

Abstract
Lysosomes are membrane-bound, acidic eukaryotic cellular organelles that play important roles in the degradation of macromolecules. Mutations that cause the loss of lysosomal protein function can lead to a group of disorders categorized as the lysosomal storage diseases (LSDs). Suspicion of LSD is frequently based on clinical and pathologic findings, but in some cases, the underlying genetic and biochemical defects remain unknown. Here, we performed whole exome sequencing (WES) on 14 suspected LSD cases to evaluate the feasibility of using WES for identifying causal mutations. By examining 2,157 candidate genes potentially associated with lysosomal function, we identified eight variants in five genes as candidate disease-causing variants in four individuals. These included both known and novel mutations. Variants were corroborated by targeted sequencing and, when possible, functional assays. In addition, we identified nonsense mutations in two individuals in genes that are not known to have lysosomal function. However, mutations in these genes could have resulted in phenotypes that were diagnosed as LSDs. This study demonstrates that WES can be used to identify causal mutations in suspected LSD cases. We also demonstrate cases where a confounding clinical phenotype may potentially reflect more than one lysosomal protein defect.
Keywords: lysosomal storage diseases, whole exome sequencing, disease-causing variants identification, metabolic disorders
Introduction
Lysosomal Storage Diseases (LSDs) are inherited metabolic multisystemic disorders caused by mutations in genes encoding resident lysosomal enzymes and transporters, as well as proteins involved in lysosome biogenesis and other lysosome related functions. These deficiencies result in lysosomal accumulation of substrates and/or catabolites and cellular dysfunction (Segatori, 2014). Multiple tissues and organs can be affected, and clinical manifestations may include bone deformities, decline in vision and hearing, organomegaly (especially in spleen and liver), cardiac disease, and other symptoms (Scriver, 2001). Most of these diseases are neuronopathic, or have neuronopathic forms, with prominent manifestation in the central nervous system.
LSDs are mostly monogenic and more than 50 genetically distinct forms have been identified (Lubke, et al., 2009), with some individual diseases having similar clinical presentations. Although individually LSDs are rare, with incidences ranging from 1:57,000 (Gaucher disease) to 1:4,200,000 (sialidosis) (Meikle, et al., 1999), collectively they have an overall prevalence about 1 in 5,000-7,700 live births (Meikle, et al., 1999; Sanderson, et al., 2006). This is likely an underestimate as cases may be undiagnosed or misdiagnosed when they present with an atypical clinical phenotype of delayed onset and attenuated progression due to partial loss-of-function mutations (Maire, 2001). In addition, there may be defects in lysosomal proteins that are not currently associated with human disease or in proteins that are not presently assigned to the lysosome (Lubke, et al., 2009; Sleat, et al., 2007). Thus, while most LSD cases are well characterized, there are patients with disease of unknown etiology that appear to be lysosomal in origin based on clinical and/or histopathological presentation.
Biochemical and genetic analyses are the classic strategies to study the etiology of LSDs (Scriver, 2001). However, these analyses have limited application in identifying defective genes in LSDs of unknown etiology because they focus on individual proteins and can only address known lysosomal disease genes. Proteomics can provide a global and unbiased approach to identify known or novel lysosomal proteins that may be defective (Jadot, et al., 2017; Sleat, et al., 2009). However, while comparative proteomic methods can help identify candidate genes in which mutations result in loss of the encoded protein, they are not effective in cases where deleterious variants result in inactive but stable mutants.
As sequencing costs decrease, whole exome sequencing (WES) is increasingly used in human genetic studies since its successful application in identifying the genetic cause of Miller Syndrome (Ng, et al., 2010). It is a cost-effective method for identifying disease-causal variants, with WES covering nearly all protein-coding regions but only requiring ~5% of the throughput needed to sequence the whole human genome. In this study, we performed WES in 14 patients diagnosed with LSD of unknown genetic etiology and evaluated this approach to identify known and novel LSDs mutations on an exome-wide scale.
Materials and Methods
Sample Information
Cell lines from 14 unrelated patients were obtained from several sources (Table 1). These patients displayed a spectrum of phenotypes suggestive of LSD, including neurodegeneration and cellular storage, but no underlying cause had been identified. Research protocols involving human subjects were approved by the Institutional Review Board of the University of Medicine and Dentistry of New Jersey.
Table 1.
Source, phenotype and sequencing coverage of the 14 LSD samples.
| Sample ID | Clinical Information | Total Bases | Total SNVs | Exonic SNVs | Mean Coverage |
|---|---|---|---|---|---|
| 00RD098 | LSD cases from Netherlands - no confirmed diagnosis. | 2,037,480,379 | 72,708 | 20,297 | 22.5 |
| 01RD492 | 2,179,163,363 | 72,137 | 20,641 | 24.1 | |
| 02RD297 | 2,023,856,437 | 70,917 | 20,280 | 22.4 | |
| 82RD265 | 2,421,322,830 | 73,687 | 20,784 | 26.7 | |
| 95RD414 | 1,584,030,773 | 63,827 | 19,471 | 17.5 | |
| 99RD299 | 1,941,261,757 | 79,121 | 22,162 | 21.4 | |
| B1278 | pycnodystosis-like, Cathepsin K positive. | 1,891,984,968 | 68,282 | 19,820 | 20.9 |
| CABMHF1 | Patient alive at 33 years. Infiltration of bone marrow by storage histiocytes (no evidence for malignancy), thrombocytopenia and splenomegaly; biochemical tests for Gaucher and Niemann-Pick diseases were negative. | 1,996,568,992 | 69,931 | 20,041 | 22.1 |
| CABMHF2 | Patient alive at 14 years. Clinical progression and ultrastructure suggestive of late-onset/attenuated form of NPC but biochemical tests were negative. | 1,652,477,376 | 69,397 | 19,882 | 18.3 |
| CABMHF3 | Patient died at 12 years. Neuronal storage resembling zebra bodies but sphingolipidoses, oligosaccharidoses, glycoproteinoses, and mucopolysaccharidoses excluded by undefined enzyme assays. | 1,835,520,789 | 70,128 | 19,962 | 20.3 |
| CABMHF4 | Patient alive at 7 years. Neurodegeneration (severe central dys- or demyelination; epilepsy, tetraparesis) and dystrophy. | 2,304,968,208 | 78,903 | 22,097 | 25.5 |
| CABMHF5 | Patient alive at 22 years. Severe neurodegeneration, mild dysmorphia and dysostosis. Storage bodies reminiscent of neuronal ceroid lipofuscinosis and mucolipidosis IV found in skin biopsy. | 1,306,118,808 | 66,253 | 18,899 | 14.4 |
| HL508Pa | Adult neuronal ceroid lipofuscinosis. | 2,108,762,505 | 71,124 | 20,384 | 23.3 |
| TC983077 | Metaphyseal acroscyphodysplasia. | 2,007,500,640 | 69,655 | 20,178 | 22.2 |
Exome Sequencing and Reads Mapping
Whole exome sequencing was performed on SOLiD platform (Life Technologies, Grand Island, NY) in 50x25 bp format. Exomes were enriched with SureSelect DNA - Human All Exon 50Mb Kit (Agilent, Santa Clara, CA). Raw sequences were aligned to the human reference genome (version hg 19) using LifeScope ™ Genomic Analysis Software (Thermo Fisher Scientific, Waltham, MA).
