

Abstract
Mitochondrial dynamics regulated by mitochondrial fusion and fission essentially maintain mitochondrial functions, whose alterations underline various human diseases. Here we show that inositol is a critical metabolite directly restricting AMPK-dependent mitochondrial fission independently of its classical mode as a precursor for phosphoinositide generation. Inositol decline by IMPA1/2 deficiency elicits AMPK activation and mitochondrial fission without affecting ATP level, while inositol accumulation prevents AMPK-dependent mitochondrial fission. Metabolic stress or mitochondrial damage causes inositol decline in cells and mice to elicit AMPK-dependent mitochondrial fission. Inositol directly binds to AMPKγ and competes with AMP for AMPKγ binding leading to restricting AMPK activation and mitochondrial fission. Our study suggests that AMP/inositol ratio is a critical determinant for AMPK activation and establishes a model where AMPK activation requires inositol decline to release AMPKγ for AMP binding. Hence, AMPK is an inositol sensor, whose inactivation by inositol serves as a mechanism to restrict mitochondrial fission.
Graphical Abstract
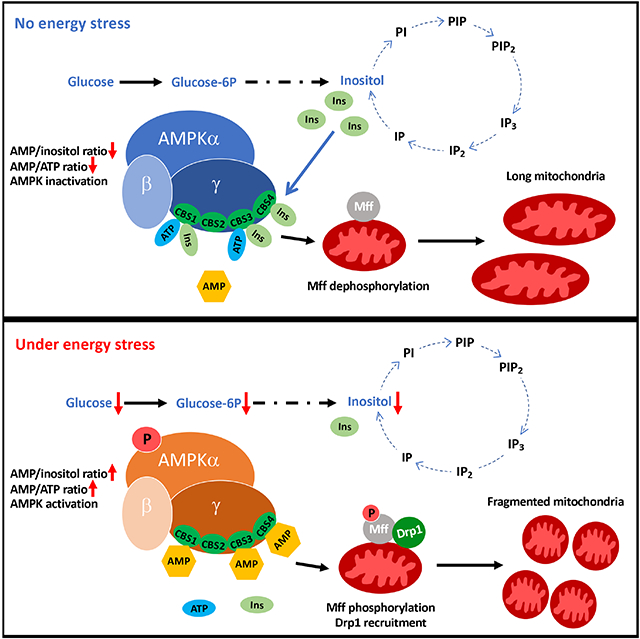
In Brief
Hsu et al. demonstrate that inositol serves as an endogenous suppressor to limit AMPK activation and mitochondrial fission through its direct binding to the AMPKγ subunit. AMP/inositol and AMP/ATP ratio serve as critical determinants for AMPK activation and mitochondrial fission. AMPK is therefore an inositol sensor orchestrating mitochondrial fission by detecting the inositol gradient in cells.
Introduction
Phosphoinositides, phosphorylated forms of phosphatidylinositol (PI), play crucial roles in diverse signaling events and biological processes (Di Paolo and De Camilli, 2006). Phosphoinositides such as PI(4)P and PI(4, 5)P2 are critical for controlling protein recruitment, membrane dynamics and ion channel activity (Fairn and Grinstein, 2012; Hammond et al., 2012). Phosphoinositide metabolism from PI(4, 5)P2 generates important second messengers such as inositol (1, 4, 5) triphosphates (IP3) and diacylglycerol (DAG), which induce calcium release (Combettes and Champeil, 1994) and protein kinase C activation (Asaoka et al., 1992), respectively, to orchestrate apoptosis and cell growth. Myo-inositol (hereafter as inositol) is a precursor used for the production of PI and phosphoinositides. The primary functions of inositol are attributed to its conversion to PI and/or distinct phosphoinositides.
The defect of inositol metabolism is associated with pathological disorders including insulin resistance (Asplin et al., 1993), kidney failure (Holub, 1986), polycystic ovarian syndrome (PCOS) (Unfer et al., 2012), neurological disorders (Chalecka-Franaszek and Chuang, 1999; Jolles et al., 1993; Shetty et al., 1995), and reproductive problems (Carlomagno et al., 2011; Ciotta et al., 2011; Condorelli et al., 2011). In animal models of inositol-related knockout mice, numerous mitochondria-related genes in the frontal cortex are upregulated similar to mice treated with lithium which attenuates phosphatidylinositol signaling in accordance with DNA microarray study (Toker et al., 2014), indicating that inositol metabolism may play an important role in modulating mitochondrial function.
Mitochondria play a central role in energy production, redox balance and metabolism control, signaling regulation, survival and proliferation (Porporato et al., 2018; Vyas et al., 2016). Mitochondrial metabolism and dynamics maintaining mitochondrial functions (Mishra and Chan, 2016) are critically regulated by two distinct processes including fission and fusion, which orchestrate the overall length and connectivity of mitochondria. Abnormal dynamics of mitochondria underline various human diseases such as cancer, neurologic diseases and cardiovascular diseases (Archer, 2013; Youle and van der Bliek, 2012). Defective energy metabolism, exercise and nitrogen starvation have been known to trigger a change in mitochondrial dynamics by regulating diverse mitochondrial fission and fusion proteins that maintain a healthy mitochondrial population (Mishra and Chan, 2016). Although earlier studies have demonstrated how mitochondrial fission and fusion proteins mechanistically modulate mitochondrial dynamic processes upon different physiological conditions (Cribbs and Strack, 2007; Dickey and Strack, 2011; Kashatus et al., 2015; Toyama et al., 2016), the crosstalk between cellular metabolites and mitochondrial dynamics under energy stress are largely unknown.
Inositol is a metabolite generated by two steps of enzymatic reactions (Figure 1A). The first reaction involves the conversion of glucose-6-phosphate (G6P), derived primarily from the first step of glucose metabolism, to inositol monophosphate (Loewus et al., 1980; Majumder et al., 1997). The second reaction gives rise to inositol by dephosphorylating inositol monophosphate through inositol monophosphatase (IMPA), which consists of two distinct isoforms, IMPA1 and IMPA2 (Figure 1A). Interestingly, our immunofluorescence (IF) assay revealed that IMPA1 is mainly localized in mitochondria (Figures 1B and S1A), indicative of the potential role of IMPA1/inositol in regulating mitochondrial functions.
Figure 1. IMPA/Inositol regulates mitochondrial fission.
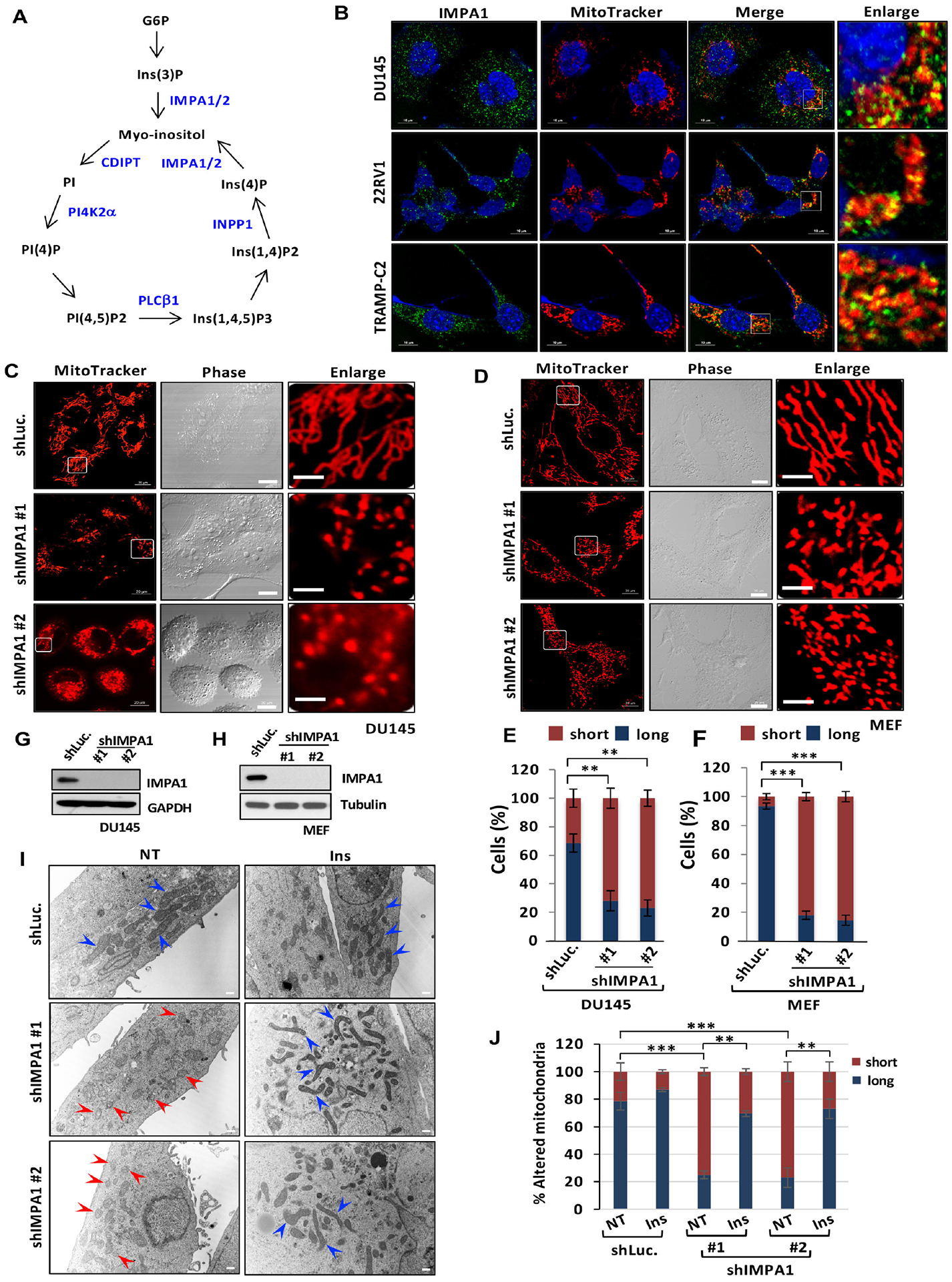
(A) Phosphoinositides metabolism with a series of enzymes. (B) Subcellular localization of IMPA1 using IMPA1 antibody (Green), MitoTracker (red) and DAPI (blue) in diverse cancer cell lines. (C and D) Mitochondrial morphology by MitoTracker (red) in shLuc. or shIMPA1 DU145 cells (C) or MEFs (D). Scale bar, 20 μm in MitoTracker and Phase; 5 μm in Enlarge. (E and F) Quantification of the mitochondrial morphology of the DU145 cells (C) and MEFs (D) shown in (E) and (F). (G and H) The shLuc. or shIMPA1 DU145 or MEFs were subjected to immunoblotting. (I) TEM was performed in shLuc. or shIMPA1 DU145 cells of upon vehicle (NT) or inositol treatment (Ins). Scale bar, 500 nm. Blue arrow, healthy/elongated mitochondria; Red arrows, fragmented mitochondria. (J) Quantification of the mitochondrial morphology (long and short) shown in (I).
Accumulating evidence reveals that energy metabolism could regulate cellular functions beyond its role in energy production and building blocks. This is achieved through the metabolites, such as α-ketoglutarate (α-KG) generated from mitochondria/TCA cycle, which can directly activate key enzymes involved in epigenetics and transcription for cell differentiation and stem cell maintenance (Carey et al., 2015; Lu and Thompson, 2012). Moreover, glycolysis-derived lactate represses RIG-I-like receptors (RLRs) signaling by directly targeting mitochondrial antiviral-signaling (MAVS), which coordinates the crosstalk between energy metabolism and antiviral signaling (Zhang et al., 2019). These studies define the unique role of the metabolites in regulating cellular functions through direct protein targeting.
Inspired by the earlier findings revealing the critical role of certain metabolites in directly targeting protein activity and/or functions, we hypothesize whether inositol can function as a second messenger to directly target certain proteins, thereby participating in unique signaling events to regulate biological outcomes beyond its known role in PI cycle metabolism. Our study not only offers the novel insight into how the specific metabolite cross-talks with AMPK-dependent mitochondrial fission, but also underscores the novel role of inositol in serving as a key metabolite directly restricting AMPK activation to maintain mitochondrial dynamics.
Results
IMPA-derived inositol regulates mitochondrial fission.
To explore a possibly new role of inositol in cellular functions, we first focused on IMPA1, which is critical for inositol production in cells (Deranieh and Greenberg, 2009). Immunofluorescence (IF) assay revealed that IMPA1 is mainly localized in mitochondria in diverse cancer cell lines (Figures 1B and S1A), indicative of the potential role of IMPA1/inositol in regulating mitochondrial functions. Because mitochondria are crucial for metabolic regulation, it is possible that specific metabolites generated from mitochondria may regulate mitochondrial dynamics. We then assessed whether IMPA1/inositol orchestrates mitochondrial dynamics. We examined the morphology of mitochondria with MitoTracker Red staining using a confocal microscope. Knockdown of IMPA1 in PC3 and DU145 and mouse embryonic fibroblasts (MEFs) markedly reduced inositol levels (Figures S1B and S1C), caused fragmented mitochondria and decreased mitochondrial aspect ratio, interconnectivity and elongation, indicative of mitochondrial fission (Figures 1C, 1E, 1G and Figures S1D–S1F). This phenomenon was also observed in MEFs with IMPA1 knockdown (Figures 1D, 1F, 1H and S1G). Transmission electron microscopy (TEM) experiments confirmed that IMPA1 knockdown induced mitochondrial fission, reflected by shrunken and fragmented mitochondria (Figures 1I and 1J). IMPA1 knockout in DU145 cells by using CRISPR/Cas9 significantly caused mitochondrial fission, similar to IMPA1 knockdown (Figures S1H–S1K). Likewise, IMPA2 knockdown cells also exhibited the reduction of inositol level, fragmented mitochondria and reduced aspect ratio, interconnectivity and elongation of mitochondria (Figures S1A–S1E).
It has been documented that W219 and D220 residues of IMPA are essential for IMPA phosphatase activity in bacteria (Goswami et al., 2018). We generated two mutants of W219A and D220A of human IMPA1 and performed the rescued experiment. Of note, restoration of IMPA1 and IMPA1 W219A, but not W220, rescued inositol levels in IMPA1 knockout cells (Figure S2F), suggesting that IMPA1 D220 is indispensable for phosphatase activity of IMPA1 in human cells. As a result, restoration of IMPA1 and IMPA1 W219A, but not IMPA1 D220A, reversed mitochondrial fission in IMPA1 knockout cells, indicative of the crucial role of IMPA1 phosphatase activity in repressing mitochondrial fission (Figures S2G–S2I). Similarly, pharmacological inactivation of IMPA1/2 activity by IMPA inhibitor lithium chloride (Cryns et al., 2008) also induced mitochondrial fission (Figures S3A and S3B). We ruled out the possibility that lithium chloride may act through GSK3β inhibition to induce mitochondrial fission, since specific GSK3β inhibitor BIO (Ding et al., 2019; Fang et al., 2019) that inhibited GSK3β kinase activity similar to lithium chloride failed to induce mitochondrial fission (Figures S3A–S3C).
Inositol directly regulates mitochondrial fission independently of its intermediates of phosphatidylinositol cycle.
Metabolomics and biochemical assays revealed that the levels of inositol and its downstream intermediates, such as inositol (Ins), phosphatidylinositol (PI), phosphatidylinositol 3-phosphate (PIP), phosphatidylinositol 4,5-bisphosphate (PIP2), phosphatidylinositol 3,4,5-bisphosphate (PIP3), inositol trisphosphate (IP3), inositol biphosphate (IP2) and inositol monophosphate (IP) were decreased in IMPA1 knockdown cells (Figures S1B and S1C). Intriguingly, knockdown of enzymes involved in PI cycles downstream of IMPA1/2 including CDP-DAG-inositol 3-phosphatidyltransferase (CDIPT), phosphatidylinositol 4-kinase type 2α (PI4K2α), phospholipase C β1 (PLCβ1) and inositol polyphosphate-1-phosphatase (INPP1), which catalyze the production of inositol downstream metabolites, PI, PI(4)P, I(1,4,5)P3 and I(4)P, did not induce mitochondrial fission (Figures 2A, 2B and Figures 2F–2J). Hence, inositol likely acts as a second messenger to directly restrict mitochondrial fission.
Figure 2. Inositol directly regulates mitochondrial fission independently of its intermediates of phosphatidylinositol cycle.
