

Abstract
Mitochondria are recognized as signaling organelles, because under stress, mitochondria can trigger various signaling pathways to coordinate the cell’s response. The specific pathway(s) engaged by mitochondria in response to mitochondrial energy defects in vivo and in high-energy tissues like the heart are not fully understood. Here, we investigated cardiac pathways activated in response to mitochondrial energy dysfunction by studying mice with cardiomyocyte-specific loss of the mitochondrial phosphate carrier (SLC25A3), an established model that develops cardiomyopathy as a result of defective mitochondrial ATP synthesis. Mitochondrial energy dysfunction induced a striking pattern of acylome remodeling, with significantly increased posttranslational acetylation and malonylation. Mass spectrometry-based proteomics further revealed that energy dysfunction-induced remodeling of the acetylome and malonylome preferentially impacts mitochondrial proteins. Acetylation and malonylation modified a highly interconnected interactome of mitochondrial proteins, and both modifications were present on the enzyme isocitrate dehydrogenase 2 (IDH2). Intriguingly, IDH2 activity was enhanced in SLC25A3-deleted mitochondria, and further study of IDH2 sites targeted by both acetylation and malonylation revealed that these modifications can have site-specific and distinct functional effects. Finally, we uncovered a novel cross talk between the two modifications, whereby mitochondrial energy dysfunction-induced acetylation of sirtuin 5 (SIRT5), inhibited its function. Because SIRT5 is a mitochondrial deacylase with demalonylase activity, this finding suggests that acetylation can modulate the malonylome. Together, our results position acylations as an arm of the mitochondrial response to energy dysfunction and suggest a mechanism by which focal disruption to the energy production machinery can have an expanded impact on global mitochondrial function.
Keywords: acylations, energy, heart, mitochondria, posttranslational modifications
INTRODUCTION
The mitochondrial oxidative phosphorylation system (OXPHOS) is the major source of fuel that drives cellular functions. The critical dependence on mitochondria as an energy source is especially evident in tissues with high-energy demands such as the heart; defects in the mitochondrial energy production machinery underlie a wide range of primary mitochondrial disorders that present with cardiac disease (1, 2), and cardiac diseases like heart failure and myocardial infarction are characterized by mitochondrial dysfunction (3–5). Yet, the mitochondrion-intrinsic mechanisms activated in response to primary defects in cardiac mitochondrial energy production are not clear.
Several pathways can communicate mitochondrial dysfunction or stress to the rest of the cell (6), and therefore are candidates for signaling cardiac mitochondrial energy stress. These include the canonical AMP kinase (AMPK) (7, 8), reactive oxygen species (ROS) (9), and the mitochondrial unfolded protein response (10) signaling pathways. More recently, metabolites derived from mitochondrial metabolism have also emerged as important mediators of mitochondrial communication (11).
An intriguing group of candidate metabolites is the acyl-coenzymes A’s (acyl-CoAs). These reactive molecules synthesized from metabolic intermediates serve as substrates for acylations, a class of protein posttranslational modifications (PTMs) that regulate proteins in pathways ranging from chromatin accessibility to metabolic protein function (12–16). Acylations are reversible covalent additions of acyl groups from reactive acyl-CoAs to the ε-amino group of target lysines (14). Acetylation, the addition of the acetyl group of acetyl-CoA to lysine residues, is among the best-studied acylations and modifies a diverse array of proteins across cellular compartments (17–19). In recent years, other acylations have been discovered; these include malonylation, succinylation, and glutarylation (derived from malonyl-CoA, succinyl-CoA, and glutaryl-CoA, respectively). These acylations also harbor the potential to modify histone and mitochondrial proteins (20–23), suggesting the tantalizing potential for this class of PTMs to coordinate cellular metabolic state with gene expression and metabolic regulation (12).
Although acetylation, malonylation, succinylation, and glutarylation are unified by their status as acylations, individually they may exert different biochemical effects. At physiological pH, acetylation neutralizes the positive charge of modified lysine residues, whereas malonylation, succinylation, and glutarylation impart a negative charge (22). In addition, these modifications differ in size and structure (22). As such, acylations may differentially affect protein structure, electrostatic interactions within a protein, or even protein-protein interactions with binding partners. Thus, understanding acylation type- and site-specific effects may be crucial for uncovering the functional consequences of acylations on target proteins.
Interestingly, mitochondrial proteins are particular targets for some acylations. Mass spectrometry studies on mice in which sirtuin 3 (SIRT3, a mitochondrial deacetylase) and sirtuin 5 (SIRT5, a mitochondrial deacylase with demalonylase, desuccinylase, and deglutarylase activity) are knocked out have revealed mitochondrial metabolic pathways as hotspots for these modifications (24, 25). However, the causes and functional consequences of acylations and their contributions to disease pathology are open areas of active interest and debate (26–32).
In this study, we sought to identify the mitochondrial stress response pathways specifically engaged by impaired mitochondrial ATP synthesis in the heart. We leveraged our previously described mouse model bearing a temporally regulated and cardiomyocyte-specific deletion of the mitochondrial phosphate carrier (SLC25A3; Slc25a3fl/flxMCM mice) (33) as a model of mitochondrial energy dysfunction. SLC25A3 is a major transporter of inorganic phosphate (Pi) into the mitochondrial matrix and an essential component of the mitochondrial ATP synthasome (34, 35). SLC25A3 deletion in adult cardiomyocytes causes reduced mitochondrial ATP synthesis and the development of mitochondrial cardiomyopathy like that observed in people with mitochondrial phosphate carrier deficiency (33, 36, 37; OMIM: 610773). Here, we report that SLC25A3 deletion-induced mitochondrial energy dysfunction drives cardiac acylome remodeling, revealing a novel pathway by which disruption to the mitochondrial energy production machinery may impose control on an expanded network of mitochondrial proteins.
MATERIALS AND METHODS
Animal Models
The Slc25a3 loxP-targeted mice (Slc25a3fl/fl) and the αMHC-MerCreMer animals expressing a tamoxifen-inducible Cre recombinase under the control of the cardiomyocyte-specific α-myosin heavy chain promoter (MCM) were described previously (33, 38). Slc25a3fl/flxMCM mice were generated by crossing Slc25a3fl/fl mice to the MCM animals, as previously described (33), with the MCM transgene exclusively maintained in the hemizygous state. Slc25a3 deletion was induced in 8-wk-old Slc25a3fl/flxMCM animals by intraperitoneal injections of tamoxifen (25 mg/kg for 5 consecutive days). As controls, 8-wk-old Slc25a3fl/fl and MCM control animals, harboring either the Slc25a3-targeted locus or MCM transgene alone, were subjected to the same tamoxifen dosing regimen. Both male and female mice were used and experiments were conducted on mice at 2, 6, and 10 wk after tamoxifen administration, as indicated. To euthanize mice for collection of heart tissue, isoflurane anesthesia was administered before cervical dislocation. All mouse experiments were approved and performed in accordance with Emory University’s Institutional Animal Care and Use Committee.
Cell Culture, Plasmid Vectors, and Transfection
Human embryonic kidney 293 (HEK293) cell lines were cultured in Dulbecco’s minimal essential media supplemented with 10% bovine growth serum and 1% penicillin/streptomycin antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin) in 5% CO2 atmosphere at 37°C. IDH2 and SIRT5 knockout (KO) HEK293 cell lines were generated by CRISPR-Cas9-mediated gene deletion (Synthego) and individual KO clonal lines were isolated by dilution cloning.
PcDNA3.1-IDH2 and pcDNA3.1-SIRT5 vectors were generated by custom DNA synthesis (Genewiz). Site-directed mutagenesis (Q5 Site Directed Mutagenesis Kit; New England Biolabs) was conducted to generate the IDH2 K48Q, K180Q, and K263Q constitutive acetylation mimics; the IDH2 K48E, K180E, and K263E constitutive malonylation mimics; and the SIRT5-K203Q constitutive acetylation mutant.
For the reexpression of IDH2 and SIRT5 constructs into IDH2 and SIRT5 KO cells, a single IDH2 or SIRT5 KO clonal line was selected for reexpression studies. Cells were transiently transfected with IDH2 and SIRT5 vectors using Lipofectamine 3000 (Invitrogen), as described by the manufacturer. Levels of IDH2 or SIRT5 overexpression were assessed by immunoblotting using specific antibodies against IDH2 and SIRT5, as described in RESULTS.
Liquid Chromatography-Tandem Mass Spectrometry and Analysis
Mouse cardiac proteins were extracted and digested using a protocol adapted from Seyfried et al. (39). Briefly, tissue was solubilized in urea lysis buffer (8 M urea, 10 mM Tris, 100 mM NaH2PO4, pH 8.5) supplemented with protease and phosphatase inhibitors (Thermo Fisher Scientific) using a Bullet Blender (Next Advance), according to the manufacturer’s protocol. Protein homogenates were sonicated and cleared by centrifugation. Subsequently, proteins were diluted with 50 mM NH4HCO3 to a final concentration of 2 M urea, reduced with 1 mM 1,4-dithiothreitol for 30 min, and alkylated with 5 mM iodoacetamide for 30 min. Protein samples were then digested with 1:100 (wt/wt) Lys-C (Wako) at room temperature (r.t.) for 4 h and then further digested overnight with 1:50 (wt/wt) trypsin (Promega) at r.t. Resulting peptides were desalted with Oasis HLB columns (Waters) and dried under vacuum.
Acylated (either acetylated or malonylated) peptides were enriched by immunoaffinity purification using the PTMScan Acetyl-Lysine or PTMScan Malonyl-Lysine Kits (Cell Signaling Technology #13416 and #93872), according to the manufacturer’s protocol. Acylated peptides were further purified by desalting using C18 StageTips. Finally, peptides were reconstituted in 0.1% trifluoroacetic acid. The protocol for data acquisition by LC-MS/MS was adapted from Seyfried et al. (39). Briefly, 2 µL of the reconstituted peptides were separated on a self-packed C18 (1.9 µm Dr. Maisch, Germany) fused silica column (25 cm × 75 µM internal diameter; New Objective, Woburn, MA) by a Dionex Ultimate 3000 RSLCNano. Elution was performed over a 120-min gradient at a rate of 300 nL/min with buffer B ranging from 3% to 80% (buffer A: 0.1% formic acid in water, buffer B: 0.1% formic in acetonitrile). Peptides were analyzed on an Orbitrap Fusion Tribrid Mass Spectrometer (Thermo Fisher Scientific). The mass spectrometer cycle was programmed to collect at the top speed for 3 s cycles. The MS scans (300–1,500 m/z range, 200,000 AGC, 50 ms maximum ion time) were collected at a resolution of 120,000 at m/z 200 in profile mode and the higher-energy collision dissociation (HCD) MS/MS spectra (15,000 resolution, 1.6 m/z isolation width, 30% collision energy, 10,000 AGC target, 35 ms maximum ion time) were detected in the Orbitrap. Dynamic exclusion was set to exclude previously sequenced precursor ions for 20 s within a 10 ppm window. Precursor ions with +1, +8, or higher charge states were excluded from sequencing.
MS/MS spectra were evaluated using Proteome Discoverer 2.1.1.21. The raw files were searched using a Uniprot Mus musculus protein database (downloaded April 2015 with 53289 target sequences). Search parameters included fully tryptic specificity, 20 ppm precursor ion tolerance, 0.05 Da product ion tolerance, static modification for carbamidomethyl cysteine (+57.02146), and dynamic modifications for oxidized methionine (+15.99492), deamidated asparagine, and glutamine (+0.98402). Additional dynamic modifications of lysine acetylation (+42.01057) and malonylation (+86.00039). All peptide spectral matches were filtered via the percolator node.
