

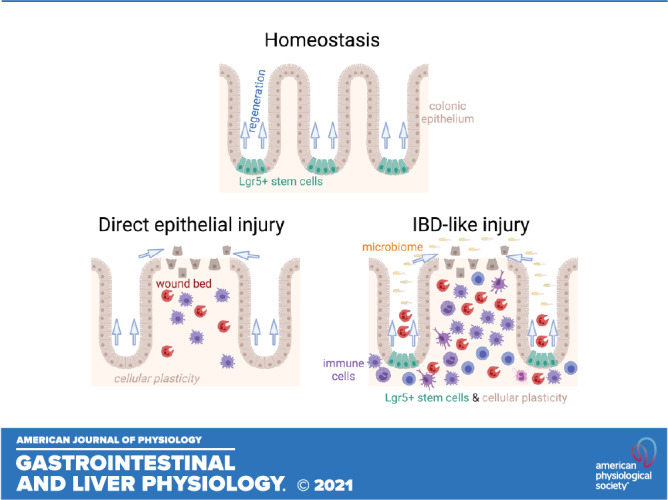
Keywords: injury, mucosal healing, regeneration, repair, wounds
Abstract
Intestinal mucosal healing is the primary therapeutic goal of medical treatments for inflammatory bowel disease (IBD). Epithelial stem cells are key players in the healing process. Lgr5+ stem cells maintain cellular turnover during homeostasis in the colonic crypt. However, they are lost and dispensable for repair in a wide variety of injury models, including dextran sulfate sodium (DSS) colitis, radiation, helminth infection, and T-cell activation. The direct loss of Lgr5+ cells activates a plasticity response in the epithelium in which other cell types can serve as stem cells. Whether this paradigm applies to mouse models of IBD remains unknown. In contrast to previously tested models, IBD models involve an inflammatory response rooted in the loss of immunologic tolerance to intestinal luminal contents including the microbiome. Here, we show the persistence of Lgr5+ cells in oxazolone, 2,4,6-trinitrobenzene sulfonic acid (TNBS), and Il10−/−, and Il10−/− Tnfr1−/− IBD models. This contrasts with results obtained from DSS-induced injury. Through high-throughput expression profiling, we find that these colitis models were associated with distinct patterns of cytokine expression. Direct exposure of colonic epithelial organoids to DSS, oxazolone, or TNBS resulted in increased apoptosis and loss of Lgr5+ cells. Targeted ablation of Lgr5+ cells resulted in severe exacerbation of chronic, antibody-induced IL-10-deficient colitis, but had only modest effects in TNBS-induced colitis. These results show that distinct mouse models of IBD-like colitis induce different patterns of Lgr5+ stem cell retention and function.
NEW & NOTEWORTHY Acute intestinal injury and epithelial repair are associated with the loss of fast-cycling Lgr5+ stem cells and plasticity in the activation of formerly quiescent cell populations. In contrast, here we show in murine inflammatory bowel disease the persistence of the Lgr5+ stem cell population and its essential role in restricting the severity of chronic colitis. This demonstrates a diversity of stem cell responses to colitis.
INTRODUCTION
Colonic epithelial injury represents a major physiological challenge that requires the coordinated responses of epithelial progenitor cells to restore the single-cell barrier. Although the mouse colonic epithelium contains multipotent Lgr5+ stem cells powering one of the fastest homeostatic turnover rates in the body (1), previous studies have documented loss of these Lgr5+ stem cells during injury and repair (2–13). Several alternate sources of progenitor cells have been proposed to mediate the repair mechanism: 1) activation of a reserve stem cell (9, 10, 12, 14), 2) reversion of partially differentiated cells to a progenitor state (2, 5, 8, 15), and 3) reprogramming of homeostatic stem cells to an injury-adapted, fetal-like (Ly6a+) state (4, 6, 11, 13, 16). These mechanisms have largely been elucidated in models in which the epithelium is directly and acutely injured, such as DSS colitis, irradiation, genetic ablation of Lgr5+ stem cells, and helminth infection and are not necessarily exclusive. However, certain studies involving radiation injury (17) or T-cell receptor ligation (6) have shown that Lgr5+ stem cells persist or that their activity may be essential for repair. Thus, whether the loss of Lgr5+ stem cells and the activation of plasticity mechanisms is a universal rule of the intestinal injury response remains in question.
Inflammatory bowel disease (IBD) represents a collection of disorders that present as chronic inflammation of the digestive tract, particularly in the colon and small intestine. In the major forms of IBD, Crohn’s disease, and ulcerative colitis, colonic crypts exhibit structural changes including bifurcation, branching, and hyperplasia that would suggest remodeling of the epithelial stem cell compartment. However, little is known about stem cell signaling in generally accepted models (18) of inflammatory bowel disease (IBD). The injury and inflammation observed in IBD models have a distinct etiology. In IBD models, the primary inflammatory response is thought to be generally directed against luminal contents, including the microbiome (19). The “cytokine storm” that results from this abnormal immune reaction damages the epithelium. This can result in chronic changes to epithelial structure and it is unclear what types of stem cell mechanisms underpin such changes. A critical question to be resolved is whether Lgr5+ stem cells have a role in the epithelial response to IBD-like injury. Decoding the key epithelial cell types involved in the injury response will help to elucidate potential therapeutic targets for the promotion of mucosal healing, which is the primary goal (20) of all medical treatments for IBD.
Here, we have surveyed the expression of primary (Lgr5) and surrogate (Ephb3, Lrig1) (1, 3, 21, 22) markers of the Lgr5+ colonic epithelial stem cells in the DSS, 2,4,6-trinitrobenzene sulfonic acid (TNBS), oxazolone, and Il10−/− colitis models. We find that, in contrast to their total depletion in the DSS model, Lgr5+ cells persist in the TNBS, oxazolone, and Il10−/− IBD models but with different spatial patterns. Through transcriptional profiling, we found that all models were associated with the profound, if differentially polarized, upregulation of cytokines. The expression of the IFNγ target Ly6a was increased in all the tested models. In colonic organoids, we found that chemical agents of colitis could directly deplete the Lgr5+ stem cell population, with varying efficacies. Lgr5+ stem cells were required for restriction of chronic colitis in a model of reduced IL-10 signaling. These results demonstrate the diversity and divergence of patterns of stem cell persistence and function in IBD models.
MATERIALS AND METHODS
Animals
Mice were maintained in accordance with regulations of the Institutional Animal Care and Use Committee (IACUC) at Children’s Hospital Los Angeles (CHLA). This study was approved by the CHLA IACUC (protocol #288). Wild-type mice were purchased from Jackson Laboratory at 6 wk of age and were cohoused for at least 8 days before experimentation. Before dissection, mice were anesthetized with isoflurane and killed via cervical dislocation. Strain and stock number for purchased mice from Jackson Laboratory were as follows: BALB/cJ #000651, SJL/J #000686, Il10−/− #002251, Tnfr1−/− #002818, C57Bl/6J #000664. Il10−/− Tnfr1−/− were generated by breeding of Il10−/− and Tnfr1−/− mice at CHLA for >10 generations. Experimental littermate mice of Il10−/− or Il10−/− Tnfr1−/− genotypes were generated by breeding of parental Il10−/− Tnfr1+− mice. Lgr5::DTR-EGFP mice (9) were a gift from Frederic de Sauvage (Genentech). Cohorts involving both male and female mice were balanced for the sex of the animals.
DSS Colitis
DSS treatment was as previously described (23, 24). Briefly, mice were trained to drink from water bottles, then exposed to water bottles containing 0.5%–3% wt/vol DSS (36–50 kDa molar mass; MP Biomedicals) in distilled water, which served as their only source of drinking water for 6 days. We examined body weight and stool consistency daily to assess colonic injury.
TNBS Colitis
A stock solution of 2,4,6-trinitrobenzenesulfonic acid (TNBS) in picrylsulfonic acid was purchased from Sigma (#P2297) and diluted in 40% ethanol to a working concentration of 3% vol/vol. Before enema administration of TNBS, 40% ethanol, or saline, SJL mice were anesthetized with isoflurane. Enemas were administered by filling a 1-mL syringe mounted with a lubricated blunt cannula (Thermo Fisher Scientific #202322) and by carefully inserting the cannula, 3 cm into the colon before slow release of a volume of 0.1 mL/mouse. After administration, mice were inverted for 2 min or until recovery from anesthesia.
Oxazolone Colitis
Oxazolone (Sigma #862207) was diluted to 6% in 50% ethanol. Enema procedures were the same as for TNBS colitis, but a volume of 50 µL was instilled into the colons of BALB/c mice.
Anti-IL10R Colitis
Cohoused Lgr5::DTR-EGFP or wild-type (WT) mice were intraperitoneally injected, on alternating sides of body, with anti-IL10R (anti-interleukin 10 receptor) antibody once per week, for a total of five injections. The dose of the anti-IL10R was titrated depending on the age of the animal: 0.5 mg/mouse: 3 wk old; 0.75 mg/mouse: 4 wk old; 1 mg/mouse: 5 wk old; 1 mg/mouse: 6 wk old; 1 mg/mouse: 7-wk old. The first injection was administered when mice were 3 wk old. Studies involving the use of diphtheria toxin were commenced at 8 wk of age. The antibody used was rat antimouse IL-10R (CD210), clone 1B1.3A (Bio X Cell #BE0050). The isotype control antibody was rat IgG1 isotype control, anti-horseradish peroxidase, clone HRPN (Bio X Cell #BE0088). For TNBS studies, Lgr5::DTR-EGFP mice, originally on a Bl/6 background, were backcrossed for two generations onto the SJL background.
