

Abstract
Respiratory syncytial virus (RSV) is a leading cause of lower respiratory tract infection in young children, and specific treatment for RSV infections remains unavailable. We herein reported a series of substituted N-(4-amino-2-chlorophenyl)-5-chloro-2-hydroxybenzamide analogues as potent RSV inhibitors. Among them, six low cytotoxic compounds (11, 12, 15, 22, 26, and 28) have been identified and selected to study associated inhibitory mechanisms. All these compounds suppressed not only the viral replication but also RSV-induced IRF3 and NF-κB activation and associated production of cytokines/chemokines. The two most potent compounds (15 and 22) were selected for further molecular mechanism studies associated with their suppression effect on RSV-activated IRF3 and NF-κB. These two molecules decreased RSV-induced IRF3 phosphorylation at serine 396 and p65 phosphorylation at serine 536 at both early and late infection phases. In addition, compound 22 also inhibited RSV-induced p65 phosphorylation at serine 276 at the late phase of RSV infection.
Keywords: Salicylamide derivatives, RSV, inflammation, RSV replication
Graphical Abstract

1. Introduction
Respiratory syncytial virus (RSV) is an enveloped non-segmented negative-sense RNA virus with two major subgroups A and B, belonging to the Orthopneumovirus genus, the Pneumoviridae family.1 It is the most significant cause of lower respiratory tract infection (LRTI) in the pediatric population.2 Almost all children are infected by RSV by the age of two and re-infection may occur through life, demonstrating incomplete immunity.3, 4 It is currently estimated that 33.1 million episodes of RSV-associated LRTI lead to about 3.2 million hospital admissions and 59,600 in-hospital deaths in children younger than 5 years old globally. In addition, in-hospital deaths, due to RSV-caused LRTI, contribute to about 45% of hospital admitted patients younger than 6 months old, demonstrating a considerable burden of RSV infection on health-care services.5, 6 RSV infection is also a common cause of upper respiratory tract infection (URTI) associated with significant outpatient visits. In high-risk populations, such as premature infants, the elderly, immunocompromised individuals, and patients with chronic medical conditions, RSV infection is also associated with an increased chance of morbidity and mortality.7–10 Moreover, RSV infection has been also associated with long-term sequelae such as wheezing and asthma in children.11–13
Despite the significant clinical impact, there currently remains no effective and specific treatment option for RSV infection.2 Palivizumab, a monoclonal antibody (mAb) targeting fusion protein (F protein) of RSV, was licensed in 1998 for prophylactic use in high-risk infants. However, it is not very cost-effective, with a limited application to high-risk infants for their first RSV season.14, 15 Ribavirin is the only approved antiviral therapy as an inhaled formulation for RSV infection, but its use in the clinic is restricted due to the very limited efficacy and significant safety concerns.16 Currently, anti-RSV drug discovery projects have mainly targeted the F protein for viral entry, the nucleoprotein (N), and RSV RNA-dependent RNA polymerase (RdRp) complex.2 In the last two decades, numerous highly potent and structurally different RSV fusion inhibitors have been reported, and some of them have successfully progressed to clinical development exemplifying compounds 1–4 (Fig. 1).2, 17–25 Presatovir (2) is one of the most advanced candidates which is orally available and reduced viral load and disease severity in phase II clinical trial, demonstrating proof of concept for fusion inhibitors in RSV infection treatment.19, 26 Replication inhibitors occupy a key position in anti-RSV drug discovery and recent efforts in developing new assays for screening have enabled the identification of a series of RdRp inhibitors with new chemotypes.27–35 Among them, nucleoside polymerase inhibitor 5 (Lumicitabine) and non-nucleoside polymerase inhibitor 6 (PC786) have progressed into clinical development.2, 28 In addition, benzodiazepine 7 was reported as potent RSV inhibitors by targeting the N protein.36, 37 While significant efforts and progress have been made in anti-RSV drug discovery, it seems that there is more, in addition to the control of viral replication and entry, to be considered for the anti-RSV therapeutic development, as accumulating data support that both direct damages from viral replication and the host immune-inflammatory response contribute to RSV-induced respiratory disease although their relative weight remains controversial.38
Fig. 1.

The representative RSV inhibitors under development.
While initial wave of innate cytokines/chemokines in response to RSV infection is essential for controlling RSV replication, accumulating evidence also supports that the host inflammatory responses to RSV infection can be too strong and long-lasting after the initial wave, leading to profound negative consequences to the host.39, 40 Especially for the infected infants, the small diameter of bronchioles makes them particularly susceptible to obstruction by edema, secretions, and exfoliated cells arising from immune-inflammatory response.38, 41, 42 Lung autopsy studies for infants who died of RSV infection revealed that in addition to the detection of RSV antigen in exfoliated alveolar cells, proinflammatory neutrophils and macrophages are abundantly present throughout the airway lumen, supporting the acute inflammatory sequelae fatal resulting from uncontrolled inflammatory disease.43, 44 Other human pathology studies, from the findings from the airways of mechanically ventilated infants and various infection models, also support significant inflammatory pathology after the infection.45, 46 In some cases, which demonstrated an effective suppression on RSV replication by antiviral treatment, the clinical impact of antiviral treatment was minimal.47, 48 Therefore, it is possible that one or more components of the inflammatory response, which is independent of ongoing virus replication, can be initiated by the viral infection and amplified, leading to an uncontrollable condition by antiviral treatment alone. Thus, developing a therapeutic strategy that can suppress both virus replication and RSV-induced inappropriate inflammatory response may serve as a unique and promising avenue for RSV infection treatment. Niclosamide, an FDA-approved anthelminthic drug, displayed broad-spectrum antiviral activities against various viruses such as coronavirus, flavivirus and Ebola virus.49, 50 Our group has been devoting to niclosamide drug repurposing and new analog optimization studies with a long-term interest51–55 and previously identified a series of N-(4-amino-2-chlorophenyl)-5-chloro-2-hydroxybenzamide derivatives as potent inhibitors of human adenovirus infection.56 Herein, we reported these novel chemotype analogues as promising candidates against not only RSV replication but also RSV infection-associated inflammatory responses.
2. Results and discussion
2.1. Chemistry
The analogues were developed as outlined in Scheme 1. Briefly, reduction of the nitro group of niclosamide 8 with zinc dust provided the amine analog 9, which was then readily converted to compounds 10–13, 16–23, 26, and 29 via acetylation followed by hydrolysis or reductive amination with responding aldehydes or ketones. Boc-deprotection of 23 under acidic conditions afforded compound 24. Alcohol analogue 25 was accessed from THP ether 29 via acidic depyranylation. Direct hydroamination of niclosamide with 2-methyl-1-pentene or 1-methyl-1-cyclohexene afforded compounds 14 and 15, respectively.57 Substitution of 2-chloro-4-fluoro-1-nitrobenzene with pyrrolidine or morpholine gave the intermediates 31 and 32, respectively, which were then reduced to provide amino derivatives 33 and 34. Condensation of amines 33, 34 with 5-chlorosalicylic acid followed by hydrolysis or 2-methoxy-5-methylbenzoic acid followed by demethylation afforded the final products 27 and 28, respectively.
Scheme 1.

Synthesis of Compounds 9–28. Reagents and conditions: (a) Zn, NH4Cl, H2O, MeOH, 0 °C to r.t, 16 h, 87–99%; (b) i. AcCl, Et3N, acetone, 50 °C, 2 h, 98%; ii. LiOH, H2O, MeOH, r.t, 1 h, 97%; (c) aldehyde or ketone, NaBH3CN, AcOH, MeOH, 0 °C to r.t, 12 h, 7–84%; (d) 1 M HCl/Et2O, MeOH, r.t, 12 h, 96%; (e) TsOH, MeOH, r.t, 12 h, 22% in two steps; (f) i. 2-methyl-1-pentene or 1-methyl-1-cyclohexene, Fe(acac)3, PhSiH3, EtOH, 60 °C, 1 h; ii. Zn, 2 M HCl (aq.), 60 °C, 1 h, 35–44%; (g) pyrrolidine or morpholine, K2CO3, DMF, 100 °C, 1 h, 88–98%; (h) i. 5-chlorosalicylic acid, EDCI, DMAP, DCM, 0 °C to r.t, overnight; ii. NaOH, H2O, MeOH, r.t, 1 h, 23% in two steps; (j) i. 5-chloro-2-methoxybenzoic acid, EDCI, DMAP, DCM, r.t, 2 h; ii. BBr3, DCM, −78 °C to r.t, 12 h, 83% in two steps.
2.2. In Vitro Evaluation of RSV Replication Inhibition
Twenty diversified substituted N-(4-amino-2-chlorophenyl)-5-chloro-2-hydroxybenzamide analogues were selected and screened for their potential inhibitory activities against RSV. Briefly, human A549 epithelial cells were infected with RSV at a multiplicity of infection (MOI) of 1 for 2 hours, and then the medium containing virus was removed and the cells were treated with the tested compound at 10 μM for 15 h. The production of progeny virus was detected by titration assays, as we previously described.58, 59 As shown in Table 1, cells treated with most substituted N-(4-amino-2-chlorophenyl)-5-chloro-2-hydroxybenzamide derivatives at 10 μM produced more than a log less infectious particles than vehicle-treated cells. Meanwhile, these compounds did not affect cell viability at 10 μM (Fig. 2), excluding the possibility of induced cell toxicity that led to a less favored environment for RSV replication. Although niclosamide displayed 1.47 log reduction in virus titer, it also showed high cytotoxicity with about 24% cell death at 10 μM. Consistent with our previous results observed for HAdV infection,56 amino substitution (9) and acetamide substitution (10) led to a complete loss of potency. However, N-alkylated derivatives 11–17 exhibited their capability in suppressing RSV replication by more than a log. Among these derivatives, Compounds 11 with n-propyl, 12 with isopropyl, and 15 with 1methylcyclohexyl possessed potent anti-RSV activities with about 1.8 log reduction in virus titer meanwhile displaying low cytotoxicity (CC50 = 69.7 μM, 82.7 μM, and 86.5 μM, respectively).
Table 1.
Antiviral Activity and Cytotoxicity of Compounds 8–28

