulin secretion and suppression on food intake [117]. GLP-1 receptor (GLP-1R)-mediated suppression on food intake is reduced by subdiaphragmatic vagotomy [122,123] and vagal deafferentation [124-126]. Rats with specific knockdown of GLP-1R in VAN have increased meal size and reduced responsiveness to intraperitoneal injection of GLP-1 to reduce food intake [127]. This evidence supports that the concept that the vagal afferent pathway responds to peripheral GLP-1 and is the major pathway transmitting the satiety signal toward the CNS.
In addition to VAN, GLP-1R is expressed in various nuclei in CNS and mediates the effect of central GLP-1 on food intake regulation, including hindbrain, hypothalamus, hippocampus, and mesolimbic system; this topic is extensively reviewed elsewhere [128]. Intracerebroventricular injection of GLP-1R agonist into fourth ventricle of mice decreases food intake and body weight [129,130]. Knockdown of GLP-1R by adeno-associated virus short hairpin RNA-interference in NTS neurons leads to an increase in food intake and an increase in meal size [131]. These data suggest that GLP-1 in the NTS neurons can also regulate food intake. Another study shows that gastric distension-induced satiety is mediated by activation of GLP-1R signaling in NTS neurons [132]. A pharmacological study to determine the second-messenger pathways activated by GLP-1 in NTS neurons, demonstrated that GLP-1 induced suppression on food intake is mediated by PKA and MAPK [133]. Furthermore, injection of a GLP-1 agonist into the fourth ventricle reduces the intake of a high palatable diet and reduced the activation of the mesolimbic system and ventral tegmental areas, suggesting GLP-1 signaling in NTS neurons not only induces satiation but also suppresses food reward [130,134]. Overall, these studies suggest that both VAN and NTS neurons respond to GLP-1 and induces satiety signaling.
In OVX rats, replacement of E2 enhances the potency of peripheral GLP-1 induced-suppression on food intake, suggesting there is a synergistic effect between estrogens and GLP-1 on food intake regulation [135,136]. The peripheral administration of conjugated molecule of estrogen and GLP-1, specifically targeting estrogen signaling within the GLP-1R positive cells, shows a larger effect on ameliorating the symptoms of metabolic syndrome, including a decrease in body weight and fat mass as well as improvement of glycemic control, than administration of GLP-1 only in both male and female diet-induced obese mice [137,138]. This metabolic effect of GLP-1–estrogen conjugate is primarily mediated through reduced food intake, with no influence on energy expenditure and physical activity. As discussed above, the peripheral GLP-1 is largely mediated by vagal-hindbrain pathway; thus, the synergism between estrogens and GLP-1 on food intake regulation is likely mediated by VAN. Further studies are needed to elucidate the interaction between estrogens and GLP-1 at the level of VAN. Other than peripheral GLP-1, several studies show that E2 enhances central GLP-1 induced suppression on food intake in various nuclei in different region of brain, including PVH, supramammillary nucleus, and medial amygdala [139-141]. Neurons in the medial amygdala have been shown to be single-minded-1 expressing neurons. Taken together, these studies demonstrate that there could well be physiologically relevant interactions between estrogen and GLP-1 in the CNS and possibly at the level of VAN, and that estradiol enhances the satiety signal induced by GLP-1.
Conclusions and Perspectives
Sex differences in the regulation of food intake has been reported in several studies, and E2 is considered as a key factor leading to these observed sex differences. The current thinking is that the synergistic effect between E2 and gut satiety signals, including CCK, GLP-1, and leptin, results in different eating patterns between males and females. This review summarizes the latest studies of vagus-hindbrain axis, the major pathway sensing endogenous gut satiety signals, and highlights the effect of E2 on this axis. There is evidence to suggest that E2 potentiates the anorexigenic action of gut satiety signals at the level of VAN and NTS neurons. The understanding of E2 action on vagus-hindbrain axis provides a promising target site for estrogen-based medication for metabolic diseases.
The majority of studies suggest that the metabolic effect of E2 is slow in onset mediated by classic nuclear receptor ERα. However, recent studies show E2 acting on mER and GPER to induce protein kinase cascade that might modulates ion channel activity raises the potential of possible crosstalk with other signaling transduction. This suggests that mER- and GPER-mediated signaling might directly modulate neuronal excitability and synergize with other signal transduction pathways. Further studies are needed to elucidate the role of membrane-bound estrogen receptors in regulation of energy balance.
Studies using the GLP-1–estrogen conjugates have revealed a potential new peptide-based medication for obesity and diabetes. Conjugation with GLP-1 eliminates the risk of estrogen-induced carcinogenesis, through targeting of GLP-1R positive cells. Similarly, estrogen enhances the anorexigenic action of GLP-1, which produces more weight loss and improvement of metabolic phenotype than GLP-1 only. Ligand conjugation might improve the specificity of estrogen effect on energy balance, by targeting the cells regulating food intake and energy expenditure. In addition, E2 has leptin-like effect on energy balance and the ability to restore leptin sensitivity in neurons, which shows a therapeutic potential of E2 replacement in treating energy imbalance caused by leptin resistance.