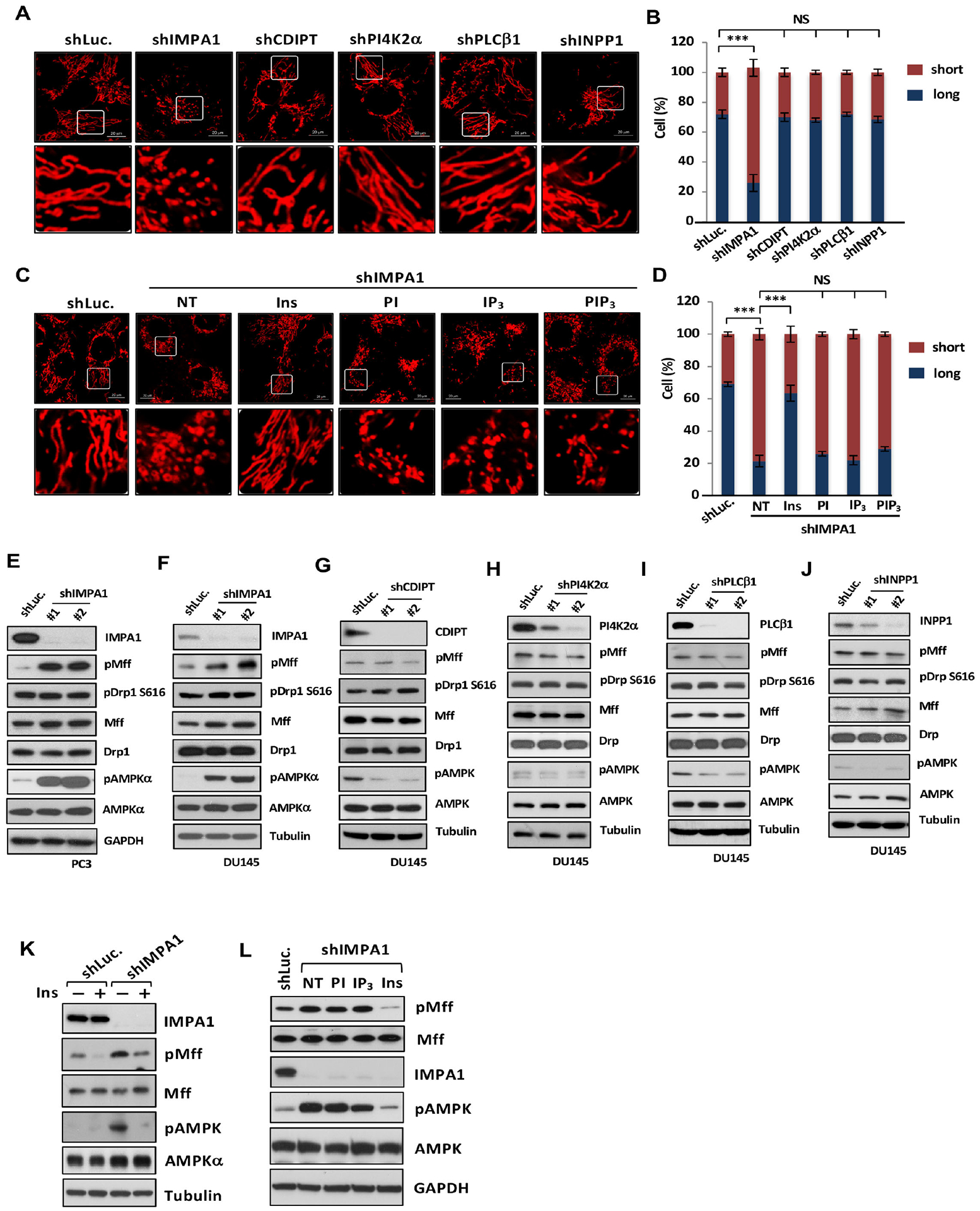
(A) Confocal images of mitochondrial morphology by MitoTracker (red) in diverse knockdown DU145 cells. Scale bar, 20 μm. (C) Confocal images of mitochondrial morphology in shLuc. or shIMPA1 DU145 cells upon indicated metabolites treatment. Vehicle, NT. (B and D) Quantification of the mitochondrial morphology of the cells shown in (A and C). (E-J) Immunoblotting of PC3 or DU145 cells stably expressing shLuc. or specific shRNAs for diverse genes. (K) Immunoblotting of shLuc. or shIMPA1 DU145 cells with or without 25 μM inositol treatment. (L) Immunoblotting of shLuc. or shIMPA1 PC3 cells upon indicated metabolites treatment.
We found that the physiological levels of inositol are about 388 μM in MEFs, 773 μM in PC3 cells, 963 μM in 22RV1 cells and 963 μM in TRAMP-C2 cells (Figure S2D). Moreover, the endogenous levels of inositol in kidney where the inositol is mainly produced is 653 μM and 179 μM in mouse serum (Figure S3D). Additionally, the physiological concentration for the metabolites such as PI, PI(3,4,5)P3 and I(1,4,5)P3 is below 10 μM (Ando et al., 2003; Liang et al., 2014; McLaughlin et al., 2002). Mass spectrometry-based analysis revealed that the treatment of inositol and its downstream metabolites in IMPA1 knockdown cells rescued intracellular inositol or phosphoinositide levels to the levels similar to control knockdown cells (Figures S3E and S3F). Of note, adding 25 μM of inositol, but not 4 μM of PI, I(1,4,5)P3, and PI(3,4,5)P3, in IMPA1 knockdown cells reversed the mitochondrial fission phenotype (Figures 2C, 2D and Figures 1I, 1J). Similarly, inositol also abrogated mitochondrial fission induced by lithium chloride (Figures S3G–S3H).
Mitochondrial fission commences upon the induction of the phosphorylation of dynamin-related protein 1 (Drp1) at S616 or mitochondrial fission factor (Mff) at S146 (Ducommun et al., 2015; Kashatus et al., 2015). By contrast, phosphorylation of Drp1 at S637 (pS637Drp1), which is induced and reduced by protein kinase A (PKA) (Chang and Blackstone, 2007), negatively regulates mitochondrial fission. We observed that the phosphorylation of Mff at S146 (pMff), but not phosphorylation of Drp1 at S616 (pS616Drp1), was markedly elevated in both IMPA1 and IMPA2 knockdown cells (Figures 2E, 2F and S2E). Knockdown of CDIPT, PI4K2α, PLCβ1 and INPP1 failed to induce either pMff or pS616Drp1 (Figures 2G–2J).
However, pS637Drp1 and CREB S133 phosphorylation, indicative of PKA activity, were not affected by IMPA1 knockdown (Figure S4A), suggesting IMAP1/inositol-mediated mitochondrial fission is unlikely to act through PKA and calcineurin signaling pathways. Moreover, proteolytic processing of OPA1, which functions as mitochondrial fusion (Head et al., 2009; Ishihara et al., 2006), was not significantly changed upon IMPA1 knockdown (Figure S4A). Remarkably, add-back of inositol, but not of its downstream metabolites, PI and IP3, reversed the pMff (S146) levels in IMPA1 knockdown cells or IMPA1 knockdown cells permeabilized with streptolysin O (SLO), although equal permeability to each metabolite was shown (Figures 2K–2L and Figures S4B–S4C). As a major receptor for Drp1, pMff (S146) recruits Drp1 to the mitochondrial outer membrane during mitochondrial fission (Loson et al., 2013; Otera et al., 2010). Consistently, Drp1 was recruited to and co-localized with mitochondria in IMPA1 knockdown cells (Figures S4D and S4E). Of note, Drp1 recruitment to mitochondria in IMPA1 knockdown cells was compromised by inositol restoration (Figures S4D and S4E). Collectively, IMPA-derived inositol serves as a repressor for pMff (S146) induction and mitochondrial fission independently of its conversion to downstream metabolites.
Aberrant mitochondrial fission leads to mitochondrial depolarization, autophagy/mitophagy and/or apoptosis (Twig et al., 2008; Twig and Shirihai, 2011; Youle and Karbowski, 2005). Consistently, mitochondrial membrane potential was decreased in IMPA1 knockdown cells (Figures S5A and S5B). Notably, restoration of IMPA1, but not IMPA1 D220A, reversed mitochondrial depolarization in IMPA1 knockout cells (Figure S5C). Mitochondrial oxygen consumption (OCR) was also reduced in IMPA1 knockdown cells, and such reduction was reversed by inositol restoration (Figure S5D). IMPA1 knockdown cells also displayed autophagy judged by the increased LC3II level, which was antagonized by inositol treatment (Figure S5E). However, IMPA1 knockdown did not affect the basal level of reactive oxygen species and cell survival, although it induced mitochondrial fission (Figures S5F–S5I). Hence, inositol plays an important role in maintaining mitochondrial health and homeostasis.
Inositol binds to and inactivates AMPK activity leading to restricting mitochondrial fission.
The finding that inositol, but not its downstream metabolites, restricts mitochondrial fission indicates that inositol may have its direct protein targets, which have not yet been identified. We sought to identify direct targets of inositol responsible for mitochondrial fission regulated by inositol. To achieve this goal, we synthesized biotin-labeled inositol and mixed biotin or biotin-labeled inositol with PC3 cell extracts, followed by biotin-streptavidin pulldown (Figure 3A). We focused on the key kinase and phosphatase involved in mitochondrial fission including PKA (Dickey and Strack, 2011), calcineurin (Cribbs and Strack, 2007), Erk1 (Kashatus et al., 2015), and AMPK (Toyama et al., 2016). Of note, inositol selectively interacted with the 5’-AMP-activated protein kinase (AMPK) complex including AMPKα, AMPKβ and AMPKγ, but not with other kinases or phosphatase involved in mitochondrial fission (Figure 3B). Moreover, inositol also failed to interact with other proteins involved in mitochondrial fission/fusion, such as Drp1, Fis1, Mff, MiD51, Opa1, Mfn1 and Mfn2, and autophagy/mitophagy, like LC3, LAMTOR2 and mTOR (Figure S6A).
Figure 3. Inositol binds to AMPK and regulates AMPK activity and mitochondrial fission.
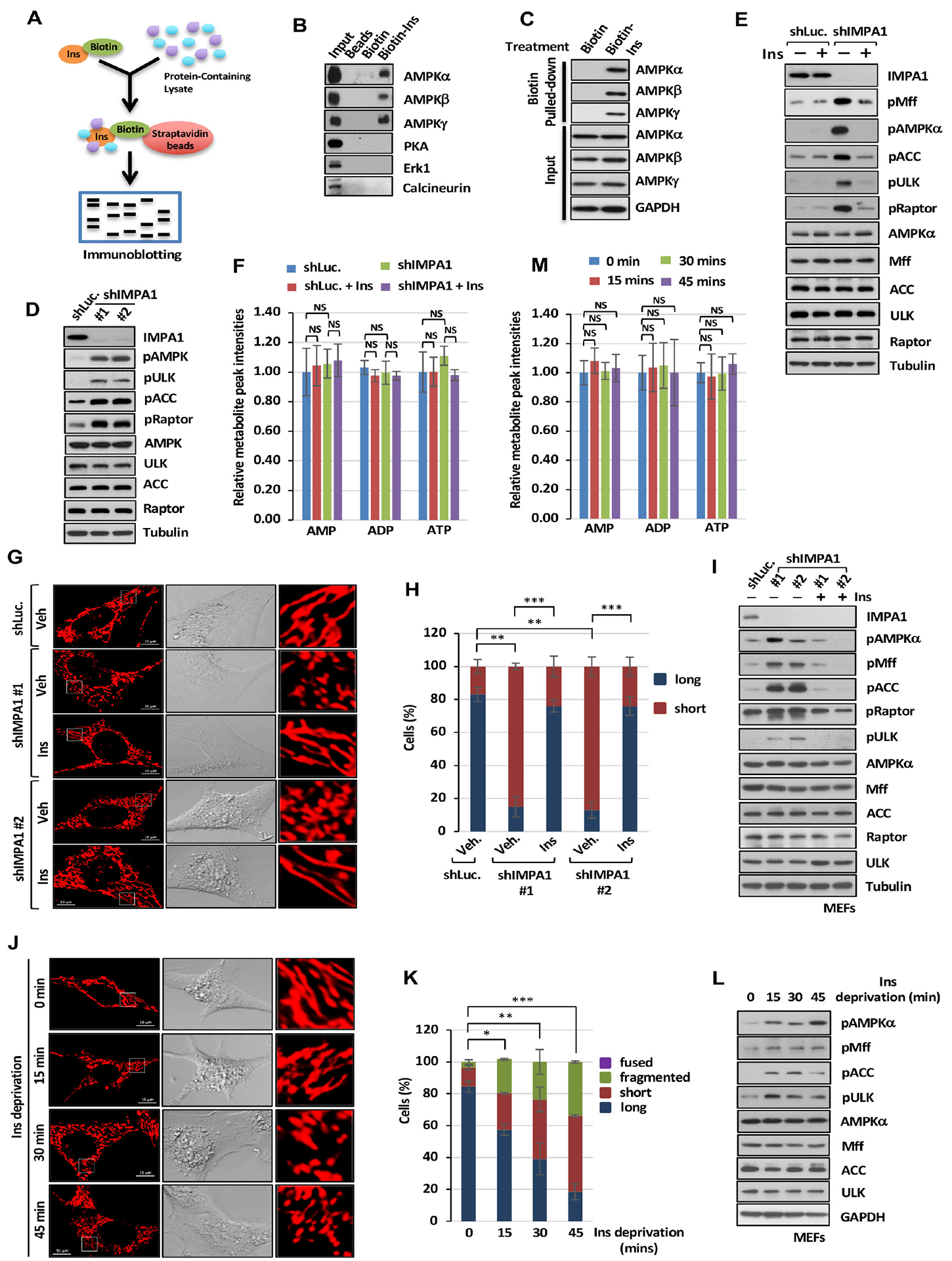
(A) Biotin-labeled inositol (Biotin-Ins) in vitro binding assay was performed by immunoblotting analysis. (B) Immunoblotting of biotin pull-down assay in PC3 cells with indicated antibodies. (C) Immunoblotting of biotin pull-down assay upon 25 μM of biotin or Biotin-Ins treatment for 1 hour. (D) Immunoblotting of shLuc. or shIMPA1 PC3 cells. (E) Immunoblotting with of shLuc. or shIMPA1 PC3 cells upon vehicle or Ins treatment. (F) Metabolic profiling of AMP, ADP and ATP in DU145 cells expressing shLuc. or shIMPA1 upon vehicle (Veh) or Ins treatment was performed by targeted mass spectrometry analysis. (G) Confocal images of mitochondrial morphology in shLuc. or shIMPA1 MEFs upon Veh. or Ins treatment. Scale bar, 10 μm. (H) Quantification of the mitochondrial morphology of the MEFs shown in (G). (I) Immunoblotting of shLuc. or shIMPA1 MEFs upon Veh. or Ins treatment. (J) Confocal images of mitochondrial morphology in MEFs upon Ins deprivation. Scale bar, 10 μm. (K) Quantification of the mitochondrial morphology of the cells shown in (J) upon Ins deprivation. (L) Immunoblotting of MEFs upon Ins deprivation. (M) Metabolic profiling of AMP, ADP and ATP in MEFs upon inositol deprivation was performed.
To determine whether the interaction between inositol and AMPK occurs in cells in vivo, we treated cells with biotin or biotin-labeled inositol. Streptavidin pull-down assay revealed that inositol could interact with AMPKα, AMPKβ and AMPKγ in cells (Figure 3C). Using the immunoprecipitation assay with AMPKα antibody under glucose-proficient or glucose-deprived conditions or with HA antibody, high levels of endogenous inositol were detected in AMPKα immunocomplex under glucose-containing conditions and in HA-AMPKα overexpression immunocomplex (Figures S6B and S6C), indicative of the in vivo interaction between inositol and AMPKα. Moreover, lower levels of endogenous inositol were detected in AMPKα immunocomplex upon glucose deprivation (−Glc.) compared with glucose-proficient conditions (+Glc.) (Figure S6B), suggesting that AMPK activation upon glucose deprivation is correlated with lower inositol binding to AMPK compared with AMPK inactivation under glucose-proficient conditions.