Gene ontology analyses were performed with the Cytoscape Cluego plugin (40). In silico interactome data were downloaded from Genemania physical interactions and REACTOME curated interactions were included in the analysis (41) and processed in Cytoscape v3.5.1 (42). Interactome connectivity graph parameters were generated in Cytoscape. Physical interactions were curated from the following sources and their strength represented by the edge thickness (43–47).
LC/MS/MS Quantitation of Acetyl-CoA and Malonyl-CoA
Working calibration standard mix was prepared for acetyl-CoA or malonyl-CoA. The calibrators and sample were spiked with a mixture of heavy isotope-labeled internal standards for CoA species to be measured. The CoAs were then extracted using Oasis HLB 1 mL (30 mg) Extraction Cartridges (Waters Corporation). The extracted samples were dried under nitrogen, reconstituted in 10 mM Ammonium carbonate, pH 9.5, and separated on an Acquity UPLC BEH C18 2.1 × 50 mm, 1.7 µm column, (Waters Corporation) using a 2.35 min linear gradient with 10 mM ammonium carbonate, pH 9.5, and ACN as eluents. Quantitation of acetyl-CoA and malonyl-CoA was achieved using multiple reaction monitoring of calibration solutions and study samples on an Agilent 1290 HPLC/6490 triple quadrupole mass spectrometer. The mass spectrometer was operated in positive ion mode using electrospray ionization with an electrospray ionization (ESI) capillary voltage of 3,500 V. The electron multiplier voltage was set to 400 V. The ion transfer tube temperature was 325°C and vaporizer temperature was 325°C. The ESI source sheath gas flow was set at 10 L/min. The mass spectrometer was operated with a mass resolution of 0.7 Da, and N2 collision gas pressure was 30 psi for the generation and detection of product ions of each metabolite. For data processing, the raw data was processed using MassHunter Quantitative Analysis software (Agilent). Calibration curves (R2 = 0.99 or greater) were either fitted with a linear or a quadratic curve with a 1/X or 1/X2 weighting.
Mitochondrial Isolation
Cardiac mitochondria used for immunoblotting and immunoprecipitation were isolated by differential centrifugation, as previously described (33), using MS-EGTA buffer (225 mM mannitol, 75 mM sucrose, 5 mM HEPES, and 1 mM EGTA, pH 7.4) supplemented with 2.4 mg/mL nicotinamide, Trichlostatin A, and a combined phosphatase and protease inhibitor cocktail (Thermo Fisher Scientific).
Electrophoresis and Immunoblotting
For Western blot analyses, total protein extracts were prepared from hearts homogenized and solubilized in radioimmunoprecipitation assay (RIPA) buffer supplemented with 2.4 mg/mL nicotinamide and a combined protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). Cardiac mitochondrial protein extracts were prepared by solubilizing heart mitochondria in the same RIPA buffer. For electrophoresis, proteins were reduced and denatured in 1X Laemmli buffer, resolved on 10% SDS-PAGE gels, transferred to PVDF membranes, immunodetected with antibodies, and imaged using a ChemiDoc system (BioRad). Primary antibodies used in the study were antiacetylated lysine (PTM Biolabs PTM-105, 1:1,000, and Cell Signaling Technology #9441, 1:1,000), antimalonylated lysine (PTM Biolabs PTM-901, 1:1,000), antisuccinylated lysine (PTM Biolabs PTM-401, 1:1,000), antiglutarylated lysine (PTM Biolabs, PTM-1151, 1:1,000), anti-pAMPK (Cell Signaling Technology #2532S 1:1,000), anti-AMPK (Cell Signaling Technology #2535S, 1:1,000) anti-SIRT5 (Cell Signaling Technology #8782, 1:1,000), anti-SIRT3 (Cell Signaling Technology #2627, 1:1,000), anti-IDH2 (Proteintech, 15932-1-AP, 1:1,000), anti-GCN5L1 [generated by Iain Scott, as described in Manning et al. (48), 1:500], anti-Porin (Abcam, ab14734, 1:1,000), anti-α-tubulin (Proteintech, 11224-1-AP, 1:1,000), and anti-GAPDH (Fitzgerald, 10 R-G109A, 1:10,000, and Cell Signaling Technologies #5174. 1:1,000). Secondary antibodies were alkaline phosphatase-linked goat antirabbit IgG (Cell Signaling Technologies #7054, 1:5,000), alkaline phosphatase-linked goat antimouse IgG (Cell Signaling Technologies #7056, 1:5,000), and horseradish peroxidase-linked goat antirabbit IgG (Proteintech, SA00001-2).
Immunoprecipitation
Mouse heart tissue was solubilized in immunoprecipitation buffer [20 mM Tris-HCl (pH 7.5) 250 mM NaCl, 1% Triton X-100, Nicotinamide 2.4 mg/mL, 0.5 mM dithiothreitol, and protease inhibitor cocktail (Roche)]. To assess IDH acetylation or malonylation, 1 mg of total heart proteins were immunoprecipitated with the following antibodies: anti-IDH2 (Bethyl Laboratories, A304-096A) or 3.5 ug rabbit IgG (Bethyl Laboratories, P120-101) for 3 h at 4°C, followed by an overnight incubation with protein A-agarose beads (Santa Cruz) at 4°C. Immunoprecipitated proteins were resolved by acrylamide gel electrophoresis and immunoblotted with antibodies against either acetylated lysine (Cell Signaling Technology, #9441, 1:1,000) or malonylated lysine (Cell Signaling Technology, #14942, 1:1,000). VeriBlot (Abcam, ab131366, 1:1,000) was used as an IP Detection Reagent; blots were visualized by chemiluminescence using the SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific) and imaged using a ChemiDoc system (BioRad). To assess SIRT5 acetylation, the same immunoprecipitation protocol, as described above, was followed, except antiacetylated lysine (Cell Signaling Technology, #9441) or control rabbit polyclonal anti-HA (Proteintech, 51064-2-AP) antibodies were used. Immunoblotting was conducted with anti-SIRT5 (Proteintech, 15122-1-AP, 1:1,000). A mouse antirabbit conformation-specific secondary antibody (Cell Signaling Technology, #3678) was used.
Real-Time PCR
Total RNA was extracted from heart tissue using the RNeasy Fibrous Tissue Mini Kit (Qiagen) and cDNA was generated using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). RT-PCR was performed on a 7500 Real-Time PCR System (Applied Biosystems) using the iTaq Universal SYBR Green Supermix (BioRad). The ΔΔC(t) method was used to quantify the fold change of the target genes. The primer sets used were Atf4 (forward, GCCGGTTTAAGTTGTGTGCT; reverse, CTGGATTCGAGGAATGTGCT) (49), Atf5 (forward, GGGTCATTTTAGCTCTGTGAGAGAA; reverse, ATTTGTGCCCATAACCCCTAGA) (50), and RPS20 (forward, AACAAGTCGGTCAGGAAGC; reverse, TCCGCACAAACCTTCTCC), Lonp1 (PrimerBank ID: 12836291a1 forward, CGGATGTGTTTCCTCACCTG; reverse, ACGCCAACATAGGGCTGTG), Hspa9 (PrimerBank ID: 6754256a1, forward, ATGGCTGGAATGGCCTTAGC; reverse, ACCCAAATCAATACCAACCACTG), and Hspd1 (PrimerBank ID: 31981679a1, forward, CACAGTCCTTCGCCAGATGAG; reverse, CTACACCTTGAAGCATTAAGGCT).
Amplex Red Assay
Isolated cardiac mitochondrial fractions were solubilized in MS-EGTA buffer, and protein concentrations were quantified by Bradford assay (BioRad). Measurements of hydrogen peroxide production was quantified using an Amplex Red assay (Invitrogen #A22188), according to manufacturer’s instructions.
IDH2 Assay and Structural Analyses
Enriched mitochondrial fractions were prepared from HEK293 cell lines by differential centrifugation using cellular mitochondrial isolation buffer (250 mM sucrose, 20 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, pH 7.5) supplemented with 2.4 mg/mL nicotinamide and a combined phosphatase and protease inhibitor cocktail (Thermo Fisher Scientific) or cardiac mitochondria prepared, as described above. Measurement of IDH2 activity on the enriched mitochondrial fractions was conducted using an IDH activity assay (Sigma Aldrich), according to the manufacturer’s instructions. Specificity for IDH2 activity was achieved by measuring activity on mitochondria, together with providing NADP+ as the substrate for the assay. Protein concentrations were quantified by Bradford assay, and IDH2 activity was normalized to the amount of mitochondrial protein used in the assay.
IDH2 [PDB ID: 5H3F (51)] structural data were retrieved from the RCSB Protein Data Bank (52) and all figures of structures were created with the Mol* software (53).
RESULTS
Canonical Pathways of Mitochondrial Energy Stress Signaling Are Not Engaged in Response to SLC25A3 Deletion in the Heart
In the Slc25a3fl/flxMCM model, Slc25a3fl/fl mice harboring a loxP-targeted Slc25a3 locus were crossed to animals expressing a Cre recombinase under the control of the tamoxifen-inducible and cardiomyocyte-specific α-myosin heavy chain promoter (MCM) (33). To induce SLC25A3 deletion in the adult heart, tamoxifen was administered to 8-wk-old adult Slc25a3fl/flxMCM mice with Slc25a3fl/fl or MCM animals serving as controls. SLC25A3 deletion recapitulated the previously reported phenotype with impaired cardiac mitochondrial ATP synthesis, sustained tissue ATP content, and subsequent severe cardiac hypertrophy (33).
Using Slc25a3fl/flxMCM mice, we investigated the stress response pathways activated during mitochondrial energy impairment. We first examined activation of AMPK, an energy-sensing kinase that is activated via phosphorylation at threonine 172 (Thr172) in response to cellular energy stress (54). We performed Western blot analyses for cardiac pAMPKα Thr172 in Slc25a3fl//flxMCM versus Slc25a3fl/fl control animals over the course of SLC25A3 deletion-induced cardiomyopathy. Total protein lysates were prepared from hearts collected at 2 (impaired ATP synthesis), 6 (development of hypertrophy), and 10 (severe cardiomyopathy) wk following tamoxifen induction. No differences were detected in the extent of AMPKα phosphorylation as compared with total AMPK expression levels (Fig. 1, A–C), suggesting that AMPK signaling is not a major pathway activated by SLC25A3 deletion-induced impairment in mitochondrial ATP synthesis.
Figure 1.
