

Abstract
Lack of adherence is a key barrier to a successful human immunodeficiency virus (HIV) treatment and prevention. We report on an ultra-long-acting (ULA) biodegradable polymeric solid implant (PSI) that can accommodate one or more antiretrovirals (e.g., dolutegravir (DTG) and rilpivirine (RPV)) at translatable human doses (65% wt.) in a single implant. PSIs are fabricated using a three-step process: (a) phase inversion of a drug/polymer solution to form an initial in-situ forming solid implant, (b) micronization of dried drug-loaded solid implants, and (c) compression of the micronized drug-loaded solid powder to generate the PSI. DTG and RPV can be pre-combined in a single PLGA-based solution to make dual-drug PSI; or formulated individually in PLGA-based solutions to generate separate micronized powders and form a bilayer dual-drug PSI. Results showed that in a single or bilayer dual-drug PSI, DTG and RPV exhibited physicochemical properties similar to their pure drug analogues. PSIs were well tolerated in vivo and effectively delivered drug(s) over 180 days with concentrations above 4 × PA-IC90 after a single subcutaneous administration. While biodegradable and do not require removal, these PSIs can safely be removed to terminate the treatment if required. The versatility of this technology makes it attractive as an ULA drug delivery platform for HIV and various therapeutic applications.
Keywords: Polymeric solid implants, long-acting drug delivery, poly(lactic-co-glycolic acid) (PLGA), dolutegravir, rilpivirine, HIV prevention
Graphical Abstract
1. Introduction
HIV is the sixth leading cause of death in the world and the third leading cause of death as a communicable disease [1, 2]. Although advances in HIV treatment (antiretroviral therapy; ART) and prevention (pre-exposure prophylaxis; PrEP) have reduced the morbidity and mortality associated with HIV, this epidemic disease continues to spread worldwide [3]. Poor patient adherence to ART/PrEP regimens has been implicated as a primary factor in determining efficacy of daily oral formulations [4, 5]. Hence, there is a need to develop PrEP/ART formulations that can provide prolonged drug delivery over several weeks or months and that can effectively prevent HIV acquisition in high-risk individuals or effectively treat HIV infected individuals.
Recent innovations introduced in the field of HIV PrEP/ART are long-acting (LA) formulations of antiretrovirals (ARVs) that offer sustained release of drugs over weeks or months either as a systemic delivery such as nano-based formulations [6, 7] and solid implants [8–10], or as a topical delivery such as gels [11, 12], vaginal films [13, 14] and intravaginal rings [15–17]. These approaches offer many benefits over the standard HIV PrEP/ART, including the ability to mitigate poor patient adherence with daily oral dosing, increase an efficacious outcome and decrease a risk of drug resistance, as well as the ability to be utilized discreetly without a partner’s knowledge. In particular, two LA injectable formulations of cabotegravir (GSK744-LA, Phase III) and rilpivirine (TMC278-LA, Phase II) have shown promising results in clinical trials [18–20]. These formulations are produced as a dense drug nano-suspension (Elan Nanocrystal® technology) and are administered intramuscularly to deliver adequate drug doses and achieve sustained plasma concentrations for 4–8 weeks [7, 10, 21]. More recently, Merck has developed a LA solid implant with islatravir (4′-ethynyl-2-fluoro-2′-deoxyadenosine, EFdA, or MK-8591) and is currently in Phase I clinical trial for HIV PrEP/ART. This solid implant is made with a non-biodegradable ethylene-vinyl acetate (EVA) polymer (Naxplanon® technology) and is projected to achieve sustained delivery of islatravir for up to one year [22–24]. Development of LA solid implants that are biodegradable is highly attractive in the field of HIV PrEP/ART since their removal after use is not required, but can be implemented if required in case of severe allergic reaction, adverse side effect, or pregnancy to terminate the treatment. To date, only three LA biodegradable solid implants are in development at the preclinical stage for HIV-PrEP/ART. These include a tenofovir alafenamide (TAF)-polycaprolactone (PCL) thin-film device [9] and MK-8591 eluting PLA (polylactide) and PCL implants [25]. These biodegradable solid implants are produced by hot-melt extrusion, solvent-casting, or compression molding, which require high temperatures and shear forces or use of large volumes of organic solvents in the fabrication process [9, 25–27].
We developed a method to fabricate biodegradable polymeric solid implants (PSIs) using phase inversion of drug-loaded polymer-based solution in combination with tablet compression technique that allows fabrication of PSIs with high drug loading (up to 85 wt%) and compact sizes. The fabrication of these PSIs is accomplished using a simple and scalable stepwise process of (a) phase inversion of a drug-loaded polymer-based solution to form an initial in-situ forming solid implant in an aqueous medium, (b) micronization of dried drug-loaded solid implants, and (c) compression of micronized drug-loaded solid powder (Fig. 1). The resulting PSIs are solvent-free and consist of only the biodegradable polymer and drug. The manufacturing process does not require high heat or high pressure and can be easily scalable to produce solid implants with high drug loading and various shapes or sizes[28]. Herein, we demonstrate that these PSIs can 1) integrate multiple ARVs, 2) achieve sustained delivery of ARVs in vitro and in vivo for ≥180 days, and 3) can be removed if required to terminate the treatment. Dolutegravir (DTG) and rilpivirine (RPV) were investigated in this study owing to their proven efficacy in HIV-PrEP/ART [20, 29–32]. Two different formulation techniques were applied to generate co-drug PSIs and investigate their physical and chemical stability and in vitro drug release kinetics. Drug PSIs were well tolerated in vivo and demonstrated the ability to maintain plasma concentrations of DTG and RPV above their protein-adjusted (PA) IC90 for 180 days. PSIs were also successfully removed from the implantation site by making a small incision in the skin of BALB/c mice. The versatility of this PSIs technology makes it highly attractive as a long-acting drug delivery platform for HIV PrEP/ART.
Figure 1.

Schematic illustration of PSI preparation process by a combination of phase inversion and tablet compression techniques. This figure was partially adapted from Figure 1 of IJP https://doi.org/10.1016/j.ijpx.2020.100068.
2. Materials and methods
2.1. Materials
50:50 Poly(DL-lactide-co-glycolide, PLGA) was purchased from LACTEL (Birmingham, AL; Cat. No. B6010–1P, Lot# 1614–09-01, Mw 27.2 kDa, i.v. 0.39, polydispersity index (PDI) 1.81, amorphous). N-methyl-2-pyrrolidone (NMP, <USP>) was received from ASHLAND (Wilmington, DE, Product Code 851263, 100% NMP). Dimethyl Sulfoxide (DMSO, ≥ 99.7%) was received from Fisher Scientific (Waltham, MA). Dolutegravir (DTG), Rilpivirine (RPV) were purchased from Selleckchem (Houston, TX; S2667-DTG, S7303-RPV). Solutol-HS 15, phosphate buffered saline (0.01M PBS, pH 7.4), HPLC grade Acetonitrile were purchased from Sigma Aldrich (St. Louis, MO).
2.2. Preparation of drug-loaded polymeric solid implants (PSIs) by phase inversion and compression
2.2.1. Preparation of single-drug PSIs
Method for preparation of polymeric solid implant (PSI) was previously optimized by varying formulation composition, phase inversion time, drying time, drug loading methods and tablet compression force in order to optimize drug content in PSIs and drug release kinetics. To fabricate a DTG PSI or RPV PSI, first in-situ forming implant (ISFI) formulations were prepared using a previously described procedure [33]. Briefly, a placebo formulation was prepared by adding 1:2 w/w PLGA:(NMP/DMSO 9:1 w/w) using an analytical balance and allowing PLGA to dissolve in NMP by continuous mixing at room temperature. Next, dolutegravir (DTG) or rilpivirine (RPV) was added to the PLGA/NMP placebo solution and allowed to stir at 37°C overnight to completely dissolve the drug and form a homogenous formulation. Second, DTG- or RPV-loaded solid depots were formed in-situ by phase inversion upon injection of the formulation solution in PBS at an optimized ratio of ISFI/PBS (25 μL:2 mL, Fig. S1). After incubation in PBS at 37°C for 24 h, the solid implants were collected and dried using a rotary evaporator (Rotavapor R-215, Buchi, Switzerland) for 1 h at 25°C to remove all residual water (Table S1). Dried depots were subsequently micronized using a mortar and pestle to obtain a fine powder of drug and PLGA as a homogenous mix (Fig. S2). To ensure homogeneity of drug distribution within the micronized DTG/PLGA or RPV/PLGA powder, samples (1–2 mg, n=4) were collected from four different areas of the solid powders and dissolved in acetonitrile. Drug concentration was subsequently quantified by high-performance liquid chromatography (HPLC). The micronized DTG/PLGA or RPV/PLGA powder was considered homogenous when the standard deviation of the average concentration in all 4 samples analyzed was ≤5% based on the USP acceptance criteria in <905> Uniformity of Dosage Units [34]. The micronized DTG/PLGA or RPV/PLGA powder was subsequently compressed using a single punch press tablet machine (Carver Hand Press model 3851, Wabash, IN). Briefly, 20 mg of micronized DTG/PLGA or RPV/PLGA powder was loaded into a 5 mm diameter cylindrical tool and pressed at 1.274 US.ton/cm2 for 10 s at room temperature. These final PSIs (DTG, RPV, DTG-RPV and S-DTG/RPV) comprised of only drugs and PLGA with no added excipients or stabilizers.