Variant Calling and Annotation
Three variant calling methods were used for variant discovery: Genome Analysis Tool Kit version 2.8-1 (GATK) (McKenna, et al., 2010), CLC Genomic Workbench (CLC) (Qiagen, Redwood City, CA) and StrandNGS (v2.3, Strand Genomics, San Francisco, CA). For the most part, variant discovery by the GATK genotyping method followed previously described recommendations (DePristo, et al., 2011). Briefly, raw sequence alignments (in binary alignment map (BAM) format) were reordered based on chromosomal coordinates, sorted, and indexed with the Picard toolset (version 1.80) (http://picard.sourceforge.net), and Samtools-0.1.19 (Li, et al., 2009). A series of GATK alignment-processing procedures were then applied to revise the alignments: indel realignment, remove duplication, and base recalibration. Multi-individual joint genotype calling was then performed on all individuals with GATK UnifiedGenotyper to generate the raw genotype calls in a single variant calling format file. Single nucleotide variants (SNVs) located within the targeted exome regions were selected based on the target-region definition provided by the SureSelect DNA - Human All Exon 50Mb Kit. Quality scores of SNVs were then recalibrated with VariantRecalibrator using recommended parameters for GATK (DePristo, et al., 2011). Variant discovery by CLC and Strand NGS were performed using default parameters as recommended by manufacturers. CLC variants with Depth of Coverage (DP) less than 10 were removed. Variant coverage was calculated by a GATK tool DepthofCoverage.
Variants were annotated using ANNOVAR (Wang, et al., 2010) with the following command: table_annovar.pl -buildver hg19 inputfile annovar/humandb/ -out outfile-remove -protocol refGene,cytoBand,genomicSuperDups,esp6500siv2_all,snp138,ljb26_all, 1000g2015aug_all,exac03,exac03nontcga,exac03nonpsych,clinvar_20160302 -operation g,r,r,f,f,f,f,f,f,f,f -nastring .-csvout
Candidate Gene Selection
A list of 2,157 candidate genes that are potentially associated with lysosome function and/or disease was compiled (Supp. Table S1). This list was based on multiple sources including: a) proteomic analyses of lysosomes and lysosomal proteins (Bagshaw, et al., 2005; Chapel, et al., 2013; Schroder, et al., 2007), b) genes involved in lysosomal related pathways (Di Fruscio, et al., 2015), c) proteins transcribed by TFEB, a master gene for lysosomal biogenesis (Palmieri, et al., 2011), d) glycoproteins that contain the mannose 6-phosphate lysosomal targeting modification (Sleat, et al., 2008; Sleat, et al., 2013), and e) global protein subcellular localization studies (Jadot, et al., 2017).
Candidate Variant Selection
Based on the ANNOVAR annotation, nonsynonymous variants and loss-of-function (LoF) variants, including stop-gain variants and canonical splicing site variants, were selected from the total SNV call sets for the 2,157 candidate genes (Supp. Table S1). For all other genes, only LoF variants were selected. The selected variants were then filtered based on their allele frequency in the population. Variants with allele frequencies ≥1% in Non-Finnish European (NFE) in the ExAC database (http://exac.broadinstitute.org/) and/or ≥5% in the 1000 Genomes Project Phase 3 dataset (1000 Genomes Project Consortium, et al., 2015) were removed. An aggregate score system (Sleat, et al., 2016) based on the results from eight functional prediction programs provided by ANNOVAR was applied to prioritize remaining nonsynonymous variants. All candidate variants in each individual are listed in the Supp. Data File.
Sanger Sequencing Validation
Candidate variants were evaluated by Sanger sequencing. Primers (Supp. Table S2) were designed with Primer3 (Steve Rozen, 1998) for PCR amplification of the genomic region covering the variants sites. The PCR amplicon containing the candidate variant was sequenced directly for amplicons less than 700 bp in size. Amplicons larger than 700 bp were first subcloned with Zero Blunt TOPO kit (Thermo Fisher Scientific, Waltham, MA) and individual clones were sequenced to determine the haplotype of heterozygous variants. Purified PCR products or plasmid DNAs were sequenced by ABI 3730 DNA Sequencer at GenScript (Piscataway, NJ). Electrophoregrams were examined with BioEdit Sequence Alignment Editor (Hall, 1999). Primers and PCR methods used for gender determination were as described previously (Hedges, et al., 2003). All novel variants that were validated by Sanger sequencing have been submitted to dbSNP and their Submitted SNP numbers (ss#) are listed in Table 3.
Table 3.
Candidate pathogenic variants.
| Case | Gene | Chr | Position | Nucleotide change | Amino acid change | Methods | ExAC NFE AF | Score | Genotype | Allele pathogenicity |
|---|---|---|---|---|---|---|---|---|---|---|
| LSD Candidate Genes | ||||||||||
| 00RD098 | GLB1 | 3 | 33099692 | rs72555366:G>A | NP_000395:p.Arg208Cys | G, C, S | 4.52E-05 | 6 | Het | known pathogenic |
| 82RD265 | SLC31A1 | 9 | 116021039 | ss2137536899:C>T | NP_001850:p.Arg90Ter | G, S | na | 1 | Het | not known |
| 82RD265 | SLC31A1 | 9 | 116022721 | ss3023056067:G>T | NP_001850:p.Val181Leu | C, S | na | −4 | Het | not known |
| 95RD414 | GLA | X | 100653420 | rs28935490:C>A | NP_000160:p.Asp313Tyr | G, C, S | 0.0044 | 5.5 | Het | known pathogenic |
| 95RD414 | SMPD1 | 11 | 6413175 | rs120074128:C>A | NP_000534:p.Gln294Lys | G, C | 3.00E-05 | 8 | Homo | known pathogenic |
| CABMHF1 | NPC1 | 18 | 21136367 | rs373751051:C>T | NP_000262:p.Arg389His | G, C | 1.50E-05 | 5.5 | Het | possible pathogenic |
| CABMHF1 | NPC1 | 18 | 21140411 | rs55680026:T>C | NP_000262:p.Asn222Ser | S | 0.0048 | 0 | Het | possible polymorphism or late-onset |
| CABMHF1 | SMPD1 | 11 | 6415259 | rs144873307:G>S | NP_000534:p.Gly492Ser | G, C, S | 0.0013 | −6 | Het | possible pathogenic |
| LoF variants in other genes | ||||||||||
| 00RD098 | KRIT1 | 7 | 91870373 | ss3023056068:G>A | NP_001013424:p.Gln66Ter | G, C, S | 8.24E-06 | na | Het | known pathogenic |
| CABMHF3 | TBCK | 4 | 107183332 | ss3023056069:G>A | NP_001156907:p.Gln102Ter | G, C, S | na | na | Homo | not known |
Methods: G: GATK, C: CLC workbench, S: Strand NGS; Score: na: not available. Validation: Het: heterozygous; Homo: homozygous.