AMPK activation induced by energy stress inducers such as mitochondria-damaging agents elicits mitochondrial fission through directly phosphorylating Mff at S146 (pMff) (Ducommun et al., 2015; Toyama et al., 2016). Increased AMP level following the decline in ATP level is thought to be a critical event for AMPK activation under distinct stress conditions (Hsu et al., 2021; Steinberg and Carling, 2019). Our finding that inositol decline upon loss of IMPA1 or IMPA2 induces pMff (S146) and mitochondrial fission and that inositol interacts with AMPK suggests that inositol is likely a direct suppressor of AMPK. Indeed, both IMPA1 and IMPA2 knockdown cells displayed markedly enhanced AMPK activation, reflected by elevated phosphorylation of AMPK at T172 and phosphorylation of its downstream substrates (Figures 3D, 2E, 2F, S1K and S6D). Inactivation IMPA1/2 by lithium chloride, but not GSK3β inhibitor BIO, also elevated phosphorylation of AMPK and its downstream substrate, Mff (Figure S6E). Moreover, restoration of IMPA1, but not IMPA1 D220A, reversed AMPK and Mff phosphorylation in IMPA1 knockout cells (Figure S2I), further supporting that phosphatase activity of IMPA1 is crucial for suppressing mitochondrial fission. Restoration of inositol, but not PI and IP3, in IMPA1 knockdown, IMPA1 knockdown SLO-permeabilized or lithium chloride-treated cells attenuated phosphorylation of AMPK and its substrate, Mff (Figures 3E, 2K, 2L and S6E), similar to the treatment of AMPK inhibitor, compound C, (Figure S6F). However, knockdown of CDIPT, PI4K2α, PLCβ1 and INPP1 failed to induce phosphorylation of AMPK and Mff (Figures 2G–2J). Surprisingly, AMP, ADP and ATP levels, AMP/ATP ratio or ADP/ATP ratio in IMPA1 knockdown cells or inositol-restored IMPA1 knockdown cells remained unchanged compared with control knockdown cells (Figure 3F and Figures S6G–S6L).
Since our above findings were conducted in an inositol-containing medium, we asked the question whether the loss of IMPA1 also triggers AMPK activation and mitochondrial fission in inositol-free medium. We indeed reproduced our findings in an inositol-free medium that inositol decline upon IMPA1 knockdown triggers mitochondrial fission and AMPK activation (Figures 3G–3I). Remarkably, restoration of inositol completely impaired aberrant mitochondrial fission, AMPK activation and phosphorylation of its downstream substrates in inositol-free medium. Of note, inositol deprivation in MEFs or IMPA1 knockdown MEFs for 15 to 45 minutes triggered phosphorylation of AMPK and its substrates and mitochondrial fission without affecting ATP levels, suggesting that inositol decline is sufficient to induce AMPK activation and mitochondrial fission independently of ATP level (Figures 3J–3M and Figure S6M). Hence, IMPA-derived inositol serves as an endogenous suppressor of AMPK activation in cells.
Consistent with the previous study (Mihaylova and Shaw, 2011), AMPK deficiency impaired the induction of mitochondrial fission as well as pMff and pULK level upon the treatment of mitochondrial electron transport chain (ETC) inhibitors, rotenone and antimycin A (Figure 4A). AMPK activation, Mff phosphorylation and mitochondrial fission upon IMPA1 knockdown, IMPA1 knockout or IMPA inactivation through lithium chloride were all compromised upon AMPK knockdown or knockout, accompanied by reduced pMff or pULK level (Figures 4B–4E; Figures 4F–4H and Figures S3G–S3H). Decreased mitochondrial membrane potential induced by loss of IMPA/inositol was also rescued upon AMPK knockdown, AMPK inhibition by Compound C or AMPK knockout (Figures S5J–S5S). Thus, IMPA/inositol restricts mitochondrial fission and maintains mitochondrial membrane potential through inhibiting AMPK activation.
Figure 4. Inositol decline upon IMPA1 loss induces mitochondrial fission in an AMPK-dependent manner.
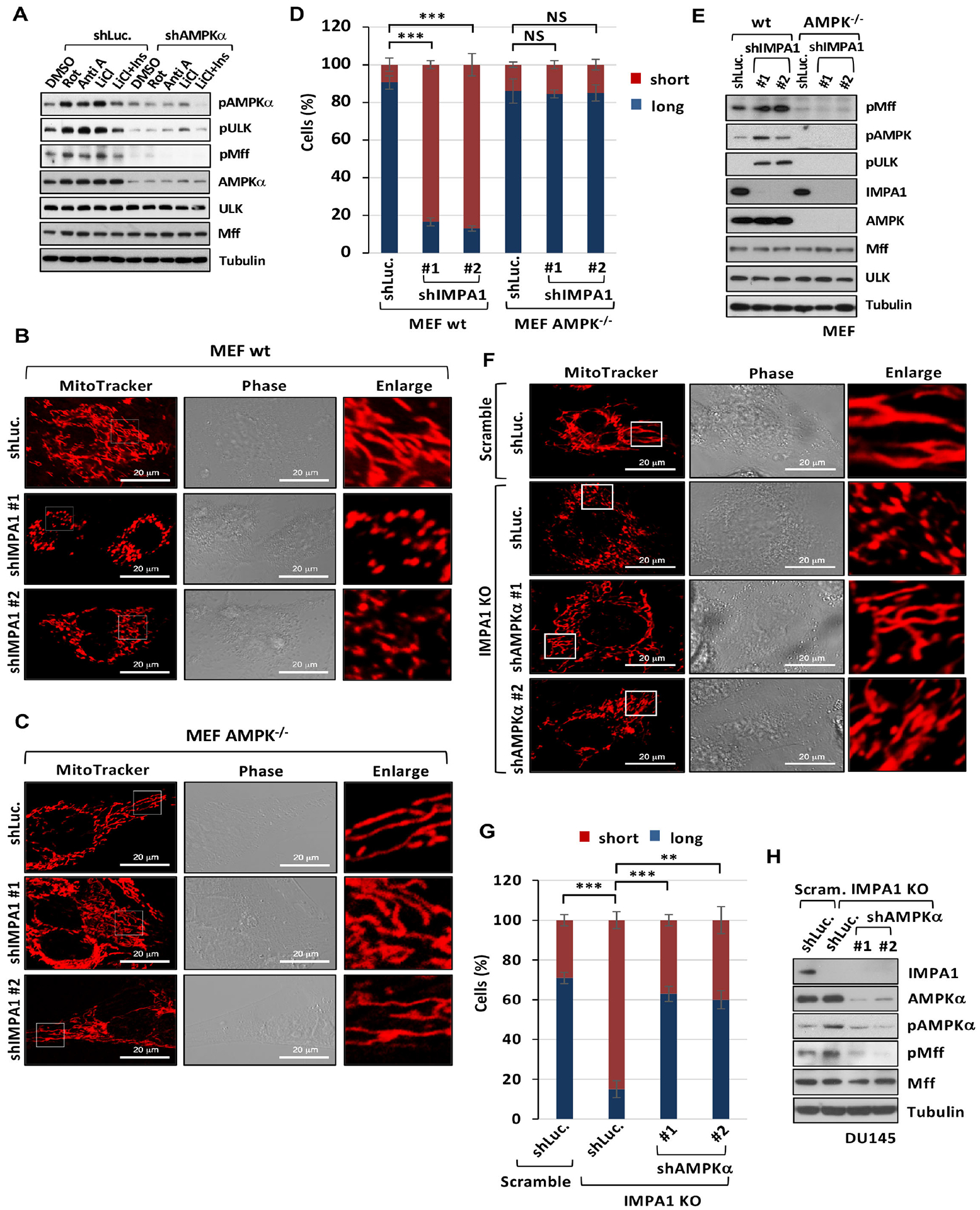
(A) Immunoblotting of shLuc. or shAMPKα DU145 cells treated with DMSO, 250 ng/ml of rotenone (Rot), 10 μM of antimycin A (Anti A), 500 μM of LiCl or 500 μM of LiCl plus 25 μM of Ins. (B and C) Confocal images of mitochondrial morphology in wild type (wt) MEFs and AMPK−/− MEFs stably transduced with shLuc. or shIMPA1. Scale bar, 20 μm. (D) Quantification of the mitochondrial morphology of MEFs shown in (B and C). (E) Immunoblotting of wt and AMPK−/− MEFs stably expressing shLuc. or shIMPA1. (F) Confocal images in CRISPR/Cas9 knockout DU145 cells stably transduced with the guide RNA pairs of scramble (Scram.), IMPA1 knockout clone #1 (IMPA1 KO), shLuc., or shAMPKα. Scale bar, 20 μm. (G) Quantification of the mitochondrial morphology of the DU145 shown in (F). (H) Immunoblotting of CRISPR/Cas9 knockout DU145 cells transduced with the guide RNA pairs of Scram., IMPA1 KO, shLuc., or shAMPKα.
Inositol decline leads to AMPK activation and mitochondrial fission under diverse stresses.
Glucose through glycolysis serves as a main source for the generation of G6P, which is a key precursor for inositol biosynthesis (Eisenberg, 1967). We rationalized that glucose would be a key nutrient for maintaining inositol levels in cells. Indeed, glucose deprivation or mitochondria-damaging agents like rotenone or antimycin A treatment reduced inositol levels, similar to IMPA1 knockdown or lithium chloride treatment (Figures S6N and Figures S1B–S1C), correlated with reduced G6P levels (Figure S6O). As a result, glucose deprivation not only decreased inositol levels but also markedly reduced inositol-AMPK binding (Figure S6B). Since inositol serves as a barrier to restrict AMPK-dependent mitochondrial fission, we determined whether glucose represents an important physiological cue to restrict mitochondrial fission. By performing time-lapse microscopic experiments, we observed the occurrence of fragmented mitochondria indicative of mitochondrial fission 10 min after glucose deprivation, similar to rotenone treatment (Figures 5A–D and movie S1, S3). This was correlated with AMPK activation (Figures 5E, 5F and Figures S6P–S6Q). Of note, inositol restoration reversed mitochondrial fragmentation induced by not only glucose deprivation but also rotenone or antimycin A treatment, which also caused the reduction of inositol levels in cells (Figures 5A–5D; Figure S6N and movie S2, S4), correlated with reduced AMPK activation (Figures 5E–5F and Figures S6P–S6Q). Thus, glucose is a key physiological cue to limit AMPK activation and mitochondrial fission by maintaining inositol levels.
Figure 5. Inositol decline leads to AMPK activation and mitochondrial fission under diverse stresses.
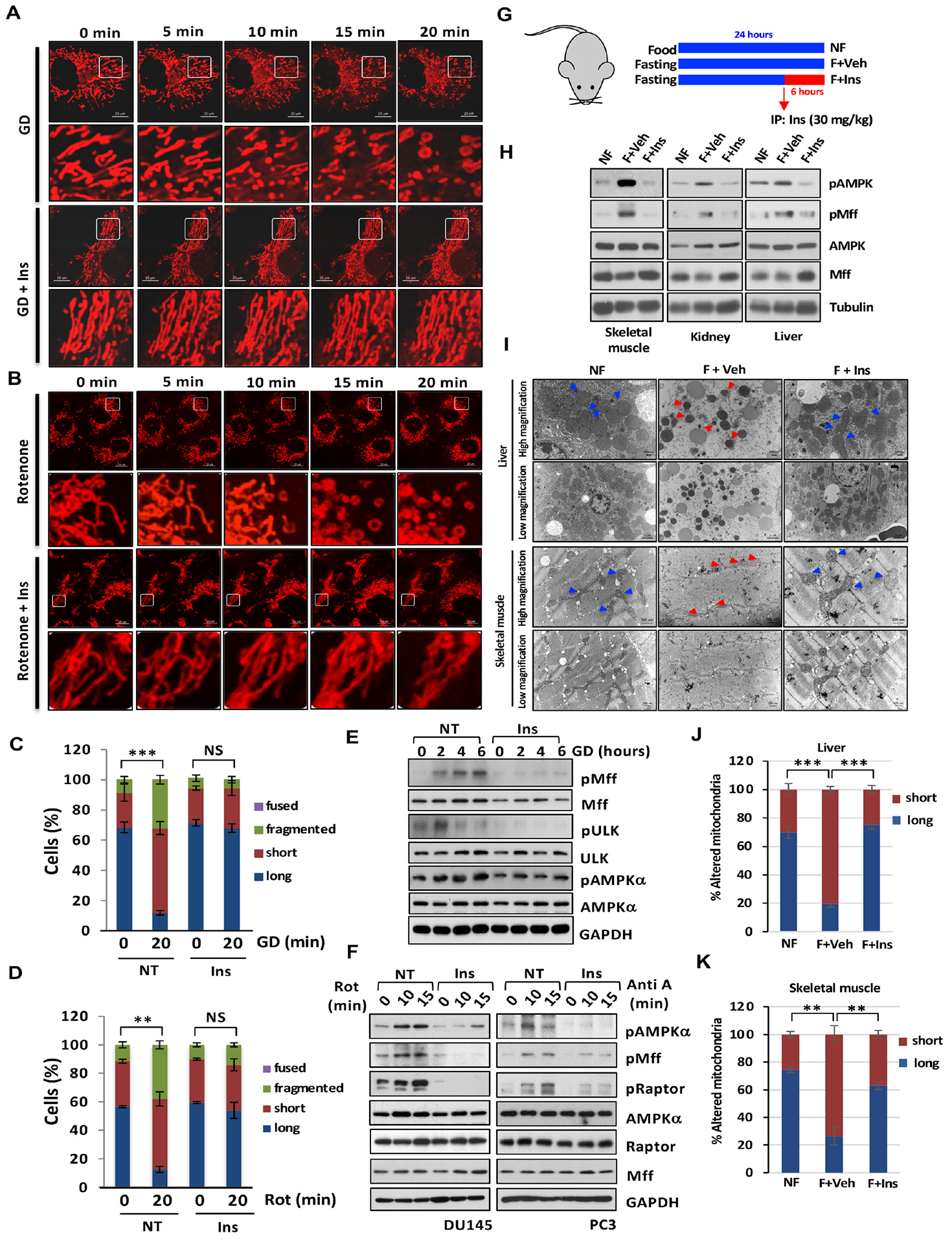
(A) Time-lapse images of DU145 cells stained with MitoTracker upon glucose deprivation (GD) or GD with Ins treatment for various time points. Scale bar, 20 μm. (B) Time-lapse images of DU145 cells stained with MitoTracker upon indicated treatments. Scale bar, 20 μm. (C) Quantification of the mitochondrial morphology of the cells shown in (A) at 20 min upon indicated treatment. (D) Quantification of the mitochondrial morphology of the cells shown in (B) at 20 min upon rotenone (Rot) with or without Ins treatment. (E) Immunoblotting of DU145 cells upon indicated treatments. (F) Immunoblotting of PC3 or DU145 cells upon indicated treatments. (G) The graphic showed mice fasted for 24 hours and Ins treatment by intraperitoneal injection (30 mg/kg) at mice fasted for 18 hours. (H) The protein extract from diverse tissues were subjected to Immunoblotting. (I) Images of TEM upon mice fed with food, no fasting (NF), fasted with vehicle (F+Veh) and fasted with Ins (F+Ins). Scale bar, 500 nm and 2 μm. (J and K) Quantification of the mitochondrial morphology (long and short) shown in (I).
To further investigate the physiological role of inositol in repressing AMPK-dependent mitochondrial fission in vivo, we conducted the animal study in which mice fasted for 24 hours to reduce inositol levels (Figures S7A–S7C and Figures S7J–S7K), followed by intraperitoneal injection or oral administration of vehicle or inositol (Figures 5G and S7D). Inositol levels in tibialis anterior, liver and kidney were partially rescued in fasted mice via either intraperitoneal injection or oral administration (Figures S7A–S7B and Figures S7J–S7K). Transmission electron microscopy revealed that fragmented mitochondria were significantly increased in liver and skeletal muscle of fasted mice, correlated with elevated phosphorylation of AMPK at T172 and phosphorylation of Mff at S146 (Figures 5H–5K and Figures S7E–S7I). Of note, inositol restoration in fasted mice abrogated the elevation of AMPK and Mff phosphorylation and mitochondrial fragmentation in skeletal muscle, kidney and liver (Figures 5H–5K and Figures S7E–S7I). Hence, inositol is an endogenous inhibitor of AMPK-dependent mitochondrial fission.
CDIPT loss leading to inositol accumulation prevents mitochondrial fission upon energy stress.