2.2.2. Preparation of dual-drug PSIs
Dual-drug PSIs were fabricated in two ways; either by adding DTG and RPV in a single ISFI formulation and forming a tablet termed DTG-RPV PSI, or by making DTG and RPV solid powders separately and generating a ‘sandwich’ (S) or bilayer tablet termed S-DTG/RPV PSI (Figure 2A–C). To prepare DTG-RPV PSIs, DTG (250 mg/mL) and RPV (250 mg/mL) were added to a 1:6 w/w PLGA/(NMP/DMSO, 9:1 w/w) placebo solution (placebo density of 1.045 g/mL) using an analytical balance and allowed to stir at 37°C overnight to completely dissolve both drugs and form a homogenous formulation (Table S2). Drug concentration in the solution formulation was determined by taking sample aliquots (~1 mg, n=4) of formulation and dissolving each sample into 1 mL of acetonitrile (ACN). Samples were analyzed by HPLC to determine drug concentration and formulation homogeneity. Subsequently, DTG-RPV loaded solid depots were formed in-situ by phase inversion upon injection of the formulation solution in PBS (0.01 M, pH 7.4, 2 mL per 25 μL ISFI). Solid depots were subsequently dried using rotary evaporation, micronized and compressed using the method described in section 2.2.1 to form the final DTG-RPV PSI. This preparation method was referred to as Method I.
Figure 2.

Fabrication of combined-drug (co-drug) PSIs as a single tablet or ‘sandwich’ bilayer tablet. A) Formulation details for DTG PSI, RPV PSI, DTG-RPV PSI, and S-DTG/RPV PSI. B) Schematic illustration of different drug-loaded PSIs. C) Images of PSIs with different drugs (DTG, RPV) and drug loading method (bilayer-tablet: S-DTG/RPV PSI or single tablet: DTG-RPV PSI).
To generate S-DTG/RPV PSIs, DTG and RPV were added separately to a 1:6 w/w PLGA/(NMP/DMSO, 9:1 w/w) placebo solution (Table S2). DTG/PLGA and RPV/PLGA solid powders were generated using the procedure described in section 2.2.1. Subsequently, 10 mg of DTG/PLGA powder was loaded into a 5 mm diameter cylindrical tool and pressed at 1.274 US.ton/cm2 for 10 s to form a DTG PSI tablet. Next, 10 mg of RPV/PLGA powder was added on top of the DTG PSI tablet and compressed at 1.274 US.ton/cm2 for 10 s at room temperature. This preparation method was referred to as Method II. Details of the final DTG-RPV and S-DTG/RPV PSIs are illustrated in Fig. 2A. All PSIs had similar dimensions and appearance (Fig. 2C).
2.3. Scanning electron microscopy (SEM) imaging of micronized powders and PSIs
SEM imaging was conducted to analyze the physical state (drug morphology) and drug distribution in the micronized drug/PLGA powders and microstructure of the final PSIs. The micronized DTG/PLGA, RPV/PLGA, DTG-RPV/PLGA powders, DTG PSI, RPV PSI, DTG-RPV PSI and S-DTG/RPV PSI samples were mounted on an aluminum stub using carbon tape, and sputter coated with 9 nm of gold-palladium alloy (60:40) (Hummer X Sputter Coater, Anatech USA, Union City, CA). The coated samples were imaged using a Zeiss Supra 25 field emission scanning electron microscope with an acceleration voltage of 5 kV, 30 μm aperture, and average working distance of 12 mm (Carl Zeiss Microscopy, LLC, Thornwood, NY).
2.4. Differential Scanning Calorimetry (DSC) analysis of drug loaded PSIs
DSC analyses of pure drug (DTG and RPV), micronized placebo PLGA powder, and micronized drug-loaded PLGA powders (DTG/PLGA, RPV/PLGA, DTG-RPV/PLGA powders) were carried out using a differential scanning calorimeter (TA Q200, USA). Samples (3–10 mg) were weighed, hermetically sealed in an aluminum pan, and placed in the differential scanning calorimeter. For DTG-loaded samples, the samples were heated from 0–250°C at a heating rate of 10°C/min under nitrogen flow rate of 20 mL/min. For RPV-loaded samples, the samples were heated from 0–300 °C at a heating rate of 10°C/min, under nitrogen flow rate of 20 mL/min. The thermograms were used to determine the melting temperature (Tm) of DTG and RPV and the physical state of DTG and RPV in PSIs (i.e., crystalline, or amorphous).
2.5. X-ray powder diffraction (XRD) analysis
XRD analyses of micronized DTG/PLGA, RPV/PLGA, DTG-RPV/PLGA powders were carried out to investigate the physical state of drugs when formulated in the micronized powder precursors to form a PSIs and compared to pure DTG and RPV. The XRD was conducted at the Chapel Hill Analytical and Nanofabrication Laboratory (CHANL) using Rigaku SmartLab system with a Cu source operated at 40 kV and 44 mA. A Kβ filter was used to remove the Kβ line from the Cu source. Data was collected in a Bragg Brentano geometry with the HyPix detector operated in 1D mode. Scans were acquired at 5 degrees/min with a step size of 0.01 degrees.
2.6. In vitro drug release studies
Drug release kinetics from DTG PSIs, RPV PSIs, DTG-RPV PSIs, and DTG/RPV PSIs were investigated by incubating of PSIs (20 mg ± 5%, Table S3) into 200 mL of release medium (0.01 M PBS pH 7.4 with 2% Solutol HS) at 37°C under static condition for up to 6 months. Sink conditions were defined as the drug concentration at or below 1/5 of maximum solubility (i.e., ≤0.12 mg/mL DTG; ≤0.03 mg/mL) in PBS/solutol. Sample aliquots (1 mL) were collected at various time points and replaced with fresh release medium. The release medium was completely removed and replaced with fresh medium every week to maintain sink conditions [28]. Drug concentration in the release samples was quantified by HPLC. A reverse-phase HPLC analysis was carried out with a Finnigan Surveyor HPLC system (Thermo Finnigan, San Jose, California, USA) with a Photodiode Array (PDA) Plus Detector, auto-sampler, and LC Pump Plus. The stationary phase utilized for the analysis was an Inertsil ODS-3 column (4 μm, 4.6 Å~ 150 mm, [GL Sciences, Torrance, CA]) maintained at 40°C. Chromatographic separation was achieved by gradient elution using a mobile phase consisting of 0.1% trifluoroacetic acid in water and ACN (H2O/ACN 95:5 v/v). The flow rate was 1.0 mL/min and the total run time was 25 min for each 25 μL injection. Cumulative drug release was calculated from the HPLC analysis and normalized to the total mass of drug in the implant. All experiments were performed in triplicate.
2.7. In vivo animal model and PSI implantation
All in vivo studies were performed in accordance with the guidelines for animal experimentation by the Institutional Animal Care and Use Committee, School of Medicine, University of North Carolina at Chapel Hill and comply with National Institutes of Health guide for the care and use of Laboratory animals (NIH Publications No. 8023, revised 1978). Eight-week (20–25 g) BALB/c mice were purchased from The Jackson Laboratory (Bar Harbor, ME). DTG-loaded PSIs (278 mg/kg; 10 mg PSI tablet), RPV-loaded PSIs (296 mg/kg; 10 mg PSI), S-DTG/RPV PSI tablets (287 and 296 mg/kg of DTG and RPV; 20 mg PSI), and placebo PSI tablets were administered subcutaneously via a 5-mm skin incision on shaved back of anesthetized BALB/c mice (n=7 per group; 4 groups, Supplementary Fig. 2 and Table 1). The skin at the incision site was then closed with clinical-grade Ethicon suture plus Vetbond between sutures or with 2–3 sterile 9 mm stainless steel wound clips.
Table 1.
Formulation details of PSIs investigated in in vivo studies.
| Implant | Dose (mg/kg) | PSI weight (mg) | Theoretical drug loaded in PSI (wt%) | Analytical drug loaded in PSI (wt%) | PSI Diameter (mm) |
|---|---|---|---|---|---|
| DTG PSI | 278 | 10 | 60 | 62.5 | 4.76 |
| RPV PSI | 296 | 10 | 60 | 66.7 | 4.76 |
| DTG/RPV PSI | 278/296 (DTG/RPV) | 20 (1:1 DTG/RPV) | 30/30 (DTG/RPV) | 31.25/33.35 (DTG/RPV) | 4.76 |
| Placebo PSI | - | 10 | - | - | 4.76 |
2.8. In vivo safety studies
A six-month in vivo study was carried out to assess the safety and in vivo biodegradation of various PSIs in female BALB/c mice (6–8 weeks, Jackson Laboratory). Local inflammation of tissues surrounding the PSI implant was evaluated using H&E histological analysis. Mice were implanted with PSIs via a skin incision and monitored for signs of inflammation (body weight, implantation site, and other signs including physical appearance, physical activity, and food consumption). On day 3, 7, 14, 30, 60, 90, and 180 days post PSI administration mice were sacrificed (n=7 at each time point), blood samples via heart puncture and implant site tissues were collected into capillary tubes and stored at −80°C to quantify pro-inflammatory cytokines including tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6) by enzyme-linked immunosorbent assay (ELISA, MAX™ Deluxe sets, BioLegend®). Mice were subsequently necropsied by intracardiac injection of 4% paraformaldehyde (pH 7.3) and the subcutaneous tissues surrounding the PSIs were harvested for histology by submerging tissue in 10% neutral buffered formalin at a ratio of 1:10 tissue-fixative at room temperature for 72 h, and then transferred to room temperature 70% ethyl alcohol. Tissues were processed, paraffin embedded, sectioned at 5-μm thick, and stained by routine hematoxylin and eosin (H&E) for histopathological examination (LCCC Animal Histopathology Core Facility at the University of North Carolina at Chapel Hill). Histopathologic evaluation was conducted by a board-certified veterinary pathologist (S. Montgomery) in an a priori blind manner in which samples within mouse groups were masked at time of scoring. Images were generated on an Olympus BX43 with a DP27 camera using CellSens software. For all tissue changes, a scoring system was developed as follows: 0, absent; 1, minimal, less than 10% of tissue affected; 2, mild, 10 to 24% of tissue affected; 3, moderate, 25 to 39% of tissue affected; 4, marked, 40 to 59% of tissue affected; 5, severe, greater than 60% of tissue affected.
