

Keywords: β2 subunit, small fiber neuropathy (SFN), sodium channel β subunits, tetrodotoxin-sensitive voltage-gated sodium channels, voltage-gated sodium (Nav) channels
Abstract
Small fiber neuropathy (SFN) is a common condition affecting thinly myelinated Aδ and unmyelinated C fibers, often resulting in excruciating pain and dysautonomia. SFN has been associated with several conditions, but a significant number of cases have no discernible cause. Recent genetic studies have identified potentially pathogenic gain-of-function mutations in several pore-forming voltage-gated sodium channel α subunits (NaV) in a subset of patients with SFN, but the auxiliary sodium channel β subunits have been less implicated in the development of the disease. β subunits modulate NaV trafficking and gating, and several mutations have been linked to epilepsy and cardiac dysfunction. Recently, we provided the first evidence for the contribution of a mutation in the β2 subunit to pain in human painful diabetic neuropathy. Here, we provide the first evidence for the involvement of a sodium channel β subunit mutation in the pathogenesis of SFN with no other known causes. We show, through current-clamp analysis, that the newly identified Y69H variant of the β2 subunit induces neuronal hyperexcitability in dorsal root ganglion neurons, lowering the threshold for action potential firing and allowing for increased repetitive action potential spiking. Underlying the hyperexcitability induced by the β2-Y69H variant, we demonstrate an upregulation in tetrodotoxin-sensitive, but not tetrodotoxin-resistant sodium currents. This provides the first evidence for the involvement of β2 subunits in SFN and strengthens the link between sodium channel β subunits and the development of neuropathic pain in humans.
NEW & NOTEWORTHY Small fiber neuropathy (SFN) often has no discernible cause, although mutations in the voltage-gated sodium channel α subunits have been implicated in some cases. We identify a patient suffering from SFN with a mutation in the auxiliary β2 subunit and no other discernible causes for SFN. Functional assessment confirms this mutation renders dorsal root ganglion neurons hyperexcitable and upregulates tetrodotoxin-sensitive sodium currents. This study strengthens a newly emerging link between sodium channel β2 subunit mutations and human pain disorders.
INTRODUCTION
Small fiber neuropathy (SFN) is a painful condition selectively affecting thinly myelinated Aδ fibers and unmyelinated C-fibers (1). Clinically, SFN often presents with chronic pain, usually described as “burning” and in a “glove and stocking” distribution, dysautonomic symptoms, no signs of large-diameter fiber involvement, and reduced intraepidermal nerve fiber density on skin biopsy (2–5). The sequelae of SFN are associated with significant reductions in patient quality of life (6). In addition, worsening SFN pain is also directly associated with worse mental health, sleep, and employment outcomes (7). Current first-line therapeutic strategies for the management of chronic pain associated with painful neuropathy, including SFN, are not very effective, in part due to adverse effects (8, 9). More effective pharmacological management is thus a high priority and a better understanding of underlying causes of pain may provide more effective treatment options.
Many causes of SFN have been identified (10–12), including autoimmune conditions, sodium channel gene mutations (1, 13–16), diabetes mellitus (17), and chemotherapy (18). However, a significant proportion of patients with SFN have no identifiable cause, termed idiopathic small fiber neuropathy (I-SFN) (10, 19). Knowledge of the etiology underlying SFN is important, as some conditions are preventable or can be targeted with a precision medicine approach (20).
Mutations in sodium channel genes are responsible for a sizable percentage of SFN cases (11.6%) (16), which is explicable as voltage-gated sodium channel α subunits (NaV) play a pivotal role in regulating neuronal excitability (21, 22). Sodium channels are composed of a pore-forming α subunit, which is associated with auxiliary β subunits (23). Nine different NaV channels (NaV1.1–NaV1.9) are expressed in humans (24). NaV1.7, NaV1.8, and NaV1.9 are preferentially expressed in the developed peripheral nervous system and have been genetically and functionally well validated as drivers of chronic pain in humans (25–27). NaV1.7 was the first NaV channel linked directly to pain, with gain-of-function mutations resulting in a variety of pain conditions, including inherited erythromelalgia (28–32), paroxysmal extreme pain disorder, and SFN (1). Loss-of-function mutations result in congenital insensitivity to pain (33–35). Subsequently, other NaV channels were found to play pivotal roles in SFN, including both NaV1.8 (13, 36) and NaV1.9 (15). However, although the contribution of NaV channels to painful neuropathies has been thoroughly investigated, the contributions of sodium channel β subunits to pain are still being elucidated.
The sodium channel β subunit family comprises four genes, SCN1B–SCN4B, encoding four distinct proteins, β1–β4, and two splice variants, β1A and β1B (37, 38). NaV channels are found as heterotrimeric complexes in vivo, consisting of one pore-forming α subunit (NaV1.1–NaV1.9) and two non-ion conducting β subunits (β1/3 and β2/4)(39, 40). β subunits play important roles in a number of cellular and molecular processes. For example, β subunits are known to alter the biophysical properties of multiple NaV α subunits (41–44). They also are important for normal NaV localization and membrane trafficking (44–49). In addition, β subunits act as cell adhesion molecules (50) and are implicated in multiple diseases of the cardiac conducting system (51, 52) and epilepsy (53–56). Given the multiple roles of sodium channel β subunits in NaV modulation, it is unsurprising that their upregulation has been linked to neuropathic pain, whereas their knockout or knockdown ameliorates pain in preclinical rodent models (57, 58). Only recently, though, has a mutation in a β subunit (β2-D109N) been linked to pain in a patient with painful diabetic neuropathy (59). Functional analysis showed that the D109N mutation caused a depolarizing shift in the voltage-dependence of NaV1.7 fast inactivation and reduced use-dependent inhibition of the NaV1.7 channel, enhancing dorsal root ganglion (DRG) neuronal action potential firing, consistent with its contribution to pain in this patient. Here, we present the case of a novel β2 subunit mutation involved in the development of small fiber neuropathy with no other underlying pathophysiology. The newly identified Y69H variant increases tetrodotoxin-sensitive (TTX-S) current density, without altering NaV gating properties, enhancing the excitability of DRG neurons. Our data expands the role of β2 subunit mutations at the sodium channel level and strengthens the evidence for a role of these subunits in human pain disorders.
MATERIALS AND METHODS
DNA Isolation from Peripheral Blood
Local medical ethical committees at each of the participating centers approved this study. Written informed consent by patients was obtained before participation in this study. A peripheral blood sample was taken from all patients in a cohort with SFN (59), and genomic DNA was extracted from peripheral blood using either a QIAamp DNA Blood Maxi Kit/Puregene Blood Core Kit (Qiagen, Hilden, Germany) or a NucleoSpin 8 Blood Isolation Kit (Macherey-Nagel, Düren, Germany). Quality and concentration of the DNA were determined by NanoDrop (Thermo Fisher Scientific, Wilmington, DE) and Qubit 2.0 Fluorometer using the Qubit dsDNA BR assay kit (Life Technologies, Bleiswijk, the Netherlands). Isolated DNA was stored with a unique numeric code in the central DNA bank at Maastricht University Medical Center and IRCCS Foundation “Carlo Besta” Neurological Institute.
Single-Molecule Molecular Inversion Probe-Next-Generation Sequencing
Genetic sequencing was conducted as previously described (60). In brief, coding exons and exon-flanking intron sequences (±20 bp) of SCN1B-4B, SCN3A, and SCN7A-11A were sequenced by single-molecule molecular inversion probe-next-generation sequencing (smMIP-NGS). Three hundred twenty-four smMIPs were designed using a modified version of MIPgen software (http://shendurelab.github.io/MIPGEN/). The gap fill length between the extension and ligation arm (region of interest) of the smMIPs was fixed to 220–230 nucleotide. Probes were synthesized by Integrated DNA Technologies (IDT, Coralville, IA).
To identify sequence variations in SCN1B-4B, SCN3A, and SCN7A-11A, patients’ coding and immediate flanking regions of these genes were compared with reference sequence GRCh37. Genetic variations detected were annotated according to the guidelines of the Human Genome Variation Society (http://www.hgvs.org/mutnomen/). Variants with a possible pathogenic effect were classified using Alamut Mutation-Interpretation Software (Interactive-Biosoftware, Rouen, France). Classification of variants was based on the practice guidelines of the Association for Clinical Genetic Science.