Functional assays
Enzyme assays were conducted essentially as described previously using the following substrates: beta-galactosidase and methylumbelliferyl-β-galactoside (Sleat, et al., 1996); tripeptidyl peptidase 1 and Ala-Ala-Phe-aminomethylcoumarin using the endpoint assay (Sohar, et al., 2000). Acid sphingomyelinase was analyzed using BODIPY-labeled C12 sphingomyelin in triplicates as previously described (He, et al., 2001). Cholesterol esterification measuring formation of [3H]cholesterol oleate from [3H]oleate and analysis of lysosomal cholesterol accumulation using filipin staining were conducted as described (Kruth, et al., 1986; Pentchev, et al., 1986).
Results
Exome Sequencing and Variant Discovery Pipelines
We performed WES on 14 unrelated patients suspected to have potential LSDs (Table 1). The average exome coverage for each individual varied from 14.4x to 26.7x, with a mean of 21.5x (Table 1). We implemented three different variant calling methods (Materials and Methods). Given that there are differences in the variants identified by each method, we performed downstream analyses on all three data sets to reduce method bias and make full use of all variant calling information. Because the 14 patients are genetically unrelated and their clinical phenotypes are diverse, it is unlikely that they share the same disease-causing mutation. Therefore, we examined each sample independently using the screening process outlined in Figure 1.
Figure 1:
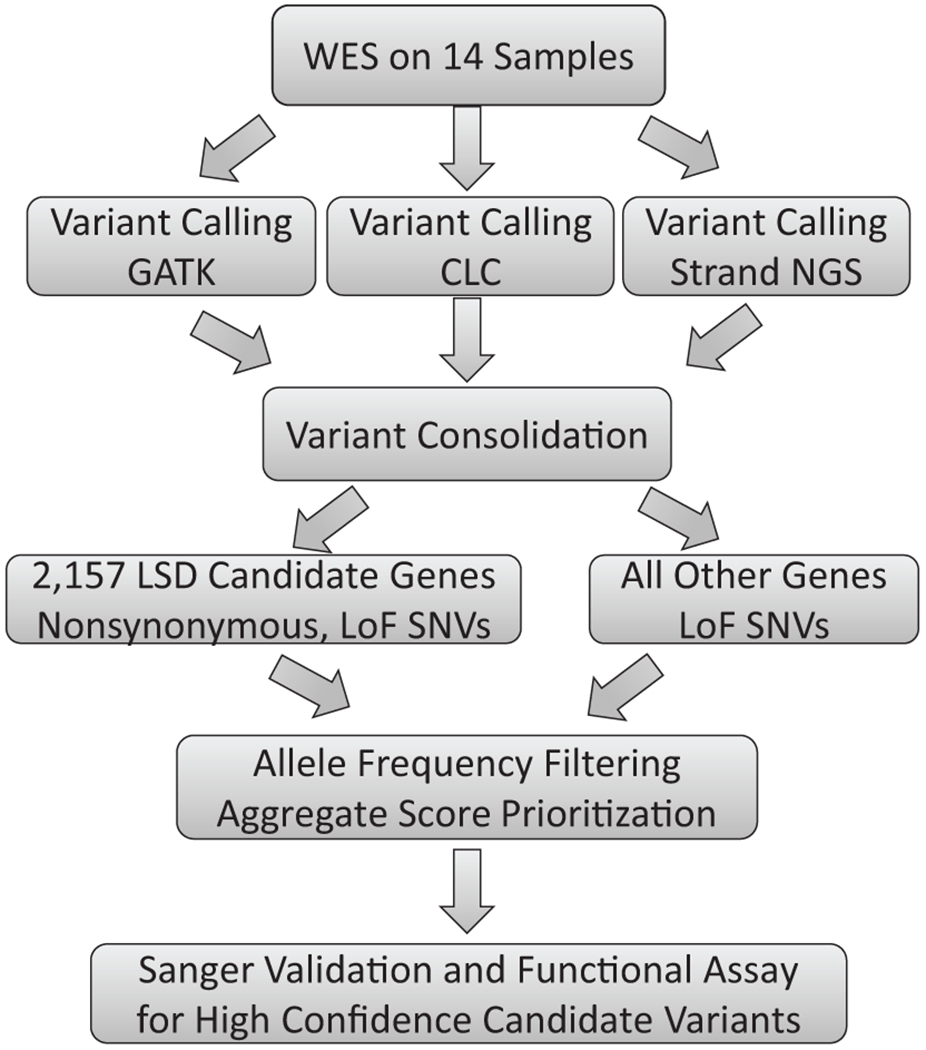
Overall analysis flow for identifying pathogenic LSD variants.
Candidate Pathogenic Variants in LSD Candidate Genes
First, we focused on a curated set of 2,157 candidate genes that are potentially associated with lysosome function (Supp. Table S1, see Materials and Methods for candidate gene selection details). Variants in these genes were extracted from the total SNV call sets and filtered as described in Figure 1. Briefly, nonsynonymous and LoF SNVs within the candidate genes were first extracted. Selected variants were then filtered based on the population minor allele frequency. Lastly, an aggregate score system (Sleat, et al., 2016) was applied to identify variants with potential functional impact. After filtering, ~ 90 variants were retained for each sample (Table 2). We then compared these variants to known pathogenic LSD variants to identify candidate variants.
Table 2.