2.9. In vivo pharmacokinetic studies
To assess drug release kinetics in vivo using the PSI formulations, pharmacokinetic studies were carried out using single-drug PSIs and dual-drug PSIs (Fig. 8). For each formulation, the indicated dose of the individual drug was administered subcutaneously into BALB/c mice (n = 7) based on their body weight, and at 1, 3, 7, 14, 21, 30, 60, 90, 120, 150, and 180 days, peripheral blood was collected from mice (n=7 at each time point) into capillary tubes coated with or without EDTA to isolate plasma or serum, respectively. All samples were stored at −80°C until analysis. Initial estimates for PK parameters were obtained through non-compartmental analysis (NCA) using WinNonlin Phoenix 6.1 (Pharsight, Mountain View, CA) on the composite median PK profile. Briefly, plasma samples were extracted by protein precipitation with methanol containing stable, isotopically labeled internal standards DTG-13C, d5 (ALSACHIM, Illkirch Graffenstaden, France) and RPV-d6 (Toronto Research Chemicals, North York, ON, Canada). Extracts were analyzed by liquid chromatography-tandem mass spectrometry (LC/MS-MS) using a Prominence HPLC system (Shimadzu, Columbia, MD, USA) on a XTerra MS C18 (50 × 2.1mm, 3.5μm particle size) analytical column (Waters, Milford, MA, USA) under reverse phase conditions with water with 0.1% formic acid and acetonitrile with 0.1% formic acid as mobile phases. Analytes were detected on an API-5000 triple quadrupole mass spectrometer (AB Sciex, Foster City, CA, USA) operated in positive ion mode. Linear regression of concentration (x) versus peak area ratio of compound to internal standard (y) using a 1/x2 weighting was used with Sciex Analyst software (version 1.6.2). Plasma concentrations were plotted over time in Fig. 8.
Figure 8.
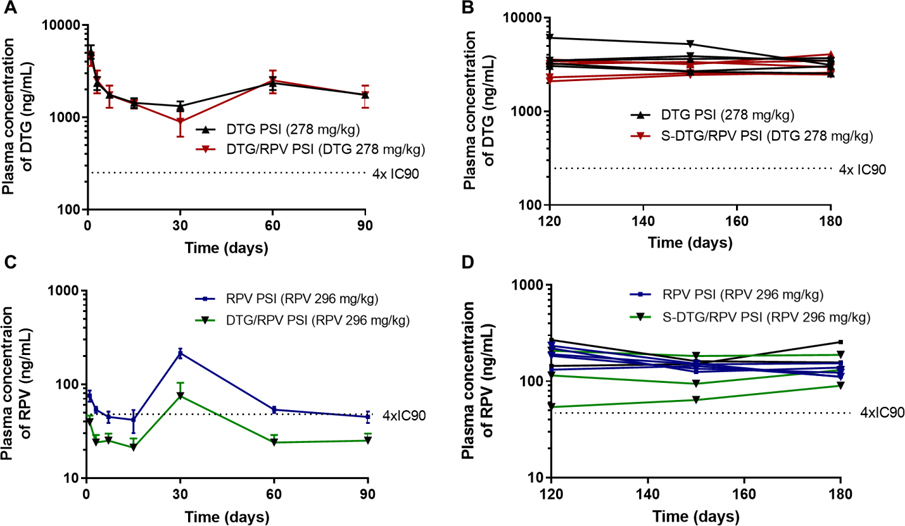
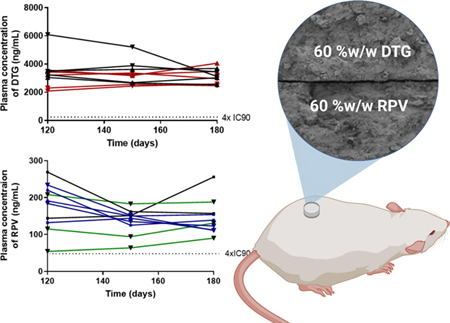
A) Plasma concentration of DTG for mice (n=5 per timepoint) implanted with DTG PSIs (278 mg/kg; 10 mg PSI tablet) or with S-DTG/RPV PSIs (278 mg/kg DTG, 296 mg/kg RPV; 20 mg PSI tablet) collected at 24h, 3d, 7d, 14d, 30d, 60d and 90d post implantation. Error bars represent standard deviation of n=5 samples. B) Plasma concentration of DTG plotted for individual mice (n=5 per group) implanted with DTG PSIs (278 mg/kg; 10 mg PSI tablet) or with S-DTG/RPV PSIs (278 mg/kg DTG, 296 mg/kg RPV; 20 mg PSI tablets) collected at 120d, 150d and180d post implantation. C) Plasma concentration of RPV for mice (n=5 per timepoint) implanted with RPV PSIs (296 mg/kg; 10 mg PSI tablet) or with S-DTG/RPV PSIs (278 mg/kg DTG, 296 mg/kg RPV; 20 mg PSI tablets) collected at 24h, 3d, 7d, 14d, 30d, 60d and 90d post implantation. D) Plasma concentration of RPV plotted for individual mouse (n=5 per group) implanted with RPV PSIs (296 mg/kg; 10 mg PSI tablet) or with S-DTG/RPV PSIs (278 mg/kg DTG, 296 mg/kg RPV; 20 mg PSI tablet) collected at 120d, 150d and180d post implantation.
2.10. PSI removal from BALB/C mice.
To investigate the ability to safely remove a PSI post administration to terminate the treatment if required due to an allergic reaction, breakthrough infection or pregnancy, placebo PSIs and drug-loaded PSIs (DTG, RPV, DTG/RPV) were implanted subcutaneously into BALB/c mice. Mice were euthanized and PSIs were removed using forceps under sterile conditions via a small cutaneous incision adjacent to the implant on day 7, 14, 30, 60, 90 and 180 post implantation (n=7 at each time point). The dimensions of retrieved implants were measured using a micro-caliper, and the estimated volumes of retrieved implants were calculated based on the formula for a cylindrical volume.
2.11. Statistical analysis
Data were summarized as means ± standard deviation (SD) and analyzed with GraphPad Prism (version 6.01) using one-way analysis of variance (ANOVA) followed with Friedman test followed by Dunn’s multiple comparison tests with no adjustment. The confidence level was set at 95%.
3. Results and Discussion
3.1. Physical state of drugs in single and co-formulated micronized PLGA powders
Here in, we demonstrate the development of biodegradable polymeric solid implants (PSIs) that can accommodate more than one drug and provide sustained drug delivery over several months after a single administration for HIV PrEP/ART applications. We developed a new engineering process to fabricate a PSI using a process that combines phase inversion of an in-situ forming implant (ISFI) formulation and compression. This process allows fabrication of biodegradable solid implants without the use of high heat, high pressure or large volumes of organic solvents, which are typically required in hot-melt extrusion or solvent casting processes to produce solid implants [9, 22, 26, 27]. DTG and RPV were co-formulated in PSIs using two different methods referred to as Method I and Method II to generate DTG-RPV PSI and S-DTG/RPV PSI, respectively. To select an appropriate technique for creating a co-formulated PSI, prior to forming a PSI, the effect of the co-formulation process on the physicochemical properties of drugs in the micronized drug/PLGA powders were investigated using DSC, XRD and SEM analysis.
The physical state of drugs (i.e. crystalline or amorphous/molecularly dispersed) is an aspect that is very important in determining the extent of burst release and release rate of the drug from the formed PSI. For drugs with poor aqueous solubility like DTG and RPV, precipitation of the drug(s) in the depot in the crystalline state can significantly slow their rate of release. In contrast, a drug that is molecularly dispersed in the depot can experience faster release [35, 36]. Factors that can influence the physical state of drugs in the micronized drug/PLGA powders prepared from phase inversion of ISFI formulation in PBS include 1) solubility of the drug in the ISFI formulation, and 2) rate of solvent diffusion and PLGA precipitation during phase inversion [37].
DSC was carried out to assess the physical state and melting temperature (Tm) of drugs in the micronized drug/PLGA powders prior to forming a PSI. As shown in Fig. 3A, pure DTG and pure RPV showed sharp endothermic peaks at 188°C and 241°C respectively, corresponding to their Tm. No endothermic peak was detected for the placebo micronized PLGA powders demonstrating the amorphous nature of PLGA [38]. The endothermic peaks of DTG and RPV (168°C and 235°C respectively) in the single-drug micronized DTG/PLGA powder and RPV/PLGA powder were significantly lower in intensity compared to their pure drug counterparts. These results indicate that DTG and RPV were partially converted to an amorphous state or had reduced size crystals and/or presence of crystal defects when formulated into the micronized drug/PLGA powders compared to the fully crystalline states of pure DTG and RPV (Fig. 3A). When DTG and RPV were co-formulated into micronized co-drug/PLGA powders (DTG-RPV), a small intensity endothermic peak was detected at 160°C corresponding to Tm of DTG. No endothermic peak was observed at the temperature corresponding to Tm of RPV. Collectively, DSC results showed no or minimal endothermic peaks corresponding to a crystalline state when DTG and RPV were co-formulated into the dual-drug micronized DTG-RPV/PLGA powder (Fig. 3A). This indicates that DTG and RPV were mostly present in an amorphous state or in a reduced crystalline state due to presence of smaller crystals and/or potential crystal defects.