Isolation and Transfection of DRG Neurons
The Y69H mutation was introduced into the plasmid encoding wild-type human-β2-IRES-GFP (61) using QuickChange Lightning site-directed mutagenesis (Agilent, Santa Clara, CA). All animal studies and procedures followed a protocol that was approved by the Veterans Administration Connecticut Healthcare System Institutional Animal Care and Use Committee. DRG neurons from 4- to 6-wk-old Sprague–Dawley rats of both sexes were harvested and dissociated, as described previously with minor differences (62). In brief, adult rat DRGs were dissociated with a 20-min incubation in 1.5 mg/mL collagenase A (Roche, Indianapolis, IN) and 0.6 mM EDTA, followed by a 17-min incubation in 1.5 mg/mL collagenase D (Roche), 0.6 mM EDTA, and 30 U/mL papain (Worthington Biochemical, Lakewood, NJ). DRGs were then centrifuged and triturated in 0.5 mL of DRG media [Dulbecco’s modified Eagle’s medium (DMEM)/F12 with 100 U/mL penicillin, 0.1 mg/mL streptomycin, and 10% fetal bovine serum] containing 1.5 mg/mL bovine serum albumin (low endotoxin) and 1.5 mg/mL trypsin inhibitor (Sigma, St. Louis, MO). After trituration, undigested tissue was filtered using a 70-μm cell strainer (Becton Dickinson, Franklin Lakes, NJ). The mesh was washed twice with 2 mL of DRG media. Neurons were then pelleted and transfected with either human-β2-Y69H or human-β2-wild-type (WT) cDNA carrying an internal ribosome entry site (IRES)-GFP to mark transfected cells using a Nucleofector IIS (Lonza, Basel, Switzerland) and Amaxa Basic Neuron SCN Nucleofector Kit (VSPI-1003).
HEK293 Stable Cell Line Transfection
Human embryonic kidney (HEK293) cells stably expressing a tetrodotoxin-resistant version of the NaV1.7 channel were transfected with either 0.5 μg/μL of human β2-Y69H or human β2-WT cDNA containing an internal ribosome entry site (IRES)-GFP tag using a LipoJet transfection kit (SignaGen Laboratories, Rockville, MD). The cells were grown and maintained under standard culture conditions (37°C, 5% CO2) in Dulbecco’s modified Eagle’s medium (DMEM/F12), supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Transfected cells were grown in 35 mm dishes, before being resuspended and plated onto PDL/laminin-coated coverslips the next day. Sodium currents were recorded 24–48 h following transfection.
Macroscopic Current Recordings
Macroscopic currents were recorded from adult rat DRG neurons in both current-clamp and voltage-clamp configurations using an EPC-10 amplifier and the PatchMaster program (HEKA Elektronik, Holliston, MA) at room temperature. Patch pipettes were pulled from borosilicate glass (1.65 mm/1.1 mm outside diameter/inside diameter; World Precision Instruments, Sarasota, FL) using a Sutter Instruments P-97 puller and had a resistance of 0.6–1.8 MΩ.
For current-clamp recordings, the extracellular solution contained (in mM) 140 NaCl, 3 KCl, 2 MgCl2, 2 CaCl2, and 10 HEPES. Patch pipettes were filled with intracellular solution containing (in mM) 140 KCl, 3 Mg-ATP, 0.5 EGTA, 5 HEPES, and 30 dextrose. Both solutions were titrated to a pH of 7.3 with NaOH and KOH, respectively, and brought to a final osmolarity (320 and 310 mosM for extracellular and intracellular solutions, respectively) using dextrose. Whole cell configuration was obtained in voltage-clamp mode before transitioning to current-clamp mode. Fluorescent small DRG neurons (<30 µm diameter) with stable (<10% variation) resting membrane potentials (RMP) and minimal leak currents at break-in were included in the analysis. Cells with an input resistance lower than 100 MΩ were excluded from analysis. Input resistance was calculated as the slope of the linear fit of the hyperpolarizing response to current steps from −5 pA to −40 pA in 5 pA increments.
In current-clamp mode, rheobase was defined as the first current injection step that resulted in action potential firing without further failure and was determined by a series of depolarizing current injections (200 ms) that increased incrementally by 5 pA. For the calculation of rheobase, action potentials were defined as rapid increases in membrane potential to >40 mV with a total amplitude >80 mV. However, as neurons often attenuate firing with repetitive action potential spiking, when examining repetitive firing, action potentials were counted if the membrane potential rapidly crossed 0 mV, regardless of overshoot or total amplitude. Action potential repetitive firing frequency was determined by quantifying the number of action potentials that a neuron fired during a 500 ms current injection at current injections between 25 and 500 pA, in 25 pA steps.
For voltage-clamp recordings, the extracellular bath solution contained (in mM) 140 NaCl, 20 TEA-Cl, 3 KCl, 1 CaCl2, 1 MgCl2, 5 CsCl, 0.1 CdCl2, and 10 HEPES [± 0.1 tetrodotoxin (TTX)] titrated to a pH of 7.3 using NaOH. Patch microelectrodes contained an intracellular solution consisting of (in mM) 140 CsF, 10 NaCl, 1.1 EGTA, and 10 HEPES titrated to a pH of 7.3 using CsOH. Both solutions were brought to final osmolarity (320 mosM for extracellular solution and 310 mosM for intracellular solution) using dextrose.
Sodium currents from fluorescent (indicating successful transfection) DRG neurons under 30 µm in diameter were recorded in the whole cell configuration. For biophysical analysis of sodium channel gating, series resistance prediction and compensation (60%–90%) were applied to reduce the voltage errors. Neurons with a peak voltage error greater than 5 mV were excluded from all biophysical analyses. The recorded currents were digitized at a rate of 50 kHz after passing through a low-pass Bessel filter setting of 10 kHz. After achieving whole cell configuration, a 5-min equilibration period was allowed before starting the recording. DRGs were held at −100 mV to minimize inactivation of NaV1.9 while maintaining neuronal cell viability.
To measure sodium channel activation, DRG neurons were pulsed to a range of potentials between −80 mV and +30 mV, in 5 mV increments, for 100 ms after being held at a holding potential of −100 mV. Peak inward currents were transformed to conductance using the equation . Reversal potentials were calculated in the FitMaster program (HEKA Elektronik). The conductance at each voltage was normalized to the maximum conductance and fit with the following Boltzmann equation to derive the activation curve: , where V0.5 is the half-maximal activation potential and k is the slope factor of the activation curve.
Use-dependent inhibition at 20 Hz stimulation was determined by holding the neurons at −80 mV for 2 ms, followed by a 10 ms step to −20 mv and measuring the charge transfer through the cell. Use-dependence curves were generated by normalizing the current passed at each sweep to the current passed at the first sweep.
To assess steady-state fast inactivation, DRG neurons were prepulsed to a range of voltages between −140 mV and 0 mV in 10 mV steps for 500 ms from a holding potential of −100 mV and then pulsed to a potential of −10 mV. Peak inward current was normalized to the maximum peak inward current and fit with the following double Boltzmann equation to derive the inactivation curve:
To assess deactivation, DRG neurons were activated by a brief 0.5 ms pulse to −10 mV from a holding potential of −120 mV. Following this brief activation step, the kinetics of deactivation were examined by 50 ms pulses at a range of potentials from −40 mV to −120 mV in −5 mV increments. The tail currents were fit with a single exponential equation to derive the time constant of decay.
To assess recovery from fast inactivation, we used a two-pulse protocol (both to −10 mV) with varied interpulse interval between 1 and 513 ms at voltage potentials between −120 mV and −70 mV. The current evoked by the test pulse at −10 mV was normalized to the current elicited by the prepulse at −10 mV to represent the fraction of recovering currents. This was then plotted against the interpulse duration to calculate the rate and extent of recovery from inactivation.
Peak current density was calculated as the peak current during the activation pulse (described in the Macroscopic Current Recordings section) normalized to the cellular capacitance. To assess tetrodotoxin-resistant (TTX-R) and TTX-S currents, 1 μM tetrodotoxin (TTX) was applied. Total sodium currents were recorded before TTX application. Extracellular bath solution containing 1 μM TTX was subsequently perfused into the chamber while continuously suctioning the chamber. To ensure full solution exchange, at least 5X the total volume of the chamber of solution was perfused. In addition, test pulses to −10 mV were conducted until the current no longer decreased. TTX-R currents were subsequently recorded. To calculate TTX-S currents, reference series subtraction in PatchMaster was utilized.
Molecular Modeling
Protein data bank (PDB) structure 6J8I from Shen et al. (39) was downloaded from RCSB.org and embedded in a heterogeneous lipid bilayer using CHARMM-GUI (63) to reflect a more physiologically relevant background. The structure was then edited in PyMol (Schrödinger, LLC).