Screening statistics for LSD candidate variants.
| Individual ID | Nonsynonymous variants in 2157 candidate genes | Variants after AF filtering * | Variants after aggregate score filtering |
|---|---|---|---|
| 00RD098 | 980 | 179 | 63 |
| 01RD492 | 993 | 220 | 102 |
| 02RD297 | 1023 | 257 | 101 |
| 82RD265 | 993 | 176 | 78 |
| 95RD414 | 912 | 130 | 57 |
| 99RD299 | 1041 | 249 | 102 |
| B1278 | 1000 | 228 | 104 |
| CABMHF1 | 963 | 175 | 90 |
| CABMHF2 | 974 | 185 | 92 |
| CABMHF3 | 979 | 195 | 100 |
| CABMHF4 | 1148 | 304 | 164 |
| CABMHF5 | 890 | 136 | 53 |
| HL508Pa | 1005 | 188 | 74 |
| TC983077 | 905 | 167 | 73 |
| Average | 986 | 199 | 90 |
ExAC Non-Finnish European: AF < 0.01; 1000 Genomes: AF < 0.05
From the filtered variant sets, we identified eight candidate pathogenic variants in five genes in four individuals (Table 3). Sanger sequencing validated seven as heterozygous and one as homozygous. Two examples are shown in Supp. Figure S1. To investigate whether these variants had been previously reported to be pathogenic, we searched the Human Gene Mutation Database (HGMD) (Stenson, et al., 2003). Three variants were previously reported to be pathogenic and three variants were predicted to be possible pathogenic. We also identified two novel mutations in the gene SLC31A1 (MIM# 603085), which encodes a protein that is potentially related to lysosomal function but which has not previously been associated with human disease (Table 3).
1). 00RD098
We found one previously reported mutation in the GLB1 (MIM# 611458) gene encoding lysosomal β-galactosidase (GLB1). Defects in GLB1 cause GM1 Gangliosidosis. The patient was heterozygous for an allele encoding NP_000395:p.Arg208Cys , which previously has been reported to be pathogenic (Boustany, et al., 1993). However, we found reduced but considerable β-galactosidase activity in 00RD098 fibroblasts (~13,000 emission/ug protein in 00RD098 vs ~21,000 -35,000 emission/ug protein in controls, Figure 2). p.Arg208Cys was previously shown to result in loss of β-galactosidase activity against synthetic substrates (Boustany, et al., 1993). One possibility is that this patient is compound heterozygous for p.Arg208Cys and another mutation which retains activity against synthetic but not natural substrates. However, we observed no other missense variant to support this hypothesis. Therefore, we conclude that this patient is a carrier for the p.Arg208Cys GLB1 mutation but diagnosis of GM1 gangliosidosis is uncertain.
Figure 2: Enzyme assays.
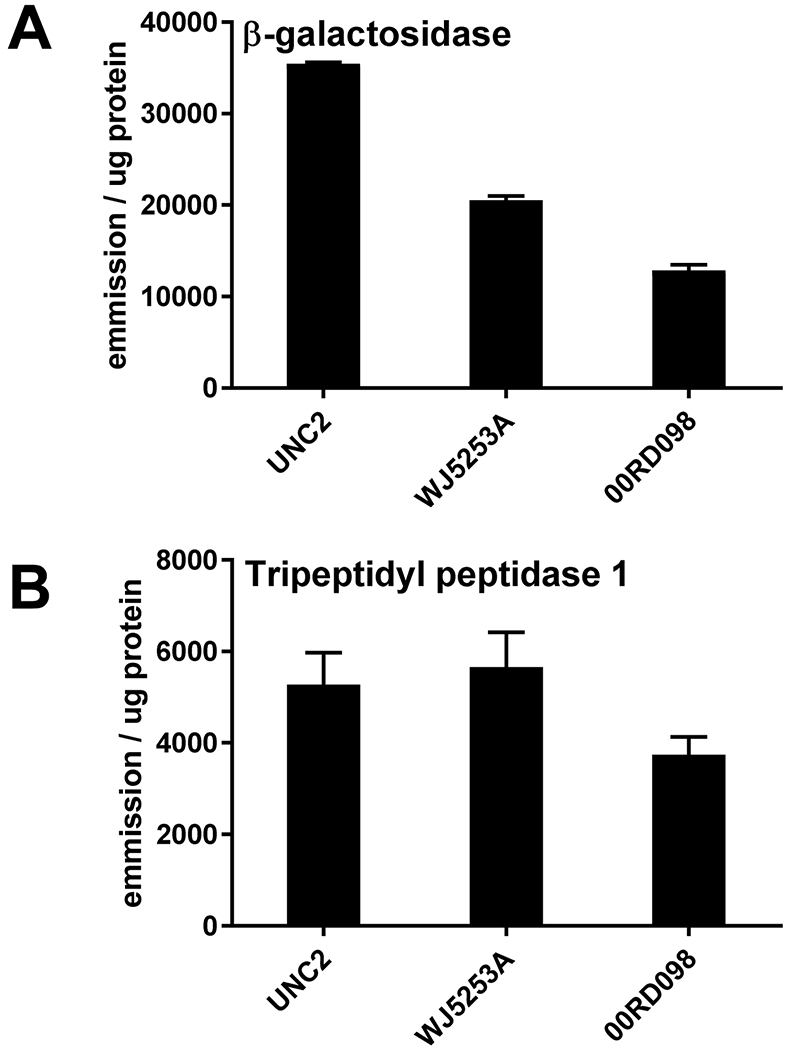
Panel A, β-galactosidase was measured as a candidate defect in 00RD098 and in two controls (UNC2 and WJ5253A). Panel B, Tripeptidyl peptidase 1 (another lysosomal protein) was measured as a positive control. Error bars indicate the standard error of the mean (SEM).
2). 95RD414
We found a known pathogenic variant that results in NP_000534:p.Gln294Lys (Pavlu and Elleder, 1997) in the gene encoding acid sphingomyelinase (SMPD1, MIM# 607608) in 95RD414 and it was confirmed to be homozygous. Note that this allele has been previously referred to as p.Gln292Lys(Pavlu and Elleder, 1997). Acid sphingomyelinase converts sphingomyelin to ceramide and mutations in the SMPD1 result in Niemann-Pick type A and B diseases. Enzyme assay showed the acid sphingomyelinase activity was greatly reduced in this patient, supporting the genetic analysis (Figure 3). Filipin staining and cholesterol esterification assays ruled out NPC disease.
Figure 3: Acid sphingomyelinase assay.
