

Abbreviations
- CLD
chronic liver disease
- LEfSe
LDA effect size
- OTU
operational taxonomic unit
- PCR
polymerase chain reaction
- PSC
primary sclerosing cholangitis
- rDNA
ribosomal DNA
To the Editor:
Extensive research on human microbiomes has largely focused on the human gut and the contribution of microbiome dysbiosis (loss of key taxa, diversity, and metabolic capacity) in the onset of diseases. Other studies have focused on additional compartments in the human body, such as the biliary system. These compartments also have an autochthonous microbiome that undergoes changes depending on the conditions of the host and vice versa. However, comprehensive studies characterizing the human bile microbiome and its relationship to liver diseases are still scarce.
Therefore, we focused on the relationship between the bile microbiome and primary sclerosing cholangitis (PSC), a difficult‐to‐treat liver disease and, as such, an indication for liver transplantation. PSC is characterized by progressive fibrosis and inflammation of the bile ducts with ulcerative lesions of the bile duct epithelia.( 1 ) Although PSC is frequently associated with bacterial infections, it can also lead to bile duct cancer and liver cirrhosis.( 1 ) To date, the pathogenesis of PSC remains unknown, and its diagnosis is challenging. Recent studies have pointed to the bile microbiome as a key player in the pathophysiology of PSC.( 2 ) Until the recent advances in microbiome research, healthy human bile had been considered sterile.( 3 ) However, microbiome studies have not only revealed the presence of a bile microbiome but also an alteration within the microbiome in patients with PSC compared with healthy individuals.( 2 ) Unlike the physical composition of bile, its microbial composition remains largely unknown. Bile is a biological fluid, mainly constituted of bile acids, cholesterol, phospholipids, and proteins. It is synthesized in the liver and stored in the gallbladder. Several bile‐related diseases modify the functionality of bile, of which the most frequent is the generation of gallstones resulting in cholestasis. Another bile‐related disease is bile duct inflammation, or cholangitis.
In previous studies, we examined different compartments of the human body that were considered to be sterile (portal, hepatic, and peripheral veins and the right atrium), as well as fluid samples of ascites patients and blood samples from patients with chronic liver disease (CLD). We found bacterial DNA and differences in the composition of the bacterial community in all compartments investigated.( 4, 5 )
Here, we aimed to characterize the biliary microbiome of PSC patients compared with control individuals and to patients with other biliary diseases, such as cholangitis and cholestasis in a cross‐sectional pilot study.
Patients and Methods
In this study, we characterized the bile microbiome of PSC patients (n = 5), cholestasis patients (n = 6), and cholangitis patients (n = 5) using 16S ribosomal DNA (rDNA) amplicon sequencing technology on bile fluid samples collected once during primary endoscopic retrograde cholangiopancreatography. We compared their microbiome to the bacterial bile microbiome of a control group (n = 3) undergoing endoscopic retrograde cholangiopancreatography for reasons other than cancer, liver disease, cholestasis, or cholangitis to exclude PSC and biliary stones. Within our control group two IBD‐patients received ECRP to exclude PSC and one patient with previous pancreatitis to exclude biliary stones. Of note these patients did not present cholestasis or cholangitis at ERCP. There were no baseline demographic differences between the patient groups. Written informed consent was obtained from all patients before enrollment under ethics approval number 203/13 from the ethics committee of the University Clinic Bonn (Bonn, Germany) in agreement with the Declaration of Helsinki.
DNA Isolation, Sequencing, and Microbiome Analysis
Bacterial DNA was isolated from bile fluid and integrity, and DNA concentrations were evaluated using NanoDrop 2000 UV spectrophotometer (Thermo Fisher Scientific, Waltham, MA) and gel electrophoresis. The amount of 16S rDNA in the samples was measured by quantitative polymerase chain reaction (qPCR). The sequence library was constructed by PCR amplification using 16S rDNA universal primers with sequencing adapters targeting the V3‐V4 region of the bacterial 16S ribosomal gene. The joint pair length was set to encompass 467 base pairs using the 2 × 300 paired‐end MiSeq Reagent kit V3 (600 cycle) (Cat# MS‐102‐3003, Illumina, San Diego, CA). Detection of the sequencing fragments was performed using the MiSeq System, Illumina, San Diego. The obtained sequences were analyzed using a bioinformatics pipeline. Briefly, after demultiplexing of the bar‐coded Illumina (Illumina, Inc, Worldwide headquarters, San Diego, CA) paired reads, single reads were obtained, cleaned, and paired for each sample. Operational taxonomic units (OTUs) were produced using single‐linkage clustering, and taxonomic assignment was done by Blast (Basic local alignment search tool) (https://blast.ncbi.nlm.nih.gov/)+ v2.2.30+ with the Silva 128 data bank in order to determine community profiles. From the obtained data, an OTU table was generated, and data were analyzed. For estimation of bacterial diversity in each patient group, the alpha diversity and taxa evenness were calculated.