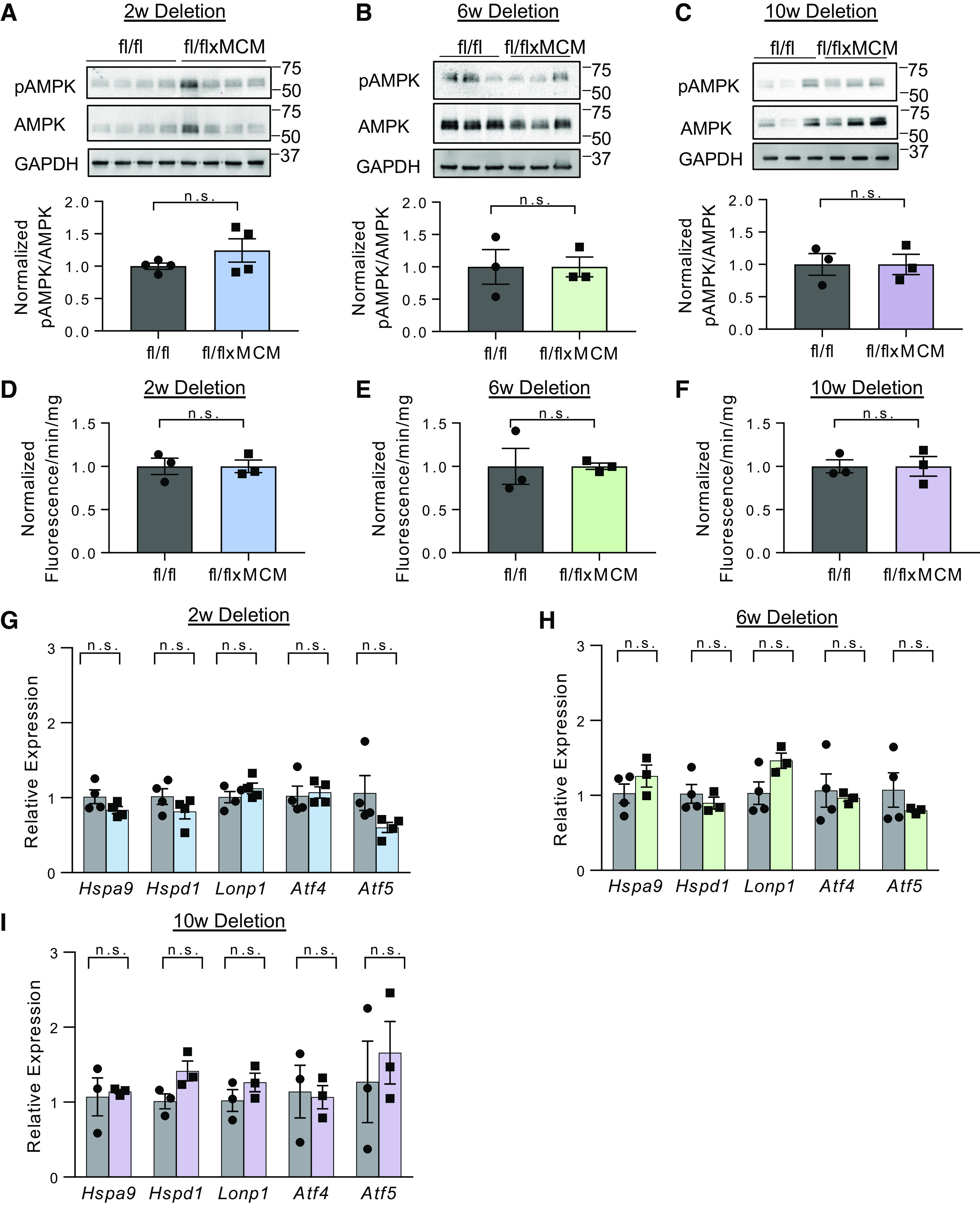
Canonical pathways of mitochondrial energy stress signaling are not activated in response to SLC25A3 deletion in the heart. A–C: Western blot analyses and quantification of phosphorylated AMPKα (pAMPK) and total AMPKα in hearts collected from Slc25a3fl/flxMCM and Slc25a3fl/fl control mice at 2, 6, and 10 wk following tamoxifen dosing. GAPDH was used as a protein loading control (2 wk: n = 4 fl/fl and n = 4 fl/flxMCM; 6 and 10 wk: n = 3 fl/fl and n = 3 fl/flxMCM animals). D–F: hydrogen peroxide production as measured by an amplex red assay on cardiac mitochondria isolated from Slc25a3fl/flxMCM and Slc25a3fl/fl mice at 2, 6, and 10 wk following tamoxifen dosing (n = 3 animals/group). G–I: RT-PCR analyses of Lonp1, Hspa9, Hspd1, Atf4, and Atf5 transcript levels in Slc25a3fl/flxMCM and Slc25a3fl/fl hearts at 2, 6, and 10 wk following tamoxifen dosing (2 wk: n = 4 fl/fl and n = 4 fl/flxMCM animals; 6 wk: n = 4 fl/fl and n = 3 fl/flxMCM animals; and 10 wk: n = 3 fl/fl and n = 3 fl/flxMCM animals). Values presented as means ± SE. Student’s t test was used for statistical analysis. P < 0.05 were considered significant. n.s., not significant.
Similarly, mitochondrial ROS production is often elevated with mitochondrial dysfunction and ROS have important signaling functions in the cell (6, 9). Using mitochondrial Amplex Red ROS production assays, we measured the rate of hydrogen peroxide synthesis from functional cardiac mitochondria isolated from Slc25a3fl//flxMCM and Slc25a3fl/fl mice 2, 6, and 10 wk following tamoxifen dosing (Fig. 1, D–F). Hydrogen peroxide production was unchanged between the two groups (Fig. 1, D–F). The absence of enhanced mitochondrial ROS production following SLC25A3 aligns with our previous findings (33), wherein SLC25A3 deletion reduces mitochondrial ATP synthesis, but does not impair respiratory chain function, a major source of mitochondrial ROS.
Finally, we examined the effects of SLC25A3 deletion on the mitochondrial unfolded protein response. This signaling pathway is implicated in mitochondria-to-nucleus retrograde communication and engaged in response to mitochondrial proteotoxic stress and mitochondrial dysfunction (10). Central pathway members are transcription factors ATF4 and ATF5, as well as canonical markers such as LONP1 (lon peptidase 1), HSPA9 (heat shock protein family A member 9), and HSPD1 (heat shock protein family D member 1) (10, 55). Slc25a3fl/flxMCM and Slc25a3fl/fl hearts displayed similar Atf4, Atf5, Lonp1, Hspa9, and Hspd1 transcript levels (Fig. 1, G–I). Taken together, our data suggest that canonical modes of mitochondrial stress signaling are not major contributors to the stress response during SLC25A3 deletion-induced mitochondrial energy defects in the heart. Thus, other response pathways must be activated in this context.
SLC25A3 Deletion Induces a Unique Signature of Cardiac Protein Hyperacylation
Among intermediate metabolites produced via mitochondrial metabolic processes and involved in mitochondrial signaling (11), acyl-CoAs are key substrates for acylations, a class of PTMs that regulate proteins in a variety of pathways and are linked to metabolic dysregulation (12–16). SLC25A3 deletion impairs mitochondrial energy production; if this alters acyl-CoA production, it could impact protein PTMs by acylations. Therefore, we assessed the effect of SLC25A3 loss on the cardiac acylome, specifically, acetylation, malonylation, succinylation, and glutarylation. We performed Western blot analyses of acetylated-, malonylated-, succinylated-, and glutarylated-lysine residues in total protein extracts from Slc25a3fl/flxMCM versus Slc25a3fl/fl hearts (Fig. 2). Hearts lacking SLC25A3 had significantly increased levels of acetylation (Fig. 2A) and malonylation (Fig. 2B); succinylation (Fig. 2C) and glutarylation (Fig. 2D) levels were unchanged. These data suggest that SLC25A3 deletion induces remodeling of the cardiac acylome with targeted alterations to the acetylome and malonylome.
Figure 2.

Cardiac SLC25A3 deletion induces a signature of elevated acetylation and malonylation. Western blot analyses of acetylated (Kac; A), malonylated (Kmal; B), succinylated (Ksucc; C), and glutarylated (Kglu; D) lysines in total protein lysates prepared from Slc25a3fl/flxMCM and Slc25a3fl/fl hearts 10 wk posttamoxifen administration.
Mitochondrial Energy Dysfunction-Induced Acylations Preferentially Target Mitochondrial Proteins
We next applied a proteomics approach to identify proteins whose acylation pattern and levels were modified in response to SLC25A3 deletion. Protein extracts from hearts collected 10 wk after tamoxifen administration (Slc25a3fl/fl and Slc25a3fl/flxMCM mice) were immunoaffinity-purified using antibody-conjugated beads to detect acetylated or malonylated lysines. Enriched peptides were analyzed by liquid chromatography tandem mass spectrometry (LC-MS/MS). Differential acylome modifications fell into one of three major categories in response to SLC25A3 deletion: 1) increased acetylation (Fig. 3A; red symbols), 2) decreased acetylation (Fig. 3A; blue symbols), and 3) increased malonylation (Fig. 3B; red symbols). Categories 1 and 2 encompassed a total of 543 peptides representing 208 unique proteins that displayed altered acetylation status in response to SLC25A3 deletion (Fig. 3A and Supplemental Table S1; all Supplemental Material is available at https://doi.org/10.6084/m9.figshare.14450097.v1). Within this set of differentially acetylated proteins, 94 proteins harbored lysines that were hyperacetylated in response to SLC25A3 deletion, whereas 153 proteins harbored lysines that were hypoacetylated (Fig. 3C; Supplemental Table S1). Interestingly, a subgroup of 39 differentially acetylated proteins reflected proteins that had some lysine sites displaying increased acetylation following SLC25A3 deletion, whereas other sites within the same protein displayed reduced acetylation [Fig. 3C (17 + 22 intersects)]. Category 3 encompassed 200 differentially malonylated peptides representing 68 proteins, all of which were hypermalonylated after SLC25A3 deletion (Fig. 3B, red symbols; Supplemental Table S2). In addition, for proteins differentially acylated in response to SLC25A3 deletion, 44 were solely hyperacetylated (category 1), 95 were solely hypoacetylated (category 2), 16 were solely hypermalonylated (category 3). Yet, 22 proteins crossed all three categories (Fig. 3C). These findings reveal a striking and unforeseen complexity of mitochondrial energy dysfunction-induced acylome remodeling.
Figure 3.

SLC25A3 deletion-induced acylome remodeling preferentially targets mitochondrial ontologies. A and B: volcano plots of acetylated proteins (Kac) and malonylated proteins (Kmal). Colored symbols depict peptide hits with a twofold increase (red symbols) or decrease (blue symbols). n = 5 fl/fl and n = 5 fl/flxMCM animals were used for acetylation MS studies, and n = 5 fl/fl and n = 4 fl/flxMCM animals were used for malonylation MS studies. Student’s t tests were used for statistical analyses with a P value < 0.05 considered significant. Isocitrate dehydrogenase 2 (IDH2)-acylated peptides are depicted in yellow with the lysine modified numbered in B. Note that the segregation of values into populations along the x-axis of the volcano plots is due to zero intensity in some of the proteins quantified (see Supplemental Tables S1 and S2 as well as Supplemental Fig. S2 for detailed information). C–E: Venn diagrams identifying different categories of acylation and their intersection with the curated Mitocarta 3.0 dataset. F: list of proteins that exhibited increases in both acylation and malonylation. G: interactome of the proteins listed in G. Node connectivity was quantified by betweenness and closeness centrality indexes using Cytoscape. H–J: ClueGo analysis of the three categories of acylome modifications: increased acetylation, decreased acetylation, and increased malonylation. The GO Biological process and KEGG databases were queried with a threshold Bonferroni-corrected P value below 0.01. H: all proteins in the three categories analyzed simultaneously. I: the percent contribution of each of the three acylation categories to the ontology term presented in H. J: P value and number of genes per ontology term presented in H.
To determine the subcellular distribution of proteins targeted by mitochondrial energy dysfunction-induced acylations, we analyzed our SLC25A3 deletion-induced acetylated and malonylated proteins according to their annotation in the Mitocarta3.0 database (56). Most hyperacetylated proteins were annotated to the mitochondrion (Fig. 3D, 91%), as were most hypermalonylated proteins (Fig. 3E, 73%). This preferential modification of mitochondrial proteins by energy dysfunction-induced acetylation and malonylation was confirmed by Western blot analyses for acetylated and malonylated lysine residues on mitochondrial protein extracts from Slc25a3fl/flxMCM versus Slc25a3fl/fl hearts (Supplemental Fig. S1). In contrast, a reduced percentage of hypoacetylated proteins were annotated to the Mitocarta 3.0 database (46%). Collectively, these results indicate that SLC25A3 deletion-induced remodeling of the cardiac acetylome and malonylome occurs predominantly in the mitochondrial compartment.