Acknowledgements:
Work was supported by NIH DDK 41004 (HR)
References
- [1].Garawi F, Devries K, Thorogood N, Uauy R, Global differences between women and men in the prevalence of obesity: is there an association with gender inequality?, Eur. J. Clin. Nutr 68 (2014) 1101–1106. doi: 10.1038/ejcn.2014.86. [DOI] [PubMed] [Google Scholar]
- [2].Gong EJ, Garrel D, Calloway DH, Menstrual cycle and voluntary food intake., Am. J. Clin. Nutr 49 (1989) 252–258. doi: 10.1093/ajcn/49.2.252. [DOI] [PubMed] [Google Scholar]
- [3].Lyons PM, Truswell AS, Mira M, Vizzard J, Abraham SF, Reduction of food intake in the ovulatory phase of the menstrual cycle., Am. J. Clin. Nutr 49 (1989) 1164–1168. doi: 10.1093/ajcn/49.6.1164. [DOI] [PubMed] [Google Scholar]
- [4].Fong AK, Kretsch MJ, Changes in dietary intake, urinary nitrogen, and urinary volume across the menstrual cycle., Am. J. Clin. Nutr 57 (1993) 43–46. doi: 10.1093/ajcn/57.1.43. [DOI] [PubMed] [Google Scholar]
- [5].Day DS, Gozansky WS, VanPelt RE, Schwartz RS, Kohrt WM, Sex hormone suppression reduces resting energy expenditure and {beta}-adrenergic support of resting energy expenditure., J. Clin. Endocrinol. Metab 90 (2005) 3312–3317. doi: 10.1210/jc.2004-1344. [DOI] [PubMed] [Google Scholar]
- [6].Simpson ER, Sources of estrogen and their importance, J. Steroid Biochem. Mol. Biol 86 (2003) 225–230. doi: 10.1016/S0960-0760(03)00360-1. [DOI] [PubMed] [Google Scholar]
- [7].Asarian L, Geary N, Cyclic estradiol treatment normalizes body weight and restores physiological patterns of spontaneous feeding and sexual receptivity in ovariectomized rats., Horm. Behav 42 (2002) 461–471. doi: 10.1006/hbeh.2002.1835. [DOI] [PubMed] [Google Scholar]
- [8].Lovejoy JC, Champagne CM, deJonge L, Xie H, Smith SR, Increased visceral fat and decreased energy expenditure during the menopausal transition., Int. J. Obes. (Lond) 32 (2008) 949–958. doi: 10.1038/ijo.2008.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rogers NH, Perfield JW 2nd, Strissel KJ, Obin MS, Greenberg AS, Reduced energy expenditure and increased inflammation are early events in the development of ovariectomy-induced obesity., Endocrinology. 150 (2009) 2161–2168. doi: 10.1210/en.2008-1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Jones MEE, Thorburn AW, Britt KL, Hewitt KN, Wreford NG, Proietto J, Oz OK, Leury BJ, Robertson KM, Yao S, Simpson ER, Aromatase-deficient (ArKO) mice have a phenotype of increased adiposity, Proc. Natl. Acad. Sci 97 (2000) 12735–12740. doi: 10.1073/pnas.97.23.12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hwang L-L, Wang C-H, Li T-L, Chang S-D, Lin L-C, Chen C-P, Chen C-T, Liang K-C, Ho I-K, Yang W-S, Chiou L-C, Sex Differences in High-fat Diet-induced Obesity, Metabolic Alterations and Learning, and Synaptic Plasticity Deficits in Mice, Obesity. 18 (2010) 463–469. doi: 10.1038/oby.2009.273. [DOI] [PubMed] [Google Scholar]
- [12].Pettersson US, Waldén TB, Carlsson P-O, Jansson L, Phillipson M, Female Mice are Protected against High-Fat Diet Induced Metabolic Syndrome and Increase the Regulatory T Cell Population in Adipose Tissue, PLoS One. 7 (2012) e46057. doi: 10.1371/journal.pone.0046057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ingvorsen C, Karp NA, Lelliott CJ, The role of sex and body weight on the metabolic effects of high-fat diet in C57BL/6N mice., Nutr. Diabetes 7 (2017) e261. doi: 10.1038/nutd.2017.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bruder-Nascimento T, Ekeledo OJ, Anderson R, Le HB, Belin de Chantemèle EJ, Long Term High Fat Diet Treatment: An Appropriate Approach to Study the Sex-Specificity of the Autonomic and Cardiovascular Responses to Obesity in Mice, Front. Physiol 8 (2017) 32. doi: 10.3389/fphys.2017.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Paruthiyil S, Hagiwara S-I, Kundassery K, Bhargava A, Sexually dimorphic metabolic responses mediated by CRF2 receptor during nutritional stress in mice., Biol. Sex Differ 9 (2018) 49. doi: 10.1186/s13293-018-0208-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Huang K-P, Ronveaux CC, Knotts TA, Rutkowsky JR, Ramsey JJ, Raybould HE, Sex Differences in Response to Short-Term High Fat Diet in Mice, Physiol. Behav (2020) 112894. doi: 10.1016/j.physbeh.2020.112894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mauvais-Jarvis F, Clegg DJ, Hevener AL, The Role of Estrogens in Control of Energy Balance and Glucose Homeostasis, Endocr. Rev 34 (2013) 309–338. doi: 10.1210/er.2012-1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].López M, Tena-Sempere M, Estrogens and the control of energy homeostasis: a brain perspective, Trends Endocrinol. Metab 26 (2015) 411–421. doi: 10.1016/j.tem.2015.06.003. [DOI] [PubMed] [Google Scholar]
- [19].Leeners B, Tobler PN, Asarian L, Geary N, Ovarian hormones and obesity, Hum. Reprod. Update 23 (2017) 300–321. doi: 10.1093/humupd/dmw045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS, Increased adipose tissue in male and female estrogen receptor-α knockout mice, Proc. Natl. Acad. Sci 97 (2000) 12729 LP–12734. doi: 10.1073/pnas.97.23.12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Geary N, Asarian L, Korach KS, Pfaff DW, Ogawa S, Deficits in E2-Dependent Control of Feeding, Weight Gain, and Cholecystokinin Satiation in ER-α Null Mice, Endocrinology. 142 (2001) 4751–4757. doi: 10.1210/endo.142.11.8504. [DOI] [PubMed] [Google Scholar]
- [22].Ohlsson C, Hellberg N, Parini P, Vidal O, Bohlooly M, Rudling M, Lindberg MK, Warner M, Angelin B, Gustafsson J-Å, Obesity and Disturbed Lipoprotein Profile in Estrogen Receptor-α-Deficient Male Mice, Biochem. Biophys. Res. Commun 278 (2000) 640–645. doi: 10.1006/bbrc.2000.3827. [DOI] [PubMed] [Google Scholar]
