

SUMMARY
Junctional coupling between endoplasmic reticulum (ER) Ca2+-sensor STIM proteins and plasma membrane (PM) Orai channels mediates Ca2+ signals in most cells. We reveal that PM-tethered, fluorescently tagged C-terminal M4x (fourth transmembrane helix contains a cytoplasmic C-terminal extension) peptides from Orai channels undergo a Leu-specific signature of direct interaction with the STIM1 Orai-activating region (SOAR), exactly mimicking STIM1 binding to gate Orai channels. The 20-amino-acid Orai3-M4x peptide associates avidly with STIM1 within ER-PM junctions, functions to competitively block native Ca2+ signals, and mediates a key modification of STIM-Orai coupling induced by 2-aminoethoxydiphenyl borate. By blocking STIM-Orai coupling, the Orai3-M4x peptide reveals the critical role of Orai channels in driving Ca2+ oscillatory signals and transcriptional control through NFAT. The M4x peptides interact independently with SOAR dimers consistent with unimolecular coupling between Orai subunits and STIM1 dimers. We reveal the critical role of M4x helices in defining the coupling interface between STIM and Orai proteins to mediate store-operated Ca2+ signals.
In brief
Baraniak et al. use plasma-membrane-tethered Orai channel C-terminal peptides to reveal a Leu signature of interaction with the active site of STIM1 exactly mimicking the STIM-Orai interaction. The Orai peptides block store-operated Ca2+ entry and reveal the critical role of Orai C-terminal helices in defining the STIM-Orai coupling interface.
Graphical abstract
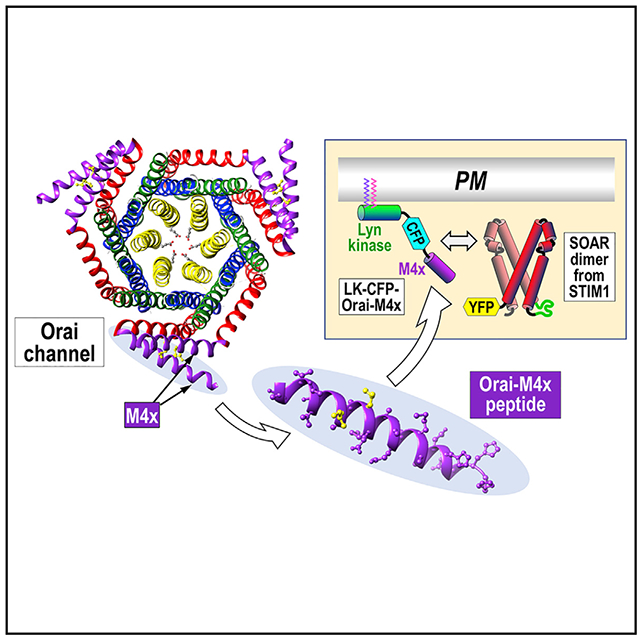
INTRODUCTION
Cytoplasmic Ca2+ signals are a combination of Ca2+ release from endoplasmic reticulum (ER) Ca2+ stores and Ca2+ entry across the plasma membrane (PM). These processes are coordinated through the following two interconnecting proteins: STIM proteins in the ER membrane that are luminal Ca2+ sensors and Orai channels in the PM that mediate “store-operated” Ca2+ entry (SOCE) signals (Derler et al., 2016; Prakriya and Lewis, 2015; Soboloff et al., 2012; Yeung et al., 2020; Zhou et al., 2019). Receptor-mediated inositol 1,4,5-trisphosphate generation activates ER Ca2+ release channels that deplete Ca2+ stores. Luminal Ca2+ dissociation from STIM proteins causes them to unfold, exposing a conserved STIM1 Orai-activating region (SOAR) (Kawasaki et al., 2009; Park et al., 2009; Wang et al., 2014; Yuan et al., 2009). A polybasic STIM C terminus binds to acidic PM phospholipids, trapping STIM proteins in ER-PM junctions (Liou et al., 2007). The exposed SOAR domains tether and activate PM Orai channels, generating Ca2+ entry signals crucial in maintaining Ca2+ oscillations and the regulation of gene expression (Derler et al., 2016; Prakriya and Lewis, 2015; Trebak and Kinet, 2019; Yeung et al., 2020; Zhou et al., 2018a)
Alterations in this fundamental Ca2+ signaling mechanism underlie a spectrum of disease states (Feske, 2019; Prakriya and Lewis, 2015); hence, the molecular understanding of the STIM-Orai interaction has been a much sought-after goal. Crystallography reveals a hexameric structure for the Drosophila Orai channel (dOrai) (Hou et al., 2012; Rothberg et al., 2013), and the closely related mammalian Orai channels (Orai1, Orai2, and Orai3) also operate as hexamers (Cai et al., 2016; Yen et al., 2016). Mammalian STIM proteins (STIM1 and STIM2) are dimers, and their SOAR domains have two symmetrical Orai-binding sites (Wang et al., 2014; Zhou et al., 2015) that bind independently to activate Orai1 channels within the dimeric SOAR framework (Li et al., 2011; Zhou et al., 2015). Two adjacent Orai subunits were thought to undergo a bimolecular interaction with the dimeric SOAR unit (Fahrner et al., 2014; Maus et al., 2015; Stathopulos et al., 2013). However, only one of the two SOAR domains is sufficient for Orai1 activation (Zhou et al., 2015). This unimolecular interaction is consistent with the observed stoichiometry of 6 STIM1 dimers interacting with a single Orai hexamer (Hoover and Lewis, 2011; Li et al., 2011; Scrimgeour et al., 2009) and allows STIM1 dimers to cross-link between Orai1 channels (Zhou et al., 2018a).
SOAR is clearly the Orai-interacting domain of STIM proteins, whereas the STIM-binding site on Orai channels has remained enigmatic. The Orai1 fourth transmembrane helix (M4) contains a cytoplasmic C-terminal extension (M4x) strongly implicated in STIM1 binding (Muik et al., 2008; Park et al., 2009; Prakriya and Lewis, 2015; Yeung et al., 2020; Zhou et al., 2019). STIM1 may also weakly interact with the N-terminal extension of the pore-forming first transmembrane domain (M1) of Orai1 (Palty and Isacoff, 2016; Park et al., 2009; Zheng et al., 2013), and it was suggested this interaction might directly gate the Orai1 channel (Derler et al., 2013; Li et al., 2007; McNally et al., 2013; Palty and Isacoff, 2016; Park et al., 2009; Zheng et al., 2013). However, such Orai1 N terminus gating has recently been questioned (Derler et al., 2018; Yeung et al., 2018; Zhou et al., 2016). Crystallography shows that two adjacent dOrai M4x helices undergo a symmetrical, antiparallel interaction mediated by two hydrophobic residues (I316 and L319) (Figure 1A). This paired M4x complex was described as a binding pocket for STIM1 (Fahrner et al., 2014; Maus et al., 2015; Stathopulos et al., 2013; Yen and Lewis, 2018; Zhou et al., 2016). Structural data suggested that the two Orai1-M4x helices remain paired together through the hydrophobic L273 and L276 residues creating the purported binding pocket (Fahrner et al., 2014; Stathopulos et al., 2013). This view has been supported by other studies suggesting that STIM1 interacts with pairs of Orai1 M4x helices (Tirado-Lee et al., 2015; Yen and Lewis, 2018). However, recent structural data have questioned the existence of paired M4x peptides, indicating that they are “unlatched” in the dOrai channel (Hou et al., 2018; Liu et al., 2019; Zhou et al., 2019)
Figure 1. PM-tethered Orai channel M4x peptides recruit cytosolic SOAR dimers and undergo Leu-specific profiles of FRET interaction.
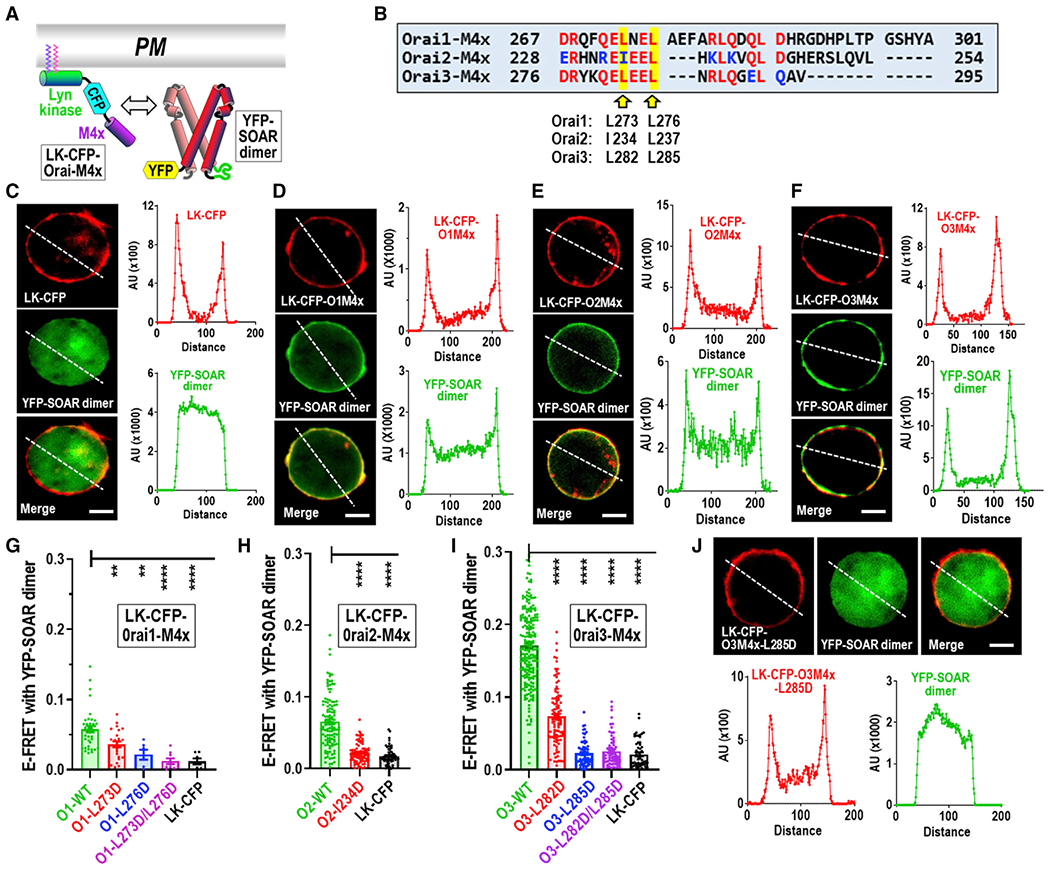
(A) Diagram of interacting constructs. LK-CFP-Orai-M4x comprises the Lyn kinase PM tether (LK), cyan fluorescent protein (CFP), and M4x peptide (controls were devoid of M4x); yellow fluorescent protein (YFP)-SOAR dimer is the concatenated dimer of SOAR tagged at the N terminus with YFP.