Bile Microbiome Compared With Other Human Compartments
The analysis of the sequencing data showed that the composition of the human bacterial bile microbiome differs strongly from the composition of the bacterial microbiome from other human samples like blood, ascites, or stool (Fig. 1A‐D).( 4, 5 ) Furthermore, the quantity and profile of bacterial DNA in the bile samples differed from those found in the negative control patients (Supporting Fig. 1), clearly demonstrating that the bacteria detected in bile fluid are not a contamination deriving from the negative control patients. Detailed microbiome analysis revealed that the bile microbiome consists of bacteria belonging to 8 different phyla, namely, Actinobacteria, Bacteroidetes, Firmicutes, Fusobacteria, Gemmatimonadetes, Proteobacteria, Saccharibacteria, and Tenericutes. However, only 5 phyla account for almost the entire bile microbiome. These are Actinobacteria, Bacteroidetes, Firmicutes, Fusobacteria, and Proteobacteria (Fig. 1E). Proteobacteria and in particular Gammaproteobacteria were the most abundant class of bacteria followed by the phylum of Firmicutes and, here in particular, bacteria belonging to the class of Bacilli.
Fig. 1.
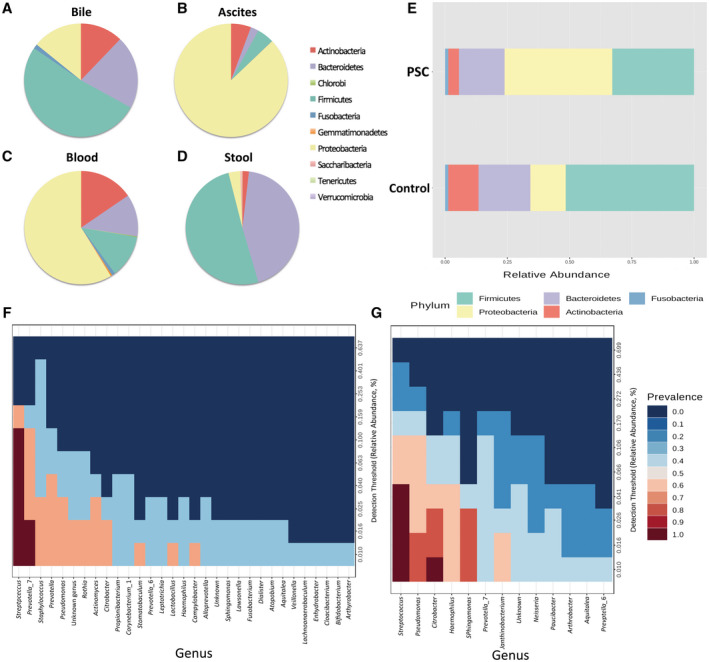
Pie charts representing the microbiota composition (relative abundance) at the phylum level from (A) human bile samples, (B) human blood samples of patients with ascites, and (C) human blood samples from different body compartments (portal vein, hepatic vein, atrium, and peripheral blood). (D) Microbiome composition of human stool samples. (E) Bar plots representing the relative abundance of bacterial phyla from human bile samples of PSC patients (n = 5) and control patients (n = 3) at the phylum level. (F) Heatmap of the core microbiome of the healthy control samples. (G) Heatmap of the core microbiome of PSC patients. Patients with PSC showed a less diverse core microbiome and a higher relative abundance of Streptococcus sp. bacteria.