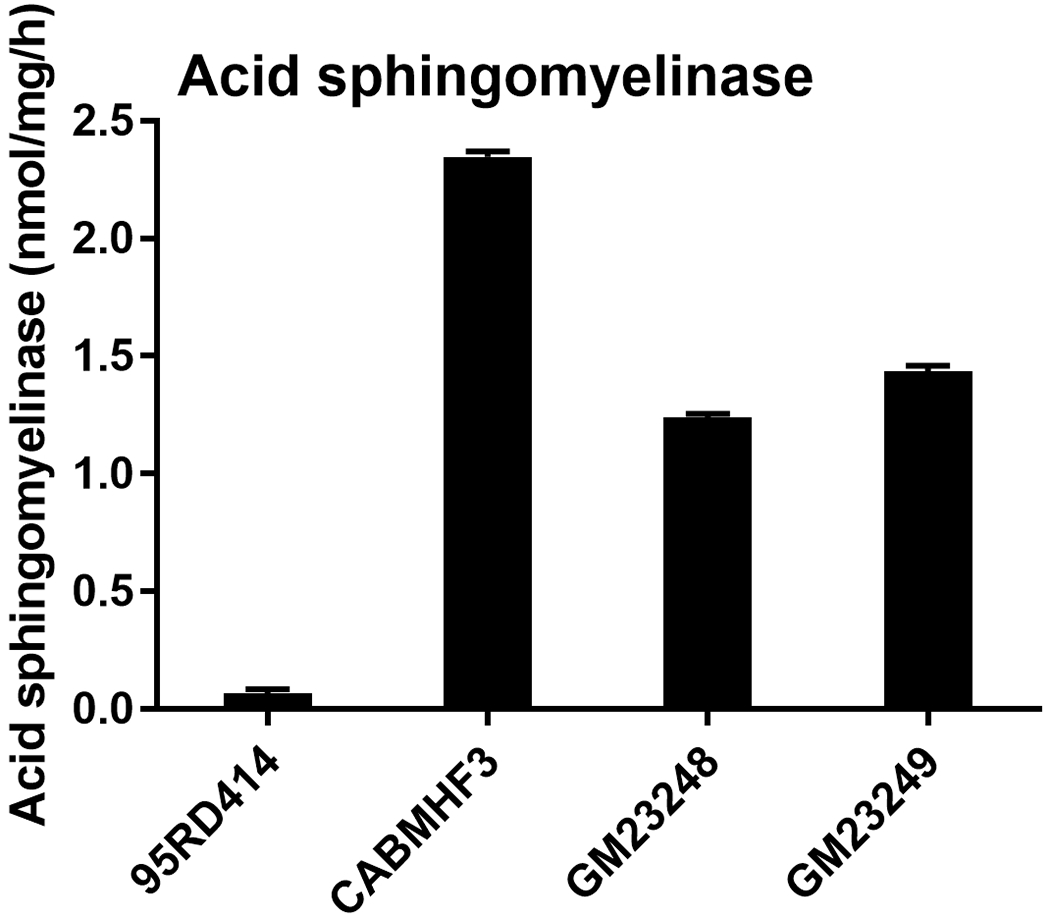
Acid sphingomyelinase was measured as candidate defect in 95RD414 and in three controls (CABMHF3, a case with no detected pathogenic changes in NPC1 and GM23248 and GM23249, fibroblasts from two apparently normal individuals). Cholesterol esterification assays and filipin staining on all four lines were within control values. Error bars indicate SEM.
Of note, this female patient (Supp. Figure S2) was also heterozygous for a previously reported NP_000160:p.Asp313Tyr disease allele (Eng, et al., 1993) in gene GLA (MIM# 300644) that encodes lysosomal alpha-galactosidase (GLA). GLA defects are associated with Fabry disease. While some heterozygotes have clinical manifestations of this X-linked disease, there is some controversy regarding the clinical relevance of p.Asp313Tyr (Niemann, et al., 2013). However, it is possible that this GLA variant contributed to the clinical phenotype, and the patient manifests a blended phenotype based on mutations in both SMPD1 and GLA. The blended phenotype can obscure the original diagnosis in this case. Increased urine globotriaosylceramide level would lend more evidence to the pathogenicity of this mutation.
3). CABMHF1
CABMHF1 was heterozygous for NP_000262:p.Asn222Ser in NPC1 (MIM# 607623), the Niemann-Pick type C disease gene (Park, et al., 2003), and a novel variant in NPC1 resulting in NP_000262:p.Arg389His. The pathogenicity of the p.Asn222Ser variant is unclear – it has been reported to be a polymorphism, with an allele frequency inconsistent with NPC1 but there also exists evidence that it may be associated with late onset forms of disease (Wassif, et al., 2016). p.Arg389His has not previously been reported in Niemann-Pick type C, but it is a very promising candidate as there are two known pathogenic mutations at this position resulting in different amino acid substitutions: p.Arg389Leu (Fancello, et al., 2009) and p.Arg389Cys (Park, et al., 2003). Sequencing of cloned NPC1 gene DNA fragments containing the two variant loci showed that the two mutant variants are located on different copies of chromosome 18 (Supp. Figure S3). Thus, the patient is compound heterozygous for these variants. We also identified a heterozygous variant in the SMPD1 gene: NP_000534:p.Gly492Ser (Table 3). Two patients with mild Niemann–Pick B disease phenotype and no acid sphingomyelinase activity were compound heterozygous for p.Gly492Ser and another SMPD1 mutation (Irun, et al., 2013). In addition, there are known pathogenic mutations very close to it: p.Thr488Ala (Rodriguez-Pascau, et al., 2009), and p.Tyr490Asn (Simonaro, et al., 2002). Similar to 95RD414, this patient could have a disease phenotype that resulted from synergistic heterozygosity of both NPC1 and SMPD1 mutations. Unfortunately, cell stocks from this case were no longer viable at the time of this study and thus we could not conduct functional validation.
4). 82RD265
We found two novel variants in case 82RD265 in the lysosome-functional related gene SLC31A1, which encodes solute carrier family 31 (copper transporters) member 1: nonsense mutation NP_001850:p.Arg90Ter and a non-synonymous NP_001850:p.Val181Leu mutation. SLC31A1 was targeted for analysis based on its dual localization to both the lysosome and to the plasma membrane, and we have reported previously on the variants in this patient (Jadot, et al., 2017). While p.Arg90Ter is a clear null, further functional analysis to test the cellular copper uptake are necessary to demonstrate the effect of these two variants together.
Loss-of-function Variants Outside of LSD Candidate Genes
We only identified potential disease-causing mutations in our lysosomal candidate list in four cases. Several reasons could account for the low identification rate, and one possibility is that these cases were misdiagnosed as LSDs. While LSDs have characteristic clinical phenotypes, many are not unique to this class of disorders. To test the possibility that disease may result from mutations encoding non-lysosomal proteins, we further examined all LoF variants (i.e., stop-gain and splicing variants) in the whole exome (Supp. Data File). In two cases, we identified potential pathogenic variants.