We focused on 30 proteins with increased acetylation and malonylation in mutant hearts. All of these proteins were annotated to mitochondria (Fig. 3, E and F). These 30 proteins belong to a high-connectivity interactome defined by protein-protein interactions and functional interactions annotated in the REACTOME database (57) (Fig. 3G). This interactome includes subunits of the respiratory chain complexes II, III, and V; all three ADP-ATP translocators (SLC25A4-6); enzymes involved in mitochondrial fatty acid oxidation such as HADHA and HADHB; and enzymes that participate in the Krebs cycle such as isocitrate dehydrogenase 2 (IDH2). IDH2 is of particular interest as an enzyme that catalyzes the NADP+-dependent conversion of isocitrate into α-ketoglutarate for use in Krebs cycle and serving as a source of NADPH needed for maintenance of mitochondrial redox balance. Importantly, in our mass-spectrometry analyses, IDH2 is an example of a protein highly acetylated and malonylated in response to SLC25A3 deletion (Fig. 3, A and B, yellow symbols).
Finally, we asked whether the three acylation categories segregated into distinct molecular mechanisms. We used the ClueGo bioinformatic tool to assess gene ontologies using the three categories as gene sets. The most significant terms identified across categories were carboxylic acid metabolic process gene ontology (GO):0019752, Bonferroni-corrected P = 3.7E-41) and the KEGG term oxidative phosphorylation (KEGG:00190, Bonferroni-corrected P = 4.3E-34; Fig. 3, H and I, gray colors and Supplemental Table S3). However, proteins with reduced acetylation in response to SLC25A3 deletion were uniquely enriched in the DNA packaging annotated term (GO:0006323, Bonferroni-corrected P = 0.0057) and cardiac muscle contraction (GO:0060048, Bonferroni-corrected P = 0.000004; Fig. 3, H and J). Malonylated proteins were enriched in the annotated term porin activity, represented by the genes Vdac1, Vdac2, and Vdac3 (GO: 0015288, Bonferroni-corrected P = 0.001). These results demonstrate that proteins newly acylated upon mitochondrial energy dysfunction center around mitochondrial annotated ontologies, whereas proteins losing acylations are distinguished by nonmitochondrial terms such as DNA packaging and muscle contraction.
Mitochondrial Acetylation and Malonylation Have Site-Specific and Functionally Distinct Effects
We next explored the functional effects of a subset of proteins targeted for acylome remodeling. As described above, IDH2 is both hyperacetylated (5 sites) and hypermalonylated (9 sites) in Slc25a3fl/flxMCM hearts when compared with Slc25a3fl/fl controls (Fig. 4, A and B). We validated the mass spectrometry findings via immunoprecipitation (IP) of IDH2 from heart protein extracts and immunoblotting with antibodies against acetylated or malonylated lysines (Fig. 4C).
Figure 4.
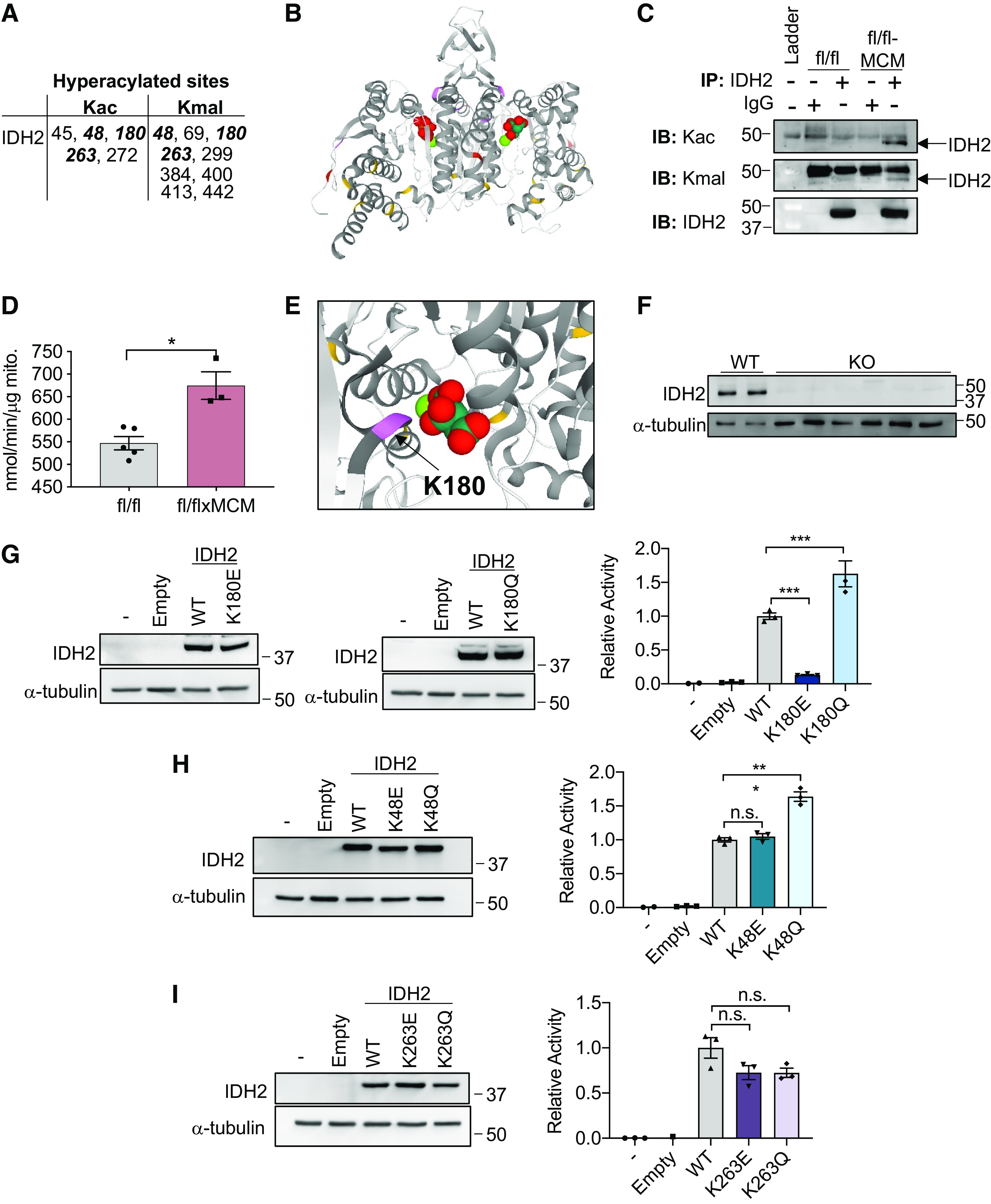
Site-specific expression and activity of isocitrate dehydrogenase 2 (IDH2) acetylation and malonylation mimics. A: IDH2 lysine residues hyperacetylated (Kac) and hypermalonylated (Kmal) in response to SLC25A3 deletion. Residues modified by both PTMs in bold. B: structure of the IDH2 dimer bound to isocitrate. Lysine resides acetylated (red), malonylated (yellow), or targeted by both posttranslational modifications (PTMs) (purple) in response to SLC25A3 deletion are indicated. C: IDH2 was immunoprecipitated from protein lysates from Slc25a3fl/fl or Slc25a3fl/flxMCM hearts and probed by immunoblotting by anti-Kac or anti-Kmal antibodies to confirm enhanced acetylation/malonylation of IDH2 in response to SLC25A3 deletion. Immunoblotting with anti-IDH2 antibodies was conducted to validate pull down of IDH2. Immunoprecipitation was also conducted with IgG antibodies as a control. D: IDH2 activity assay performed on cardiac mitochondria isolated from Slc25a3fl/fl and Slc25a3fl/flxMCM animals 10 wk posttamoxifen administration (n = 5 fl/fl and n = 3 fl/flxMCM animals). E: representation of K180 (purple) and isocitrate in the IDH2 binding pocket. F: Western blot analyses of IDH2 expression in wild-type (WT) and IDH2 KO clones. Α-tubulin was used as a protein loading control. G: Western blot analyses of IDH2 expression in IDH2 KO cells reexpressing either the pcDNA3.1 vector alone (empty), WT IDH2, IDH2 K180E, or IDH2 K180Q constructs, or the IDH2 activity assays on mitochondria isolated from these cell lines (n = 3 replicates/group). H: Western blot analyses of IDH2 expression in IDH2 KO cells reexpressing either the empty vector, WT IDH2, IDH2 K48E, or IDH2 K48Q constructs or the IDH2 activity assays on mitochondria isolated from these cell lines (n = 3 replicates/group). I: Western blot analyses of IDH2 expression in IDH2 KO cells reexpressing either the empty vector, WT IDH2, IDH2 K263E, or IDH2 K263Q constructs or the IDH2 activity assays on mitochondria isolated from these cell lines (n = 3 replicates/group). For all graphs, values presented as means ± SE. For D–F, one-way ANOVA followed by Welch’s test was used for statistical analysis with P < 0.05 considered significant. For G, a Student’s t test was used for statistical analysis. P < 0.05 were considered significant. *P < 0.05, ***P < 0.001, n.s., not significant.
Acylations of mitochondrial metabolic enzymes, including IDH2, are reported to be inhibitory (51, 58, 59). We therefore hypothesized that hyperacylations of IDH2 would suppress its function. To test this, we examined IDH2 expression and activity in mitochondria isolated from Slc25a3fl/flxMCM versus Slc25a3fl/fl control hearts. Although SLC25A3 deletion had no effect on overall cardiac mitochondrial IDH2 protein levels (Supplemental Fig. S3), surprisingly, mitochondria isolated from Slc25a3fl/flxMCM hearts had significantly elevated IDH2 activity as compared with mitochondria from Slc25a3fl/fl controls (Fig. 4D).
This striking finding prompted us to examine the structural positioning of the hyperacylated lysines (Fig. 4B). We determined that K180, a lysine targeted by both acetylation and malonylation, lies in close proximity to the isocitrate substrate-binding pocket (Fig. 4E), suggesting that modification at this site may modulate enzyme function. Two other sites, K48 and K263, were also hyperacetylated and hypermalonylated in response to SLC25A3 deletion. We leveraged these three modification targets to determine whether functional effects of acylations are site specific. We generated mutant IDH2 constructs simulating constitutive acetylation (mutation of lysine to glutamine; Q) (60) or constitutive malonylation (mutation of the target lysine to glutamic acid; E) (25). Wild-type (WT) IDH2 or the acylation mutant constructs were transiently reexpressed into an HEK293 IDH2 knockout (KO) cell line generated by CRISPR-Cas9-mediated gene editing (Fig. 4F), which were then assayed for IDH2 activity on mitochondria. IDH2 KO mitochondria, as well as mitochondria from KO cells expressing the pcDNA3.1 vector alone (empty), displayed minimal IDH2 activity; in contrast, mitochondria isolated from KO cells reexpressing WT IDH2 displayed significantly elevated IDH2 function (Fig. 4, G–I). In line with previous reports that K263 acetylation has no impact on IDH2 function (59), mitochondria isolated from KO cells reexpressing the IDH2 K263Q acetylation mimic displayed IDH2 activity levels similar to mitochondria from WT reexpressing cells (Fig. 4I). Mitochondria isolated from KO cells reexpressing the K263E constitutive malonylation mutant also displayed activity levels similar to WT mitochondria (Fig. 4I). These findings collectively suggest that acetylation and malonylation at K263 does not impact IDH2 function.