- [23].Windahl SH, Hollberg K, Vidal O, Gustafsson J-Å, Ohlsson C, Andersson G, Female Estrogen Receptor β−/− Mice Are Partially Protected Against Age-Related Trabecular Bone Loss, J. Bone Miner. Res 16 (2001) 1388–1398. doi: 10.1359/jbmr.2001.16.8.1388. [DOI] [PubMed] [Google Scholar]
- [24].Foryst-Ludwig A, Clemenz M, Hohmann S, Hartge M, Sprang C, Frost N, Krikov M, Bhanot S, Barros R, Morani A, Gustafsson J-Å, Unger T, Kintscher U, Metabolic Actions of Estrogen Receptor Beta (ERβ) are Mediated by a Negative Cross-Talk with PPARγ, PLOS Genet. 4 (2008) e1000108. 10.1371/journal.pgen.1000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yepuru M, Eswaraka J, Kearbey JD, Barrett CM, Raghow S, Veverka KA, Miller DD, Dalton JT, Narayanan R, Estrogen Receptor-β-selective Ligands Alleviate High-fat Diet- and Ovariectomy-induced Obesity in Mice, J. Biol. Chem . 285 (2010) 31292–31303. doi: 10.1074/jbc.M110.147850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Micevych P, Dominguez R, Membrane estradiol signaling in the brain, Front. Neuroendocrinol 30 (2009) 315–327. doi: 10.1016/j.yfrne.2009.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tonn Eisinger KR, Gross KS, Head BP, Mermelstein PG, Interactions between estrogen receptors and metabotropic glutamate receptors and their impact on drug addiction in females, Horm. Behav 104 (2018) 130–137. doi: 10.1016/j.yhbeh.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Arnal J-F, Lenfant F, Metivier R, Flouriot G, Henrion D, Adlanmerini M, Fontaine C, Gourdy P, Chambon P, Katzenellenbogen B, Katzenellenbogen J, Membrane and Nuclear Estrogen Receptor Alpha Actions: From Tissue Specificity to Medical Implications, Physiol. Rev 97 (2017) 1045–1087. doi: 10.1152/physrev.00024.2016. [DOI] [PubMed] [Google Scholar]
- [29].Ueda K, Takimoto E, Lu Q, Liu P, Fukuma N, Adachi Y, Suzuki R, Chou S, Baur W, Aronovitz MJ, Greenberg AS, Komuro I, Karas RH, Membrane-Initiated Estrogen Receptor Signaling Mediates Metabolic Homeostasis via Central Activation of Protein Phosphatase 2A, Diabetes. 67 (2018) 1524 LP–1537. doi: 10.2337/db17-1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ, Estrogen Signaling through the Transmembrane G Protein–Coupled Receptor GPR30, Annu. Rev. Physiol 70 (2008) 165–190. doi: 10.1146/annurev.physiol.70.113006.100518. [DOI] [PubMed] [Google Scholar]
- [31].Mårtensson UEA, Salehi SA, Windahl S, Gomez MF, Swärd K, Daszkiewicz-Nilsson J, Wendt A, Andersson N, Hellstrand P, Grände P-O, Owman C, Rosen CJ, Adamo ML, Lundquist I, Rorsman P, Nilsson B-O, Ohlsson C, Olde B, Leeb-Lundberg LMF, Deletion of the G Protein-Coupled Receptor 30 Impairs Glucose Tolerance, Reduces Bone Growth, Increases Blood Pressure, and Eliminates Estradiol-Stimulated Insulin Release in Female Mice, Endocrinology. 150 (2009) 687–698. doi: 10.1210/en.2008-0623. [DOI] [PubMed] [Google Scholar]
- [32].Otto C, Fuchs I, Kauselmann G, Kern H, Zevnik B, Andreasen P, Schwarz G, Altmann H, Klewer M, Schoor M, Vonk R, Fritzemeier K-H, GPR30 Does Not Mediate Estrogenic Responses in Reproductive Organs in Mice, Biol. Reprod 80 (2009) 34–41. doi: 10.1095/biolreprod.108.071175. [DOI] [PubMed] [Google Scholar]
- [33].Isensee J, Meoli L, Zazzu V, Nabzdyk C, Witt H, Soewarto D, Effertz K, Fuchs H, Gailus-Durner V, Busch D, Adler T, deAngelis MH, Irgang M, Otto C, Noppinger PR, Expression Pattern of G Protein-Coupled Receptor 30 in LacZ Reporter Mice, Endocrinology. 150 (2009) 1722–1730. doi: 10.1210/en.2008-1488. [DOI] [PubMed] [Google Scholar]
- [34].Kastenberger I, Schwarzer C, GPER1 (GPR30) knockout mice display reduced anxiety and altered stress response in a sex and paradigm dependent manner, Horm. Behav 66 (2014) 628–636. doi: 10.1016/j.yhbeh.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wang A, Luo J, Moore W, Alkhalidy H, Wu L, Zhang J, Zhen W, Wang Y, Clegg DJ, Xu B, Cheng Z, McMillan RP, Hulver MW, Liu D, GPR30 regulates diet-induced adiposity in female mice and adipogenesis in vitro, Sci. Rep 6 (2016) 34302. doi: 10.1038/srep34302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Haas E, Bhattacharya I, Brailoiu E, Damjanović M, Brailoiu GC, Gao X, Mueller-Guerre L, Marjon NA, Gut A, Minotti R, Meyer MR, Amann K, Ammann E, Perez-Dominguez A, Genoni M, Clegg DJ, Dun NJ, Resta TC, Prossnitz ER, Barton M, Regulatory role of G protein-coupled estrogen receptor for vascular function and obesity, Circ. Res 104 (2009) 288–291. doi: 10.1161/CIRCRESAHA.108.190892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sharma G, Hu C, Brigman JL, Zhu G, Hathaway HJ, Prossnitz ER, GPER Deficiency in Male Mice Results in Insulin Resistance, Dyslipidemia, and a Proinflammatory State, Endocrinology. 154 (2013) 4136–4145. doi: 10.1210/en.2013-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Davis KE, Carstens EJ, Irani BG, Gent LM, Hahner LM, Clegg DJ, Sexually dimorphic role of G protein-coupled estrogen receptor (GPER) in modulating energy homeostasis, Horm. Behav 66 (2014) 196–207. doi: 10.1016/j.yhbeh.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sharma G, Hu C, Staquicini DI, Brigman JL, Liu M, Mauvais-Jarvis F, Pasqualini R, Arap W, Arterburn JB, Hathaway HJ, Prossnitz ER, Preclinical efficacy of the GPER-selective agonist G-1 in mouse models of obesity and diabetes, Sci. Transl. Med 12 (2020) eaau5956. doi: 10.1126/scitranslmed.aau5956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chang RB, Strochlic DE, Williams EK, Umans BD, Liberles SD, Vagal Sensory Neuron Subtypes that Differentially Control Breathing, Cell. 161 (2015) 622–633. doi: 10.1016/j.cell.2015.