(B) The M4x sequences of Orai channel subtypes highlighting key hydrophobic residues.
(C) Cell localization images (left) and intensity plots (right) of control LK-CFP with YFP-SOAR dimer co-expressed in HEK-Orai1/2/3TKO cells. (AUs, arbitrary units).
(D–F) Cells as in (C) but co-expressing LK-CFP-O1M4x (containing the Orai1 267-301 M4x peptide) (D), LK-CFP-O2M4x (containing the Orai2 228-254 M4x peptide) (E), or LK-CFP-O3M4x (containing the Orai3 276-295 M4x peptide) (F), each with YFP-SOAR dimer.
(G) E-FRET measurements for the interaction of YFP-SOAR dimer with LK-CFP-Orai1-M4x peptide, containing either wild-type Orai1-M4x (O1-WT); Orai1-M4x with the L273D, L276D, or combined L273D/L276D mutations; or the control (LK-CFP).
(H) E-FRET between YFP-SOAR dimer and LK-CFP-Orai2-M4x peptide, with either WT Orai2-M4x (O2-WT), Orai2-M4x-I234D (O2-I234D), or the control (LK-CFP).
(I) E-FRET between YFP-SOAR dimer and LK-CFP-Orai3-M4x peptide, with either WT Orai3-M4x (O3-WT), Orai3-M4x-L282D (O3-L282D), Orai3-M4x-L285D (O3-L285D), Orai3-M4x-L282D/L285D double mutant (O3-L282D/L285D), or the control (LK-CFP).
(J) Cells as in (C) but co-expressing LK-CFP-O3M4x-L285D mutant with YFP-SOAR dimer. Images and intensity plots are representative of three independently repeated experiments. Scale bars represent 5 μm. One-way ANOVA analysis was performed on E-FRET results (**p < 0.01, ***p < 0.001, ****p < 0.0001). Results are means ± SEM, representative of at least three independent experiments. E-FRET analyses were on cells expressing a narrow range of LK-CFP-Orai-M4x fluorescence to assure accuracy of E-FRET values. The CFP levels are shown in Figure S3.
Given this controversy, we evaluated the role of the Orai C terminus by engineering fluorescently tagged, PM-tethered M4x peptides from each Orai channel subtype. Remarkably, the M4x peptide constructs undergo a Leu-specific signature of direct interaction with concatenated SOAR dimers or whole STIM1 proteins mimicking intact Orai channels. The M4x Leu residues, critical for Orai1 activation, mediate direct STIM1 binding and not peptide self-association, militating against the model of dimerized M4x peptides creating a STIM1-binding pocket. Interestingly, we reveal that the tethered Orai3-M4x peptide is a powerful in situ cellular blocker of STIM-Orai coupling, allowing us to define the crucial role of Orai channels in mediating receptor-driven Ca2+ oscillatory responses and their exclusive requirement in downstream transcriptional control through NFAT activation. Our studies reveal the critical role of the short, highly specific Orai channel M4x helices in defining the coupling interface between STIM and Orai proteins. These peptides may provide molecular templates for the design of specific coupling modifiers with potentially high therapeutic value.
RESULTS
The PM-tethered M4x peptides recruit SOAR dimers from the cytosol
Our studies focused on the C-terminal M4x peptides of Orai channels strongly implicated in STIM protein coupling (Muik et al., 2008; Park et al., 2009; Prakriya and Lewis, 2015; Yeung et al., 2020; Zhou et al., 2019). Crystal structures suggested the M4x helices in adjacent dOrai subunits are dimers linked through symmetrical, antiparallel interactions between I316 and L319 residues (Hou et al., 2012), giving the hexameric channel 3-fold outer symmetry (Figure S1A). We addressed whether the M4x helices of Orai channels must interact as dimers to form the active STIM-Orai complex. We created constructs in which cyan fluorescent protein (CFP)-labeled M4x peptides from Orai1, Orai2, or Orai3 (Figure S1B) were linked to Lyn kinase that becomes fatty acylated and strongly anchored to the PM (Figure S1C; Inoue et al., 2005). These M4x constructs were co-expressed with yellow fluorescent protein (YFP)-tagged concatenated SOAR dimers (Wang et al., 2009, 2014; Figures 1A and 1B) in HEK cell lines in which expression of all three Orai channels was eliminated by CRISPR-Cas9 gene editing (HEK-Orai1/2/3TKO cells) (Yoast et al., 2020; Zhou et al., 2018a). This assured that YFP-SOAR did not undergo association with endogenous Orai proteins. In controls using LK-CFP without any Orai-derived sequence, there was no localization of the YFP-SOAR dimer to the PM (Figure 1C). In contrast, expression of the Orai1-M4x construct (LK-CFP-O1M4x) resulted in YFP-SOAR dimer localization to the PM (Figure 1D). The corresponding Orai2-M4x and Orai3-M4x constructs gave stronger YFP-SOAR dimer co-localization (Figures 1E and 1F), with little remaining in the cytosol.
A specific Leu signature defines the M4x-peptide-SOAR interaction
We undertook E-FRET analysis using the LK-CFP-Orai-M4x constructs derived from Orai1, Orai2, or Orai3, comparing E-FRET with the YFP-SOAR dimer derived from STIM1 co-expressed in HEK-Orai1/2/3TKO cells. We mutated the two leucines shown in dOrai to be mediating interconnections between the C-terminal M4x peptides (Figures S1A and S1B; Hou et al., 2012). For the LK-CFP-O1M4x construct, E-FRET with YFP-SOAR was relatively small but was reduced with the L273D Orai1-M4x mutant and further reduced with the L276D mutant (Figure 1G). The L273D/L276D double mutant reduced E-FRET down to the background E-FRET observed with LK-CFP lacking the M4x peptide. Using the LK-CFP-O2M4x construct, we found that the E-FRET value with YFP-SOAR was higher than that of the Orai1 construct and was much reduced with the I234D mutation (Figure 1H). The E-FRET value for LK-CFP-O3M4x with YFP-SOAR was much higher than that for the Orai1- or Orai2-derived constructs (Figure 1I). The robust E-FRET with the Orai3 peptide was reduced >50% with the L282D mutation and completely blocked to background levels with either the L285D or the L282D/L285D mutations. Imaging revealed a corresponding complete reduction in co-localization of LK-CFP-O3M4x-L285D with YFP-SOAR dimer (Figure 1J). The high E-FRET value for the Orai3-M4x peptide interaction with SOAR indicated a much more robust binding than that of Orai1 or Orai2. We were surprised that the short 20-amino-acid (aa) Orai3-M4x peptide had a much higher apparent affinity for SOAR than the 35-aa Orai1 peptide. Also interesting was the consistency of the Leu mutational profiles for association of the Orai1 and Orai3 M4x peptides with SOAR. Clearly, the first Leu mutation in either Orai1-M4x or Orai3-M4x (L273 or L282, respectively) was less effective, and the second Leu mutation (L276 or L285, respectively) was more effective. Indeed, the L285D mutation almost completely eliminated Orai3-M4x binding to SOAR and the double L282D/L285D mutation was little more effective. The avidity of Orai3-M4x peptide binding and the clear Leu signature of its interaction with SOAR allowed us to study the role of the M4x peptide.
Mutation of these Leu residues to Ser in the M4x region of intact Orai channels was shown earlier to reduce their STIM1 coupling, which was thought to be due to perturbation of M4x helicity (Frischauf et al., 2009; Muik et al., 2008). We examined this finding further by using LK-CFP-O3M4x, substituting the L285 residue with other hydrophobic residues not expected to destabilize helicity. Substitution of Leu-285 with the aromatic hydrophobic residues Phe or Tyr substantially reduced E-FRET with YFP-SOAR (Figure 2A). Met substitution was a little less effective at reducing E-FRET. Thus, mutation of L285 to other helical-preserving hydrophobic residues still largely diminished STIM1 interaction with Orai3-M4x. Thus, the loss of the STIM1 interaction by L285 mutation is not due to helical disruption but instead indicates that the Leu residue plays an important allosteric role in STIM1 binding to the M4x peptide.