Data Analysis
All data are represented as means ± standard error (SE), unless otherwise noted. All Boltzmann fits and graphical representation were conducted in GraphPad Prism. Significance in figures is noted as *P ≤ 0.05, **P ≤ 0.01, or ***P ≤ 0.001.
RESULTS
Identification of a Novel Variant in the Coding Region of the SCN2B Gene
In our cohort of 548 patients diagnosed with painful SFN and no other underlying causes, MIPs-NGS identified two potentially pathogenic novel SCN2B gene variants that were found only in patients with painful SFN and not in any patients with painless conditions. Of these variants, one (c.502G > T; pG168C) was in the transmembrane segment of β2, whereas the second (c.205T > C; pY69H), a tyrosine-to-histidine missense mutation at position 69 resides in the immunoglobulin domain of the protein (Fig. 1). The rare Y69H variant, classified as a variant of unknown significance (64) with an allele frequency of 4.95 × 10−5, was initially chosen for investigation because it had also been identified in patients with Brugada syndrome and one patient with chronic atrial fibrillation (65). In addition, the only previously characterized mutation in the sodium channel β2 subunit associated with a painful neuropathy was also located in the immunoglobulin domain (59).
Figure 1.

Molecular modeling of the β2-Y69H variant. The immunoglobulin domain of the human β2 subunit (cyan) is linked to the NaV1.7 channel α subunit (green) via a disulfide bond with the cystine at position 55 (C55, yellow). The sidechain of the tyrosine at position 69 (Y69, magenta) is shown, facing away from the α subunit. The star in the cartoon diagram of the β2 subunit (top right) indicates the relative position of the mutation. The sidechain of the aspartic acid at position 109 (D109, orange) is also shown. The α subunit is embedded in a lipid bilayer membrane (red, white, and green spheres). A small portion of the β1 subunit (red, top left) may be seen, as well.
The β2-Y69H variant was identified in a patient with painful idiopathic SFN presenting as a stabbing and burning pain in both legs, as well as red discoloration of the soles of her feet, beginning at the age of 53. She was assessed clinically and her neurological physical exam was normal. She also had normal nerve conduction studies, consistent with unaffected large, myelinated nerve fibers. Thermal testing showed abnormal warmth and cold sensation and reduced intraepidermal nerve fiber density (1.1 per mm, with a lower limit of normal accepted at 4.3 per mm). There was no other relevant medical history that could potentially explain her peripheral neuropathy. Cardiovascular workup was normal and no prior history of cardiovascular disease was noted. Electrocardiogram conducted at the time showed sinus rhythm with normal conduction times; the PR interval spanned 154 ms (normal limits: 120–200 ms), the QRS complex spanned 86 ms (normal limits: 60–100 ms), and the QT interval spanned 464 ms, with a corrected QT (QTc) interval of 401 ms (99th percentile QTc in postpubertal females: 480 ms). No other SCN1B-4B, SCN3A, or SCN7A-11A variants, others than the SCN2B Y69H variant, were detected by MIPs-NGS; she was heterozygous for the SCN2B Y69H variant.
Human β2-Y69H Confers Hyperexcitability on DRG Neurons
To test whether the Y69H variant contributes to enhanced neuronal excitability, DRG neurons were isolated from adult rats and transfected with either wild-type human β2 or the β2-Y69H variant. Excitability of DRG neurons was analyzed by current-clamp recordings. We examined passive membrane properties, such as the neuronal RMP (Fig. 2A). The RMP of neurons expressing the β2-Y69H variant (−48.21 ± 1.99 mV, n = 11) were not statistically different from that of neurons expressing the wild-type β2 subunit (−50.18 ± 2.48 mV, n = 11, two-tailed Student’s t test P = 0.54). In addition, input resistances of nonspontaneously firing DRG neurons were comparable between those expressing the wild-type β2 subunit (732.95 ± 130.13 MΩ, n = 11) and those expressing the Y69H variant (959.70 ± 97.82 MΩ, n = 11, Student’s t test, P = 0.18). Neurons expressing the β2-Y69H variant were more excitable at rest than neurons exhibiting the wild-type β2 subunit (Fig. 2B), with a numerically larger percentage of neurons firing spontaneous action potentials; 47.62% of neurons expressing the Y69H variant fired action potentials spontaneously compared with only 21.42% of neurons expressing the wild-type β2 subunit. However, this study was not powered to detect spontaneous action potential firing and was therefore not statistically significant (Fisher’s exact test, P = 0.16).
Figure 2.

The Y69H variant does not alter resting membrane potential of dorsal root ganglion (DRG) neurons. A: comparison of resting membrane potentials for DRG neurons expressing either the wild-type human β2 subunit (monochrome) or the Y69H variant (orange) illustrates no change in neuronal resting membrane potentials (Em, RMP). B: a numerically larger, but not statistically significant, proportion of DRG neurons expressing the Y69H variant fire spontaneous action potentials at rest than DRG neurons expressing the wild-type human β2 subunit. WT, wild type.
Neurons expressing the β2-Y69H variant are significantly more excitable in response to current injection stimuli than neurons expressing the wild-type β2 subunit. We examined the current injection necessary to evoke an action potential in adult rat DRG neurons (Fig. 3, A and B). Rheobase (Fig. 3B) was significantly lower in neurons expressing the Y69H variant (51.36 ± 16.22 pA, n = 11) compared with neurons expressing the wild-type β2 subunit (163.64 ± 47.85 pA, n = 11, Student’s t test, P = 0.038). Furthermore, neurons expressing the Y69H variant were capable of significantly increased repetitive action potential firing compared with neurons expressing the wild-type β2 variant (Fig. 3, C–E). When each neuron was injected with a 1-s current stimulus at the amplitude that was sufficient to stimulate an action potential at 200-ms duration, to the nearest 25 pA, neurons expressing the wild-type β2 subunit fired significantly fewer action potentials (0.73 ± 0.14, n = 11) than did neurons expressing the Y69H variant (3.82 ± 1.01, n = 11, two-tailed Student’s t test, P = 0.0065; Fig. 3C). In response to graded current injections from 25 to 500 pA in 25 pA increments, neurons expressing the Y69H variant fired significantly more action potentials across the entire stimulus range (2-way repeated-measures ANOVA, P = 0.004). This difference was statistically significant after correcting for multiple comparisons using the Holm-Šídák method at 150, 175, 200, 225, and 275 pA current injections (Fig. 3E).
Figure 3.

The Y69H variant reduces the threshold of action potential firing and enhances repetitive spiking in dorsal root ganglion (DRG) neurons. A: representative traces depicting the action potential response of DRG neurons expressing the wild-type (black) or Y69H variant (orange) human β2 subunit during stimulation with current injections of 200 ms duration. The 0 mV membrane potential is indicated by the dashed line. B: DRG neurons expressing the Y69H variant displayed a significantly lower threshold to action potential firing than did neurons expressing the wild-type β2 subunit. C: when neurons were stimulated with a 1,000-ms pulse at the threshold amplitude as determined by a 200-ms injection in B, DRG neurons expressing the Y69H variant fired significantly more action potentials than those expressing the wild-type β2 subunit. D: representative traces depicting repetitive action potential firing of DRG neurons expressing the Y69H variant (orange) compared with those expressing the wild-type β2 subunit (black). E: neurons expressing the Y69H variant (orange) fired significantly more action potentials during a 1,000-ms stimulus of graded amplitude than did neurons expressing the wild-type β2 subunit (monochrome). WT, wild type. *P ≤ 0.05, **P ≤ 0.01.
Biophysical Analysis of the β2-Y69H Variant on Total Sodium Current
We investigated mechanisms underlying the hyperexcitability of DRG neurons expressing the β2-Y69H variant using voltage-clamp recordings to study voltage-dependent sodium currents. We examined the biophysical properties of the total sodium current passed by DRG neurons transfected with either wild-type β2 or β2-Y69H plasmids (Fig. 4). The Y69H variant did not significantly alter the half-maximal voltage of activation (V1/2, −21.04 ± 1.40 mV, n = 18) for total sodium current compared with neurons expressing the wild-type β2 subunits (−16.78 ± 2.18 mV, n = 15, Student’s t test, P = 0.10), although the point estimates showed an ∼4.3 mV hyperpolarization (Fig. 4A). Similarly, the slope of the activation curves showed a trend toward faster activation of sodium current in neurons expressing the Y69H variant (6.37 ± 0.45 ms, n = 18) compared with wild-type (8.72 ± 1.20 ms, n = 15, Student’s t test, P = 0.059). In addition to activation, we also examined the effect of the variant β2 subunit on sodium current inactivation (Fig. 4A). Current traces were best fit with a double Boltzmann equation and showed no difference in the V1/2 of fast inactivation or the slope for the development of fast inactivation between the Y69H variant and the wild-type subunit (Table 1).