| ||||
|---|---|---|---|---|
| Compd | R1 | R2 | Virus titer log reductiona | CC50 (μM)b |
| Niclosamide | 1.47 ± 0.04 | NTC | ||
| 9 | H | H | −0.01 ± 0.20 | NT |
| 10 | H |

|
0.09 ± 0.08 | NT |
| 11 | H |

|
1.82 ± 0.10 | 69.7 ± 0.9 |
| 12 | H |

|
1.88 ± 0.43 | 82.7 ± 1.6 |
| 13 | H |
|
1.16 ± 0.08 | 18.6 ± 0.5 |
| 14 | H |

|
1.81 ± 0.22 | 23.7 ± 0.8 |
| 15 | H |

|
1.82 ± 0.14 | 86.5 ± 3.7 |
| 16 | H |

|
1.42 ± 0.21 | 141.4 ± 3.9 |
| 17 | H |

|
1.44 ± 0.23 | NT |
| 18 | H |

|
1.53 ± 0.32 | 81.3 ± 5.6 |
| 19 | H |

|
0.54 ± 0.09 | NT |
| 20 | H |

|
1.54 ± 0.31 | NT |
| 21 | H |
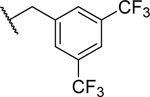
|
2.59 ± 0.15 | 22.9 ± 0.3 |
| 22 | H |

|
2.13 ± 0.23 | 76.3 ± 2.3 |
| 23 | H |

|
1.53 ± 0.37 | NT |
| 24 | H |

|
−0.07 ± 0.37 | NT |
| 25 | H |
|
−0.16 ± 0.88 | NT |
| 26 |

|

|
1.91 ± 0.33 | 322.6 ± 45.8 |
| 27 |
|
1.51 ± 0.08 | NT | |
| 28 |
|
1.74 ± 0.33 | 89.9 ± 4.7 | |
Virus titer log reduction was calculated by subtracting the log10 means of the production of progeny virus in the presence of 10 μM compounds from the log10 means of the production of progeny virus in the vehicle-treated cells.
Cytotoxic concentration 50% (CC50).
NT: not tested.
Fig. 2.

Cytotoxicity assay of compounds. A549 cells were treated with the tested compounds at 10 μM for 15 h. Cell damage was determined by the LDH release assay. DMSO was used as a vehicle control. P < 0.05 (*) and P < 0.0001 (****) relative to the DMSO group.
Long length alkyl groups seemed not to be favorable as shown by the activity of compound 13. Derivatives 18–22 were selected to investigate the effect of various aryl groups. Compounds 18 with benzyl and 20 with 4-flurobenzyl exhibited similar potency with about 1.5 log reduction in virus titer, while 3,5-bis(trifluoromethyl) substituted derivative 21 displayed the most potent antiRSV activity, reducing viral titer by 2.59 log, compared with vehicle treatment. However, it also showed moderate cytotoxicity (CC50 = 22.9 μM). Unexpectedly, 3-pyridyl substitution (19) led to a significant loss of potency. Notably, 2-furanyl substitution (22) reduced viral titer by 2.13 log. This compound also showed low cytotoxicity (CC50 = 76.3 μM). We also found that polar substitutions were not tolerated, as N-Boc protected derivative 23 was effective in viral replication suppression, while its corresponding amine 24 and hydroxyalkyl analogue 25 were completely incapable of inhibiting replication at 10 μM. Intriguingly, N,N-dialkylated derivatives 26–28 all showed potent inhibitory activities against RSV with reduction in virus titer ranging from 1.51 to 1.91 log, while compounds 26 and 28 also displayed very low cytotoxicity (CC50 = 322.6 μM and 89.9 μM, respectively).
2.3. Modulation of RSV-activated IRF and NF-κB by derivatives
Cell intrinsic RIG-I-like receptors (RLRs) can detect RSV double-stranded (ds) RNA and stimulate a signaling cascade, leading to the activation of interferon regulatory factor (IRF) and NF-κB. The activated IRFs and NF-κB would translocate to nuclear and initiate the transcription of numerous genes involved in the immune/inflammatory responses to viral infections. Moreover, Toll-like receptors (TLRs) on the cell membrane can also sense RSV ssRNA, dsRNA, and viral envelope protein, resulting in the activation of IRFs and NF-κB.60 To study the effect of compounds on RSV-induced IRF and NF-κB, six potent compounds (11, 12, 15, 22, 26 and 28) with low cytotoxicity and one negative compound (19) were selected. A549 cells were transfected with luciferase reporter plasmids harboring the binding sites of transcriptional factors IRF3 or NF-κB. After 20 h, cells were infected with RSV at a MOI of 1, and the viruses were removed 2 h later, followed by treatment with 10 μM compounds for 15 h. As shown in Fig. 3A and 3B, five compounds (11, 12, 22, 26 and 28) dramatically suppressed both RSV-induced IRF3 and NF-κB activation. However, compound 15 only suppressed RSV-activated IRF3 with no effect on NF-κB. Compound 19 as a negative control, which did not affect the production of progeny RSV, showed no effect on RSV-induced IRF3 and NF-κB activities.
Fig. 3.

The effect of compounds on the activation of IRF and NF-κB by RSV. (A-B) A549 cells were transiently transfected with IRF or NF-κB luciferase reporter plasmids. At 20 h post-transfection, the cells were infected with RSV (MOI = 1), and then treated with 10 μM compounds. At 15 h posttreatment, the cells were harvested to measure luciferase expression to assess the activation of IRF-3 (A) or NF-κB (B). (C) A549 cells were infected with RSV (MOI = 5), and then treated with 10 μM compounds. At 4 h posttreatment, RSV genome copies in the cells were detected by RT-qPCR. (D-E) The experiments were carried out similarly to what is described for A and B, except that the cells were infected with RSV (MOI = 5), and the activation of IRF-3 (C) and NF-κB (D) were investigated at 4 h posttreatment. P < 0.05 (*), P < 0.01 (**), P < 0.001(***) and P < 0.0001 (****) relative to the DMSO group.
As discussed, besides the replication-dependent inflammation, there are also inflammatory components which are independent of the replication. To investigate whether there is replication-independent inflammation and whether the compounds suppress such inflammation, we assessed the reporter activity at 4 h posttreatment when the significant replication has not started yet. As shown in Fig. 3C, RSV genome copies were comparable between each group at 4 h posttreatment. Interestingly, all six active compounds (11, 12, 15, 22, 26 and 28) significantly inhibited IRF3 and NF-κB activation, indicating that they also could suppress RSV replication independent signaling (Figs. 3D and 3E). The negative control (19), as expected, displayed no suppression on either RSV-induced IRF3 or NF-κB activity. Intriguingly, compound 15, which failed to inhibit NF-κB activation at 15 h post-infection (p.i., Fig. 3B.), could suppress RSV-induced NF-κB activation at the early phase of RSV infection (Fig. 3E). Overall, these data showed the compounds’ suppression on viral replication-independent inflammatory responses. In addition, the cells were treated with the compounds after RSV entry, suggesting that these compounds inhibit RSV-induced IRF3 and NF-κB activity independent from viral entry.
2.4. The impact of derivatives on RSV-induced inflammatory response
The impact of compounds on the early inflammation was also investigated in small airway epithelial cells (SAECs), the primary human airway cells, by investigating their effect on RSV-induced cytokines/chemokines in SAECs. The SAECs were infected by RSV at MOI = 5. At 2 h post infection, viruses were removed, followed by the treatment with 10 μM compounds individually. At 4 h posttreatment, the cytokines/chemokines in the supernatant were detected. As shown in Fig. 4A, RSV-induced MCP-1, IP-10, and RANTES were significantly decreased by these six active compounds. Regarding the effect of compounds on RSV-induced MIP-1β, IL-6 and TNF-α, compounds 11 and 15 were capable to inhibit MIP-1β induction, only compound 15 could inhibit IL-6 induction, and compound 26 and 15 were able to inhibit TNF-α. Our negative control 19 did not show any inhibitory effect on RSV-induced cytokines/chemokines. Given the result showing that RSV genome copies were comparable among groups (Fig. 4B), the study supported that the active compounds could be a regulator of induced early inflammation which was not RSV replication dependent.
Fig. 4.