Figure 2. Defining the Leu signature and mechanistic role of the Orai3 M4x peptide interaction with SOAR dimer.

E-FRET analysis of transiently co-expressed LK-CFP-Orai3-M4x peptide with the YFP-SOAR dimer in HEK-Orai1/2/3TKO cells.
(A) E-FRET between YFP-SOAR and LK-CFP-Orai-M4x peptide, with either O3-WT, O3-L285D, Orai3-M4x-L285M (O3-L285M), Orai3-M4x-L285F (O3-L285F), Orai3-M4x-L285Y (O3-L285Y), or the control (LK-CFP) with no M4x peptide.
(B) E-FRET between YFP-SOAR and LK-CFP-Orai-M4x peptide, with either O3-WT, O3-L282D, O3-L285D, Orai3-M4x-L288D (O3-L288D), or the control (LK-CFP).
(C) Predicted structure of the hOrai3-M4x peptide to show positions of the four Leu residues (L282, L285, L288, and L292) mutated in (B).
(D and E) Measurement of self-interactions between M4x peptides by E-FRET-analysis between CFP- and YFP-tagged M4x peptides in HEK-Orai1/2/3TKO cells. E-FRET between LK-YFP-O1M4x and either LK-CFP-O1M4x WT (teal), LK-CFP-O1M4x-L273D/L276D double mutant (magenta), or LK-CFP control (black) (D). E-FRET measurements between LK-YFP-O3M4x and either LK-CFP-OM4x WT (green), LK-CFP-O3M4x-L282D/L285D double mutant (purple), or LK-CFP control (black) (E). E-FRET analyses for (E) and (D) were undertaken on cells expressing a narrow range of both LK-CFP-Orai-M4x and LK-YFP-Orai-M4x fluorescence levels to assure accuracy of E-FRET values. The CFP and YFP levels of cells are shown in Figure S4.
(F) E-FRET interaction between YFP-SOAR dimer and full-length CFP-Orai3 WT (green), CFP-Orai3-L282D (red), CFP-Orai3-L285D (blue), or CFP-Orai3-L282D/L285D double mutation (purple). Orai constructs and YFP-SOAR dimer were transiently expressed in HEK-Orai1/2/3TKO cells.
One-way ANOVA analysis was performed on E-FRET results (****p < 0.0001). Results are means ± SEM of at least three independent experiments.
The L282/L285 Leu pair in the Orai3 M4x peptide is followed by a second Leu pair, L288/L292, creating an amphipathic helix with all four leucines aligned adjacently on the helix (Figures 2C). We compared the mutation of each of the four Leu residues to aspartates (L282D, L285D, L288D, and L292D, respectively) on the ability of LK-CFP-Orai3-M4x to undergo E-FRET with YFP-SOAR dimer (Figure 2B). Mutation of each of the second Leu pair (L288D or L292D) substantially reduced E-FRET with YFP-SOAR. L288D was more effective than L292D but neither were as effective as the L285D mutation. Thus, all four Leu residues contribute to SOAR binding but the middle two leucines are more significant. L285 is the most critical, and its replacement with a charged residue eliminates SOAR binding.
These results reveal the critical STIM-binding role of the small hydrophobic Leu residues aligned along the Orai3-M4x helix. The 7-aa longer Orai2-M4x peptide has a similar Leu distribution but lower E-FRET with SOAR. The Orai1-M4x has the lowest E-FRET with SOAR and has an insertion of three residues between the first and second Leu pairs (Figure 1B). For all three Orai subtypes, the second Leu residue in the M4x is most critical for a SOAR interaction. The intervening residues between the leucines are mostly charged or polar; thus, the peptides are strongly amphipathic and likely critical to their STIM1 interactions.
Do M4x peptides undergo interactions to form an Orai channel binding pocket?
Based on earlier structural data for M4x peptide interactions with STIM (Fahrner et al., 2014; Maus et al., 2015; Stathopulos et al., 2013; Yen and Lewis, 2018; Zhou et al., 2016), we considered the model that two M4x peptides might combine to form a SOAR binding pocket. We therefore investigated self-association between M4x peptides. We made both CFP- and YFP-labeled LK-Orai3-M4x and LK-Orai1-M4x constructs and performed E-FRET to analyze potential inter-dimer formation. We focused on whether mutations in the critical Leu residues in Orai1-M4x (L273 and L276) or Orai3-M4x (L282 and L285) had effects on inter-dimer formation. We first compared E-FRET between LK-YFP-O1M4x and either wild-type (WT) LK-CFP-O1M4x, the double L273D/L276D mutant of LK-CFP-O1M4x, or LK-CFP without the M4x peptide as a negative control (Figure 2D). Strikingly, we found no significant differences in E-FRET association between any of the Orai1-M4x constructs nor any differences from background E-FRET levels with the control LK-CFP. Similarly, using Orai3-M4x constructs or their double mutant counterparts (L282D/L285D), we detected no inter-dimer interactions for Orai3-M4x (Figure 2E). We also checked whether dimerization of GFP-based tags (Zacharias et al., 2002) might interfere with M4x peptide interactions. Using non-dimerizing A206K mutants of CFP and YFP in our M4x and SOAR dimer constructs, we found that E-FRET measurements on either M4x-M4x or M4x-SOAR interactions were not discernibly different (Figure S2). Thus, we have no evidence that the M4x peptides undergo direct interactions with each other. Although we cannot rule out weak M4x-M4x interactions within resting Orai channels, M4x peptides clearly undergo strong interactions with SOAR, with a clear and consistent Leu signature, conserved across Orai channel subtypes. Our results reveal the aligned Leu residues are critical for mediating direct STIM1 interactions and do not support earlier models (Fahrner et al., 2014; Maus et al., 2015; Stathopulos et al., 2013; Zhou et al., 2016) that Leu-mediated crosslinking of M4x peptides forms a STIM-binding pocket.
The M4x peptide mimics the intact Orai3 channel in binding to SOAR
Key was whether binding M4x peptide constructs with SOAR reflected the binding properties of intact Orai channels. The robust interaction and clear Leu signature of the Orai3-M4x peptide provided a powerful means with which to assess this question. We co-expressed full-length N-labeled CFP-Orai3 constructs in HEK-Orai1/2/3TKO cells with the YFP-SOAR dimer (Figure 2F). E-FRET between WT CFP-Orai3 and YFP-SOAR was robust and considerably higher than that observed for WT CFP-Orai1 (Zhou et al., 2018a). The L282D mutation in CFP-Orai3 almost halved E-FRET, and the L285D mutation almost completely abolished E-FRET with SOAR, as did the mutation of both leucines. These results reveal that the Leu signature of SOAR binding to the full-length Orai3 channel is almost identical to its isolated M4x peptide (Figure 2F). Thus, the M4x peptide is a predominant determinant of Orai3 binding to SOAR.
Store-activated full-length STIM1 recruits the Orai3 M4x peptide into ER-PM junctions
We questioned whether full-length STIM1 would show the same Leu binding signature for the M4x peptide as the SOAR dimer. We assessed the time course of the E-FRET interaction of LK-CFP-O3M4x with full-length STIM1-YFP following ionomycin-induced store depletion in HEK-Orai1/2/3TKO cells. WT LK-CFP-O3M4x underwent a rapid and strong E-FRET interaction with STIM1-YFP (Figures 3A and 3B). This E-FRET was reduced by more than one-third by using the L282D-mutated LK-CFP-O3M4x. The L285D-mutated LK-CFP-O3M4x had substantially lower E-FRET, and this finding was almost the same seen with the L282D/L285D mutation. Thus, the M4x Leu signature for STIM1 is highly consistent with that for SOAR. Important to assess was whether the PM-tethered M4x peptide co-localized with STIM1 in ER-PM junctions. Using HEK-Orai1/2/3TKO cells expressing STIM1-YFP, we used high-resolution imaging to compare colocalization of LK-CFP-O3M4x-WT (Figure 3C) and LK-CFP-O3M4x-L285D (Figure 3D), following store depletion. Deconvolved planar images of the PM adjacent to the coverslip revealed that the WT-peptide construct (red) co-localized into the same junctional areas as STIM1-YFP (green), giving a clearly defined coincidence in the merged image. In contrast, the L285D single-point-mutated peptide remained randomly distributed in the PM, with no visible STIM1 colocalization. Thus, the M4x peptide localizes into junctions with STIM1 analogously to the intact Orai channel and with the same M4x Leu dependence as with E-FRET. The lack of endogenous Orai channels indicates that the M4x domain is sufficient for recruitment into STIM-mediated junctions.
Figure 3. Store-activated full-length STIM1 recruits the Orai3 M4x peptide into ER-PM junctions.