1). 00RD098
While we identified promising candidates in this case (see above), functional validation experiments failed to verify lysosomal defects. However, we identified and validated a heterozygous stop-gain variant NP_001013424:p.Gln66Ter in the Krev interaction trapped protein 1 (KRIT1) gene (MIM# 604214). The variant was detected in one individual as heterozygous among 60,660 sequenced individuals in the ExAC database (http://exac.broadinstitute.org/variant/7-91870373-G-A). Homozygous mouse KRIT1 mutants die prenatally due to cerebral cavernous malformations (Whitehead, et al., 2004). A previous study also reported a human family with this p.Gln66Ter variant (Gianfrancesco, et al., 2007). Family members carrying this variant exhibited a high variability in the penetrance and phenotypes (Gianfrancesco, et al., 2007). Given 00RD098 carries one GLB1 variant and showed reduced β-galactosidase activity, this KRIT1 mutation and associated nervous system angiomas may have contributed to a blend phenotype that leads to the LSD diagnosis.
2). CABMHF3
In case CABMHF3, we identified a homozygous stop-gain variant NP_001156907:p.Gln102Ter in the TBC1 Domain Containing Kinase (TBCK) gene (MIM# 616899). The mutation is not observed in the ExAC database. CABMHF3 was diagnosed as a potential LSD case by the presence of neuronal storage. Interestingly, in a previous study of patients who are homozygous for a 4-bp deletion in TBCK at amino acid 205, the affected siblings (two boys and one girl) presented with profound hypotonia, global developmental delay, and slow motor development with no progress beyond the ability to sit independently (Guerreiro, et al., 2016). They also had epilepsy and similar distinctive facial features, and the two youngest siblings had signs of precocious puberty. The older sibling died at 9 years of age, and the youngest died at 12 years (Guerreiro, et al., 2016). There was no report of analysis of neuronal pathology in these cases so it is unclear whether TBCK defects were associated with neuronal storage, but the clinical presentation appears to be consistent with the diagnosis of LSD. In another recent study of nine unrelated families with LoF mutations in TBCK, the patients presented with intellectual disability and hypotonia, and their suspected diagnoses included LSD (Bhoj, et al., 2016). In addition, TBCK was implicated in the mTOR pathway (Liu, et al., 2013), which regulates lysosomal biogenesis and function (Kinghorn, et al., 2017). Therefore, although subcellular localization analysis (Jadot, et al., 2017) indicates that TBCK does not reside within the lysosome, these studies suggest that TBCK may control some aspects of lysosomal function and could result in phenotypes in CABMHF3.
Discussion
In this study, we explored the feasibility of identifying pathogenic alleles using WES in 14 patients with disease that was suspected to be lysosomal in origin. In an initial targeted approach that focused on proteins that have lysosomal location or function, we identified probable disease-causing mutations in three cases in genes encoding proteins that are known to be associated with lysosomal disease. In another case, we found novel mutations in the gene encoding SLC31A1 – a protein that may have lysosomal function but has not previously been associated with human disease.
Several studies have also applied next generation sequencing technology to identify LSD pathogenic variants, although these studies have focused on lysosomal proteins in their sequencing surveys. For example, one study used a panel of 57 lysosomal genes to identify potential causal mutations in ~40% (26/66) of the cases (Fernandez-Marmiesse, et al., 2014). Another recent study used a gene panel that includes 891 genes involved in autophagy-lysosomal pathways (Di Fruscio, et al., 2015). A diagnosis rate of ~60% (29/48) was reported for patients with the clinical phenotype of neuronal ceroid lipofuscinosis (Di Fruscio, et al., 2015). Because we started from WES, we were able to examine LoF variants outside of the lysosomal gene list. Using this unbiased approach, we identified potentially pathogenic variants in two cases in genes that encode proteins that do not appear to have lysosomal function. These results support the use of WES or whole genome sequencing as a genetic diagnostic method for suspected LSD cases. Under a clinical setting, pedigree analysis would provide further weight to assignment of potential pathogenic alleles.
We only identified potential causal mutations in gene encoding lysosomal proteins in four cases and several reasons may account for this. First, our study was based on the hypothesis that the diseases were caused by monogenic coding variants. If the diseases were caused by non-coding variants (e.g., intronic or promoter variants), WES will be likely to miss the causal variant. Second, although we applied several bioinformatic methods for variant identification, sensitivity is limited by the technology: with variability in exome coverage, we do not have 100% sensitivity for all coding variants. Thirdly, the diagnosis of a likely LSD case based on clinical criteria may not be correct. This may be illustrated by the two cases that appear to have pathogenic variants in genes encoding non-lysosomal proteins, KRIT1 and TBCK. Another limitation of our study is that because we analyzed historical archived cases, we cannot conduct additional phenotyping on the probands or obtain more detailed clinical and family histories. Nevertheless, our results demonstrated the feasibility of using WES to identify potential causal mutations and can potentially benefit LSD diagnosis.
It is worth noting that in several cases we not only identified probable mutations (homozygous or compound heterozygous) in genes encoding lysosomal proteins that are known to be associated with human diseases, but we also find heterozygosity for pathogenic variants in genes encoding other lysosomal proteins. These observations raise the intriguing possibility that clinical presentation in these cases may reflect synergistic effects of multigenic defects, which is possibly why initial diagnosis was complicated. For example, case 95RD414 was homozygous for a known pathogenic allele in SMPD1 (p.Gln294Lys), and this was validated by functional assay, but we also identified a known pathogenic allele in GLA (p.Asp313Tyr), which may produce disease manifestations in this female patient, depending on the pattern of lyonization of this X-linked gene. Case CABMHF1 was compound heterozygous for a possible pathogenic allele in NPC1 (p.Arg389His), as well as another allele of unclear significance (p.Asn222Ser), but was also heterozygous for a known mutation in SMPD1 (p.Gly492Ser). It is possible that the combination of mutations in NPC1 and SMPD1 deficiency manifested in a complex presentation. These cases may be examples of synergistic heterozygosity and blended phenotypes, where disease was resulted from multiple partial defects in one or more metabolic pathways (Li, et al., 2016; Vockley, et al., 2000). Case 00RD098 was heterozygous for a known pathogenic allele in GLB1 and while we did not identify a marked deficiency of this enzyme, it is possible that this complicated the phenotype of an as yet undiscovered mutation in another gene. The concept that haploinsufficiency for a lysosomal protein may influence the phenotype of a genetically distinct defect is not without precedent: for example, glucocerebrosidase defects as a risk factor for Parkinson disease are well established (reviewed in (Aflaki, et al., 2017)).