For modification at K48, mitochondria from KO cells reexpressing the IDH2 K48E mutant displayed similar IDH2 activity levels as WT IDH2 reexpressing mitochondria, suggesting that malonylation at this site has no functional effects (Fig. 4H). Surprisingly, however, mitochondria from KO cells expressing the constitutive acetylation K48Q mutant displayed elevated IDH2 activity levels as compared with WT reexpressing mitochondria (Fig. 4H), suggesting that acetylation at this site enhances function. For SLC25A3-deletion responsive acylations at K180, mitochondria reexpressing the constitutive malonylation K180E mutant displayed significantly reduced IDH2 activity, whereas reexpression of the constitutive acetylation K180Q mutant at this resulted in elevated activity (Fig. 4G), suggesting that acetylation and malonylation at K180 have opposing effects.
Our data suggest that although acetylation and malonylation at individual sites may have functionally distinct effects, with acetylation of IDH2 enhancing function and malonylation largely having the opposing effect, cumulatively, SLC25A3 deletion-induced hyperacylations of IDH2 enhance enzyme activity.
Mitochondrial Malonylation Is Modulated by Acetylation
Our analyses of the pathways engaged in response to selective ablation of SLC25A3 in the heart revealed that defective mitochondrial ATP synthesis causes specific hyperacetylation and hypermalonylation of mitochondrial proteins (Fig. 3). Although alterations in mitochondrial protein acetylation are associated with cardiac mitochondrial energy dysfunction and various models of cardiac disease (18, 28, 58), our discovery that mitochondrial ATP synthesis deficiency induces a concomitant increase in mitochondrial protein malonylation was striking and unanticipated.
To investigate the mechanisms underlying energy dysfunction-induced acylations, we examined the effects of SLC25A3 deletion on pathways that contribute to the regulation of mitochondrial protein acetylation and malonylation. The mitochondrial acylome may be regulated by pathways controlling protein acetylation and deacetylation. Mitochondrial protein acetylation is thought to occur by acyltransferase-independent chemical reactions facilitated by permissive conditions within the mitochondrial matrix (61, 62) (e.g., increased acetyl-CoA substrate supply), as well as acyltransferase-dependent mechanisms via the mitochondria-localized acetyltransferase GCN5L1 (63, 64). Mitochondrial protein deacetylation is thought to be regulated by the mitochondria-localized deacetylase sirtuin 3 (SIRT3) (24). To investigate the contribution of these pathways to the increased mitochondrial acetylation observed with SLC25A3 deletion, we first measured acetyl-CoA levels in Slc25a3fl/flxMCM and Slc25a3fl/fl control hearts by targeted liquid chromatography-tandem mass spectrometry (LC-MS). SLC25A3 had no effect on acetyl-CoA levels in the heart (Fig. 5A), suggesting that SLC25A3 deletion does not promote nonenzymatic chemical acetylation. We next examined the effects of SLC25A3 deletion on GCN5L1 expression. GCN5L1 protein levels were surprisingly decreased in Slc25a3fl/flxMCM hearts as compared with Slc25a3fl/fl controls (Fig. 5, B and C), suggesting that GCN5L1 does not contribute to the observed SLC25A3 deletion-induced increases in mitochondrial protein acetylation. Unexpectedly, however, we found that mitochondria from Slc25a3fl/flxMCM hearts displayed a small but significant decrease of the deacetylase SIRT3 as compared with Slc25a3fl/fl controls (Fig. 5, D and E). Taken together, these data suggest that mitochondrial acetylation may be enhanced in SLC25A3-deleted hearts due to reduced mitochondrial SIRT3 levels.
Figure 5.
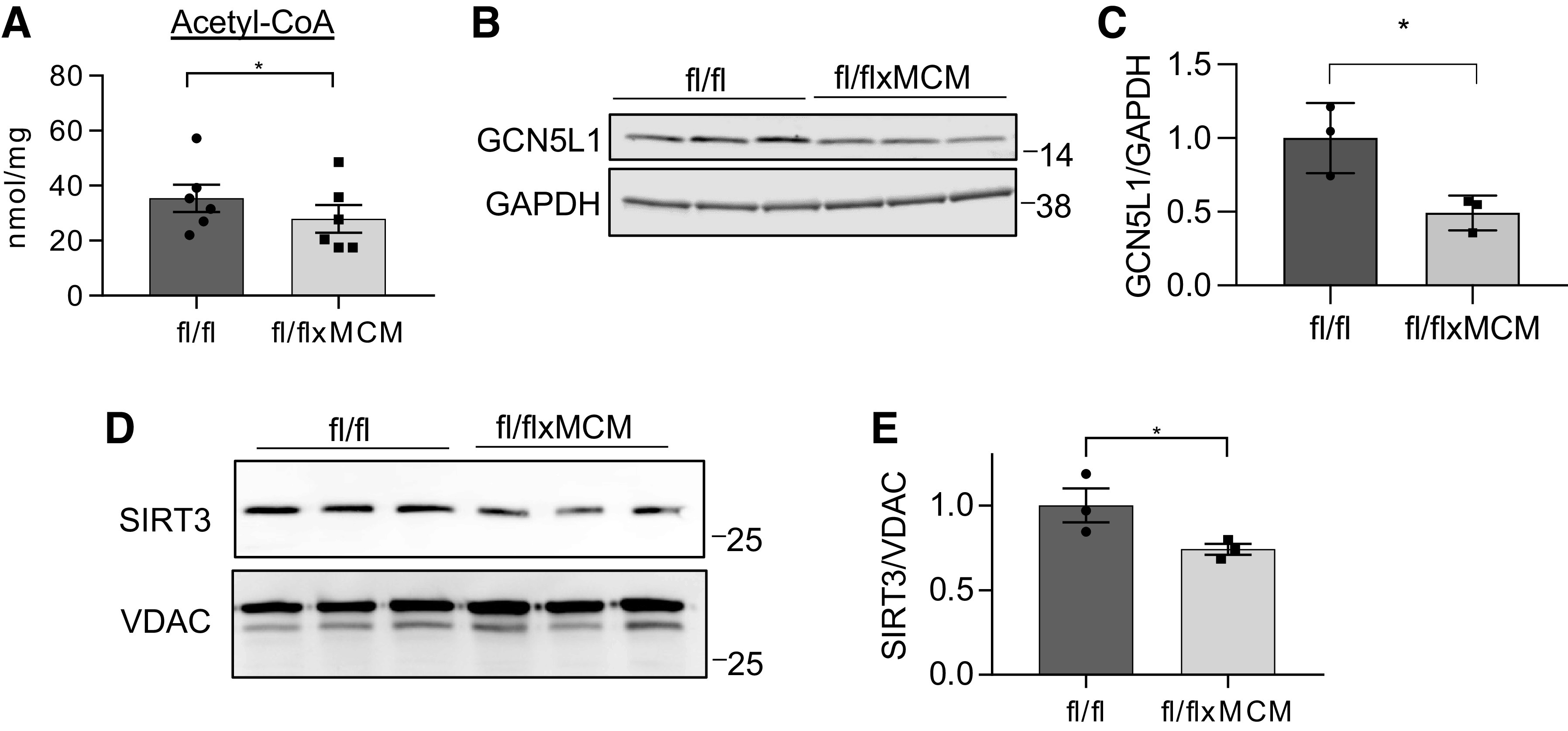
SLC25A3 deletion decreases levels of sirtuin 3 (SIRT3) in mitochondria. A: cardiac acetyl-CoA levels in Slc25a3fl/fl vs. Slc25a3fl/flxMCM mice at 10 wk posttamoxifen administration (n = 6 fl/fl and n = 6 fl/flxMCM animals). One-way ANOVA followed by Welch’s test was used for statistical analysis, with P < 0.05 considered significant. *P < 0.05 B: Western blot analysis of GCN5L1 expression in total heart protein lysates from Slc25a3fl/fl vs. Slc25a3fl/flxMCM animals. GAPDH was used as a loading control. C: quantification of cardiac GCN5L1 levels in the indicated groups (n = 3 animals/group). Student’s t test was used for statistical analysis, with P < 0.05 considered significant. *P < 0.05. D: Western blot analysis of SIRT3 expression in cardiac mitochondria isolated from Slc25a3fl/fl vs. Slc25a3fl/flxMCM animals. Voltage dependent anion channel (VDAC) was used as a mitochondrial loading control. E: quantification of mitochondrial SIRT3 levels in cardiac mitochondria from the indicated groups (n = 3 animals/group). Student’s t test was used for statistical analysis with P < 0.05 considered significant. *P < 0.05.
We employed similar approaches to characterize the mechanisms underlying energy dysfunction-induced malonylation. Like acetylation, malonylation of mitochondrial proteins occurs by acyltransferase-independent chemical reactions (61, 62); however, unlike acetylation, chemical malonylation is thought to be the main pathway of mitochondrial malonylation, mitochondrial malonyltransferases have not been identified. Furthermore, chemical malonylation can be modulated by malonyl-CoA substrate availability. We therefore conducted LC-MS to measure malonyl-CoA levels in Slc25a3fl/flxMCM and Slc25a3fl/fl control hearts. Remarkably, SLC25A3 deletion promoted significantly elevated malonyl-CoA levels in the heart (Fig. 6A). Because the mitochondrial malonylome is also regulated by the SIRT5, a mitochondrial demalonylase (62), we examined the effects of SLC25A3 deletion on SIRT5. Total levels of SIRT5 were unaltered by SLC25A3 deletion (Fig. 6B). However, further examination of our mass spectrometry data revealed that SIRT5 is hyperacetylated at K203 in response to SLC25A3 deletion. This finding was confirmed by IP and immunoblotting (Fig. 6C).
Figure 6.