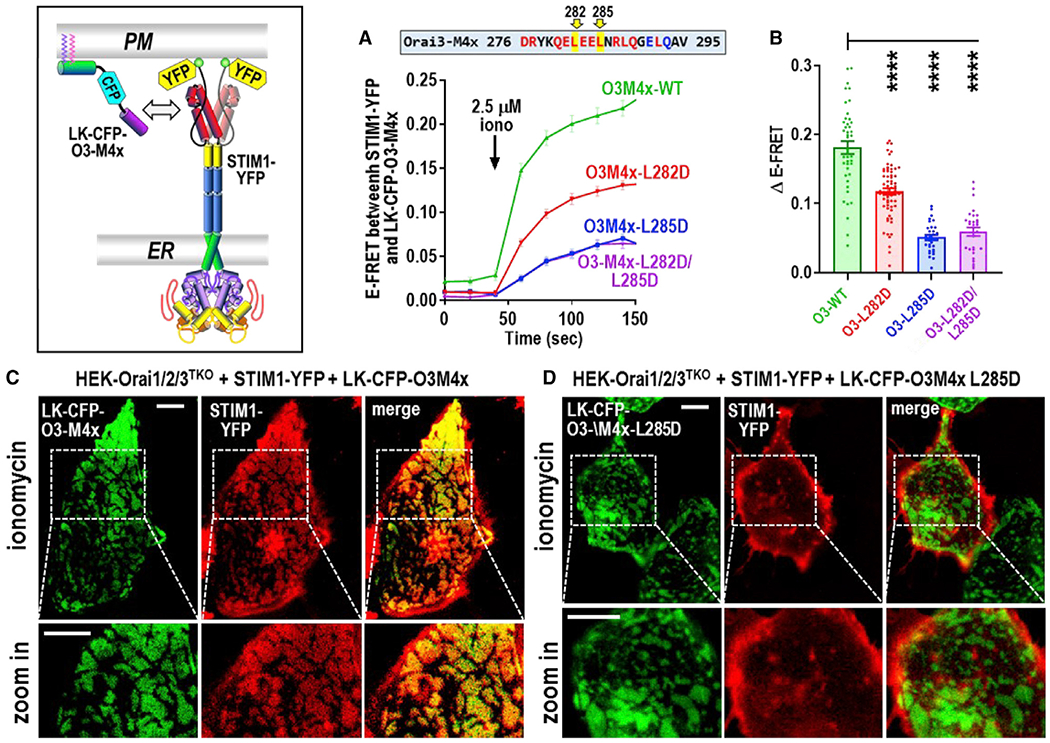
(A) Time course of E-FRET interactions between transiently expressed STIM1-YFP with LK-CFP-O3M4x constructs (either WT M4x, single L282D or L285D M4x mutants, or the L282D/L285D double mutant) in HEK-Orai1/2/3TKO cells. Store depletion was initiated by 2.5 μM ionomycin.
(B) Summary data for experiments in (A) showing the change in E-FRET from baseline (0 s) to peak after ionomycin (140 s).
(C and D) High-resolution fluorescence imaging of transiently expressed LK-CFP-O3M4x constructs with STIM1-YFP in ER-PM junctions measured at the PM layer adjacent to the coverslip in HEK-Orai1/2/3TKO cells. Images were taken 5 min after 2.5 μM ionomycin addition to deplete Ca2+ stores. In (C), LK-CFP-O3M4x WT peptide was highly localized with STIM1-YFP, and image magnification reveals a complete overlap of fluorescence in the punctal areas. In contrast, LK-CFP-O3M4x bearing the single M4x L285D point mutation (D) showed no obvious overlap with STIM1-YFP fluorescence, and magnified STIM1 punctal areas reveal no co-localized M4x construct. Images are representative of at least three independent experiments with 5-μm scale bars. One-way ANOVA was performed on E-FRET results (****p < 0.0001). Results are means ± SEM of at least three independent experiments.
The M4x Leu signature translates into functional Orai3 channel activation
To complete our assessment of the role of the Orai3-M4x peptide, we examined the Leu dependence of the interaction of full-length Orai3 with full-length STIM1 (Figures 4A and 4B). Compared to WT Orai3, the E-FRET for the Orai3-L282D mutant was reduced by almost 50%, and the Orai3-L285D and Orai3-L282D/285D mutants were reduced to almost background levels. Thus, this Leu specificity reveals that the M4x peptide of Orai3 is the defining site mediating the STIM1 interaction. Although the M4x peptide defines tethering of Orai3 to STIM1, we questioned whether this defines the allosteric trigger for channel activation. Ca2+-imaging experiments using fura-2-loaded HEK-Orai1/2/3TKO cells expressing STIM1 and Orai3-WT revealed robust SOCE following store depletion. This SOCE was greatly reduced with the Orai3-L282D mutant and almost abolished with the Orai3-L285D or Orai3-L282D/L285D mutants (Figures 4C and 4D). Measuring Orai3-mediated currents, the same Orai3 species gave similar results, but differences between the mutant Orai3-mediated currents were too small to be resolved (Figure 4E). The similar Leu mutation profile for Orai3-mediated Ca2+ entry and E-FRET for Orai3-STIM1 or Orai3-M4x-STIM1 interactions together reveal the same crucial differential dependence on the L282 and L285 residues. Thus, the M4x peptide defines the Orai3-STIM1 coupling interface that initiates Orai3 channel gating.
Figure 4. The Orai3 M4x Leu profile reveals the peptide mediates STIM1-induced Orai3 channel activation.
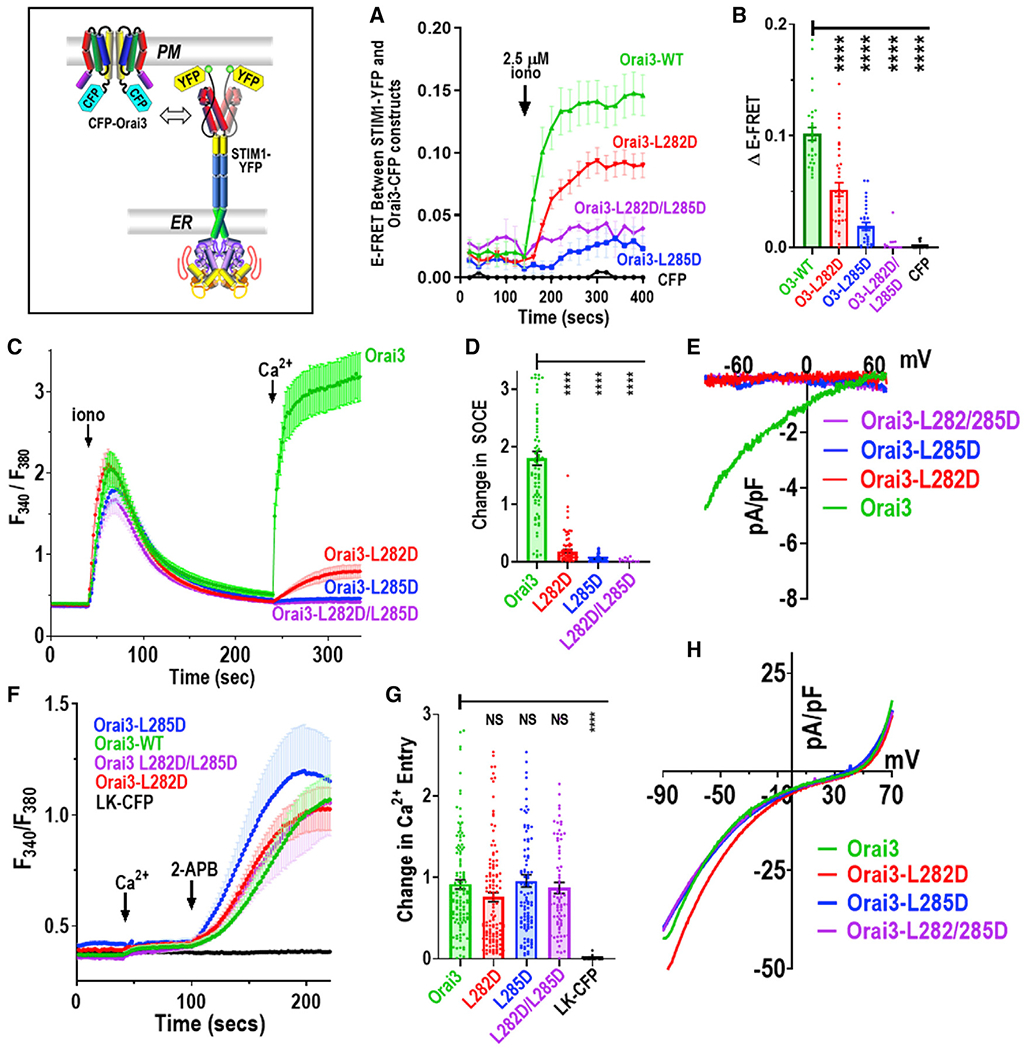
(A) Time course of E-FRET interactions between STIM1-YFP co-expressed with either full-length WT CFP-Orai3, CFP-Orai3 L282D or L285D single mutants, CFP-Orai3 L282D/L285D double mutant, or CFP control, before and after store depletion with 2.5 μM ionomycin.
(B) Summary data for E-FRET analyses in (A), showing the change in E-FRET from baseline (0 s) to the peak after ionomycin addition (400 s).
(C) Cytosolic Ca2+ signals measured by fura-2 ratiometric Ca2+ imaging in HEK-Orai1/2/3TKO transiently expressing STIM1-mCherry together with either WT CFP-Orai3 (green) or L282D (red), L285D (blue), or L282D/L285D (purple) mutants of CFP-Orai3 (purple). Cells in Ca2+-free medium were treated with 2.5 μM ionomycin followed by 1 mM Ca2+ add back.
(D) Summary statistics for the average peak of store-operated Ca2+ entry in (C).
(E) I/V relationship of whole-cell ICRAC measurements in HEK-Orai1/2/3TKO cells transfected with the same CFP-Orai3 constructs shown in (C) together with STIM1-YFP.