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Williams EK, Chang RB, Strochlic DE, Umans BD, Lowell BB, Liberles SD, Sensory Neurons that Detect Stretch and Nutrients in the Digestive System, Cell. 166 (2016) 209–221. doi: 10.1016/j.cell.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bai L, Mesgarzadeh S, Ramesh KS, Huey EL, Liu Y, Gray LA, Aitken TJ, Chen Y, Beutler LR, Ahn JS, Madisen L, Zeng H, Krasnow MA, Knight ZA, Genetic Identification of Vagal Sensory Neurons That Control Feeding, Cell. 179 (2019) 1129–1143.e23. doi: 10.1016/j.cell.2019.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kupari J, Häring M, Agirre E, Castelo-Branco G, Ernfors P, An Atlas of Vagal Sensory Neurons and Their Molecular Specialization, Cell Rep. 27 (2019) 2508–2523.e4. doi: 10.1016/j.celrep.2019.04.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zhu JX, Wu XY, Owyang C, Li Y, Intestinal serotonin acts as a paracrine substance to mediate vagal signal transmission evoked by luminal factors in the rat, J. Physiol 530 (2001) 431–442. doi: 10.1111/j.1469-7793.2001.0431k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Brookes SJH, Spencer NJ, Costa M, Zagorodnyuk VP, Extrinsic primary afferent signalling in the gut, Nat. Rev. Gastroenterol. Hepatol 10 (2013) 286–296. doi: 10.1038/nrgastro.2013.29. [DOI] [PubMed] [Google Scholar]
- [46].Mazzone SB, Undem BJ, Vagal Afferent Innervation of the Airways in Health and Disease, Physiol. Rev 96 (2016) 975–1024. doi: 10.1152/physrev.00039.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Waise TMZ, Dranse HJ, Lam TKT, The metabolic role of vagal afferent innervation, Nat. Rev. Gastroenterol. Hepatol 15 (2018) 625–636. doi: 10.1038/s41575-018-0062-1. [DOI] [PubMed] [Google Scholar]
- [48].Dun NJ, Wu SY, Lin CW, Excitatory effects of cholecystokinin octapeptide on rat nodose ganglion cells in vitro, Brain Res. 556 (1991) 161–164. doi: 10.1016/0006-8993(91)90562-A. [DOI] [PubMed] [Google Scholar]
- [49].Gaisano GG, Park SJ, Daly DM, Beyak MJ, Glucagon-like peptide-1 inhibits voltage-gated potassium currents in mouse nodose ganglion neurons, Neurogastroenterol. Motil 22 (2010) 470–e111. doi: 10.1111/j.1365-2982.2009.01430.x. [DOI] [PubMed] [Google Scholar]
- [50].Koda S, Date Y, Murakami N, Shimbara T, Hanada T, Toshinai K, Niijima A, Furuya M, Inomata N, Osuye K, Nakazato M, The Role of the Vagal Nerve in Peripheral PYY3–36-Induced Feeding Reduction in Rats, Endocrinology. 146 (2005) 2369–2375. doi: 10.1210/en.2004-1266. [DOI] [PubMed] [Google Scholar]
- [51].Peters JH, Ritter RC, Simasko SM, Leptin and CCK modulate complementary background conductances to depolarize cultured nodose neurons, Am. J. Physiol. Physiol 290 (2006) C427–C432. doi: 10.1152/ajpcell.00439.2005. [DOI] [PubMed] [Google Scholar]
- [52].deLartigue G, Putative roles of neuropeptides in vagal afferent signaling, Physiol. Behav 136 (2014) 155–169. doi: 10.1016/j.physbeh.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lee SJ, Krieger J-P, Vergara M, Quinn D, McDougle M, deAraujo A, Darling R, Zollinger B, Anderson S, Pan A, Simonnet EJ, Pignalosa A, Arnold M, Singh A, Langhans W, Raybould HE, deLartigue G, Blunted Vagal Cocaine- and Amphetamine-Regulated Transcript Promotes Hyperphagia and Weight Gain, Cell Rep. 30 (2020) 2028–2039.e4. doi: 10.1016/j.celrep.2020.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Grill HJ, Hayes MR, Hindbrain Neurons as an Essential Hub in the Neuroanatomically Distributed Control of Energy Balance, Cell Metab. 16 (2012) 296–309. doi: 10.1016/j.cmet.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kentish SJ, Frisby CA, Wittert GA, Page AJ, OP291 Modulation of gastric vagal afferent satiety signals by the mouse oesterus cycle and 17B-oestradiol: UEG Week 2014 Oral Presentations, United Eur. Gastroenterol. Conf (2014) A92–A93. doi: 10.1177/2050640614548974. [DOI] [Google Scholar]
- [56].Huang K-P, Ronveaux CC, deLartigue G, Geary N, Asarian L, Raybould HE, Deletion of leptin receptors in vagal afferent neurons disrupts estrogen signaling, body weight, food intake and hormonal controls of feeding in female mice., Am. J. Physiol. Endocrinol. Metab 316 (2019) E568–E577. doi: 10.1152/ajpendo.00296.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ciriello J, Caverson MM, Effect of estrogen on vagal afferent projections to the brainstem in the female, Brain Res. 1636 (2016) 21–42. doi: 10.1016/j.brainres.2016.01.041. [DOI] [PubMed] [Google Scholar]
- [58].Qiao G-F, Li B-Y, Lu Y-J, Fu Y-L, Schild JH, 17β-Estradiol restores excitability of a sexually dimorphic subset of myelinated vagal afferents in ovariectomized rats, Am. J. Physiol. Physiol 297 (2009) C654–C664. doi: 10.1152/ajpcell.00059.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Schlenker EH, Hansen SN, Sex-specific densities of estrogen receptors alpha and beta in the subnuclei of the nucleus tractus solitarius, hypoglossal nucleus and dorsal vagal motor nucleus weanling rats, Brain Res. 1123 (2006) 89–100. doi: 10.1016/j.brainres.2006.09.035. [DOI] [PubMed] [Google Scholar]
- [60].Thammacharoen S, Lutz TA, Geary N, Asarian L, Hindbrain Administration of Estradiol Inhibits Feeding and Activates Estrogen Receptor-α-Expressing Cells in the Nucleus Tractus Solitarius of Ovariectomized Rats, Endocrinology. 149 (2008) 1609–1617. doi: 10.1210/en.2007-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Thammacharoen S, Kitchanukitwattana P, Suwanapaporn P, Chaiyabutr N, Effects of Hindbrain Infusion of an Estrogen Receptor Antagonist on Estrogenic Modulation of Eating Behavior, Neurophysiology. 49 (2017) 72–77. doi: 10.1007/s11062-017-9631-0. [DOI] [Google Scholar]
