

Clonal ZBTB33 mutations are detected within blood sequencing datasets of healthy donors and in several MDS patients, identifying ZBTB33 as a candidate driver of clonal hematopoiesis. Mouse hematopoietic stem cells with Zbtb33 mutations exhibit clonal expansion and changes in RNA splicing.
Abstract
Clonal hematopoiesis results from somatic mutations in cancer driver genes in hematopoietic stem cells. We sought to identify novel drivers of clonal expansion using an unbiased analysis of sequencing data from 84,683 persons and identified common mutations in the 5-methylcytosine reader ZBTB33 as well as in YLPM1, SRCAP, and ZNF318. We also identified these mutations at low frequency in patients with myelodysplastic syndrome. Zbtb33-edited mouse hematopoietic stem and progenitor cells exhibited a competitive advantage in vivo and increased genome-wide intron retention. ZBTB33 mutations potentially link DNA methylation and RNA splicing, the two most commonly mutated pathways in clonal hematopoiesis and myelodysplastic syndromes.
Significance:
Mutations in known driver genes can be found in only about half of individuals with clonal hematopoiesis. Here, we performed a somatic mutation discovery effort in nonmalignant blood samples, which identified novel candidate genes that may play biological roles in hematopoietic stem cell expansion and hematologic malignancies.
Introduction
Clonal hematopoiesis is an age-associated process in which a hematopoietic stem cell (HSC) acquires a mutation that promotes clonal expansion, resulting in a hematopoietic system disproportionately derived from a single clone (1,2,3). Individuals with clonal hematopoiesis are at increased risk of developing hematologic malignancies such as myelodysplastic syndromes (MDS) or acute myeloid leukemia (AML; refs. 1, 4). Clonal hematopoiesis of indeterminate potential (CHIP) is defined by the presence of a somatic mutation in a known blood cancer–associated gene with a variant allele fraction (VAF) greater than 2% in persons without evidence of hematologic malignancy (4). However, only approximately 50% of persons with a clonal expansion state in their blood, which can be identified by the presence of multiple passenger mutations at a similar VAF, have a mutation in a known driver, suggesting that the catalog of mutations causing clonal hematopoiesis is incomplete (2, 5, 6).
Somatic driver mutation discovery efforts have focused on malignant rather than premalignant states due in part to the availability of cancer biopsy samples to analyze and the high mutational burden in these samples. The availability of sequencing data from tens of thousands of individuals from peripheral blood provides an opportunity for a broad gene-discovery effort for a premalignant condition. We leveraged 45,676 whole exomes from the Exome Aggregation Consortium (ExAC) and 39,007 whole genomes from the Trans-omics for Precision Medicine (TOPMed) data sets to identify prevalent mutations in four genes not previously recognized to drive clonal expansion of hematopoietic cells (7). To identify whether mutations in the identified genes were also present in myeloid malignancies, we sequenced these genes in a large cohort of patients with MDS by targeted sequencing. We performed functional studies on the gene that was most frequently mutated in CHIP and MDS, ZBTB33.
Results
Identification of Recurrent Somatic Mutations in Large Blood Exome Sequencing Data Sets
To discover novel candidate drivers of clonal hematopoiesis, we analyzed 45,676 exomes from the ExAC (Supplementary Table S1) for the presence of recurrent somatic mutations using two methodologies. The first approach identified genes with nonsense variants that were enriched for somatic mutations by systematically depleting likely germline variants and artifacts (Supplementary Fig. S1A; see Methods for details). Nonsense mutations were chosen because they have a clear functional consequence (truncating the protein product). Reassuringly, the four genes with the highest prevalence of candidate somatic nonsense mutations were DNMT3A, ASXL1, TET2, and PPM1D, which are all well-established drivers of clonal hematopoiesis (Supplementary Table S2). Also among the most commonly mutated genes were other known drivers of clonal hematopoiesis or hematologic cancer such as BRCC3, NF1, PHIP, NXF1, RAD21, and BCORL1 (Supplementary Table S2). Several genes not previously appreciated to be drivers of clonal expansion were also detected.
To further refine the set of candidate genes, we developed an alternative approach to identify somatic drivers. We relied on the principle that driver mutations leading to clonal expansion are likely to occur in cells that have already accumulated multiple passenger mutations. We utilized PhylogicNDT to perform Dirichlet clustering to identify clusters of somatic mutations, including single-nucleotide variants (SNV) and indels (see Methods section), at a similar VAF, indicative of a clonal population (Supplementary Fig. S1B and S1C; ref. 8). We defined the cluster with the largest VAF that was distinct from the germline heterozygous cluster as the “clonal somatic heterozygous cluster.” Considering only such clusters with at least four mutations (n = 2,266 cases), we identified 40 significantly mutated genes with a false discovery rate (FDR) less than 0.1 (Supplementary Fig. S1D). This included commonly mutated genes in CHIP (DNMT3A, ASXL1, PPM1D, TET2, SF3B1, JAK2, CBL, TP53, GNB1, and PRPF8), as well as several novel candidates.
Among the novel candidate genes, four (SRCAP, YLPM1, ZBTB33, and ZNF318) were identified by both methods. Truncating mutations, including nonsense, frameshift, and splice mutations, were found in all four of these genes (Fig. 1A; Supplementary Table S3). The only gene with a nonrandom pattern of putative somatic missense variants was ZBTB33, and we noted that these mutations clustered in the protein's functional domains, described below (Fig. 1A; Supplementary Table S3). Mutations in some of these genes, such as ZBTB33, have been detected below the threshold for statistically significant recurrence in previously published sequencing studies of myeloid malignancies (9,10,11,12,13). Notably, a study of 10 patients with AML identified a ZBTB33 mutation that was determined to be preleukemic based on its presence in both leukemia cells and preleukemic HSCs but not T cells from the patient (14). Expansion of ZNF318 and SRCAP mutant clones has been reported following cytotoxic therapy (15, 16).
Figure 1.

Detection of recurrent somatic mutations in large blood exome sequencing data sets. A, Lollipop plots showing the specific mutations identified in SRCAP, YLPM1, ZBTB33, and ZNF318 in 45,676 exomes from ExAC. Missense mutations are shown as green squares, truncating mutations (including nonsense mutations, frameshift insertions/deletions, and splice-site mutations) are shown as black circles, and in-frame mutations are shown as brown hexagons. BTB, broad-complex, tramtrack, and bric a brac protein–protein interaction domain; NLS, nuclear localization signal; SA, spindle-associated domain. B, Graph comparing the number of mutations identified in specific genes in 45,676 ExAC samples versus in 39,007 samples from the TOPMed cohort. Novel candidate CHIP genes are labeled in red. C–F, Graphs showing the prevalence of mutation in ZBTB33 (C), ZNF318 (D), YLPM1 (E), and SRCAP (F) among individuals from ExAC and TOPMed in different age groups. Error bars represent 95% confidence intervals.
To validate these previously uncharacterized CHIP drivers in an independent data set, we evaluated whole-genome sequencing (WGS) of 39,007 individuals from TOPMed (ref. 17; Supplementary Table S4) for mutations in SRCAP, YLPM1, ZBTB33, and ZNF318. Mutations in these genes were found at similar frequencies in both TOPMed and ExAC, and these genes were also among the most commonly mutated genes in CHIP (overall mutation frequencies: SRCAP: 0.06%, YLPM1: 0.07%, ZNF318: 0.12%, ZBTB33: 0.18%; Fig. 1B). ZBTB33 was the sixth most commonly mutated gene overall and was more frequently mutated than the canonical MDS-associated genes SF3B1, SRSF2, and TP53. Finally, we assessed whether these mutations were age associated, as would be expected for bona fide somatic mutations. The presence of a mutation in these four genes was associated with age in linear regression models, strongly suggesting that the identified variants were not germline polymorphisms or technical artifacts (Fig. 1C; Supplementary Tables S5 and S6).
New Candidate CHIP Drivers Are Also Mutated in Patients with MDS
Because CHIP can progress to MDS, we examined whether the candidate CHIP drivers we identified were also mutated in patients with MDS. We sequenced the four candidate CHIP genes in a cohort of 1,206 MDS cases and found 16 ZBTB33 (1.3%), 16 YLPM1 (1.3%), 11 SRCAP (0.9%), and 4 ZNF318 (0.3%) mutation carriers (Fig. 2A; Supplementary Table S7). These findings are consistent with mutations that are recurrent drivers of MDS but at frequencies that are too low to have been detected in previous exome or genome sequencing studies of myeloid malignancies. The observation that these mutations are found at higher prevalence in MDS cases than in CHIP supports the hypothesis that these are bona fide drivers of MDS.
Figure 2.

Identification of mutations in ZBTB33, YLPM1, SRCAP, and ZNF318 in a cohort of 1,206 patients with MDS. A, Graph depicting the number of mutations in potential new CHIP genes identified by targeted exome sequencing of 1,206 patients with MDS. B, Pie chart showing the sex for the 16 cases with ZBTB33 mutations. C, The VAF of each ZBTB33 mutation is plotted as an individual point, with bars representing mean and SEM. D, Lollipop plot showing the specific mutations identified in ZBTB33 relative to ZBTB33's functional domains in 1,206 MDS samples. Missense mutations are shown as green squares and truncating mutations are shown as black circles. BTB, broad-complex, tramtrack, and bric a brac protein–protein interaction domain; NLS, nuclear localization signal; SA, spindle-associated domain. E, Comutation plot showing mutations in MDS-associated genes identified in the 16 cases with ZBTB33 mutations. Genes encoding splicing factors and genes with three or more mutations are shown.
The most frequently mutated of these new genes, ZBTB33, is notable due to its proposed role as a reader of methylated DNA, linking it to the biology of the two most common CHIP genes, DNMT3A and TET2. DNMT3A and TET2 alter DNA methylation through distinct and complementary mechanisms, as the two genes are commonly comutated in the same clone (18,19,20,21). Consistent with ZBTB33 being an X-linked gene, the majority of mutations in this gene were found in male patients (Fig. 2B), and the mean ZBTB33 VAF was 0.63 for males and 0.23 for females (Fig. 2C).
In both CHIP and MDS, the majority of variants identified were missense mutations that clustered in ZBTB33's functional domains, suggesting that disruption of these domains' function may be important for CHIP and/or MDS pathogenesis (Figs. 1A and 2D). ZBTB33 belongs to the BTB/POZ subfamily of zinc finger (ZF) transcription factors, which each contain an N-terminal BTB protein–protein interaction domain and several C-terminal ZF DNA-binding domains, of which ZBTB33 contains three (18). The missense mutations we identified mapped to ZBTB33's BTB and ZF domains, as well as to two regions necessary for ZBTB33's association with centrosomes and the mitotic spindle (SA1 and SA2; refs. 22, 23) and its two sumo-interacting motifs (SIM; ref. 24; Figs. 1A and 2D).