Organoid Culture
Distal colonic tissue isolated from Lgr5::DTR-EGFP mice was processed for digestion and crypt isolation and cultured in Matrigel and Intesticult (Stem cell Technologies), as per previously established protocols (25, 26). Tissue was plated on glass-bottomed dishes to facilitate monitoring of living cells via microscopy. Colonic epithelial organoids (colonoids) were passaged at least two times, seeded at 1 × 103 cells/mL per well and were treated at 4 days after passage with increasing concentrations of DSS (0.01%–1%), TNBS (0.0005%–0.5%), oxazolone (0.00001%–0.001%), or ethanol (0.00001%–0.001%). After 3 h (DSS or oxazolone) or 5 h (TNBS) of treatment, colonoids were imaged for green fluorescent protein (GFP) signals (corresponding to Lgr5 expression) using a Zeiss LSM 710 Inverted Confocal Microscope (Zeiss, Oberkochen, Germany). Subsequently, the colonoids were treated with 10 µM EDU for 30 min and fixed thereafter with 2% paraformaldehyde for 20 min. Experiments were performed three times, and the data representing >30 organoids per experiment and per condition were aggregated.
Histological Processing
After euthanasia and dissection, the colon was removed from the abdominal cavity, opened longitudinally, and cleaned of feces. A small (1-mm wide) sliver of hindgut-derived colon cut along the longitudinal axis was saved for tissue RNA extraction. Colon were flattened and fixed in 4% paraformaldehyde at room temperature for 16 h. Paraffin embedding, sectioning, and histological stains such as hematoxylin and eosin (H&E) were performed according to standard protocols.
Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling
For dUTP nick end labeling (TUNEL), we performed assays according to protocols from the ApopTag Peroxidase In Situ Apoptosis Detection Kit (Millipore #S7100) on paraffin-embedded tissue sections. A diaminobenzidine (DAB) staining kit (Vector Labs #SK4105) was used for the chromogenic reaction. Tissues were counterstained with hematoxylin for 45 s and blued for 10 s in 0.006 N ammonium hydroxide. Slides were dehydrated, cleared in xylenes, and mounted with Permount.
In Situ Hybridization
In situ hybridization was performed with the RNAscope.5 HD Reagent Kit Brown-Mm Kit (ACDBio #322371), in accordance with the manufacturer’s instructions. Signals were detected with DAB incubation (Vector Labs #SK4105) for 10 min. The following probes from ACDBio were acquired and used: RNAscope Probe-Mm-Lgr5 (#312171), RNAscope Probe-Mm-Ephb3 (#510251), and RNAscope Probe-Mm-Lrig1 (#310521). For Ly6a and Cdh1 double staining, we used the RNAscope 2.5 HD Duplex Reagent Kit (ACDBio #322430). Probes used for this kit were RNAscope Probe-Mm-Cdh1 (#408651) and RNAscope Probe-Mm-Ly6a-C2 (#427571-C2).
Immunohistochemistry
Immunohistochemical labeling on formalin-fixed paraffin-embedded sections was performed, according to standard procedures and as previously described (23, 27). Staining signal was revealed using the Immpress HRP reagent kits (Vector Labs) in conjunction with DAB reagents (Vector Labs). Primary antibodies used are rabbit anti-MUC2 (Santa Cruz Biotechnology #sc15334, 1:100 dilution), rabbit anti-CHGA (Immunostar #20085, 1:100 dilution), rabbit anti-DCLK1 (Abcepta #AP7219B, 1:200 dilution), rabbit anti-ANXA1 (Proteintech #21990-1-AP, 1:100 dilution), rabbit anti-GFP (Novus Biologicals #NB600-308(AF488), 1:200 dilution), and rabbit anti-cleaved caspase-3 (CC3) (Cell Signaling Technology #9661S, 1:200 dilution).
EdU Staining
Whole mounts of organoids treated with increasing concentrations of DSS, TNBS, and oxazolone were stained with the Click-iT Plus EdU Alexa Fluor488 Imaging Kit (Thermo Fisher Scientific, #C10632), as per the manufacturer’s recommendations.
Diphtheria Toxin
Diphtheria toxin was purchased from Sigma (#D0564). Each 1-mg vial of toxin was reconstituted with 0.5 mL of sterile distilled water to form a buffered solution in 0.01 M Tris and 1 µM Na2EDTA, pH 7.5. DT was additionally diluted in saline and was administered via intraperitoneal injection to mice at a dose of 50 mg/kg and a volume of 0.1 mL per animal.
Cytokine Expression Profiling
RNA was profiled for expression of immune-related genes using the nCounter Mouse Immunology Panel (Nanostring, 561 genes). A previously published dataset (28) with analysis performed using the same methods and reagents was included to obtain expression values from colons of Il10−/− and Il10−/− Tnfr1−/− mice. Abundance values per sample were normalized to the mean expression of the panel of housekeeping genes. The normalized expression values were merged, and principal component analysis was performed on the merged dataset in R. To generate heatmaps, expression values were compared with controls (e.g., uninjured mice) within the same injury model. The ratios of expression between injured animals and controls were merged across models. Genes selected for display include interleukins (“Il*”), chemokines (“Cxcl*” and “Ccl*”), Tnf pathway members (“Tnf*”), and interferons (“Ifn*”).
Analysis
Images were obtained using a bright-field microscope with tilescan capabilities to capture a large field of view. Histologic assessment of colitis severity was performed in blind fashion by a gastrointestinal pathologist (M.K.W.), similarly as previously described (29, 30). Statistical tests were performed in Prism (Graphpad).
For counting of organoids, the numbers of Lgr5-EGFP+ stem cells, EdU+ proliferating cells, and CC3+ (cleaved caspase 3-positive) apoptotic cells were quantified in the crypt-like budding structures of colonoids. The budding structures were identified based on the three-dimensional structure of the organoids, which could be reconstructed after computational processing of whole mount z-stack imaging in ImageJ. These buds were typically of at least 50 × 50 × 50 µm3 volume. To count the number of Lgr5+ stem cells, the z-plane showing the maximum cross-sectional area of the bud was identified. The number of cells was counted in this z-plane and expressed relative to this cross-sectional area. To count the number of proliferative or apoptotic cells, the raw number of stained cells was obtained throughout the entire volume of the bud. These numbers per bud were averaged across the organoid to obtain a per-organoid average. This average was then merged across all experimental replicates. The data shown therefore aggregates all organoids from three different experiments. The number of organoids sampled per experiment was equally balanced. Statistical comparisons were performed using a one-way ANOVA, with a Tukey’s post hoc test.
To analyze densitometric profiles of gene expression, we used the Integrated Density tool available in ImageJ. Multichannel images were split into individual RGB color channels, and the red channel was used for analysis. The region of the crypt was manually traced from the histology image. The Integrated Density tool calculates the sum of all stained pixels within the region of interest. The sum is then divided by the area of the selection. In each mouse, we counted 10 contiguous crypts from a representative affected region. The density values of each of the crypts were averaged to obtain a per-animal density estimate of gene expression. The per-animal estimates were then compared across all the animals in the cohort to obtain the graphs shown in the figures.
RESULTS
We compared the expression level and spatial distribution of colonic stem cell markers in DSS (epithelial injury and inflammation), TNBS (Th1 responses), oxazolone (Th2 responses), and Il10−/− (spontaneous colitis) models of IBD. Application of these models induced a mosaic pattern of injury and inflammation in the mouse colon. Regions of crypt remodeling, ulceration, and inflammatory infiltration were bordered by histologically normal mucosa; we refer to these regions as “affected” and “unaffected” areas, respectively and show example images in Supplemental Fig. S1 (https://doi.org/10.6084/m9.figshare.14870211). The expression profiling was focused on the gene Lgr5, which marks a population of crypt base columnar cells present in every colonic gland; lineage tracing of these cells has demonstrated their ability to regenerate every cell in the crypt during homeostatic epithelial turnover (1). To confirm findings, we second analyzed the expression of Ephb3, a gene highly enriched in colonic Lgr5+ stem cells during homeostatic conditions (3). Third, we profiled Lrig1 expression in these IBD models as Lrig1 is believed to mark colonic epithelial progenitors (21). Finally, in all models, we tested whether the epithelial response to injury was characterized by upregulation of Ly6a (SCA-1), an interferon-inducible gene that has been proposed to mark injury-associated epithelial reprogramming (6, 11).
DSS Colitis
We first tested whether we could validate previous findings of decreased Lgr5 expression and increased Ly6a in DSS-induced colitis. To induce colonic epithelial ulceration and mucosal immune infiltration, we treated Bl/6 mice with 3% DSS for 6 days, beginning on experimental day (exp day) 0. Mice were euthanized, dissected, and analyzed on exp day 6. Mice exposed to DSS lost ∼5% of their body weight (Fig. 1A), exhibited thicker colons, a hallmark of injury (Fig. 1B), and showed elevated histological injury scores (Fig. 1C) compared with water-treated controls (Fig. 1D).