If inositol reduction in cells elicits spontaneous AMPK activation and mitochondrial fission, inositol accumulation in cells should exhibit an opposite effect on AMPK activation and mitochondrial fission under energy stress conditions. We tested this hypothesis by establishing cells with inositol accumulation. Since CDIPT is the enzyme converting inositol to PI, loss of CDIPT may lead to inositol accumulation. Indeed, loss of CDIPT caused inositol accumulation and abrogated AMPK activation, pMff induction and mitochondrial fission upon glucose deprivation or antimycin A treatment (Figures 6A–6E). Of note, treatment of IMPA1/2 inhibitor lithium chloride could partially rescue the defects in AMPK activation, pMff induction and mitochondrial fission in CDIPT knockdown cells upon glucose deprivation (Figures 6F, 6G and Figure S6R). While AMPK activation upon the treatment of AMPK activators, such as AICAR or A769662, induced pMff and mitochondrial fission, inositol treatment inhibited AMPK activation, pMff induction and mitochondrial fission induced by AICAR or A769662 (Figures S8A–S8E and movie S5, S6). Collectively, inositol decline triggers AMPK activation and mitochondrial fission under energy stress conditions.
Figure 6. CDIPT loss leading to inositol accumulation prevents mitochondrial fission upon energy stress.
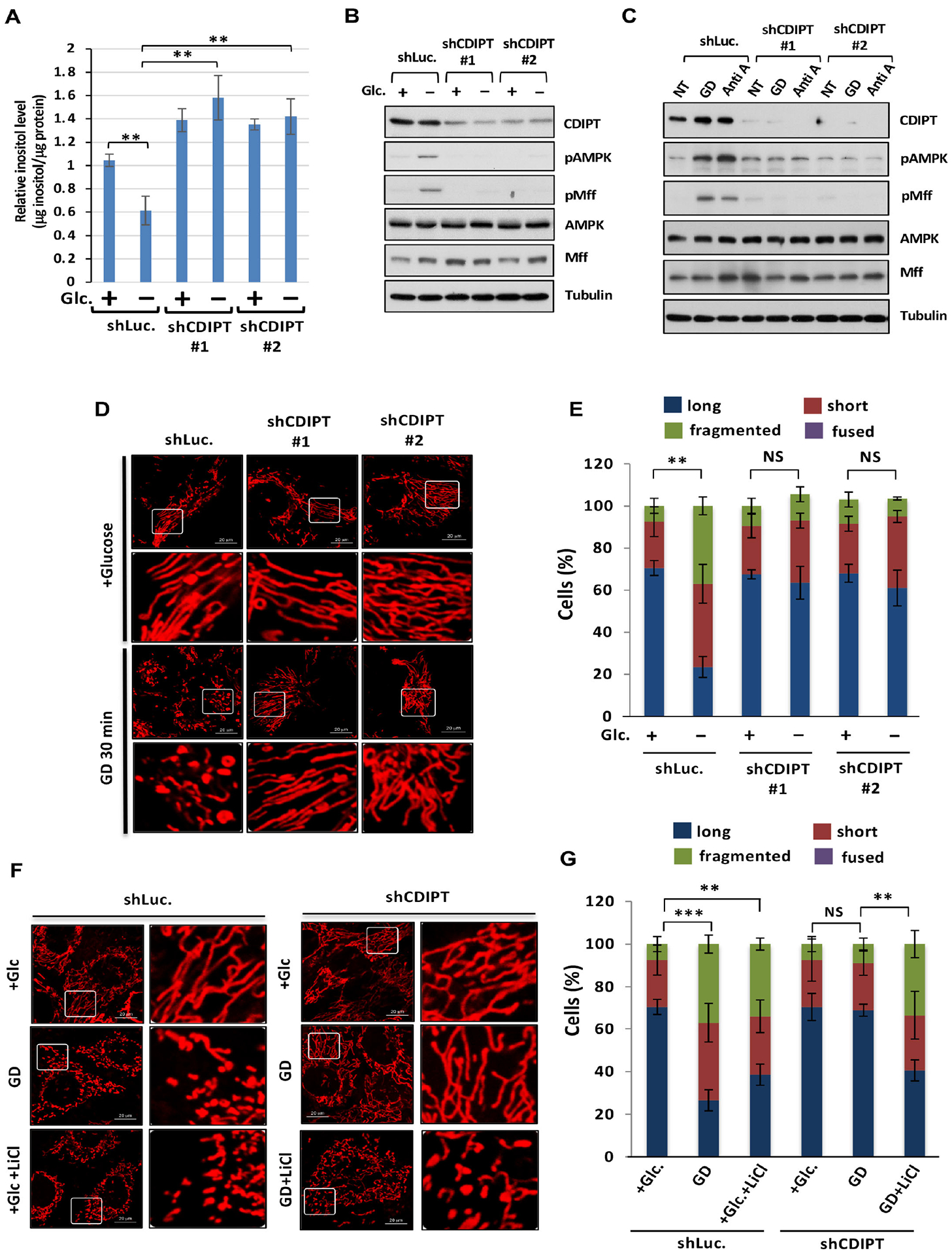
(A) The levels of inositol in shLuc. or shCDIPT DU145 with or without glucose for 30 minutes is determined by K-INOSL assay kit. (B) Immunoblotting of shLuc. or shCDIPT DU145 cells with or without glucose deprivation (GD) for 30 minutes. (C) Immunoblotting of shLuc. or shCDIPT PC3 cells upon vehicle (NT), GD or Anti A. (D) Confocal images of mitochondrial morphology in shLuc. or shCDIPT DU145 cells upon GD for 30 minutes. Scale bar, 20 μm. (E) Quantification of the mitochondrial morphology of the cells shown in (D). (F) Confocal images of mitochondrial morphology in shLuc. or shCDIPT DU145 cells upon GD or lithium chloride treatment for 30 minutes. (G) Quantification of the mitochondrial morphology of the cells shown in (F).
AMPK activation plays a role in maintaining cell survival under metabolic stress (Jones et al., 2005). We speculated that IMPA1 knockdown cells with elevated AMPK activation may exhibit less cell death feature under metabolic stress compared with control knockdown cells. Indeed, the impairment of apoptosis, accompanied by marked reduction in cleaved PARP, caspase 3 and elevated AMPK activation, upon glucose deprivation was observed in IMPA1 knockdown cells (Figures S9A–S9C), indicating that IMPA/inositol regulates AMPK-mediated mitochondrial fission and cell survival under metabolic stress.
Inositol competes with AMP in binding to AMPKγ subunit and represses AMPK kinase activity.
AMP serves as a physiological activation of AMPK complex by directly binding to AMPKγ subunit, while ATP acts as a suppressor of AMPK by competing with AMP for binding to AMPKγ (Gowans et al., 2013). Hence, increase of AMP/ATP ratio results in AMPK activation during energy stress (Gowans et al., 2013). As inositol interacts with AMPK and serves as a key metabolite to restrict AMPK activation and mitochondrial fission in cells and in mice, we assessed whether inositol could directly inhibit AMPK kinase activity. The in vitro kinase assay showed that inositol inhibits kinase AMPK kinase activity towards phosphorylation of two AMPK substrates, GST-Mff or ULK protein (Figures 7A–7C and Figure S10A). We also performed AMPK enzymatic kinetic curve under different concentrations of SAMS peptide and inositol. Ki (519.8 μM) for inositol on AMPK activity was obtained by fitting the Michaelis-Menten equation (Figures 7D and 7E), and inositol displayed non-competitive inhibition according to Lineweaver-Burk plot (Figures 7D and S10B), suggesting that inositol potentially serves as an allosteric inhibitor of AMPK. Given AMPK has multiple isoforms, we also examine the effects of inositol on activity of different AMPK isoform complexes. The results revealed that inositol inhibits the activity of AMPKα1β1γ1, AMPKα1β2γ1, AMPKα2β2γ1 and AMPK α1β1γ3 complexes, but not the activity of AMPKα2β1γ1 and AMPK α1β1γ2 complexes. (Figures 7B–7C and Figures S10C–S10F). Remarkably, inositol, but not PI, IP3 and inositol monophosphate (IP), impaired AMPK kinase activity (Figures S10G and S10H). Moreover, AMP directly enhances AMPK kinase activity in a dose-dependent manner (Figures 7G and S10I).
Figure 7. Inositol competes with AMP in binding to AMPKγ and represses AMPK kinase activity.
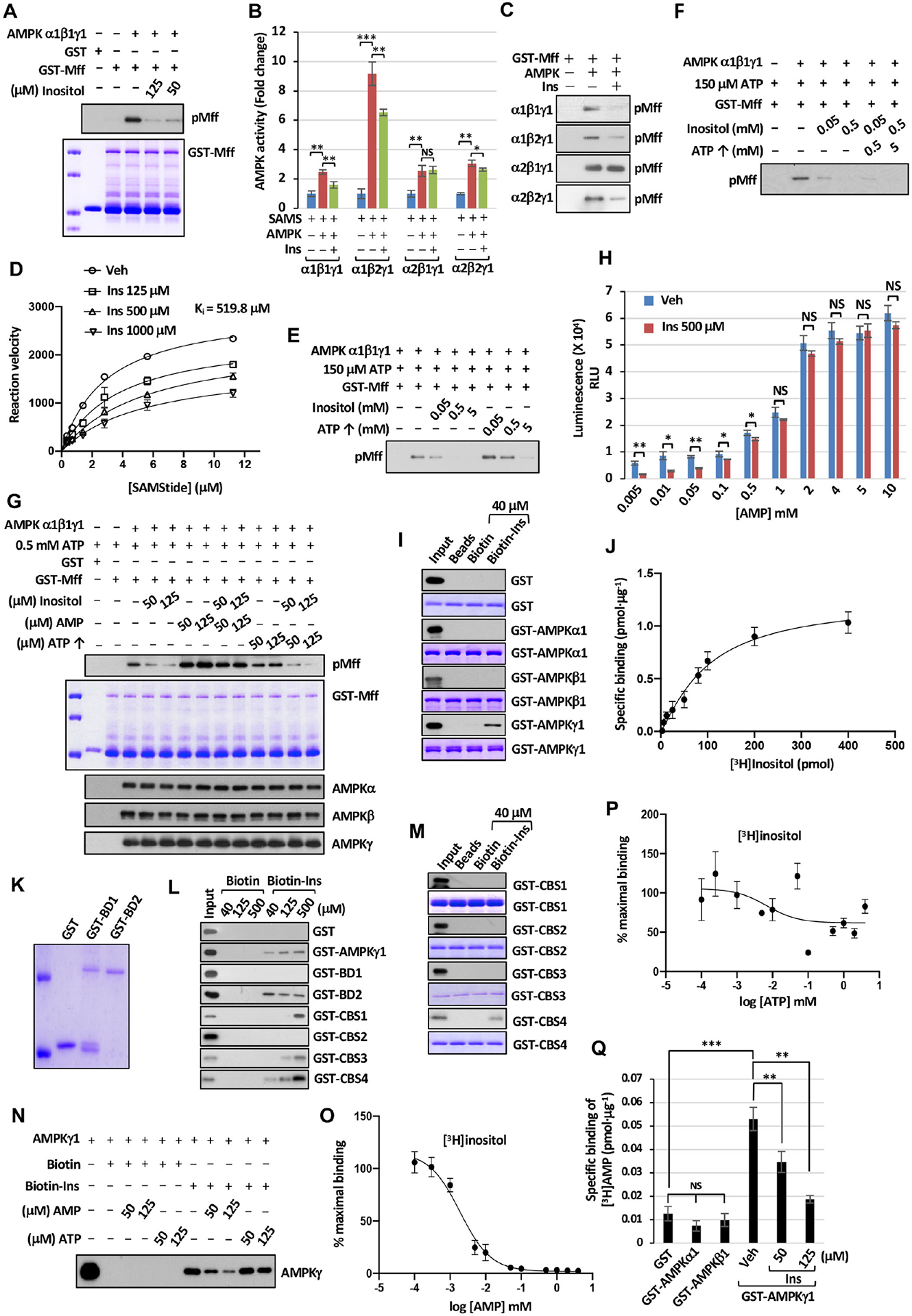
(A) AMPK kinase activity upon Ins treatment was determined by immunoblotting with pMff. (B) AMPK kinase activity was determined by ADP-Glo™ kinase assay. (C) AMPK kinase activity was determined by immunoblotting with pMff. (D) The effects of Ins on AMPK activity using SAMS peptide were determined by fitting Michaelis-Menten equation. Ki for inositol was calculated by using Michaelis-Menten equation. (E) AMPK kinase activity with fixed amount of ATP upon Ins or ATP treatment was determined by immunoblotting with pMff. (F) AMPK kinase activity with fixed amount of ATP upon adding Ins with or without ATP was determined by immunoblotting with pMff. (G) AMPK kinase activity upon adding Ins, AMP or ATP was determined by immunoblotting with pMff. (H) Bar graphs represent each concentration of AMP with or without Ins in AMPK enzymatic kinetic curve. (I and M) Immunoblotting of biotin pull-down assay incubated with recombinant fusion proteins. (J) Saturation binding of [3H]inositol with recombinant GST-AMPKγ1 proteins. Dissociation constant (Kd) value of 98.2 pmol ± 2.8 pmol was determined for binding affinity between Ins and AMPKγ1. (K) Coomassie blue staining of the indicated recombinant proteins. GST-Bateman domain 1 (BD1) and Bateman domain 2 (BD2). (L) The binding between Biotin-Ins and indicated recombinant proteins was determined by immunoblotting. (N) The competitive binding assay was subjected to immunoblotting with specific AMPKγ antibody. (O) Competition binding of [3H]inositol (3.75 μM) and AMP with recombinant GST-AMPKγ1 protein (100 ng). IC50 = 1.9 μM ± 0.5 μM. (P) Competition binding of [3H]inositol (3.75 μM) and ATP with recombinant GST-AMPKγ1 protein. (Q) The competitive binding assay between AMP and Ins was determined upon incubation of 3.33 μM of tritium-labeled AMP (3H-AMP) and indicated recombinant proteins.
To further elucidate whether both inositol and ATP are essential for inhibition of AMPK activity, we examined in vitro AMPK activity upon different concentrations of inositol and ATP. While 50 and 500 μM of inositol was sufficient to inhibit AMPK kinase activity, ATP displayed no significant inhibitory effect at these two concentrations on AMPK kinase activity (Figure 7E and Figures S10J–S10K). However, ATP at physiological concentration (5 mM) achieved the inhibitory effect on AMPK kinase activity (Figure 7E). Under physiological concentrations of both inositol and ATP, inositol could cooperate with ATP to inhibit AMPK kinase activity (Figure 7F). Importantly, the inhibitory effect on AMPK kinase activity by inositol could be rescued by adding AMP, but not ATP (Figure 7G). Inositol also attenuated AMPK activity and AMPK kinase activation induced by low concentration of AMP, while high concentration (1 mM or above) of AMP impaired the inhibitory effect of inositol (0.5 mM) on AMPK kinase activity (Figures 7H and S10L), suggesting that inositol and ATP represent endogenous inhibitors of AMPK, but inositol requires much lower concentration than ATP to achieve its inhibitory effect on AMPK activation. Collectively, inositol not only serves as an allosteric inhibitor of AMPK via non-competitive inhibition with the substrate, SAMS peptide, but also represses AMP-mediated AMPK activation via competing with AMP for binding to AMPKγ.
In vitro binding assay using recombinant proteins revealed that inositol could directly bind to AMPKγ1, but not AMPKα1 and AMPKβ1 (Figure 7I). Saturation binding assay using tritium-labeled inositol revealed that inositol directly binds to AMPKγ1 in a dose-dependent manner (Figure 7J). Direct binding of inositol to AMPKγ1 was also demonstrated by isothermal titration calorimetry (ITC) (Figures S10M–S10O). Inositol binds to Bateman domain 2 composed of cystathionine-β synthase (CBS) 3 and CBS4 motifs, but not Bateman domain 1 composed of CBS1 and CBS2 motifs. Moreover, inositol could bind to single CBS1, CBS3 and CBS4 motifs of AMPKγ1, but not CBS2 motif within its physiological concentration in cells (Figures 7K–7M). However, inositol bound to the CBS4 motif of AMPKγ1 with much higher affinity than CBS1 and CBS3 motifs of AMPKγ1, as it at 40 μM could bind to the CBS4 motif of AMPKγ1, but not CBS1 and CBS3 motifs of AMPKγ1 (Figures 7L and 7M).