ZBTB33 mutations co-occurred with mutations in splicing factor genes in MDS in 14/16 cases (Fig. 2E; Supplementary Fig. S2A). The co-occurrence of ZBTB33 and SF3B1 mutations was statistically significant (Fisher exact text, P = 0.03; Supplementary Fig. S2B). Although SF3B1 mutations are powerfully associated with ring sideroblasts, iron-laden mitochondria that surround the nucleus, we did not find an association between ZBTB33 mutations and the presence of ring sideroblasts (Supplementary Fig. S2C). Based on analysis of the VAF, ZBTB33 mutations were in the founding clone in 11/14 splicing factor gene-mutated cases, whereas they appeared to be secondary events or unrelated to the MDS clone in the other 3 cases (Supplementary Table S8). To validate the co-occurrence of ZBTB33 and SF3B1 mutations, we sequenced ZBTB33 in an additional cohort of 127 SF3B1 mutant MDS cases. ZBTB33 mutations were present in 2.36% of this cohort, which is consistent with the ZBTB33 mutation frequency in SF3B1 mutant cases in the initial cohort (2.83%; Supplementary Table S9).
CRISPR/Cas9 Editing of Zbtb33 Results in Clonal Expansion and a Competitive Advantage of Mouse Hematopoietic Stem and Progenitor Cells In Vivo
Dnmt3a or Tet2 loss in mouse hematopoietic stem and progenitor cells (HSPC) confers increased self-renewal potential in vitro and clonal dominance in vivo (25,26,27). The identification of clonal somatic ZBTB33 mutations in healthy people and patients with MDS led us to hypothesize that ZBTB33 loss may similarly lead to clonal hematopoiesis in murine models. To test the ability of Zbtb33-mutated mouse HSPCs to expand relative to wild-type (WT) cells, we performed bone marrow (BM) transplantation experiments in mice.
We used CRISPR/Cas9 editing in male HSPCs to introduce loss-of-function mutations into the single Zbtb33 allele in Cas9 transgenic mice (28). We transduced c-kit+ HSPCs from MxCre Cas9 mice with a single-guide RNA (sgRNA) targeting Zbtb33 (at a sequence corresponding to the SA1 domain in human ZBTB33) or a control sgRNA targeting noncoding sequence, followed by transplantation into lethally irradiated CD45.1+ mice (n = 7 per group; Fig. 3A). We verified that donor cells were edited by sequencing peripheral blood (PB) drawn from the recipient mice every 4 to 6 weeks following transplant and quantifying the percentage of reads with insertions or deletions (indels) near the CRISPR cut sites (Supplementary Fig. S3A; Fig. 3B). In parallel, donor cell engraftment was validated by flow cytometry (Supplementary Fig. S3B). We observed significant expansion of Zbtb33-edited cells over 32 weeks (linear regression nonzero slope test, P = 0.0038), whereas the percentage of cells edited by the control sgRNA in noncoding sequence was not significantly different over time (linear regression nonzero slope test, P = 0.80; Fig. 3B). The majority of Zbtb33 indels were frameshift mutations (Supplementary Fig. S3C), suggesting that the cells that expanded inactivated Zbtb33 protein function. At harvest, there was no difference in spleen weight between mice transplanted with Zbtb33 or control noncoding sequence edited cells (Supplementary Fig. S3D). To assess editing in an HSC-enriched population, we sequenced Zbtb33 from BM donor LSK cells (CD45.2+Lin−Sca+Kit+) of mice that were sacrificed at 44 weeks. We observed 35% to 85% indels in Zbtb33, which confirmed that LSKs were edited and that an edited population persisted over the course of 44 weeks (Supplementary Fig. S3E).
Figure 3.

Expansion of Zbtb33-edited HSPCs in mouse transplant models. A, Schematic of noncompetitive transplant setup. HSPCs from male mice expressing Cas9 were lentivirally transduced with an sgRNA targeting Zbtb33 or a negative control sgRNA targeting a noncoding region and transplanted into lethally irradiated mice (n = 7 per group). B, PB was drawn every 4 to 6 weeks; DNA was extracted, PCR amplified, and sequenced; and the percentage of reads with indels near the CRISPR cut site was measured. For each mouse, the indel percentage at each time point was normalized to that at week 4. Data, mean ± SEM. Prism was used to perform a linear regression for each group of mice and compute whether the slope was significantly nonzero. P = 0.0038 for mice transduced with Zbtb33 sgRNA and P = 0.80 for control sgRNA. C, Schematic of competitive transplant setup. n = 8 recipients. D and E, The percentage of cells expressing RFP or BFP in the CD45.2+ (D) or CD45.2+ CD11b+ (E) PB at each time point, as measured by flow cytometry. Data, mean ± SEM. The ratio of percentage of RFP+ to percentage of BFP+ cells was calculated for each mouse at each time point, and Prism was used to perform a linear regression and compute whether the slope was significantly nonzero. P = 0.00006 for the CD45.2+ PB and P = 0.0002 for the CD45.2+ CD11b+ PB. F, The percentage of RFP- or BFP-expressing cells in the c-kit+–enriched BM 44 weeks after transplant, as measured by flow cytometry. Data are plotted as individual mice (n = 7), with bars representing the mean and SEM. P = 0.019, computed using a two-tailed paired t test.
To test directly whether Zbtb33-edited cells have a competitive advantage compared with control-edited cells, we performed competitive transplant experiments. We transplanted recipient mice (n = 8) with a 1:1 mix of c-kit+ cells lentivirally transduced with an sgRNA targeting Zbtb33 or a control sgRNA (Fig. 3C). We utilized lentiviral sgRNA plasmids that also strongly express tagRFP or tagBFP, allowing us to measure the percentage of cells expressing each sgRNA by flow cytometry. We verified in the input c-kit+ donor cells that we could detect both editing of the sgRNA target sites by sequencing and expression of red fluorescent protein (RFP) and blue fluorescent protein (BFP) by FACS (Supplementary Fig. S3F). We also confirmed engraftment of CD45.2+ donor cells (Supplementary Fig. S3G). We observed increasing ratios of RFP+ to BFP+ cells over time (linear regression nonzero slope test, P = 0.00006; Fig. 3D), indicating a competitive advantage of Zbtb33-edited cells. This effect was also observed in the Cd11b+ population (linear regression nonzero slope test, P = 0.0002; Fig. 3E), consistent with expansion of myeloid cells. We also observed a significantly higher percentage of cells expressing RFP than BFP in c-kit+ cells enriched from BM harvested at the end of the experiment (two-tailed paired t test, P = 0.019; Fig. 3F), indicating an expansion of Zbtb33-edited HSPCs.
ZBTB33 Interacts with Splicing-Associated and Mitochondrial Proteins in Hematopoietic Cells
To explore ZBTB33's cellular role in hematopoietic cells, we sought to identify proteins with which it interacts. Because ZBTB33 missense mutations cluster in ZBTB33's functional domains, including the BTB domain that mediates protein–protein interactions, we also examined the interactome of ZBTB33 R26C, the most commonly observed missense mutation in the BTB domain. We expressed V5-tagged WT ZBTB33 or mutant ZBTB33 R26C in the TF-1 hematopoietic cell line, which expresses WT ZBTB33 (Supplementary Fig. S4A). Overexpression of ZBTB33 R26C or other mutations frequently observed in the clonal hematopoiesis exome sequencing data (e.g., SIM2 mutant ZBTB33 G438D or ZF mutant ZBTB33 C552R) did not appear to affect stability of the protein (Supplementary Fig. S4A). We performed immunoprecipitation (IP) using V5 antibody, followed by mass spectrometry (IP/MS; Supplementary Fig. S4B and S4C).
Comparisons were made between the WT ZBTB33 IP and untransduced control IP, or between the ZBTB33 R26C IP and WT ZBTB33 IP. Significant interactors were determined based on log fold change and adjusted P value (Supplementary Fig. S5A and S5B). We identified significant interactions between ZBTB33 and multiple proteins involved in RNA splicing (Fig. 4A; Supplementary Table S10), and we validated several of these interactions by IP/Western blot (Supplementary Fig. S5C–S5F). Several of these proteins were differentially enriched between WT ZBTB33 and the R26C mutant (Fig. 4B; Supplementary Table S11; Supplementary Fig. S5E and S5F). Additionally, many mitochondrial proteins were enriched in the WT and R26C ZBTB33 interactomes compared with the untransduced control (Supplementary Fig. S5G and S5H; Supplementary Tables S10 and S11). ZBTB33 has never been described to localize to the mitochondria previously, but it was identified using an RNA interference screen for genes that affect mitochondrial abundance and function and was validated as having a functional role in mitochondrial respiration (29). Using cellular fractionation, we confirmed that exogenously expressed, epitope-tagged as well as endogenous ZBTB33 are present in the mitochondrial fractions of TF-1 cells (Supplementary Fig. S5I).
Figure 4.

Interaction of ZBTB33 with splicing-associated proteins and increased IR upon Zbtb33 loss. A and B, Volcano plots visualizing significant protein interacting partners enriched in the WT ZBTB33-V5 IP compared with control (A) and differentially enriched in the WT ZBTB33-V5 and ZBTB33 R26C-V5 IPs (B). Proteins involved in RNA splicing are colored red. C, Schematic depicting experimental setup for transplant to isolate mouse LSKs for RNA-seq experiment. Thirty-eight weeks after transplant, BM was harvested from recipient mice (n = 5). RFP+ and RFP− recipient LSKs were isolated by FACS, followed by RNA extraction and RNA-seq. D, Scatterplot comparing constitutive IR in RFP− LSKs (n = 5) versus RFP+ LSKs (n = 5). Axes measure the fraction of mRNAs with spliced introns. Red and blue dots represent introns that met the thresholds for significance (P ≤ 0.05) and effect size (absolute percentage change in isoform usage of ≥10% or log fold change ≥ 2) and were retained less or more frequently, respectively, in RFP+ compared with RFP− cells. E, Bar graphs plotting the percentage of significant IR events that were increased (blue) or decreased (red) in SF3B1 single-mutation MDS samples (n = 4) versus healthy controls (n = 3; left) and in ZBTB33/SF3B1 comutation MDS samples (n = 3) versus SF3B1 single-mutation MDS samples (n = 4; right).
Zbtb33 Loss Leads to Increased Constitutive Intron Retention in Mouse HSPCs
The co-occurrence of ZBTB33 and SF3B1 mutations in patients with MDS and our finding that ZBTB33 interacts with spliceosome proteins provide two lines of evidence suggesting an interaction between ZBTB33 and RNA splicing. We evaluated the impact of Zbtb33 loss on alternative splicing in mouse HSPCs. We performed competitive transplant experiments as described previously except that we used FACS-sorted LSKs (Lin−Sca+Kit+) as donor cells to minimize heterogeneity (Fig. 4C). We transplanted 30,000 LSKs per recipient mouse (n = 5). Thirty-eight weeks after transplant, we FACS sorted RFP+ and RFP− LSKs from harvested BM, which confirmed long-term expression of the Zbtb33 sgRNA 38 weeks after transplant (Supplementary Fig. S6). To query whether there are differences in alternative splicing in Zbtb33-edited LSKs compared with control LSKs, we performed RNA sequencing (RNA-seq) on FACS-sorted LSK cells competitively transplanted with Zbtb33 or control sgRNA.