Figure 1.

Validation of Lgr5+ stem cell loss in dextran sulfate sodium (DSS) colitis. Compared with uninjured water-treated mice, those treated with 3% DSS for 6 days exhibit body weight loss (A), thickening of the colon (B), and histological damage as scored by a pathologist (C) and demonstrated in hematoxylin-eosin (H&E) stained images (D) (n = 5 male mice per group). In situ hybridization (ISH) with probes targeted to stem cell marker genes Lgr5 (E), Ephb3 (F), and Lrig1 (G) shows localization of expression to the crypt base during homeostasis. However, after DSS treatment, the expression of these markers appears greatly reduced, in both atrophic regions (“affected”) and in regions of the distal colon that appear histologically normal (“unaffected”). ISH with probes targeted to the interferon-responsive gene Ly6a (H) shows the highest levels of expression in the affected region. A counterstain with Cdh1 is provided because Ly6a expression is not exclusive to the epithelium. I: densitometry of the ISH stainings in the epithelial layer of colonic crypts quantifies their epithelial-specific expression patterns. Ten contiguous distal colonic crypts were chosen for image quantification. Lgr5, Ephb3, and Lrig1 exhibit near-total loss of expression, whereas Lrig1 shows strong epithelial upregulation. J: the density of ISH-recorded Lgr5 staining depended on the dose of DSS given (n = 5 mice per dose). At lower doses (e.g., 0.5% vs. 3% DSS), many Lgr5+ stem cells persisted. Scale bars: 50 µm. Statistics: *P < 0.05; **P < 0.01; ***P < 0.001. Two-tailed t test. Error bars: means ± SE.
To visualize the expression and localization of stem cell marker transcripts, we profiled Lgr5 (Fig. 1E), Ephb3 (Fig. 1F), and Lrig1 (Fig. 1G) expression using in situ hybridization. In water-treated control animals, the expression of Lgr5 and Ephb3 was evident from dense punctate staining in cells located at the bases of colonic crypts. However, in samples from DSS-treated mice, no labeling was observed in surviving (“unaffected”) distal crypts or in regions of ulceration and crypt dropout (“affected”). In contrast, Lgr5 expression persisted in the proximal colon, which is less affected by DSS injury (data not shown). Hybridization signals for Lrig1 were located in the cells occupying the lower third of colonic crypts and were weakened by treatment with DSS. Crypts in both affected and unaffected regions in DSS-treated colons expressed Ly6a (Fig. 1H), which was not found in epithelia of water-treated colons. These qualitative observations were confirmed with densitometry (Fig. 1I), which enabled quantification of marker expression levels specifically in epithelium. Thus, DSS-induced injury is associated with the loss of Lgr5+ stem cells and the epithelial upregulation of the interferon-responsive Ly6a gene.
To determine whether the loss of adult stem cell markers was linked to altered differentiation, we counted the abundance of mature secretory cell types in the epithelium. We found decreased goblet (MUC2+) and increased tuft (DCLK1+) cells, but no change in CHGA+ enteroendocrine cells, in the affected regions (Supplemental Fig. S2A; see https://doi.org/10.6084/m9.figshare.14870220). Apoptotic (TUNEL+) cells were found near the luminal surface in unaffected regions, consistent with cell shedding, and also deep inside the crypt in affected regions (Supplemental Fig. S2B). These results support a fundamental change to progenitor cell function and differentiation in DSS-induced injury, which may be related to the apoptosis of cells deep within the crypt.
We asked whether the complete loss of Lgr5 was necessary to observe Ly6a upregulation, a proposed marker of epithelial cell reprogramming, in the injured colon. We therefore tested lower doses of DSS to determine whether Lgr5+ cell persistence and Ly6a upregulation could be simultaneously observed. We examined that mice given 0.5% or 1.5% DSS. In both of these lower doses, Lgr5+ stem cells were detectable by in situ hybridization (Supplemental Fig. S2C), albeit at reduced levels of Lgr5 transcript expression (quantification of density of punctate staining, Fig. 1J). Ly6a expression was also detected in adjacent sections (Supplemental Fig. S2D); however, the intensity of the signals was also qualitatively reduced compared with findings from the 3% dose. Furthermore, whereas Ly6a signals were present in the whole crypt upon treatment with 3% DSS, these signals were generally restricted to upper crypt cells at the 0.5% and 1.5% levels of DSS. Thus, although the magnitude of Lgr5 loss and Ly6a upregulation depends strongly on the administered dose of DSS, both the relative persistence of Lgr5 and Ly6a upregulation can occur together. Thus, complete loss of Lgr5+ cells is not a prerequisite for Ly6a upregulation in the injured colon.
TNBS and Oxazolone Colitis
To examine whether similar results could be found in hapten-induced colitis, we analyzed the TNBS (nominally Th1 T-cell driven) (31–34) and oxazolone (nominally Th2 T-cell driven) (28, 35–37) colitis models. To apply the TNBS model, we treated SJL mice with enemas of PBS, ethanol diluent (“ethanol”), or 3% TNBS/ethanol (“TNBS”) and euthanized the mice at 1 (Fig. 2, A–D) or 4 days (Fig. 2, E–H) after treatment. No topical presensitization with haptens was performed, as in pilot experiments this resulted in either rapid death or complete absence of disease. TNBS-treated animals exhibited sustained loss of body weight, beginning 1 day after treatment (Fig. 2, A and E) and continuing up to 4 days after treatment (Fig. 2E). At 1 day after treatment, mild injury indicated by increased colonic thickness (Fig. 2B) and histological scoring (Fig. 2, C and D) was observed in the ethanol-treated group, and severe injury was found in the TNBS-treated group. At 4 days after treatment, the colons of TNBS-treated animals still exhibited crypt alterations and infiltration, and residual injury was also observed in ethanol-treated samples (Fig. 2, F and H).
Figure 2.

Lgr5+ cells persist in 2,4,6-trinitrobenzene sulfonic acid (TNBS) colitis. SJL mice were treated with rectal enemas of TNBS. Body weights (A and E), colonic thickening (B and F), histological injury score (C and G), and representative hematoxylin-eosin (H&E) images (D and H) of mice treated with TNBS, ethanol vehicle, or saline (PBS) for 1 day (A–D) or for 4 days (E–H) (n ≥ 5 male mice per group). Photomicrographs demonstrating outcomes of in situ hybridization (ISH) experiments to localize the expression of Lgr5 (I), Ephb3 (J), Lrig1 (K), and Ly6a (L) at day 1 or day 4 of treatment with either saline, ethanol vehicle, or TNBS. After treatment of TNBS, there was heterogeneity in the presentation of crypts with regard to the localization of stem cell markers Lgr5, Ephb3, and Lrig1: some crypts showed strong staining for these markers, whereas in other crypts no cells were stained. The expression of Ly6a appeared highest during the acute phase of injury (i.e., in TNBS-treated day 1 samples). M: densitometry of 10 contiguous representative crypts for the ISH staining of Lgr5, Ephb3, Lrig1, and Ly6a mRNA. Although some modest reductions in Ephb3 and Lrig1 expression were observed in TNBS-treated day 1 samples, the overall quantitative pattern supports the persistence of Lgr5+ cells during TNBS-induced colitis. Scale bars: (D and H): 50 µm. (I–L): 25 µm. Statistics: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Pairwise two-tailed t tests with Benjamini-Hochberg multiple testing correction. Error bars: means ± SE.
The results of in situ hybridization studies were in marked contrast to those obtained in the DSS model. In both affected and unaffected regions of TNBS-treated colons, we observed strong hybridization signals for Lgr5 (Fig. 2I), Ephb3 (Fig. 2J), and Lrig1 (Fig. 2K) at the crypt base. The epithelial expression of Ly6a underwent a marked increase in TNBS-treated samples at exp day 1 but was at reduced levels at exp day 4 and in the ethanol-treated or PBS-treated samples (Fig. 2L). However, we note that in TNBS-treated samples, at exp day 1, there was marked crypt-to-crypt variability in hybridization signals of stem cell markers. Within the affected regions of the same sample (Supplemental Fig. S3A; see https://doi.org/10.6084/m9.figshare.14870226), we detected crypts that were completely devoid of signal, whereas neighboring crypts appeared to have enriched signals marking an expanded progenitor cell domain. By exp day 4, this variability was still observed in the affected region. Quantification of staining density showed no overall change in Lgr5 expression (Fig. 2M). We stained for IRF1, a marker of interferon signaling, to determine whether these differences were due to local variability of inflammation. However, neither the staining pattern of IRF1 nor the morphology of crypts appeared to correlate with the density of Lgr5 hybridization signals (data not shown). We found TUNEL+ cells at the crypt base only in TNBS-treated conditions (Supplemental Fig. S3). We did not detect broad changes in the census of secretory epithelial cells (Supplementary Fig. S3C); however, the number of goblet cells trended downward with TNBS treatment at exp day 1. In total, these results suggest the persistence of Lgr5+ stem cells in TNBS-induced colitis despite apoptotic events in the crypt and rapid upregulation of interferon-responsive Ly6a.