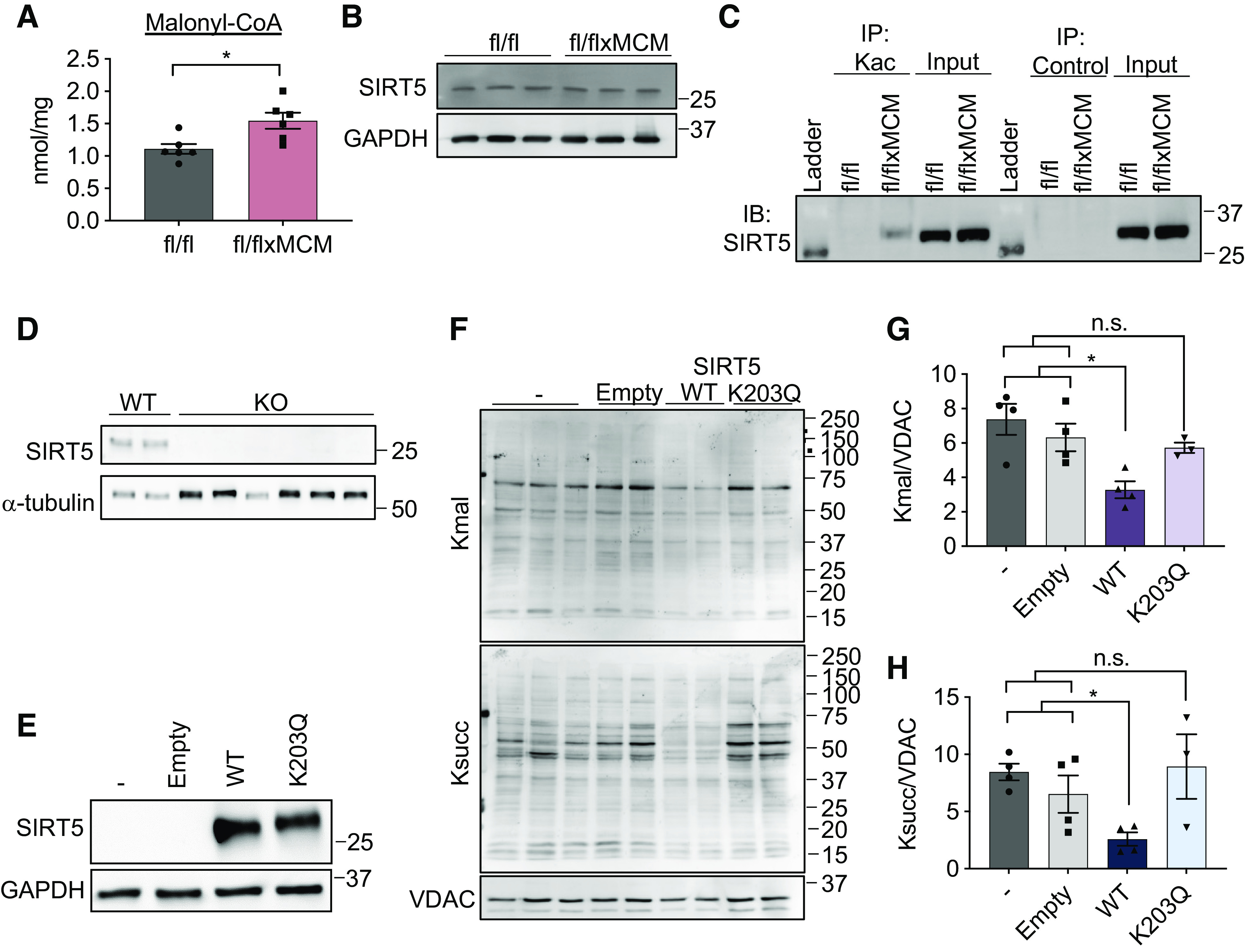
Acetylation of sirtuin 5 (SIRT5) modulates malonylation status of mitochondrial proteins. A: cardiac malonyl-CoA levels in Slc25a3fl/fl vs. Slc25a3fl/flxMCM mice at 10 wk post-tamoxifen administration (n = 5 fl/fl and n = 6 fl/flxMCM animals). B: Western blot analysis of SIRT5 expression in total heart protein lysates from Slc25a3fl/fl vs. Slc25a3fl/flxMCM animals. GAPDH was used as a loading control. C: immunoprecipitation confirmation of enhanced SIRT5 acetylation in Slc25a3fl/flxMCM vs. Slc25a3fl/fl hearts. Acetylated proteins were immunoprecipitated with anti-Kac antibodies followed by immunoblotting with an antibody against SIRT5. An anti-HA antibody was used as an immunoprecipitation control. D: Western blot analysis of SIRT5 expression in HEK293 WT vs. SIRT5 KO clones. α-Tubulin was used as a protein loading control. E: Western blot analysis of SIRT5 expression in SIRT5 KO cells reexpressing a pcDNA3.1 empty vector, WT SIRT5, or the SIRT5 K203Q mutant. GAPDH was used as a loading control. F: representative Western blot of protein lysine malonylation (Kmal) and succinylation (Ksucc) of mitochondria isolated from SIRT5 KO cells, or SIRT5 KO cells reexpressing the empty vector, WT SIRT5, or SIRT5 K203Q. VDAC was used as a loading control. G: quantification of protein malonylation in SIRT5 KO cells reexpressing the wild-type (WT) and K203Q SIRT5 constructs (n = 3–4 replicates/group). H: quantification of protein succinylation in SIRT5 KO cells reexpressing the WT and K203Q SIRT5 constructs (n = 3–4 replicates/group). One-way ANOVA followed by Welch’s test was used for statistical analysis with P < 0.05 considered significant. *P < 0.05, n.s., not significant.
To investigate the functional impact of SIRT5 K203 acetylation, we generated SIRT5 KO HEK293 cell lines by CRISPR/Cas9-mediated gene deletion (Fig. 6D) and reexpressed either WT SIRT5 (pcDNA3.1-SIRT5-WT) or a SIRT5 K203Q mutant (pcDNA3.1-SIRT5-K203Q) to mimic enforced lysine acetylation (Fig. 6E). We then examined the effects of these constructs on global protein malonylation. As anticipated, expression of a pcDNA3.1 empty vector control in SIRT5 KO cells had no effect on malonylation levels, whereas overexpression of WT SIRT5 reduced the degree of malonylation observed in total cell lysates (Fig. 6, F–H). Surprisingly, overexpression of the SIRT5 K203Q-enforced acetylation mutant had no effect on the extent of malonylation observed in SIRT5 KO cells (Fig. 6, F–G), suggesting that acetylation at K203 inhibits SIRT5 function. Because SIRT5 also possesses desuccinylase activity, we examined the effects of WT SIRT5 and the K203Q-enforced acetylation mutant on protein succinylation. Cells overexpressing WT SIRT5 displayed reduced total succinylation as compared with untransfected and empty vector controls, but overexpression of the K203Q mutant did not alter total protein succinylation (Fig. 6, F and H). Taken together, these data suggest that acetylation of SIRT5 at K203 impairs the ability of SIRT5 to function as a deacylase.
DISCUSSION
Acylations like acetylation and malonylation are well documented to target mitochondrial proteins (58, 65, 66) and are linked to metabolism. In fact, metabolic pathways like the tricarboxylic acid cycle and β-oxidation are highly acylated when mitochondrial deacylation is disrupted, suggesting that acylations may play a critical role in modulating mitochondrial metabolic function (22, 24, 25, 67). Here, we found that the converse can also occur that primary defects in mitochondrial energetics (via induction of impaired mitochondrial ATP production through SLC25A3 deletion) can dictate specific changes to the cardiac acylome.
Our initial efforts to identify mitochondria-mediated response pathways engaged by mitochondrial energy dysfunction led us to examine AMPK, ROS production, and mitochondrial unfolded protein response pathways over the course of the development of SLC25A3 deletion-induced mitochondrial cardiomyopathy. Our analyses, performed at 2-, 6-, and 10- wk posttamoxifen, did not reveal alterations/activation of these pathways in response to SLC25A3 deletion. These pathways are dynamic and thus our analyses may only represent pathway status at individual time points. Yet, our findings suggest that in vivo and in the context of the heart, mitochondrial energy dysfunction caused by impaired mitochondrial ATP synthesis may not engage these canonical modes of mitochondrial stress signaling. Instead, we found that impaired mitochondrial ATP synthesis induces cardiac acylome remodeling with a specific signature of elevated mitochondrial acetylation and malonylation (summarized in Fig. 7). Acylations present an attractive potential link between cellular metabolism and gene regulation (16). Acetylation, for example, can modify histone proteins to regulate gene expression by modulating chromatin accessibility (17, 20, 21, 23). Our observations of elevated acetylation and malonylation of cardiac proteins led us to initially postulate that histone proteins could be a major target of mitochondrial energy dysfunction-induced acylations. Surprisingly, however, mass spectrometry analyses revealed that these energy dysfunction-responsive PTMs were concentrated in the mitochondrial compartment, with proteins involved in metabolism (carboxylic acid metabolism and oxidative phosphorylation) representing some of the top modified pathways.
Figure 7.

Model of mitochondrial energy dysfunction-induced enhancement of mitochondrial protein acylation. Deletion of SLC25A3 deletion in the heart causes impaired mitochondrial ATP synthesis resulting in mitochondrial energy dysfunction. This leads to mitochondrial protein hyperacetylation and hypermalonylation. Loss of SLC25A3 decreases sirtuin 3 (SIRT3) in cardiac mitochondria, which can lead to increases in mitochondrial protein acetylation. Loss of SLC25A3 also increases cardiac malonyl-CoA levels, which can support enhanced malonylation. In addition, SLC25A3 deletion results in SIRT5 acetylation, which inhibits SIRT5 function as a second mechanism to potentiate mitochondrial malonylation. Finally, while mitochondrial energy dysfunction-induced malonylation decreases IDH2 activity, acetylation increases IDH2 activity. The cumulative effect of mitochondrial energy dysfunction-induced acetylation and malonylation of IDH2 increases IDH2 activity. Created using BioRender.com.
Although acylations can exert different regulatory consequences (68), in the context of metabolism, acetylation and malonylation of mitochondrial proteins are largely thought to be inhibitory (58). Here, we found that SLC25A3 deletion enhances both acetylation and malonylation of IDH2 and that mitochondrial IDH2 activity is actually increased in the absence of SLC25A3. Intriguingly, we found that the subset of IDH2 lysine residues (K48, K180, and K263) targeted by both PTMs exerted different functional consequences. Mutagenesis studies indicated that IDH2 acylation at some sites can enhance function. Given that acetylation neutralizes the positive charge of target lysines, whereas malonylation imparts a negative charge and is a structurally larger modification (22), it is not surprising that we found that acetylation and malonylation may elicit differing effects on target proteins, even within the same protein target. At present, we do not know how all of the identified energy dysfunction-induced acylations of IDH2 are integrated to produce the increased activity that we observed in Slc25a3fl/flxMCM hearts. However, the ultimate functional consequences of these PTMs may depend in part on the type of modification, the specific sites modified, the occupancy rates of each modification, and the cooperative effects of these modifications. Thus, future work must deploy quantitative methods to gain a more complete picture of how energy dysfunction-induced acylations modify target proteins.
Enhanced acetylation occurs in a number of different of cardiac disease models (18, 28), as well as in a model of impaired complex I assembly (69). We anticipated that mitochondrial energy dysfunction due to SLC25A3 deletion would induce similar acetylome changes. Our studies suggest that although SLC25A3 deletion does not enhance pathways that would promote mitochondrial acetylation (acetyl-CoA/GCN5L1), mitochondrial acetylation may be enhanced due to reduced mitochondrial protein deacetylation via decreased SIRT3 in mitochondria. Ongoing studies are aimed at dissecting this intriguing phenomenon. We were surprised, however, to find that SLC25A3 deletion resulted in a high degree of mitochondrial protein malonylation. Our data point to two major pathways that contribute to energy dysfunction-induced malonylation (Fig. 7). First, our observation that SLC25A3-deleted hearts display significantly elevated levels of malonyl-CoA suggests that increased malonyl-CoA availability could facilitate chemical additions that lead to the observed increases in malonylation. Second, acetylation of SIRT5 at a novel site (K203) reduced this enzyme’s ability to demalonylate proteins, suggesting that impaired SIRT5 function could also contribute to the observed hypermalonylation. How SIRT5 acetylation inhibits function is unclear, as K203 lies outside of the enzyme’s catalytic domain (70). Structurally, however, SIRT5 is composed of two globular domains, the smaller of which harbors a zinc binding site that is essential for enzyme structure/function and substrate recognition (70, 71). K203 resides within this smaller globular domain, and acetylation at this site could impact substrate recognition and zinc binding. Technically, glutamine acetyl-mimetics may not fully recapitulate the effects of endogenous protein lysine acetylation, and mutations at K203 may have the primary effect of impacting/impairing SIRT5 folding and structure. Future studies to investigate such possibilities are needed to uncover the structural importance of SIRT5 acetylation. Importantly, however, our findings suggest a novel cross talk between acetylation and malonylation, whereby acetylation can modulate the malonylome through SIRT5 inhibition.