Figure 3.
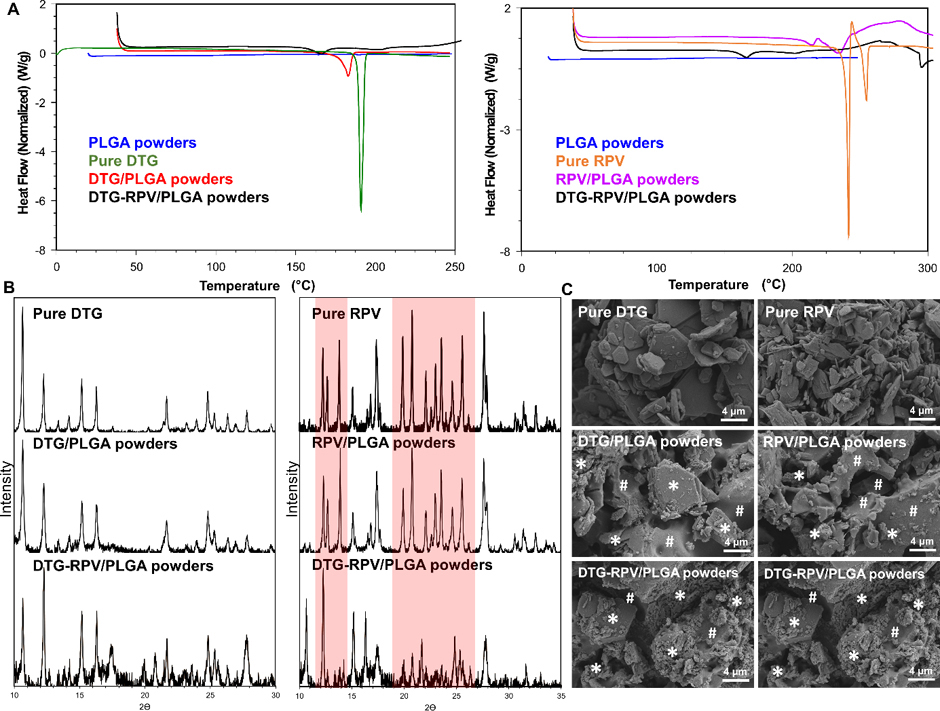
Characterization of physical state of DTG and RPV when formulated individually or in combination in micronized PLGA powders. A) DSC thermograms of DTG/PLGA, RPV/PLGA, and DTG-RPV PLGA micronized powders compared to pure DTG and RPV and placebo PLGA powder. B) XRD patterns of DTG/PLGA, RPV/PLGA, and DTG-RPV PLGA micronized powders compared to pure DTG and RPV. Red highlights represent the different crystallinity patterns and relative ratios of peak intensity of RPV in DTG-RPV PLGA compared to pure RPV. C) SEM images representing microstructure of DTG/PLGA, RPV/PLGA, and DTG-RPV PLGA micronized powders compared to pure DTG and RPV. The white * symbols represent drugs (DTG or RPV) and the white # symbol represents PLGA in the DTG/PLGA, RPV/PLGA or DTG-RPV/PLGA micronized powders.
XRD analysis was further used to investigate the effect of the method of drug co-formulation on the crystalline patterns of drugs that retained predominant or partial crystalline state in the micronized drug/PLGA powders. As shown in Fig. 3B, the crystalline pattern of DTG did not change in the micronized PLGA powders, when formulated alone or in combination with RPV, as demonstrated by no change or shift in peak positions or peak intensities compared to pure DTG (Fig. 3B left). Similarly, RPV’s XRD peaks retained their positions and did not shift when formulated in the micronized RPV/PLGA powders compared to pure RPV. However, some peak intensities were relatively lower for formulated RPV compared to its pure analogue due to possible reduction in crystal size and/or presence of crystal defects when formulated in the micronized RPV/PLGA powders. Moreover, when DTG and RPV were co-formulated in the micronized co-drug/PLGA powders (DTG-RPV), both peak positions and intensities were substantially different and reduced respectively for RPV compared to its pure analogue (Fig. 3B right). This could also be due to significant reduction in crystal size and/or presence of crystal defects when formulated in the micronized co-drug/PLGA powder.
SEM imaging analyses of micronized drug/PLGA powders were also used to investigate the effect of drug co-formulation methods on the physical state of drugs and drug distribution in the micronized drug/PLGA powders. As shown in Fig. 3C, both DTG and RPV when formulated individually in micronized PLGA powders, were present as a mixture of crystalline and molecularly dispersed drug within PLGA. Consistently with the DSC and XRD results, SEM images of micronized DTG-RPV/PLGA powders showed that both DTG and RPV were amorphous and molecularly dispersed in PLGA (Fig. 3C). DTG and RPV were uniformly distributed in all micronized powders (DTG/PLGA, RPV/PLGA, and DTG-RPV/PLGA), which was in agreement with the homogeneity data of drug concentration in PSIs determined by HPLC analysis (Fig. 5). Collectively, results from DSC, SEM, and XRD demonstrated that when co-formulated in a single micronized PLGA powder, DTG and RPV became less crystalline and more molecularly dispersed within the formulation.
Figure 5.
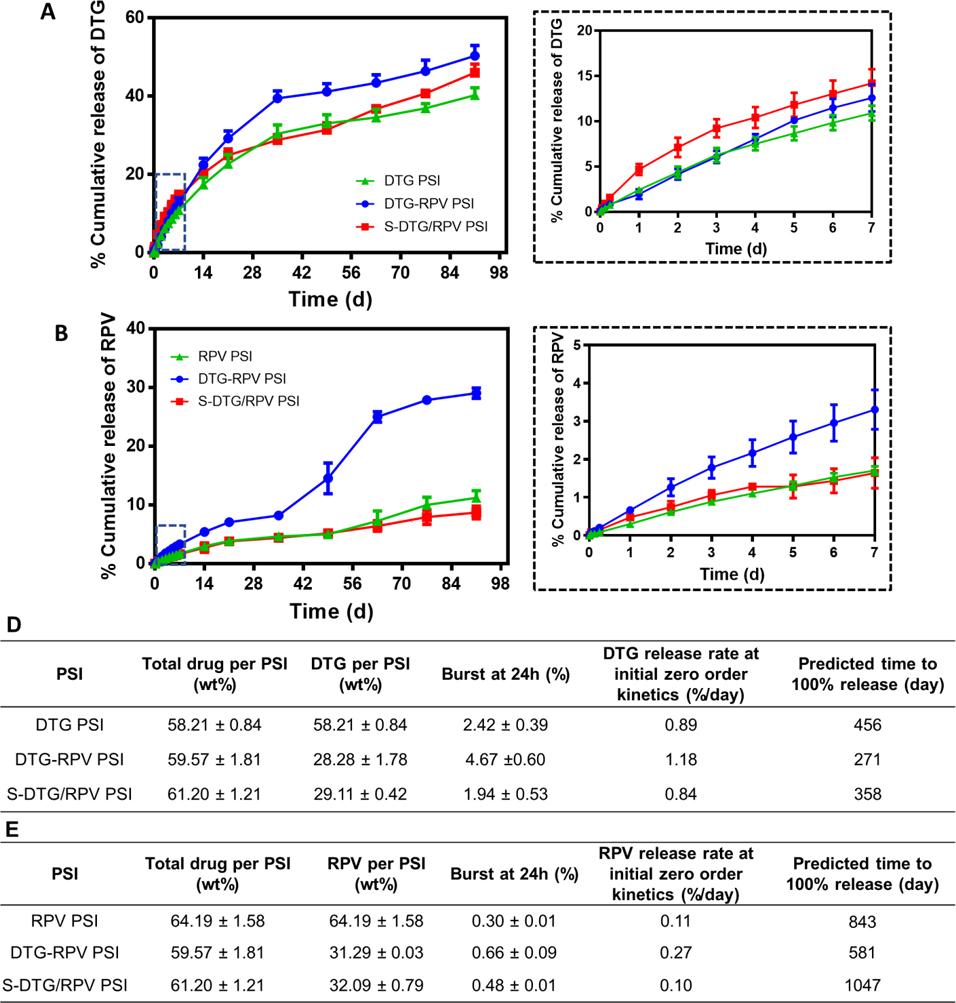
In vitro release kinetics of DTG (A) and RPV (B) from single-drug DTG PSIs, RPV PSIs, and dual-drug DTG-RPV PSIs, S-DTG/RPV PSIs. Error bars represent standard deviation of n=3 samples. Insets represent the release kinetics of DTG (A) and RPV (B) during the first 7 days. C) Summary of in vitro release kinetics parameters of DTG from DTG PSIs, DTG-RPV PSIs and S-DTG/RPV PSIs (n=3). D) Summary of in vitro release kinetics parameters of RPV from RPV PSIs, DTG-RPV PSIs and S-DTG/RPV PSIs (n=3).