As the entire coding sequence of Zbtb33 is located in one exon, the edited Zbtb33 transcript is not expected to undergo nonsense-mediated decay via the exon junction complex, and a truncated or mutant nonfunctional protein is therefore more likely than full knockout. Consistent with this, we confirmed DNA frameshift editing in Zbtb33, that Zbtb33 was still expressed in Zbtb33-edited cells, and that the frameshift edits were present in the RNA as well (Supplementary Fig. S7).
To evaluate splicing changes, we performed paired parametric tests comparing RFP+ (Zbtb33 sgRNA+) and RFP− (Zbtb33 sgRNA−) LSKs and restricted to events that had sufficient (≥20) informative reads and met our thresholds for significance (P ≤ 0.05) and effect size (≥10% absolute change in isoform usage or a log fold increase of ≥2). We observed minimal changes in usage of competing 5′ or 3′ splice sites or splicing efficiency of introns that are frequently retained (Supplementary Fig. S8A–S8C). However, we observed an increase in retention of normally constitutively spliced introns in RFP+ cells (Fig. 4D). Further limiting to events in which the intron retention (IR) rate was increased by at least 5% (absolute scale) in Zbtb33-edited cells, we detected 908 differentially retained constitutive introns with P ≤ 0.05 (Supplementary Table S12). Using RNA-seq read coverage plots, we manually confirmed IR for selected events with the highest log fold change in IR (Supplementary Fig. S9). We also investigated motif enrichment at IR events (Supplementary Fig. S10A–S10C). Although IR was widespread and did not appear to act specifically on transcripts corresponding to one class of genes, gene ontology (GO) analysis revealed that the list of IR events was enriched for splicing factors, which have been reported to be regulated by IR previously (Supplementary Table S13). We also noted that retained introns were observed in transcripts corresponding to two of our other candidate CHIP genes, Ylpm1 and Srcap (Supplementary Table S12).
To investigate whether similar splicing changes were observed in patients with MDS, we evaluated IR in CD34+ BM cells from ZBTB33/SF3B1-comutated patients with MDS (n = 3) and quantified events that were significantly increased or decreased compared with patients with MDS with SF3B1 single mutation (n = 4) or healthy donor controls (n = 3). Consistent with previous reports, we observed significantly decreased IR in SF3B1 mutant cases compared with control samples. Three hundred forty-two significant IR events were identified with FDR <0.1 and delta psi > |0.1|, and 254/342 (74%) exhibited decreased IR. However, in cases with ZBTB33 and SF3B1 mutations, we observed significant differential IR events that were both increased [57/110 (52%)] and decreased [53/110 (48%)] compared with SF3B1 single-mutation cases (Fig. 4E; Supplementary Fig. S11). Of the 57 genes in which increased IR was observed, only two (CCNL2/Ccln2 and DHRS4L2/Dhrs4) overlapped with genes in which increased IR was observed in mice.
Discussion
Sequencing studies of blood from healthy individuals have revealed that mutations in specific genes associated with hematologic malignancies drive clonal hematopoiesis in a large proportion of the population (1, 2). Because many individuals with evidence of clonal hematopoiesis do not have a mutation in a known cancer gene (2, 5, 6), the focus of this study was to discover mutations in previously uncharacterized genes that drive clonal expansion of HSCs. By utilizing very large sequencing data sets and developing an algorithm to distinguish low VAF somatic mutations, we identified low frequency mutations and discovered new drivers of clonal hematopoiesis, including ZBTB33, YLPM1, SRCAP, and ZNF318.
Our work nominates genes that may regulate the self-renewal or survival of HSCs and that may be implicated in hematologic cancers. This is supported by our findings that ZBTB33, YLPM1, SRCAP, and ZNF318 are mutated in patients with MDS. Mutations have previously been detected in several of these genes at frequencies that did not reach statistical significance, which supports our findings and highlights the need for mutation discovery in large cohorts. Notably, the relative frequency of driver gene mutations in CHIP does not always correlate with that in MDS or AML. For example, mutations in DNMT3A and PPM1D are relatively more common in CHIP than MDS, and mutations in genes encoding splicing factors are relatively more common in MDS than CHIP (30, 31). Future work studying the biological role and function of these genes is likely to provide insight into mechanisms important for normal HSC function, clonal hematopoiesis, and MDS and other hematologic malignancies. For example, SRCAP is involved in chromatin remodeling, and ZNF318 is a putative RNA-binding protein, suggesting these pathways may also contribute to clonal hematopoiesis and MDS. Chromatin remodeling, transcription, RNA transport, and RNA splicing all influence the transcriptional state of hematopoietic stem cells and cellular differentiation, indicating how these mutations could potentially converge on downstream effects.
We chose to focus our validation efforts on ZBTB33, as it was the most frequently mutated of the novel genes in multiple cohorts of individuals with clonal hematopoiesis and MDS, scored by multiple methodologies. Furthermore, we were intrigued by its connection to DNA methylation, a process known to be perturbed in hematologic malignancies. As functional validation of a role for ZBTB33 in HSCs, we have demonstrated that mouse HSPCs with inactivated Zbtb33 undergo expansion over time and have a competitive advantage in the transplant setting. Dnmt3a or Tet2 mutations confer similar effects in mouse HSPCs, indicating that Zbtb33 mutant cells exhibit phenotypes characteristic of well-studied CHIP mutations.
Although it was previously appreciated that DNA methylation regulates alternative splicing, specifically exon skipping/inclusion (32,33,34,35) and IR (36), we have identified an additional 5-methylcytosine (5mC) reader, ZBTB33, that appears to modulate IR. Although our findings warrant more work and validation, our discoveries that Zbtb33-edited mouse LSK cells exhibit increased IR and that IR is also altered in ZBTB33/SF3B1-comutated MDS samples compared with SF3B1-mutated samples are consistent with established roles of DNA methylation and 5mC binding proteins in regulating alternative splicing. Known mechanisms of modulation of alternative splicing by 5mC binding proteins include altered recruitment of splicing factors and modulation of RNA polymerase II (RNA Pol II) kinetics (32,33,34,35,36). Our IP/MS results indicate that ZBTB33 binds components of the RNA spliceosome and subunits of the RNA Pol II complex, providing rationale for a similar role for ZBTB33. Future studies will be necessary to determine the precise mechanisms associated with ZBTB33-mediated IR, including whether this process is directly mediated by ZBTB33 binding to 5mC.
Our work provides a precedent for future mutation discovery efforts in other premalignant states. Recent studies have revealed a number of corresponding premalignant states in other organs and tissues, including the esophagus and skin (37,38,39,40). This study provides a framework for performing unbiased mutation identification studies in nonmalignant tissue, which could be adapted to discover potential uncharacterized driver genes of other premalignancies.
Methods
Human studies were approved by ethics committees at all involved institutions and were conducted in accordance with ethical guidelines and the U.S. Common Rule.
Cohorts with Genetic Sequencing Data
For the discovery set, we used whole-exome sequencing data from 45,676 persons included as part of the ExAC. These included 18,040 persons from 22 cohorts in the T2D-GENES and GoT2D consortia previously analyzed by us (1), 12,404 persons who were part of a case–control study for schizophrenia and bipolar disease (2), and 15,232 persons who were part of the Myocardial Infarction Genetics Consortium that included several cohorts previously described (41). Numbers of individuals from each cohort, mean age, and CHIP prevalence are shown in Supplementary Table S1. CHIP variant calls were produced using MuTect and Indelocator as previously described (42). The protocols for these studies were approved by the ethics committees at all involved institutions and were conducted in accordance with ethical guidelines. Written informed consent was obtained from all participants.
For the replication set, we used WGS data from 39,007 persons in five cohorts within the TOPMed project (BioMe, ref. 43; Cardiovascular Health Study, ref. 44; Framingham Heart Study, ref. 45; COPDGene, ref. 46; and Women's Health Initiative, ref. 47; Supplementary Table S14). Numbers of individuals from each cohort, mean age, and CHIP prevalence are shown in Supplementary Table S4. CHIP variant calls were produced using Mutect2 as previously described (17). Written informed consent was obtained from all human participants by each of the studies that contributed to TOPMed with approval of study protocols by ethics committees at participating institutions, as described previously (17).
Nonsense Mutation Screen for Clonal Expansion
Clonal hematopoiesis in most studies is defined by the presence of a cancer-associated variant in the blood of a person without a known hematologic malignancy. We hypothesized that clonal hematopoiesis may also arise due to mutations in genes not currently appreciated to be cancer drivers. We first focused on nonsense variants in the whole exome to test this hypothesis. Nonsense variants were chosen because they have a clear functional consequence (truncation of the protein product) and they are less subject to alignment artifacts or sequencing errors than frameshifts caused by small insertions/deletions. Nonetheless, it is still challenging to distinguish true somatic variants from germline alleles or technical artifacts from data from a single tissue source. To do this, we implemented a strict filtering approach that incorporated several steps (Supplementary Fig. S1A).
Step 1: Removal of Variants that Are OxoG Artifacts.
Guanine bases in DNA may be oxidized to 8-oxoguanine (OxoG) ex vivo, which leads to a common artifact in sequencing data (G > T or C > A) that can be identified by strand bias. We used the D-ToxoG package (48) to eliminate putative OxoG artifacts from the full set of nonsense variants using an adjusted P-value cutoff of 0.05.
Step 2: Removal of Genes with Many Variants at Low VAF.
Heterozygous germline variants should have VAF distribution that centers around 0.5. However, some genes have a large number of variants that are at low VAF. With rare exceptions, these are very likely to represent genes with recurrent sequencing artifacts or alignment artifacts rather than true driver somatic variants. To eliminate these low VAF genes, we first created a file with the full list of variant calls output from MuTect (“callstats.txt”), which includes known germline SNPs, for 2,314 random exomes in the data set. We excluded variants that were homozygous for a nonreference allele (VAF >0.8) and required three or more alternate reads. We then calculated the mean VAF for each unique variant (excluding variants present less than five times or with VAF <0.05) in each protein coding gene across all 2,314 samples. If a gene had an overall mean VAF for all its variants of less than 0.4, it was excluded.
Step 3: Removal of Genes with Many Likely Germline Nonsense Variants.
Genes in which somatic nonsense driver mutations occur are not expected to have a large number of germline nonsense variants also; if the latter is true, it is more likely that loss of function in the gene is well tolerated and unlikely to have large fitness effects on clones. Thus, these genes are not likely to have true somatic drivers and instead are enriched for germline events and artifacts. To eliminate these genes, we defined a variant to be likely germline if the VAF was greater than 0.35. We then calculated the proportion of nonsense variants that had VAF below or above this cutoff in each gene. We excluded all genes where the proportion of nonsense variants with VAF >0.35 was more than 40%.