- [62].Dufresne M, Seva C, Fourmy D, Cholecystokinin and Gastrin Receptors, Physiol. Rev 86 (2006) 805–847. doi: 10.1152/physrev.00014.2005. [DOI] [PubMed] [Google Scholar]
- [63].GIBBS J, YOUNG RC, SMITH GP, Cholecystokinin elicits Satiety in Rats with Open Gastric Fistulas, Nature. 245 (1973) 323–325. doi: 10.1038/245323a0. [DOI] [PubMed] [Google Scholar]
- [64].Smith GP, Jerome C, Cushin BJ, Eterno R, Simansky KJ, Abdominal vagotomy blocks the satiety effect of cholecystokinin in the rat, Science (80-. ) 213 (1981) 1036 LP–1037. doi: 10.1126/science.7268408. [DOI] [PubMed] [Google Scholar]
- [65].Berthoud H-R, Patterson LM, Anatomical Relationship between Vagal Afferent Fibers and CCK-Immunoreactive Entero-Endocrine Cells in the Rat Small Intestinal Mucosa, Cells Tissues Organs. 156 (1996) 123–131. doi: 10.1159/000147837. [DOI] [PubMed] [Google Scholar]
- [66].Brenner L, Yox DP, Ritter RC, Suppression of sham feeding by intraintestinal nutrients is not correlated with plasma cholecystokinin elevation, Am. J. Physiol. Integr. Comp. Physiol 264 (1993) R972–R976. doi: 10.1152/ajpregu.1993.264.5.R972. [DOI] [PubMed] [Google Scholar]
- [67].Sternini C, Wong H, Pham T, deGiorgio R, Miller LJ, Kuntz SM, Reeve JR Jr., Walsh JH, Raybould HE, Expression of cholecystokinin a receptors in neurons innervating the rat stomach and intestine, Gastroenterology. 117 (1999) 1136–1146. doi: 10.1016/S0016-5085(99)70399-9. [DOI] [PubMed] [Google Scholar]
- [68].Patterson LM, Zheng H, Berthoud H-R, Vagal afferents innervating the gastrointestinal tract and CCKA-receptor immunoreactivity, Anat. Rec 266 (2002) 10–20. doi: 10.1002/ar.10026. [DOI] [PubMed] [Google Scholar]
- [69].Cooper SJ, Dourish CT, Clifton PG, CCK antagonists and CCK-monoamine interactions in the control of satiety, Am. J. Clin. Nutr 55 (1992) 291S–295S. doi: 10.1093/ajcn/55.1.291s. [DOI] [PubMed] [Google Scholar]
- [70].Reidelberger RD, Hernandez J, Fritzsch B, Hulce M, Abdominal vagal mediation of the satiety effects of CCK in rats, Am. J. Physiol. Integr. Comp. Physiol 286 (2004) R1005–R1012. doi: 10.1152/ajpregu.00646.2003. [DOI] [PubMed] [Google Scholar]
- [71].Lankisch TO, Tsunoda Y, Lu Y, Owyang C, Characterization of CCKA receptor affinity states and Ca2+ signal transduction in vagal nodose ganglia, Am. J. Physiol. Liver Physiol 282 (2002) G1002–G1008. doi: 10.1152/ajpgi.00313.2001. [DOI] [PubMed] [Google Scholar]
- [72].Burdakov D, Ashcroft FM, Cholecystokinin Tunes Firing of an Electrically Distinct Subset of Arcuate Nucleus Neurons by Activating A-Type Potassium Channels, J. Neurosci 22 (2002) 6380 LP–6387. doi: 10.1523/JNEUROSCI.22-15-06380.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Zhao H, Simasko SM, Role of Transient Receptor Potential Channels in Cholecystokinin-Induced Activation of Cultured Vagal Afferent Neurons, Endocrinology. 151 (2010) 5237–5246. doi: 10.1210/en.2010-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Wang R, Lu Y, Cicha MZ, VSingh M, Benson CJ, Madden CJ, Chapleau MW, Abboud FM, TMEM16B determines cholecystokinin sensitivity of intestinal vagal afferents of nodose neurons, JCI Insight. 4 (2019). doi: 10.1172/jci.insight.122058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Dulawa SC, Vanderweele DA, Cholecystokinin and estradiol synergistically potentiate satiety in rats, Peptides. 15 (1994) 913–918. doi: 10.1016/0196-9781(94)90050-7. [DOI] [PubMed] [Google Scholar]
- [76].Lindén A, Uvnäs-Moberg K, Forsberg G, Bednar I, Södersten P, Involvement of Cholecystokinin in Food Intake: III. Oestradiol Potentiates the Inhibitory Effect of Cholecystokinin Octapeptide on Food Intake in Ovariectomized Rats, J. Neuroendocrinol 2 (1990) 797–801. doi: 10.1111/j.1365-2826.1990.tb00643.x. [DOI] [PubMed] [Google Scholar]
- [77].Butera PC, Bradway DM, Cataldo NJ, Modulation of the satiety effect of cholecystokinin by estradiol, Physiol. Behav 53 (1993) 1235–1238. doi: 10.1016/0031-9384(93)90387-U. [DOI] [PubMed] [Google Scholar]
- [78].Geary N, Trace D, McEwen B, Smith GP, Cyclic estradiol replacement increases the satiety effect of CCK-8 in ovariectomized rats, Physiol. Behav 56 (1994) 281–289. doi: 10.1016/0031-9384(94)90196-1. [DOI] [PubMed] [Google Scholar]
- [79].Eckel LA, Geary N, Endogenous cholecystokinin’s satiating action increases during estrus in female rats, Peptides. 20 (1999) 451–456. doi: 10.1016/S0196-9781(99)00025-X. [DOI] [PubMed] [Google Scholar]
- [80].Asarian L, Geary N, Cyclic estradiol treatment phasically potentiates endogenous cholecystokinin’s satiating action in ovariectomized rats, Peptides. 20 (1999) 445–450. doi: 10.1016/S0196-9781(99)00024-8. [DOI] [PubMed] [Google Scholar]
- [81].Asarian L, Geary N, Estradiol Enhances Cholecystokinin-Dependent Lipid-Induced Satiation and Activates Estrogen Receptor-α-Expressing Cells in the Nucleus Tractus Solitarius of Ovariectomized Rats, Endocrinology. 148 (2007) 5656–5666. doi: 10.1210/en.2007-0341. [DOI] [PubMed] [Google Scholar]