3.2. Microstructure of single-drug and dual-drug PSIs
The microstructure of a biodegradable solid implants is one of the important physical properties that influence drug release kinetics. Solid implant microstructure can be modulated by different parameters including polymer properties (e.g., types, MW, functional groups), drug properties (e.g., pKa, Log P, MW), additives (e.g., binders, solvents, porogens), and fabrication techniques (e.g., extrusion, solvent casting, compression) [39–42]. SEM imaging analyses were carried out to investigate the effect of co-formulation techniques by Method I (co-formulating DTG and RPV in a single micronized PLGA powder followed by compression to form a PSI, DTG-RPV PSI) and Method II (iterative compression of single-drug PLGA powders to form a ‘sandwich’ bilayer PSI, S-DTG/RPV PSI) on drug distribution and microstructure of the resulting PSIs. As shown in Fig. 4, the microstructures of both dual-drug PSIs (DTG-RPV PSI, S-DTG/RPV PSI) and single-drug PSIs (DTG PSI, RPV PSI) were similar and highly compact. In S-DRG/RPV PSI, a clear separation between DTG and RPV layers was observed as a result of the stepwise compression procedure to form the ‘sandwich’ bilayer PSI tablet.
Figure 4.

SEM images representing surface and cross-section images (at 200x and 600x magnification) of single-drug PSIs (DTG PSI, RPV PSI) and dual-drug PSIs (DTG-RPV PSI and DTG/RPV PSI).
3.3. In vitro release kinetics of single-drug and dual-drug PSIs
In vitro release studies of DTG and RPV were carried out to investigate the effect co-formulation techniques (Method I, Method II) on drug release kinetics. It has been shown that drug release from PLGA solid implants can take place via diffusion of drugs from the implant and/or by degradation/erosion of PLGA matrix [37, 43, 44]. For DTG, all PSIs (DTG PSI, DTG-RPV PSI, S-DTG/RPV PSI) exhibited minimum initial burst release (< 5%) in the first 24 h (Fig. 5A, 5C). The burst release of DTG at 24h was 2.42%, 1.94%, and 4.67% for DTG, DTG-RPV, and S-DTG/RPV PSIs, respectively (n=3, p > 0.05). DTG exhibited similar release kinetics from all PSIs with a biphasic zero-order release with Phase 1 lasting from 0–35 days and phase 2 lasting from 36–90 days post-incubation in PBS at 37°C. In the initial fast zero-order release between day 0–35, and prior to the initiation of PLGA bulk degradation via ester hydrolysis, DTG was released mainly through diffusion from the outer layer of the PSI. In phase 2, a slow sustained release of DTG was achieved via bulk degradation of the PLGA matrix by hydrolysis of ester linkages in the presence of water [37]. The release rate of DTG from DTG-RPV PSIs was 1.18%/day in phase I (day 0–35), and was significantly faster than its release rate from single-drug PSIs (DTG PSI) and ‘sandwich’ dual-drug drug PSIs (S-DTG/RPV PSI) (0.89%, 0.84% per day, respectively, p < 0.05). The significant increase in release rate of DTG from DTG-RPV PSIs was likely attributed to the amorphous state of DTG in the micronized DTG-RPV powder, as confirmed by DSC, XRD and SEM analyses, compared to a more crystalline state in the single-drug micronized PLGA powder.
Compared to DTG, RPV exhibited slower release kinetics from PSIs due to differences in drug properties (i.e., Log P, pKa as shown in Table S4). In the initial 24h, single-drug PSI (RPV PSIs) and dual-drug PSIs (DTG-RPV PSIs, S-DTG/RPV PSIs) had minimum burst release of 0.3, 0.66 and 0.48% RPV, respectively (p > 0.05, Fig. 5B, 5D). The release kinetics of RPV from single-drug PSIs (RPV PSIs) and ‘sandwich’ dual-drug PSIs (S-DTG/RPV PSIs) had a similar zero-order profile throughout the study duration (0.12% and 0.10% per day, respectively, p > 0.05). In comparison, as shown in Fig. 5B, the release of RPV from dual-drug PSIs (DRG-RPV PSIs, Method I) exhibited triphasic kinetics with an initial slow zero-order release (0.25% per day, day 0–35), followed by faster zero-order kinetics (0.60% per day, day 35–63), and finally slower zero-order kinetics (0.13% per day, day 63–90). The sharp increase in RPV release from DTG-RPV PSIs at day 49 was attributed to swelling of the PSIs ~7 weeks post-incubation in PBS at 37°C. These results demonstrate that when RPV was co-formulated with DTG in PSIs using Method I, its amorphous physical state contributed to the different release kinetics observed compared to when formulated alone (RPV PSIs) or in combination with DTG using Method II (S-DTG/RPV PSIs) (Fig. 5B). On the other hand, when co-formulated with DTG using Method II (S-DTG/RPV bilayer PSIs), RPV exhibited similar release kinetics compared to single-drug RPV PSIs. Based on these results, the bilayer tableting technique (Method II) was used to co-formulate DTG and RPV in PSIs and test in subsequent in vivo safety and PK studies.
3.4. In vivo safety of single-drug and dual-drug PSIs.
Safety and biocompatibility of implantable drug delivery devices are essential in development of a LA drug delivery system. Results from a long-term in vivo safety study showed that all PSIs were well tolerated and mice did not show any signs of overt toxicity, behavioral changes, water consumption or weight loss (Fig. S4). Histological staining analysis (H&E) of excised subcutaneous tissues surrounding the PSIs showed that all PSI treatment groups exhibited similar mild-to-moderate presence of lesions in the first week post PSI administration as shown by multiple tracts of inflammation and moderate diffused immune cells (purple-stained cells/areas indicated by arrows in Fig. 6A). The median skin microscopic inflammation scores of excised subcutaneous tissues surrounding the PSIs in all PSI treatment groups were between 2–3 at day 3, and 2 at day 7 due to the incision process (Fig. 6A–B, Fig. S3). Substantial decrease in inflammation was observed at later time points (day 14 and day 30; median of scores 2 and 1) in all groups as shown by small numbers of infiltrated immune cells (purple-stained cells/areas indicated by arrows Fig. 6A). No inflammation was observed beyond day 30 in all treatment groups (day 60–180) with median scores of 0 comparable to the control no treatment group (Fig. 6A–B, Fig. S5). Results from the ELISA assay, quantifying levels of TNF-α and IL-6 pro-inflammatory cytokines in plasma from peripheral blood collections, showed that no systemic acute, or chronic inflammation was present in all PSI treatment groups. IL-6 proinflammatory cytokines in plasma were in the range of 0–9 pg/mL and were comparable to IL-6 levels in plasma of sham mice (p=0.169–0.994, n=4) (Fig. 6C). Similarly, TNF-α proinflammatory cytokine levels in plasma were the range of 0–0.1 pg/mL in all PSI groups at day 3 to day 90 post-administration and were comparable to sham mice (p=0.142–0.999, n=4, Fig. 6D). These results demonstrate that all PSIs are well-tolerated in vivo with no significant inflammation, weight loss or other signs of toxicity. Notably, at day 30, TNF-α proinflammatory cytokine levels in RPV PSI groups exhibited some variability compared to other groups possibly due to inter-individual variability or hormone-cycle variability in mice [45]. Additional immunotoxicity evaluations according to S8 guidance documents for preclinical filing of pharmaceuticals (S8: Immunotoxicity Studies for Human Pharmaceuticals [46]), such as immune cell function in vitro, in vivo immune challenges, as well as extended histopathological examination of lymphoid tissue should be considered for future animal toxicity studies to ascertain the safety of PSIs.
Figure 6.
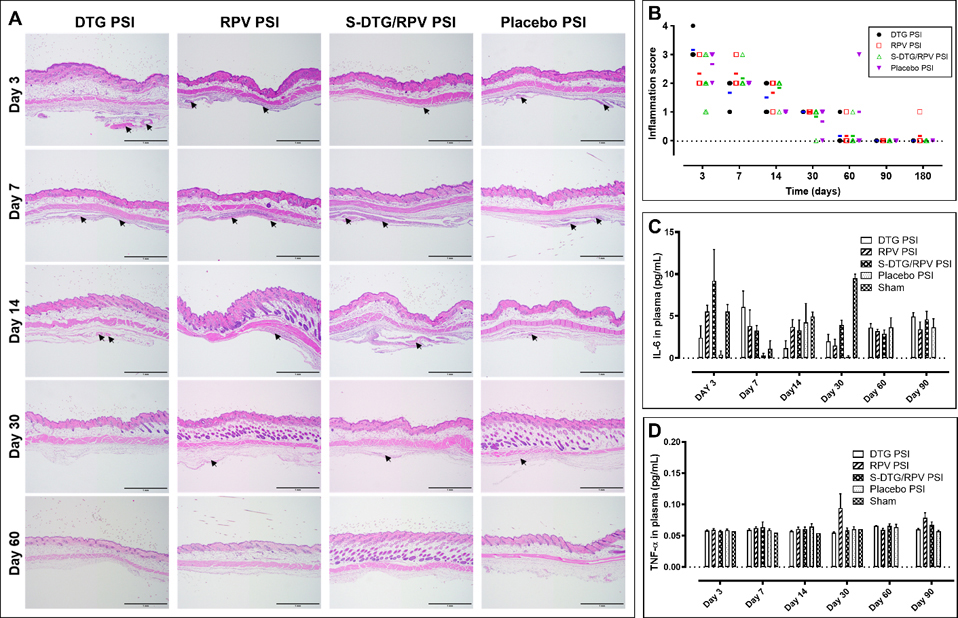
In vivo safety evaluation of PSIs in BALB/c mice (n=7 per timepoint). A) Local inflammation of implanted subcutaneous tissues collected at day 3, 7, 14, 30, and 60 post-implantation and stained with H&E. Arrows indicate areas of inflammation identified by a certified anatomic pathologist. All scale bars represent 1 mm. B) Inflammatory scores of implanted subcutaneous tissues evaluated using light microscope, and blindly scored by a certified pathologist. The bars represent the median of inflammation scores in each group at each timepoint (n=7 per group). C) Concentration of IL-6 (pg/mL) in plasma post-PSI implantation quantified by ELISA (n=4 per group). D) Concentration of TNF-α (pg/mL) in plasma post-PSI implantation quantified by ELISA (n=4 per group).