Step 4: Removal of Variants that Are Highly Recurrent.
True somatic truncating variants are less likely to be highly recurrent compared with germline variants or artifacts. Therefore, we removed all nonsense variants present seven or more times in the data set.
Step 5: Removal of Variants that Segregate by Ancestry.
Unlike germline variants, somatic variants should have weak or no association to self-reported ancestry. Within our data set, persons self-reported as African (15.9%), East Asian (4.7%), European (46.3%), Hispanic (12.2%), or South Asian (20.9%) ancestry. A true somatic variant would be expected to have a frequency distribution similar to the overall composition of the data set. We used chi-square tests to calculate whether the actual ancestry distribution for a given variant deviated from the expected distribution and excluded variants that had an FDR-adjusted P value <0.05.
Step 6: Removal of Variants that Have a VAF that Is Not Significantly Different from Germline.
True somatic variants are less likely to have a VAF of around 0.5, which is what we expect for germline variants. We calculated a binomial probability of VAF deviating from an expected VAF of 0.5 and retained only those variants that had an FDR-adjusted P value <0.05.
After these filtering steps, the remaining variants were considered to be putatively somatic, and the top 40 genes with the most variants are listed in Supplementary Table S2.
Detection of Significantly Mutated Genes Using MutSig
Mutation Calling Workflow.
We identified mutations in 49,291 individuals in the ExAC consortium that had known age annotation (7). We called somatic mutations using the Cancer Genome Analysis Team mutation calling pipeline in FireHose including OxoG filtering and Panel of Normals filtering with a likelihood cutoff of −5 to account for the heterogeneity of sequencing cohorts included as well as the lack of a matched normal control. We used an earlier iteration of this pipeline in which MuTect1 was used for SNV calling, and Indelocator was used for calling indels. In addition, given that the expected mutation rate of CHIP is 0.2/Mb instead of 1/Mb, we adjusted the tumor_lod threshold accordingly such that we accepted mutations if their “i_t_lod_fstar” was greater than 8.6 (equation 1; ref. 49).
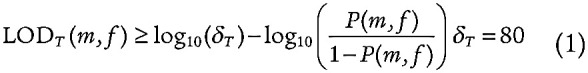 |
Given that CHIP can be found in healthy blood, we included 1,493 individuals younger than 30 years old and who lacked the common somatic hotspots DNMT3A R882 and JAK2 V617, as evidence in the literature suggests these mutations are among the most common drivers of CHIP and their recurrence suggests a true somatic event as opposed to a common artifact (1). We filtered out indels 10 base pairs (bp) or longer given that most of our filtering steps demanded an accurate determination of allele fraction and that Indelocator underestimates allele fractions by including all split reads. In addition, we filtered out indels found in homopolymer runs greater than 6 bp long as well as variants that had depth greater than the 99% confidence interval of SNV depth. We imposed cutoffs on homopolymer length and read depth similar to those imposed by Strelka: filtering out variants in homopolymer runs greater than 6 and those whose depth had a poisson probability greater than 0.99 given the median depth of a sample's SNVs (50).
Although the Panel of Normals was designed to eliminate common sequencing artifacts, we noticed common bleed-through artifacts in sequencing cohorts that were not well represented in the Panel of Normals, indicated by a preponderance of low allele fraction mutations whose alternate allele was shared with its reference trinucleotide context as a result of adjacent fluorescence. Given that the only mutational signature we expect to see in control blood samples is aging signature (COSMIC1), we identified the expected 99% quantile of mutation frequencies at each trinucleotide context based on a set of 1,000 individuals well represented in the Panel of Normals and showed evidence of aging signature (51). For each sample we analyzed, if there were more mutations found in a given context than expected by the set of 1,000 individuals with aging signature and the alternate allele was shared with the reference context, we filtered out all mutations found in this trinucleotide context for that given sample with a VAF less than 50%.
Given the sequencing heterogeneity, we observed orientation bias artifacts that were not filtered out by the Panel of Normals. To account for these, we filtered out mutations whose FDR-corrected binomial probability of F1R2 alternate allele counts or F2R1 alternate allele counts at 0.96 was greater than 0.99. A corrected binomial probability greater than 0.99 suggests that the mutation is more consistent with orientation bias artifact than a typical mutational process; hence, these mutations were filtered out.
To account for mismapping artifacts, we simulated reads containing the mutation of interest within a sliding window of 75 bp, excluding the 10 reads on either end of the sequence. We eliminated all mutations whose next best mapping score was within 60 units of the maximum 76. In addition, we eliminated variants that overlapped with segmental duplications, common germline copy-number gains and losses as per 1000 Genomes, variants excluded in the strict mask of 1000 Genomes, variants that were present at greater than 1% frequency in the ExAC population, and sites that failed Hardy–Weinberg equilibrium in 1000 Genomes (2).
Dirichlet Clustering Identifies Clonal Somatic Mutations in Blood Samples without Known Hematologic Malignancies.
Methods for identifying somatic mutations in a tumor without a matched normal sample rely on an estimation of the tumor purity: the proportion of a sample described by tumor-derived cells (52). We define purity using the PhylogicNDT clustering tool on the filtered SNV calls, which utilizes Dirichlet clustering to resolve the clonal structure of a sample given the VAFs of the sample's SNVs and the sample's ploidy, determined by its absolute somatic copy number. Because we are using control blood samples, the somatic copy-number ratio is 1, giving us a ploidy of 2; this observation means that the purity is equal to two times the VAF at which the largest cluster distinct from germline heterozygous mutations is found. We define clusters “distinct from germline heterozygous mutations” to be clusters not found within 5% VAF above or below 50% and it must be less than 50%. To ensure that the clusters we identified were representative of clonal processes, we stipulated that the largest clone must have at least four mutations, consistent with the definition CHIP samples as whole-exome outliers in Zink and colleagues (5). We included indels for only these samples. To ensure that these indels were somatic, we stipulated that the mutations had to pass a likelihood ratio test scaled by 0.4/θGermline in order to account for Indelocator's counting split reads as part of its depth calculation, where θGermline is the allele fraction where the sample's germline heterozygous mutations were found (equation 2).
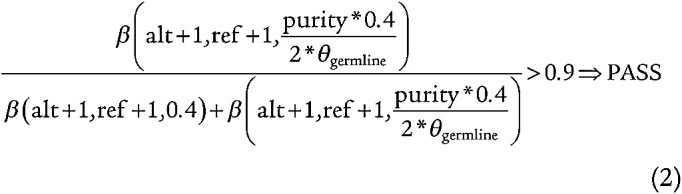 |
Samples Were Selected Based on True-Positive Rate and FDR of Simulated Reads Given Purity, Depth, and the Germline Cluster.
To evaluate our ability to discover true driver mutations, we simulated 10,000 alternate reads on a range of purities from [0,1] using two binomial distributions: one whose probability was the VAF corresponding to the somatic cluster (S) and a second whose probability was the VAF corresponding to the germline heterozygous cluster (G) as calculated by PhylogicNDT clustering (equation 3). The number of trials in the binomial distribution corresponded to the median depth of the sample (equation 3).
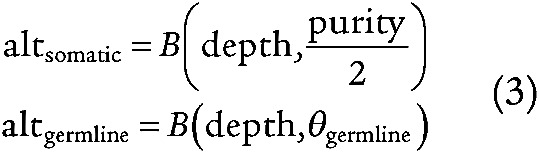 |
For each simulated read, we calculated the beta likelihood given the VAF of the somatic cluster and median depth of coverage to be the likelihood of the alternate read count belonging to the somatic cluster (equation 4). We performed the same calculation given the VAF of the germline cluster and the median depth of coverage and determined the log odds ratio between the germline and somatic models (equation 4).
 |
For the range of calculated log likelihoods, we test each value in steps defined by the length of the vector divided by 100 (equation 5). If the somatic ratio exceeds the tested likelihood and has greater than three simulated alternate counts, the value is a true positive; otherwise, the value is considered a false positive (equation 5). If the germline ratio is less than the tested ratio or has a simulated alternate read count less than or equal to three, the value is a true negative and is otherwise a false negative (equation 5). Once we have calculated the true-positive, false-positive, true-negative, and false-negative counts, we determine the true-positive rate (TPR) and the FDR for each sample, and exclude those with TPR less than 0.9 and FDR greater than 0.1. For our FDR calculation, we applied prior probabilities to the false-positive and true-positive counts given the expected germline mutation rate (1 × 10−3) and the expected somatic mutation rate (1 × 10−6), respectively (equation 5).
 |
Rare Germline Events Were Removed by Training a support Vector Regression Model to Predict the Expected Allele Fraction of Germline Mutations at Given Genomic Loci.
Although we removed common germline mutations in our call set, there were a number of rare germline mutations that were reported at low allele fractions due to bait bias and mappability issues that could not be readily modeled. As a result, we trained a support vector regression model (SVM) implemented in scikit-learn using 30,000 mutations rejected by MuTect for the following reasons: “germline_risk,” “normal_lod,” and “alt_allele_in_normal” and that were present in greater than 1% of the ExAC population (53). The SVM regression was used to predict the expected germline allele fraction of an SNP at a given genomic locus was determined from the allele fractions of the adjacent SNPs, distance in genomic space to the nearest SNP, distance to Agilent bait boundaries, and whether the site overlaps with a segmental duplication, copy-number variation (CNV) gain, or CNV loss. We applied the model to the test data after k-fold validation, observing a median L1SmoothLoss less than 0.002. Once applying the model to test data, we then calculated a likelihood ratio of beta distributions comparing whether a variant was more consistent with the estimated purity or the site-specific germline SNP allele fraction, stipulating that a given site's likelihood ratio should be greater than 0.9 (equation 6).
 |
Significantly Mutated Genes Were Identified Using MutSig2CV.
After filtering for common and rare germline mutations, we ran MutSig2CV to identify significantly mutated genes. To ensure that we were modeling hematopoietic drivers, we ran multiple hypothesis correction only using genes that were expressed in HSCs using the single-cell expression from Corces and colleagues, selecting only genes whose median expression was greater than 2 across hematopoietic stem cells (54).