The results obtained from the oxazolone colitis model administered to BALB/c mice were similar. The gross clinical characteristics of oxazolone colitis were sustained weight loss (Fig. 3, A and E), increased colonic size (Fig. 3, B and F), and severe disease with mucosal bleeding in oxazolone-treated versus ethanol-treated controls (Fig. 3, C and D, G and H). In tissue hybridization experiments (Fig. 3, I–L), we observed overall persistence but intercrypt variability (Supplemental Fig. S4A; see https://doi.org/10.6084/m9.figshare.14870223) in labeling with Lgr5 (Fig. 3I), Ephb3 (Fig. 3J), and Lrig1 (Fig. 3K) markers. Ly6a levels were highest in oxazolone-treated specimens at exp day 1 (Fig. 3, L and M). TUNEL+ cells were found inside the crypt in oxazolone-affected regions (Supplemental Fig. 4B). The secretory cell census revealed a decline in the number of goblet cells, whereas the numbers of tuft and enteroendocrine cells did not appear to be grossly affected (Supplemental Fig. S4C). Thus, despite causing severe injury, oxazolone colitis is associated with retention of the Lgr5+ stem cell population.
Figure 3.

Lgr5+ cells persist in oxazolone colitis. Oxazolone (OXA) was intrarectally distilled into BALB/c mice. Outcomes were compared against ethanol- or PBS-treated mice (n ≥ 5 male mice per group). Body weights (A and E), colonic size (B and F), pathology score (C and G), and hematoxylin-eosin (H&E)-stained images (D and H) of mice treated with oxazolone, ethanol (vehicle), or PBS for 1 day (A–D) or for 5 days (E–H). Oxazolone-treated mice exhibit significant exacerbation of injury vs. ethanol-treated animals. Histological images showing the localization of Lgr5 (I), Ephb3 (J), Lrig1 (K), and Ly6a (L), through in situ hybridization (ISH), at day 1 or day 5 of treatment with either saline, ethanol vehicle, or oxazolone. A similar crypt-to-crypt variability in retention of Lgr5 staining was observed, as in 2,4,6-trinitrobenzene sulfonic acid (TNBS)-treated animals. The expression of Ly6a was highest in oxazolone-treated day 1 samples). M: densitometry of the staining signal of Lgr5, Ephb3, Lrig1, and Ly6a in 10 contiguous representative crypts labeled by ISH. The overall pattern was consistent with retention of Lgr5 expression during oxazolone-induced colitis. Scale bars: D and H: 100 µm; I–L: 25 µm. Statistics: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Pairwise two-tailed t tests with Benjamini-Hochberg multiple testing correction. Error bars: means ± SE.
We next tested whether the observed retention of Lgr5+ cells in TNBS- or oxazolone-induced disease was related to the reliance of these models on alternate (i.e., non-Bl/6) mouse strains. Despite attempts to replicate the extended disease course shown in Figs. 2 and3 in the Bl/6 background, we could only achieve a transient loss in body weight lasting 1–2 days (Supplemental Fig. S5A; see https://doi.org/10.6084/m9.figshare.14870217). Nonetheless, during the weight loss period, we could still find Lgr5+ cells in crypts in the affected mucosa (Supplemental Fig. S5B). Thus, the persistence of Lgr5+ stem cells during inflammation and injury is a feature of hapten-induced colitis in both disease-susceptible (BALB/c, SJL) and resistant (Bl/6) mouse lines.
Il10−/− Colitis
To assess patterns of epithelial stem cell marker expression in a genetic model of spontaneous and chronic IBD (38), we profiled stem cell marker expression in Il10−/− mice. Il10−/− mice in the Bl/6 background are relatively resistant to colitis (39–42), with many samples being histologically normal, up to and after 24 wk old in our facilities. To compare results to accelerated colitis in Il10−/− background, we compared Il10−/− mice with doubly mutant Il10−/− Tnfr1−/− [Tnfrsf1a−/− (43)] mice, which develop a severe pancolitis at 4 wk of age. This accelerated colitis is associated with upregulation of Tnf and is caused by an inability to establish mucosal tolerance during development (27). The independent 8-wk-old cohorts isolated for this study demonstrated significant histological injury in Il10−/− Tnfr1−/− mice (Fig. 4, A and B) and normal colon in Il10−/− mice (Fig. 4B). We note that due to our breeding scheme prioritizing the analysis of littermate animals, we could not explicitly compare stem cell marker expression between Il10−/− and WT littermate mice.
Figure 4.

Retention of stem cell populations in Il10−/− and Il10−/− Tnfr1−/− colitis. Il10−/− Tnfr1−/− (IL10R1) mice (n = 5 mixed-sex mice per group) exhibited worsened colitis and infiltration, abscess, and hyerplasia, as demonstrated by histological scoring (A) and photomicrographs of hematoxylin-eosin (H&E)-stained sections (B). In situ hybridization: positively stained cells for Lgr5 (C), Ephb3 (D), and Lrig1 (E) are located at the crypt base in unaffected, hyperplastic, and severely affected colonic regions from Il10−/− (IL10) and Il10−/− Tnfr1−/− (IL10R1) mice. Strong upregulation (arrows) of Ly6a (F) was observed in Il10−/− Tnfr1−/− mice (F). G: densitometry of 10 representative crypts in the affected regions of the colon. No significant changes were observed in the overall epithelial abundance of Lgr5, Ephb3, or Lrig1. However, Ly6a was highly upregulated in Il10−/− Tnfr1−/− mice. H: summary of overall changes in the density of Lgr5 staining in the different injury models. Control measurements were obtained from water-treated mice in the cohort shown in Fig. 1. I: summary of the overall proportion of crypts that contain at least one Lgr5+ cell in the different injury models. Thirty crypts were sampled from the affected regions of each mouse. Scale bars: B: 100 µm, C–F: 50 µm. *P < 0.05. Statistics: ns P > 0.05; *P < 0.05; **P < 0.01. Two-tailed t tests. Error bars: means ± SE.
In situ hybridization analyses of Lgr5 (Fig. 4C), Ephb3 (Fig. 4D), and Lrig1 (Fig. 4E) expression confirmed the presence of these positively stained populations in all regions in both genotypes of mice. Whereas Ly6a expression was largely absent from Il10−/− samples, it was strongly upregulated in the affected regions demonstrating hyperplasia and crypt abscess in Il10−/− Tnfr1−/− samples (Fig. 4, F and G). We observed decreased abundance of tuft and enteroendocrine cells (Supplemental Fig. 6; see https://doi.org/10.6084/m9.figshare.14870214). Thus, severe inflammation in Il10−/− Tnfr1−/−- mice was associated with changes in secretory cell differentiation and epithelial upregulation of Ly6a, but nonetheless with persistence of the Lgr5+ cell population.
To compare patterns of Lgr5+ stem cell retention across the tested IBD models, we merged the quantitative results together. We found that administration of DSS induced a profound loss of Lgr5+ cells. This loss was not observed in TNBS, oxazolone, or Il10-deficient models (Fig. 4H). Moreover, only the TNBS and oxazolone models showed crypt-to-crypt variability in the persistence of Lgr5+ cells (Fig. 4I). In DSS and Il10-deficient models, Lgr5+ cells were either fully lost or fully retained within macroscopic regions of colon. These results summarize the variability in the patterns of Lgr5+ cell retention across IBD models.
Comparison of Cytokine Expression between Colitis Models
We asked whether changes in stem cell responses to different colitis models could be linked to distinct patterns of cytokine regulation. We therefore profiled cytokine expression in these models using an array of 561 immune-related genes (Nanostring mouse immunology panel). Merged principal component analysis of all samples demonstrated significant differences between chemical and genetic colitis models (Fig. 5A). Although water-treated or PBS-treated samples in DSS or hapten models had similar immunological profiles, Il10−/− mice, by their displaced position on the principal component plot, demonstrated significant immunological changes, despite largely appearing histologically normal. These differences were more profound in Il10−/− Tnfr1−/− mice. In contrast, oxazolone and TNBS induced similar displacement on the principal component plot reflecting overall similarity in the pattern of expression of immune-related genes. These changes in immunological profile were much more pronounced at exp day 1 than at later timepoints. DSS-induced changes were most similar to changes induced by ethanol enemas, which nonetheless were still distinguishable from the baseline condition (PBS enemas).
Figure 5.

Distinct patterns of cytokine expression in chemical and genetic colitis models. RNA obtained from full-thickness samples (n ≥ 5 male or mixed-sex mice per group, see Figs. 1, 2, 3, and 4) of the distal colon was measured for immunological gene expression using the Nanostring Mouse Immunology Panel (561 genes). A: merged data (all samples from all models) were reduced to principal components (PC) and plotted. The Il10−/− and Il10−/− Tnfr1−/− models occupied a distinct region (left side) of the PC plot compared with the chemical [oxazolone, dextran sulfate sodium (DSS), 2,4,6-trinitrobenzene sulfonic acid (TNBS)] models (right). B: all models were associated with the upregulation of classical interferon or interferon-responsive genes (Ifng, Ifitm1, Irf1, and Socs1). Shown is the fold change response of the abundance value. In TNBS and oxazolone models, the interferon changes were most pronounced on day 1. C: heatmap of individual cytokine or chemokine changes associated with exacerbated injury in each of the colitis models. Each model is compared with its own control, but the data are merged to facilitate comparisons across models. Each row represents a mouse and each column a gene. Cytokine profiles of oxazolone and TNBS models were overall similar to each other, but changing with time. Columns are grouped together by overall similarity and reveal groups of cytokines as they covary across models. Statistics: ***P < 0.001. Pairwise t tests with Holm-Bonferroni correction.