The impact of compounds on the cytokine/chemokine induction in SAECs by RSV. (A) SAECs were infected with RSV (MOI = 5), and then treated with 10 μM compounds. At 4 h posttreatment, the supernatant was collected, and the level of cytokines/chemokines was measured by Bio-Plex. (B) RSV genomic copies were also measured by RT-qPCR for samples from panels A. P < 0.05 (*), P < 0.01 (**), P < 0.001(***), or P < 0.0001 (****) shown on the bars is relative to the DMSO group. A P-value of <0.05 was considered significant. Mean ± SE is shown.
The nature of inflammation seen in severe bronchiolitis indicates that enhanced disease in infants is associated with an imbalanced or dysregulated immune response to viral infection.43, 61 MCP-1, MIP-1β, IP-10, and RANTES are important proinflammatory chemokines and involved in the up-regulation of inflammatory response. The increase of nasal MCP-1 and MIP-1β in RSV patients is positively associated with severity.62 The administration of IP-10 in RSV infected mice enhanced the severity of pneumonia.63 RSV‐induced RANTES contributes to the exacerbation of allergic airway inflammation. The selected active compounds significantly suppressed the induction of these four proinflammatory chemokines, implying their potential roles in easing RSV-induced inflammation. TNF-α and IL-6 are well known proinflammatory cytokines. TNF-a levels are highest during the acute phase of infection and then decline during recovery. The association of IL-6 with RSV severity is elusive and controversial. Some groups reported high IL-6 nasopharyngeal concentrations are associated with the severity of RSV infection.64 In contrast, in a cohort of children with RSV bronchiolitis, higher nasal IL-6 was associated with a shorter requirement for supplemental oxygen.65 It was also reported that IL-6 in BAL fluid of preterm infants with RSV bronchiolitis was lower than infants born at term and the low IL-6 may reflect the prolonged clinical course.66 Although all six active compounds can suppress MCP-1, IP-10 and RANTES, they exhibit different impacts on MIP-1β, IL-6 and TNF-α induction. Together with the results showing different impacts of compounds on the activation of IRF-3 and NF-κB, these data suggest that the compounds likely employ substantially different antiviral and anti-inflammatory strategies.
2.5. The impact of compounds 22 and 15 on RSV replication and RSV-activated IRF and NF-κB pathway
As shown in Fig. 4, compound 15 inhibited the induction of all tested immune mediators, while the other five active compounds showed inhibitory effects on several tested cytokines/chemokines. Compounds 12, 22, and 28 displayed no suppression on MIP-1α, Il-6, and TNF-α, while compound 22 indeed enhanced RSV-induced IL-6. As compounds 15 and 22 displayed intriguing effects on RSV-induced cytokines/chemokines in SAECs, we further explored their effect on RSV replication as well as IRF3 and NF-κB pathways. Various doses of compounds 15 or 22 were used to treat RSV-infected cells for 15 h, and then the production of infectious progeny virus was detected. As shown in Fig. 5A and 5B, compounds 15 at 2.5 μM and 22 at 7.5 μM still led to a 1.57 and 1.22 log reduction in virus titer, respectively. To explore the impact of compounds on viral RNA replication at different times, the RSV genome copies in the infected cells were detected at 6 h, 15 h and 24 h posttreatment. JNJ-53718678, a fusion inhibitor under clinical development, was employed as a control.22 The viral genome copies in each group were comparable at 6 h posttreatment, and compounds 15 and 22 significantly decreased viral genome copies at 15 h and 24 h posttreatment (Fig. 5C). JNJ-53718678 did not affect viral genome copies at 15 h posttreatment and slightly decreased viral genome copies at 24 h posttreatment, possibly due to the entry inhibition of progeny virus. Moreover, compounds 15 and 22 decreased the expressions of viral phosphoprotein (P), matrix (M) and N proteins, and fusion protein (F) (Fig. 5D). These results indicated that compounds 15 and 22 significantly suppressed the generation of progeny viruses, viral RNA replication and viral protein expression.
Fig. 5.

Dose- and time-dependent suppression of compounds 15 and 22 in RSV replication. (A) and (B) A549 cells were infected with RSV (MOI = 1) and treated with compound 15 (A) or compound 22 (B) at various doses as indicated for 15 h. Total viruses were harvested, and titers determined. (C) A549 cells were infected with RSV (MOI = 1), and treated with 10 μM compound 15, compound 22 or JNJ-53718678 for 6, 15 or 24 h. RSV genomic copies were measured by RTq-PCR. (D) Viral proteins were detected by western blot using anti-RSV (upper panel) or anti-F (middle panel) for 15 h post-treatment samples from panels C. The loading control β-actin was also investigated. P < 0.05 (*) and P < 0.01 (**) relative to the DMSO group.
NF-κB is a dimeric transcription factor composed of various combinations of the Rel family of proteins, including RelA/p65, RelB, c-Rel, p50/p105, and p52/p100. Owing to its abundance in most cell types and the presence of a strong transactivation domain, p65 is thought to be responsible for most transcriptional activity of NF-κB.67 Phosphorylation and nuclear translocation are critical to IRF3 and p65 transcriptional activity.68 To investigate whether compounds 15 and/or 22 affected RSV-induced IRF3 or/and NF-κB activity via influencing the phosphorylation and nuclear translocation of IRF3 and p65, we prepared the nucleus fractions for cells after the treatment of compound 15 or 22. Consistent with comparable RSV replication, determined by the genome copies shown Fig. 4B, we found RSV protein expression was also comparable in control cells and cells treated with compound 15 or 22, at early time point of infection (4 h p.i., Fig. 6A). We also found that compounds 15 and 22 significantly decreased RSV-induced IRF3 phosphorylation at serine 396 (Ser396) and nuclear translocation of IRF3 at 4 and 15 h posttreatment, shown in Figs. 6B and 6D, respectively. The suppression of Ser396 phosphorylation observed for compounds 15 and 22 was consistent with their inhibitory effects on RSV-induced IRF3 activity at the early and late viral infection (Figs. 3D and 3A). Inducible phosphorylation on Ser276 and Ser536 can regulate p65 transcriptional activity without modification of nuclear translocation or DNA-binding activity.69 Thus, we investigated the phosphorylation levels of Ser276 and Ser536 of p65 in treated cells. At 4 h posttreatment, RSV-induced Ser536 phosphorylation and nuclear translocation of p65 were suppressed by both compounds, while RSV-induced Ser276 phosphorylation was not affected by either compound (Figs. 6C and 6E). The suppression of Ser536 phosphorylation observed for compounds 15 and 22 was consistent with their inhibitory effects on RSV-induced NF-κB activity at the early viral infection (Fig. 3E), suggesting both compounds suppressed the early activation of NF-κB by inhibiting Ser536 phosphorylation. At 15 h posttreatment, compound 22 robustly suppressed RSV-induced Ser276 and Ser536 phosphorylation and nuclear translocation of p65 (Fig. 6E). In contrast, compound 15 only impaired RSV-induced Ser536 phosphorylation of p65, and decreased nuclear translocation of p65, but with no effect on Ser276 phosphorylation of p65 (Fig. 6E). Together with the findings on the distinct impact of compounds 15 and 22 on the later activation of NF-κB (Fig. 3B), the result of Fig. 6E suggested that, at the late infection phase, compound 22 suppressed NF-κB activation by impairing the phosphorylation of Ser276 and Ser536, while compound 15 targeted only Ser536 phosphorylation. We also found that compound 22 or 15 had undetectable or minimal impact on p65 or IRF-3 total protein levels in the cell (Fig. 6F).
Fig. 6.

The impact of compounds 15 and 22 on RSV-induced IRF and NF-κB modifications. (A)-(C) A549 cells were infected with or without RSV (MOI = 5), and then treated with 10 μM compound 15 or 22 for 4 h. (A) Viral proteins were detected by western blot. (B) and (C) Nuclear extracts of cells were prepared and subjected to Western blotting. (D and E) A549 cells were infected with or without RSV (MOI = 1), and then treated with 10 μM compound 15 or 22 for 15 h. Nuclear extracts of cells were prepared and subjected to Western blotting. (F) The total IRF-3 and p65 from the cell lysates at 4 h (upper panel) or 15 h (lower panel) of infection were also investigated. In all blots, β-actin was used as loading controls.
In this study, we also investigated the effects of compounds 22 and 15 on Poly I:C and TNF-α induced NF-κB and IRF-3 activation to confirm the suppression role of both compounds in viral replication-independent cellular signaling. We found that Poly I:C-activated IRF-3 was suppressed by both compounds 22 and 15 (Fig. 7A). However, Poly I:C-induced NF-κB activation was only impacted by compound 22, not 15 (Fig. 7B). We also used TNF-α to activate NF-κB, which again was not impacted by compound 15 (Fig. 7C). These results were consistent with the effects of compounds on RSV-induced NF-κB and IRF-3 activation at late infection phases and meanwhile demonstrated the suppression role of both compounds on viral replication-independent inflammatory response.
Fig. 7.