Figure 4.

The Y69H variant does not alter total NaV channel gating properties. A: the voltage-dependence of channel inactivation (left; diamonds) are not statistically different between dorsal root ganglion (DRG) neurons expressing the wild-type and Y69H variant subunits. In addition, the voltage-dependence of activation (right, circles) is also not statistically different. B: there is no difference in the use-dependent inhibition of sodium channels in DRG neurons expressing either the wild-type or the Y69H β2 subunit. C: the extent of sodium channel recovery from inactivation at voltages between −70 and −120 mV is comparable for DRG neurons expressing either the wild-type or the variant β2 subunits. D: the rate of recovery from channel inactivation is also comparable in both groups. E: there is no difference in the kinetics of entry into deactivation between DRG neurons expressing the wild-type or Y69H variant. WT, wild type.
Table 1.
Fast-inactivation properties in DRG neurons expressing either the wild-type or Y69H β2 subunit
| Wild Type, n = 14 | Y69H, n = 12 | Student’s t Test P Value | |
|---|---|---|---|
| TTX-R V1/2 | −45.10 ± 1.81 | −43.61 ± 2.14 | 0.60 |
| TTX-R slope | 6.18 ± 0.32 | 6.035 ± 0.83 | 0.86 |
| TTX-S V1/2 | −75.48 ± 1.76 | −79.22 ± 1.87 | 0.16 |
| TTX-S slope | 5.31 ± 0.59 | 5.89 ± 0.78 | 0.55 |
DRG, dorsal root ganglion; TTX-R, tetrodotoxin-resistant; TTX-S, tetrodotoxin-sensitive.
The Y69H variant caused no reduction in total sodium current use-dependent inhibition (Fig. 4B); after 1.5 s of 20 Hz pulses to −20 mV, neurons expressing the wild-type human β2 subunit passed 76.12% of the current passed during the first stimulus, which was comparable to neurons expressing the Y69H variant (74.85%). Likewise, neurons expressing either construct showed a similar extent of channel recovery (Fig. 4C) and there was no change in the rate of recovery from fast inactivation between neurons expressing the wild-type and the Y69H variant (Fig. 4D). Finally, we assessed the rate of channel deactivation (Fig. 4E) and found no change between DRG neurons expressing the wild-type or Y69H variant β2 subunit.
Tetrodotoxin-Sensitive Currents Are Upregulated by the β2-Y69H Variant
Given that there were no major biophysical changes conferred on sodium channels by the Y69H variant β2 subunit, we then examined sodium channel current density, as the β subunits are known to enhance channel trafficking to the plasma membrane (44, 66). DRG neurons were held at −100 mV and then pulsed to a range of potentials from −80 mV to +30 mV for 100 ms in 5 mV increments and the current density was calculated at each step pulse. Neurons expressing the Y69H variant exhibited a significantly larger peak total sodium current density (−590.90 ± 46.21 pA/pF, n = 21) after a 100 ms pulse compared with DRG neurons expressing the wild-type β2 subunit (−359.11 ± 36.11 pA/pF, n = 17, Student’s t test, P = 0.00052, Fig. 5B). In addition, as current density is derived from both the amplitude of the currents, as well as the capacitance of the neurons, we sought to confirm that there was no difference in the capacitance of the cells recorded. The capacitance of DRG neurons expressing the Y69H variant β2 subunit (22.91 ± 2.05 pF, n = 21) was similar to that of DRG neurons expressing the wild-type subunit (21.80 ± 2.42 pF, n = 17, Student’s t test, P = 0.73).
Figure 5.

Tetrodotoxin-sensitive (TTX-S) current is upregulated in dorsal root ganglion (DRG) neurons expressing Y69H β2 subunits. A: representative traces illustrating reference series subtraction to determine TTX-S current (right) from the difference between total sodium current (left) and tetrodotoxin-resistant (TTX-R) current (middle) recorded in 1 μM TTX. Example traces are shown for sodium currents in both neurons expressing the wild-type β2 variant (top) and the Y69H variant (bottom, orange). B: there is a statistically significant increase in total sodium current passed by DRG neurons expressing the Y69H variant compared with DRG neurons expressing the wild-type β2 subunit. C: TTX-S sodium currents in DRG neurons are upregulated in the presence of the Y69H variant (orange), relative to the wild-type (monochrome) β2 subunit. However, there is no change in TTX-R sodium current. D: the voltage dependence of NaV1.7 channel inactivation (left; diamonds) is not statistically different between HEK293 cells expressing the wild-type β2 (monochromatic) and the Y69H variant (orange) subunits. In addition, the voltage-dependence of activation (right, circles) is also not statistically different. E: the Y69H variant does not significantly upregulate.7 current density in HEK293 cells stably expressing NaV1.7 channels when compared with the wild-type β2 subunit. WT, wild type. *P ≤ 0.05, ***P ≤ 0.001.
We next sought to determine whether the upregulated sodium currents were TTX-S or TTX-R. The same voltage protocol to calculate current density, was applied first in the absence of TTX, then after perfusion of 1 µM into the recording chamber. TTX-S current was calculated by reference series subtraction of TTX-R current from the total sodium current (Fig. 5A). It is important to note that a voltage error greater than 5 mV was an exclusion criterion in the first set of experiments to minimize uncertainty in biophysical analysis of channel gating. In this second set of experiments, however, voltage error was not considered. This was a decision consciously made to avoid artificially limiting any difference between the wild-type and Y69H-expressing neurons by excluding cells with large currents. Crucially, voltage error would not affect the determination of sodium current density. We were able to confirm again, in this second set of experiments, an upregulation of total NaV current in DRG neurons expressing the Y69H variant compared with DRG neurons expressing the wild-type variant (Y69H: −879.82 ± 261.94 pA/pF, n = 15; wild type: −435.02 ± 69.42 pA/pF, n = 26; Student’s t test, P = 0.048). However, we did not detect a statistically significant difference in TTX-R current in cells expressing the YH variant (−214.273 ± 44.81 pA/pF, n = 15) compared with DRG neurons expressing the wild-type variant (−189.30 ± 32.47 pA/pF, n = 26, Student’s t test, P = 0.65). By contrast, TTX-S current was more than doubled (Fig. 5C) in DRG neurons expressing the Y69H variant (−768.21 ± 234.99 pA/pF, n = 15), relative to TTX-S current in DRG neurons expressing the wild-type β2 subunit (−330.23 ± 66.27 pA/pF, n = 26, Student’s t test, P = 0.032).
As DRG neurons express a multiple NaV channel isoforms, we sought to investigate the Y69H variant in a heterologous system expressing only NaV1.7, which is responsible for the majority of TTX-S sodium currents in rat DRG neurons (67). Consistent with our previous results in DRG neurons, we confirmed that the β2-Y69H variant does not alter gating properties (Fig. 5D) of NaV1.7 channels stably expressed in HEK293 cells. Specifically, the V1/2 of activation of NaV1.7 in HEK293 cells transfected with the wild-type β2 subunit was −22.97 ± 1.58 mV (n = 16), which is comparable to those transfected with the Y69H variant (−21.79 ± 1.32, n = 20, Student’s t test, P = 0.57). Similarly, we did not observe a difference in the V1/2 of fast inactivation of NaV1.7 channels in HEK293 cells transfected with the wild-type β2 subunit (−79.60 ± 1.54 mV, n = 17) compared with those transfected with the Y69H variant (−79.73 ± 1.67 mV, n = 20, Student’s t test, P = 0.96). Interestingly, we observed no statistically significant difference in the NaV1.7 current density (Fig. 5E) in these cells, either current density of NaV1.7 in cells transfected with the wild-type β2 subunit (−572.67 ± 64.71 pA/pF, n = 31) was not different from that in cells transfected with the Y69H variant (−686.57 ± 57.84 pA/pF, n = 30, Student’s t test, P = 0.20).