The current model proposed that the binding of AMP to AMPKγ subunit causes the conformational change of AMPK complex that exposes AMPKα for the phosphorylation of AMPKα at T172 by its upstream kinases leading to AMPK activation (Gowans et al., 2013). Thus, AMP serves as an allosteric activator for AMPK activation. Given AMP, but not ATP, rescued impaired of AMPK kinase activity by inositol upon the addition of the same doses (50 and 125 μM) in vitro (Figure 7G), we examine whether AMP abrogates the binding of inositol to AMPKγ1. The result revealed that increased amount of AMP, but not ATP, disrupted inositol-AMPKγ binding (Figure 7N). The competition binding assay showed that AMP, but not ATP, inhibits inositol binding to AMPKγ1 in a dose-dependent manner (Figures 7O and 7P). Reciprocally, we observed AMP bound to AMPKγ1, but not AMPK α1 and AMPKβ1, and such binding was markedly abolished by inositol (Figure 7Q). We conclude that inositol is a natural inhibitor of AMPKγ by directly binding to AMPKγ and preventing AMP from binding to AMPKγ.
Discussion
Mitochondrial fission and fusion are tightly regulated to maintain normal mitochondrial functions and cellular homeostasis. Defect in these processes is linked to diverse human diseases (Archer, 2013; Youle and van der Bliek, 2012). While numerous proteins are identified in regulating mitochondrial fission or fusion (Cardenas et al., 2010; Cogliati et al., 2013; Loson et al., 2013; Richter et al., 2014), the specific metabolites impinging on mitochondrial dynamics are largely unknown. Our study identifies inositol, generated from IMPA1 and/or IMPA2 localized in mitochondria, as a key metabolite to orchestrate mitochondrial dynamics. We show that decline in inositol levels during metabolic stress drives mitochondrial fission in cells and mice leading to muscle atrophy (Figure 5 and Figure S7). Hence, imbalance of inositol level leading to defect of mitochondrial dynamics provides great insight into the pathogenesis of mitochondrial fission and fusion in human disease (Archer, 2013).
Earlier studies suggest that the functional role of inositol primarily attributes to the generation of its downstream metabolites. However, the direct role of inositol in serving as a second messenger to regulate signaling and biological outcomes has not been reported. We present convincing evidence that inositol serves as an allosteric inhibitor that represses AMPK activation to restrict mitochondrial fission independently of its known role in serving as a precursor for PI and phosphoinositides. In addition, inositol also regulates AMPK activation to regulate cell death feature under metabolic stress (Figures S9A–S9C), suggesting IMPA/inositol modulates diversely biological functions controlled by AMPK. Our study unveils that inositol is a previously uncharacterized metabolite directly restricting AMPK-dependent mitochondrial fission.
AMPK is activated in cells under energy stresses and inhibition of mitochondrial electron transport chain through increased AMP level (Hardie, 2011). Our study reveals that in addition to AMP elevation, inositol decline is also a prerequisite step for AMPK activation upon the stimulation of these stress conditions. Inositol reduction results in unoccupied AMPKγ CBS1, CBS3 and CBS4 motifs, thereby allowing for AMP-AMPKγ binding to facilitate AMPK activation and mitochondrial fission (Figure S11). Hence, AMP/inositol ratio is a critical determinant for AMPK activation. Notably, we identify IMPA1 and IMPA2 as key enzymes to maintain inositol biosynthesis, whose deficiency or inactivation is sufficient for AMPK activation and mitochondrial fission without affecting ATP level. It is important to note that the loss of CDIPT, a key enzyme depleting inositol level in cells by converting inositol to PI, causes inositol accumulation leading to the prevention of AMPK-dependent mitochondrial fission during energy stress conditions. Thus, inositol is a key rate-limiting metabolite that serves as an endogenous suppressor to restrict AMPK activation and mitochondrial fission in normal steady-state conditions.
AMPKγ subunit contains four tandem repeats of sequence motif termed the CBS repeat. There are three AMP binding sites (site-1, site-2 and site-3) where AMP binds to aspartic acid located on CBS1, CBS3 and CBS4 motifs, respectively (Xiao et al., 2007). Two AMP-1 and AMP-2 sites in that AMP binds to CBS1 and CBS3 motifs can be competed by ATP and is therefore involved in AMP/ATP sensing. However, since there is no competition for ATP in the AMP binding site-3 in which AMP tightly binds to CBS4 domain, ATP is not involved in AMP/ATP sensing (Xiao et al., 2007). Interestingly, AMP binding site-3 is tightly bound between CBS4 motif and AMP due to a serine residue (315) interacting with phosphate group which does not occur in the other two sites (Xiao et al., 2007). Our results further uncover that inositol binds to the CBS4 motif of AMPKγ with much higher affinity than CBS1 and CBS3 motifs of AMPKγ. As inositol binds to CBS4 motif of AMPKγ and competes with AMP for AMPKγ binding, it might also compete at AMP binding site-3. Moreover, since 50 and 125 μM of inositol is sufficient to repress AMPK kinase activity and inhibition of AMPK activity by ATP requires up to 5 mM, it is plausible that AMPK may sense the inositol gradient more efficiently and rapidly than ATP in response to diverse energy stresses. As both inositol and ATP could repress AMPK kinase activity under their physiological concentrations, AMPK could sense both inositol and ATP gradient for its activation in response to diverse energy stresses and mitochondrial damage (Figure S11).
In summary, our study provides the compelling evidence that inositol is a second messenger that directly binds to AMPKγ to limit AMPK activation for mitochondrial fission induction. This study expands our current understanding of how inositol regulates novel signaling and biological processes beyond its classic role in PI cycle regulation. We identify AMPK as an inositol sensor detecting the inositol gradient in cells to adjust its activity for regulating mitochondrial dynamics. As deregulation of mitochondrial dynamics underlies various human diseases, we speculate that certain human diseases associated with IMPA1/2 or CDIPT mutations (Figueiredo et al., 2016; Murphy et al., 2011; Ohnishi et al., 2014; Thakur et al., 2011) may result from aberrant AMPK-dependent mitochondrial fission caused by the alteration of inositol level. We propose that inositol treatment, activating IMPA1/2 and/or targeting CDIPT may be a promising strategy to mitigate various human diseases associated with abnormal AMPK-dependent mitochondrial dynamics.
Limitation of study
Although our in vitro results suggest that inositol allosterically inhibits AMPK enzymatic activity via directly competing with AMP for binding to AMPKγ subunit, it is not crystal clear how this competition indeed occurs. To gain deeper insight into how inositol competes with AMP for binding to AMPKγ, identification of the crystal structure of inositol-AMPKγ complex will be necessary. Another limitation concerning the role of IMPA1/inosiotl in restricting AMPK-mediated mitochondrial fission in our study is mostly conducted in cell-based models, although the role of inositol in suppressing AMPK-mediated mitochondrial fission was verified in in vivo animal experiments. Further in vivo experiments using conditional Impa1 knockout mice to examine elevated AMPK activation and mitochondrial fission in diverse tissues will be required to further strengthen the conclusion.
STAR★METHODS
Resource availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Hui-Kuan Lin (hulin@wakehealth.edu).
Materials Availability
The plasmids, stable cell lines and antibodies generated in this study have not been deposited to any repositories yet. These materials will be available upon request.
Data and Code Availability
The unprocessed data of immunoblotting have been deposited to Mendeley Data: http://dx.doi.org/10.17632/nvf6fgc77g.1 https://data.mendeley.com/datasets/nvf6fgc77g/draft?a=3c37aafc-0338-44f4-8619-183d8fc4e67f. The software utilized in this study can be found in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| SOURCE | ||
|---|---|---|
| IMPA1 (clone H-7) | Santa Cruz Biotechnology | Cat#sc-374234; RRID: AB_10988246 |
| Mff (clone B-2) | Santa Cruz Biotechnology | Cat# sc-398617; RRID: AB_2744543 |
| phospho-Mff Ser146 (polyclone) | Cell Signaling Tech | Cat#49281; RRID: AB_2799354 |
| IMPA2 (polyclone) | GeneTex | Cat#GTX103636; RRID: AB_1950587 |
| AMPKα (polyclone) | Cell Signaling Tech | Cat#2532; RRID: AB_330331 |
| AMPKβ (polyclone) | Cell Signaling Tech | Cat#12063; RRID: AB_ 2797812 |
| AMPKγ (polyclone) | Cell Signaling Tech | Cat#4187; RRID: AB_10695248 |
| phospho-AMPKα T172 (clone D4D6D) | Cell Signaling Tech | Cat#50081; RRID: AB_2799368 |
| PKA (clone A-2) | Santa Cruz, sc-28315 | Cat#sc-28315; RRID: AB_628136 |
| phospho-Drp1 S616 (polyclone) | Cell Signaling Tech | Cat#3455; RRID: AB_2085352 |
| phospho-Drp1 S635 (clone D3A4) | Cell Signaling Tech | Cat#6319; RRID: AB_10971640 |
| Fis (clone B-5) | Santa Cruz Biotechnology | Cat#sc-376447; RRID: AB_11149382 |
| Mfn1 (clone D-10) | Santa Cruz Biotechnology | Cat#sc-166644; RRID: AB_2142616 |
| Mfn2 (clone F-5) | Santa Cruz Biotechnology | Cat#sc-515647; RRID: AB_2811176 |
| ULK (clone D8H5) | Cell Signaling Tech | Cat#8054; RRID: AB_11178668 |
| Raptor (clone 24C12) | Cell Signaling Tech | Cat#2280; RRID: AB_561245 |
| phospho-ULK S555 (clone D1H4) | Cell Signaling Tech | Cat#5869; RRID: AB_10707365 |
| phospho-Raptor S792 (polyclone) | Cell Signaling Tech | Cat#2083, RRID: AB_2249475 |
| Actin (clone AC-15) | Sigma-Aldrich | Cat#A5441, RRID: AB_476744 |
| GAPDH (clone GAPDH-71.1) | Sigma-Aldrich | Cat#G8795, RRID: AB_1078991 |
| Tubulin (clone AA13) | Sigma-Aldrich | Cat#T8203, RRID: AB_1841230 |
| CDIPT (clone C-2) | Santa Cruz Biotechnology | Cat#sc-514255 |
| GST (clone B-14) | Santa Cruz Biotechnology | Cat#sc-138, RRID: AB_627677 |
| LC3 α/β (polyclone) | Novus | Cat#NB100-2220, RRID: AB_10003146 |
| Calcineurin (D-1) | Santa Cruz Biotechnology | Cat#sc-373803, RRID: AB_10917925 |
| MiD51 (clone A-6) | Santa Cruz Biotechnology | Cat#sc-514135 |
| CREB (clone 24H2B) | Santa Cruz Biotechnology | Cat#sc-271, RRID: AB_627300 |
| phospho-CREB S133 (clone 10E9) | Santa Cruz Biotechnology | Cat#sc-81486, RRID: AB_1125727 |
| choactase (clone E-7) | Santa Cruz Biotechnology | Cat#sc-55557, RRID: AB_2291743 |
| TOM20 (monoclone) | Abcam | Cat#ab56783, RRID: AB_945896 |
| HRP-coupled antibodies to mouse | Santa Cruz Biotechnology | Cat#sc-2055, RRID: AB_631738 |
| HRP-coupled antibodies to rabbit | Thermo Fisher | Cat#31480, RRID: AB_228457 |
| Alexa Fluor 488 anti-mouse IgG | Thermo Fisher | Cat#A-21202, RRID: AB_141607 |
| Rabbit normal IgG | Santa Cruz Biotechnology | Cat#sc-2027; RRID: AB_737197 |
| Mouse Anti-rabbit IgG | Cell Signaling Tech | Cat#45262; RRID: AB_2799281 |
| lithium chloride | Millipore | Cat#5910 |
| AICAR | Sigma-Aldrich | Cat#A9978 |
| A769662 | Reagents Direct | Cat#A769662 |
| Compound C | Sigma-Aldrich | Cat#P5499 |
| Myo-inositol | Sigma-Aldrich | Cat#I7508 |
| AMP | Sigma-Aldrich | Cat#A1752 |
| ATP | Sigma-Aldrich | Cat#A2383 |
| BIO | Selleckchem.com | Cat#S7198 |
| L-α-Phosphatidylinositol | Sigma-Aldrich | Cat#P6636 |
| myo-Inositol 1,4,5-trisphosphate hexapotassium salt | Tocris | Cat#1482 |
| phosphatidylinositol 3,4,5-triphosphate | Echelon® | Cat#P-3924 |
| AMPKα1, β1, γ1 protein | Millipore | Cat#14-840 |
| AMPKα1, β1, γ1 protein | Promega | Cat#V1921 |
| AMPKα1, β2, γ1 protein | Thermo Fisher | Cat#PV6244 |
| AMPKα2, β1, γ1 protein | BPS Bioscience | Cat#40024 |
| AMPKα2, β2, γ1 protein | Thermo Fisher | Cat#PV6247 |
| AMPKα1 | Novus Biologicals | Cat#NBP2-51992 |
| AMPKβ1 | Novus Biologicals | Cat#NBP2-23375 |
| AMPKγ1 | Novus Biologicals | Cat#NBP-23377 |
| ULK protein | Thermo Fisher | Cat# PV6430 |
| SAMS peptide | Promega | Cat#V1921 |
| Tetramethylrhodamine | Thermo Fisher | Cat# T669 |
| Dulbecco’s Modified Eagle’s Medium w/o inositol | MP Biomedicals | Cat# 091642954 |
| [3H]AMP | American Radiolabeled Chemicals Inc. | Cat#ART0843 |
| [3H]inositol | American Radiolabeled Chemicals Inc. | Cat#ART0261 |
| K-INOSL Assay Kit | Megazyme | Cat#820517 |
| Glucose-6-Phosphate Colorimetric Assay Kit | BioVision | Cat#K657 |
| ATP Determination Kit | Thermal Fisher | Cat#A22066 |
| ADP-Glo™ kinase assay | Promega | Cat#V6930 |
| Human: PC3 | ATCC | Cat#CRL-1435 |
| Human: DU145 | ATCC | Cat#HTB-81 |
| Mouse: mouse embryonic fibroblasts | ATCC | Cat#SCRC-1008 |
| Mouse: AMPK−/− mouse embryonic fibroblasts | Dr. Kun-Liang Guan (The University of California, San Diego) | N/A |
| Mouse: B6SJL.SOD1-G93A | The Jackson Laboratory | JAX: 002726 |
| Mouse: C57BL/6J | The Jackson Laboratory | JAX: 000664 |
| ShRNA: human IMPA1-#1 | Sigma-Aldrich | Cat#TRCN0000310192 |
| ShRNA: human IMPA1-#2 | Sigma-Aldrich | Cat#TRCN0000296135 |
| ShRNA: mouse IMPA1-#1 | Sigma-Aldrich | Cat#TRCN0000081409 |
| ShRNA: mouse IMPA1-#2 | Sigma-Aldrich | Cat#TRCN0000306604 |
| ShRNA: human IMPA2-#1 | Sigma-Aldrich | Cat#TRCN0000050318 |
| ShRNA: human IMPA2-#2 | Sigma-Aldrich | Cat#TRCN0000050320 |
| ShRNA: human CDIPT-#1 | Sigma-Aldrich | Cat#TRCN0000035987 |
| ShRNA: human CDIPT-#2 | Sigma-Aldrich | Cat#TRCN0000035988 |
| ShRNA: human INPP1-#1 | Sigma-Aldrich | Cat#TRCN0000051600 |
| ShRNA: human INPP1-#2 | Sigma-Aldrich | Cat#TRCN0000051601 |
| ShRNA: human PI4K2α-#1 | Sigma-Aldrich | Cat#TRCN0000195093 |
| ShRNA: human PI4K2α-#2 | Sigma-Aldrich | Cat#TRCN0000037605 |
| ShRNA: human PLCβ1-#1 | Sigma-Aldrich | Cat#TRCN0000226441 |
| ShRNA: human PLCβ1-#2 | Sigma-Aldrich | Cat#TRCN0000226442 |
| ShRNA: human AMPKα1 | Sigma-Aldrich | Cat#TRCN0000199831 |
| ShRNA: human AMPKα2 | Sigma-Aldrich | Cat#TRCN0000219690 |
| pGEX6P-1-AMPKα | This paper | N/A |
| pGEX6P-1-AMPKβ | This paper | N/A |
| pGEX6P-1-AMPKγ | This paper | N/A |
| pGEX6P-1-Bateman domain 1 | This paper | N/A |
| pGEX6P-1-Bateman domain 2 | This paper | N/A |
| pGEX6P-1-CBS1 | This paper | N/A |
| pGEX6P-1-CBS2 | This paper | N/A |
| pGEX6P-1-CBS3 | This paper | N/A |
| pGEX6P-1-CBS4 | This paper | N/A |
| pFLAG-CMV-2-IMPA1 | This paper | N/A |
| pFLAG-CMV-2-IMPA1 D220A | This paper | N/A |
| pFLAG-CMV-2-IMPA1 W219A | This paper | N/A |
| ImageJ | ImageJ software | https://imagej-nih-gov.go.libproxy.wakehealth.edu/ij/index.html |
| Fiji | Fiji software | https://imagej.net/Fiji |
| Graphpad Prism 8 | Graphpad software | N/A |
| CRISPR Design tool | Zhang Lab | https://zlab.bio/guide-design-resources |
| Unprocessed images | This paper | http://dx.doi.org/10.17632/nvf6fgc77g.1 |
Experimental model and subject details
Animal Studies
C57BL/6J mice (Male, 14 weeks) were conducted for fasting and inositol treatment by intraperitoneal injection (30 mg/kg) or oral administration (8 mg/g) via drinking water. All the related protocols were approved animal protocol by Institutional Animal Care and Use Committee (IACUC) at Wake Forest School of Medicine.