Validation of Novel Drivers of Clonal Hematopoiesis
Four genes overlapped from the list of variants identified from the nonsense mutation screen and those identified from MutSig (ZBTB33, ZNF318, YLPM1, and SRCAP). Therefore, we focused on these four genes for further analyses. We first queried whether frameshift and splice mutations could be found in these genes, which was true in all cases. For ZBTB33, YLPM1, and SRCAP, we observed truncating mutations at nongermline VAFs throughout the open reading frame. For ZNF318, mutations after amino acid position 1531 were found to have high VAFs, suggesting that these were likely rare germline mutations with uncertain functional consequence. We therefore included only mutations from amino acid position 1 to 1531 for this gene. We then asked whether there were recurrent missense or hotspot mutations in any of these genes. The only gene with a nonrandom pattern of putative somatic missense variants was ZBTB33, with mutations clustered in several functional domains (Fig. 1A). A full list of the putative somatic variants in these genes can be found in Supplementary Table S3. A list of variants for all previously known CHIP genes in the ExAC data set (plotted in Fig. 1B) can be found in Supplementary Table S15. Variants in known CHIP genes in the TOPMed data set were previously published (17).
To further validate that mutations in the four genes were truly somatic, we assessed whether carrying these mutations was associated with age, as neither germline variants nor technical artifacts should have any association with age. After excluding those with >1 driver mutation, we used age as the dependent variable in a linear regression model and mutated CHIP gene as the explanatory variable (with no CHIP mutations as the referent group). The results of this analysis can be found in Supplementary Table S5.
Finally, we sought replication of these genes as novel drivers of clonal hematopoiesis in an independent data set. We used WGS data from TOPMed and identified mutations in these four genes using the same approach as in the test set (nonsense, frameshift, and splice-site for all four genes, as well as missense mutations in functional domains for ZBTB33). We then used a linear regression model, as described above for the test set, to test whether carrying each driver gene was associated with age. The results of this analysis can be found in Supplementary Table S6.
MDS Patient Data Cohort and Analysis
Mutation status for candidate CHIP genes was acquired for a cohort of 1,206 samples from the University of Pavia and Karolinska University Hospital, consisting primarily of MDS samples with a small number of AML samples included. Mutations were discovered by targeted sequencing (covering known MDS driver genes and ZBTB33, YLPM1, SRCAP, and ZNF318). ZBTB33 was subsequently sequenced in an additional 127 SF3B1 mutant MDS cases.
To determine whether there was an association of ring sideroblasts with ZBTB33 and/or SF3B1 mutations, we considered only patients with a confirmed diagnosis of MDS (n = 1,056). A total of 150 patients with World Health Organization 2016 diagnoses of AML, AML-MRC (AML with myelodysplasia-related changes), AML T-33 [AML with inv(3)(q21.3q26.2 or t(3;3)(q21.3;q26.2)], NA (no diagnosis recorded), or NO-MDS (sample did not support MDS diagnosis) were excluded from this analysis. Additionally, we excluded 86 patients for whom ring sideroblast status was not reported.
Cell Lines and Plasmids
TF-1 cells were obtained from the ATCC and were cultured in RPMI-1640 (Corning), supplemented with 10% heat-inactivated fetal bovine serum (Omega Scientific) and 1% penicillin, streptomycin, and l-glutamine (Gibco), plus 2 ng/mL recombinant human GM-CSF (Miltenyi Biotec). Cells were not authenticated or tested for Mycoplasma. Cells were used within 6 months of thawing.
The pLKO5.sgRNA.EFS.tRFP (Addgene, Ebert Lab), pLKO5.sgRNA.SFFV.tRFP (James Kennedy, Ebert Lab), and pLKO5.sgRNA.SFFV.tBFP (James Kennedy, Ebert Lab) vectors were used for CRISPR/Cas9 experiments, and guide RNAs (sgRNAs) were cloned into the vectors using a BsmBI restriction site. sgRNA sequences are listed in Supplementary Table S16.
A human ZBTB33 cDNA was obtained from the Harvard Medical School PlasmID Repository and cloned into the plx307 backbone (Addgene) by Gateway cloning (Life Technologies), followed by site-directed mutagenesis to replace one amino acid residue that differed from the ZBTB33 reference sequence. Site-directed mutagenesis was also performed to generate the ZBTB33 point mutants R26C, G438D, and C552R (QuickChange II XL, Agilent). These point mutations were chosen as they correspond to the most common missense mutations observed in the BTB, SIM2, and ZF domains in the clonal hematopoiesis exome sequencing data. TF-1s were lentivirally transduced with WT and mutant ZBTB33 plx307 plasmids and selected using 2 μg/mL puromycin (Gibco).
Mouse Transplant Experiments and Analysis
Experiments were executed in compliance with institutional guidelines and were approved by institutional review boards at Boston Children's Hospital (Protocol Number: 16-04-3156R) and Brigham and Women's Hospital (Protocol Number: 2017N000060). Male donor mice, ages 6 to 10 weeks (Cas9 Mx1Cre1+; ref. 28), were treated with 3 doses of 200 mg poly(I:C) HMW (high molecular weight; Invivogen). Two to four weeks following poly(I:C) injection, BM was harvested from long bones and spine. C-kit+ cells were isolated using magnetic beads (Miltenyi Biotec), stimulated overnight with recombinant mouse SCF and TPO (PeproTech), and lentivirally transduced the following day with plasmids containing the sgRNA of interest. For competitive transplants, bulk cell populations transduced with Zbtb33 sgRNA (targeting sequence corresponding to the human ZBTB33 SA1 domain) or control Gapdh sgRNA (targeting noncoding sequence within an intron of Gapdh; Supplementary Table S16) were mixed 1:1. At least 350,000 c-kit+ cells were transplanted by retro-orbital injection into lethally irradiated (2 × 4.5 Gy) CD45.1+ recipient mice. PB was obtained from the recipient mice every 4 to 6 weeks following transplant, red blood cell lysis was performed twice (RBC lysis solution, Qiagen), and editing was assessed by flow cytometry for RFP and BFP or sequencing to detect indels near the CRISPR cut sites as described below. Mice with low engraftment of donor cells (<30% CD45.2+ cells measured by FACS) were excluded from analysis. Some animals died or became moribund and were euthanized before the conclusion of the experiment, and later time points were analyzed using only remaining live mice. Mice were sacrificed between 30 and 45 weeks after transplant, and BM and spleens were harvested. For the RNA-seq experiment, transplants were performed as described above but using sorted LSK cells (30,000 cells per recipient) as the donor population.
To evaluate expansion over time, we performed linear regression analyses in GraphPad Prism and tested whether slopes were significantly nonzero. For noncompetitive transplants (Fig. 3B), we performed separate analyses for each group of mice. For competitive transplants (Fig. 3D and E), we calculated the ratio of % RFP+/% BFP+ cells for each mouse at each time point and performed linear regression analysis on the ratios.
Confirmation of CRISPR/Cas9 Editing by Next-Generation Sequencing
Assessment of editing was performed by extracting genomic DNA (DNA blood mini kit, Qiagen) and PCR amplifying 150 to 400 bp of genomic sequence spanning the predicted Cas9 cut site, followed by deep sequencing performed by the Massachusetts General Hospital CCIB DNA Core Facility (Cambridge, MA). PCR primers are listed in Supplementary Table S16. The percentage of reads with indels was calculated using the CRISPR workflow on Basepair (www.basepairtech.com).
Flow Cytometry
Flow cytometry was performed using a FACSCanto II (BD Biosciences) and analyzed using FlowJo. FACS sorting was performed using a FACSAria II (BD Biosciences).
Mouse PB cells were stained with antibodies against CD45.1 (PerCP/Cy5.5, A20, BioLegend), CD45.2 (APC/Cy7, 104, BioLegend), Ly-6G (APC, 1A8, eBioscience), and Cd11b (PE/Cy7, M1/70, BioLegend). RFP and BFP expression were also measured to assess the percentage of cells expressing sgRNAs.
For the noncompetitive transplant, BM cells harvested at the end of the experiment were FACS sorted to isolate the CD45.2+ LSK population. C-kit+ cells were first isolated using magnetic beads (Miltenyi Biotec) and then were stained using antibodies against Ly-6A/E (Sca-1; PE/Cy7, D7, BioLegend), CD45.1 (APC, A20, BioLegend), CD45.2 (APC/Cy7, 104, BioLegend), CD3 (PerCP-eFluor710, 17A2, eBioscience), B220 (PerCP-eFluor 710, RA3-6B2, eBioscience), CD11b (PerCP-Cy5.5, M1/70, eBioscience), Ly-6G (PerCP-eFluor 710, 1A8, eBioscience), and TER-119 (PerCP-Cy5.5, TER-119, eBioscience). DAPI (Roche Diagnostics) was used for a live/dead stain.
For the RNA-seq experiment, LSKs were sorted from donor mouse BM by first isolating c-kit+ cells and then staining cells with antibodies against Ly-6A/E (Sca-1; PE/Cy7, D7, BioLegend), CD117 (c-Kit; APC, 2B8, BioLegend), CD3 (Pacific Blue, 17A2, BioLegend), B220 (Pacific Blue, RA3-6B2, BioLegend), CD11b (Pacific Blue, M1/70, BioLegend), Ly-6G (Pacific Blue, RB6-8C5, BioLegend), and TER-119 (Pacific Blue, TER-119, BioLegend). The PE+ population was used to gate out autofluorescent dead cells. Following transplant, BM was harvested, and c-kit+ cells were enriched. CD45.2+ RFP+ and CD45.2+ RFP− LSKs were sorted after staining cells with antibodies against Ly-6A/E (Sca-1; PE/Cy7, D7, BioLegend), CD117 (c-Kit; APC, 2B8, BioLegend), CD45.1 (PerCP/Cy5.5, A20, BioLegend), CD45.2 (APC/Cy7, 104, BioLegend), CD3 (PerCP-eFluor710, 17A2, eBioscience), B220 (PerCP-eFluor 710, RA3-6B2, eBioscience), CD11b (PerCP-Cy5.5, M1/70, eBioscience), Ly-6G (PerCP-eFluor 710, 1A8, eBioscience), and TER-119 (PerCP-Cy5.5, TER-119, eBioscience).
IP/MS Experiments
TF-1 parental and overexpression cell lines were harvested in RIPA lysis buffer (Sigma-Aldrich) supplemented with Halt protease inhibitor cocktail (Life Technologies), and protein concentration was quantified by BCA assay (Pierce). Protein lysate (10 mg) was incubated overnight with V5-tag magnetic beads (MBL #167-11), followed by four washes in lysis buffer (150 mmol/L NaCl, 50 mmol/L Tris pH 7.5), supplemented with Halt protease inhibitor cocktail (Life Technologies), using a magnetic rack.
On-Bead Protein Digestion.
The beads from immunopurification were washed once with IP lysis buffer and then three times with PBS. Two different lysates of each replicate were resuspended in 90 μL digestion buffer (2M Urea, 50 mmol/L Tris-HCl), 2 μg of sequencing grade trypsin was added, and then the lysates were shaken for 1 hour at 800 rpm. The supernatant was removed and placed in a fresh tube. The beads were washed twice with 50 μL digestion buffer and combined with the supernatant. The supernatants were reduced (2 μL 500 mmol/L DTT, 30 minutes, RT), alkylated (4 μL 500 mmol/L IAA, 45 minutes, dark), and a longer overnight digestion was performed: 2 μg (4 μL) trypsin, shake overnight. The samples were then quenched with 20 μL 10% formic acid and desalted on 1 cc 10 mg Oasis HLB cartridges.