Bile Microbiome in PSC Patients
In our first analysis, we compared the bile microbiome of PSC patients (n = 4) with the bacterial microbiome of the control group (n = 3). The analysis revealed that the microbiome composition differed between PSC patients and that of the control group, similar to the results published by Liwinski et al.( 2 ) Although the abundance of Actinobacteria, Bacteroidetes, Firmicutes, and Fusobacteria was lower in PSC patients, the abundance of Proteobacteria was greater compared with patients from the control group (Fig. 1E). Core microbiome analysis revealed that both groups harbored a diverse core microbiome, with Streptococcus as the predominant genus in the core microbiome of the control group and that of PSC patients (Fig. 1F,G).
Interestingly, on the bacterial species level, patients with PSC showed a lower abundance of several bacterial species, and their core microbiome consisted of fewer bacterial species than the core microbiome of the control group (shown in Fig. 1G), reinforcing the concept of biliary dysbiosis of the bile fluid being pathophysiologically relevant for the formation of PSC. Pairwise analysis between PSC patients and the control group using Linear discriminant analysis effect size (LEfSe) revealed differences in the bacterial microbiome composition at the bacterial family level. Bacteria belonging to the family of Pasteurellaceae and Pseudomonadaceae were more abundant in the bile microbiome of PSC patients, whereas, bacteria belonging to the families of Prevotellaceae and Bifidobacteriaceae were more abundant in the control group, indicating dysbiosis of the bile microbiome in PSC patients (Fig. 2E). Alpha diversity analysis showed that PSC patients had a significantly lower alpha diversity (at the phylum level) than the control group (P < 0.05) (data not shown).
Fig. 2.
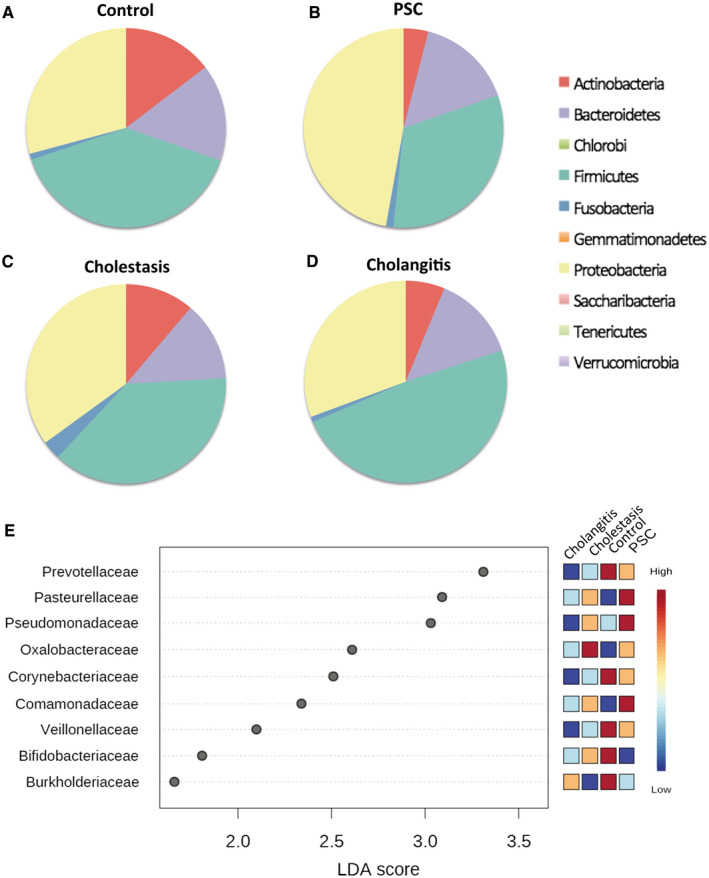
Pie charts representing the microbiota composition (relative abundance) at the phylum level from (A) the control bile samples (n = 3), (B) patients with PSC (n = 5), (C) patients with cholestasis (n = 6), and (D) patients with cholangitis (n = 5). (E) Heatmaps of the LEfSe analysis results (at the family level) of the bile microbiome.
Bile Microbiome in Cholestasis and Cholangitis
In a second analysis, we investigated the microbiome composition of cholestasis (n = 6) and cholangitis (n = 5) patients and compared it with the microbiome composition found in PSC patients and the control group. Microbiome analysis on the phylum level revealed that the bacterial bile microbiome composition differed between these patient groups (Fig. 2A‐D).