Cell Culture and Viral Packaging
Wild type (WT) and AMPK knockout (AMPK−/−) mouse embryonic fibroblasts (MEFs) were cultured in DMEM medium supplied with 10% FBS, 2 mM glutamine or inositol-free DMEM medium (MP Biomedicals) supplied with 10% FBS, 2 mM glutamine. DU145 and PC3 cells were cultured in RPMI-1640 medium supplied with 10% FBS, 2mM glutamine. To generate shRNA knockdown cells, HEK293T cells were prepared and co-transfected with either luciferase (shLuc.) or target gene shRNA with packaging plasmid (pPAX) and envelope plasmid (pMD.2G) by using the calcium phosphate transfection method. Medium was changed 6 hours later and virus particles were harvested after another 48 hours to infect parental cells, then selected by puromycin.
Method details
Immunoblotting, Immunoprecipitation and Immunofluorescence
Cells were lysed in RIPA buffer supplemented with proteinase inhibitor cocktail and subjected to immunoblotting (IB) by indicated antibodies. Immunoblotting assays were repeated at least three times for each dataset. For immunoprecipitation assays, primary antibodies were incubated with magic protein agarose A/G beads for 30 minutes at room temperature (RT), followed by incubation with cell lysates rotated at room temperature for 3 hours. The beads were washed four times with lysis buffer and analyzed by IB. Vehicle (NT), 25 μM of inositol (Ins), 4 μM of PI, IP3 or PIP3 treatment. For immunofluorescence, the cells were stained with 200 nM MitoTracker Red for 15 minutes, fixed for 10 min with 4% paraformaldehyde in phosphate buffered saline (PBS) and then permeabilized with 0.4% Triton X-100. The cells were then incubated with anti-Drp1 antibody (cell signaling, #14647) for 2~3 hours at room temperature and washed three times with phosphate-buffered saline, followed by incubation with Alexa 488 fluorescein-conjugated anti-mouse IgG. Nuclei were visualized by ProlongGold (Life Technologies). The stained cells were observed by Olympus FV1200 Spectral Laser Scanning Confocal Microscope.
Quantification of Mitochondrial Morphology
For quantification of mitochondrial morphology of MitoTracker-stained cells, scoring was done blindly to different genotypes and treatments. “Fragmented” was defined as cells with spherical mitochondria majority; “Short” was defined as cells with a majority of mitochondria less than about 10 μm; “Long” represents cells which the majority of mitochondria was more than about 10 μm, and “Fused” was defined as cells with highly interconnected mitochondria of less than 4–5 free ends. For the statistic of colocalization of Drp1 and MitoTracker, images were acquired in ImageJ using colocalization analysis. Colocalization were counted using particle analyzed and normalized to total mitochondrial area in each picture. Three independent experiments for each sample were included in statistics consisting of at least five pictures representing at least 100 cells in every condition. For quantification of mitochondrial morphology of transmission electron microscope, scoring was done blindly to different genotypes and treatments. “Short” was defined as the length of mitochondria less than about 500 nm; “Long” represents the length of mitochondria more than about 1 μm. At least 50 mitochondria from the liver and skeletal muscle in each male mouse and three male mice for each group were counted and analyzed by an observer who is blind to the experimental conditions. To quantify the aspect ratio, elongation and interconnectivity of individual mitochondria, confocal images stained by MitoTracker Red FM using Fiji/ImageJ software were processed, binarized and subjected to particle analysis to generate mitochondrial morphology characteristics for each cell. Aspect ratio was employed as an index of major and minor axis of ellipse equivalent to the mitochondria. Mitochondrial elongation was measured by inverse circularity and mitochondrial interconnectivity was measured by the mean area to perimeter ratio according to the published protocol (Dagda et al., 2009; Wiemerslage and Lee, 2016). The mean area/mean perimeter ratio was employed as an index of mitochondrial interconnectivity. Inverse circularity was validated using the equation of 1/4× π × (mean area/mean perimeter2) as a measure of mitochondrial elongation.
Biotin Pull-Down Assay, [3H]AMP Binding Assay and In Vitro Kinase Assay
The full length recombinant proteins of GST-AMPK α, β, γ or GST-CBS1, CBS2, CBS3, CBS4 of AMPKγ were expressed in Escherichia coli BL21 and purified. The purity of the recombinant proteins was resolved by SDS-PAGE, followed by Coomassie blue staining. For biotin-inositol pull-down assays, magnetic Dynabeads MyOne Streptavidin T1 (Thermal Fisher) was pre-incubated with free biotin or biotin-labeled inositol for 30 minutes at room temperature (RT), and then incubated with cell lysates or recombinant proteins rotated for 2 hours at 4°C. The beads were washed with PBS 3–4 times and analyzed by Immunoblotting. For competition assay, 50 mM and 125 mM of ATP and AMP were incubated with AMPKγ recombinant protein and biotin-labeled inositol for 1 hour at 4°C, washed and analyzed by IB. For in vitro AMP and AMPK binding assay, [3H]AMP obtained from American Radiolabeled Chemicals Inc. was incubated with the GST fusion recombinant proteins on glutathione-sepharose beads (GE Healthcare) or 50 mM and 125 mM of inositol, for competitive binding with AMP, at 4°C for 1 hour in binding buffer (10 mM Hepes pH 7.5, 50 mM NaCl, 0.1% Nonidet P-40, 0.5 mM dithiothreitol, and 0.5 mM EDTA), washed two times with PBS, and radioactivity was determined using a liquid scintillation counter (LS6500; Beckman Coulter). For in vitro kinase assay, the recombinant protein GST-Mff was expressed in Escherichia coli BL21 and purified. Recombinant protein ULK was obtained from Thermo Fisher, active AMPK from Millipore (#14-840). 100 ng of active AMPK incubated with 100 ng of recombinant proteins and 0.5 mM of ATP in kinase reaction buffer (50 mM Hepes pH 7.4, 1 mM MgCl2, 0.1 mM dithiothreitol, and 0.1 mM EGTA) upon adding 50 and 125 μM of inositol, 50 and 125 μM of AMP or 50 and 125 μM of ATP. The reaction was incubated at 30°C for 15 minutes and subjected to immunoblotting using phospho-Mff S146 or phospho-ULK S555 antibody. For in vitro SAMS peptide phosphorylation assay, the recombinant AMPKα1β1γ1 (Promega, V1921), α1β2γ1 (Thermo Fisher, PV6244), α2β1γ1 (BPS Bioscience, 40024) or α2β2γ1 (Thermo Fisher, PV6247) complex was obtained from Thermo fisher or BPS Biosceience. 100 ng of AMPKα1β1γ1, α1β2γ1, α2β1γ1 or α2β2γ1 (Thermo Fisher, PV6247) complex was incubated with fixed amount of ATP (150 μM) and 400 ng of SAMS peptide (HMRSAMSGLHLVKRR) (Promega, V1921) in kinase reaction buffer (8 mM Tris pH7.5, 4 mM MgCl2, 20 μg/ml BSA, 10 μM dithiothreitol) upon adding 125 μM of inositol at room temperature for 30 minutes. Flag vector control, Flag-AMPKγ2 and Flag-AMPKγ3 complexes were overexpressed in 293T cells followed by immunoprecipitation with Flag antibody and elution by Flag peptide. Active 100 ng of heterotrimer AMPKα1 (Novus Biologicals, NBP2-51992), AMPKβ1 (Novus Biologicals, NBP2-23375) and Flag-AMPKγ2 (α1β1γ2) /Flag-AMPKγ3 (α1β1γ3) complex, Flag-AMPKγ2 (AMPKγ2) or Flag-AMPKγ3 (AMPKγ3) complexes was incubated with fixed amount of ATP (150 μM) and 100 ng of GST-Mff or 400 ng of SAMS peptide, followed by adding 125 μM of inositol at 30°C for 15 or 30 minutes. The AMPK kinase activity was determined by ADP-Glo™ kinase assay (Promega, V6930) according to manufacturer’s procedures. The values of AMPK enzymatic kinetic curve were calculated based on the equation: Y = basal + {[(activation × basal − basal) × X] / (EC50 + X)} and then normalized with basal, where Y is kinase activity and X is AMP, ATP or inositol concentrations. All data are representative of three independent experiments and means ± SEM of triplicate values.
AMPK Enzyme Kinetics
The velocity of AMPK enzymatic kinetics was calculated using the Michaelis-Menten Equation using a substrate concentration range of between 200 nM and 11 μM with vehicle, 125, 500, 1000 μM of inositol treatment. Lineweaver–Burk plots were generated from 1/velocity and 1/concentrations of SAMS peptide. The inhibition constant (Ki) of inositol was determined from Michaelis-Menten saturation plot versus the concentrations of SAMS peptide. Graphs were plotted by using Prism 8 (GraphPad Software Inc.) The values of the correlation coefficient, slope and standard errors were obtained by linear and non-linear regression analysis using these programs. Each condition is shown from three independent experiments (mean ± SEM, n = 3).
Saturation and Competition Binding Assay Using Tritium-labeled inositol and Isothermal Titration Calorimetry (ITC)
Recombinant GST-AMPKγ proteins and [3H]inositol were incubated at room temperature (RT) for 1 hour in binding buffer (10 mM Hepes pH 7.5, 50 mM NaCl, 0.1% Nonidet P-40, 0.5 mM dithiothreitol, and 0.5 mM EDTA), followed by glutathione-sepharose beads (GE Healthcare) pull-down for 3 hours. 7–9 different concentrations of [3H]inositol in a range of 50 μM to 4 mM were employed. After glutathione-sepharose beads were washed three times with PBS, radioactivity was determined using a liquid scintillation counter (LS6500; Beckman Coulter). For competition assay, recombinant GST-AMPKγ proteins and [3H]inositol were incubated at room temperature (RT) for 1 hour in binding buffer upon adding AMP or ATP. 9–11 different concentrations of AMP or ATP in a range of 0.1 μM to 4 mM were employed. After glutathione-sepharose beads were washed three times with PBS, radioactivity was determined using a liquid scintillation counter. All data are representative of three independent experiments and means ± SEM of triplicate values. [3H] inositol obtained from American Radiolabeled Chemicals Inc. Equilibrium dissociation constant (Kd) was determined from saturation binding plot versus the concentrations of [3H]inositol. Graphs were plotted by using Prism 8 (GraphPad Software Inc.) The values of the correlation coefficient, slope and standard errors were obtained by linear and non-linear regression analysis using these programs. ITC experiments were performed using a MicroCal PEAQ-ITC calorimeter at 25 °C. The sample cell was filled with 300 μL of recombinant protein GST or GST-AMPKγ (7.8 μM in 100 mM Tris buffer) and stirred at 750 rpm. The syringe was filled with 90 μL inositol (780 μM in 100 mM Tris buffer). Inositol solution was injected into the sample cell with a single injection of 0.4 μL, followed by nineteen 1.5 μL injections at 150 s intervals. The data were analyzed with the MicroCal PEAQ-ITC Analysis Software using the single binding site model.
Time-lapse Microscopy
DU145 cells were seeded in the chambered coverslips (LabTeK®II) and stained with 200 nM MitoTracker Red for 15 minutes before treatment of 25 μM inositol (Sigma), 300 μM A769662 (Selleckchem) or 2 mM AICAR. Image was collected and acquired every minute for 30 minutes by Olympus FV1200 Spectral Laser Scanning Confocal Microscope at 37°C and 5% CO2. Each image was acquired, exported and uncompressed in AVI format with 2 times speed.
Lentiviral Infection and Sequences of shRNAs
To generate lentivirus-producing cell lines, calcium phosphate was used to transfect with luciferase (control), IMPA1, IMPA2, CDIPT, INPP1, PI4K2α and PLC β1 shRNA, virus Gag-Pol packing vector (pPAX) and virus envelope glycoprotein expression vector (pMD.2G) in 293T cells. Supernatants were collected 48 hours after transfection, filtered (0.45 μm), purified by centrifugation, and stored at −80°C. Infections were carried out overnight in the presence of 8 μg/mL Polybrene (Sigma). After viral infection, cells were selected with puromycin (2 μg/ml). The individual mission shRNAs kept in frozen bacterial glycerol were purchased from Sigma Aldrich. The following shRNAs sequences were used:
IMPA1 for human #1 TRCN0000310192: CCGGCAACGAGACGACGAAGATTAACTCGAGTTAATCTTCGTCGTCTCGTTGT TTTTG; IMPA1 for human #2 TRCN0000296135: CCGGCATCAACTGAAATAGTCAATTCTCGAGAATTGACTATTTCAGTTGATGT TTTTG; IMPA1 for mouse #1 TRCN0000081409: CCGGCGGAGAATAATTGCCGCAAATCTCGAGATTTGCGGCAATTATTCTCCGT TTTTG; IMPA1 for mouse #2 TRCN0000306604: CCGGATGTTCCAGAAGCCATATTTGCTCGAGCAAATATGGCTTCTGGAACATT TTTTG; IMPA2 #1 TRCN0000050318: CCGGCCTGAAGCTGTTCCTGAGTAACTCGAGTTACTCAGGAACAGCTTCAGG TTTTTG; IMPA2 #2 TRCN0000050320: CCGGGCTGTTCGACAAGAGCTTGAACTCGAGTTCAAGCTCTTGTCGAACAGC TTTTTG; CDIPT #1 TRCN0000035987: CCGGGAGTCACAAGATGATCGACTTCTCGAGAAGTCGATCATCTTGTGACTCT TTTTG; CDIPT #2 TRCN0000035988: CCGGCTCGCGCTCTTAATCAAGGAACTCGAGTTCCTTGATTAAGAGCGCGAG TTTTTG; INPP1 #1 TRCN0000051600: CCGGGCTTAGAAAGAAATCCAGAAACTCGAGTTTCTGGATTTCTTTCTAAGCT TTTTG; INPP1 #2 TRCN0000051601: CCGGCCACATTCAAATGGGACTCTTCTCGAGAAGAGTCCCATTTGAATGTGGT TTTTG; PI4K2α #1 TRCN0000195093: CCGGCAATGACAACTGGCTGATTAACTCGAGTTAATCAGCCAGTTGTCATTGT TTTTTG; PI4K2α #2 TRCN0000037605: CCGGCCATAAGCAGATTGCTGTCATCTCGAGATGACAGCAATCTGCTTATGGT TTTTG; PLCβ1 #1 TRCN0000226441: CCGGCAGCGAGATCCTCGGCTTAATCTCGAGATTAAGCCGAGGATCTCGCTG TTTTTG; PLCβ1 #2 TRCN0000226442: CCGGTCTAATCTGGTGAACTATATTCTCGAGAATATAGTTCACCAGATTAGAT TTTTG; AMPKα1 #1 TRCN0000199831: CCGGGTGACCTCACTTGACTCTTCTCTCGAGAGAAGAGTCAAGTGAGGTCAC TTTTTTG; AMPKα1 #2 TRCN0000219690: CCGGGAAGGTTGTAAACCCATATTACTCGAGTAATATGGGTTTACAACCTTCT TTTTG
Knockout Cell Generation Using CRISPR
Knockout cells were generated using the Cas9 nickase strategy as described previously (Ran et al., 2013). Two pairs of guide RNAs (gRNA) targeting the exon 3 and exon 4 respectively were designed for IMPA1 gene using the online design tool at crispr.mit.edu. Each gRNA duplex was cloned into pX462 vector encoding SpCas9n-2A-puro (Addgene # 48141). The lentivirus of two pairs of guide RNAs was produced and used to infect cells, which were selected by puromycin (2 μg/ml) and the single colony was picked. Individual clones with IMPA1 knockout were verified by immunoblotting.