DISCUSSION
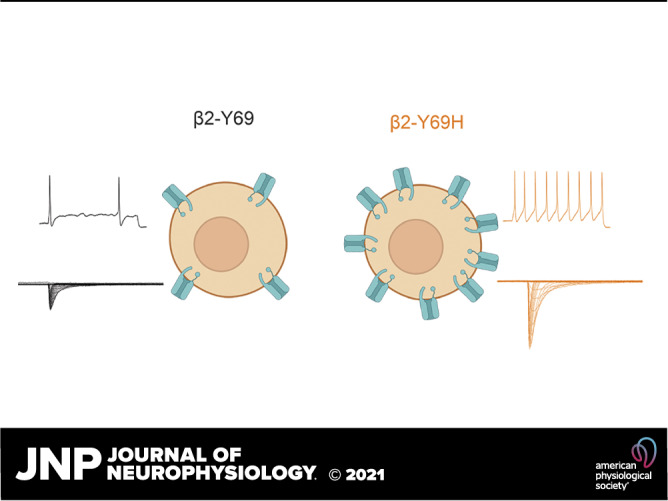
Although the contribution of β subunits to sodium channel physiology and neuronal excitability has long been studied, only recently have mutations in β subunits, and specifically the β2 subunit, been linked to human pain disorders. Here, we document the first case of idiopathic small fiber neuropathy associated with a mutation in the SCN2B gene. The expression of the β2-Y69H mutation in small diameter DRG neurons increases current density of TTX-S channels and renders these neurons hyperexcitable compared with the wild-type human β2 subunit (Fig. 6). The expression of the β2-Y69H subunit reduces the threshold to action potential firing, allowing for enhanced repetitive action potential firing. Interestingly, the mechanism underlying this hyperexcitability is a significant increase in TTX-S current density, rather than a major change in channel gating properties. We also report that the coexpression of the β2-Y69H mutant with NaV1.7 channels in HEK293 cells does not recapitulate the increased current density that we observe in DRG neurons, suggesting a cell background-specific mechanism to unmask the effect of this β2 subunit mutation.
Figure 6.

Schematic illustrating the proposed mechanism of action of the β2-Y69H (right) on small-diameter dorsal root ganglion neurons. Compared to the wild-type β2 subunit (left), neurons expressing the Y69H variant are hyperexcitable (top traces) and display a larger tetrodotoxin-sensitive sodium current density (bottom traces), suggesting increased trafficking of sodium channels to the neuronal membrane.
The association of β subunits with the pore-forming α subunits has been shown to regulate channel trafficking to the plasma membrane and gating properties (38). Although gain-of-function mutations in α subunits leading to neuropathic pain typically result in biophysical shifts in voltage dependences of activation or inactivation, or altered recovery from inactivation, changes in channel current density alone are sufficient to alter the excitability of DRG neurons (67); increasing the current density of NaV1.7, which accounts for ∼70% of TTX-S current density in small diameter DRG neurons, both reduces the current threshold to action potential firing and enhances repetitive action potential spiking (67). Vasylyev et al. (67) used dynamic clamp to precisely modulate the levels of NaV1.7 current in small diameter DRG neurons and showed that increasing NaV1.7 current density by 100% resulted in a near 30% reduction in current threshold. Although it would be interesting to compare our results directly to the results from that study, it is not feasible for multiple reasons. Primarily, dynamic clamp injects current using an equation modeling a specific ion channel, but the levels of the physical channel are unchanged. Because we presumably had an increase in the number of TTX-S channels at the cell surface, this may also have affected some change to neuronal excitability, possibly by an interaction with one or more of their many channel-binding partners (68). In addition, Vasylyev et al. (67) only modulated up to a 100% change in NaV1.7 levels, whereas we recorded an ∼130% increase in TTX-S current density, so a comparison would both require an extrapolation of their generated fit and an assumption that the Y69H variant increases NaV1.7 current density exclusively; indeed, we were unable to confirm in a heterologous expression system that the Y69H variant upregulates NaV1.7 currents. This may imply that the Y69H variant upregulates other TTX-S NaV channels, such as NaV1.6, which has also been implicated in pain disorders (69) and shown to regulate sensory neuronal excitability (70). Alternatively, NaV1.7 may be a target of upregulation by the Y69H variant, but differences in trafficking mechanisms between DRG neurons and HEK293 cells obscured our ability to detect an effect in the latter system. Regardless, it remains clear that the increase in TTX-S current density conferred by the β2-Y69H variant is consistent with neuronal hyperexcitability.
Previously, we reported and characterized the first β subunit mutation, an aspartate-to-asparagine substitution at position 109 in the β2 subunit, in a patient with a pain syndrome, specifically, diabetic neuropathy (59). Although the D109N variant was discovered in a patient with underlying longstanding diabetes, functional validation of that variant confirmed that it induces neuronal hyperexcitability. Although it is not clear why pain and neuropathy in the patient described here were not manifested until the fifth decade, the discovery of the Y69H β2 variant, which increased the current density of the TTX-S currents, further strengthens a contributory link between the β2 subunit and neuropathic pain, as no other putative cause of pain was identified.
The purported mechanism of action of both the newly identified Y69H variant and the previously identified D109N variant is concordant with established effects of the β2 subunit in DRG neurons. All β subunits, with the exception of the β1b splice variant, share the common structure of an extracellular immunoglobulin domain and a transmembrane domain with a short cytoplasmic tail. The Y69H variant is located in the extracellular immunoglobulin domain, which binds to NaV channels within the DII-S5-6 extracellular linker via a disulfide bond (39, 71) and enhances channel trafficking to the plasma membrane. This is consistent with the known role of β2 subunits in small-diameter DRG neurons; Lopez-Santiago et al. (48) showed that β2 null mice DRG neurons exhibited approximately a 50% reduction in TTX-S current and NaV1.7 protein levels.
The β2-Y69H variant was also identified in two patients with Brugada syndrome, and, when coexpressed with NaV1.5 in HEK293 cells, resulted in a reduction in current density (72). The data from this early study have only been available in a meeting abstract and is thus difficult to evaluate. By contrast, the present studies were done in DRG neurons where a full complement of additional factors could be important for uncovering the true effects of this mutation on the TTX-S channels. These data suggest that the Y69H variant may exert its effect in an isoform-specific manner or in a cell background-dependent manner.
Both the D109N and Y69H variants are located in the extracellular immunoglobulin domain and both act on TTX-S but not TTX-R channels; however, the mechanism of action for the Y69H variant is distinct from that of the D109N variant. This different mode of action may be influenced by the position of the two residues in the folded structure of the β2 subunit. The side chain of the Y69H variant points away from the NaV α subunit (Fig. 1), whereas the D109N mutant residue is located in a spatially separate and distinct location within the β2 subunit. We postulate that mutations in residues at different loci in the β2 subunit may perturb their respective local structures and interfere differentially with normal subunit functioning. For example, based on the data presented in this manuscript, we postulate that mutations facing away from the channel may be more likely to modulate channel trafficking to the cell surface or enhance channel stability at the plasma membrane via interactions with other membrane proteins or extracellular matrix proteins. On the other hand, mutations within local environments closer to the α subunits of TTX-S channels may engage in interactions with the channel itself and cause biophysical alterations in their gating properties. These modes of action of mutations in the β2 subunit are difficult to predict using static modeling even with the advances in determining structures of NaV subunits at the atomic level because these structures do not capture the dynamic nature of conformational changes of the channel within the plasma membrane during gating steps.
There is a paucity of publications of functional characterization of β2 mutations associated with excitability disorders, highlighting the need to fill in this knowledge gap to better understand the contribution of these subunits to normal neuronal physiology and to the effects of mutations of this subunit in the pathophysiology of human pain disorders. The data that we present here, together with our previous publication of a β2 mutation in a patient with painful diabetic neuropathy (59), strengthens the evidence for a role of these subunits in the pathophysiology of human pain disorders.
GRANTS
This work was supported by Award B9253-C from the U.S. Department of Veterans Affairs Rehabilitation Research and Development Service (S.G.W., M.A., J.I.R.L., D.S., P.Z., S.D.H). M.A. is supported by the NIH/NIGMS Medical Scientist Training Program T32GM007205 and the Lo Graduate Fellowship for Excellence in Stem Cell Research from the Yale Stem Cell Center. J.I.R.L., G.L., C.G.F., S.G.W., and S.D.H. received funding from the Molecule-to-Man Pain Network, from the European Union's Horizon 2020 research and innovation program under grant agreement No. 721841. R.A., M.G., J.G.J.H., G.L., C.G.F., S.G.W., and S.D.H received funding through the European Union Seventh Framework Programme grant 602273. The Center for Neuroscience and Regeneration Research is a Collaboration of the Paralyzed Veterans of America with Yale University.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.A., R.A., S.G.W., and S.D.-H. conceived and designed research; M.A., J.I.R.L., D.S., and P.Z. performed experiments; M.A. and J.I.R.L. analyzed data; M.A. interpreted results of experiments; M.A. prepared figures; M.A. drafted manuscript; M.A., R.A., M.G., J.G.J.H., G.L., C.G.F., S.G.W., and S.D.-H. edited and revised manuscript; M.A., D.S., P.Z., R.A., M.G., J.G.J.H., G.L., C.G.F., S.G.W., and S.D.-H. approved final version of manuscript.