3.5. In vivo PSI removability and biodegradation
An important consideration when developing a LA drug delivery system for HIV PrEP/ART applications is the ability to remove the solid implant to terminate the treatment in case of toxicity, breakthrough infection, allergic response, pregnancy or any other adverse event. To demonstrate the ability to safely remove PSIs post administration, PSIs were removed at different time points using a small incision (5 mm) at euthanasia. PSIs were successfully removed from the implantation site with no excessive fibrosis tissue surrounding the implants, or any PSI breaking during the removal process. DTG, RPV, S-DTG/RPV PSIs appeared slightly softer and swollen when removed at day 14 or beyond post implantation. In vivo degradation of PSIs was qualitatively measured by SEM imaging the microstructure at different time points. Results showed that for drug PSIs, a substantial degradation was observed after 60 days post administration (Figure 7A–B, Table S5–6). Results also showed that DTG and RPV PSIs exhibited different swelling properties. Specifically, explanted RPV PSIs were swollen as early as day 3 relative to the original RPV PSIs on day 0 (p=0.041, Table S5–6) and remained swollen at day 30 followed by substantial degradation observed at day 60 and 90 (Fig. 6B). On the other hand, DTG PSIs removed at day 7, 14 and 30 post administration had similar dimensions and volumes followed by substantial degradation observed in DTG PSIs removed at day 60 and 90 post administration (Fig. 6B). The dual-drug S-DTG/RPV PSIs exhibited swelling on day 14 and 30 relative to day 0 (p<0.005, Table S5–6) and minimal degradation observed in PSIs removed at day 90 post administration. Placebo PSIs exhibited faster degradation compared to all drug PSIs and were substantially smaller in size at day 7 compared to day 0 and were fully degraded at day 60 post administration (Fig. 7B). Dimensions (diameter, thickness) of explanted PSIs were measured and PSI volumes were estimated at each time point and presented in Table S5–6. These results demonstrate that presence of drug and drug physical and chemical properties play a major role in the swelling and degradation rates of the PLGA matrix [47–50]. The hydrophobic nature of DTG and RPV resulted in slower degradation of PLGA matrix due to limited water diffusion into the PLGA matrix. For DTG PSIs, the basic nature of DTG (pKa 8.2) could be a significant contributing factor in the observed slower degradation compared to placebo and RPV PSIs (RPV pKa 5.6). As a basic drug, DTG (pKa 8.2) can neutralize the acidic microenvironment of PLGA’s degradation by-products (lactic acid and glycolic acid) and as a result slow the degradation of PLGA.
Figure 7.

Explanted PSIs retrieved from subcutaneous skin of BALB/C mice at euthanasia. A) SEM images of explanted DTG PSIs, RPV PSIs, and S-DTG/RPV PSIs removed at day 7, 14, 30, 60 and 90 post administration. B) Images of PSIs removed at day 7, 14, 30, 60 and 90 post administration.
3.6. In vivo pharmacokinetics
To investigate drug release kinetics in vivo using single-drug and dual-drug, pharmacokinetic studies were carried out in BALB/c mice over 6 months. DTG and RPV plasma concentrations were quantitated using a validated LC/MS–MS method [51, 52]. Non-compartmental analysis of the median composite pharmacokinetic (PK) profile of DTG demonstrated a biexponential decay. The mean (standard error) area under the concentration time curve (AUC) was estimated for each treatment group using the linear trapezoidal rule and the sparse sampling function in Phoenix 64 Build 8.1 (Fig. S6G). The median composite concentration vs time profiles suggested a multiphasic elimination for single-drug and dual-drug PSI formulations. After an initial first order decline in plasma concentrations (0–7 days), the release of DTG approached zero-order kinetics [30, 33]. Plasma concentrations of DTG when formulated alone (DTG PSIs) or in combination with RPV (S-DTG/RPV PSIs) were similar and reached levels that were 10 × greater than the protein-adjusted IC90 of DTG (DTG PA-IC90 64 ng/mL [53–55]) for at least 6 months post-administration (Fig. 8A–B, Fig. S6A–C, G). Notably, the plasma levels of DTG in both DTG PSIs and S-DTG/RPV PSIs were slightly higher at day 120 and beyond (Fig. 8B) compared to their early timepoints (Fig. 8A) due to PSI swelling and degradation. In the first 30 days, drug release is mainly governed by a diffusion process. The plasma levels of both DTG and RPV after day 30 were slightly higher compared to day 7 and 14. This is likely attributed to higher drug release due to PSI swelling and degradation. When comparing the PSI dimensions for DTG and RPV groups, RPV PSIs exhibited significantly greater swelling at day 14 compared to DTG PSIs (Supplementary Tables S5 and S6). At day 14, DTG PSIs had an average volume of 7.59 ± 0.37 mm3 compared to 13.59 ± 0.45 mm3 for RPV PSIs (Fig. 7B). This increase in PSI volume due to swelling and degradation resulted in an increase in plasma RPV at d14 versus d30 for DTG (Fig. 8C). This difference is mainly attributed to drug physical/chemical properties of RPV (pKa 5.6) and DTG (pKa 8.2). This difference can influence the degradation kinetics of the PLGA-based PSIs by promoting (RPV) or slowing (DTG) PLGA degradation via ester hydrolysis, a phenomenon that we have previously observed with other drugs [56].
Non-compartmental analysis of the median composite pharmacokinetic (PK) profile of RPV showed a biexponential decay with minimum initial first order decline compared to DTG PSIs. Plasma concentration of RPV when formulated alone (RPV PSI) was at or slightly above 4 × PA-IC90 of RPV (RPV PA-IC90 12 ng/mL [57]) for the duration of the study (180 days). Plasma RPV levels were slightly below 4 × PA-IC90 levels when co-formulated with DTG (S-DTG/RPV PSIs) in the first 90 days (Fig. 8C, Fig. S6D). Interestingly, RPV plasma concentrations slightly increased after day 30 and 120 in both single-drug PSIs (RPV PSIs) and dual-drug PSIs (S-DTG/RPV PSIs) and were maintained at levels 4 × PA-IC90 up to day 180 post administration (Fig. 8D, Fig. S6E, F). The increase in RPV plasma levels at day 30 can be attributed to the swelling properties and matrix degradation of RPV PSIs that were observed from the explanted RPV and S-DTG/RPV PSIs at day 30 and 120 (Fig. 7A–B, Table S5–6). Collectively, these PK results demonstrate that PSIs can sustain the release of DTG and RPV for at least 180 days with drug levels at or above their 4 × PA-IC90, a known benchmark concentration for antiretroviral drugs to achieve efficacy in HIV prevention. DTG from single-drug and dual-drug PSIs had higher plasma concentrations compared to RPV at equivalent dose (296 mg/kg, p<0.0001). These results are in agreement with data obtained from in vitro release studies (Fig. S7), demonstrating a good in vivo-in vitro correlation and that in vivo PK and in vitro drug release kinetics from a PSIs is drug-dependent and exhibit different profiles for drugs with different physical and chemical properties.
Collectively, these results demonstrate a new engineering process to fabricate biodegradable solid implants utilizing phase inversion and compression techniques with the ability to co-formulate two drugs or more in a single compact implant, combined with the ability to achieve high plasma concentrations of drugs for at least 6 months with a single administration. To our best knowledge, this is the first report on a biodegradable and removable solid polymeric implant that can accommodate two drugs or more and provide ultra-long-acting delivery of drugs for 6 months or longer with sustained high plasma concentrations.
4. Conclusion
We demonstrate a new fabrication process of polymeric solid implants (PSIs) using by a phase inversion and compression process that can integrate one or more antiretrovirals (ARVs) without compromising drug potency in vitro [28] and the ability to sustain release of ARVs independently to achieve the target plasma levels over 6 months in vivo. The ultra-long-acting PSIs were safe and well tolerated in BALB/c mice without signs of severe incision site reactions or systemic inflammation over 6 months. Importantly, the ultra-long-acting PSIs are biodegradable, but can be safely removed via a small incision under the skin as demonstrated in BALB/c mice. This is the first report on a multi-drug PLGA-based solid implant that is fabricated by a phase inversion and compression process. This fabrication process does not require high heat, high pressure or use of high volumes of organic solvents, and is simple and scalable to produce solid implants with high drug loading to provide translatable to human doses for HIV PrEP/ART. Therefore, the PSI technology reported in this study represents a highly tunable ultra-long-acting drug delivery system for delivery of antiretroviral drugs for HIV PrEP/ART and for other drugs and indications.