Finally, in addition to the energy dysfunction-induced increases in mitochondrial protein acetylation and malonylation, our proteomics study revealed a surprising and unexpected decrease in acetylation of a small group of nonmitochondrial proteins implicated in DNA packaging and cardiac muscle contraction. At present, the functional impact and mechanisms underlying this energy dysfunction-responsive reduction in acetylation are unknown. However, alterations to these pathways may contribute to the overall cardiac response to mitochondrial energy deficits.
In conclusion, we found that mitochondrial energy dysfunction due to defective mitochondrial ATP synthesis initiates a novel stress response independent of canonical AMPK, ROS, and mitochondrial unfolded protein response signaling. Hyperacylation of mitochondrial proteins has been implicated in a number of disease contexts (18, 28, 58, 72–74), and the expanded network of mitochondrial proteins targeted by mitochondrial energy dysfunction-induced acylations that we uncovered suggest a mechanism whereby focal defects in the mitochondrial ATP synthasome (via SLC25A3 deletion) can control an expanded network of mitochondrial proteins. Although we have in this study focused on two foundational aspects of energy dysfunction-induced acylation: cross talk between acetylation and malonylation via SIRT5, and the differential effects of acetylation versus malonylation using IDH2 as a model, future work will be focused on understanding the integrated functions of energy dysfunction-induced acylations on cardiac function. Notably, recent work using a striated muscle-specific double-knockout model of SIRT3 and carnitine acetyltransferase has called in to question the role of enhanced mitochondrial acetylation, if any, in the transverse aortic constriction surgical model of cardiac pressure overload-induced heart failure (27); thus, future work should determine the functional consequences of hyperacetylation and hypermalonylation in the context of primary mitochondrial dysfunction and cardiac mitochondrial ATP synthesis defects. Understanding whether acylations merely serve as posttranslational biomarkers/signatures of altered metabolism or whether they have context-specific and disease-specific roles of promoting pathology will critical next steps as we move to a greater understanding of how mitochondria communicate dysfunction and aim to leverage these pathways for future therapies.
SUPPLEMENTAL DATA
Supplemental Figs. S1–S3 and Supplemental Tables S1–S3: https://doi.org/10.6084/m9.figshare.14450097.v1.
GRANTS
J. Q. Kwong is supported by a Pilot & Feasibility Award from the Southeast Center for Integrated Metabolomics (SECIM), which was used for the metabolomics assays (U24 DK097209; NIH/Common Fund). V. Faundez is supported by NIH 1RF1AG060285. Research reported in this publication was supported in part by the Emory Integrated Proteomics shared resource of Winship Cancer Institute of Emory University and NIH/NCI under Award No. P30CA138292.
DISCLAIMERS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The acyl-CoA measurements were performed by the Southeast Center for Integrated Metabolomics (SECIM) at the Sanford Burnham Prebys Medical Research Institute, Orlando, Florida.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.Q.K. conceived and designed research; J.N.P., N.G., D.M.D., K.R.H., J.R.M., and J.Q.K. performed experiments; J.N.P., N.G., D.M.D., V.F., and J.Q.K. analyzed data; J.N.P., N.T.S., V.F., and J.Q.K. interpreted results of experiments; J.N.P. and J.Q.K. prepared figures; J.N.P. and J.Q.K. drafted manuscript; J.Q.K. edited and revised manuscript; D.M.D., K.R.H., J.R.M., N.T.S., V.F., and J.Q.K. approved final version of manuscript.
REFERENCES
- 1.Bates MGD, Bourke JP, Giordano C, d'Amati G, Turnbull DM, Taylor RW. Cardiac involvement in mitochondrial DNA disease: clinical spectrum, diagnosis, and management. Eur Heart J 33: 3023–3033, 2012. doi: 10.1093/eurheartj/ehs275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duran J, Martinez A, Adler E. Cardiovascular manifestations of mitochondrial disease. Biology (Basel) 8, 34, 2019. doi: 10.3390/biology8020034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lesnefsky EJ, Chen Q, Tandler B, Hoppel CL. Mitochondrial dysfunction and myocardial ischemia-reperfusion: implications for novel therapies. Annu Rev Pharmacol Toxicol 57: 535–565, 2017. doi: 10.1146/annurev-pharmtox-010715-103335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramachandra CJA, Hernandez-Resendiz S, Crespo-Avilan GE, Lin YH, Hausenloy DJ. Mitochondria in acute myocardial infarction and cardioprotection. EBioMedicine 57: 102884, 2020. doi: 10.1016/j.ebiom.2020.102884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou B, Tian R. Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Invest 128: 3716–3726, 2018. doi: 10.1172/JCI120849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chandel NS. Mitochondria as signaling organelles. BMC Biol 12: 34, 2014. doi: 10.1186/1741-7007-12-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol 19: 121–135, 2018. doi: 10.1038/nrm.2017.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Toyama EQ, Herzig S, Courchet J, Lewis TL Jr, Loson OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC, Shaw RJ. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351: 275–281, 2016. doi: 10.1126/science.aab4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shadel GS, Horvath TL. Mitochondrial ROS signaling in organismal homeostasis. Cell 163: 560–569, 2015. doi: 10.1016/j.cell.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Melber A, Haynes CM. UPR(mt) regulation and output: a stress response mediated by mitochondrial-nuclear communication. Cell Res 28: 281–295, 2018. doi: 10.1038/cr.2018.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martinez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun 11: 102, 2020. doi: 10.1038/s41467-019-13668-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol 15: 536–550, 2014. doi: 10.1038/nrm3841. [DOI] [PubMed] [Google Scholar]
- 13.Pougovkina O, Te Brinke H, Ofman R, van Cruchten AG, Kulik W, Wanders RJ, Houten SM, de Boer VC. Mitochondrial protein acetylation is driven by acetyl-CoA from fatty acid oxidation. Hum Mol Genet 23: 3513–3522, 2014. doi: 10.1093/hmg/ddu059. [DOI] [PubMed] [Google Scholar]
- 14.Ringel AE, Tucker SA, Haigis MC. Chemical and physiological features of mitochondrial acylation. Mol Cell 72: 610–624, 2018. doi: 10.1016/j.molcel.2018.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sabari BR, Zhang D, Allis CD, Zhao Y. Metabolic regulation of gene expression through histone acylations. Nat Rev Mol Cell Biol 18: 90–101, 2017. doi: 10.1038/nrm.2016.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simithy J, Sidoli S, Yuan ZF, Coradin M, Bhanu NV, Marchione DM, Klein BJ, Bazilevsky GA, McCullough CE, Magin RS, Kutateladze TG, Snyder NW, Marmorstein R, Garcia BA. Characterization of histone acylations links chromatin modifications with metabolism. Nat Commun 8: 1141, 2017. doi: 10.1038/s41467-017-01384-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eberharter A, Becker PB. Histone acetylation: a switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep 3: 224–229, 2002. doi: 10.1093/embo-reports/kvf053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horton JL, Martin OJ, Lai L, Riley NM, Richards AL, Vega RB, Leone TC, Pagliarini DJ, Muoio DM, Bedi KC Jr, Margulies KB, Coon JJ, Kelly DP. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight 2: e84897, 2016. doi: 10.1172/jci.insight.84897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Janke C, Montagnac G. Causes and consequences of microtubule acetylation. Curr Biol 27: R1287–R1292, 2017. doi: 10.1016/j.cub.2017.10.044. [DOI] [PubMed] [Google Scholar]
- 20.Bao X, Liu Z, Zhang W, Gladysz K, Fung YME, Tian G, Xiong Y, Wong JWH, Yuen KWY, Li XD. Glutarylation of histone H4 lysine 91 regulates chromatin dynamics. Mol Cell 76: 660–675, 2019. doi: 10.1016/j.molcel.2019.08.018. [DOI] [PubMed] [Google Scholar]
- 21.Chen Y, Sprung R, Tang Y, Ball H, Sangras B, Kim SC, Falck JR, Peng J, Gu W, Zhao Y. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol Cell Proteomics 6: 812–819, 2007. doi: 10.1074/mcp.M700021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirschey MD, Zhao Y. Metabolic regulation by lysine malonylation, succinylation, and glutarylation. Mol Cell Proteomics 14: 2308–2315, 2015. doi: 10.1074/mcp.R114.046664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie Z, Dai J, Dai L, Tan M, Cheng Z, Wu Y, Boeke JD, Zhao Y. Lysine succinylation and lysine malonylation in histones. Mol Cell Proteomics 11: 100–107, 2012. doi: 10.1074/mcp.M111.015875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, Yang Y, Chen Y, Hirschey MD, Bronson RT, Haigis M, Guarente LP, Farese RV Jr, Weissman S, Verdin E, Schwer B. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol 27: 8807–8814, 2007. doi: 10.1128/MCB.01636-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishida Y, Rardin MJ, Carrico C, He W, Sahu AK, Gut P, Najjar R, Fitch M, Hellerstein M, Gibson BW, Verdin E. SIRT5 regulates both cytosolic and mitochondrial protein malonylation with glycolysis as a major target. Mol Cell 59: 321–332, 2015. doi: 10.1016/j.molcel.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alrob OA, Sankaralingam S, Ma C, Wagg CS, Fillmore N, Jaswal JS, Sack MN, Lehner R, Gupta MP, Michelakis ED, Padwal RS, Johnstone DE, Sharma AM, Lopaschuk GD. Obesity-induced lysine acetylation increases cardiac fatty acid oxidation and impairs insulin signalling. Cardiovasc Res 103: 485–497, 2014. doi: 10.1093/cvr/cvu156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davidson MT, Grimsrud PA, Lai L, Draper JA, Fisher-Wellman KH, Narowski TM, Abraham DM, Koves TR, Kelly DP, Muoio DM. Extreme acetylation of the cardiac mitochondrial proteome does not promote heart failure. Circ Res 127: 1094–1108, 2020. doi: 10.1161/CIRCRESAHA.120.317293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grillon JM, Johnson KR, Kotlo K, Danziger RS. Non-histone lysine acetylated proteins in heart failure. Biochim Biophys Acta 1822: 607–614, 2012. doi: 10.1016/j.bbadis.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hershberger KA, Abraham DM, Liu J, Locasale JW, Grimsrud PA, Hirschey MD. Ablation of Sirtuin5 in the postnatal mouse heart results in protein succinylation and normal survival in response to chronic pressure overload. J Biol Chem 293: 10630–10645, 2018. doi: 10.1074/jbc.RA118.002187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hershberger KA, Abraham DM, Martin AS, Mao L, Liu J, Gu H, Locasale JW, Hirschey MD. Sirtuin 5 is required for mouse survival in response to cardiac pressure overload. J Biol Chem 292: 19767–19781, 2017. doi: 10.1074/jbc.M117.809897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koentges C, Pfeil K, Schnick T, Wiese S, Dahlbock R, Cimolai MC, Meyer-Steenbuck M, Cenkerova K, Hoffmann MM, Jaeger C, Odening KE, Kammerer B, Hein L, Bode C, Bugger H. SIRT3 deficiency impairs mitochondrial and contractile function in the heart. Basic Res Cardiol 110: 36, 2015. doi: 10.1007/s00395-015-0493-6. [DOI] [PubMed] [Google Scholar]