Non targeting scramble gRNA sequence:
guide A: CGAGGCTTAACGCCAGATTC; guide B: GAACTCGTTAGGCCGTGAAG
IMPA1 gRNA sequence:
guide A: AATAGGGTCAATGATCCATG; guide B: CCTTTTGTAGCTGTTTCAAT
Seahorse XFp Cell Mito Stress Test
Seahorse XF Cell Mito Stress Tests (Seahorse Bioscience, Agilent Technologies) were performed according to the manufacturer’s protocol on an XF instrument. Briefly, one day prior to assay, the sensor cartridge was hydrated with Seahorse XF Calibrant buffer pH 7.4 (Seahorse Bioscience) at 37°C without CO2 overnight. PC3 and DU145 (15,000 cells /well) were plated in Seahorse XF Cell Culture microplates (Seahorse Bioscience) in RPMI-1640 medium overnight. At day of assay, XF assay medium was supplemented with 10 mM glucose, 1 mM pyruvate, and 2 mM glutamine with pH adjusted to 7.4. Cells were rinsed twice with XF assay medium and incubated with the fresh XF assay medium for 1 hour in 37 °C without CO2. After placing the calibration plate with loaded sensor of cartridge, 1 μM of Oligomycin, 0.5 μM of FCCP and 1 μM of Rotenone/Antimycin A were added in the ports respectively, and oxygen consumption rate (OCR) was measured by Seahorse XF Analyzer. Data analysis was performed using the Seahorse XF Wave software.
The Synthesis of Biotin-inositol
A solution of inositol (180 mg, 1.0 mmol), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) (384 mg, 2.0 mmol) and 4-dimethylaminopyridine (DMAP) (11 mg, 0.01 mmol) in dry dichloromethane (DCM) (10 mL) was stirred at room temperature. Then, biotin (244 mg, 1.0 mmol) in 10 mL DCM was added dropwise for 5 min. The mixture was stirred at room temperature overnight. The solvent was removed and the product was precipitated from 20% ethyl acetate/hexanes. 210 mg of wax solid was obtained and the yield was 52%. LC/MS calculated for C16H27N2O8S [M+H]+, 407; found 407.
Inositol level, ATP level, G6P level, Mitochondrial ROS and Membrane Potential Measurement
5×106–107 cells were harvested and extracted by 80% methanol. The inositol, glucose-6-phosphate (G6P) levels and ATP levels in cells were determined by the K-INOSL Assay Kit (Megazyme, 820517), Glucose-6-Phosphate Colorimetric Assay Kit (BioVision, K657) and ATP Determination Kit (Thermal Fisher, A22066), respectively, according to manufacturer’s protocol. The inositol, G6P and ATP levels were normalized by protein concentration in each experimental group. The inositol, G6P and ATP levels are shown as mean ± SEM of three independent experiments. Mitochondrial ROS levels were measured using the MitoSOX probe (Life Technologies) according to the manufacturer’s protocol. Following treatment with 2.5 μM of MitoSOX for 30 minutes, cells were trypsinized, resuspended in PBS and immediately analyzed by flow cytometry. Mitochondrial membrane potential levels were measured using Tetramethylrhodamine (TMRM) (ThermoFisher) according to the manufacturer’s protocol. Following treatment with 200 nM of TMRM for 30 minutes at 37°C, cells were trypsinized, resuspended in PBS and immediately analyzed by flow cytometry.
Extraction of Metabolites and LC-MS/MS Measurement
For the extraction of Ins, AMP, ADP, ATP, IP, 1 × 106 cells were harvested from the 10 cm dish. After culture media were aspirated, cells were washed twice in ice-cold PBS and lysed in 1 ml of ice-cold 80% methanol with the plates placed on dry ice to quench metabolism. After 5 minutes, cells were scraped and transferred into 1.5 ml tubes and incubated in −80°C for 30 minutes. Lysates were centrifuged at 13,000 rpm for 10 minutes and supernatants were transferred to new vials and dried at 30°C in a Speed-Vac for 3 hours. For the extraction of PI, PIP, PIP2, PIP3, IP2, IP3, 1 × 106 cells were harvested from the 10 cm dish. After culture media were aspirated, cells were washed twice in ice-cold PBS and lysed in 1 ml of ice-cold 25% methanol containing 20 μM PC (17:0) and PG (17:0) with the plates placed on dry ice to quench metabolism. cells were scraped and transferred into 1.5 ml tubes and kept in MTBE (MTBE:methanol:H2O = 10:2:5). After sonicating in ice bath for 15 minutes, cells were incubated in −20°C for 30 minutes, followed by centrifuged at 3,000 rpm for 5 minutes. Supernatants evaporated the MTBE layer and were dried in 100 μL of chloroform:MeOH (1:1). The targeted LC-MS/MS method was modeled and developed as previously described (Carroll et al., 2015; Eghlimi et al., 2020). Briefly, all LC-MS/MS experiments were performed on an Agilent 1290 UPLC-6490 QQQ-MS (Santa Clara, CA) system. Each sample was injected twice, 10 μL for analysis using negative ionization mode and 4 μL for analysis using positive ionization mode. Both chromatographic separations were performed in hydrophilic interaction chromatography (HILIC) mode on a Waters XBridge BEH Amide column (150 × 2.1 mm, 2.5 μm particle size, Waters Corporation, Milford, MA). The flow rate was 0.3 mL/min, auto-sampler temperature was kept at 4 °C, and the column compartment was set at 40 °C. The mobile phase was composed of Solvents A (10 mM ammonium acetate, 10 mM ammonium hydroxide in 95% H2O/5% ACN) and B (10 mM ammonium acetate, 10 mM ammonium hydroxide in 95% ACN/5% H2O). After the initial 1 min isocratic elution of 90% B, the percentage of Solvent B decreased to 40% at t=11 min. The composition of Solvent B maintained at 40% for 4 min (t=15 min), and then the percentage of B gradually went back to 90%, to prepare for the next injection. The mass spectrometer is equipped with an electrospray ionization (ESI) source. Targeted data acquisition was performed in multiple-reaction-monitoring (MRM) mode. The whole LC-MS system was controlled by Agilent Masshunter Workstation software (Santa Clara, CA). The extracted MRM peaks were integrated using Agilent MassHunter Quantitative Data Analysis (Santa Clara, CA).
Quantification and Statistical Analysis
Statistical significance was identified by Student’s t test using Prism 8 software. P-values of less than 0.05 were considered statistically significant; *, p < 0.05; **, p < 0.001; ***, P < 0.0001; NS, non-significant.
Supplementary Material
Video S1. DU145 stained with MitoTracker Red upon glucose deprivation, Related to Figure 5.
Video S2. DU145 stained with MitoTracker Red upon glucose deprivation and 25 μM of inositol treatment, Related to Figure 5.
Video S3. DU145 stained with MitoTracker Red upon 250 ng/ml of Rotenone treatment, Related to Figure 5.
Video S4. DU145 stained with MitoTracker Red upon 250 ng/ml of Rotenone and 25 μM of inositol treatment, Related to Figure 5.
Video S5. DU145 stained with MitoTracker Red upon 300 μM of A769662 treatment, Related to Figures 5 and S8.
Video S6. DU145 stained with MitoTracker Red upon 300 μM of A769662 and 25 μM of inositol treatment, Related to Figures 5 and S8.
Highlights.
IMPA-derived inositol modulates mitochondrial fission.
Inositol restricts aberrant AMPK activation and mitochondrial fission.
Inositol decline leads to AMPK-dependent mitochondrial fission upon energy stress.
Inositol directly binds to AMPKγ and competes with AMP for AMPKγ binding.
Acknowledgements
This research was supported by Postdoctoral Research Abroad Program from Ministry of Science and Technology in Taiwan to Che-Chia Hsu (105-2917-I-564-067) and by Startups funds and Cancer Center Pilot grant award from Wake Forest Baptist Comprehensive Cancer Center, NIH grants (R01CA182424, R01CA193813 and R01CA248037) and Anderson Endowed Professorship fund to H.K.L. We thank the technical supports and discussion of TEM and confocal microscope from Ken Grant and Steven Kridel for using of Seahorse XF Analyzer (Wake Forest School of Medicine). We acknowledge the support of Wake Forest Baptist Comprehensive Cancer Center Cell & Cellular Imaging and Flow cytometry Share Resources, supported by the National Cancer Institute’s Cancer Center Support Grant (P30CA012197).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests.
References
- Ando H, Mizutani A, Matsu-ura T, and Mikoshiba K (2003). IRBIT, a novel inositol 1,4,5-trisphosphate (IP3) receptor-binding protein, is released from the IP3 receptor upon IP3 binding to the receptor. J Biol Chem 278, 10602–10612. [DOI] [PubMed] [Google Scholar]
- Archer SL (2013). Mitochondrial dynamics--mitochondrial fission and fusion in human diseases. N Engl J Med 369, 2236–2251. [DOI] [PubMed] [Google Scholar]
- Asaoka Y, Nakamura S, Yoshida K, and Nishizuka Y (1992). Protein kinase C, calcium and phospholipid degradation. Trends in biochemical sciences 17, 414–417. [DOI] [PubMed] [Google Scholar]
- Asplin I, Galasko G, and Larner J (1993). chiro-inositol deficiency and insulin resistance: a comparison of the chiro-inositol- and the myo-inositol-containing insulin mediators isolated from urine, hemodialysate, and muscle of control and type II diabetic subjects. Proc Natl Acad Sci U S A 90, 5924–5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, Vais H, Cheung KH, Yang J, Parker I, et al. (2010). Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142, 270–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey BW, Finley LW, Cross JR, Allis CD, and Thompson CB (2015). Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518, 413–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlomagno G, Nordio M, Chiu TT, and Unfer V (2011). Contribution of myo-inositol and melatonin to human reproduction. Eur J Obstet Gynecol Reprod Biol 159, 267–272. [DOI] [PubMed] [Google Scholar]
- Carroll PA, Diolaiti D, McFerrin L, Gu H, Djukovic D, Du J, Cheng PF, Anderson S, Ulrich M, Hurley JB, et al. (2015). Deregulated Myc requires MondoA/Mlx for metabolic reprogramming and tumorigenesis. Cancer Cell 27, 271–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalecka-Franaszek E, and Chuang DM (1999). Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc Natl Acad Sci U S A 96, 8745–8750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CR, and Blackstone C (2007). Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. The Journal of biological chemistry 282, 21583–21587. [DOI] [PubMed] [Google Scholar]
- Ciotta L, Stracquadanio M, Pagano I, Carbonaro A, Palumbo M, and Gulino F (2011). Effects of myo-inositol supplementation on oocyte’s quality in PCOS patients: a double blind trial. Eur Rev Med Pharmacol Sci 15, 509–514. [PubMed] [Google Scholar]
- Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC, et al. (2013). Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155, 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combettes L, and Champeil P (1994). Calcium and inositol 1,4,5-trisphosphate-induced Ca2+ release. Science 265, 813–815. [DOI] [PubMed] [Google Scholar]
- Condorelli RA, La Vignera S, Di Bari F, Unfer V, and Calogero AE (2011). Effects of myoinositol on sperm mitochondrial function in-vitro. Eur Rev Med Pharmacol Sci 15, 129–134. [PubMed] [Google Scholar]
- Cribbs JT, and Strack S (2007). Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO reports 8, 939–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryns K, Shamir A, Van Acker N, Levi I, Daneels G, Goris I, Bouwknecht JA, Andries L, Kass S, Agam G, et al. (2008). IMPA1 is essential for embryonic development and lithium-like pilocarpine sensitivity. Neuropsychopharmacology 33, 674–684. [DOI] [PubMed] [Google Scholar]
- Dagda RK, Cherra SJ 3rd, Kulich SM, Tandon A, Park D, and Chu CT (2009). Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem 284, 13843–13855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deranieh RM, and Greenberg ML (2009). Cellular consequences of inositol depletion. Biochemical Society transactions 37, 1099–1103. [DOI] [PubMed] [Google Scholar]
- Di Paolo G, and De Camilli P (2006). Phosphoinositides in cell regulation and membrane dynamics. Nature 443, 651–657. [DOI] [PubMed] [Google Scholar]
- Dickey AS, and Strack S (2011). PKA/AKAP1 and PP2A/Bbeta2 regulate neuronal morphogenesis via Drp1 phosphorylation and mitochondrial bioenergetics. The Journal of neuroscience : the official journal of the Society for Neuroscience 31, 15716–15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Madamsetty VS, Kiers S, Alekhina O, Ugolkov A, Dube J, Zhang Y, Zhang JS, Wang E, Dutta SK, et al. (2019). Glycogen Synthase Kinase-3 Inhibition Sensitizes Pancreatic Cancer Cells to Chemotherapy by Abrogating the TopBP1/ATR-Mediated DNA Damage Response. Clin Cancer Res 25, 6452–6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducommun S, Deak M, Sumpton D, Ford RJ, Nunez Galindo A, Kussmann M, Viollet B, Steinberg GR, Foretz M, Dayon L, et al. (2015). Motif affinity and mass spectrometry proteomic approach for the discovery of cellular AMPK targets: identification of mitochondrial fission factor as a new AMPK substrate. Cell Signal 27, 978–988. [DOI] [PubMed] [Google Scholar]
- Eghlimi R, Shi X, Hrovat J, Xi B, and Gu H (2020). Triple Negative Breast Cancer Detection Using LC-MS/MS Lipidomic Profiling. J Proteome Res 19, 2367–2378. [DOI] [PubMed] [Google Scholar]
- Eisenberg F Jr. (1967). D-myoinositol 1-phosphate as product of cyclization of glucose 6-phosphate and substrate for a specific phosphatase in rat testis. The Journal of biological chemistry 242, 1375–1382. [PubMed] [Google Scholar]
- Fairn GD, and Grinstein S (2012). Cell biology. Precursor or charge supplier? Science 337, 653–654. [DOI] [PubMed] [Google Scholar]
- Fang G, Zhang P, Liu J, Zhang X, Zhu X, Li R, and Wang H (2019). Inhibition of GSK-3beta activity suppresses HCC malignant phenotype by inhibiting glycolysis via activating AMPK/mTOR signaling. Cancer Lett 463, 11–26. [DOI] [PubMed] [Google Scholar]
- Figueiredo T, Melo US, Pessoa AL, Nobrega PR, Kitajima JP, Rusch H, Vaz F, Lucato LT, Zatz M, Kok F, et al. (2016). A homozygous loss-of-function mutation in inositol monophosphatase 1 (IMPA1) causes severe intellectual disability. Molecular psychiatry 21, 1125–1129. [DOI] [PubMed] [Google Scholar]
- Goswami R, Bondoc JMG, Wheeler PR, Jafari A, Gonzalez T, Mehboob S, and Movahedzadeh F (2018). Inositol Monophosphatase: A Bifunctional Enzyme in Mycobacterium smegmatis. ACS Omega 3, 13876–13881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowans GJ, Hawley SA, Ross FA, and Hardie DG (2013). AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab 18, 556–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond GR, Fischer MJ, Anderson KE, Holdich J, Koteci A, Balla T, and Irvine RF (2012). PI4P and PI(4,5)P2 are essential but independent lipid determinants of membrane identity. Science 337, 727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG (2011). AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev 25, 1895–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head B, Griparic L, Amiri M, Gandre-Babbe S, and van der Bliek AM (2009). Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. The Journal of cell biology 187, 959–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holub BJ (1986). Metabolism and function of myo-inositol and inositol phospholipids. Annu Rev Nutr 6, 563–597. [DOI] [PubMed] [Google Scholar]