(F) Ca2+ levels in fura2-loaded HEK-Orai1/2/3TKO cells transiently expressing either WT CFP-Orai3 (green); L282D (red), L285D (blue), or L282D/L285D (purple) mutants; or negative control LK-CFP (black). Constitutive Ca2+ entry and 2-APB responses were in Ca2+-free solution, replaced with 1 mM Ca2+ followed by 50 μM 2-APB, as shown.
(G) Summary statistics for the 2-APB-induced Ca2+ entry peak in the experiments in (F).
(H) I/V relationship of 2-APB-induced whole-cell current measurements for HEK-Orai1/2/3TKO cells transfected with the same CFP-Orai3 constructs shown in (F). One-way ANOVA analyses were under taken on results in (B), (D), and (G). Ca2+ entry shown are means ± SEM (****p < 0.0001) and representative results from at least three independent experiments.
Direct Orai3 channel opening by 2-APB is independent of the M4x Leu profile
Orai3 is unique among Orai channels in being directly activated by 2-aminoethoxydiphenyl borate (2-APB) (DeHaven et al., 2008; Lis et al., 2007; Peinelt et al., 2008; Schindl et al., 2008; Zhang et al., 2008), an effect independent of stores. We examined the effects of M4x mutations on 2-APB-induced Orai3 activation. In fura-2-loaded HEK-Orai1/2/3TKO cells, 2-APB-induced Ca2+ entry activated by Orai3-CFP L282D, L285D, and L282D/L285D mutations was not significantly different from that of Orai3-WT (Figures 4F and 4G). The action of 2-APB on Orai3 reflects an interesting permissive effect on the pore (Amcheslavsky et al., 2013; Peinelt et al., 2008; Schindl et al., 2008; Zhang et al., 2008), and our patch clamp measurements reveal the typical I/V profile of altered ion selectivity and significant outward current induced by 2-APB (Figure 4H). The M4x mutations do not alter the current density or I/V profile mediated by 2-APB, and thus, the M4x peptides do not mediate 2-APB-induced opening of the Orai3 pore.
The Orai3 M4x peptide is a powerful SOCE modulator and probe for physiological downstream transcriptional control
The high affinity of the Orai3-M4x peptide led us to investigate how it might modulate endogenous SOCE when expressed in HEK cells. Transfection of LK-CFP-O3M4x almost completely blocked native SOCE compared to the control LK-CFP construct (Figures 5A and 5C). In contrast, the lower affinity LK-CFP-O1M4x had little effect. The L285D point mutation in the Orai3-L285D M4x peptide completely prevented its SOCE-blocking action (Figures 5B and 5C). In Jurkat T cells, which have high endogenous SOCE levels, the Orai3 M4x peptide was similarly effective (Figure S5). Thus, the Orai3 M4x peptide is a strong and specific competitive inhibitor of endogenous STIM-Orai coupling to mediate SOCE and its downstream role. In HeLa cells stably expressing GFP-NFAT1, transfected Orai3-M4x peptide blocked >80% of thapsigargin-induced SOCE (Figure S6). In cells expressing the M4x peptide, nuclear translocation of GFP-NFAT1 was suppressed by 80%, whereas adjacent cells either without an expressed peptide or expressing the M4x-L285D mutant peptide had full GFP-NFAT nuclear translocation.
Figure 5. The Orai3 M4x peptide is a powerful SOCE blocker and defines the physiological role of Orai channels in Ca2+ oscillation generation and downstream NFAT1 activation.

(A) Endogenous SOCE in WT HEK293 cells transiently expressing LK-CFP-O3M4x (red), LK-CFP-O1M4x (green), or LK-CFP control (black).
(B) SOCE as in (A) in cells transiently expressing LK-CFP-O3M4x (red), LK-CFP-O3M4x-L285D (blue), or LK-CFP control (black).
(C) Summary statistics of endogenous SOCE responses shown in (A) and (B).
(D) Receptor-activated Ca2+ responses induced by 100 μM carbachol (CCh) measured in fura-2-loaded WT HEK293 cells transiently expressing either LK-CFP-O3M4x (red), LK-CFP-O3M4x-L285D mutant (blue), or LK-CFP control (black).
(E–G) Representative single-cell Ca2+ responses to CCh from each of the traces shown in (D) and the fraction of oscillating (OSC) and non-oscillating (NO) cells unde reach condition. Cells were transfected with LK-CFP(77.5% OSC, 22.5 NO; 44 cells) (E), LK-O3M4x-WT (19.7% OSC, 80.3% NO; 71 cells) (F), and LK-CFP-O3M4x-L285D (64.4% OSC, 35.6 NO; 59 cells) (G).
(H–J) Time-dependent nuclear localization of NFAT1 in WT HEK293 cells transiently expressing NFAT1-GFP together with either LK-CFP control (H), LK-CFP-O3M4x (I), or LK-CFP-O3M4x-L285D mutant (J). Images of CFP, CFP/GFP, and GFP were taken before CCh (0 min) and at 3, 5, 10, and 15min after 100 μM CCh addition. Fluorescence of LK-CFP constructs and NFAT1-GFP are shown in red and green, respectively.
(K) Summary of NFAT1-GFP translocation imaged in cells co-expressing LK-CFP (black), LK-CFP-O3M4x-WT (red), and LK-CFP-O3M4x-L285D (blue). Nuclear/cytosolic GFP-NFAT1 intensity ratios were measured in imaged cells treated as in (H)—(J) by using images taken before and after CCh addition, at the times shown. Oscillating cells were defined as having >4 peaks/10 min after initial Ca2+ release (400 s), with each peak at >0.03 F340/F380. One-way ANOVA analyses were undertaken on results in (C). Ca2+ entry results shown are means ± SEM (****p < 0.0001). All results shown are representative of at least three independent experiments. Scale bars represent 10 μm.
The Orai3-M4x peptide provided a powerful means with which to probe the physiological role of receptor-activated SOCE. Receptors induce InsP3R-mediated Ca2+ release and subsequent Ca2+ oscillations, which are considered to require a combination of store release and SOCE. Subsequent NFAT1 nuclear translocation is thought to depend on SOCE, but without specific channel blockers, the contribution of Orai channels remains uncertain. Orai channel knockout can lead to uncertain pleiotropic effects on Ca2+ homeostasis and ER-PM junction formation. Expression of the PM-tethered Orai3-M4x peptide in WT-HEK cells completely blocked the longer-term muscarinic-receptor-induced Ca2+ response following the large initial Ca2+ release phase (Figure 5D). In single cells expressing control LK-CFP, the longer-term Ca2+ responses were clearly oscillatory (Figure 5E). LK-M4x expression eliminated these oscillations (Figure 5F), whereas with LK-M4x-L285D, oscillations were retained (Figure 5G). The onset of oscillations after 5 min correlates with nuclear NFAT1-GFP translocation in control cells (Figure 5H). The M4x-mediated elimination of Ca2+ oscillations completely blocked GFP-NFAT1 nuclear translocation (Figure 5I), whereas the L285D mutant was without effect. These striking results reveal the essential role of Orai channels in generating physiological receptor-induced Ca2+ oscillations and thereafter the critical downstream activation of transcription through nuclear translocation of NFAT.
The Orai3 M4x avidity for STIM1 overcomes a powerful STIM1 coupling mutant
We previously defined a critical activating locus in the SOAR domain of STIM1, the F394 residue (Wang et al., 2014; Zhou et al., 2015, 2016). The F394H mutation abolishes coupling and activation of Orai1. We wondered if the inherently strong Orai3-STIM1 interaction was similarly sensitive to this SOAR mutation. Using STIM null HEK-STIM1/2DKO cells, we transiently co-expressed CFP-Orai3 with either the WT YFP-SOAR1 dimer, the YFP-SOAR-F394H homodimer, or YFP as a control (Figures 6A and 6B). Unexpectedly, the F394H mutation in SOAR reduced but did not eliminate activation of Orai3-mediated Ca2+ entry. We examined the YFP-SOAR-F394H mutation further by measuring E-FRET with the LK-CFP-Orai3-M4x peptide (Figure 6C). Compared to the WT-SOAR dimer, E-FRET of the SOAR-F394H homodimer with the M4x peptide was reduced but still measurable. The M4x-L285D mutated peptide reduced E-FRET to background levels with both WT-SOAR and F394H-SOAR dimers. The similar SOAR mutant specificity of Orai3 channel activation and Orai3-M4x binding indicates that the M4x peptide in Orai3 defines the coupling with STIM1 that mediates Orai3 channel activation. Thus, the more extreme STIM1-binding affinity of the Orai3-M4x peptide partially overcomes the F394H mutation that completely ablates Orai1 channel activation. This finding indicates again that the functional coupling between Orai3 and STIM1 is defined by the Orai3 M4x peptide.
Figure 6. The Orai3 M4x peptide overcomes a powerful STIM1 coupling mutant and mediates an important action of 2-APB.

(A) Constitutive Ca2+ entry in fura-2-loaded HEK-STIM1/2DKO cells transiently expressing CFP-Orai3 with either WT YFP-SOAR dimer (red), the YFP-SOAR-F394H homodimer mutant (blue), or YFP vector alone (black). A total of 1 mM Ca2+ was added to cells in Ca2+-free solution, as shown.
(B) Summary statistics for the Ca2+ entry shown in (A).
(C) E-FRET analysis of HEK293 cells transiently expressing either WT YFP-SOAR dimer (red) or the YFP-SOAR-F394H homodimer mutant (blue), together with either WT LK-CFP-O3M4x or its L285D mutant.