REFERENCES
- 1.Faber CG, Hoeijmakers JG, Ahn HS, Cheng X, Han C, Choi JS, Estacion M, Lauria G, Vanhoutte EK, Gerrits MM, Dib-Hajj S, Drenth JP, Waxman SG, Merkies IS. Gain of function Naν1.7 mutations in idiopathic small fiber neuropathy. Ann Neurol 71: 26–39, 2012. doi: 10.1002/ana.22485. [DOI] [PubMed] [Google Scholar]
- 2.Sopacua M, Hoeijmakers JGJ, Merkies ISJ, Lauria G, Waxman SG, Faber CG. Small-fiber neuropathy: expanding the clinical pain universe. J Peripher Nerv Syst 24: 19–33, 2019. doi: 10.1111/jns.12298. [DOI] [PubMed] [Google Scholar]
- 3.Tesfaye S, Boulton AJ, Dyck PJ, Freeman R, Horowitz M, Kempler P, Lauria G, Malik RA, Spallone V, Vinik A, Bernardi L, Valensi P; Toronto Diabetic Neuropathy Expert Group. Diabetic neuropathies: update on definitions, diagnostic criteria, estimation of severity, and treatments. Diabetes Care 33: 2285–2293, 2010[Erratum inDiabetes Care33: 2725, 2010]. doi: 10.2337/dc10-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Han Y, Smith MT. Pathobiology of cancer chemotherapy-induced peripheral neuropathy (CIPN). Front Pharmacol 4: 156, 2013. doi: 10.3389/fphar.2013.00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Devigili G, Rinaldo S, Lombardi R, Cazzato D, Marchi M, Salvi E, Eleopra R, Lauria G. Diagnostic criteria for small fibre neuropathy in clinical practice and research. Brain 142: 3728–3736, 2019. doi: 10.1093/brain/awz333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bakkers M, Faber CG, Hoeijmakers JG, Lauria G, Merkies IS. Small fibers, large impact: quality of life in small-fiber neuropathy. Muscle Nerve 49: 329–336, 2014. doi: 10.1002/mus.23910. [DOI] [PubMed] [Google Scholar]
- 7.Schaefer C, Mann R, Sadosky A, Daniel S, Parsons B, Nalamachu S, Stacey BR, Tuchman M, Anschel A, Nieshoff E. Health status, function, productivity, and costs among individuals with idiopathic painful peripheral neuropathy with small fiber involvement in the United States: results from a retrospective chart review and cross-sectional survey. J Med Econ 17: 394–407, 2014. doi: 10.3111/13696998.2014.909439. [DOI] [PubMed] [Google Scholar]
- 8.Nishikawa N, Nomoto M. Management of neuropathic pain. J Gen Fam Med 18: 56–60, 2017. doi: 10.1002/jgf2.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, Gilron I, Haanpaa M, Hansson P, Jensen TS, Kamerman PR, Lund K, Moore A, Raja SN, Rice AS, Rowbotham M, Sena E, Siddall P, Smith BH, Wallace M. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol 14: 162–173, 2015. doi: 10.1016/S1474-4422(14)70251-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Greef BTA, Hoeijmakers JGJ, Gorissen-Brouwers CML, Geerts M, Faber CG, Merkies ISJ. Associated conditions in small fiber neuropathy—a large cohort study and review of the literature. Eur J Neurol 25: 348–355, 2018. doi: 10.1111/ene.13508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brouwer BA, Merkies IS, Gerrits MM, Waxman SG, Hoeijmakers JG, Faber CG. Painful neuropathies: the emerging role of sodium channelopathies. J Peripher Nerv Syst 19: 53–65, 2014. doi: 10.1111/jns5.12071. [DOI] [PubMed] [Google Scholar]
- 12.Lehmann HC, Wunderlich G, Fink GR, Sommer C. Diagnosis of peripheral neuropathy. Neurol Res Pract 2: 20, 2020. doi: 10.1186/s42466-020-00064-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang J, Yang Y, Zhao P, Gerrits MM, Hoeijmakers JG, Bekelaar K, Merkies IS, Faber CG, Dib-Hajj SD, Waxman SG. Small-fiber neuropathy Nav1.8 mutation shifts activation to hyperpolarized potentials and increases excitability of dorsal root ganglion neurons. J Neurosci 33: 14087–14097, 2013. doi: 10.1523/JNEUROSCI.2710-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Faber CG, Lauria G, Merkies IS, Cheng X, Han C, Ahn HS, Persson AK, Hoeijmakers JG, Gerrits MM, Pierro T, Lombardi R, Kapetis D, Dib-Hajj SD, Waxman SG. Gain-of-function Nav1.8 mutations in painful neuropathy. Proc Natl Acad Sci USA 109: 19444–19449, 2012. doi: 10.1073/pnas.1216080109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang J, Han C, Estacion M, Vasylyev D, Hoeijmakers JG, Gerrits MM, Tyrrell L, Lauria G, Faber CG, Dib-Hajj SD, Merkies IS, Waxman SG; PROPANE Study Group. Gain-of-function mutations in sodium channel Na(v)1.9 in painful neuropathy. Brain 137: 1627–1642, 2014. doi: 10.1093/brain/awu079. [DOI] [PubMed] [Google Scholar]
- 16.Eijkenboom I, Sopacua M, Hoeijmakers JGJ, de Greef BTA, Lindsey P, Almomani R, Marchi M, Vanoevelen J, Smeets HJM, Waxman SG, Lauria G, Merkies ISJ, Faber CG, Gerrits MM. Yield of peripheral sodium channels gene screening in pure small fibre neuropathy. J Neurol Neurosurg Psychiatry 90: 342–352, 2019. doi: 10.1136/jnnp-2018-319042. [DOI] [PubMed] [Google Scholar]
- 17.Vlckova-Moravcova E, Bednarik J, Belobradkova J, Sommer C. Small-fibre involvement in diabetic patients with neuropathic foot pain. Diabet Med 25: 692–699, 2008. doi: 10.1111/j.1464-5491.2008.02446.x. [DOI] [PubMed] [Google Scholar]
- 18.Brewer JR, Morrison G, Dolan ME, Fleming GF. Chemotherapy-induced peripheral neuropathy: current status and progress. Gynecol Oncol 140: 176–183, 2016. doi: 10.1016/j.ygyno.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bednarik J, Vlckova-Moravcova E, Bursova S, Belobradkova J, Dusek L, Sommer C. Etiology of small-fiber neuropathy. J Peripher Nerv Syst 14: 177–183, 2009. doi: 10.1111/j.1529-8027.2009.00229.x. [DOI] [PubMed] [Google Scholar]
- 20.de Greef BTA, Hoeijmakers JGJ, Geerts M, Oakes M, Church TJE, Waxman SG, Dib-Hajj SD, Faber CG, Merkies ISJ. Lacosamide in patients with Nav1.7 mutations-related small fibre neuropathy: a randomized controlled trial. Brain 142: 263–275, 2019. doi: 10.1093/brain/awy329. [DOI] [PubMed] [Google Scholar]
- 21.Waxman SG. The neuron as a dynamic electrogenic machine: modulation of sodium-channel expression as a basis for functional plasticity in neurons. Philos Trans R Soc Lond B Biol Sci 355: 199–213, 2000. doi: 10.1098/rstb.2000.0559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol 117: 500–544, 1952. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Catterall WA, Lenaeus MJ, Gamal El-Din TM. Structure and pharmacology of voltage-gated sodium and calcium channels. Annu Rev Pharmacol Toxicol 60: 133–154, 2020. doi: 10.1146/annurev-pharmtox-010818-021757. [DOI] [PubMed] [Google Scholar]
- 24.Yu FH, Catterall WA. Overview of the voltage-gated sodium channel family. Genome Biol 4: 207, 2003. doi: 10.1186/gb-2003-4-3-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dib-Hajj SD, Yang Y, Black JA, Waxman SG. The Na(V)1.7 sodium channel: from molecule to man. Nat Rev Neurosci 14: 49–62, 2013. doi: 10.1038/nrn3404. [DOI] [PubMed] [Google Scholar]
- 26.Han C, Huang J, Waxman SG. Sodium channel Nav1.8: emerging links to human disease. Neurology 86: 473–483, 2016. doi: 10.1212/WNL.0000000000002333. [DOI] [PubMed] [Google Scholar]
- 27.Bennett DL, Clark AJ, Huang J, Waxman SG, Dib-Hajj SD. The role of voltage-gated sodium channels in pain signaling. Physiol Rev 99: 1079–1151, 2019. doi: 10.1152/physrev.00052.2017. [DOI] [PubMed] [Google Scholar]