- 32.Scott I, Sack MN. Rethinking protein acetylation in pressure overload-induced heart failure. Circ Res 127: 1109–1111, 2020. doi: 10.1161/CIRCRESAHA.120.317910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwong JQ, Davis J, Baines CP, Sargent MA, Karch J, Wang X, Huang T, Molkentin JD. Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell Death Differ 21: 1209–1217, 2014. doi: 10.1038/cdd.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ko YH, Delannoy M, Hullihen J, Chiu W, Pedersen PL. Mitochondrial ATP synthasome. Cristae-enriched membranes and a multiwell detergent screening assay yield dispersed single complexes containing the ATP synthase and carriers for Pi and ADP/ATP. J Biol Chem 278: 12305–12309, 2003. doi: 10.1074/jbc.C200703200. [DOI] [PubMed] [Google Scholar]
- 35.Kolbe HV, Costello D, Wong A, Lu RC, Wohlrab H. Mitochondrial phosphate transport. Large scale isolation and characterization of the phosphate transport protein from beef heart mitochondria. J Biol Chem 259: 9115–9120, 1984. doi: 10.1016/S0021-9258(17)47273-5. [DOI] [PubMed] [Google Scholar]
- 36.Mayr JA, Merkel O, Kohlwein SD, Gebhardt BR, Bohles H, Fotschl U, Koch J, Jaksch M, Lochmuller H, Horvath R, Freisinger P, Sperl W. Mitochondrial phosphate-carrier deficiency: a novel disorder of oxidative phosphorylation. Am J Hum Genet 80: 478–484, 2007. doi: 10.1086/511788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mayr JA, Zimmermann FA, Horvath R, Schneider HC, Schoser B, Holinski-Feder E, Czermin B, Freisinger P, Sperl W. Deficiency of the mitochondrial phosphate carrier presenting as myopathy and cardiomyopathy in a family with three affected children. Neuromuscul Disord 21: 803–808, 2011. doi: 10.1016/j.nmd.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 38.Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res 89: 20–25, 2001. doi: 10.1161/hh1301.092687. [DOI] [PubMed] [Google Scholar]
- 39.Seyfried NT, Dammer EB, Swarup V, Nandakumar D, Duong DM, Yin L, Deng Q, Nguyen T, Hales CM, Wingo T, Glass J, Gearing M, Thambisetty M, Troncoso JC, Geschwind DH, Lah JJ, Levey AI. A multi-network approach identifies protein-specific co-expression in asymptomatic and symptomatic Alzheimer's disease. Cell Syst 4: 60–72, 2017. doi: 10.1016/j.cels.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pages F, Trajanoski Z, Galon J. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25: 1091–1093, 2009. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT, Maitland A, Mostafavi S, Montojo J, Shao Q, Wright G, Bader GD, Morris Q. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res 38: W214–W220, 2010. doi: 10.1093/nar/gkq537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504, 2003. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Havugimana PC, Hart GT, Nepusz T, Yang H, Turinsky AL, Li Z, Wang PI, Boutz DR, Fong V, Phanse S, Babu M, Craig SA, Hu P, Wan C, Vlasblom J, Dar VU, Bezginov A, Clark GW, Wu GC, Wodak SJ, Tillier ER, Paccanaro A, Marcotte EM, Emili A. A census of human soluble protein complexes. Cell 150: 1068–1081, 2012. doi: 10.1016/j.cell.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, Toyoda Y, Gak IA, Weisswange I, Mansfeld J, Buchholz F, Hyman AA, Mann M. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 163: 712–723, 2015. doi: 10.1016/j.cell.2015.09.053. [DOI] [PubMed] [Google Scholar]
- 45.Narayan SB, Master SR, Sireci AN, Bierl C, Stanley PE, Li C, Stanley CA, Bennett MJ. Short-chain 3-hydroxyacyl-coenzyme A dehydrogenase associates with a protein super-complex integrating multiple metabolic pathways. PLoS One 7: e35048, 2012. doi: 10.1371/journal.pone.0035048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rolland T, Taşan M, Charloteaux B, Pevzner SJ, Zhong Q, Sahni N, et al. A proteome-scale map of the human interactome network. Cell 159: 1212–1226, 2014. doi: 10.1016/j.cell.2014.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wan C, Borgeson B, Phanse S, Tu F, Drew K, Clark G, Xiong X, Kagan O, Kwan J, Bezginov A, Chessman K, Pal S, Cromar G, Papoulas O, Ni Z, Boutz DR, Stoilova S, Havugimana PC, Guo X, Malty RH, Sarov M, Greenblatt J, Babu M, Derry WB, Tillier ER, Wallingford JB, Parkinson J, Marcotte EM, Emili A. Panorama of ancient metazoan macromolecular complexes. Nature 525: 339–344, 2015. doi: 10.1038/nature14877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Manning JR, Thapa D, Zhang M, Stoner MW, Traba J, McTiernan CF, Corey C, Shiva S, Sack MN, Scott I. Cardiac-specific deletion of GCN5L1 restricts recovery from ischemia-reperfusion injury. J Mol Cell Cardiol 129: 69–78, 2019. doi: 10.1016/j.yjmcc.2019.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quiros PM, Prado MA, Zamboni N, D'Amico D, Williams RW, Finley D, Gygi SP, Auwerx J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J Cell Biol 216: 2027–2045, 2017. doi: 10.1083/jcb.201702058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang S, Chen JJ. Requirement of activating transcription factor 5 for murine fetal liver erythropoiesis. Br J Haematol 188: 582–585, 2020. doi: 10.1111/bjh.16202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu Y, Liu L, Nakamura A, Someya S, Miyakawa T, Tanokura M. Studies on the regulatory mechanism of isocitrate dehydrogenase 2 using acetylation mimics. Sci Rep 7: 9785, 2017. doi: 10.1038/s41598-017-10337-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burley SK, Bhikadiya C, Bi C, Bittrich S, Chen L, Crichlow GV, et al. RCSB Protein Data Bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res 49: D437–D451, 2021. doi: 10.1093/nar/gkaa1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sehnal D, Rose AS, Koča J, Burley SK, Velankar S. Mol*: towards a common library and tools for web molecular graphics. In: Proceedings of the Workshop on Molecular Graphics and Visual Analysis of Molecular Data. Brno, Czech Republic: Eurographics Association, 2018, p. 29–33. [Google Scholar]
- 54.Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem 271: 27879–27887, 1996. doi: 10.1074/jbc.271.44.27879. [DOI] [PubMed] [Google Scholar]
- 55.Pareek G, Pallanck LJ. Inactivation of Lon protease reveals a link between mitochondrial unfolded protein stress and mitochondrial translation inhibition. Cell Death Dis 9: 1168, 2018. doi: 10.1038/s41419-018-1213-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rath S, Sharma R, Gupta R, Ast T, Chan C, Durham TJ, Goodman RP, et al. MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res 49: D1541–D1547, 2021. doi: 10.1093/nar/gkaa1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A, Sidiropoulos K, Cook J, Gillespie M, Haw R, Loney F, May B, Milacic M, Rothfels K, Sevilla C, Shamovsky V, Shorser S, Varusai T, Weiser J, Wu G, Stein L, Hermjakob H, D'Eustachio P. The reactome pathway knowledgebase. Nucleic Acids Res 48: D498–D503, 2020. doi: 10.1093/nar/gkv1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carrico C, Meyer JG, He W, Gibson BW, Verdin E. The mitochondrial acylome emerges: proteomics, regulation by sirtuins, and metabolic and disease implications. Cell Metab 27: 497–512, 2018. doi: 10.1016/j.cmet.2018.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yu W, Dittenhafer-Reed KE, Denu JM. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J Biol Chem 287: 14078–14086, 2012. doi: 10.1074/jbc.M112.355206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kamieniarz K, Schneider R. Tools to tackle protein acetylation. Chem Biol 16: 1027–1029, 2009. doi: 10.1016/j.chembiol.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 61.Wagner GR, Bhatt DP, O'Connell TM, Thompson JW, Dubois LG, Backos DS, Yang H, Mitchell GA, Ilkayeva OR, Stevens RD, Grimsrud PA, Hirschey MD. A class of reactive acyl-CoA species reveals the non-enzymatic origins of protein acylation. Cell Metab 25: 823–837, 2017. doi: 10.1016/j.cmet.2017.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wagner GR, Hirschey MD. Nonenzymatic protein acylation as a carbon stress regulated by sirtuin deacylases. Mol Cell 54: 5–16, 2014. doi: 10.1016/j.molcel.2014.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Scott I, Wang L, Wu K, Thapa D, Sack MN. GCN5L1/BLOS1 links acetylation, organelle remodeling, and metabolism. Trends Cell Biol 28: 346–355, 2018. doi: 10.1016/j.tcb.2018.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Scott I, Webster BR, Li JH, Sack MN. Identification of a molecular component of the mitochondrial acetyltransferase programme: a novel role for GCN5L1. Biochem J 443: 655–661, 2012. doi: 10.1042/BJ20120118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Colak G, Pougovkina O, Dai L, Tan M, Te Brinke H, Huang H, Cheng Z, Park J, Wan X, Liu X, Yue WW, Wanders RJ, Locasale JW, Lombard DB, de Boer VC, Zhao Y. Proteomic and biochemical studies of lysine malonylation suggest its malonic aciduria-associated regulatory role in mitochondrial function and fatty acid oxidation. Mol Cell Proteomics 14: 3056–3071, 2015. doi: 10.1074/mcp.M115.048850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, Grishin NV, White M, Yang XJ, Zhao Y. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell 23: 607–618, 2006. doi: 10.1016/j.molcel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 67.Zhang M, Wu J, Sun R, Tao X, Wang X, Kang Q, Wang H, Zhang L, Liu P, Zhang J, Xia Y, Zhao Y, Yang Y, Xiong Y, Guan KL, Zou Y, Ye D. SIRT5 deficiency suppresses mitochondrial ATP production and promotes AMPK activation in response to energy stress. PLoS One 14: e0211796, 2019. doi: 10.1371/journal.pone.0211796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Caron C, Boyault C, Khochbin S. Regulatory cross-talk between lysine acetylation and ubiquitination: role in the control of protein stability. Bioessays 27: 408–415, 2005. doi: 10.1002/bies.20210. [DOI] [PubMed] [Google Scholar]
- 69.Karamanlidis G, Lee CF, Garcia-Menendez L, Kolwicz SC Jr, Suthammarak W, Gong G, Sedensky MM, Morgan PG, Wang W, Tian R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab 18: 239–250, 2013. doi: 10.1016/j.cmet.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yuan H, Marmorstein R. Structural basis for sirtuin activity and inhibition. J Biol Chem 287: 42428–42435, 2012. doi: 10.1074/jbc.R112.372300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chakrabarty SP, Balaram H. Reversible binding of zinc in Plasmodium falciparum Sir2: structure and activity of the apoenzyme. Biochim Biophys Acta 1804: 1743–1750, 2010. doi: 10.1093/nar/gkv1351. [DOI] [PubMed] [Google Scholar]
- 72.Du Y, Cai T, Li T, Xue P, Zhou B, He X, Wei P, Liu P, Yang F, Wei T. Lysine malonylation is elevated in type 2 diabetic mouse models and enriched in metabolic associated proteins. Mol Cell Proteomics 14: 227–236, 2015. doi: 10.1074/mcp.M114.041947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sadhukhan S, Liu X, Ryu D, Nelson OD, Stupinski JA, Li Z, Chen W, Zhang S, Weiss RS, Locasale JW, Auwerx J, Lin H. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc Natl Acad Sci USA 113: 4320–4325, 2016. doi: 10.1073/pnas.1519858113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vadvalkar SS, Baily CN, Matsuzaki S, West M, Tesiram YA, Humphries KM. Metabolic inflexibility and protein lysine acetylation in heart mitochondria of a chronic model of type 1 diabetes. Biochem J 449: 253–261, 2013. doi: 10.1042/BJ20121038. [DOI] [PMC free article] [PubMed] [Google Scholar]