(D) SOCE in fura-2-loaded HEK cells transiently expressing CFP-Orai3 with either WT STIMI-YFP (red), the STIM1-YFP-F394H homodimer mutant (blue), or YFP control (black). Store depletion was with 2.5 μM ionomycin, followed by 1 mM Ca2+ addback, and 50 μM 2-APB, as shown.
(E) Enlarged detail for the 2-APB-mediated Ca2+ responses shown in (D) in cells expressing either STIM1-YFP-F394H or YFP control.
(F) Summary statistics for Ca2+ entry experiments represented in (D).
(G) Time course E-FRET measurements for HEK-Orai1/2/3TKO cells transiently co-expressing CFP-Orai3 with either WT STIM1-YFP (red) or STIM1-YFP F394H mutant (blue). Additions of 2.5 μM ionomycin and 50 μM 2-APB were as shown.
(H) Average change in E-FRET from baseline (before Ca2+) to store-depleted peak (iono), and additional E-FRET peak change after the 2-APB addition, from experiments in (G).
(I) E-FRET time course measured in HEK-Orai1/2/3TKO cells transiently co-expressing the Orai3-M4x peptide with either STIM1-YFP (red) or STIM1-YFP-F394H mutant (blue). Additions of 2.5 μM ionomycin and 50 μM 2-APB were as shown.
(J) Summary statistics for E-FRET results shown in (I). Changes in FRET are shown from baseline (Ca2+-free) to store-depleted peak (iono) and additional FRET peak after 2-APB.
One-way ANOVA analysis was performed in (B) and (F). Ca2+ entry results are means ± SEM (****p < 0.0001). Results are representative of at least three independent experiments
The M4x peptide mediates an intriguing pharmacological parameter
We investigated the role of the Orai3 M4x peptide in an intriguing action of 2-APB on the STIM1-Orai3 interaction, an effect distinct from its permissive action on the Orai3 pore described above. We examined the actions of full-length STIM1-WT or the STIM1-F394H mutant on SOCE and E-FRET in HEK-STIM1/2DKO cells transiently expressing CFP-Orai3 (Figures 6D–6F). As with the SOAR dimer, STIM1-mediated SOCE was greatly reduced by using the STIM1-F394H mutant. Addition of 2-APB induced a rapid increase in Ca2+ entry with STIM1-F394H, bringing it to slightly above the level with STIM1-WT. Also, 2-APB induced a small enhancement of the already large SOCE with STIM1-WT. Control cells not expressing STIM1 showed no SOCE (Figure 6D, black line), but 2-APB still induced a large Ca2+ entry. The latter effect is much slower (see expanded timescale, Figure 6E) and represents the store-independent, direct action of 2-APB on Orai3 described above (Figure 5D). The rapid action of 2-APB to reverse the inhibitory effect of the STIM1-F394H mutant on Orai3 (Figure 6E) is consistent with earlier results (Wang et al., 2014). This 2-APB action was further characterized by measuring E-FRET between STIM1-YFP and CFP-Orai3 (Figures 6G and 6H). Store depletion induced a rapid and robust STIM1-WT/Orai3 interaction. The Orai3 interaction was substantially reduced with the STIM1-F394H mutant, consistent with the effect on Ca2+ entry. Importantly, the E-FRET between STIM1-F394H-YFP and CFP-Orai3 was rapidly (albeit partially) restored by 2-APB. Thus, as for Orai1 (Wang et al., 2014), the action of 2-APB is to restore not only the defective binding seen between the STIM1-F394H and Orai3 but also coupling to activate the channel.
These results prompted some key questions on the site of action of 2-APB. Is it targeting the SOAR-M4x interface to reverse the critical Orai-binding defect on the STIM1 protein? Or, is this effect a manifestation of 2-APB binding to a downstream site within the channel, perhaps mediating the Orai3 opening? Indeed, the 2-APB activating site on Orai3 may be the cytoplasmic 2-3 loop (Amcheslavsky et al., 2013, 2014; Zhang et al., 2008) that is close to the C-terminal M4x helix in the dOrai structure (Hou et al., 2012). Our experiments using the Orai3 M4x peptide give a clear answer to this question. Thus, surprisingly, the M4x helix of Orai3 exactly recapitulates the 2-APB-mediated recovery of Orai3 binding to the mutated STIM1 (Figures 6I and 6J). We measured E-FRET between LK-CFP-O3-M4x and full-length STIM1-YFP co-expressed in HEK-Orai1/2/3TKO cells. Store depletion resulted in a rapid and large increase in E-FRET between WT STIM1-YFP and LK-CFP-O3-M4x and little change in E-FRET with 2-APB. Using STIM1-F394H-YFP, we found that the increase in E-FRET following store depletion was greatly reduced, but addition of 2-APB induced an almost complete recovery of E-FRET. Thus, the pattern of these M4x results (Figure 6I) is strikingly similar to that for full-length Orai3 (Figure 6G). From this finding, we conclude that 2-APB is working directly on the M4x-STIM1 interaction.
The profound action(s) of 2-APB on SOCE has been the subject of much interest and confusion for many years (Amcheslavsky et al., 2013, 2014; DeHaven et al., 2008; Lis et al., 2007; Peinelt et al., 2008; Schindl et al., 2008; Wang et al., 2014; Zhang et al., 2008). Whether its actions are mediated by distinct targets on Orai channels and STIM proteins is unresolved. We show unequivocally that 2-APB activates Orai3 in the absence of STIM1 and STIM2 (Figure 5D) and that Orai3 M4x mutations preventing any STIM binding have no effect on this activation. Thus, there is clearly a direct, slow-onset activating action of 2-APB on Orai3 itself. The M4x peptide has allowed us to define a very different action of 2-APB to rapidly target the interaction between STIM1 and Orai3, allowing the coupling-defective STIM1 mutant to both engage with and activate Orai3. This fast action is also seen with Orai1 (Wang et al., 2014). This is yet further evidence that the SOAR-M4x interaction is a localized, avid, and critical step in the STIM-Orai pathway and one that is pharmacologically targeted by the small 2-APB molecule.
DISCUSSION
Our results provide new understanding on the STIM-Orai coupling interface, revealing that the short C-terminal M4x peptides of Orai channels define the critical interaction with STIM proteins that triggers channel gating. The STIM-Orai interface involves two surprisingly small domains, namely, the 99-aa SOAR domain in STIM proteins and the 20- to 35-aa M4x peptides in human Orai channels. The SOAR unit in STIM1 remains as a tightly folded dimer with two independent Orai binding sites (Zhou et al., 2015). The M4x peptides in adjacent Orai subunits have been thought to undergo pairing to create a STIM-binding site (Fahrner et al., 2014; Maus et al., 2015; Stathopulos et al., 2013; Yen and Lewis, 2018; Zhou et al., 2016), but our results indicate they interact independently with STIM1 as monomers. Indeed, we reveal that the critical leucines thought to mediate M4x dimerization are instead mediating the M4x-SOAR interaction itself. Most significantly, the observed signature for the requirements of the highly conserved M4x Leu residues provides compelling evidence for the critical role of the M4x peptides. This signature is faithfully preserved in both FRET binding and functional channel activation measurements by using all combinations of M4x peptides or intact Orai channels together with SOAR dimers or whole STIM1 proteins. Moreover, we show that the Orai3-M4x peptide associates avidly with STIM1 within ER-PM junctions, functions as a competitive blocker of native SOCE signals, provides a powerful probe to reveal the physiological role of Orai channels in Ca2+ oscillations and downstream transcriptional control, and mimics a key pharmacological modification of the STIM-Orai interaction induced by 2-APB.
The M4x helical peptides of Orai channels have a highly conserved pattern of repeating hydrophobic residues (Figure 7A). In both Orai2 and Orai3, four Leu/Ile residues are interposed by pairs or triplets of polar or charged residues forming an amphipathic helix (Figure 2C) that binds strongly with the SOAR dimer. In the Orai1 M4x, the pattern of four repeating leucines is interrupted by three additional residues giving an extra helical turn that separates the leucines into two pairs, disrupting the otherwise highly amphipathic nature of the M4x region and likely accounting for the lower affinity of the Orai1 M4x peptide. Interestingly, a recent study using very different in vitro biochemical approaches on isolated, soluble Orai channel C-terminal peptides also reveals a major difference in STIM1 binding between Orai1 and Orai3 (Niu et al., 2020), providing strong support for these conclusions. Indeed, we recently revealed that full-length Orai2 and Orai3 both undergo a considerably higher basal E-FRET interaction with full-length STIM1 (Yoast et al., 2020), which is supported by earlier work involving STIM1 fragments (Frischauf et al., 2009), likely reflecting the enhanced M4x binding of the peptides described here.
Figure 7. Models depicting the role of Orai channel M4x in coupling with the SOAR domain of STIM proteins.