- 28.Yang Y, Wang Y, Li S, Xu Z, Li H, Ma L, Fan J, Bu D, Liu B, Fan Z, Wu G, Jin J, Ding B, Zhu X, Shen Y. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet 41: 171–174, 2004. doi: 10.1136/jmg.2003.012153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cummins TR, Dib-Hajj SD, Waxman SG. Electrophysiological properties of mutant Nav1.7 sodium channels in a painful inherited neuropathy. J Neurosci 24: 8232–8236, 2004. doi: 10.1523/JNEUROSCI.2695-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dib-Hajj SD, Rush AM, Cummins TR, Hisama FM, Novella S, Tyrrell L, Marshall L, Waxman SG. Gain-of-function mutation in Nav1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain 128: 1847–1854, 2005. doi: 10.1093/brain/awh514. [DOI] [PubMed] [Google Scholar]
- 31.Han C, Rush AM, Dib-Hajj SD, Li S, Xu Z, Wang Y, Tyrrell L, Wang X, Yang Y, Waxman SG. Sporadic onset of erythermalgia: a gain-of-function mutation in Nav1.7. Ann Neurol 59: 553–558, 2006. doi: 10.1002/ana.20776. [DOI] [PubMed] [Google Scholar]
- 32.Lampert A, Dib-Hajj SD, Tyrrell L, Waxman SG. Size matters: erythromelalgia mutation S241T in Nav1.7 alters channel gating. J Biol Chem 281: 36029–36035, 2006. doi: 10.1074/jbc.M607637200. [DOI] [PubMed] [Google Scholar]
- 33.Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, Al-Gazali L, Hamamy H, Valente EM, Gorman S, Williams R, McHale DP, Wood JN, Gribble FM, Woods CG. An SCN9A channelopathy causes congenital inability to experience pain. Nature 444: 894–898, 2006. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goldberg YP, MacFarlane J, MacDonald ML, Thompson J, Dube MP, Mattice M, Fraser R, Young C, Hossain S, Pape T, Payne B, Radomski C, Donaldson G, Ives E, Cox J, Younghusband HB, Green R, Duff A, Boltshauser E, Grinspan GA, Dimon JH, Sibley BG, Andria G, Toscano E, Kerdraon J, Bowsher D, Pimstone SN, Samuels ME, Sherrington R, Hayden MR. Loss-of-function mutations in the Nav1.7 gene underlie congenital indifference to pain in multiple human populations. Clin Genet 71: 311–319, 2007. doi: 10.1111/j.1399-0004.2007.00790.x. [DOI] [PubMed] [Google Scholar]
- 35.Ahmad S, Dahllund L, Eriksson AB, Hellgren D, Karlsson U, Lund PE, Meijer IA, Meury L, Mills T, Moody A, Morinville A, Morten J, O'Donnell D, Raynoschek C, Salter H, Rouleau GA, Krupp JJ. A stop codon mutation in SCN9A causes lack of pain sensation. Hum Mol Genet 16: 2114–2121, 2007. doi: 10.1093/hmg/ddm160. [DOI] [PubMed] [Google Scholar]
- 36.Han C, Vasylyev D, Macala LJ, Gerrits MM, Hoeijmakers JG, Bekelaar KJ, Dib-Hajj SD, Faber CG, Merkies IS, Waxman SG. The G1662S NaV1.8 mutation in small fibre neuropathy: impaired inactivation underlying DRG neuron hyperexcitability. J Neurol Neurosurg Psychiatry 85: 499–505, 2014. doi: 10.1136/jnnp-2013-306095. [DOI] [PubMed] [Google Scholar]
- 37.Chahine M, O'Leary ME. Regulatory role of voltage-gated Na channel beta subunits in sensory neurons. Front Pharmacol 2: 70, 2011. doi: 10.3389/fphar.2011.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bouza AA, Isom LL. Voltage-gated sodium channel β subunits and their related diseases. Handb Exp Pharmacol 246: 423–450, 2018. doi: 10.1007/164_2017_48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shen H, Liu D, Wu K, Lei J, Yan N. Structures of human Na(v)1.7 channel in complex with auxiliary subunits and animal toxins. Science 363: 1303–1308, 2019. doi: 10.1126/science.aaw2493. [DOI] [PubMed] [Google Scholar]
- 40.Mantegazza M, Catterall WA. Voltage-gated Na+ channels: structure, function, and pathophysiology. In: Jasper's Basic Mechanisms of the Epilepsies (4th ed.), edited by Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV.. New York: Oxford University Press, 2012, p. 41–54. [Google Scholar]
- 41.Sokolov MV, Henrich-Noack P, Raynoschek C, Franzén B, Larsson O, Main M, Dabrowski M. Co-expression of β subunits with the voltage-gated sodium channel Na(V)1.7: the importance of subunit association and phosphorylation and their effects on channel pharmacology and biophysics. J Mol Neurosci 65: 154–166, 2018. doi: 10.1007/s12031-018-1082-6. [DOI] [PubMed] [Google Scholar]
- 42.Cusdin FS, Nietlispach D, Maman J, Dale TJ, Powell AJ, Clare JJ, Jackson AP. The sodium channel β3-subunit induces multiphasic gating in NaV1.3 and affects fast inactivation via distinct intracellular regions. J Biol Chem 285: 33404–33412, 2010. doi: 10.1074/jbc.M110.114058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johnson D, Montpetit ML, Stocker PJ, Bennett ES. The sialic acid component of the beta1 subunit modulates voltage-gated sodium channel function. J Biol Chem 279: 44303–44310, 2004. doi: 10.1074/jbc.M408900200. [DOI] [PubMed] [Google Scholar]
- 44.Isom LL, De Jongh KS, Patton DE, Reber BF, Offord J, Charbonneau H, Walsh K, Goldin AL, Catterall WA. Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel. Science 256: 839–842, 1992. doi: 10.1126/science.1375395. [DOI] [PubMed] [Google Scholar]
- 45.Brackenbury WJ, Calhoun JD, Chen C, Miyazaki H, Nukina N, Oyama F, Ranscht B, Isom LL. Functional reciprocity between Na+ channel Nav1.6 and beta1 subunits in the coordinated regulation of excitability and neurite outgrowth. Proc Natl Acad Sci USA 107: 2283–2288, 2010. doi: 10.1073/pnas.0909434107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Laedermann CJ, Syam N, Pertin M, Decosterd I, Abriel H. β1- and β3- voltage-gated sodium channel subunits modulate cell surface expression and glycosylation of Nav1.7 in HEK293 cells. Front Cell Neurosci 7: 137, 2013. doi: 10.3389/fncel.2013.00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cummins TR, Aglieco F, Renganathan M, Herzog RI, Dib-Hajj SD, Waxman SG. Nav1.3 sodium channels: rapid repriming and slow closed-state inactivation display quantitative differences after expression in a mammalian cell line and in spinal sensory neurons. J Neurosci 21: 5952–5961, 2001. doi: 10.1523/JNEUROSCI.21-16-05952.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lopez-Santiago LF, Pertin M, Morisod X, Chen C, Hong S, Wiley J, Decosterd I, Isom LL. Sodium channel beta2 subunits regulate tetrodotoxin-sensitive sodium channels in small dorsal root ganglion neurons and modulate the response to pain. J Neurosci 26: 7984–7994, 2006. doi: 10.1523/JNEUROSCI.2211-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen C, Bharucha V, Chen Y, Westenbroek RE, Brown A, Malhotra JD, Jones D, Avery C, Gillespie PJ 3rd, Kazen-Gillespie KA, Kazarinova-Noyes K, Shrager P, Saunders TL, Macdonald RL, Ransom BR, Scheuer T, Catterall WA, Isom LL. Reduced sodium channel density, altered voltage dependence of inactivation, and increased susceptibility to seizures in mice lacking sodium channel beta 2-subunits. Proc Natl Acad Sci USA 99: 17072–17077, 2002. doi: 10.1073/pnas.212638099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shimizu H, Tosaki A, Ohsawa N, Ishizuka-Katsura Y, Shoji S, Miyazaki H, Oyama F, Terada T, Shirouzu M, Sekine SI, Nukina N, Yokoyama S. Parallel homodimer structures of the extracellular domains of the voltage-gated sodium channel beta4 subunit explain its role in cell-cell adhesion. J Biol Chem 292: 13428–13440, 2017. doi: 10.1074/jbc.M117.786509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiong H, Yang Q, Zhang X, Wang P, Chen F, Liu Y, Wang P, Zhao Y, Li S, Huang Y, Chen S, Wang X, Zhang H, Yu D, Tan C, Fang C, Huang Y, Wu G, Wu Y, Cheng X, Liao Y, Zhang R, Yang Y, Ke T, Ren X, Li H, Tu X, Xia Y, Xu C, Chen Q, Wang QK. Significant association of rare variant p.Gly8Ser in cardiac sodium channel β4-subunit SCN4B with atrial fibrillation. Ann Hum Genet 83: 239–248, 2019. doi: 10.1111/ahg.12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Riuró H, Beltran-Alvarez P, Tarradas A, Selga E, Campuzano O, Vergés M, Pagans S, Iglesias A, Brugada J, Brugada P, Vázquez FM, Pérez GJ, Scornik FS, Brugada R. A missense mutation in the sodium channel β2 subunit reveals SCN2B as a new candidate gene for Brugada syndrome. Hum Mutat 34: 961–966, 2013. doi: 10.1002/humu.22328. [DOI] [PubMed] [Google Scholar]