We next examined individual cytokines and chemokines that were differentially regulated in the tested injury models. Consistent with the common upregulation of the interferon-responsive epithelial Ly6a, canonical target genes of the interferon pathway were upregulated in all models (Fig. 5B). The heatmap shown in Fig. 5C clusters cytokine modules by similarity across the experimental models. Intriguingly, we did not observe significant differences in cytokines between TNBS and oxazolone models, which may be consistent with the similar stem cell responses seen in histological analyses. However, the TNBS, oxazolone, and Il10−/− Tnfr1 models, which are associated with severe colitis but retention of the Lgr5+ stem cell population, exhibit a polarization to the cytokine response that is not found in the DSS samples. Whereas the DSS model was associated with broad increases in many cytokines, decreases in mRNA expression of key IL-17 and IL-22 genes, IL-7, IL-18, and IL-15 were observed in the acute phases of the other models. Thus, retention of Lgr5+ stem cells is associated with distinct and broad patterns of cytokine expression across the tested models of experimental colitis.
Organoid Culture with Agents of Chemical Colitis
Because DSS induces near-total loss of Lgr5 expression in vivo, we asked whether this effect, as well as the effects of the TNBS and oxazolone molecules on Lgr5 depletion in some crypts, could be due to their direct chemical effect on the colonic epithelium. To test this, we grew colonic epithelial organoids (colonoids) isolated from Lgr5::DTR-EGFP mice (9) and tested their acute responses (within 5 h) to DSS, oxazolone, or TNBS added to the culture media. Imaging of organoids enabled identification of Lgr5+ cells through their expression of GFP (Fig. 6, A–D). DSS induced loss of fluorescent Lgr5+ cells from the tips of the crypt-like budding structures in colonoids (Fig. 6A). These responses were monotonically dose dependent. In contrast, the lowest tested dose of TNBS or oxazolone spared the Lgr5+ cell population (Fig. 6, B and C). At higher doses, the Lgr5+ cell population was depleted, although studies of exposure to the ethanol diluent also supported a partial depletion of the Lgr5+ cells (Fig. 6D). The organoids were then stained for incorporation of 5-ethynyl-2′-deoxyuridine (EdU, marking DNA synthesis or proliferation) and cleaved caspase-3 (CC3, marking apoptosis) (Fig. 6, E–H). DSS (Fig. 6E), TNBS (Fig. 6F), oxazolone (Fig. 6G), or ethanol diluent (Fig. 6G) had minimal effect on EdU incorporation. However, DSS, TNBS, and oxazolone could all induce CC3 staining through the full range of doses, which ethanol did not do (Fig. 6H). We note that we were not able to reliably expand colonoids from Il10−/− Tnfr1−/− mice, but could do so from Il10−/− mice, perhaps due to increased epithelial apoptosis in the double-knockout epithelium (27). In total, these results suggest that chemical colitis agents can acutely ablate Lgr5+ stem cells and induce apoptotic signaling in colonic epithelium.
Figure 6.

Agents of chemical colitis directly induce apoptosis of colonic epithelium. Epithelial organoids isolated from the distal colon of Lgr5::DTR-EGFP (Lgr5-GFP) mixed-sex mice were grown and passaged for >5 rounds. Organoids were exposed to chemical colitis agents at different concentrations for 3–5 h, fixed, stained, and imaged on a confocal microscope. The results shown are aggregated from three independent experiments, each of which involved the analysis of >30 organoids per group. The presence of Lgr5+ stem cells within the crypt-like buds of the organoids was assessed by imaging the GFP label, as shown in photomicrographs (A–C). Living cells uniformly express GFP on the cell membrane; in contrast, dying or dead cells exhibit blebs of signal. The basolateral layer of epithelium is highlighted by the dashed white line. The apical layer adjacent to the lumen (“L”) is highlighted by the dashed red line. White arrows within zoomed insets indicate examples of visually normal Lgr5+ cells; magenta arrows indicate examples of blebs. Exposure to low doses of dextran sulfate sodium (DSS) induced blebbing in Lgr5+ cells (A). Exposure to TNBS (B) or oxazolone (C) induced abnormalities in Lgr5+ cells only at higher doses. Some of these effects were attributable to the ethanol (EtOH) solvent (C) for the haptenating agents. Quantification (D) of the density of visually normal Lgr5+ cells confirms the dose-dependent loss of cells with DSS exposure and loss at higher doses of TNBS, oxazolone, or ethanol. E–H: organoids were exposed to 10 µM 5-ethynyl-2′-deoxyuridine (EdU) for 30 min before fixation. Fixed organoids were stained for the EdU incorporation (proliferation, magenta) and cleaved caspase 3 (apoptosis, green). The blue signal represents Hoechst staining (cellular nuclei). Shown are images associated with exposure to DSS (E), TNBS (F), oxazolone (G), or EtOH (G), as well as the quantification (H) of the density of positively stained cells per organoid bud. The results support only very minor proliferative changes associated with treatments. However, the all agents increased the number of apoptotic cells at low (DSS) or higher (TNBS, oxazolone, EtOH) doses. Scale bars: 50 µm. Statistics: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. One-way ANOVA, Tukey’s post hoc test. Error bars: means ± SE.
Role of Lgr5+ Stem Cells in Epithelial Regeneration in Experimental Colitis
To determine whether the retention of Lgr5+ stem cells in hapten-induced or IL-10-deficient models of colitis is essential for health, we ablated these cells in Lgr5::DTR-EGFP mice, in which the Lgr5+ cells are targetable through injection of diphtheria toxin (DT). A previous study has demonstrated using these transgenic mice that Lgr5+ stem cells are dispensable for epithelial hyperproliferation in DSS-induced colitis (17). To test whether Lgr5+ stem cells were necessary to maintain epithelium during IL-10 deficiency, we injected mice with an antibody targeted against the IL-10 receptor (Fig. 7A). Five weekly injections, beginning at 3 wk of age, with this antibody induces colitis resembling the disease found in susceptible Il10−/− mice (27, 44–47). At 8 wk of age, anti-IL10R-injected mice exhibited colitis (Fig. 7B). Similar to findings in Il10−/− mice (Fig. 4), colonic crypts in anti-IL10R-injected colitic mice harbored Lgr5+ stem cells marked by GFP expression (Fig. 7C). Two intraperitoneal injections of DT over a span of 4 days ablated Lgr5+ stem cells from colonic crypts (Fig. 7C). During this time period, DT-injected transgenic mice lost significant body weight, while PBS-injected controls and DT-injected wild-type mice appeared unaffected (Fig. 7D). Consistent with previous studies (9, 17) demonstrating the dispensability of Lgr5+ stem cells in intestinal homeostasis, injection of DT to uninjured Lgr5::DTR-EGFP mice did not result in weight loss (Fig. 7D). Postmortem analyses showed that DT treatment to transgenic colitic mice resulted in increased colonic thickness (Fig. 7E). Although the overall histological scores between DT-injected and PBS-injected colitic mice were similar (Fig. 7F), the disease in DT-injected mice involved more-proximal regions of the colon (Fig. 7, G and H). Histological scoring of noncolitic DT-injected transgenic mice confirmed the lack of pathology associated with Lgr5+ stem cell ablation in homeostasis (Fig. 7F). Thus, we conclude that Lgr5+ stem cells are essential for restriction of disease in IL-10-deficient colitis.
Figure 7.

Ablation of Lgr5+ stem cells exacerbates IL-10-deficiency colitis but has only modest effects on 2,4,6-trinitrobenzene sulfonic acid (TNBS) colitis. A: experimental interventions performed on Lgr5::DTR-EGFP (Lgr5-DTR) and wild-type (WT) mice. Data shown are representative from three independent experiments, each involving five mixed-sex mice per group. Mice were injected weekly with an antibody against the IL-10 receptor (IL10R) or an isotype control antibody, beginning at 3-wk old. At 7-wk old, mice were injected with diphtheria toxin (DT) to ablate Lgr5+ stem cells, or PBS control. The clinical course was acutely monitored for 4 days. B: hematoxylin-eosin (H&E)-stained photomicrographs show epithelial hyperplasia and mucosal immune infiltration in anti-IL10R-treated animals. C: immuostaining for GFP marking Lgr5+ stem cells validates the persistence of Lgr5+ stem cells in the crypts of anti-IL10R-treated mice. However, injection of diphtheria toxin (DT) ablates the staining signal consistent with the DT-induced loss of Lgr5+ stem cells. D: body weight curve of mice with colitis (anti-IL10R) or in homeostasis (IgG) with or without DT treatment. Specifically, colitic mice with DT treatment (ablated Lgr5+ stem cells) exhibited a rapid loss of body weight. E: ablation of Lgr5+ cells increased the colonic thickness of mice with colitis. This is consistent with exacerbation of injury. Overall histological colitis score (F) was unchanged among the sample groups. No histological damage was found to be associated with Lgr5+ stem cell ablation in homeostasis. However, subcategory scoring (G) revealed a higher proximal-to-distal percentage of the mucosa involved in colitic mice with ablated Lgr5+ stem cells, which could be seen in histological images (H). I: summary schematic of the experimental interventions performed on Lgr5::DTR-EGFP (Lgr5-DTR) mice in the TNBS colitis model. Data shown are representative of two independent experiments, each involving the analysis of four mice per group. No significant differences in body weight (J), colonic thickness (K), or overall histological score (L) were observed to be associated with DT treatment of mice with TNBS colitis. Subcategory histological scoring (M) showed a higher index of crypt inflammation. Displayed is an example image (O) of the qualitatively increased mucosal infiltrate. Scale bars: 50 µm. Statistics: *P < 0.05; **P < 0.01; ***P < 0.001. Two-tailed t test. Error bars: means ± SE.