Conclusion
Using WES data from 14 patients suspected to have LSDs, we identified both known pathogenic variants and novel potential pathogenic variants in five genes in four cases. In addition, in two cases we identified LoF variants in genes that are not thought to be associated with LSDs, although mutations in these genes could have resulted in phenotypes that were diagnosed as LSDs. This study demonstrates that WES can be used to study the genetic bases of LSDs with unknown etiology and has potential as an approach for genetic diagnosis of LSDs. In addition, we identify cases where a confounding clinical phenotype may potentially reflect more than one lysosomal protein defect. This possibility should be borne in mind in apparent cases of lysosomal disease that defy traditional methods of diagnosis.
Supplementary Material
Acknowledgements
We would like to thank the following for generously providing clinical samples for this study: Dr. Otto van Diggelen (Erasmus University Medical Center); Dr. Klaus Harzer (University of Tubingen); Dr. Bruce Gelb (Mount Sinai Medical Center); Dr. Sara Mole (University College London), and; Dr. George Thomas (Kennedy Krieger Institute). We would also like to thank the three anonymous reviewers for their instructive comments.
References
- 1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR. 2015. A global reference for human genetic variation. Nature 526(7571):68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aflaki E, Westbroek W, Sidransky E. 2017. The Complicated Relationship between Gaucher Disease and Parkinsonism: Insights from a Rare Disease. Neuron 93(4):737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagshaw RD, Mahuran DJ, Callahan JW. 2005. A proteomic analysis of lysosomal integral membrane proteins reveals the diverse composition of the organelle. Mol Cell Proteomics 4(2):133–43. [DOI] [PubMed] [Google Scholar]
- Bhoj EJ, Li D, Harr M, Edvardson S, Elpeleg O, Chisholm E, Juusola J, Douglas G, Guillen Sacoto MJ, Siquier-Pernet K, Saadi A, Bole-Feysot C, Nitschke P, Narravula A, Walke M, Horner MB, Day-Salvatore DL, Jayakar P, Vergano SA, Tarnopolsky MA, Hegde M, Colleaux L, Crino P, Hakonarson H. 2016. Mutations in TBCK, Encoding TBC1-Domain-Containing Kinase, Lead to a Recognizable Syndrome of Intellectual Disability and Hypotonia. Am J Hum Genet 98(4):782–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boustany RM, Qian WH, Suzuki K. 1993. Mutations in acid beta-galactosidase cause GM1-gangliosidosis in American patients. Am J Hum Genet 53(4):881–8. [PMC free article] [PubMed] [Google Scholar]
- Chapel A, Kieffer-Jaquinod S, Sagne C, Verdon Q, Ivaldi C, Mellal M, Thirion J, Jadot M, Bruley C, Garin J, Gasnier B, Journet A. 2013. An extended proteome map of the lysosomal membrane reveals novel potential transporters. Mol Cell Proteomics 12(6):1572–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43(5):491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fruscio G, Schulz A, De Cegli R, Savarese M, Mutarelli M, Parenti G, Banfi S, Braulke T, Nigro V, Ballabio A. 2015. Lysoplex: An efficient toolkit to detect DNA sequence variations in the autophagy-lysosomal pathway. Autophagy 11(6):928–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng CM, Resnick-Silverman LA, Niehaus DJ, Astrin KH, Desnick RJ. 1993. Nature and frequency of mutations in the alpha-galactosidase A gene that cause Fabry disease. Am J Hum Genet 53(6):1186–97. [PMC free article] [PubMed] [Google Scholar]
- Fancello T, Dardis A, Rosano C, Tarugi P, Tappino B, Zampieri S, Pinotti E, Corsolini F, Fecarotta S, D’Amico A, Di Rocco M, Uziel G, Calandra S, Bembi B, Filocamo M. 2009. Molecular analysis of NPC1 and NPC2 gene in 34 Niemann-Pick C Italian patients: identification and structural modeling of novel mutations. Neurogenetics 10(3):229–39. [DOI] [PubMed] [Google Scholar]
- Fernandez-Marmiesse A, Morey M, Pineda M, Eiris J, Couce ML, Castro-Gago M, Fraga JM, Lacerda L, Gouveia S, Perez-Poyato MS, Armstrong J, Castineiras D, Cocho JA. 2014. Assessment of a targeted resequencing assay as a support tool in the diagnosis of lysosomal storage disorders. Orphanet J Rare Dis 9:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianfrancesco F, Cannella M, Martino T, Maglione V, Esposito T, Innocenzi G, Vitale E, Liquori CL, Marchuk DA, Squitieri F. 2007. Highly variable penetrance in subjects affected with cavernous cerebral angiomas (CCM) carrying novel CCM1 and CCM2 mutations. Am J Med Genet B Neuropsychiatr Genet 144B(5):691–5. [DOI] [PubMed] [Google Scholar]
- Guerreiro RJ, Brown R, Dian D, de Goede C, Bras J, Mole SE. 2016. Mutation of TBCK causes a rare recessive developmental disorder. Neurol Genet 2(3):e76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT Nucleic Acids Symposium Series 41:95–98. [Google Scholar]
- He X, Chen F, Gatt S, Schuchman EH. 2001. An enzymatic assay for quantifying sphingomyelin in tissues and plasma from humans and mice with Niemann-Pick disease. Anal Biochem 293(2):204–11. [DOI] [PubMed] [Google Scholar]
- Hedges DJ, Walker JA, Callinan PA, Shewale JG, Sinha SK, Batzer MA. 2003. Mobile element-based assay for human gender determination. Anal Biochem 312(1):77–9. [DOI] [PubMed] [Google Scholar]
- Irun P, Mallen M, Dominguez C, Rodriguez-Sureda V, Alvarez-Sala LA, Arslan N, Bermejo N, Guerrero C, Perez de Soto I, Villalon L, Giraldo P, Pocovi M. 2013. Identification of seven novel SMPD1 mutations causing Niemann-Pick disease types A and B. Clin Genet 84(4):356–61. [DOI] [PubMed] [Google Scholar]
- Jadot M, Boonen M, Thirion J, Wang N, Xing J, Zhao C, Tannous A, Qian M, Zheng H, Everett JK, Moore DF, Sleat DE, Lobel P. 2017. Accounting for Protein Subcellular Localization: A Compartmental Map of the Rat Liver Proteome. Mol Cell Proteomics 16(2):194–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinghorn KJ, Asghari AM, Castillo-Quan JI. 2017. The emerging role of autophagic-lysosomal dysfunction in Gaucher disease and Parkinson’s disease. Neural Regen Res 12(3):380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruth HS, Comly ME, Butler JD, Vanier MT, Fink JK, Wenger DA, Patel S, Pentchev PG. 1986. Type C Niemann-Pick disease. Abnormal metabolism of low density lipoprotein in homozygous and heterozygous fibroblasts. J Biol Chem 261(35):16769–74. [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16):2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Salfelder A, Schwab KO, Grunert SC, Velten T, Lutjohann D, Villavicencio-Lorini P, Matysiak-Scholze U, Zabel B, Kottgen A, Lausch E. 2016. Against all odds: blended phenotypes of three single-gene defects. Eur J Hum Genet 24(9):1274–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Yan X, Zhou T. 2013. TBCK influences cell proliferation, cell size and mTOR signaling pathway. PLoS One 8(8):e71349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubke T, Lobel P, Sleat DE. 2009. Proteomics of the lysosome. Biochim Biophys Acta 1793(4):625–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maire I 2001. Is genotype determination useful in predicting the clinical phenotype in lysosomal storage diseases? J Inherit Metab Dis 24Suppl 2:57–61; discussion 45-6. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20(9):1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meikle PJ, Hopwood JJ, Clague AE, Carey WF. 1999. Prevalence of lysosomal storage disorders. JAMA 281(3):249–54. [DOI] [PubMed] [Google Scholar]
- Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, Shendure J, Bamshad MJ. 2010. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet 42(1):30–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemann M, Rolfs A, Giese A, Mascher H, Breunig F, Ertl G, Wanner C, Weidemann F. 2013. Lyso-Gb3 Indicates that the Alpha-Galactosidase A Mutation D313Y is not Clinically Relevant for Fabry Disease. JIMD Rep 7:99–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, Ballabio A. 2011. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet 20(19):3852–66. [DOI] [PubMed] [Google Scholar]
- Park WD, O’Brien JF, Lundquist PA, Kraft DL, Vockley CW, Karnes PS, Patterson MC, Snow K. 2003. Identification of 58 novel mutations in Niemann-Pick disease type C: correlation with biochemical phenotype and importance of PTC1-like domains in NPC1. Hum Mutat 22(4):313–25. [DOI] [PubMed] [Google Scholar]
- Pavlu H, Elleder M. 1997. Two novel mutations in patients with atypical phenotypes of acid sphingomyelinase deficiency. J Inherit Metab Dis 20(4):615–6. [DOI] [PubMed] [Google Scholar]
- Pentchev PG, Kruth HS, Comly ME, Butler JD, Vanier MT, Wenger DA, Patel S. 1986. Type C Niemann-Pick disease. A parallel loss of regulatory responses in both the uptake and esterification of low density lipoprotein-derived cholesterol in cultured fibroblasts. J Biol Chem 261(35):16775–80. [PubMed] [Google Scholar]
- Rodriguez-Pascau L, Gort L, Schuchman EH, Vilageliu L, Grinberg D, Chabas A. 2009. Identification and characterization of SMPD1 mutations causing Niemann-Pick types A and B in Spanish patients. Hum Mutat 30(7):1117–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson S, Green A, Preece MA, Burton H. 2006. The incidence of inherited metabolic disorders in the West Midlands, UK. Arch Dis Child 91(11):896–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder B, Wrocklage C, Pan C, Jager R, Kosters B, Schafer H, Elsasser HP, Mann M, Hasilik A. 2007. Integral and associated lysosomal membrane proteins. Traffic 8(12):1676–86. [DOI] [PubMed] [Google Scholar]
- Scriver CR. 2001. The metabolic & molecular bases of inherited disease. New York: McGraw-Hill. [Google Scholar]
- Segatori L 2014. Impairment of homeostasis in lysosomal storage disorders. IUBMB Life 66(7):472–7. [DOI] [PubMed] [Google Scholar]
- Simonaro CM, Desnick RJ, McGovern MM, Wasserstein MP, Schuchman EH. 2002. The demographics and distribution of type B Niemann-Pick disease: novel mutations lead to new genotype/phenotype correlations. Am J Hum Genet 71(6):1413–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleat DE, Della Valle MC, Zheng H, Moore DF, Lobel P. 2008. The mannose 6-phosphate glycoprotein proteome. J Proteome Res 7(7):3010–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleat DE, Ding L, Wang S, Zhao C, Wang Y, Xin W, Zheng H, Moore DF, Sims KB, Lobel P. 2009. Mass spectrometry-based protein profiling to determine the cause of lysosomal storage diseases of unknown etiology. Mol Cell Proteomics 8(7):1708–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleat DE, Gedvilaite E, Zhang Y, Lobel P, Xing J. 2016. Analysis of large-scale whole exome sequencing data to determine the prevalence of genetically-distinct forms of neuronal ceroid lipofuscinosis. Gene 593(2):284–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleat DE, Jadot M, Lobel P. 2007. Lysosomal proteomics and disease. Proteomics Clin Appl 1(9):1134–46. [DOI] [PubMed] [Google Scholar]
- Sleat DE, Sohar I, Lackland H, Majercak J, Lobel P. 1996. Rat brain contains high levels of mannose-6-phosphorylated glycoproteins including lysosomal enzymes and palmitoyl-protein thioesterase, an enzyme implicated in infantile neuronal lipofuscinosis. J Biol Chem 271(32):19191–8. [DOI] [PubMed] [Google Scholar]
- Sleat DE, Sun P, Wiseman JA, Huang L, El-Banna M, Zheng H, Moore DF, Lobel P. 2013. Extending the mannose 6-phosphate glycoproteome by high resolution/accuracy mass spectrometry analysis of control and acid phosphatase 5-deficient mice. Mol Cell Proteomics 12(7):1806–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohar I, Lin L, Lobel P. 2000. Enzyme-based diagnosis of classical late infantile neuronal ceroid lipofuscinosis: comparison of tripeptidyl peptidase I and pepstatin-insensitive protease assays. Clin Chem 46(7):1005–8. [PubMed] [Google Scholar]
- Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S, Krawczak M, Cooper DN. 2003. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat 21(6):577–81. [DOI] [PubMed] [Google Scholar]
- Steve Rozen HJS. 1998. Primer3. [Google Scholar]
- Vockley J, Rinaldo P, Bennett MJ, Matern D, Vladutiu GD. 2000. Synergistic heterozygosity: disease resulting from multiple partial defects in one or more metabolic pathways. Mol Genet Metab 71(1-2):10–8. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. 2010. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38(16):e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassif CA, Cross JL, Iben J, Sanchez-Pulido L, Cougnoux A, Platt FM, Ory DS, Ponting CP, Bailey-Wilson JE, Biesecker LG, Porter FD. 2016. High incidence of unrecognized visceral/neurological late-onset Niemann-Pick disease, type C1, predicted by analysis of massively parallel sequencing data sets. Genet Med 18(1):41–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead KJ, Plummer NW, Adams JA, Marchuk DA, Li DY. 2004. Ccm1 is required for arterial morphogenesis: implications for the etiology of human cavernous malformations. Development 131(6):1437–48. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.