- [82].Roman CW, Sloat SR, Palmiter RD, A tale of two circuits: CCKNTS neuron stimulation controls appetite and induces opposing motivational states by projections to distinct brain regions, Neuroscience. 358 (2017) 316–324. doi: 10.1016/j.neuroscience.2017.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].D’Agostino G, Lyons DJ, Cristiano C, Burke LK, Madara JC, Campbell JN, Garcia AP, Land BB, Lowell BB, Dileone RJ, Heisler LK, Appetite controlled by a cholecystokinin nucleus of the solitary tract to hypothalamus neurocircuit, Elife. 5 (2016) e12225. doi: 10.7554/eLife.12225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Roman CW, Derkach VA, Palmiter RD, Genetically and functionally defined NTS to PBN brain circuits mediating anorexia, Nat. Commun 7 (2016) 11905. doi: 10.1038/ncomms11905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Butera PC, Xiong M, Davis RJ, Platania SP, Central implants of dilute estradiol enhance the satiety effect of CCK-8., Behav. Neurosci 110 (1996) 823–830. doi: 10.1037//0735-7044.110.4.823. [DOI] [PubMed] [Google Scholar]
- [86].Cammisotto P, Bendayan M, A review on gastric leptin: the exocrine secretion of a gastric hormone, Anat Cell Biol. 45 (2012) 1–16. doi: 10.5115/acb.2012.45.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Allison MB, Myers MG, 20 YEARS OF LEPTIN: Connecting leptin signaling to biological function, J. Endocrinol 223 (2014) T25–T35. doi: 10.1530/JOE-14-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Coppari R, Bjørbæk C, Leptin revisited: its mechanism of action and potential for treating diabetes, Nat. Rev. Drug Discov 11 (2012) 692–708. doi: 10.1038/nrd3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Pan WW, Myers MG Jr, Leptin and the maintenance of elevated body weight, Nat. Rev. Neurosci 19 (2018) 95. doi: 10.1038/nrn.2017.168. [DOI] [PubMed] [Google Scholar]
- [90].Grill HJ, Schwartz MW, Kaplan JM, Foxhall JS, Breininger J, Baskin DG, Evidence That the Caudal Brainstem Is a Target for the Inhibitory Effect of Leptin on Food Intake, Endocrinology. 143 (2002) 239–246. doi: 10.1210/endo.143.1.8589. [DOI] [PubMed] [Google Scholar]
- [91].Kanoski SE, Zhao S, Guarnieri DJ, DiLeone RJ, Yan J, DeJonghe BC, Bence KK, Hayes MR, Grill HJ, Endogenous leptin receptor signaling in the medial nucleus tractus solitarius affects meal size and potentiates intestinal satiation signals, Am. J. Physiol. Metab 303 (2012) E496–E503. doi: 10.1152/ajpendo.00205.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Hayes MR, Skibicka KP, Leichner TM, Guarnieri DJ, DiLeone RJ, Bence KK, Grill HJ, Endogenous Leptin Signaling in the Caudal Nucleus Tractus Solitarius and Area Postrema Is Required for Energy Balance Regulation, Cell Metab. 11 (2010) 77–83. doi: 10.1016/j.cmet.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Peiser C, Springer J, Groneberg DA, McGregor GP, Fischer A, Lang RE, Leptin receptor expression in nodose ganglion cells projecting to the rat gastric fundus, Neurosci. Lett 320 (2002) 41–44. doi: 10.1016/S0304-3940(02)00023-X. [DOI] [PubMed] [Google Scholar]
- [94].Peters JH, Ritter RC, Simasko SM, Leptin and CCK selectively activate vagal afferent neurons innervating the stomach and duodenum, Am. J. Physiol. Integr. Comp. Physiol 290 (2006) R1544–R1549. doi: 10.1152/ajpregu.00811.2005. [DOI] [PubMed] [Google Scholar]
- [95].deLartigue G, Ronveaux CC, Raybould HE, Deletion of leptin signaling in vagal afferent neurons results in hyperphagia and obesity, Mol. Metab 3 (2014) 595–607. doi: 10.1016/j.molmet.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Kievit P, Howard JK, Badman MK, Balthasar N, Coppari R, Mori H, Lee CE, Elmquist JK, Yoshimura A, Flier JS, Enhanced leptin sensitivity and improved glucose homeostasis in mice lacking suppressor of cytokine signaling-3 in POMC-expressing cells, Cell Metab. 4 (2006) 123–132. doi: 10.1016/j.cmet.2006.06.010. [DOI] [PubMed] [Google Scholar]
- [97].deLa Serre CB, deLartigue G, Raybould HE, Chronic exposure to Low dose bacterial lipopolysaccharide inhibits leptin signaling in vagal afferent neurons, Physiol. Behav 139 (2015) 188–194. doi: 10.1016/j.physbeh.2014.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].White CL, Whittington A, Barnes MJ, Wang Z, Bray GA, Morrison CD, HF diets increase hypothalamic PTP1B and induce leptin resistance through both leptin-dependent and -independent mechanisms, Am. J. Physiol. Metab 296 (2009) E291–E299. doi: 10.1152/ajpendo.90513.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Loh K, Fukushima A, Zhang X, Galic S, Briggs D, Enriori PJ, Simonds S, Wiede F, Reichenbach A, Hauser C, Sims NA, Bence KK, Zhang S, Zhang Z-Y, Kahn BB, Neel BG, Andrews ZB, Cowley MA, Tiganis T, Elevated Hypothalamic TCPTP in Obesity Contributes to Cellular Leptin Resistance, Cell Metab. 14 (2011) 684–699. doi: 10.1016/j.cmet.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Münzberg H, Flier JS, Bjørbæk C, Region-Specific Leptin Resistance within the Hypothalamus of Diet-Induced Obese Mice, Endocrinology. 145 (2004) 4880–4889. doi: 10.1210/en.2004-0726. [DOI] [PubMed] [Google Scholar]
- [101].deLartigue G, Barbier de la Serre C, Espero E, Lee J, Raybould HE, Leptin resistance in vagal afferent neurons inhibits cholecystokinin signaling and satiation in diet induced obese rats., PLoS One. 7 (2012) e32967. doi: 10.1371/journal.pone.0032967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Eckel LA, Langhans W, Kahler A, Campfield LA, Smith FJ, Geary N, Chronic administration of OB protein decreases food intake by selectively reducing meal size in female rats, Am. J. Physiol. Integr. Comp. Physiol 275 (1998) R186–R193. doi: 10.1152/ajpregu.1998.275.1.R186. [DOI] [PubMed] [Google Scholar]
- [103].Pelleymounter MA, Baker MB, McCaleb M, Does estradiol mediate leptin’s effects on adiposity and body weight?, Am. J. Physiol. Metab 276 (1999) E955–E963. doi: 10.1152/ajpendo.1999.276.5.E955. [DOI] [PubMed] [Google Scholar]