Bile Microbiome in Cholestasis
Comparisons of the microbiome from patients with cholestasis with the microbiome of the control group revealed that bacteria belonging to the families of Corynebacteriaceae and Ruminococcaceae were more abundant in samples from the control group. Additionally, bacteria belonging to the families of Oxalobactereaceae and Burkholderiaceae were more abundant in the control group (Fig. 2E). Comparison of cholestasis patients with PSC patients revealed that bacteria belonging to the families of Comamonadaceae and Burkholderiaceae were more abundant in PSC patients. Comparison at the phylum level revealed that the proportion of bacteria belonging to the phylum Proteobacteria was lower, whereas the proportion of Actinobacteria and Firmicutes was higher (Fig. 2C). Interestingly, cholestasis patients had the highest alpha diversity.
Bile Microbiome in Cholangitis
Analysis on the phylum level showed that in cholangitis patients, the proportion of Bacteroidetes was decreased, while the proportion of Proteobacteria was higher than in the control group (Fig. 2D). Bacteria belonging to the families of Microbacteriaceae, Veillonellaceae, and Burkholderiaceae were more abundant in the control group patients than in patients with cholangitis (Fig. 2E). The comparison of the bile microbiome of cholangitis patients with that of cholestasis patients revealed bacteria belonging to the family of Burkholderiacea to be more abundant in cholangitis patients. Comparisons of cholestasis patients with cholangitis patients showed that bacteria belonging to the families of Staphylococcaceae and Microbacteriaceae were more abundant in cholestasis patients (Fig. 2E).
Analysis on OTU numbers revealed significant differences in OTU richness between the 4 patient groups (P < 0.05). These results indicate that the bacterial diversity of the bile microbiome depends on the biliary pathology. Additionally, there was no major difference or variance within the 5 cholangitis patients and in the other patient groups, indicating that the microbiome composition within the groups was reliable.
This study has several limitations. Because of the small sample size of the data set, we cannot account for antibiotics or Proton‐pump inhibitor use. However, statistical analysis between the study patients revealed that this had no significant effect on their microbiome composition at the phylum level (P > 0.05).
In summary, our results confirm recently published data on the bile microbiome composition in PSC patients, emphasizing the reproducibility of bile microbiome assessment. Moreover, we show that the composition of the bile microbiome differed between patients exhibiting different biliary pathologies. However, additional work is needed to address any causality of the microbiome composition and its pathogenesis in biliary diseases.
Supporting information
Fig S1
Acknowledgments
This study was funded by the MICROB‐PREDICT project, which has received funding from the European Union’s Horizon 2020 Research and Innovation Programme under grant 825694. The authors would like to thank Sabine Dentler for English proofreading.
Stefan Zeuzem advises for Janssen Pharmaceutica, Luksogst, AbbVie, and Gilead Sciences; consults for Janssen Pharmaceutica, AbbVie, and Gilead Sciences; and is on the speakers’ bureau for Merck, AbbVie, and Gilead Sciences. Benjamin Lelouvier is employed by Vaiomer SAS.
This article reflects only the authors’ views, and the European Commission is not responsible for any use that may be made of the information it contains.
See Editorial on Page 1571
References
- 1.Karlsen TH, Folseraas T, Thorburn D, Vesterhus M. Primary sclerosing cholangitis—a comprehensive review. J Hepatol 2017;67:1298‐1323. [DOI] [PubMed] [Google Scholar]
- 2.Liwinski T, Zenouzi R, John C, Ehlken H, Rühlemann MC, Bang C, et al. Alterations of the bile microbiome in primary sclerosing cholangitis. Gut 2020;69:665‐672. [DOI] [PubMed] [Google Scholar]
- 3.Warburton RC, Gelson W, Harper S, Rushbrook S, Narbad A. Human bile retrieved from the normal biliary system is not sterile. J Hepatol 2017;66:S65. [Google Scholar]
- 4.Alvarez‐Silva C, Schierwagen R, Pohlmann A, Magdaleno F, Uschner FE, Ryan P, et al. Compartmentalization of immune response and microbial translocation in decompensated cirrhosis. Front Immunol 2019;10:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schierwagen R, Alvarez‐Silva C, Madsen MSA, Kolbe CC, Meyer C, Thomas D, et al. Circulating microbiome in blood of different circulatory compartments. Gut 2019;68:578‐580. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1