The impact of compounds 15 and 22 on Poly I:C and TNF-α induced IRF-3 and/or NF-κB activation. HEK-293 cells were transiently transfected with IRF-3 (A) or NF-κB (B and C) luciferase reporter plasmids. At 15 h post-transfection, the cells were transfected with Poly I:C (A and B) or treated with 10 ng/ml TNF-α (C). At 4 h post-treatment, the luciferase activity was investigated. P < 0.01 (**) and P < 0.001(***) relative to the DMSO group.
The overall suppressive role of compound 22 in p65 and IRF-3 could not explain why the IL-6 induction was enhanced by compound 22 at the early time point post-infection. Since many inflammatory/immune mediators are regulated by other transcription factors, such as C/EBP and AP-1, in addition to p65 and IRF-3, compound 22 was possibly unable to suppress the activation of other transcription factors controlling the IL-6 induction. Nevertheless, our results identified several candidates that can suppress inflammatory responses to RSV at various levels.
3. Conclusion
In summary, a series of substituted N-(4-amino-2-chlorophenyl)-5-chloro-2-hydroxybenzamide analogues was discovered as potent RSV inhibitors. Six selected potent compounds 11 (JMX0439–2), 12 (JMX0447), 15 (JMX0509), 22 (JMX0457), 26 (JMX0439–1) and 28 (JMX0490) with low cytotoxicity were found to inhibit RSV replication substantially and effectively, supported by their suppression on the generation of progeny viruses, viral genome copies, and/or viral proteins expression. We also found that these compounds can decrease replication-independent and -dependent cytokine/chemokine induction by RSV, highlighting their potential acting as anti-inflammatory agents to alleviate RSV-associated symptoms. The potential controller, at the transcriptional factor level, was also investigated and discussed. Overall, this is the first proof-of-concept description on using N-(4-amino-2-chlorophenyl)-5-chloro-2-hydroxybenzamide analogues as a therapeutic agent to potentially treat RSV infection and associated inflammation. In the future, we will 1) investigate whether substituted N-(4-amino-2-chlorophenyl)-5-chloro-2-hydroxybenzamide analogues also play a role in RSV-induced host response of immune cells; 2) identify the molecular mechanisms used by compounds for their suppressive role in viral replication and associated inflammation; and 3) test these potent compound candidates in inhibiting RSV replication and pulmonary inflammation in the animal models of RSV.
4. Experimental section
4.1. Chemistry.
4.1.1. General Chemistry Methods
All commercially available starting materials and solvents were reagent grade and used without further purification. Reactions were performed under a nitrogen atmosphere in dry glassware with magnetic stirring. Preparative column chromatography was performed using silica gel 60, particle size 0.063–0.200 mm (70–230 mesh, flash). Analytical TLC was carried out employing silica gel 60 F254 plates (Merck, Darmstadt). Visualization of the developed chromatograms was performed with detection by UV (254 nm). NMR spectra were recorded on a Bruker-300 (1H, 300 MHz; 13C, 75 MHz) spectrometer. 1H and 13C NMR spectra were recorded with TMS as an internal reference. Chemical shifts were expressed in ppm, and J values were given in Hz. High-resolution mass spectra (HRMS) were obtained from Thermo Fisher LTQ Orbitrap Elite mass spectrometer. Parameters include the following: Nano ESI spray voltage was 1.8 kV; Capillary temperature was 275 °C and the resolution was 60,000; Ionization was achieved by positive mode. The most intense molecular ion peaks of these derivatives shown in ESI-MS matched well with the calculated mass values by using the 35Cl isotope. Purities of final compounds were established by analytical HPLC, which was carried out on a Shimadzu HPLC system (model: CBM-20A LC-20AD SPD-20A UV/VIS). HPLC analysis conditions: Waters μBondapak C18 (300 × 3.9 mm); flow rate 0.5 mL/min; UV detection at 270 and 254 nm; linear gradient from 10% acetonitrile in water to 100% acetonitrile in water in 20 min followed by 30 min of the last-named solvent (0.1% TFA was added into both acetonitrile and water). All biologically evaluated compounds are >95% pure. The detailed syntheses of compounds 9–13, 16–18, 20, 21 and 23–26 were reported in our previous publication.56
4.1.2. 5-Chloro-N-(2-chloro-4-((2-methylpentan-2-yl)amino)phenyl)-2-hydroxybenzamide (14).
To a solution of niclosamide (150 mg, 0.46 mmol) and Fe(acac)3 (49 mg, 0.14 mmol) in EtOH (4 mL) was added donor olefin 2-methyl-1-pentene (113 mg, 1.38 mmol), and PhSiH3 (99 mg, 0.92 mmol). The resulting mixture was heated in an oil bath preheated to 60 °C with stirring for 1 h. The reaction mixture was then cooled to room temperature and Zn (598 mg, 9.20 mmol) and 2 N HCl (2 mL) was added to the reaction mixture. After stirring at 60 °C for another 1 h, the reaction mixture was cooled to room temperature and filtered through Celite®. After the filter cake was washed with EtOAc, the filtrate was neutralized with sat. NaHCO3 (aq.) and extracted with EtOAc for three times. The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude product was then purified on SiO2 to furnish compound 14 (62 mg, 35%) as a yellow solid. HPLC purity 98.5% (tR = 16.70 min). 1H NMR (300 MHz, CDCl3) δ 8.14 (s, 1H), 7.88 (d, J = 9.0 Hz, 1H), 7.50 (d, J = 2.4 Hz, 1H), 7.37 (dd, J = 9.0, 2.4 Hz, 1H), 6.96 (d, J = 8.7 Hz, 1H), 6.76 (d, J = 2.4 Hz, 1H), 6.62 (dd, J = 9.0, 2.4 Hz, 1H), 1.66 – 1.57 (m, 2H), 1.41 – 1.24 (m, 8H), 0.92 (t, J = 7.2 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 166.8, 160.3, 145.6, 134.5, 126.1, 125.2, 124.4, 123.8, 123.1, 120.4, 115.8, 115.8, 115.3, 54.2, 44.0, 28.2 (2C), 17.4, 14.6. HRMS (ESI) calcd for C19H23Cl2N2O2, 381.1137 (M + H)+; found, 381.1130.
4.1.3. 5-Chloro-N-(2-chloro-4-((1-methylcyclohexyl)amino)phenyl)-2-hydroxybenzamide (15).
Compound 15 was prepared by a procedure similar to that used to prepare compound 14, starting from niclosamide and 1-methyl-1-cyclohexene. The title compound (80 mg, 44%) was obtained as a light-yellow solid. HPLC purity 98.9% (tR = 24.32 min). 1H NMR (300 MHz, CDCl3) δ 11.92 (br s, 1H), 8.15 (s, 1H), 7.87 (d, J = 9.0 Hz, 1H), 7.50 (d, J = 2.4 Hz, 1H), 7.37 (dd, J = 9.0, 2.4 Hz, 1H), 6.96 (d, J = 8.7 Hz, 1H), 6.78 (d, J = 2.4 Hz, 1H), 6.64 (dd, J = 9.0, 2.7 Hz, 1H), 3.61 (br s, 1H), 1.90 – 1.77 (m, 2H), 1.59 – 1.42 (m, 8H), 1.33 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 166.8, 160.3, 145.4, 134.5, 126.1, 125.2, 124.4, 123.8, 123.0, 120.4, 115.9, 115.9, 115.5, 53.5, 38.1 (2C), 26.8, 25.7, 22.1 (2C). HRMS (ESI) calcd for C20H23Cl2N2O2, 393.1137 (M + H)+; found, 393.1130.
4.1.4. 5-Chloro-N-(2-chloro-4-((pyridin-3-ylmethyl)amino)phenyl)-2-hydroxybenzamide (19).
Compound 9 (100 mg, 0.34 mmol) and 3-pyridinecarboxaldehyde (54 mg, 0.51 mmol) were suspended in DCE (20 mL) and treated with AcOH (61 mg, 1.01 mmol). NaBH(OAc)3 (179 mg, 0.84 mmol) was added in portions at 0 °C, and the mixture was stirred at r.t overnight. The pH of the mixture was adjusted to 9~10 with NaHCO3 (aq.) at 0 °C. The yellow solid was isolated by filtration to afford compound 19 (52 mg, 40%). HPLC purity 99.2% (tR = 15.66 min). 1H NMR (300 MHz, DMSO-d6) δ 12.20 (s, 1H), 10.43 (s, 1H), 8.59 (d, J = 1.2 Hz, 1H), 8.46 (dd, J = 4.5, 1.2 Hz, 1H), 7.99 (d, J = 2.7 Hz, 1H), 7.79 – 7.69 (m, 2H), 7.46 (dd, J = 8.7, 2.7 Hz, 1H), 7.36 (dd, J = 7.8, 4.8 Hz, 1H), 7.01 (d, J = 8.7 Hz, 1H), 6.73 (d, J = 2.4 Hz, 1H), 6.66 – 6.53 (m, 2H), 4.33 (d, J = 6.0 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) δ 163.7, 156.6, 148.9, 148.1, 146.9, 135.0 (2C), 133.1, 128.9, 126.8, 126.0, 123.5, 123.2, 122.9, 119.1, 118.8, 112.0, 111.5, 43.9. HRMS (ESI) calcd for C19H16Cl2N3O2, 388.0620 (M + H)+; found, 388.0612.