Supplementary Material
Acknowledgements
This work was supported by National Institute of Allergy and Infectious Diseases (grant number R01AI131430 to RB). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases. This research was supported by the University of North Carolina at Chapel Hill Center for AIDS Research (CFAR), an NIH funded program P30 AI050410. We would like to thank the UNC Animal Histopathology Core who performed the histopathology and the Animal Histopathology & Laboratory Medicine Core at the University of North Carolina, which is supported in part by an NCI Center Core Support Grant (5P30CA016086-41) to the UNC Lineberger Comprehensive Cancer Center. We would also like to thank Katie Mollan, senior biostatistician in the UNC Center for AIDS Research, for her input on statistical analysis reported for in vitro drug release studies and in vivo PK studies.
Abbreviations
- ARV
antiretroviral
- DMSO
dimethyl
- DSC
differential scanning calorimetry
- DTG
dolutegravir
- DTG-RPV PSI
dual-drug loaded PSI prepared by adding DTG and RPV in a single ISFI formulation and forming a tablet (referred as Method I)
- EVA
ethylene-vinyl acetate
- HIV PrEP/ART
human immunodeficiency virus pre-exposure prophylaxis/ antiretroviral therapy
- HPLC
high-performance liquid chromatography
- ISFI
in-situ forming implant
- LC/MS-MS
liquid chromatography-tandem mass spectrometry
- NMP
N-methyl pyrrolidone
- (PA) IC90
protein-adjusted concentration required for 90% viral inhibition
- PBS
phosphate buffer saline
- PSI
polymeric solid implant
- PCL
polycaprolactone
- PLA
poly-lactide
- PLGA
poly(DL-lactide-co-glycolide)
- RPV
rilpiviline
- S-DTG/RPV PSI
dual-drug loaded PSI prepared by making DTG and RPV solid powders separately and generating a ‘sandwich’ (S) or bilayer tablet (referred as Method II)
- SEM
scanning electron microscopy
- XRD
x-ray powder diffraction
- ULA
ultra-long acting
Footnotes
Conflict of Interest Disclosure
SRB and PM are inventors on a patent application related to this work filed by the University of North Carolina, Office of Technology Commercialization (UNC OTC) (PCT International Application PCT/US20/16061, filed on January 31, 2020 and U.S. Provisional Patent Application No. 62/800087 filed on February 6, 2019). The authors declare no competing financial interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Research B, Diagnostics and Therapeutics for HIV: Global Markets, www.bccresearch.com (April 2014).
- [2].Ritchie P, Cause of Death, Black & White Publishing Ltd; 2017. [Google Scholar]
- [3].J.U.N.P.o.H.A. (UNAIDS), AIDS Data, 2016.
- [4].Keller SB, Smith DM, The price of tenofovir-emtricitabine undermines the cost-effectiveness and advancement of pre-exposure prophylaxis, AIDS (London, England) 25(18) (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].McCormack S, Dunn DT, Desai M, Dolling DI, Gafos M, Gilson R, Sullivan AK, Clarke A, Reeves I, Schembri G, Pre-exposure prophylaxis to prevent the acquisition of HIV-1 infection (PROUD): effectiveness results from the pilot phase of a pragmatic open-label randomised trial, The Lancet 387(10013) (2016) 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Baert L, van ‘t Klooster G, Dries W, François M, Wouters A, Basstanie E, Iterbeke K, Stappers F, Stevens P, Schueller L, Van Remoortere P, Kraus G, Wigerinck P, Rosier J, Development of a long-acting injectable formulation with nanoparticles of rilpivirine (TMC278) for HIV treatment, Eur. J. Pharm. Biopharm. 72(3) (2009) 502–508. [DOI] [PubMed] [Google Scholar]
- [7].Landovitz RJ, Kofron R, McCauley M, The Promise and Pitfalls of Long Acting Injectable Agents for HIV Prevention, Current opinion in HIV and AIDS 11(1) (2016) 122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gunawardana M, Remedios-Chan M, Miller CS, Fanter R, Yang F, Marzinke MA, Hendrix CW, Beliveau M, Moss JA, Smith TJ, Baum MM, Pharmacokinetics of Long-Acting Tenofovir Alafenamide (GS-7340) Subdermal Implant for HIV Prophylaxis, Antimicrob. Agents Chemother. 59(7) (2015) 3913–3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Schlesinger E, Johengen D, Luecke E, Rothrock G, McGowan I, van der Straten A, Desai T, A Tunable, Biodegradable, Thin-Film Polymer Device as a Long-Acting Implant Delivering Tenofovir Alafenamide Fumarate for HIV Pre-exposure Prophylaxis, Pharmaceutical Research 33(7) (2016) 1649–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Barnhart M, Long-Acting HIV Treatment and Prevention: Closer to the Threshold, Global Health: Science and Practice 5(2) (2017) 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Date AA, Shibata A, Goede M, Sanford B, La Bruzzo K, Belshan M, Destache CJ, Development and evaluation of a thermosensitive vaginal gel containing raltegravir+ efavirenz loaded nanoparticles for HIV prophylaxis, Antiviral Res. 96(3) (2012) 430–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Grammen C, Ariën KK, Venkatraj M, Joossens J, Van der Veken P, Heeres J, Lewi PJ, Haenen S, Augustyns K, Vanham G, Development and in vitro evaluation of a vaginal microbicide gel formulation for UAMC01398, a novel diaryltriazine NNRTI against HIV-1, Antiviral Res. 101 (2014) 113–121. [DOI] [PubMed] [Google Scholar]
- [13].Guthrie KM, Rohan L, Rosen RK, Vargas SE, Shaw JG, Katz D, Kojic EM, Ham AS, Friend D, Buckheit KW, Buckheit RW Jr., Vaginal film for prevention of HIV: using visual and tactile evaluations among potential users to inform product design, Pharmaceutical development and technology 23(3) (2018) 311–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bunge KE, Dezzutti CS, Rohan LC, Hendrix CW, Marzinke MA, Richardson-Harman N, Moncla BJ, Devlin B, Meyn LA, Spiegel HML, Hillier SL, A Phase 1 Trial to Assess the Safety, Acceptability, Pharmacokinetics, and Pharmacodynamics of a Novel Dapivirine Vaginal Film, Journal of acquired immune deficiency syndromes (1999) 71(5) (2016) 498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].García-Lerma JG, Heneine W, Animal models of antiretroviral prophylaxis for HIV prevention, Current opinion in HIV and AIDS 7(6) (2012) 505–513. [DOI] [PubMed] [Google Scholar]
- [16].Devlin B, Nuttall J, Wilder S, Woodsong C, Rosenberg Z, Development of dapivirine vaginal ring for HIV prevention, Antiviral Res. 100 (2013) S3–S8. [DOI] [PubMed] [Google Scholar]
- [17].Fetherston SM, Boyd P, McCoy CF, McBride MC, Edwards K-L, Ampofo S, Malcolm RK, A silicone elastomer vaginal ring for HIV prevention containing two microbicides with different mechanisms of action, Eur. J. Pharm. Sci. 48(3) (2013) 406–415. [DOI] [PubMed] [Google Scholar]
- [18].Spreen WR, Margolis DA, Pottage JC Jr, Long-acting injectable antiretrovirals for HIV treatment and prevention, Current opinion in HIV and AIDS 8(6) (2013) 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hoeben E, Borghys H, Looszova A, Bouche M-P, van Velsen F, Baert L, Pharmacokinetics and disposition of rilpivirine (TMC278) nanosuspension as a long-acting injectable antiretroviral formulation, Antimicrob. Agents Chemother. 54(5) (2010) 2042–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Azijn H, Tirry I, Vingerhoets J, de Béthune M-P, Kraus G, Boven K, Jochmans D, Van Craenenbroeck E, Picchio G, Rimsky LT, TMC278, a next-generation nonnucleoside reverse transcriptase inhibitor (NNRTI), active against wild-type and NNRTI-resistant HIV-1, Antimicrob. Agents Chemother. 54(2) (2010) 718–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Penrose KJ, Parikh UM, Hamanishi KA, Else L, Back D, Boffito M, Jackson A, Mellors JW, Selection of Rilpivirine-Resistant HIV-1 in a Seroconverter From the SSAT 040 Trial Who Received the 300-mg Dose of Long-Acting Rilpivirine (TMC278LA), The Journal of Infectious Diseases 213(6) (2015) 1013–1017. [DOI] [PubMed] [Google Scholar]
- [22].Barrett SE, Teller RS, Forster SP, Li L, Mackey MA, Skomski D, Yang Z, Fillgrove KL, Doto GJ, Wood SL, Lebron J, Grobler JA, Sanchez RI, Liu Z, Lu B, Niu T, Sun L, Gindy ME, Extended-Duration MK-8591-Eluting Implant as a Candidate for HIV Treatment and Prevention, Antimicrobial Agents and Chemotherapy 62(10) (2018) e01058–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Carr A, DYBUL M, ROSS HEWITT M, HICKS C, MOYLE G, YOULE M, 10th conference on retroviruses and opportunistic infections, Conf Retrovir Opportunistic Infect, Citeseer, 2006, p. 13. [Google Scholar]
- [24].Jacobson JM, Flexner CW, Universal antiretroviral regimens: thinking beyond one-pill-once-a-day, Current opinion in HIV and AIDS 12(4) (2017) 343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liu L, Gao Q, Lu X, Zhou H, In situ forming hydrogels based on chitosan for drug delivery and tissue regeneration, Asian Journal of Pharmaceutical Sciences 11(6) (2016) 673–683. [Google Scholar]
- [26].Zajc N, Obreza A, Bele M, Srcic S, Physical properties and dissolution behaviour of nifedipine/mannitol solid dispersions prepared by hot melt method, Int. J. Pharm. 291(1–2) (2005) 51–8. [DOI] [PubMed] [Google Scholar]
- [27].Dinunzio JC, Brough C, Hughey JR, Miller DA, Williams RO 3rd, McGinity JW, Fusion production of solid dispersions containing a heat-sensitive active ingredient by hot melt extrusion and Kinetisol dispersing, Eur. J. Pharm. Biopharm. 74(2) (2010) 340–51. [DOI] [PubMed] [Google Scholar]
- [28].Maturavongsadit P, Paravyan G, Kovarova M, Garcia JV, Benhabbour SR, A New Engineering Process of Biodegradable Polymeric Solid Implants for Ultra-Long-Acting Drug Delivery, International Journal of Pharmaceutics Accepted (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Nelson M, Elion R, Cohen C, Mills A, Hodder S, Segal-Maurer S, Bloch M, Garner W, Guyer B, Williams S, Rilpivirine versus efavirenz in HIV-1–infected subjects receiving emtricitabine/tenofovir DF: Pooled 96-week data from ECHO and THRIVE studies, HIV clinical trials 14(3) (2013) 81–91. [DOI] [PubMed] [Google Scholar]
- [30].Kovarova M, Benhabbour SR, Massud I, Spagnuolo RA, Skinner B, Baker CE, Sykes C, Mollan KR, Kashuba ADM, Garcia-Lerma JG, Mumper RJ, Garcia JV, Ultra-long-acting removable drug delivery system for HIV treatment and prevention, Nat Commun 9(1) (2018) 4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Cholewa K, Poleszak J, Wysokińska O, Gędek A, Zaremba B, Szabat P, Milanowska J, JULUCA-a new therapeutic opportunity for patients infected with HIV-1, World Scientific News; 123 (2019) 181–191. [Google Scholar]
- [32].Ribera E, New Dual Combination of Dolutegravir-Rilpivirine for Switching to Maintenance Antiretroviral Therapy, (2018). [DOI] [PubMed]
- [33].Royals MA, Fujita SM, Yewey GL, Rodriguez J, Schultheiss PC, Dunn RL, Biocompatibility of a biodegradable in situ forming implant system in rhesus monkeys, J. Biomed. Mater. Res. 45(3) (1999) 231–9. [DOI] [PubMed] [Google Scholar]
- [34].<905>Uniformity of Dosage Units, The UnitedStates Pharmacopeia 30 and the National Formulary 25, USP NF 1 (2007) 378–384.