We performed a similar experiment using the TNBS model in Lgr5::DTR-EGFP mice (Fig. 7I). These mice were backcrossed for two generations onto the susceptible SJL background. However, administration of DT to transgenic mice did not alter the pattern of weight loss (Fig. 7J). Colonic thickness (Fig. 7K) and histological scores (Fig. 7L) were indistinguishable between the conditions. However, subcategory scoring of the samples revealed a slight increase in the inflammatory infiltrate in DT-treated samples (Fig. 7, M and N). The overall effects of Lgr5+ cell ablation in TNBS colitis were much reduced when compared with those in the anti-IL10R model. Thus, Lgr5+ stem cells appear more dispensable for health during TNBS-induced colitis.
DISCUSSION
We have surveyed the fates of the colonic epithelial Lgr5+ stem cells across representative mouse models of IBD. We found divergent responses of epithelium depending on the injury model. These are summarized in Table 1. Although all tested models were associated with upregulation of Ly6a, there was a spectrum of phenotypes in regard to the retention of the Lgr5+ stem cell population. A total loss of Lgr5+ cells was observed in the DSS colitis model (Fig. 1). Spatially varying expansion and dropout of the Lgr5+ population characterized hapten-induced colitis (Figs. 2 and 3). The persistence of Lgr5+ stem cells was associated with exacerbations in Il10−/− colitis induced by the codeletion of Tnfr1, which causes improper development of mucosal immunity and tolerance (27) (Fig. 4). These differences in fates parallel the results of functional studies (Fig. 7). In models where Lgr5+ cells are lost from all or some crypts (i.e., the acute DSS and TNBS/oxazolone models), these cells play only minor roles in outcomes of disease. However, in the tested model of chronic (IL10R inhibited) colitis, which is associated with the retention of Lgr5+ stem cells, the presence of this cell population restricted disease. Thus, in this chronic colitis model, the retention of the Lgr5+ stem cell population underscores their physiological necessity.
Table 1.
Comparison of stem cell responses in different models of mouse IBD
| DSS | TNBS | Oxazolone | Exacerbated Il10 Deficiency | |
|---|---|---|---|---|
| Putative mechanism of injury | Direct epithelial injury with inflammation | Haptenation of luminal contents | Haptenation of luminal contents | Loss of immunoregulatory signaling |
| Lgr5+ stem cell persistence | − | + (most crypts) | + (most crypts) | + |
| Ly6a upregulation | + | + | + | + |
| Secretory cell census | Reduced goblet cells, increased tuft cells | Not grossly affected, perhaps small decrease in goblet cells | Reduced goblet cells | Reduced secretory cell density due to hyperplasia of crypts/expansion of progenitor cell zone |
| Effect on Lgr5+ stem cells and overall cell survival in organoids | Dose-dependent depletion | Depletion at higher concentrations | Depletion at higher concentrations | Il10−/− Tnfr1−/− organoids did not survive |
| Functional reliance on Lgr5+ cells | Not tested but previously found to be not reliant (17) | Minor reliance | Not tested | Major reliance |
Although a simple rule of “presence equals necessity” may be an attractive explanation, it applies neither to homeostasis, in which Lgr5+ cell loss has no major histological effect on epithelium (for example, Fig. 7), nor to the TNBS/oxazolone models, in which Lgr5+ stem cells are nonetheless retained in many crypts but are not required for health. In these situations, we believe the activation of cellular plasticity mechanisms compensates for the loss of Lgr5+ stem cells. The chronicity of the IL-10-deficient injury models may be a key factor in determining the requirement of Lgr5+ stem cells. Future studies will need to examine stem cell patterns at different timepoints in the physiological history of injury. Key questions include whether Lgr5+ cells are depleted at the acute onset of a chronic colitis, or if a dependence on Lgr5+ cells can be uncovered with repetitive administration of chemically induced injury models.
Why are the results so different between models? One explanation may be the differential polarization of cytokines and their different kinetics of elevation (Fig. 5). We note that the lack of a presensitization step in the hapten models could have affected the immune profile. Cytokines have direct effects on intestinal epithelial stem cell function (48). Another possibility is that the pattern of Lgr5 expression is not the result of inflammation, but rather a direct consequence of the chemical irritant used to induce injury and inflammation. This is partially supported by the findings of direct impact of chemical agents on Lgr5+ stem cells and apoptotic signaling in organoids (Fig. 6). The loss of Lgr5+ cells from histologically normal crypts in the DSS model (Fig. 1) suggests that the chemical agent, and not the overt secondary inflammation, is causing this loss directly. In the hapten models, we cannot discriminate between these two mechanisms (inflammation vs. direct injury) based on their kinetics of onset alone. Both the elevation in cytokines and effects on the epithelium are rapid, evident within 1 day after treatment (Figs. 2, 3, and 5) in vivo. We would propose, however, that the intercrypt variability of Lgr5+ cell retention may result from local variations in TNBS/oxazolone concentration causing direct epithelial injury, as small volumes of these chemical agents are challenging to disperse uniformly throughout the lumen through enema. Because the inflammatory response results from the haptenation and dispersal of luminal contents, the histological injury pattern will appear more widespread than the initial distribution of TNBS/oxazolone and will involve both Lgr5-positive and Lgr5-negative crypts. This explanation would suggest that inflammation does not fundamentally alter the overall pattern of Lgr5 persistence or absence, which is dictated by the unique mechanism of initial injury. However, this and other competing hypotheses deserve further study.
The epithelial compartment in human IBD undergoes significant transcriptional remodeling (49–54). Reports performed in small sets of patients suggest retention of Lgr5+ cells in human ulcerative colitis and colitis-associated cancer (55, 56), supporting the relevance of the preclinical findings here. However, the diversity of individualized pathophysiological mechanisms comprising the umbrella diagnosis of human IBD [e.g., (57)] may challenge any proposed universal and unequivocal answer to how human epithelial stem cells respond in IBD. No preclinical model of IBD is a perfect replica of the human disease. Based on the different types of stem cell responses observed here, in models that each replicate certain aspects of IBD physiology, there may not be a single answer to what happens to stem cells in human IBD. Rather, the answer may depend on the specific type of injury and location of disease. Further elucidation of stem cell responses in a variety of preclinical intestinal injury models may help define the range of key mechanisms at play.
SUPPLEMENTAL DATA
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.14870211;
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.14870220;
Supplemental Fig. S3, https://doi.org/10.6084/m9.figshare.14870226;
Supplemental Fig. S4, https://doi.org/10.6084/m9.figshare.14870223;
Supplemental Fig. S5, https://doi.org/10.6084/m9.figshare.14870217;
Supplemental Fig. S6, https://doi.org/10.6084/m9.figshare.14870214.
GRANTS
This work was funded by the National Institutes of Health (R01-DK108648 to DBP, R01-DK056008 to D.B. Polk). CYL was funded by a Career Development Award (#694110) from the Crohn’s and Colitis Foundation and by the National Institutes of Health (P30-DK042086). Y. Huang and Z. Sharifkhodaei acknowledge funding from The Saban Research Institute postdoctoral fellowship program at Children’s Hospital Los Angeles.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
N.G., C.Y.L., and D.B.P. conceived and designed research; N.G., C.Y.L., S.G., M.L.G., Y.H., and Z.S. performed experiments; N.G., C.Y.L., S.G., Y.H., Z.S., and M.K.W. analyzed data; N.G., C.Y.L., S.G., Y.H., M.K.W., and D.B.P. interpreted results of experiments; N.G., C.Y.L., and S.G. prepared figures; C.Y.L. drafted manuscript; D.B.P. edited and revised manuscript; N.G., C.Y.L., S.G., M.L.G., Y.H., Z.S., M.K.W., and D.B.P. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank G. Esteban Fernandez for technical assistance with microscopy, Gricelda Vasquez for animal husbandry, and Ivan Fuss for helpful discussions about TNBS and oxazolone mouse models.