4.1.5. 5-Chloro-N-(2-chloro-4-((furan-2-ylmethyl)amino)phenyl)-2-hydroxybenzamide (22).
To a solution of compound 9 (100 mg, 0.34 mmol) and 2-furaldehyde (48 mg, 0.51 mmol) in 10 mL of MeOH was added AcOH (81 mg, 1.35 mmol) at 0 °C. The mixture was stirred at 0 °C for 30 min, then NaBH3CN (85 mg, 1.35 mmol) was added. The resulting mixture was stirred at r.t. overnight. The pH of the mixture was adjusted to 9~10 with NaHCO3 (aq.) at 0 °C. The mixture was extracted with DCM, dried (Na2SO4) and concentrated. The residue was purified by column chromatography (Hex/EtOAc) to give compound 22 (105 mg, 82%) as a white solid. HPLC purity 99.2% (tR = 18.88 min). 1H NMR (300 MHz, CDCl3) δ 11.90 (s, 1H), 8.10 (s, 1H), 7.94 (d, J = 9.0 Hz, 1H), 7.48 (d, J = 2.1 Hz, 1H), 7.43 – 7.32 (m, 2H), 6.97 (d, J = 8.7 Hz, 1H), 6.73 (d, J = 2.4 Hz, 1H), 6.61 (dd, J = 8.7, 2.4 Hz, 1H), 6.33 (s, 1H), 6.25 (d, J = 2.7 Hz, 1H), 4.30 (s, 2H), 4.17 (s, 1H). 13C NMR (75 MHz, CDCl3) δ 166.9, 160.4, 151.9, 146.2, 142.3, 134.6, 126.6, 125.2, 124.8, 123.8, 123.6, 120.5, 115.8, 113.0, 112.5, 110.6, 107.5, 41.4. HRMS (ESI) calcd for C18H15Cl2N2O3, 377.0460 (M + H)+; found, 377.0456.
4.1.6. 5-Chloro-N-(2-chloro-4-(pyrrolidin-1-yl)phenyl)-2-hydroxybenzamide (27).
To a solution of 2-chloro-4-fluoronitrobenzene (520 mg, 2.96 mmol) and pyrrolidine (256 mg, 3.55 mmol) in 10 mL of DMF was added K2CO3 (818 mg, 5.92 mmol). The resulting mixture was stirred at 100 °C for 1 h. Then the mixture was cooled to r.t and poured into 50 mL of H2O. Yellow precipitate was isolated by filtration and dried to afford 1-(3-chloro-4-nitrophenyl)pyrrolidine (31) (660 mg, 98%). 1H NMR (300 MHz, CDCl3) δ 8.06 (d, J = 9.3 Hz, 1H), 6.52 (d, J = 2.4 Hz, 1H), 6.39 (dd, J = 9.3, 2.4 Hz, 1H), 3.41 – 3.33 (m, 4H), 2.10 – 2.04 (m, 4H).
To a solution of compound 31 (660 mg, 2.91 mmol) in 20 mL of MeOH was added 4 mL of saturated NH4Cl (aq.). Zinc dust (946 mg, 14.56 mmol) was added to the solution at 0 °C. The reaction was stirred at r.t for 16 h. TLC indicated that the starting material was gone. 200 mL of EtOAc was added to the solution. The Zinc solid was filtered, and the filtrate was washed with 30 mL of brine, dried (Na2SO4) and concentrated under vacuum. The residue was purified by column chromatography to afford 2-chloro-4-(pyrrolidin-1-yl) (33) (500 mg, 87%) as a brown solid. 1H NMR (300 MHz, CDCl3) δ 6.72 (d, J = 8.7 Hz, 1H), 6.53 (d, J = 2.7 Hz, 1H), 6.39 (dd, J = 8.7, 2.7 Hz, 1H), 3.56 (s, 2H), 3.23 – 3.15 (m, 4H), 2.01 – 1.94 (m, 4H).
To a solution of compound 33 (129 mg, 0.66 mmol), 5-chlorosalicylic acid (119 mg, 0.69 mmol) and DMAP (10 mg, 0.082 mmol) in 30 mL of DCM was added EDCI (251 mg, 1.31 mmol) at 0 °C. The resulting mixture was stirred at r.t overnight. The mixture was concentrated and then to the residue was added H2O (10 mL) and 2 N NaOH (aq., 3 mL). The mixture was stirred at r.t for 1 h. The pH of the mixture was adjusted to 6~7 with 2 N HCl (aq.). Then the mixture was extracted with EtOAc (2 × 80 mL) and dried (Na2SO4) and concentrated under vacuum. The residue was purified by column chromatography (Hex/EtOAc = 5/1 to 3/1) followed by crystallization in MeOH to give compound 27 (50 mg, 23%) as a yellow solid. HPLC purity 97.4% (tR = 20.09 min). 1H NMR (300 MHz, CDCl3) δ 12.01 (s, 1H), 8.08 (s, 1H), 7.91 (d, J = 9.0 Hz, 1H), 7.49 (d, J = 2.1 Hz, 1H), 7.37 (dd, J = 8.7, 2.1 Hz, 1H), 6.96 (d, J = 8.7 Hz, 1H), 6.57 (s, 1H), 6.48 (d, J = 8.7 Hz, 1H), 3.33 – 3.19 (m, 4H), 2.08 – 1.94 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 166.9, 160.4, 146.6, 134.4, 126.8, 125.2, 124.9, 123.7, 121.2, 120.4, 115.9, 111.5, 110.7, 47.8 (2C), 25.6 (2C). HRMS (ESI) calcd for C17H17Cl2N2O2 351.0667 (M + H)+, found 351.0662.
4.1.7. 5-Chloro-N-(2-chloro-4-morpholinophenyl)-2-hydroxybenzamide (28).
To a solution of 2-chloro-4-fluoronitrobenzene (640 mg, 3.65 mmol) and morpholine (381 mg, 4.38 mmol) in 10 mL of DMF was added K2CO3 (1007 mg, 7.29 mmol). The resulting mixture was stirred at 100 °C for 1 h. Then the mixture was cooled to r.t and poured into 50 mL of H2O. Yellow precipitate was isolated by filtration and dried to afford 4-(3-chloro-4-nitrophenyl)morpholine (32) (780 mg, 88%). 1H NMR (300 MHz, CDCl3) δ 8.03 (d, J = 9.3 Hz, 1H), 6.86 (d, J = 2.7 Hz, 1H), 6.73 (dd, J = 9.3, 2.7 Hz, 1H), 3.89 – 3.82 (m, 4H), 3.37 – 3.31 (m, 4H).
To a solution of compound 32 (780 mg, 3.21 mmol) in 20 mL of MeOH was added 4 mL of saturated NH4Cl (aq.). Zinc dust (1040 mg, 16.07 mmol) was added to the solution at 0 °C. The reaction was stirred at r.t for 16 h. TLC indicated that the starting material was gone. 200 mL of EtOAc was added to the solution. The Zinc solid was filtered, and the filtrate was washed with 30 mL of brine, dried (Na2SO4) and concentrated under vacuum. The residue was purified by column chromatography to afford 2-chloro-4-morpholinoaniline (34) (650 mg, 95%) as a yellow solid. 1H NMR (300 MHz, CDCl3) δ 6.86 (t, J = 1.5 Hz, 1H), 6.74 – 6.71 (m, 2H), 3.86 – 3.81 (m, 4H), 3.77 (s, 2H), 3.03 – 2.98 (m, 4H).
To a solution of compound 34 (166 mg, 0.78 mmol), 5-chloro-2-methoxybenzoic acid (218 mg, 1.17 mmol) and DMAP (10 mg, 0.082 mmol) in 20 mL of DCM was added EDCI (449 mg, 2.34 mmol) at 0 °C. The resulting mixture was stirred at r.t overnight. The mixture was diluted with 100 mL of DCM, washed with H2O (2 × 30 mL), dried (Na2SO4) and concentrated to afford the crude product, which was used for the next step without further purification. The crude product was dissolved in 20 mL of DCM, and then BBr3 (3.91 mL, 3.91 mmol, 1 M in DCM) was added dropwise at 0 °C. The resulting mixture was stirred at r.t for 1h, and then the mixture was poured into 50 mL of ice water. The pH of mixture was adjusted to 6~7 with Na2CO3 (aq.). The mixture was extracted with DCM (2 × 80 mL), dried (Na2SO4) and concentrated. The residue was purified by column chromatography to afford compound 28 (240 mg, 83% in two steps) as a brown solid. HPLC purity 98.1% (tR = 18.12 min). 1H NMR (300 MHz, CDCl3) δ 8.23 (s, 1H), 8.10 (d, J = 9.3 Hz, 1H), 7.50 (d, J = 2.4 Hz, 1H), 7.39 (dd, J = 8.7, 2.4 Hz, 1H), 6.98 (d, J = 9.0 Hz, 1H), 6.95 (d, J = 2.7 Hz, 1H), 6.86 (dd, J = 9.0, 2.7 Hz, 1H), 3.91 – 3.81 (m, 4H), 3.20 – 3.11 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 167.0, 160.5, 149.5, 134.7, 126.0, 125.4, 125.2, 124.1, 123.9, 120.6, 115.9, 115.8, 114.8, 66.8 (2C), 49.1 (2C). HRMS (ESI) calcd for C17H17Cl2N2O3 367.0616 (M + H)+, found 367.0614.
4.2. Biology
4.2.1. Cell lines, virus, and antibodies
HEp-2 and A549 cells were purchased from the ATCC (Manassas, VA), and were maintained in F12K and MEM medium respectively, containing 10% (v/v) FBS. SAECs, isolated from the normal human lung distal portion, were purchased from Lonza (Pittsburgh, PA), and were maintained according to the manufacturer’s protocol. RSV long strain was propagated in HEp-2 cells at 37 °C and purified by sucrose gradient as previously described.58, 70 Viral titer was determined by immunostaining in HEp-2 cells using polyclonal biotin-conjugated goat anti-RSV antibody (7950–0104; Bio-Rad, Hercules, CA) and streptavidin peroxidase polymer (S2438; Sigma-Aldrich, St. Louis, MO) sequentially, as previously described.58, 70 The polyclonal biotin-conjugated goat anti-RSV antibody was also used for Western blot to detect viral protein expression. The monoclonal antibody against β-actin was from Sigma (A1978). Primary antibodies against IRF-3(CST#4302), phospho-IRF3(Ser396)(#29047), p65(CST#4764), phospho-p65(Ser276)(CST#3037), and phospho-p65(Ser536)(CST# 3033) were purchased from Cell Signaling Technology (Denvers, MA), and goat anti-rabbit IgG-HRP (4050–05) was purchased from SouthernBiotech (Birmingham, AL).