Labeling with Tandem Mass Tag Isobaric Mass Tags and Basic Reverse Phase Fractionation.
Desalted peptides were labeled with TMT10 reagents (Thermo Fisher Scientific, lot # QL228730A) according to the manufacturer's instructions. Peptides were resuspended in 25 μL of fresh 100 mmol/L HEPES buffer. The labeling reagent was resuspended in 42 μL of acetonitrile and 10 μL added to each sample (126: untransduced replicate 1, 127N: untransduced replicate 2, 127C: WT replicate 1, 128N: WT replicate 2, 128C: C552R replicate 1, 129N: C552R replicate 2, 129C: G438D replicate 1, 130N: G438D replicate 2, 130C: R26C replicate 1, and 131: R26C replicate 2).
After 1-hour incubation, the reaction was quenched with 8 μL of 5% hydroxylamine. Differentially labeled peptides were subsequently mixed and prepared for basic reverse phase fractionation on 10 mg SepPak columns according to the following protocol: cartridges were prepared for desalting by equilibrating with methanol, then 50% acetonitrile (ACN), 1% formic acid, and three washes with 0.1% TFA. Samples were loaded on to the cartridge and washed three times with 1% formic acid. A pH switch was performed with 5 mmol/L ammonium formate at pH 10, collected, and saved. Subsequent fractions were collected at the following ACN concentrations: 10% ACN in 5 mmol/L ammonium formate; 15% ACN in 5 mmol/L ammonium formate; 20% ACN in 5 mmol/L ammonium formate; 30% ACN in 5 mmol/L ammonium formate; 40% ACN in 5 mmol/L ammonium formate; and 50% ACN in 5 mmol/L ammonium formate.
Protein Identification with Nanolc–MS System.
Reconstituted peptides were separated on an online nanoflow EASY-nLC 1000 UHPLC system (Thermo Fisher Scientific) and analyzed on a benchtop Orbitrap Q Exactive plus mass spectrometer (Thermo Fisher Scientific). The peptide samples were injected onto a capillary column (Picofrit with 10-μm tip opening/75 μm diameter, New Objective, PF360-75-10-N-5) packed in-house with 20-cm C18 silica material (1.9-μm ReproSil-Pur C18-AQ medium, Dr. Maisch GmbH, r119.aq). The UHPLC setup was connected with a custom-fit microadapting tee (360 μm, IDEX Health and Science, UH-753), and capillary columns were heated to 50°C in column heater sleeves (Phoenix-ST) to reduce backpressure during UHPLC separation. Injected peptides were separated at a flow rate of 200 nL/min with a linear 50-minute gradient from 100% solvent A (3% acetonitrile, 0.1% formic acid) to 30% solvent B (90% acetonitrile, 0.1% formic acid), followed by a linear 9-minute gradient from 30% solvent B to 60% solvent B and a 1-minute ramp to 90% solvent B. The Q Exactive instrument was operated in the data-dependent mode acquiring HCD MS/MS scans (R = 17,500) after each MS1 scan (R = 70,000) on the 12 top most abundant ions using an MS1 ion target of 3 × 106 ions and an MS2 target of 5 × 104 ions. The maximum ion time utilized for the MS/MS scans was 120 ms; the HCD-normalized collision energy was set to 31; the dynamic exclusion time was set to 20 seconds; and the peptide match and isotope exclusion functions were enabled.
Database Search and Data Processing.
All mass spectra were processed using the Spectrum Mill software package v7.0 prerelease (Broad Institute, https://proteomics.broadinstitute.org/), which includes modules for tandem mass tag (TMT)–based quantification. For peptide identification, MS/MS spectra were searched against human Uniprot database, to which a set of common laboratory contaminant proteins and V5-tagged ZBTB33 WT and mutants was appended. Search parameters included ESI-QEXACTIVE-HCD scoring parameters, trypsin enzyme specificity with a maximum of two missed cleavages, 40% minimum matched peak intensity, ± 20 ppm precursor mass tolerance, ± 20 ppm product mass tolerance, carbamidomethylation of cysteines, and TMT6-Full labeling of lysines and peptide n-termini as fixed modifications. Allowed variable modifications were oxidation of methionine (M), acetyl (ProtN-term), and deamidated (N), with a precursor MH+ shift range of −18 to 64 Da. Identities interpreted for individual spectra were automatically designated as valid by optimizing score and delta rank1–rank2 score thresholds separately for each precursor charge state in each LC-MS/MS while allowing a maximum target-decoy–based FDR of 1.0% at the peptide spectrum match (PSM) level.
TMT10 ratios were obtained from the protein-comparisons export table in Spectrum Mill. Rather than use a physical common control, that is, one TMT channel containing a physical mixture of all the samples, we created a virtual one during data analysis. Each PSM-level TMT ratio was calculated with a single reporter ion intensity in the numerator, whereas the denominator was the median intensity of all 10 reporter ions. This was done in Spectrum Mill with the control ion set to MedianMulti and all 10 reporter ions selected. To obtain TMT10 protein-level ratios, Spectrum Mill calculated the median over all PSMs assigned to a protein subgroup in each replicate. PSMs with a precursor isolation purity <50% were excluded from protein-level calculations. Before calculating ratios, Spectrum Mill corrected the reporter ion intensities for isotopic impurities using its afRICA correction method, which implements determinant calculations according to Cramer's rule and correction factors obtained from the TMT reagent manufacturer's certificate of analysis. IP/MS data are available in Supplementary Table S17.
Mitochondrial Fractionation
Fractionation was performed using the Mitochondria Isolation Kit for Cultured Cells (Thermo Scientific). Cytoplasm and mitochondrial pellets were boiled with SDS loading buffer, followed by Western blotting. Western blots were also probed with an antibody against the mitochondrial-specific protein AIF (Cell Signaling Technologies) to assess efficiency of fractionation.
Western Blots
Protein lysates were run on Criterion Tris-HCl 4% to 15% gels (Bio-Rad) or Nupage 4% to 12% Bis Tris gels (Life Technologies) at constant voltage and transferred either to Immobilon-P transfer membranes (Millipore) or to Trans-Blot Turbo PVDF transfer membranes (Bio-Rad) at constant amperage. Blots were blocked in 5% Blotto nonfat dry milk (Santa-Cruz) dissolved in Tris-buffered saline with 1% Tween 20. Primary antibodies against ZBTB33 (Bethyl #A303-558A, Santa-Cruz #98589, Sigma #HPA005732), V5 (MBL), AIF (Cell Signaling Technologies), actin (Abcam), HSPA8 (Abcam), SRSF5 (MBL), SRSF 9 (MBL), POLR2E (Abcam), HNRNPDL (Sigma), and PPIL1 (Abcam) were used. Secondary antibodies used were horseradish peroxidase–linked goat anti-mouse and goat anti-rabbit (Prometheus Protein Biology Products). SuperSignal Chemiluminescent Substrate (Thermo Fisher) and a ChemiDoc (Bio-Rad) were used for protein detection.
RNA-seq Experiments and Data Analysis
RNA-seq of Mouse LSK Cells.
RNA was isolated using the RNeasy mini kit (Qiagen). Library preparation and sequencing were performed by Novogene, using the SMARTseq v4 (Takara) + Nextera XT kit (Illumina). We used poly(A) selection and unstranded libraries. More than 50 million paired-end read pairs (2 × 150 bp) were obtained per sample.
Alternative Splicing Analysis of Mouse LSK Cells.
RNA-seq reads were aligned to a gene annotation for the human genome assembly hg19/GRCh37. This annotation was composed of a merge of the Ensembl v.71.1 gene annotation (55), the UCSC knownGene gene annotation (56), and the MISO v.2.0 isoform annotation (57). Read alignment was performed using RSEM v.1.2.4 (58), Bowtie v.1.0.0 (59), and TopHat v.2.1.14 (60) as previously described (61). Isoform ratios for annotated alternative splicing events were estimated with MISO v.2.0 (57); isoform ratios for changes in constitutive intron splicing were estimated with junction-spanning reads as previously described (61). Gene-expression estimates produced by RSEM were normalized using the TMM method (62), with all coding genes used as a reference set.
Significantly differentially spliced isoforms were identified by comparing isoform ratios between groups with a two-sided paired t test. Splicing events were classified as differentially spliced if at least one isoform had a difference between sample groups of ≥10% (absolute, not relative, scale) or a fold change ≥2 with at least 20 informative (distinguishing between isoforms) reads and P ≤ 0.05.
GO enrichment analysis was performed on the list of genes with retained constitutive intron events that had an increased rate of retention in the RFP+ versus RFP− cells of at least 5%, were statistically significant with P ≤ 0.05, and had at least 20 informative reads each. The analysis was restricted to “biological process” GO terms.
RNA-seq analysis figures were plotted using the ggplot2 package in R (63).
We determined the enrichment of three previously described motifs for ZBTB33—the minimal core sequence CTGCNA from Daniel and colleagues (2002) and the full DNA-binding site TCTCGCGAGA and core sequence CGCG from Raghav and colleagues (2012)—across the regions flanking the 5′ and 3′ splice sites of constitutive exons. Only U2 introns were included as there was not a sufficient number of U12 introns retained. The enrichment was calculated by dividing the motif coverage in the target sequences plus a pseudocount (25% of the expected coverage assuming equal nucleotide frequencies) by the coverage in the background sequences plus the same pseudocount (Supplementary Fig. S10A).
We also performed ab initio motif enrichment to look for motifs of length 4, 5, or 6 nucleotides in three regions of the retained introns: 5′ splice-site adjacent [+1, +100], 3′ splice-site adjacent [−100, −1], and the full intron [+1, −1]. The enrichment was calculated similarly, except a per-locus pseudocount of 10% of the expected observations of each k-mer was used (Supplementary Fig. S10B).
Differential Gene-Expression Analysis of Mouse LSK Cells.
Paired FASTQ files were uploaded to the Broad Institute Google Bucket (FireCloud by Terra), and in FireCloud, publicly available workflows from the Broad Methods Repository were utilized. Specifically, reads from FASTQ files were aligned to a reference genome (mouse mm10_M17) using the STAR aligner (v2.6.1c) workflow. Following the alignment, duplicates were marked using the Mark Duplicates workflow. Finally, the reads aligned to the genome were quantified to generate counts for each gene using the RNASEQC2 quantification workflow (mouse gencode.vM17.GRCm38p6). Read alignments for specific samples were visualized by Integrative Genomics Viewer (IGV; Version 2.5.3).