- [104].Chen Y, Heiman ML, Increased weight gain after ovariectomy is not a consequence of leptin resistance, Am. J. Physiol. Metab 280 (2001) E315–E322. doi: 10.1152/ajpendo.2001.280.2.E315. [DOI] [PubMed] [Google Scholar]
- [105].Fujitani M, Mizushige T, Bhattarai K, Iwahara A, Aida R, Kishida T, The daidzein- and estradiol- induced anorectic action in CCK or leptin receptor deficiency rats, Biosci. Biotechnol. Biochem 79 (2015) 1164–1171. doi: 10.1080/09168451.2015.1018123. [DOI] [PubMed] [Google Scholar]
- [106].Kim JS, Rizwan MZ, Clegg DJ, Anderson GM, Leptin Signaling Is Not Required for Anorexigenic Estradiol Effects in Female Mice., Endocrinology. 157 (2016) 1991–2001. doi: 10.1210/en.2015-1594. [DOI] [PubMed] [Google Scholar]
- [107].Gao Q, Mezei G, Nie Y, Rao Y, Choi CS, Bechmann I, Leranth C, Toran-Allerand D, Priest CA, Roberts JL, Gao X-B, Mobbs C, Shulman GI, Diano S, Horvath TL, Anorectic estrogen mimics leptin’s effect on the rewiring of melanocortin cells and Stat3 signaling in obese animals, Nat. Med 13 (2007) 89–94. doi: 10.1038/nm1525. [DOI] [PubMed] [Google Scholar]
- [108].Gao Q, Horvath TL, Cross-talk between estrogen and leptin signaling in the hypothalamus, Am. J. Physiol. Endocrinol. Metab 294 (2008) E817–E826. doi: 10.1152/ajpendo.00733.2007. [DOI] [PubMed] [Google Scholar]
- [109].Kwon O, Kang ES, Kim I, Shin S, Kim M, Kwon S, Oh SR, Ahn YS, Kim CH, GPR30 mediates anorectic estrogen-induced STAT3 signaling in the hypothalamus, Metabolism. 63 (2014) 1455–1461. doi: 10.1016/j.metabol.2014.07.015. [DOI] [PubMed] [Google Scholar]
- [110].Ainslie DA, Morris MJ, Wittert G, Turnbull H, Proietto J, Thorburn AW, Estrogen deficiency causes central leptin insensitivity and increased hypothalamic neuropeptide Y, Int. J. Obes 25 (2001) 1680–1688. doi: 10.1038/sj.ijo.0801806. [DOI] [PubMed] [Google Scholar]
- [111].Clegg DJ, Riedy CA, Smith KAB, Benoit SC, Woods SC, Differential Sensitivity to Central Leptin and Insulin in Male and Female Rats, Diabetes. 52 (2003) 682 LP–687. doi: 10.2337/diabetes.52.3.682. [DOI] [PubMed] [Google Scholar]
- [112].Clegg DJ, Brown LM, Woods SC, Benoit SC, Gonadal Hormones Determine Sensitivity to Central Leptin and Insulin, Diabetes. 55 (2006) 978–987. doi: 10.2337/diabetes.55.04.06.db05-1339. [DOI] [PubMed] [Google Scholar]
- [113].Litwak SA, Wilson JL, Chen W, Garcia-Rudaz C, Khaksari M, Cowley MA, Enriori PJ, Estradiol prevents fat accumulation and overcomes leptin resistance in female high-fat diet mice., Endocrinology. 155 (2014) 4447–4460. doi: 10.1210/en.2014-1342. [DOI] [PubMed] [Google Scholar]
- [114].Meli R, Pacilio M, Raso GM, Esposito E, Coppola A, Nasti A, DiCarlo C, Nappi C, DiCarlo R, Estrogen and Raloxifene Modulate Leptin and Its Receptor in Hypothalamus and Adipose Tissue from Ovariectomized Rats, Endocrinology. 145 (2004) 3115–3121. doi: 10.1210/en.2004-0129. [DOI] [PubMed] [Google Scholar]
- [115].Rocha M, Bing C, Williams G, Puerta M, Physiologic estradiol levels enhance hypothalamic expression of the long form of the leptin receptor in intact rats, J. Nutr. Biochem 15 (2004) 328–334. doi: 10.1016/j.jnutbio.2004.01.003. [DOI] [PubMed] [Google Scholar]
- [116].He Z, Zhang SS, Meng Q, Li S, Zhu HH, Raquil M-A, Alderson N, Zhang H, Wu J, Rui L, Cai D, Feng G-S, Shp2 Controls Female Body Weight and Energy Balance by Integrating Leptin and Estrogen Signals, Mol. Cell. Biol 32 (2012) 1867 LP–1878. doi: 10.1128/MCB.06712-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Holst JJ, The Physiology of Glucagon-like Peptide 1, Physiol. Rev 87 (2007) 1409–1439. doi: 10.1152/physrev.00034.2006. [DOI] [PubMed] [Google Scholar]
- [118].Kastin AJ, Akerstrom V, Pan W, Interactions of glucagon-like peptide-1 (GLP-1) with the blood-brain barrier, J. Mol. Neurosci 18 (2002) 7–14. doi: 10.1385/JMN:18:1-2:07. [DOI] [PubMed] [Google Scholar]
- [119].Kastin AJ, Akerstrom V, Entry of exendin-4 into brain is rapid but may be limited at high doses, Int. J. Obes 27 (2003) 313–318. doi: 10.1038/sj.ijo.0802206. [DOI] [PubMed] [Google Scholar]
- [120].Holt MK, Richards JE, Cook DR, Brierley DI, Williams DL, Reimann F, Gribble FM, Trapp S, Preproglucagon Neurons in the Nucleus of the Solitary Tract Are the Main Source of Brain GLP-1, Mediate Stress-Induced Hypophagia, and Limit Unusually Large Intakes of Food, Diabetes. 68 (2019) 21 LP–33. doi: 10.2337/db18-0729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Varin EM, Mulvihill EE, Baggio LL, Koehler JA, Cao X, Seeley RJ, Drucker DJ, Distinct Neural Sites of GLP-1R Expression Mediate Physiological versus Pharmacological Control of Incretin Action, Cell Rep. 27 (2019) 3371–3384.e3. doi: 10.1016/j.celrep.2019.05.055. [DOI] [PubMed] [Google Scholar]
- [122].Abbott CR, Monteiro M, Small CJ, Sajedi A, Smith KL, Parkinson JRC, Ghatei MA, Bloom SR, The inhibitory effects of peripheral administration of peptide YY3–36 and glucagon-like peptide-1 on food intake are attenuated by ablation of the vagal–brainstem–hypothalamic pathway, Brain Res. 1044 (2005) 127–131. doi: 10.1016/j.brainres.2005.03.011. [DOI] [PubMed] [Google Scholar]
- [123].Iwasaki Y, Sendo M, Dezaki K, Hira T, Sato T, Nakata M, Goswami C, Aoki R, Arai T, Kumari P, Hayakawa M, Masuda C, Okada T, Hara H, Drucker DJ, Yamada Y, Tokuda M, Yada T, GLP-1 release and vagal afferent activation mediate the beneficial metabolic and chronotherapeutic effects of D-allulose, Nat. Commun 9 (2018) 113. doi: 10.1038/s41467-017-02488-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Talsania T, Anini Y, Siu S, Drucker DJ, Brubaker PL, Peripheral Exendin-4 and Peptide YY3–36 Synergistically Reduce Food Intake through Different Mechanisms in Mice, Endocrinology. 146 (2005) 3748–3756. doi: 10.1210/en.2005-0473. [DOI] [PubMed] [Google Scholar]