- 53.Wallace RH, Wang DW, Singh R, Scheffer IE, George AL Jr, Phillips HA, Saar K, Reis A, Johnson EW, Sutherland GR, Berkovic SF, Mulley JC. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat Genet 19: 366–370, 1998. doi: 10.1038/1252. [DOI] [PubMed] [Google Scholar]
- 54.Meadows LS, Malhotra J, Loukas A, Thyagarajan V, Kazen-Gillespie KA, Koopman MC, Kriegler S, Isom LL, Ragsdale DS. Functional and biochemical analysis of a sodium channel beta1 subunit mutation responsible for generalized epilepsy with febrile seizures plus type 1. J Neurosci 22: 10699–10709, 2002. doi: 10.1523/JNEUROSCI.22-24-10699.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Audenaert D, Claes L, Ceulemans B, Löfgren A, Van Broeckhoven C, De Jonghe P. A deletion in SCN1B is associated with febrile seizures and early-onset absence epilepsy. Neurology 61: 854–856, 2003. doi: 10.1212/01.wnl.0000080362.55784.1c. [DOI] [PubMed] [Google Scholar]
- 56.Patino GA, Claes LR, Lopez-Santiago LF, Slat EA, Dondeti RS, Chen C, O'Malley HA, Gray CB, Miyazaki H, Nukina N, Oyama F, De Jonghe P, Isom LL. A functional null mutation of SCN1B in a patient with Dravet syndrome. J Neurosci 29: 10764–10778, 2009. doi: 10.1523/JNEUROSCI.2475-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pertin M, Ji RR, Berta T, Powell AJ, Karchewski L, Tate SN, Isom LL, Woolf CJ, Gilliard N, Spahn DR, Decosterd I. Upregulation of the voltage-gated sodium channel beta2 subunit in neuropathic pain models: characterization of expression in injured and non-injured primary sensory neurons. J Neurosci 25: 10970–10980, 2005. doi: 10.1523/JNEUROSCI.3066-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xie W, Zhang J, Strong JA, Zhang JM. Role of Na(V)1.6 and Na(V)β4 sodium channel subunits in a rat model of low back pain induced by compression of the dorsal root ganglia. Neuroscience 402: 51–65, 2019. doi: 10.1016/j.neuroscience.2019.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alsaloum M, Estacion M, Almomani R, Gerrits MM, Bönhof GJ, Ziegler D, Malik R, Ferdousi M, Lauria G, Merkies IS, Faber CG, Dib-Hajj S, Waxman SG, Propane Study Group. A gain-of-function sodium channel β2-subunit mutation in painful diabetic neuropathy. Mol Pain 15: 1744806919849802, 2019. doi: 10.1177/1744806919849802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Almomani R, Marchi M, Sopacua M, Lindsey P, Salvi E, Koning B, Santoro S, Magri S, Smeets HJM, Martinelli Boneschi F, Malik RR, Ziegler D, Hoeijmakers JGJ, Bonhof G, Dib-Hajj S, Waxman SG, Merkies ISJ, Lauria G, Faber CG, Gerrits MM; PROPANE Study Group.. Evaluation of molecular inversion probe versus TruSeq(R) custom methods for targeted next-generation sequencing. PLoS One 15: e0238467, 2020. e0238467 doi: 10.1371/journal.pone.0238467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lossin C, Wang DW, Rhodes TH, Vanoye CG, George AL Jr.. Molecular basis of an inherited epilepsy. Neuron 34: 877–884, 2002. doi: 10.1016/S0896-6273(02)00714-6. [DOI] [PubMed] [Google Scholar]
- 62.Dib-Hajj SD, Choi JS, Macala LJ, Tyrrell L, Black JA, Cummins TR, Waxman SG. Transfection of rat or mouse neurons by biolistics or electroporation. Nat Protoc 4: 1118–1126, 2009. doi: 10.1038/nprot.2009.90. [DOI] [PubMed] [Google Scholar]
- 63.Jo S, Kim T, Iyer VG, Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem 29: 1859–1865, 2008. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- 64.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Genome Aggregation Database Consortium, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581: 434–443, 2020. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Olesen MS, Andreasen L, Jabbari J, Refsgaard L, Haunsø S, Olesen SP, Nielsen JB, Schmitt N, Svendsen JH. Very early-onset lone atrial fibrillation patients have a high prevalence of rare variants in genes previously associated with atrial fibrillation. Heart Rhythm 11: 246–251, 2014. doi: 10.1016/j.hrthm.2013.10.034. [DOI] [PubMed] [Google Scholar]
- 66.Isom LL, Ragsdale DS, De Jongh KS, Westenbroek RE, Reber BF, Scheuer T, Catterall WA. Structure and function of the beta 2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell 83: 433–442, 1995. doi: 10.1016/0092-8674(95)90121-3. [DOI] [PubMed] [Google Scholar]
- 67.Vasylyev DV, Han C, Zhao P, Dib-Hajj S, Waxman SG. Dynamic-clamp analysis of wild-type human Nav1.7 and erythromelalgia mutant channel L858H. J Neurophysiol 111: 1429–1443, 2014. doi: 10.1152/jn.00763.2013. [DOI] [PubMed] [Google Scholar]
- 68.Kanellopoulos AH, Koenig J, Huang H, Pyrski M, Millet Q, Lolignier S, Morohashi T, Gossage SJ, Jay M, Linley JE, Baskozos G, Kessler BM, Cox JJ, Dolphin AC, Zufall F, Wood JN, Zhao J. Mapping protein interactions of sodium channel NaV1.7 using epitope-tagged gene-targeted mice. EMBO J 37: 427–445, 2018. doi: 10.15252/embj.201796692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tanaka BS, Zhao P, Dib-Hajj FB, Morisset V, Tate S, Waxman SG, Dib-Hajj SD. A gain-of-function mutation in Nav1.6 in a case of trigeminal neuralgia. Mol Med 22: 338–348, 2016. doi: 10.2119/molmed.2016.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Israel MR, Tanaka BS, Castro J, Thongyoo P, Robinson SD, Zhao P, Deuis JR, Craik DJ, Durek T, Brierley SM, Waxman SG, Dib-Hajj SD, Vetter I. Na(V) 1.6 regulates excitability of mechanosensitive sensory neurons. J Physiol 597: 3751–3768, 2019. doi: 10.1113/JP278148. [DOI] [PubMed] [Google Scholar]
- 71.Chen C, Calhoun JD, Zhang Y, Lopez-Santiago L, Zhou N, Davis TH, Salzer JL, Isom LL. Identification of the cysteine residue responsible for disulfide linkage of Na+ channel α and β2 subunits. J Biol Chem 287: 39061–39069, 2012. doi: 10.1074/jbc.M112.397646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hu D, Barajas-Martinez H, Medeiros-Domingo A, Crotti L, Tester D, Veltmann C, Schimpf R, Pfeiffer R, Dezi F, Liu Y, Burashnikov E, Guidicessi JR, Ye D, Wolpert C, Borggrefe M, Schwartz P, Ackerman MJ, Antzelevitch C. Novel mutations in the sodium channel 2 subunit gene (SCN2B) associated with Brugada syndrome and atrial fibrillation (Abstract). Circulation 126 Suppl 21: A16521, 2012. https://www.ahajournals.org/doi/10.1161/circ.126.suppl_21.A16521. [Google Scholar]