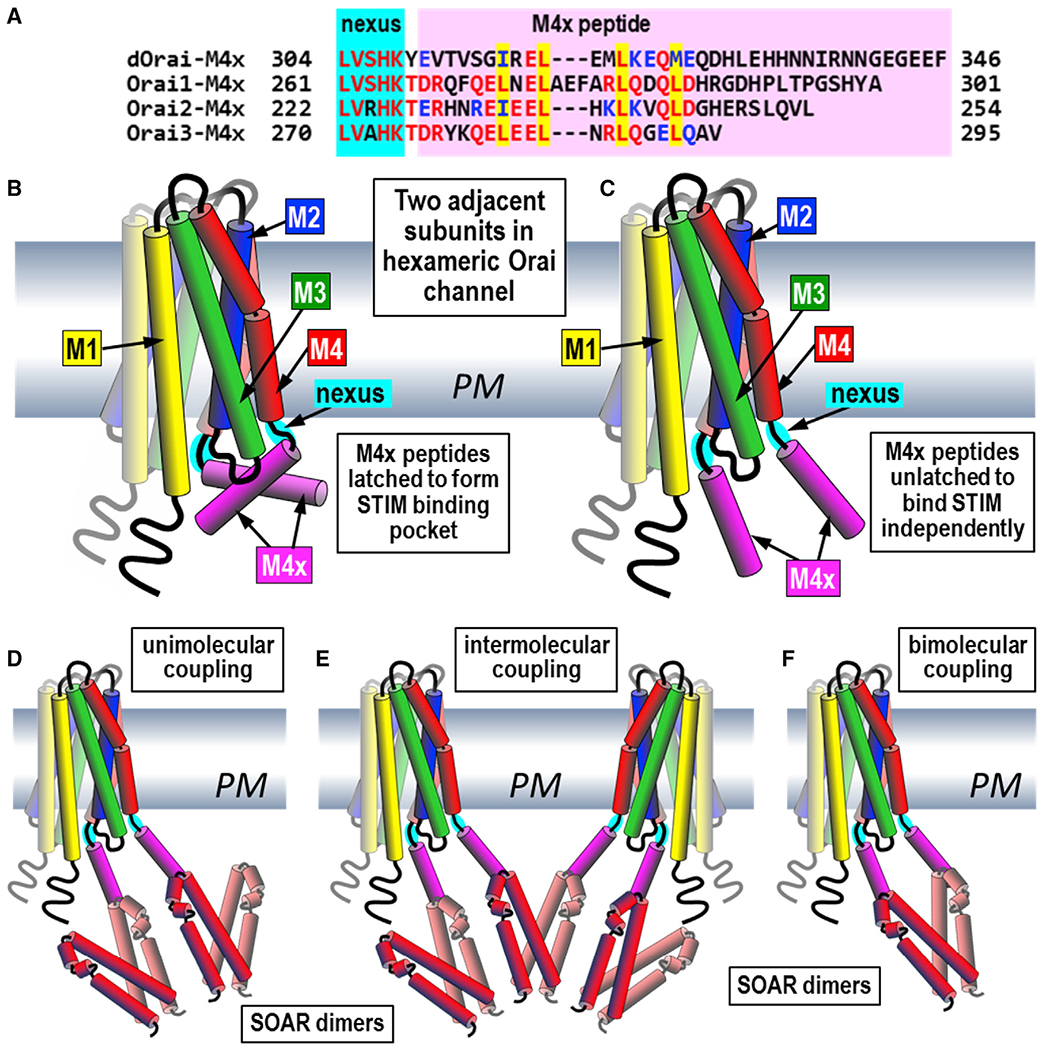
(A) C-terminal sequences of Orai channels showing the highly conserved, flexible 5-aa “nexus” that attaches the M4 helix to the helical M4x extension of 35, 27, and 20 aa, in Orai1, Orai2, and Orai3, respectively. The M4x peptides contain four conserved hydrophobic residues (yellow highlighted); the first two have been thought to mediate antiparallel pairing between M4x peptides in adjoining Orai channel subunits.
(B and C) Structural models of two adjacent subunits from the hexameric Orai channel. The orientation of the four transmembrane helices (M1–M4) are based on the dOrai crystal structure shown in Figure S1A. The central pore-forming M1 helices are surrounded by the M2, M3, and outermost M4 helices. In (B), the M4x peptides are shown in a paired or “latched” configuration predicted from the dOrai crystal structure (Hou et al., 2012) and suggested to form a STIM-binding pocket in Orai (Fahrner et al., 2014; Maus et al., 2015; Stathopulos et al., 2013; Yen and Lewis, 2018; Zhou et al., 2016). In (C), the two M4x peptides are shown as “unlatched” helices, with each able to undergo independent interactions with STIM as indicated by the current studies.
(D–F) Predicted models of coupling between the active, dimeric SOAR units of STIM proteins, and M4x peptides in Orai channels. In (D), “unimolecular” coupling is shown between each of the two independent Orai-binding sites on the SOAR dimer (Zhou et al., 2015) and single M4x peptides. This model is consistent with the optimal 2:1 stoichiometry for STIM-Orai coupling (Hoover and Lewis, 2011). In (E), the unimolecular coupling between SOAR and M4x peptides allows for “intermolecular” coupling, consistent with the observed cross-linking and clustering of Orai channels by STIM1 (Zhou et al., 2018a). In (F), a “bimolecular” interaction between independent M4x peptides and SOAR dimers cannot be excluded, but this is distinct from formation of abinding pocket formed through M4x interactions and is not the stoichiometrically preferred coupling configuration.
Previously, binding of STIM1 to the Orai1 C terminus was thought to facilitate weak STIM1 interactions with the cytosolic extension of the N-terminal pore-forming M1 helix (M1x), thus directly gating the pore (Derler et al., 2013; Li et al., 2007; McNally et al., 2013; Palty and Isacoff, 2016; Park et al., 2009; Zheng et al., 2013). We recently questioned any role of an N-terminal STIM interaction in channel gating, revealing that mutation of just the Orai1 C terminus was sufficient to emulate STIM1-induced gating (Zhou et al., 2016). The C-terminal M4x peptides in each Orai channel are linked to the M4 helix through a highly conserved 5-aa “nexus” region (Figures 7A–7C). The hinged nexus plays a vital role in transducing allosteric control from STIM binding to activate the Orai channel (Yeung et al., 2020; Zhou et al., 2016, 2019). Mutation of the Orai1 nexus induces constitutive channel activation almost identical to that activated by STIM1, even without the remaining M4x sequence (Zhou et al., 2016, 2019). The simple, consistent M4x Leu profile of STIM interactions and channel activation we report here indicates that the M4x peptide is the critical STIM-targeted allosteric trigger for gating Orai channels by STIM proteins. We conclude that the M4x helix is a STIM-operated “handle” that exerts the precise conformational change on the hinged nexus and triggers allosteric channel activation.
The concept that M4x peptides from adjacent Orai channel subunits join and function as a Leu-linked binding pocket for STIM proteins (Figure 7B) originated from the dOrai crystal structure (Hou et al., 2012). This showed pairing of the amphipathic dOrai M4x helices through the I316 and L319 residues (Figure 7A). This view was reinforced by nuclear magnetic resonance (NMR) studies showing an 11-aa isolated peptide from Orai1-M4x associated with isolated SOAR fragments (Stathopulos et al., 2013). However, the SOAR peptides used were non-functional and the purported Orai-binding site is not exposed in the functional SOAR dimer (Zhou et al., 2015, 2017). We recently questioned whether adjacent M4x peptides undergo any cointeraction in Orai channels (Zhou et al., 2019). Thus, although the dOrai structure revealed anti-parallel pairing of M4x helices (Hou et al., 2012), this structure was stabilized in the crystal through Zn2+ binding to pairs of histidines at the far C-terminal end of the dOrai M4x. Thus, the pairing of the amphipathic dOrai M4x helices through the I316 and L319 residues may be an artifact of crystallization conditions (Zhou et al., 2019). The confusion has been confounded because Leu replacement (for example, L273D and/or L275D in Orai1), which blocks STIM binding and channel activation, was assumed to be breaking the M4x interactions and disturbing the purported STIM-binding pocket (Cai et al., 2016; Fahrner et al., 2014; Stathopulos et al., 2013; Zhou et al., 2016). Our data here showing no measurable self-association between these peptides argue strongly against this assumption. Moreover, our data showing strong direct interactions of M4x peptides (especially Orai3-M4x) with SOAR and STIM1, which is prevented by the same Leu mutations thought to mediate M4x-M4x interactions, reveal these leucines are needed to bind directly to SOAR.
The conclusion that M4x peptides do not form a binding pocket allows us to reinterpret experiments we undertook earlier with concatenated Orai1 dimers (Cai et al., 2018). We expressed Orai1 dimers with L273D mutations in either one, both, or neither of the M4x peptides. The mutated homodimer had no STIM1 binding or STIM1 activation, but the heterodimers had strong STIM1-binding activity and almost no channel function. We concluded this lack of heterodimer function reflected the inability of the M4x peptides to bind together to form the STIM-binding pocket (Fahrner et al., 2014; Maus et al., 2015; Stathopulos et al., 2013; Zhou et al., 2016). Instead, our results here indicate that the M4x peptides operate independently in their interaction with STIM1. Thus, in the Orai1 heterodimer, the unpaired M4x peptides were still able to bind STIM; however, only three Orai1 subunits in the hexamer would be active, which is insufficient to open the pore that needs at least four or five active subunits in the hexamer (Cai et al., 2016; Yen and Lewis, 2018).