Gene-level differential expression analyses were performed using R software (Version 3.6.1), R Studio (Version 1.1.463), and DESeq2 (Version 1.24.0). For the mouse LSK experiment, a paired analysis was performed. Prefiltering removed genes with average counts below 10 counts across samples. Shrinkage of log2 fold change (LFC) estimates was performed to account for genes that had low counts and high dispersion across samples. A fold-change threshold was not used. Genes with P-adjusted values below 0.05 were considered significantly differentially expressed (Supplementary Table S18).
Gene Set Enrichment Analysis.
Functional analysis was performed using LFC results obtained from the differential expression analysis and R software (Version 3.6.1), R Studio (Version 1.1.463). The genome build used for the differential expression analysis was mouse gencode.vM17.GRCm38p6.genes.gtf. AnnotationDbi (Version 1.46.1) and genome-wide annotations (Release 3.10: mouse org.Mm.eg.db) were used to map Entrez Gene Identifiers to genes. Enrichment of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (mouse mmu; https://www.genome.jp/kegg/catalog/org_list.html) was tested with 1,000 permutations (default) and P value cutoff set at 0.2 (Supplementary Table S19).
Alternative Splicing Analysis of MDS Patient Samples.
We explored alternative splicing events in a previously published MDS cohort from IRCCS Ospedale San Matteo, Pavia, Italy (64), and these investigations were approved by the Ethics Committee of the Fondazione IRCCS Proclinico San Matteo, Pavia, and conducted in accordance with ethical guidelines. Written informed consent was obtained from all patients. We identified three patients with concomitant ZBTB33 and SF3B1 mutation, four with single-hit SF3B1 mutation, and three healthy normal controls. BM CD34+ cells were obtained at the time of diagnosis.
RNA-seq libraries were prepared using the TruSeq RNA library Illumina kit according to the manufacturer's recommendations. Libraries were sequenced using the HiSeq 2500 platform with 100-bp paired-end read protocol. Quality control was performed on raw RNA-seq data using MultiQC and nf-core RNA-seq pipeline outputs (65, 66). Samples were aligned to the GRCh37 reference genome.
Five types of splicing events were analyzed, namely, IR, alternative 3′ splice site (A3′SS), alternative 5′ splice site (A5′SS), exon skipping (SE), and mutually exclusive exons (MXE). These splicing event subtypes were quantified using PSI (Percent Spliced In) values in each sample according to Mixture of Isoforms Tools (57).
Differentially spliced events were identified using rMATS tool (v 4.1.1). More specifically, three pairwise comparisons between SF3B1-single–driven MDS, SF3B1/ZBTB33-comutated MDS, and healthy normal controls were performed using paired-end mode (67). The differentially spliced events for each comparison were filtered with FDR <0.05.
Data Availability
Mouse RNA-seq.
RNA-seq data have been deposited in the NCBI Sequence Read Archive with accession code “PRJNA681911” (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA681911/).
Proteomics.
The original mass spectra and the protein sequence database used for searches have been deposited in the public proteomics repository MassIVE (http://massive.ucsd.edu/ProteoSAFe/static/massive.jsp) and are accessible at ftp://massive.ucsd.edu/MSV000087304/.
Supplementary Material
Acknowledgments
This work was supported by the NIH (R01HL082945, P01CA108631, and P50CA206963), the Howard Hughes Medical Institute, the Edward P. Evans Foundation, the Adelson Medical Research Foundation, and the Leukemia & Lymphoma Society to B.L. Ebert and by the NIH (1DP2HL15754001), the Edward P. Evans Foundation, the Burroughs Wellcome Foundation, and the Ludwig Cancer Research to S. Jaiswal. E.M. Beauchamp was supported by the NIH (T32GM007226 and F31HL143844). E. Bernard was supported by the Edward P. Evans Foundation. L. Malcovati was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC, Milan, Italy) International Accelerator Program—Project Code: 22796; 5 × 1000 program—Project Code: 21267; and Investigator Grant 2017—Project Code: 20125. R.K. Bradley is a Scholar of the Leukemia & Lymphoma Society (1344-18). R.K. Bradley was supported in part by the Edward P. Evans Foundation; the Blood Cancer Discoveries Grant program (8023-20) through the Leukemia & Lymphoma Society, Mark Foundation for Cancer Research, and Paul G. Allen Frontiers Group; and the NIH/National Institute of Diabetes and Digestive and Kidney Diseases (R01 DK103854), NIH/NHLBI (R01 HL128239 and R01 HL151651), and NIH/NCI (R01 CA251138).
Molecular data for the Trans-Omics in Precision Medicine (TOPMed) program were supported by the NHLBI. WGS for “NHLBI TOPMed: Whole Genome Sequencing and Related Phenotypes in the Framingham Heart Study” (phs000974.v1.p1) was performed at the Broad Institute of MIT and Harvard (HHSN268201500014C). WGS for “NHLBI TOPMed: Women's Health Initiative (phs001237.v1.p1) was performed at the Broad Institute of MIT and Harvard (HHSN268201500014C). WGS for NHLBI TOPMed: BioME (phs001644.v1.p1) was performed at McDonnell Genome Institute at Washington University (HHSN268201600037I) and Baylor (HHSN268201600033I). WGS for NHLBI TOPMed: Cardiovascular Health Study (phs001368.v1.p1) was performed at Baylor (HHSN268201600033I). WGS for NHLBI TOPMed: Women's Health Initiative (phs001237.v1.p1) was performed at the Broad Institute of MIT and Harvard (HHSN268201500014C) and Northwest Genomics Center (HHSN268201600032I). Core support including centralized read mapping and genotype calling, along with variant quality metrics and filtering, were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1; contract HHSN268201800002I). Phenotype harmonization, data management, sample-identity quality control, and general study coordination were provided by the TOPMed Data Coordinating Center (3R01HL-120393-02S1; contract HHSN268201800001I). The authors gratefully acknowledge the studies and participants who provided biological samples and data for TOPMed.
The Mount Sinai BioMe Biobank has been supported by The Andrea and Charles Bronfman Philanthropies and in part by federal funds from the NHLBI and National Human Genome Research Institute (U01HG00638001, U01HG007417, and X01HL134588). The authors thank all participants in the Mount Sinai Biobank. They also thank all our recruiters who have assisted and continue to assist in data collection and management, and are grateful for the computational resources and staff expertise provided by Scientific Computing at the Icahn School of Medicine at Mount Sinai.
The Cardiovascular Health Study was supported by contracts HHSN268201200036C, HHSN268200800007C, HHSN268201800001C, N01HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, and N01HC85086, and grants U01HL080295 and U01HL130114 from the NHLBI, with additional contribution from the National Institute of Neurological Disorders and Stroke. Additional support was provided by R01AG023629 from the National Institute on Aging. A full list of principal Cardiovascular Health Study investigators and institutions can be found at https://chs-nhlbi.org/. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NHLBI or the NIH.
The COPDGene project described was supported by award numbers U01 HL089897 and U01 HL089856 from the NHLBI. The COPDGene project is also supported by the COPD Foundation through contributions made to an Industry Advisory Board composed of AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, Novartis, Pfizer, Siemens, and Sunovion. A full listing of COPDGene investigators can be found at http://www.copdgene.org/directory.
The Framingham Heart Study acknowledges the support of contracts NO1-HC-25195 and HHSN268201500001I from the NHLBI and grant supplement R01 HL092577-06S1 for this research. The authors also acknowledge the dedication of the Framingham Heart Study participants without whom this research would not have been possible.
The Women's Health Initiative program is funded by the NHLBI, NIH, U.S. Department of Health and Human Services through contracts HHSN268201600018C, HHSN268201600001C, HHSN268201600002C, HHSN268201600003C, and HHSN268201600004C.
Footnotes
Note: Supplementary data for this article are available at Blood Cancer Discovery Online (https://bloodcancerdiscov.aacrjournals.org/).
#S. Jaiswal and B.L. Ebert contributed equally to this article.
Blood Cancer Discov 2021;2:500–17
Authors' Disclosures
E.M. Beauchamp reports grants from NIH during the conduct of the study. E. Bernard reports grants from Edward P. Evans Foundation during the conduct of the study. A. Niroula reports grants from Knut and Alice Wallenberg Foundation during the conduct of the study. B.M. Psaty reports grants from NIH grants during the conduct of the study and serves on the Steering Committee of the Yale Open Data Access Project funded by Johnson & Johnson. P. Desai reports other support from Agios Pharmaceuticals, Kura Oncology, Bristol Meyers Squib/Celgene, Sanofi, and Cellerant Pharmaceuticals and grants from Astex Pharmaceuticals and Janssen Research outside the submitted work, as well as a patent for methods for detecting AML pending to Cornell University. M.H. Cho reports grants from National Heart, Lung, and Blood Institute (NHLBI) during the conduct of the study, as well as personal fees from AstraZeneca, Illumina, and Genentech, and grants from Bayer and GlaxoSmithKline outside the submitted work. D.G. MacArthur reports personal fees from Goldfinch Bio, GlaxoSmithKline, Variant Bio, and Insitro outside the submitted work. D.S. Neuberg reports grants from NCI during the conduct of the study. L. Malcovati reports grants from Associazione Italiana per la Ricerca sul Cancro (AIRC) during the conduct of the study. E. Papaemmanuil reports grants from MDS Foundation/Celgene during the conduct of the study, as well as other support from Isabl Inc. outside the submitted work. G. Getz reports research funds from IBM and Pharmacyclics; is an inventor on patent applications related to ABSOLUTE, MutSig, MSMuTect, MSMutSig, MSIDetect, POLYSOLVER, and TensorQTL; and is a founder of, is a consultant for, and holds privately held equity in Scorpion Therapeutics. S. Jaiswal reports personal fees from Genentech, Novartis, AVRO Bio, and Foresite Labs outside the submitted work. B.L. Ebert reports grants from Celgene, Deerfield, Novartis, and Calico and personal fees from Neomorph Therapeutics, Skyhawk Therapeutics, and Exo Therapeutics outside the submitted work. No disclosures were reported by the other authors.