4.2.2. Lactate dehydrogenase (LDH) assay
A549 cells were seeded into 96-well plates at a density of 1.5 × 104 cells/well. After 24 h, the whole medium was replaced with fresh medium containing 10 μM compounds and 2% FBS. At 15 h posttreatment, the cells were harvested and subjected to LDH assay using LDH-Glo™ Cytotoxicity Assay Kit (Promega, Madison, WI) according to the manufacturer’s protocol. Three biological replicates were evaluated.
4.2.3. Cell growth assay
A549 cells were seeded into 96-well plates at a density of 1.2 × 104 cells/well. After 24 h, the whole medium was replaced with fresh medium containing compounds (10~200 μM) and 2% FBS. At 15 h posttreatment, the cells were washed with PBS 3 times, followed by staining with trypan blue. For each sample, four non-overlapping fields at 100× magnification were randomly captured using a video camera, and the number of the unstained (viable) cells was counted in a blinded fashion. Two biological replicates were evaluated.
4.2.4. Cytokine and chemokine quantification
RSV-induced chemokines and cytokines were quantified by using a multi-analytic human (M50–0KCAF0Y) cytokine/chemokine profiling kit from Bio-Rad (Hercules, CA) according to the manufacturer’s instructions. Data were analyzed using the multiplex analysis software from Bio-Rad.
4.2.5. qRT-PCR
Total cellular RNA was extracted using TRIzol reagents (Thermo Fisher Scientific, Waltham, MA). qRT-PCR, used to examine viral replication, was performed using SYBR as we previously described for RSV or its family member human metapeumovirus.70, 71 In brief, to quantify viral antigenomic copies in the context of RSV infection, synthetic transcripts of the genome were generated from Topo plasmid containing N-P-M genes, using the T7 MegaScript kit, following the digestion with PmeI. The reaction mixture was then treated with Turbo DNase and purified using the MegaScript kit. Primers were designed to span the N and P regions of the viral genome and incorporated a Cmr tag. First-strand cDNA was transcribed with a P-specific primer, 5′-CTGCGATGAGTGGCAGGCACTACAGTGTATTAGACTTRACAGCAGAAG-3′. The underlined letters indicate the Cmr tag sequence. QPCRs were performed using the following primers: forward, 5′-CTGCGATGAGTGGCAGGC-3′, and reverse, 5′-GCATCTTCTCCATGRAATTCAGG-3′. RT-PCRs and QPCRs were performed as described above.
4.2.6. Western blot analysis
Total cellular lysates or cytosol and nuclear extracts were prepared for uninfected or infected cells as previously described.70, 72 Proteins were then quantified with a protein quantification kit from Bio-Rad, followed by fractionation using SDS-PAGE denaturing gels and protein transfer to polyvinylidene difluoride membranes as previously described. Membranes were blocked with 5% milk in TBS-Tween 20 and incubated with the proper primary antibodies according to the manufacturer’s instructions.
4.2.7. Reporter gene assays
The cells were transfected in triplicate with luciferase reporter gene plasmids containing multiple copies of NF-κB binding sites (NF-κB-Luc) or the IRF-3 binding site (PRDIII-I-Luc and IRF-3-Luc) using FuGene 6 (Roche, Indianapolis, IN), as previously described.70, 72, 73 At 20 h post-transfection, cells were infected with RSV for various times and lysed to measure luciferase reporter activity.
4.2.8. Statistical analysis
All experiments were carried out at least twice. The test results were analyzed by Graphpad Prism 5 software. Two groups comparison was evaluated using an unpaired two-tailed t-test. A one-way ANOVA test was performed to analyze differences among multiple groups. A p-value of less than 0.05 was considered significant. Means ± standard errors (SE) are shown.
Supplementary Material
Highlights:
A series of 5-chloro-2-hydroxybenzamides was discovered as potent RSV inhibitors.
Several molecules suppressed both RSV viral replication and associated cytokines.
Two selected compounds inhibited RSV-induced IRF3 and NF-κB activation.
These potential anti-inflammatory agents may alleviate RSV-associated symptoms.
Acknowledgments
This work was supported by the grants R01 AI116812 and R21 AG069226 from the National Institutes of Health, and FAMRI Clinical Innovator Award (#160020) to X.B. J.Z. is also partly supported by the John D. Stobo, M. D. Distinguished Chair Endowment Fund, and the John Sealy Memorial Endowment Fund at UTMB.
Abbreviations
- RSV
respiratory syncytial virus
- LRTI
lower respiratory tract infection
- URTI
upper respiratory tract infection
- mAb
monoclonal antibody
- F protein
fusion protein
- N protein
nucleoprotein
- G protein
glycoprotein
- P protein
phosphoprotein
- M protein
matrix protein
- RdRp
RNA-dependent RNA polymerase
- Boc
t-butyloxy carbonyl
- THP
tetrahydropyran
- CC50
cytotoxicity concentration 50%
- MOI
multiplicity of infection
- RLRs
RIG-I-like receptors
- ds
double-stranded
- IRF
interferon regulatory factor
- TLRs
Toll-like receptors
- SAECs
small airway epithelial cells
- MIP
macrophage inflammatory proteins
- MCP-1
monocyte chemoattractant protein 1
- IP-10
interferon gamma-induced protein 10
- Ser
serine
- RANTES
regulated on activation, normal T cell expressed and secreted
- TNF
tumor necrosis factor
- p.i.
post-infection
- TLC
thin layer chromatography
- UV
ultraviolet
- TMS
tetramethylsilane
- HRMS
high-resolution mass spectrometry
- HPLC
high-performance liquid chromatography
- DCE
1,2-dichloroethane
- DCM
dichloromethane
- DMAP
4-(dimethylamino)pyridine
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- EDCI
1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride
- EtOAc
ethyl acetate
Footnotes
The authors declare no competing financial interest.
Declaration of Interest Statement
The authors have no conflict of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Afonso CL; Amarasinghe GK; Bányai K, et al. Taxonomy of the order Mononegavirales: update 2016. Arch Virol. 2016; 161(8): 2351–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cockerill GS; Good JAD.Mathews N State of the art in respiratory syncytial virus drug discovery and development. J Med Chem. 2019; 62(7): 3206–3227. [DOI] [PubMed] [Google Scholar]
- 3.Glezen WP; Taber LH; Frank AL.Kasel JA Risk of primary infection and reinfection with respiratory syncytial virus. Am J Dis Child. 1986; 140(6): 543–546. [DOI] [PubMed] [Google Scholar]
- 4.Henderson FW; Collier AM; Clyde WA Jr..Denny FW Respiratory-syncytial-virus infections, reinfections and immunity. A prospective, longitudinal study in young children. N Engl J Med. 1979; 300(10): 530–534. [DOI] [PubMed] [Google Scholar]
- 5.Shi T; McAllister DA; O’Brien KL, et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: a systematic review and modelling study. Lancet. 2017; 390(10098): 946–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kramer R; Duclos A; Lina B.Casalegno JS Cost and burden of RSV related hospitalisation from 2012 to 2017 in the first year of life in Lyon, France. Vaccine. 2018; 36(45): 6591–6593. [DOI] [PubMed] [Google Scholar]
- 7.Falsey AR; Hennessey PA; Formica MA; Cox C.Walsh EE Respiratory syncytial virus infection in elderly and high-risk adults. N Engl J Med. 2005; 352(17): 1749–1759. [DOI] [PubMed] [Google Scholar]