- [35].Andrews GP, Abu-Diak O, Kusmanto F, Hornsby P, Hui Z, Jones DS, Physicochemical characterization and drug-release properties of celecoxib hot-melt extruded glass solutions, J. Pharm. Pharmacol. 62(11) (2010) 1580–1590. [DOI] [PubMed] [Google Scholar]
- [36].Coceani N, Magarotto L, Ceschia D, Colombo I, Grassi M, Theoretical and experimental analysis of drug release from an ensemble of polymeric particles containing amorphous and nano-crystalline drug, Chem. Eng. Sci. 71 (2012) 345–355. [Google Scholar]
- [37].Fredenberg S, Wahlgren M, Reslow M, Axelsson A, The mechanisms of drug release in poly(lactic-co-glycolic acid)-based drug delivery systems--a review, Int. J. Pharm. 415(1–2) (2011) 34–52. [DOI] [PubMed] [Google Scholar]
- [38].Nikkola L, Viitanen P, Ashammakhi N, Temporal control of drug release from biodegradable polymer: multicomponent diclofenac sodium releasing PLGA 80/20 rod, Journal of Biomedical Materials Research Part B: Applied Biomaterials: An Official Journal of The Society for Biomaterials, The Japanese Society for Biomaterials, and The Australian Society for Biomaterials and the Korean Society for Biomaterials 89(2) (2009) 518–526. [DOI] [PubMed] [Google Scholar]
- [39].Solorio L, Olear AM, Hamilton JI, Patel RB, Beiswenger AC, Wallace JE, Zhou H, Exner AA, Noninvasive Characterization of the Effect of Varying PLGA Molecular Weight Blends on In Situ Forming Implant Behavior Using Ultrasound Imaging, Theranostics 2(11) (2012) 1064–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Astaneh R, Erfan M, Moghimi H, Mobedi H, Changes in morphology of in situ forming PLGA implant prepared by different polymer molecular weight and its effect on release behavior, J. Pharm. Sci. 98(1) (2009) 135–145. [DOI] [PubMed] [Google Scholar]
- [41].Ravi PR, Kotreka UK, Saha RN, Controlled release matrix tablets of zidovudine: effect of formulation variables on the in vitro drug release kinetics, AAPS PharmSciTech 9(1) (2008) 302–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hamoudi-Ben Yelles MC, Tran Tan V, Danede F, Willart JF, Siepmann J, PLGA implants: How Poloxamer/PEO addition slows down or accelerates polymer degradation and drug release, J. Controlled Release 253 (2017) 19–29. [DOI] [PubMed] [Google Scholar]
- [43].Parent M, Nouvel C, Koerber M, Sapin A, Maincent P, Boudier A, PLGA in situ implants formed by phase inversion: Critical physicochemical parameters to modulate drug release, J. Controlled Release 172(1) (2013) 292–304. [DOI] [PubMed] [Google Scholar]
- [44].Clark JT, Clark MR, Shelke NB, Johnson TJ, Smith EM, Andreasen AK, Nebeker JS, Fabian J, Friend DR, Kiser PF, Engineering a Segmented Dual-Reservoir Polyurethane Intravaginal Ring for Simultaneous Prevention of HIV Transmission and Unwanted Pregnancy, PLoS One 9(3) (2014) e88509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].LaMarca BD, Chandler DL, Grubbs L, Bain J, McLemore GR Jr., Granger JP, Ryan MJ, Role of sex steroids in modulating tumor necrosis factor alpha-induced changes in vascular function and blood pressure, Am. J. Hypertens. 20(11) (2007) 1216–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Guideline IHT, Immunotoxicity studies for human pharmaceuticals, Guideline, 2005. [Google Scholar]
- [47].Tang Y, Singh J, Controlled delivery of aspirin: effect of aspirin on polymer degradation and in vitro release from PLGA based phase sensitive systems, Int. J. Pharm. 357(1–2) (2008) 119–125. [DOI] [PubMed] [Google Scholar]
- [48].Packhaeuser C, Schnieders J, Oster C, Kissel T, In situ forming parenteral drug delivery systems: an overview, Eur. J. Pharm. Biopharm. 58(2) (2004) 445–455. [DOI] [PubMed] [Google Scholar]
- [49].Zlomke C, Barth M, Mäder K, Polymer degradation induced drug precipitation in PLGA implants – Why less is sometimes more, Eur. J. Pharm. Biopharm. 139 (2019) 142–152. [DOI] [PubMed] [Google Scholar]
- [50].Klose D, Siepmann F, Elkharraz K, Siepmann J, PLGA-based drug delivery systems: importance of the type of drug and device geometry, Int. J. Pharm. 354(1–2) (2008) 95–103. [DOI] [PubMed] [Google Scholar]
- [51].Adams JL, Patterson KB, Prince HM, Sykes C, Greener BN, Dumond JB, Kashuba AD, Single and multiple dose pharmacokinetics of dolutegravir in the genital tract of HIV negative women, Antiviral therapy 18(8) (2013) 1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Else LJ, Tjia J, Jackson A, Penchala SD, Egan D, Boffito M, Khoo SH, Back DJ, Quantification of rilpivirine in human plasma, cervicovaginal fluid, rectal fluid and genital/rectal mucosal tissues using liquid chromatography–tandem mass spectrometry, Bioanalysis 6(14) (2014) 1907–1921. [DOI] [PubMed] [Google Scholar]
- [53].the state of the antiretroviral drug market in low- and middle-income countries, ARV market report, 2016. [Google Scholar]
- [54].Kaplan G, Casoy J, Zummo J, Impact of long-acting injectable antipsychotics on medication adherence and clinical, functional, and economic outcomes of schizophrenia, Patient preference and adherence 7 (2013) 1171–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Mansur AH, Srivastava S, Mitchell V, Sullivan J, Kasujee I, Longterm clinical outcomes of omalizumab therapy in severe allergic asthma: Study of efficacy and safety, Respir. Med. 124 (2017) 36–43. [DOI] [PubMed] [Google Scholar]
- [56].Benhabbour SR, Kovarova M, Jones C, Copeland DJ, Shrivastava R, Swanson MD, Sykes C, Ho PT, Cottrell ML, Sridharan A, Fix SM, Thayer O, Long JM, Hazuda DJ, Dayton PA, Mumper RJ, Kashuba ADM, Victor Garcia J, Ultra-long-acting tunable biodegradable and removable controlled release implants for drug delivery, Nature Communications 10(1) (2019) 4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Aouri M, Barcelo C, Guidi M, Rotger M, Cavassini M, Hizrel C, Buclin T, Decosterd LA, Csajka C, Swiss HIVCS, Population Pharmacokinetics and Pharmacogenetics Analysis of Rilpivirine in HIV-1-Infected Individuals, Antimicrob. Agents Chemother. 61(1) (2016) e00899–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.