Authors' Contributions
E.M. Beauchamp: Conceptualization, formal analysis, funding acquisition, validation, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing. M. Leventhal: Software, formal analysis, visualization, methodology, writing–review and editing. E. Bernard: Resources, data curation, formal analysis. E.R. Hoppe: Formal analysis, visualization. G. Todisco: Resources, formal analysis, investigation, visualization, methodology. M. Creignou: Resources, formal analysis, investigation, methodology. A. Gallì: Formal analysis, investigation. C.A. Castellano: Investigation. M. McConkey: Investigation. A. Tarun: Software, formal analysis. W. Wong: Formal analysis. M. Schenone: Formal analysis, investigation, visualization. C. Stanclift: Formal analysis, investigation, visualization. B. Tanenbaum: Formal analysis, investigation, visualization. E. Malolepsza: Formal analysis. B. Nilsson: Formal analysis, methodology. A.G. Bick: Resources, software, formal analysis, methodology. J.S. Weinstock: Resources, software, formal analysis, methodology. M. Miller: Writing–review and editing. A. Niroula: Formal analysis, methodology. A. Dunford: Formal analysis, methodology. A. Taylor-Weiner: Formal analysis, methodology. T. Wood: Formal analysis, methodology. A. Barbera: Formal analysis, methodology. S. Anand: Formal analysis, methodology. B.M. Psaty: Resources, data curation. P. Desai: Resources, data curation. M.H. Cho: Resources, data curation. A.D. Johnson: Resources, data curation. R. Loos: Resources, data curation. D.G. MacArthur: Resources, data curation. M. Lek: Resources, data curation. D.S. Neuberg: Formal analysis, supervision, methodology, writing–review and editing. K. Lage: Supervision. S.A. Carr: Supervision. E. Hellstrom-Lindberg: Resources, data curation, formal analysis, supervision. L. Malcovati: Resources, data curation, formal analysis, supervision. E. Papaemmanuil: Resources, data curation, formal analysis, supervision. C. Stewart: Formal analysis, supervision, methodology. G. Getz: Supervision, methodology. R.K. Bradley: Software, formal analysis, supervision, visualization. S. Jaiswal: Conceptualization, resources, software, formal analysis, supervision, funding acquisition, validation, investigation, visualization, methodology, project administration, writing–review and editing. B.L. Ebert: Conceptualization, resources, supervision, funding acquisition, methodology, project administration, writing–review and editing.
References
- 1.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371:2488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014;371:2477–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 2014;20:1472–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015;126:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zink F, Stacey SN, Norddahl GL, Frigge ML, Magnusson OT, Jonsdottir I, et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 2017;130:742–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holstege H, Pfeiffer W, Sie D, Hulsman M, Nicholas TJ, Lee CC, et al. Somatic mutations found in the healthy blood compartment of a 115-yr-old woman demonstrate oligoclonal hematopoiesis. Genome Res 2014;24:733–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leshchiner I, Livitz D, Gainor JF, Rosebrock D, Spiro O, Martinez A, et al. Comprehensive analysis of tumour initiation, spatial and temporal progression under multiple lines of treatment. bioRxiv 508127 [Preprint]. 2019. Available from: https://doi.org/10.1101/508127. [Google Scholar]
- 9.The Cancer Genome Atlas Research Network.Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013;368:2059–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell 2014;25:91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med 2013;369:2391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uy GL, Duncavage EJ, Chang GS, Jacoby MA, Miller CA, Shao J, et al. Dynamic changes in the clonal structure of MDS and AML in response to epigenetic therapy. Leukemia 2017;31:872–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018;562:526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corces-Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R.Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci U S A 2014;111:2548–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wong TN, Miller CA, Klco JM, Petti A, Demeter R, Helton NM, et al. Rapid expansion of preexisting nonleukemic hematopoietic clones frequently follows induction therapy for de novo AML. Blood 2016;127:893–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong TN, Miller CA, Jotte MRM, Bagegni N, Baty JD, Schmidt AP, et al. Cellular stressors contribute to the expansion of hematopoietic clones of varying leukemic potential. Nat Commun 2018;9:455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bick AG, Weinstock JS, Nandakumar SK, Fulco CP, Bao EL, Zekavat SM, et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 2020;586:763–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Roy FM, McCrea PD.A role for Kaiso-p120ctn complexes in cancer? Nat Rev Cancer 2005;5:956–64. [DOI] [PubMed] [Google Scholar]
- 19.Daniel JM, Spring CM, Crawford HC, Reynolds AB, Baig A.The p120(ctn)-binding partner Kaiso is a bi-modal DNA-binding protein that recognizes both a sequence-specific consensus and methylated CpG dinucleotides. Nucleic Acids Res 2002;30:2911–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoon HG, Chan DW, Reynolds AB, Qin J, Wong J.N-CoR mediates DNA methylation-dependent repression through a methyl CpG binding protein Kaiso. Mol Cell 2003;12:723–34. [DOI] [PubMed] [Google Scholar]
- 21.Prokhortchouk A, Hendrich B, Jørgensen H, Ruzov A, Wilm M, Georgiev G, et al. The p120 catenin partner Kaiso is a DNA methylation-dependent transcriptional repressor. Genes Dev 2001;15:1613–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soubry A, Staes K, Parthoens E, Noppen S, Stove C, Bogaert P, et al. The transcriptional repressor kaiso localizes at the mitotic spindle and is a constituent of the pericentriolar material. PLoS One 2010;5: e9203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kantidze OL, Kamalyukova IM, Razin SV.Association of the mammalian transcriptional regulator kaiso with centrosomes and the midbody. Cell Cycle 2009;8:2303–4. [DOI] [PubMed] [Google Scholar]
- 24.Aguilar-Martinez E, Chen X, Webber A, Mould AP, Seifert A, Hay RT, et al. Screen for multi-SUMO-binding proteins reveals a multi-SIM-binding mechanism for recruitment of the transcriptional regulator ZMYM2 to chromatin. Proc Natl Acad Sci U S A 2015;112:E4854–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet 2012;44:23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 2011;20:11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guryanova OA, Lieu YK, Garrett-Bakelman FE, Spitzer B, Glass JL, Shank K, et al. Dnmt3a regulates myeloproliferation and liver-specific expansion of hematopoietic stem and progenitor cells. Leukemia 2016;30:1133–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 2014;159:440–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoon JC, Ng A, Kim BH, Bianco A, Xavier RJ, Elledge SJ.Wnt signaling regulates mitochondrial physiology and insulin sensitivity. Genes Dev 2010;24:1507–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sperling AS, Gibson CJ, Ebert BL.The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat Rev Cancer 2017;17:5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ogawa S.Genetics of MDS. Blood 2019;133:1049–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lev Maor G, Yearim A, Ast G.The alternative role of DNA methylation in splicing regulation. Trends Genet 2015;31:274–80. [DOI] [PubMed] [Google Scholar]
- 33.Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M, et al. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 2011;479:74–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maunakea AK, Chepelev I, Cui K, Zhao K.Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res 2013;23:1256–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yearim A, Gelfman S, Shayevitch R, Melcer S, Glaich O, Mallm JP, et al. HP1 is involved in regulating the global impact of DNA methylation on alternative splicing. Cell Rep 2015;10:1122–34. [DOI] [PubMed] [Google Scholar]
- 36.Wong JJL, Gao D, Nguyen TV, Kwok CT, van Geldermalsen M, Middleton R, et al. Intron retention is regulated by altered MeCP2-mediated splicing factor recruitment. Nat Commun 2017;8:15134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015;348:880–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blokzijl F, de Ligt J, Jager M, Sasselli V, Roerink S, Sasaki N, et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 2016;538:260–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F, Hall MWJ, et al. Somatic mutant clones colonize the human esophagus with age. Science 2018;362:911–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yokoyama A, Kakiuchi N, Yoshizato T, Nannya Y, Suzuki H, Takeuchi Y, et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature 2019;565:312–7. [DOI] [PubMed] [Google Scholar]
- 41.Stitziel NO, Khera AV, Wang X, Bierhals AJ, Vourakis AC, Sperry AE, et al. ANGPTL3 deficiency and protection against coronary artery disease. J Am Coll Cardiol 2017;69:2054–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med 2017;377:111–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Belbin GM, Odgis J, Sorokin EP, Yee M-C, Kohli S, Glicksberg BS, et al. Genetic identification of a common collagen disease in puerto ricans via identity-by-descent mapping in a health system. Elife 2017;6:e25060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fried LP, Borhani NO, Enright P, Furberg CD, Gardin JM, Kronmal RA, et al. The Cardiovascular Health Study: design and rationale. Ann Epidemiol 1991;1:263–76. [DOI] [PubMed] [Google Scholar]
- 45.Feinleib M, Kannel WB, Garrison RJ, McNamara PM, Castelli WP.The Framingham Offspring Study. Design and preliminary data. Prev Med 1975;4:518–25. [DOI] [PubMed] [Google Scholar]
- 46.Regan EA, Hokanson JE, Murphy JR, Make B, Lynch DA, Beaty TH, et al. Genetic epidemiology of COPD (COPDGene) study design. COPD. 2010;7:32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Design of the Women's Health Initiative clinical trial and observational study. The Women's Health Initiative Study Group. Control Clin Trials 1998;19:61–109. [DOI] [PubMed] [Google Scholar]
- 48.Costello M, Pugh TJ, Fennell TJ, Stewart C, Lichtenstein L, Meldrim JC, et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res 2013;41:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31:213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, Cheetham RK.Strelka: accurate somatic small-variant calling from sequenced tumor–normal sample pairs. Bioinformatics 2012;28:1811–7. [DOI] [PubMed] [Google Scholar]
- 51.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med 2018;24:679–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, et al. Scikit-learn: machine learning in python. J Mach Learn Res 2011;12:2825–30. [Google Scholar]
- 54.Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, Koenig JL, et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet 2016;48:1193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, Brent S, et al. Ensembl 2013. Nucleic Acids Res 2013;41:D48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meyer LR, Zweig AS, Hinrichs AS, Karolchik D, Kuhn RM, Wong M, et al. The UCSC Genome Browser database: extensions and updates 2013. Nucleic Acids Res 2013;41:D64–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Katz Y, Wang ET, Airoldi EM, Burge CB.Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods 2010;7:1009–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li B, Dewey CN.RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Langmead B, Trapnell C, Pop M, Salzberg SL.Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009;10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Trapnell C, Pachter L, Salzberg SL.TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009;25:1105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ilagan JO, Ramakrishnan A, Hayes B, Murphy ME, Zebari AS, Bradley P, et al. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res 2015;25:14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Robinson MD, Oshlack A.A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 2010;11:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wickham H. ggplot2: elegant graphics for data analysis.New York:Springer;2016. [Google Scholar]
- 64.Shiozawa Y, Malcovati L, Gallì A, Pellagatti A, Karimi M, Sato-Otsubo A, et al. Gene expression and risk of leukemic transformation in myelodysplasia. Blood 2017;130:2642–53. [DOI] [PubMed] [Google Scholar]
- 65.Ewels P, Magnusson M, Lundin S, Käller M.MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016;32:3047–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ewels PA, Peltzer A, Fillinger S, Patel H, Alneberg J, Wilm A, et al. The nf-core framework for community-curated bioinformatics pipelines. Nat Biotechnol 2020;38:276–8. [DOI] [PubMed] [Google Scholar]
- 67.Shen S, Park JW, Lu Z, Lin L, Henry MD, Wu YN, et al. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-seq data. Proc Natl Acad Sci U S A 2014;111:E5593–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mouse RNA-seq.
RNA-seq data have been deposited in the NCBI Sequence Read Archive with accession code “PRJNA681911” (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA681911/).
Proteomics.
The original mass spectra and the protein sequence database used for searches have been deposited in the public proteomics repository MassIVE (http://massive.ucsd.edu/ProteoSAFe/static/massive.jsp) and are accessible at ftp://massive.ucsd.edu/MSV000087304/.