- 8.Falsey AR Respiratory syncytial virus infection in adults. Semin Respir Crit Care Med. 2007; 28(2): 171–181. [DOI] [PubMed] [Google Scholar]
- 9.Jensen TO; Stelzer-Braid S; Willenborg C, et al. Outbreak of respiratory syncytial virus (RSV) infection in immunocompromised adults on a hematology ward. J Med Virol. 2016; 88(10): 1827–1831. [DOI] [PubMed] [Google Scholar]
- 10.Nair H; Nokes DJ; Gessner BD, et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet. 2010; 375(9725): 1545–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blanken MO; Rovers MM; Molenaar JM, et al. Respiratory syncytial virus and recurrent wheeze in healthy preterm infants. N Engl J Med. 2013; 368(19): 1791–1799. [DOI] [PubMed] [Google Scholar]
- 12.Perez-Yarza EG; Moreno A; Lazaro P; Mejias A.Ramilo O The association between respiratory syncytial virus infection and the development of childhood asthma: a systematic review of the literature. Pediatr Infect Dis J. 2007; 26(8): 733–739. [DOI] [PubMed] [Google Scholar]
- 13.Sigurs N Epidemiologic and clinical evidence of a respiratory syncytial virus-reactive airway disease link. Am J Respir Crit Care Med. 2001; 163(3 Pt 2): 2–6. [DOI] [PubMed] [Google Scholar]
- 14.Resch B Product review on the monoclonal antibody palivizumab for prevention of respiratory syncytial virus infection. Hum Vaccines Immunother. 2017; 13(9): 2138–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Committee On Infectious Diseases and Bronchiolitis Guidelines Committee. Updated guidance for palivizumab prophylaxis among infants and young children at increased risk of hospitalization for respiratory syncytial virus infection. Pediatrics. 2014; 134(2): e620. [DOI] [PubMed] [Google Scholar]
- 16.Ventre K.Randolph AG Ribavirin for respiratory syncytial virus infection of the lower respiratory tract in infants and young children. Cochrane Database Syst Rev. 2007; (1): CD000181. [DOI] [PubMed] [Google Scholar]
- 17.Bonfanti J-F; Doublet F; Fortin J, et al. Selection of a respiratory syncytial virus fusion inhibitor clinical candidate, part 1: Improving the pharmacokinetic profile using the structure−property relationship. J Med Chem. 2007; 50(19): 4572–4584. [DOI] [PubMed] [Google Scholar]
- 18.Bonfanti J-F; Meyer C; Doublet F, et al. Selection of a respiratory syncytial virus fusion inhibitor clinical candidate. 2. Discovery of a morpholinopropylaminobenzimidazole derivative (TMC353121). J Med Chem. 2008; 51(4): 875–896. [DOI] [PubMed] [Google Scholar]
- 19.Mackman RL; Sangi M; Sperandio D, et al. Discovery of an oral respiratory syncytial virus (RSV) fusion inhibitor (GS-5806) and clinical proof of concept in a human RSV challenge study. J Med Chem. 2015; 58(4): 1630–1643. [DOI] [PubMed] [Google Scholar]
- 20.Feng S; Hong D; Wang B, et al. Discovery of imidazopyridine derivatives as highly potent respiratory syncytial virus fusion inhibitors. ACS Med Chem Lett. 2015; 6(3): 359–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng X; Wang L; Wang B, et al. Discovery of piperazinylquinoline derivatives as novel respiratory syncytial virus fusion inhibitors. ACS Med Chem Lett. 2016; 7(6): 558–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roymans D; Alnajjar SS; Battles MB, et al. Therapeutic efficacy of a respiratory syncytial virus fusion inhibitor. Nat Commun. 2017; 8(1): 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng X; Liang C; Wang L, et al. Discovery of benzoazepinequinoline (BAQ) derivatives as novel, potent, orally bioavailable respiratory syncytial virus fusion inhibitors. J Med Chem. 2018; 61(22): 10228–10241. [DOI] [PubMed] [Google Scholar]
- 24.Shi W; Jiang Z; He H, et al. Discovery of 3,3’-spiro[azetidine]-2-oxo-indoline derivatives as fusion inhibitors for treatment of RSV infection. ACS Med Chem Lett. 2018; 9(2): 94–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng X; Gao L; Wang L, et al. Discovery of ziresovir as a potent, selective, and orally bioavailable respiratory syncytial virus fusion protein inhibitor. J Med Chem. 2019; 62(13): 6003–6014. [DOI] [PubMed] [Google Scholar]
- 26.DeVincenzo JP; Whitley RJ; Mackman RL, et al. Oral GS-5806 activity in a respiratory syncytial virus challenge study. N Engl J Med. 2014; 371(8): 711–722. [DOI] [PubMed] [Google Scholar]
- 27.Jiménez-Somarribas A; Mao S; Yoon J-J, et al. Identification of non-nucleoside inhibitors of the respiratory syncytial virus polymerase complex. J Med Chem. 2017; 60(6): 2305–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang G; Deval J; Hong J, et al. Discovery of 4’-chloromethyl-2’-deoxy-3’,5’-di-O-isobutyryl-2’-fluorocytidine (ALS-8176), a first-in-class RSV polymerase inhibitor for treatment of human respiratory syncytial virus infection. J Med Chem. 2015; 58(4): 1862–1878. [DOI] [PubMed] [Google Scholar]
- 29.Matharu DS; Flaherty DP; Simpson DS, et al. Optimization of potent and selective quinazolinediones: Inhibitors of respiratory syncytial virus that block RNA-dependent RNA-polymerase complex activity. J Med Chem. 2014; 57(24): 10314–10328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deval J; Hong J; Wang G, et al. Molecular basis for the selective inhibition of respiratory syncytial virus RNA polymerase by 2’-fluoro-4’-chloromethyl-cytidine triphosphate. PLoS Pathog. 2015; 11(6): e1004995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clarke MO; Mackman R; Byun D, et al. Discovery of β-D-2’-deoxy-2’-α-fluoro-4’-α-cyano-5-aza-7,9-dideaza adenosine as a potent nucleoside inhibitor of respiratory syncytial virus with excellent selectivity over mitochondrial RNA and DNA polymerases. Bioorg Med Chem Lett. 2015; 25(12): 2484–2487. [DOI] [PubMed] [Google Scholar]
- 32.Sudo K; Miyazaki Y; Kojima N, et al. YM-53403, a unique anti-respiratory syncytial virus agent with a novel mechanism of action. Antiviral Res. 2005; 65(2): 125–131. [DOI] [PubMed] [Google Scholar]
- 33.Xiong H; Foulk M; Aschenbrenner L, et al. Discovery of a potent respiratory syncytial virus RNA polymerase inhibitor. Bioorg Med Chem Lett. 2013; 23(24): 6789–6793. [DOI] [PubMed] [Google Scholar]
- 34.Fordyce EAF; Brookes DW; Lise-Ciana C, et al. Discovery of novel benzothienoazepine derivatives as potent inhibitors of respiratory syncytial virus. Bioorg Med Chem Lett. 2017; 27(10): 2201–2206. [DOI] [PubMed] [Google Scholar]
- 35.Fordyce EAF; Fraser Hunt S; Crepin D, et al. Conformationally restricted benzothienoazepine respiratory syncytial virus inhibitors: their synthesis, structural analysis and biological activities. MedChemComm. 2018; 9(3): 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shook BC; Kim IJ; Blaisdell TP; Yu J; Panarese J.Or YS Preparation of Benzodiazepine Derivatives for Use as RSV Inhibitors. WO2017015449A1, 2017. [Google Scholar]
- 37.Shook BC; Kim IJ; Blaisdell TP, et al. Aminobenzodiazepinones as RSV Inhibitors for the Use in Combinations with Other Pharmaceutical Agents. WO2019067864A1, 2019. [Google Scholar]
- 38.Collins PL.Melero JA Progress in understanding and controlling respiratory syncytial virus: still crazy after all these years. Virus Res. 2011; 162(1–2): 80–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Piedra FA; Mei M; Avadhanula V, et al. The interdependencies of viral load, the innate immune response, and clinical outcome in children presenting to the emergency department with respiratory syncytial virus-associated bronchiolitis. PLoS ONE. 2017; 12(3): e0172953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thwaites RS; Coates M; Ito K, et al. Reduced nasal viral load and IFN responses in infants with respiratory syncytial virus bronchiolitis and respiratory failure. Am J Respir Crit Care Med. 2018; 198(8): 1074–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Domachowske JB.Rosenberg HF Respiratory syncytial virus infection: immune response, immunopathogenesis, and treatment. Clin Microbiol Rev. 1999; 12(2): 298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hogg JC; Williams J; Richardson JB; Macklem PT.Thurlbeck WM Age as a factor in the distribution of lower-airway conductance and in the pathologic anatomy of obstructive lung disease. N Engl J Med. 1970; 282(23): 1283–1287. [DOI] [PubMed] [Google Scholar]