REFERENCES
- 1.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, Clevers H. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449: 1003–1007, 2007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 2.Buczacki SJ, Zecchini HI, Nicholson AM, Russell R, Vermeulen L, Kemp R, Winton DJ. Intestinal label-retaining cells are secretory precursors expressing Lgr5. Nature 495: 65–69, 2013. doi: 10.1038/nature11965. [DOI] [PubMed] [Google Scholar]
- 3.Davidson LA, Goldsby JS, Callaway ES, Shah MS, Barker N, Chapkin RS. Alteration of colonic stem cell gene signatures during the regenerative response to injury. Biochim Biophys Acta 1822: 1600–1607, 2012. doi: 10.1016/j.bbadis.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gregorieff A, Liu Y, Inanlou MR, Khomchuk Y, Wrana JL. Yap-dependent reprogramming of Lgr5(+) stem cells drives intestinal regeneration and cancer. Nature 526: 715–718, 2015. doi: 10.1038/nature15382. [DOI] [PubMed] [Google Scholar]
- 5.Murata K, Jadhav U, Madha S, van Es J, Dean J, Cavazza A, Wucherpfennig K, Michor F, Clevers H, Shivdasani RA. Ascl2-dependent cell dedifferentiation drives regeneration of ablated intestinal stem cells. Cell Stem Cell 26: 377–390.e6,2020. doi: 10.1016/j.stem.2019.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nusse YM, Savage AK, Marangoni P, Rosendahl-Huber AKM, Landman TA, de Sauvage FJ, Locksley RM, Klein OD. Parasitic helminths induce fetal-like reversion in the intestinal stem cell niche. Nature 559: 109–113, 2018. [Erratum inNature562: E22, 2018]. doi: 10.1038/s41586-018-0257-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ritsma L, Ellenbroek SIJ, Zomer A, Snippert HJ, de Sauvage FJ, Simons BD, Clevers H, van Rheenen J. Intestinal crypt homeostasis revealed at single-stem-cell level by in vivo live imaging. Nature 507: 362–365, 2014. doi: 10.1038/nature12972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tetteh PW, Basak O, Farin HF, Wiebrands K, Kretzschmar K, Begthel H, van den Born M, Korving J, de Sauvage F, van Es JH, van Oudenaarden A, Clevers H. Replacement of lost Lgr5-positive stem cells through plasticity of their enterocyte-lineage daughters. Cell Stem Cell 18: 203–213, 2016. doi: 10.1016/j.stem.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 9.Tian H, Biehs B, Warming S, Leong KG, Rangell L, Klein OD, de Sauvage FJ. A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature 478: 255–259, 2011. [Erratum inNature482: 120, 2011]. doi: 10.1038/nature10408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yan KS, Chia LA, Li X, Ootani A, Su J, Lee JY, Su N, Luo Y, Heilshorn SC, Amieva MR, Sangiorgi E, Capecchi MR, Kuo CJ. The intestinal stem cell markers Bmi1 and Lgr5 identify two functionally distinct populations. Proc Natl Acad Sci USA 109: 466–471, 2012. doi: 10.1073/pnas.1118857109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yui S, Azzolin L, Maimets M, Pedersen MT, Fordham RP, Hansen SL, Larsen HL, Guiu J, Alves MRP, Rundsten CF, Johansen JV, Li Y, Madsen CD, Nakamura T, Watanabe M, Nielsen OH, Schweiger PJ, Piccolo S, Jensen KB. YAP/TAZ-dependent reprogramming of colonic epithelium links ECM remodeling to tissue regeneration. Cell Stem Cell 22: 35–49.e7,2018. doi: 10.1016/j.stem.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ayyaz A, Kumar S, Sangiorgi B, Ghoshal B, Gosio J, Ouladan S, Fink M, Barutcu S, Trcka D, Shen J, Chan K, Wrana JL, Gregorieff A. Single-cell transcriptomes of the regenerating intestine reveal a revival stem cell. Nature 569: 121–125, 2019. doi: 10.1038/s41586-019-1154-y. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Chiang IL, Ohara TE, Fujii S, Cheng J, Muegge BD, Ver Heul A, Han ND, Lu Q, Xiong S, Chen F, Lai CW, Janova H, Wu R, Whitehurst CE, VanDussen KL, Liu TC, Gordon JI, Sibley LD, Stappenbeck TS. Long-term culture captures injury-repair cycles of colonic stem cells. Cell 179: 1144–1159.e15,2019. doi: 10.1016/j.cell.2019.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sangiorgi E, Capecchi MR. Bmi1 is expressed in vivo in intestinal stem cells. Nat Genet 40: 915–920, 2008. doi: 10.1038/ng.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmitt M, Schewe M, Sacchetti A, Feijtel D, van de Geer WS, Teeuwssen M, Sleddens HF, Joosten R, van Royen ME, van de Werken HJG, van Es J, Clevers H, Fodde R. Paneth cells respond to inflammation and contribute to tissue regeneration by acquiring stem-like features through SCF/c-kit signaling. Cell Rep 24: 2312–2328,2018. e2317 doi: 10.1016/j.celrep.2018.07.085. [DOI] [PubMed] [Google Scholar]
- 16.Serra D, Mayr U, Boni A, Lukonin I, Rempfler M, Challet Meylan L, Stadler MB, Strnad P, Papasaikas P, Vischi D, Waldt A, Roma G, Liberali P. Self-organization and symmetry breaking in intestinal organoid development. Nature 569: 66–72, 2019. doi: 10.1038/s41586-019-1146-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Metcalfe C, Kljavin NM, Ybarra R, de Sauvage FJ. Lgr5+ stem cells are indispensable for radiation-induced intestinal regeneration. Cell Stem Cell 14: 149–159, 2014. doi: 10.1016/j.stem.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 18.Kiesler P, Fuss IJ, Strober W. Experimental models of inflammatory bowel diseases. Cell Mol Gastroenterol Hepatol 1: 154–170, 2015. doi: 10.1016/j.jcmgh.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, International IBDGC, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491: 119–124, 2012. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neurath MF, Travis SP. Mucosal healing in inflammatory bowel diseases: a systematic review. Gut 61: 1619–1635, 2012. doi: 10.1136/gutjnl-2012-302830. [DOI] [PubMed] [Google Scholar]
- 21.Powell AE, Wang Y, Li Y, Poulin EJ, Means AL, Washington MK, Higginbotham JN, Juchheim A, Prasad N, Levy SE, Guo Y, Shyr Y, Aronow BJ, Haigis KM, Franklin JL, Coffey RJ. The pan-ErbB negative regulator Lrig1 is an intestinal stem cell marker that functions as a tumor suppressor. Cell 149: 146–158, 2012. doi: 10.1016/j.cell.2012.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong VW, Stange DE, Page ME, Buczacki S, Wabik A, Itami S, van de Wetering M, Poulsom R, Wright NA, Trotter MW, Watt FM, Winton DJ, Clevers H, Jensen KB. Lrig1 controls intestinal stem-cell homeostasis by negative regulation of ErbB signalling. Nat Cell Biol 14: 401–408, 2012. doi: 10.1038/ncb2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scheiffele F, Fuss IJ. Induction of TNBS colitis in mice. Curr Protoc Immunol, 2002. doi: 10.1002/0471142735.im1519s49. [DOI] [PubMed] [Google Scholar]
- 24.Antoniou E, Margonis GA, Angelou A, Pikouli A, Argiri P, Karavokyros I, Papalois A, Pikoulis E. The TNBS-induced colitis animal model: an overview. Ann Med Surg (Lond) 11: 9–15, 2016. doi: 10.1016/j.amsu.2016.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morris GP, Beck PL, Herridge MS, Depew WT, Szewczuk MR, Wallace JL. Hapten-induced model of chronic inflammation and ulceration in the rat colon. Gastroenterology 96: 795–803, 1989. [PubMed] [Google Scholar]
- 26.Elson CO, Sartor RB, Tennyson GS, Riddell RH. Experimental models of inflammatory bowel disease. Gastroenterology 109: 1344–1367, 1995. doi: 10.1016/0016-5085(95)90599-5. [DOI] [PubMed] [Google Scholar]
- 27.Kojima R, Kuroda S, Ohkishi T, Nakamaru K, Hatakeyama S. Oxazolone-induced colitis in BALB/C mice: a new method to evaluate the efficacy of therapeutic agents for ulcerative colitis. J Pharmacol Sci 96: 307–313, 2004. doi: 10.1254/jphs.fp0040214. [DOI] [PubMed] [Google Scholar]
- 28.Liu CY, Tam SS, Huang Y, Dube PE, Alhosh R, Girish N, Punit S, Nataneli S, Li F, Bender JM, Washington MK, Polk DB. TNF receptor 1 promotes early-life immunity and protects against colitis in mice. Cell Rep 33: 108275, 2020. doi: 10.1016/j.celrep.2020.108275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boirivant M, Fuss IJ, Chu A, Strober W. Oxazolone colitis: a murine model of T helper cell type 2 colitis treatable with antibodies to interleukin 4. J Exp Med 188: 1929–1939, 1998. doi: 10.1084/jem.188.10.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heller F, Fuss IJ, Nieuwenhuis EE, Blumberg RS, Strober W. Oxazolone colitis, a Th2 colitis model resembling ulcerative colitis, is mediated by IL-13-producing NK-T cells. Immunity 17: 629–638, 2002. doi: 10.1016/s1074-7613(02)00453-3. [DOI] [PubMed] [Google Scholar]