- [125].Rüttimann EB, Arnold M, Hillebrand JJ, Geary N, Langhans W, Intrameal Hepatic Portal and Intraperitoneal Infusions of Glucagon-Like Peptide-1 Reduce Spontaneous Meal Size in the Rat via Different Mechanisms, Endocrinology. 150 (2009) 1174–1181. doi: 10.1210/en.2008-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Labouesse MA, Stadlbauer U, Weber E, Arnold M, Langhans W, Pacheco-López G, Vagal Afferents Mediate Early Satiation and Prevent Flavour Avoidance Learning in Response to Intraperitoneally Infused Exendin-4, J. Neuroendocrinol 24 (2012) 1505–1516. doi: 10.1111/j.1365-2826.2012.02364.x. [DOI] [PubMed] [Google Scholar]
- [127].Krieger J-P, Arnold M, Pettersen KG, Lossel P, Langhans W, Lee SJ, Knockdown of GLP-1 Receptors in Vagal Afferents Affects Normal Food Intake and Glycemia, Diabetes. 65 (2016) 34 LP–43. doi: 10.2337/db15-0973. [DOI] [PubMed] [Google Scholar]
- [128].Kanoski SE, Hayes MR, Skibicka KP, GLP-1 and weight loss: unraveling the diverse neural circuitry, Am. J. Physiol. Integr. Comp. Physiol 310 (2016) R885–R895. doi: 10.1152/ajpregu.00520.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Hayes MR, Skibicka KP, Grill HJ, Caudal Brainstem Processing Is Sufficient for Behavioral, Sympathetic, and Parasympathetic Responses Driven by Peripheral and Hindbrain Glucagon-Like-Peptide-1 Receptor Stimulation, Endocrinology. 149 (2008) 4059–4068. doi: 10.1210/en.2007-1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Richard JE, Anderberg RH, Göteson A, Gribble FM, Reimann F, Skibicka KP, Activation of the GLP-1 Receptors in the Nucleus of the Solitary Tract Reduces Food Reward Behavior and Targets the Mesolimbic System, PLoS One. 10 (2015) e0119034. doi: 10.1371/journal.pone.0119034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Alhadeff AL, Mergler BD, Zimmer DJ, Turner CA, Reiner DJ, Schmidt HD, Grill HJ, Hayes MR, Endogenous Glucagon-like Peptide-1 Receptor Signaling in the Nucleus Tractus Solitarius is Required for Food Intake Control, Neuropsychopharmacology. 42 (2017) 1471–1479. doi: 10.1038/npp.2016.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Hayes MR, Bradley L, Grill HJ, Endogenous Hindbrain Glucagon-Like Peptide-1 Receptor Activation Contributes to the Control of Food Intake by Mediating Gastric Satiation Signaling, Endocrinology. 150 (2009) 2654–2659. doi: 10.1210/en.2008-1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Hayes MR, Leichner TM, Zhao S, Lee GS, Chowansky A, Zimmer D, DeJonghe BC, Kanoski SE, Grill HJ, Bence KK, Intracellular Signals Mediating the Food Intake-Suppressive Effects of Hindbrain Glucagon-like Peptide-1 Receptor Activation, Cell Metab. 13 (2011) 320–330. doi: 10.1016/j.cmet.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Alhadeff AL, Grill HJ, Hindbrain nucleus tractus solitarius glucagon-like peptide-1 receptor signaling reduces appetitive and motivational aspects of feeding, Am. J. Physiol. Integr. Comp. Physiol 307 (2014) R465–R470. doi: 10.1152/ajpregu.00179.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Asarian L, Abegg K, Geary N, Schiesser M, Lutz TA, Bueter M, Estradiol Increases Body Weight Loss and Gut-Peptide Satiation After Roux-en-Y Gastric Bypass in Ovariectomized Rats, Gastroenterology. 143 (2012) 325–327.e2. doi: 10.1053/j.gastro.2012.05.008. [DOI] [PubMed] [Google Scholar]
- [136].Asarian L, Geary N, Sex differences in the physiology of eating., Am. J. Physiol. Regul. Integr. Comp. Physiol 305 (2013) R1215–67. doi: 10.1152/ajpregu.00446.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Finan B, Yang B, Ottaway N, Stemmer K, Müller TD, Yi C-X, Habegger K, Schriever SC, García-Cáceres C, Kabra DG, Hembree J, Holland J, Raver C, Seeley RJ, Hans W, Irmler M, Beckers J, deAngelis MH, Tiano JP, Mauvais-Jarvis F, Perez-Tilve D, Pfluger P, Zhang L, Gelfanov V, DiMarchi RD, Tschöp MH, Targeted estrogen delivery reverses the metabolic syndrome, Nat. Med 18 (2012) 1847–1856. doi: 10.1038/nm.3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Schwenk RW, Baumeier C, Finan B, Kluth O, Brauer C, Joost H-G, DiMarchi RD, Tschöp MH, Schürmann A, GLP-1–oestrogen attenuates hyperphagia and protects from beta cell failure in diabetes-prone New Zealand obese (NZO) mice, Diabetologia. 58 (2015) 604–614. doi: 10.1007/s00125-014-3478-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Xu P, Cao X, He Y, Zhu L, Yang Y, Saito K, Wang C, Yan X, Hinton AO Jr., Zou F, Ding H, Xia Y, Yan C, Shu G, Wu S-P, Yang B, Feng Y, Clegg DJ, DeMarchi R, Khan SA, Tsai SY, DeMayo FJ, Wu Q, Tong Q, Xu Y, Estrogen receptor–α in medial amygdala neurons regulates body weight, J. Clin. Invest 125 (2015) 2861–2876. doi: 10.1172/JCI80941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Vogel H, Wolf S, Rabasa C, Rodriguez-Pacheco F, Babaei CS, Stöber F, Goldschmidt J, DiMarchi RD, Finan B, Tschöp MH, Dickson SL, Schürmann A, Skibicka KP, GLP-1 and estrogen conjugate acts in the supramammillary nucleus to reduce food-reward and body weight, Neuropharmacology. 110 (2016) 396–406. doi: 10.1016/j.neuropharm.2016.07.039. [DOI] [PubMed] [Google Scholar]
- [141].Maske CB, Jackson CM, Terrill SJ, Eckel LA, Williams DL, Estradiol modulates the anorexic response to central glucagon-like peptide 1, Horm. Behav 93 (2017) 109–117. doi: 10.1016/j.yhbeh.2017.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]