- 43.Welliver TP; Reed JL.Welliver RC Sr. Respiratory syncytial virus and influenza virus infections: observations from tissues of fatal infant cases. Pediatr Infect Dis J. 2008; 27(10 Suppl): 92–96. [DOI] [PubMed] [Google Scholar]
- 44.Johnson JE; Gonzales RA; Olson SJ; Wright PF.Graham BS The histopathology of fatal untreated human respiratory syncytial virus infection. Mod Pathol. 2007; 20(1): 108–119. [DOI] [PubMed] [Google Scholar]
- 45.McNamara PS; Flanagan BF; Hart CA.Smyth RL Production of chemokines in the lungs of infants with severe respiratory syncytial virus bronchiolitis. J Infect Dis. 2005; 191(8): 1225–1232. [DOI] [PubMed] [Google Scholar]
- 46.Bueno SM; González PA; Riedel CA; Carreño LJ; Vásquez AE.Kalergis AM Local cytokine response upon respiratory syncytial virus infection. Immunol Lett. 2011; 136(2): 122–129. [DOI] [PubMed] [Google Scholar]
- 47.Van Woensel JB; Kimpen JL.Brand PL Respiratory tract infections caused by respiratory syncytial virus in children. Diagnosis and treatment. Minerva Pediatr. 2001; 53(2): 99–106. [PubMed] [Google Scholar]
- 48.Reassessment of the indications for ribavirin therapy in respiratory syncytial virus infections. American Academy of Pediatrics Committee on Infectious Diseases. Pediatrics. 1996; 97(1): 137–140. [PubMed] [Google Scholar]
- 49.Xu J; Shi P-Y; Li H.Zhou J Broad spectrum antiviral agent niclosamide and its therapeutic potential. ACS Infect Dis. 2020; 6(5): 909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu J; Xue Y; Zhou R; Shi P-Y; Li H.Zhou J Drug repurposing approach to combating coronavirus: Potential drugs and drug targets. Med Res Rev. 2020: 10.1002/med.21763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu J; Berastegui-Cabrera J; Chen H; Pachon J; Zhou J.Sanchez-Cespedes J Structure-Activity Relationship Studies on Diversified Salicylamide Derivatives as Potent Inhibitors of Human Adenovirus Infection. J Med Chem. 2020; 63(6): 3142–3160. [DOI] [PubMed] [Google Scholar]
- 52.Fan X; Xu J; Files M, et al. Dual activity of niclosamide to suppress replication of integrated HIV-1 and Mycobacterium tuberculosis (Beijing). Tuberculosis. 2019; 116: 28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu J; Pachón-Ibáñez ME; Cebrero-Cangueiro T; Chen H; Sánchez-Céspedes J.Zhou J Discovery of niclosamide and its O-alkylamino-tethered derivatives as potent antibacterial agents against carbapenemase-producing and/or colistin resistant Enterobacteriaceae isolates. Bioorg Med Chem Lett. 2019; 29(11): 1399–1402. [DOI] [PubMed] [Google Scholar]
- 54.Chen H; Yang Z; Ding C, et al. Discovery of O-alkylamino-tethered niclosamide derivatives as potent and orally bioavailable anticancer agents. ACS Med Chem Lett. 2013; 4(2): 180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li Z; Xu J; Lang Y, et al. JMX0207, a niclosamide derivative with improved pharmacokinetics, suppresses ZIKA virus infection both in vitro and in vivo. ACS Infect Dis. 2020; 6(10): 2616–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu J; Berastegui-Cabrera J; Ye N, et al. Discovery of novel substituted N-(4-Amino2-chlorophenyl)-5-chloro-2-hydroxybenzamide analogues as potent human adenovirus inhibitors. J Med Chem. 2020; 63(21): 12830–12852. [DOI] [PubMed] [Google Scholar]
- 57.Gui J; Pan C-M; Jin Y, et al. Practical olefin hydroamination with nitroarenes. Science. 2015; 348(6237): 886–891. [DOI] [PubMed] [Google Scholar]
- 58.Deng J; Ptashkin RN; Chen Y, et al. Respiratory syncytial virus utilizes a tRNA fragment to suppress antiviral responses through a novel targeting mechanism. Mol Ther. 2015; 23(10): 1622–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Q; Lee I; Ren J; Ajay SS; Lee YS.Bao X Identification and functional characterization of tRNA-derived RNA fragments (tRFs) in respiratory syncytial virus infection. Mol Ther. 2013; 21(2): 368–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu P; Jamaluddin M; Li K; Garofalo RP; Casola A.Brasier AR Retinoic acidinducible gene I mediates early antiviral response and Toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells. J Virol. 2007; 81(3): 1401–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Openshaw PJ.Chiu C Protective and dysregulated T cell immunity in RSV infection. Curr Opin Virol. 2013; 3(4): 468–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tabarani CM; Bonville CA; Suryadevara M, et al. Novel inflammatory markers, clinical risk factors and virus type associated with severe respiratory syncytial virus infection. Pediatr Infect Dis J. 2013; 32(12): e437–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Luo H; Wang D; Che HL; Zhao Y.Jin H Pathological observations of lung inflammation after administration of IP-10 in influenza virus- and respiratory syncytial virus-infected mice. Exp Ther Med. 2012; 3(1): 76–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brand HK; Ferwerda G; Preijers F, et al. CD4+ T-cell counts and interleukin-8 and CCL-5 plasma concentrations discriminate disease severity in children with RSV infection. Pediatr Res. 2013; 73(2): 187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bennett BL; Garofalo RP; Cron SG, et al. Immunopathogenesis of respiratory syncytial virus bronchiolitis. J Infect Dis. 2007; 195(10): 1532–1540. [DOI] [PubMed] [Google Scholar]
- 66.McNamara PS; Flanagan BF; Selby AM; Hart CA.Smyth RL Pro- and anti-inflammatory responses in respiratory syncytial virus bronchiolitis. Eur Respir J. 2004; 23(1): 106–112. [DOI] [PubMed] [Google Scholar]
- 67.Hu J; Nakano H; Sakurai H.Colburn NH Insufficient p65 phosphorylation at S536 specifically contributes to the lack of NF-κB activation and transformation in resistant JB6 cells. Carcinogenesis. 2004; 25(10): 1991–2003. [DOI] [PubMed] [Google Scholar]
- 68.Bao X; Indukuri H; Liu T, et al. IKKε modulates RSV-induced NF-κB-dependent gene transcription. Virology. 2010; 408(2): 224–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhong H; Voll RE.Ghosh S Phosphorylation of NF-κB p65 by PKA Stimulates Transcriptional Activity by Promoting a Novel Bivalent Interaction with the Coactivator CBP/p300. Mol Cell. 1998; 1(5): 661–671. [DOI] [PubMed] [Google Scholar]
- 70.Choi EJ; Ren Y; Chen Y, et al. Exchange proteins directly activated by cAMP and their roles in respiratory syncytial virus infection. J Virol. 2018; 92(22): e01200–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ren J; Wang Q; Kolli D, et al. Human metapneumovirus M2–2 protein inhibits innate cellular signaling by targeting MAVS. J Virol. 2012; 86(23): 13049–13061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bao X; Kolli D; Liu T; Shan Y; Garofalo RP.Casola A Human metapneumovirus small hydrophobic protein inhibits NF-κB transcriptional activity. J Virol. 2008; 82(16): 8224–8229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen Y; Deng X; Deng J, et al. Functional motifs responsible for human metapneumovirus M2–2-mediated innate immune evasion. Virology. 2016; 499: 361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.