- 31.Wang X, Ouyang Q, Luo WJ. Oxazolone-induced murine model of ulcerative colitis. Chin J Dig Dis 5: 165–168, 2004. doi: 10.1111/j.1443-9573.2004.00173.x. [DOI] [PubMed] [Google Scholar]
- 32.Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 75: 263–274, 1993. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 33.Bristol IJ, Farmer MA, Cong Y, Zheng XX, Strom TB, Elson CO, Sundberg JP, Leiter EH. Heritable susceptibility for colitis in mice induced by IL-10 deficiency. Inflamm Bowel Dis 6: 290–302, 2000. doi: 10.1002/ibd.3780060407. [DOI] [PubMed] [Google Scholar]
- 34.Farmer MA, Sundberg JP, Bristol IJ, Churchill GA, Li R, Elson CO, Leiter EH. A major quantitative trait locus on chromosome 3 controls colitis severity in IL-10-deficient mice. Proc Natl Acad Sci USA 98: 13820–13825, 2001. doi: 10.1073/pnas.241258698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mahler M, Most C, Schmidtke S, Sundberg JP, Li R, Hedrich HJ, Churchill GA. Genetics of colitis susceptibility in IL-10-deficient mice: backcross versus F2 results contrasted by principal component analysis. Genomics 80: 274–282, 2002. doi: 10.1006/geno.2002.6840. [DOI] [PubMed] [Google Scholar]
- 36.Punit S, Dube PE, Liu CY, Girish N, Washington MK, Polk DB. Tumor necrosis factor receptor 2 restricts the pathogenicity of CD8(+) T cells in mice with colitis. Gastroenterology 149: 993–1005.e2,2015. doi: 10.1053/j.gastro.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pfeffer K, Matsuyama T, Kundig TM, Wakeham A, Kishihara K, Shahinian A, Wiegmann K, Ohashi PS, Kronke M, Mak TW. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell 73: 457–467, 1993. doi: 10.1016/0092-8674(93)90134-C. [DOI] [PubMed] [Google Scholar]
- 38.Singh V, Kumar M, San Yeoh B, Xiao X, Saha P, Kennett MJ, Vijay-Kumar M. Inhibition of interleukin-10 signaling induces microbiota-dependent chronic colitis in apolipoprotein E deficient mice. Inflamm Bowel Dis 22: 841–852, 2016. doi: 10.1097/MIB.0000000000000699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kullberg MC, Jankovic D, Feng CG, Hue S, Gorelick PL, McKenzie BS, Cua DJ, Powrie F, Cheever AW, Maloy KJ, Sher A. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J Exp Med 203: 2485–2494, 2006. doi: 10.1084/jem.20061082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carvalho FA, Nalbantoglu I, Ortega-Fernandez S, Aitken JD, Su Y, Koren O, Walters WA, Knight R, Ley RE, Vijay-Kumar M, Gewirtz AT. Interleukin-1beta (IL-1beta) promotes susceptibility of Toll-like receptor 5 (TLR5) deficient mice to colitis. Gut 61: 373–384, 2012. doi: 10.1136/gut.2011.240556. [DOI] [PubMed] [Google Scholar]
- 41.Asseman C, Mauze S, Leach MW, Coffman RL, Powrie F. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J Exp Med 190: 995–1004, 1999. doi: 10.1084/jem.190.7.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Biton M, Haber AL, Rogel N, Burgin G, Beyaz S, Schnell A, Ashenberg O, Su CW, Smillie C, Shekhar K, Chen Z, Wu C, Ordovas-Montanes J, Alvarez D, Herbst RH, Zhang M, Tirosh I, Dionne D, Nguyen LT, Xifaras ME, Shalek AK, von Andrian UH, Graham DB, Rozenblatt-Rosen O, Shi HN, Kuchroo V, Yilmaz OH, Regev A, Xavier RJ. T helper cell cytokines modulate intestinal stem cell renewal and differentiation. Cell 175: 1307–1320.e22,2018. doi: 10.1016/j.cell.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang B, Chen Z, Geng L, Wang J, Liang H, Cao Y, et al. Mucosal profiling of pediatric-onset colitis and IBD reveals common pathogenics and therapeutic pathways. Cell 179: 1160–1176.e24, 2019. doi: 10.1016/j.cell.2019.10.027. [DOI] [PubMed] [Google Scholar]
- 44.Martin JC, Chang C, Boschetti G, Ungaro R, Giri M, Grout JA, Gettler K, Chuang L-S, Nayar S, Greenstein AJ, Dubinsky M, Walker L, Leader A, Fine JS, Whitehurst CE, Mbow ML, Kugathasan S, Denson LA, Hyams JS, Friedman JR, Desai PT, Ko HM, Laface I, Akturk G, Schadt EE, Salmon H, Gnjatic S, Rahman AH, Merad M, Cho JH, Kenigsberg EK. Single-cell analysis of Crohn’s disease lesions identifies a pathogenic cellular module associated with resistance to anti-TNF therapy. Cell 178: 1493–1508.e20, 2019. doi: 10.1016/j.cell.2019.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parikh K, Antanaviciute A, Fawkner-Corbett D, Jagielowicz M, Aulicino A, Lagerholm C, Davis S, Kinchen J, Chen HH, Alham NK, Ashley N, Johnson E, Hublitz P, Bao L, Lukomska J, Andev RS, Björklund E, Kessler BM, Fischer R, Goldin R, Koohy H, Simmons A. Colonic epithelial cell diversity in health and inflammatory bowel disease. Nature 567: 49–55, 2019. doi: 10.1038/s41586-019-0992-y. [DOI] [PubMed] [Google Scholar]
- 46.Haberman Y, Karns R, Dexheimer PJ, Schirmer M, Somekh J, Jurickova I, Braun T,. et al. Ulcerative colitis mucosal transcriptomes reveal mitochondriopathy and personalized mechanisms underlying disease severity and treatment response. Nat Commun 10: 38, 2019. doi: 10.1038/s41467-018-07841-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Howell KJ, Kraiczy J, Nayak KM, Gasparetto M, Ross A, Lee C, Mak TN, Koo BK, Kumar N, Lawley T, Sinha A, Rosenstiel P, Heuschkel R, Stegle O, Zilbauer M. DNA methylation and transcription patterns in intestinal epithelial cells from pediatric patients with inflammatory bowel diseases differentiate disease subtypes and associate with outcome. Gastroenterology 154: 585–598, 2018. doi: 10.1053/j.gastro.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sarvestani SK, Signs SA, Lefebvre V, Mack S, Ni Y, Morton A, Chan ER, Li X, Fox P, Ting A, Kalady MF, Cruise M, Ashburn J, Stiene J, Lai W, Liska D, Xiang S, Huang EH. Cancer-predicting transcriptomic and epigenetic signatures revealed for ulcerative colitis in patient-derived epithelial organoids. Oncotarget 9: 28717–28730, 2018. doi: 10.18632/oncotarget.25617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dube PE, Liu CY, Girish N, Washington MK, Polk DB. Pharmacological activation of epidermal growth factor receptor signaling inhibits colitis-associated cancer in mice. Sci Rep 8: 9119, 2018. doi: 10.1038/s41598-018-27353-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu CY, Dube PE, Girish N, Reddy AT, Polk DB. Optical reconstruction of murine colorectal mucosa at cellular resolution. Am J Physiol Gastrointest Liver Physiol 308: G721–G735, 2015. doi: 10.1152/ajpgi.00310.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sugimoto S, Sato T. Establishment of 3D intestinal organoid cultures from intestinal stem cells. Methods Mol Biol 1612: 97–105, 2017. doi: 10.1007/978-1-4939-7021-6_7. [DOI] [PubMed] [Google Scholar]
- 52.Kazama S, Kishikawa J, Tanaka T, Hata K, Kawai K, Nozawa H, Ishihara S. Immunohistochemical expression of CD133 and LGR5 in ulcerative colitis-associated colorectal cancer and dysplasia. In Vivo 33: 1279–1284, 2019. doi: 10.21873/invivo.11600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shimokawa M, Ohta Y, Nishikori S, Matano M, Takano A, Fujii M, Date S, Sugimoto S, Kanai T, Sato T. Visualization and targeting of LGR5(+) human colon cancer stem cells. Nature 545: 187–192, 2017. doi: 10.1038/nature22081. [DOI] [PubMed] [Google Scholar]
- 54.Czarnewski P, Parigi SM, Sorini C, Diaz OE, Das S, Gagliani N, Villablanca EJ. Conserved transcriptomic profile between mouse and human colitis allows unsupervised patient stratification. Nat Commun 10: 2892, 2019. doi: 10.1038/s41467-019-10769-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, Van Houdt WJ, Pronk A, Van Gorp J, Siersema PD, Clevers H. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology 141: 1762–1772, 2011. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 56.Kennedy RJ, Hoper M, Deodhar K, Erwin PJ, Kirk SJ, Gardiner KR. Interleukin 10-deficient colitis: new similarities to human inflammatory bowel disease. Br J Surg 87: 1346–1351, 2000. doi: 10.1046/j.1365-2168.2000.01615.x. [DOI] [PubMed] [Google Scholar]
- 57.Zhang Y, Dube PE, Washington MK, Yan F, Polk DB. ErbB2 and ErbB3 regulate recovery from dextran sulfate sodium-induced colitis by promoting mouse colon epithelial cell survival. Lab Invest 92: 437–450, 2012. doi: 10.1038/labinvest.2011.192. [DOI] [PMC free article] [PubMed] [Google Scholar]