- Hsu CC, Peng D, Cai Z, and Lin HK (2021). AMPK signaling and its targeting in cancer progression and treatment. Semin Cancer Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N, Fujita Y, Oka T, and Mihara K (2006). Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. The EMBO journal 25, 2966–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolles J, Bothmer J, Markerink M, and Ravid R (1993). Reduced phosphatidylinositol kinase activity in Alzheimer’s disease: effects of age and onset. Dementia 4, 81–86. [DOI] [PubMed] [Google Scholar]
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, and Thompson CB (2005). AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell 18, 283–293. [DOI] [PubMed] [Google Scholar]
- Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, Counter CM, and Kashatus DF (2015). Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Molecular cell 57, 537–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang T, Xie L, Chao C, Kang Y, Lin X, Qin T, Xie H, Feng ZP, and Gaisano HY (2014). Phosphatidylinositol 4,5-biphosphate (PIP2) modulates interaction of syntaxin-1A with sulfonylurea receptor 1 to regulate pancreatic beta-cell ATP-sensitive potassium channels. J Biol Chem 289, 6028–6040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewus MW, Loewus FA, Brillinger GU, Otsuka H, and Floss HG (1980). Stereochemistry of the myo-inositol-1-phosphate synthase reaction. J Biol Chem 255, 11710–11712. [PubMed] [Google Scholar]
- Loson OC, Song Z, Chen H, and Chan DC (2013). Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell 24, 659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, and Thompson CB (2012). Metabolic regulation of epigenetics. Cell metabolism 16, 9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder AL, Johnson MD, and Henry SA (1997). 1L-myo-inositol-1-phosphate synthase. Biochim Biophys Acta 1348, 245–256. [DOI] [PubMed] [Google Scholar]
- McLaughlin S, Wang J, Gambhir A, and Murray D (2002). PIP(2) and proteins: interactions, organization, and information flow. Annu Rev Biophys Biomol Struct 31, 151–175. [DOI] [PubMed] [Google Scholar]
- Mihaylova MM, and Shaw RJ (2011). The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nature cell biology 13, 1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra P, and Chan DC (2016). Metabolic regulation of mitochondrial dynamics. J Cell Biol 212, 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TR, Vihtelic TS, Ile KE, Watson CT, Willer GB, Gregg RG, Bankaitis VA, and Hyde DR (2011). Phosphatidylinositol synthase is required for lens structural integrity and photoreceptor cell survival in the zebrafish eye. Experimental eye research 93, 460–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi T, Murata T, Watanabe A, Hida A, Ohba H, Iwayama Y, Mishima K, Gondo Y, and Yoshikawa T (2014). Defective craniofacial development and brain function in a mouse model for depletion of intracellular inositol synthesis. The Journal of biological chemistry 289, 10785–10796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, and Mihara K (2010). Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. The Journal of cell biology 191, 1141–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porporato PE, Filigheddu N, Pedro JMB, Kroemer G, and Galluzzi L (2018). Mitochondrial metabolism and cancer. Cell Res 28, 265–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nature protocols 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter V, Palmer CS, Osellame LD, Singh AP, Elgass K, Stroud DA, Sesaki H, Kvansakul M, and Ryan MT (2014). Structural and functional analysis of MiD51, a dynamin receptor required for mitochondrial fission. J Cell Biol 204, 477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty HU, Schapiro MB, Holloway HW, and Rapoport SI (1995). Polyol profiles in Down syndrome. myo-Inositol, specifically, is elevated in the cerebrospinal fluid. J Clin Invest 95, 542–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg GR, and Carling D (2019). AMP-activated protein kinase: the current landscape for drug development. Nat Rev Drug Discov 18, 527–551. [DOI] [PubMed] [Google Scholar]
- Thakur PC, Stuckenholz C, Rivera MR, Davison JM, Yao JK, Amsterdam A, Sadler KC, and Bahary N (2011). Lack of de novo phosphatidylinositol synthesis leads to endoplasmic reticulum stress and hepatic steatosis in cdipt-deficient zebrafish. Hepatology 54, 452–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toker L, Bersudsky Y, Plaschkes I, Chalifa-Caspi V, Berry GT, Buccafusca R, Moechars D, Belmaker RH, and Agam G (2014). Inositol-related gene knockouts mimic lithium’s effect on mitochondrial function. Neuropsychopharmacology 39, 319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama EQ, Herzig S, Courchet J, Lewis TL Jr., Loson OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC, et al. (2016). Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351, 275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Hyde B, and Shirihai OS (2008). Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochimica et biophysica acta 1777, 1092–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, and Shirihai OS (2011). The interplay between mitochondrial dynamics and mitophagy. Antioxidants & redox signaling 14, 1939–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unfer V, Carlomagno G, Dante G, and Facchinetti F (2012). Effects of myo-inositol in women with PCOS: a systematic review of randomized controlled trials. Gynecol Endocrinol 28, 509–515. [DOI] [PubMed] [Google Scholar]
- Vyas S, Zaganjor E, and Haigis MC (2016). Mitochondria and Cancer. Cell 166, 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiemerslage L, and Lee D (2016). Quantification of mitochondrial morphology in neurites of dopaminergic neurons using multiple parameters. J Neurosci Methods 262, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B, Heath R, Saiu P, Leiper FC, Leone P, Jing C, Walker PA, Haire L, Eccleston JF, Davis CT, et al. (2007). Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 449, 496–500. [DOI] [PubMed] [Google Scholar]
- Youle RJ, and Karbowski M (2005). Mitochondrial fission in apoptosis. Nature reviews. Molecular cell biology 6, 657–663. [DOI] [PubMed] [Google Scholar]
- Youle RJ, and van der Bliek AM (2012). Mitochondrial fission, fusion, and stress. Science 337, 1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wang G, Xu ZG, Tu H, Hu F, Dai J, Chang Y, Chen Y, Lu Y, Zeng H, et al. (2019). Lactate Is a Natural Suppressor of RLR Signaling by Targeting MAVS. Cell 178, 176–189 e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. DU145 stained with MitoTracker Red upon glucose deprivation, Related to Figure 5.
Video S2. DU145 stained with MitoTracker Red upon glucose deprivation and 25 μM of inositol treatment, Related to Figure 5.
Video S3. DU145 stained with MitoTracker Red upon 250 ng/ml of Rotenone treatment, Related to Figure 5.
Video S4. DU145 stained with MitoTracker Red upon 250 ng/ml of Rotenone and 25 μM of inositol treatment, Related to Figure 5.
Video S5. DU145 stained with MitoTracker Red upon 300 μM of A769662 treatment, Related to Figures 5 and S8.
Video S6. DU145 stained with MitoTracker Red upon 300 μM of A769662 and 25 μM of inositol treatment, Related to Figures 5 and S8.
Data Availability Statement
The unprocessed data of immunoblotting have been deposited to Mendeley Data: http://dx.doi.org/10.17632/nvf6fgc77g.1 https://data.mendeley.com/datasets/nvf6fgc77g/draft?a=3c37aafc-0338-44f4-8619-183d8fc4e67f. The software utilized in this study can be found in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| SOURCE | ||
|---|---|---|
| IMPA1 (clone H-7) | Santa Cruz Biotechnology | Cat#sc-374234; RRID: AB_10988246 |
| Mff (clone B-2) | Santa Cruz Biotechnology | Cat# sc-398617; RRID: AB_2744543 |
| phospho-Mff Ser146 (polyclone) | Cell Signaling Tech | Cat#49281; RRID: AB_2799354 |
| IMPA2 (polyclone) | GeneTex | Cat#GTX103636; RRID: AB_1950587 |
| AMPKα (polyclone) | Cell Signaling Tech | Cat#2532; RRID: AB_330331 |
| AMPKβ (polyclone) | Cell Signaling Tech | Cat#12063; RRID: AB_ 2797812 |
| AMPKγ (polyclone) | Cell Signaling Tech | Cat#4187; RRID: AB_10695248 |
| phospho-AMPKα T172 (clone D4D6D) | Cell Signaling Tech | Cat#50081; RRID: AB_2799368 |
| PKA (clone A-2) | Santa Cruz, sc-28315 | Cat#sc-28315; RRID: AB_628136 |
| phospho-Drp1 S616 (polyclone) | Cell Signaling Tech | Cat#3455; RRID: AB_2085352 |
| phospho-Drp1 S635 (clone D3A4) | Cell Signaling Tech | Cat#6319; RRID: AB_10971640 |
| Fis (clone B-5) | Santa Cruz Biotechnology | Cat#sc-376447; RRID: AB_11149382 |
| Mfn1 (clone D-10) | Santa Cruz Biotechnology | Cat#sc-166644; RRID: AB_2142616 |
| Mfn2 (clone F-5) | Santa Cruz Biotechnology | Cat#sc-515647; RRID: AB_2811176 |
| ULK (clone D8H5) | Cell Signaling Tech | Cat#8054; RRID: AB_11178668 |
| Raptor (clone 24C12) | Cell Signaling Tech | Cat#2280; RRID: AB_561245 |
| phospho-ULK S555 (clone D1H4) | Cell Signaling Tech | Cat#5869; RRID: AB_10707365 |
| phospho-Raptor S792 (polyclone) | Cell Signaling Tech | Cat#2083, RRID: AB_2249475 |
| Actin (clone AC-15) | Sigma-Aldrich | Cat#A5441, RRID: AB_476744 |
| GAPDH (clone GAPDH-71.1) | Sigma-Aldrich | Cat#G8795, RRID: AB_1078991 |
| Tubulin (clone AA13) | Sigma-Aldrich | Cat#T8203, RRID: AB_1841230 |
| CDIPT (clone C-2) | Santa Cruz Biotechnology | Cat#sc-514255 |
| GST (clone B-14) | Santa Cruz Biotechnology | Cat#sc-138, RRID: AB_627677 |
| LC3 α/β (polyclone) | Novus | Cat#NB100-2220, RRID: AB_10003146 |
| Calcineurin (D-1) | Santa Cruz Biotechnology | Cat#sc-373803, RRID: AB_10917925 |
| MiD51 (clone A-6) | Santa Cruz Biotechnology | Cat#sc-514135 |
| CREB (clone 24H2B) | Santa Cruz Biotechnology | Cat#sc-271, RRID: AB_627300 |
| phospho-CREB S133 (clone 10E9) | Santa Cruz Biotechnology | Cat#sc-81486, RRID: AB_1125727 |
| choactase (clone E-7) | Santa Cruz Biotechnology | Cat#sc-55557, RRID: AB_2291743 |
| TOM20 (monoclone) | Abcam | Cat#ab56783, RRID: AB_945896 |
| HRP-coupled antibodies to mouse | Santa Cruz Biotechnology | Cat#sc-2055, RRID: AB_631738 |
| HRP-coupled antibodies to rabbit | Thermo Fisher | Cat#31480, RRID: AB_228457 |
| Alexa Fluor 488 anti-mouse IgG | Thermo Fisher | Cat#A-21202, RRID: AB_141607 |
| Rabbit normal IgG | Santa Cruz Biotechnology | Cat#sc-2027; RRID: AB_737197 |
| Mouse Anti-rabbit IgG | Cell Signaling Tech | Cat#45262; RRID: AB_2799281 |
| lithium chloride | Millipore | Cat#5910 |
| AICAR | Sigma-Aldrich | Cat#A9978 |
| A769662 | Reagents Direct | Cat#A769662 |
| Compound C | Sigma-Aldrich | Cat#P5499 |
| Myo-inositol | Sigma-Aldrich | Cat#I7508 |
| AMP | Sigma-Aldrich | Cat#A1752 |
| ATP | Sigma-Aldrich | Cat#A2383 |
| BIO | Selleckchem.com | Cat#S7198 |
| L-α-Phosphatidylinositol | Sigma-Aldrich | Cat#P6636 |
| myo-Inositol 1,4,5-trisphosphate hexapotassium salt | Tocris | Cat#1482 |
| phosphatidylinositol 3,4,5-triphosphate | Echelon® | Cat#P-3924 |
| AMPKα1, β1, γ1 protein | Millipore | Cat#14-840 |
| AMPKα1, β1, γ1 protein | Promega | Cat#V1921 |
| AMPKα1, β2, γ1 protein | Thermo Fisher | Cat#PV6244 |
| AMPKα2, β1, γ1 protein | BPS Bioscience | Cat#40024 |
| AMPKα2, β2, γ1 protein | Thermo Fisher | Cat#PV6247 |
| AMPKα1 | Novus Biologicals | Cat#NBP2-51992 |
| AMPKβ1 | Novus Biologicals | Cat#NBP2-23375 |
| AMPKγ1 | Novus Biologicals | Cat#NBP-23377 |
| ULK protein | Thermo Fisher | Cat# PV6430 |
| SAMS peptide | Promega | Cat#V1921 |
| Tetramethylrhodamine | Thermo Fisher | Cat# T669 |
| Dulbecco’s Modified Eagle’s Medium w/o inositol | MP Biomedicals | Cat# 091642954 |
| [3H]AMP | American Radiolabeled Chemicals Inc. | Cat#ART0843 |
| [3H]inositol | American Radiolabeled Chemicals Inc. | Cat#ART0261 |
| K-INOSL Assay Kit | Megazyme | Cat#820517 |
| Glucose-6-Phosphate Colorimetric Assay Kit | BioVision | Cat#K657 |
| ATP Determination Kit | Thermal Fisher | Cat#A22066 |
| ADP-Glo™ kinase assay | Promega | Cat#V6930 |
| Human: PC3 | ATCC | Cat#CRL-1435 |
| Human: DU145 | ATCC | Cat#HTB-81 |
| Mouse: mouse embryonic fibroblasts | ATCC | Cat#SCRC-1008 |
| Mouse: AMPK−/− mouse embryonic fibroblasts | Dr. Kun-Liang Guan (The University of California, San Diego) | N/A |
| Mouse: B6SJL.SOD1-G93A | The Jackson Laboratory | JAX: 002726 |
| Mouse: C57BL/6J | The Jackson Laboratory | JAX: 000664 |
| ShRNA: human IMPA1-#1 | Sigma-Aldrich | Cat#TRCN0000310192 |
| ShRNA: human IMPA1-#2 | Sigma-Aldrich | Cat#TRCN0000296135 |
| ShRNA: mouse IMPA1-#1 | Sigma-Aldrich | Cat#TRCN0000081409 |
| ShRNA: mouse IMPA1-#2 | Sigma-Aldrich | Cat#TRCN0000306604 |
| ShRNA: human IMPA2-#1 | Sigma-Aldrich | Cat#TRCN0000050318 |
| ShRNA: human IMPA2-#2 | Sigma-Aldrich | Cat#TRCN0000050320 |
| ShRNA: human CDIPT-#1 | Sigma-Aldrich | Cat#TRCN0000035987 |
| ShRNA: human CDIPT-#2 | Sigma-Aldrich | Cat#TRCN0000035988 |
| ShRNA: human INPP1-#1 | Sigma-Aldrich | Cat#TRCN0000051600 |
| ShRNA: human INPP1-#2 | Sigma-Aldrich | Cat#TRCN0000051601 |
| ShRNA: human PI4K2α-#1 | Sigma-Aldrich | Cat#TRCN0000195093 |
| ShRNA: human PI4K2α-#2 | Sigma-Aldrich | Cat#TRCN0000037605 |
| ShRNA: human PLCβ1-#1 | Sigma-Aldrich | Cat#TRCN0000226441 |
| ShRNA: human PLCβ1-#2 | Sigma-Aldrich | Cat#TRCN0000226442 |
| ShRNA: human AMPKα1 | Sigma-Aldrich | Cat#TRCN0000199831 |
| ShRNA: human AMPKα2 | Sigma-Aldrich | Cat#TRCN0000219690 |
| pGEX6P-1-AMPKα | This paper | N/A |
| pGEX6P-1-AMPKβ | This paper | N/A |
| pGEX6P-1-AMPKγ | This paper | N/A |
| pGEX6P-1-Bateman domain 1 | This paper | N/A |
| pGEX6P-1-Bateman domain 2 | This paper | N/A |
| pGEX6P-1-CBS1 | This paper | N/A |
| pGEX6P-1-CBS2 | This paper | N/A |
| pGEX6P-1-CBS3 | This paper | N/A |
| pGEX6P-1-CBS4 | This paper | N/A |
| pFLAG-CMV-2-IMPA1 | This paper | N/A |
| pFLAG-CMV-2-IMPA1 D220A | This paper | N/A |
| pFLAG-CMV-2-IMPA1 W219A | This paper | N/A |
| ImageJ | ImageJ software | https://imagej-nih-gov.go.libproxy.wakehealth.edu/ij/index.html |
| Fiji | Fiji software | https://imagej.net/Fiji |
| Graphpad Prism 8 | Graphpad software | N/A |
| CRISPR Design tool | Zhang Lab | https://zlab.bio/guide-design-resources |
| Unprocessed images | This paper | http://dx.doi.org/10.17632/nvf6fgc77g.1 |