Clearly, the Leu residues in the M4x peptides have a critical role in binding to STIM proteins and initiating the STIM-triggered allosteric change that induces gating. We do not support the earlier concept of a STIM-binding pocket formed by adjacent M4x peptides (Figure 7B). Instead, we conclude each M4x peptide in the hexamer functions independently to bind STIM1 (Figure 7C). This model fits with our conclusions on the unimolecular coupling of SOAR dimers that we showed have two independent binding sites for Orai1 subunits (Zhou et al., 2015). Thus, the unimolecular coupling of SOAR dimers can independently occur on each M4x peptide in the Orai channel hexamer (Figure 7D). This finding is consistent with the optimal stoichiometry for STIM1-Orai1 coupling that is observed to be 2:1, that is, one dimer of STIM1 interacting with each monomer of Orai1 (Hoover and Lewis, 2011). Our findings here on the independence of M4x peptides in binding STIM proteins are also consistent with the model for intermolecular binding of STIM proteins between separate Orai channel hexamers (Figure 7E). Thus, we recently revealed that the unimolecular coupling between STIM and Orai allows STIM1 to cross-link and mediate clustering of Orai1 channels (Zhou et al., 2018a). Our data on the independent binding of M4x peptides do not exclude a bimolecular model for STIM-Orai interactions (Figure 7F). However, such a model is quite distinct from the formation of a binding pocket mediated by M4x interactions and is inconsistent with the observed 2:1 stoichiometry for STIM-Orai coupling.
Interestingly, despite the original dOrai crystallography indicating M4x pairing, recent dOrai structural data reveal that the M4x peptides may not be held in the antiparallel, Leu-paired configuration (Hou et al., 2018; Liu et al., 2019). It was argued that unpairing of the M4x helices was required during activation of the channel and that “unlatching” allowed for dilation of the M1 pore-forming helices and channel opening (Hou et al., 2018). However, open channel structures were shown for constitutively active dOrai channel mutations that were independent of STIM and not necessarily authentic models for STIM-induced gating (Zhou et al., 2019). Enigmatically, these studies also revealed a similar unlatched M4x configuration for the WT closed dOrai channel, which provides support for the concept that the M4x helices are not paired. Determining the structures for the authentic STIM-Orai complex that show the exact configuration of the M4x peptides bound to SOAR is an important goal for understanding the allosteric control of channel gating.
Overall, our studies pinpoint the pivotal role of the M4x peptides in recognizing STIM proteins and initiating the allosteric activation of Orai channel gating. Intriguing is the considerable difference in STIM1 affinity between the Orai1, Orai2, and Orai3 M4x peptides. Clearly, the length and Leu configuration of the M4x peptides determine these differences. However, differences in the unfolding, availability, and exposure of M4x peptides may play an important role in the efficacy of STIM interaction for each Orai subtype. The simplicity of the M4x peptide as a critical STIM-binding site allows for structural modeling and the design of small molecule modifiers of the STIM-Orai coupling pathway. The stark differences in SOAR-binding affinity between the Orai1, Orai2, and Orai3 M4x peptides predicts that there may be critical differences in the efficacy of such drugs for each Orai subtype. These differences are pharmacologically crucial since vastly different expressions of Orai channel subtypes are observed between tissues and in a range of immunological and cardiovascular pathologies and cancer (Feske, 2019; Fiorio Pla et al., 2016; Prakriya and Lewis, 2015). We recently exposed major differences in the functional consequences of the interactions between endogenously expressed Orai channel subtypes and the intricacies of hetero-hexameric diversity of Orai channels (Yoast et al., 2020). Structural understanding of the STIM-Orai interaction will provide insights into the targeted design of new pharmacological modifiers of store-operated Ca2+ signals.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed toward and fulfilled by the lead contact, Donald Gill (dongill@psu.edu).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
The datasets supporting the current study are available from the corresponding authors upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
A majority of the experiments were carried out on HEK293 cells, referred to in this manuscript as “HEK.” HEK-Orai1/2/3TKOand HEK-STIM1/2DKO lines cells were generated using the CRISPR-Cas9 nickase system (Yoast et al., 2020; Zhou et al., 2018a), and cultured in DMEM (Corning Cellgro) supplemented with 10% fetal bovine serum, plasmocin (InvivoGen), penicillin and streptomycin (Gemini Bioproducts) at 37°C with 5% CO2. Additional physiological experiments were performed on HeLa cells stably expressing GFP-NFAT1 (HeLa-GFP-NFAT1 cells) that were developed earlier (Zhou et al., 2016), and cultured in the same medium as above, supplemented with G418 (Corning, 100 mg/ml). Further supplemental experiments were carried out on Jurkat T cell lines, which were cultured in RPMI 1640 (Corning Cellgro) supplemented with 10% fetal bovine serum, plasmocin (InvivoGen), penicillin and streptomycin (Gemini Bioproducts) at 37°C with 5% CO2.
METHOD DETAILS
DNA constructs
The LK-CFP-Orai1-M4x (including M4x sequence, 267-301), LK-CFP-Orai2-M4x (including Orai2 M4x sequence, 228-254), and LK-CFP-Orai3-M4x constructs (including Orai3 M4x sequence, 276-295) were made using the overlap extension PCR technique (Bryksin and Matsumura, 2010) with the following primer pairs: (Orai1-M4x) 5′-GGCATGGACGAG’ with 5′-ACAGCTCATCCT’, (Orai2-M4x) 5′-GGCATGGACGAGCTGTACAAGAAGCTTwith 5′-GGCCCGC’, (Orai3-M4x) 5′-GGCATGGACGAGCTGTACAA’ with 5′-GGCCCG CGGTACCGTCGA’ and the LK-CFP-FRB template (Várnai et al., 2007). Point mutations were performed in the M4x region of the peptides and full-length Orai counterparts (O1-M4x: L273, L276; O2-M4x: I234D; O3-M4x: L282, L285, L288, L292) and CFP-Orai3 (L282, L285) using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent, 210518). For generation of LK-YFP-Orai-M4x peptides, the ECFP tag in LK-CFP-Orai1-M4x and LK-CFP-Orai3-M4x were changed to EYFP (originating from the EYFP-C1 vector that was excised and inserted into a pCR-TOPO 2.1 vector) with PvuI and EcoRI restriction sites. All concatemeric-dimer SOAR constructs (WT or F394H) were made by Mutagenex, NJ as described earlier (Zhou et al., 2015).
Cell transfection
All transfections were undertaken by electroporation at 180 V, 25 ms in 4-mm cuvettes (Molecular Bioproducts) using the Bio-Rad gene Pulser Xcell system in Optimem media. Post-transfection, cells are plated on microscope coverslips (Fisher Scientific). For Jurkat-based experiments, poly-l-lysine (Sigma-Aldrich) was used to coat the coverslips before application of cells. Experiments were all performed 18-24 hours after transfection.
Enhanced fluorescence image analysis
Enhanced images of cellular distribution of YFP-tagged SOAR-dimers and either the LK-CFP-Orai M4x peptides or LK-CFP control constructs were obtained after brief treatment (10-15 s) with 0.05% trypsin (Cellgro). Images to provide clear delineation of the PM boundary (Figure 1) were obtained within 30 min of application to coverslips (Fisher Scientific), to obtain cells still remaining in a rounded state. Other cell images (Figure 3) were obtained with cells that had remained in culture 18-24 hr on coverslips. Cellular images were collected in stacks of 10-20 three-dimensional z axis image planes at 1-μm steps. Slidebook 6.0 software (Intelligent Imaging Solutions) constrained iterative deconvolution function was used to analyze image stacks and derive enhanced deconvolved images. Images shown here are representative of at least three independent experiments.
Förster resonance energy transfer measurements
FRET signals between our CFP-labeled (donor fluorophore) M4x peptides or Orai3 protein and either YFP-labeled (acceptor fluorophore) STIM1, SOAR dimers (WT or F394H), or M4x peptides, were measured using a Leica DMI 6000B inverted automated fluorescence microscope equipped with CFP (438 nm excited/483 nm emission), YFP (500 nm excited/542 nm emission), and FRET (438 nm excited/542 nm emission) filter cubes. Three sets of images (CFP, YFP, and FRET) were captured at room temperature using a 40x oil objective (numerical aperture 1.35; Leica) and processed using Slidebook 6.0 software (Intelligent Imaging Innovations). For time course FRET, images were collected at 20 s intervals. Exposure times for the CFP, YFP, and FRET channels were 1,000 ms, 500 ms, and 1000, ms respectively. YFP exposure time was decreased to account for the greater fluorescence intensity of YFP compared to CFP. Three-channel corrected FRET was calculated using the formula:
where: IDA, IAA, and IDA represents the background subtracted CFP, YFP, and FRET images respectively; FC represents the corrected energy transfer; Fd/Dd representative of the CFP measured bleed-through into the FRET filter; Fa/Da is the bleed-through of YFP into the FRET filter. We used the E-FRET method to analyze 3-cube FRET images as described by Zal and Gascoigne (2004), using the formula:
in which, G represents the instrument specific constant. The value of G (1.87 ± 0.1) was measured as described earlier (Zhou et al., 2018b). E-FRET analyses were conducted on cells expressing a narrow range of CFP and/or YFP intensity (see Figures S3 and S4). Regions of interest were selected in the immediate area of the PM for E-FRET analysis.
Cytosolic Ca2+ measurements
Cytosolic Ca2+ measurements were undertaken by ratiometric imaging on fura-2-loaded (Molecular Probes) cells, 18-24 hours post transfection as described earlier (Wang et al., 2010). Cells were incubated in 2 μM fura-2/AM in buffer containing the following: 107 mM NaCl, 7.2 mM KCl, 1.2 mM MgCl2. 1 mM CaCl2, 11.5 mM glucose, 0.1% BSA, 20 mM HEPES at pH 7.2 for 60 min at room temperature, followed by treatment with fura-2 free solution for another 30 min. Constitutive Ca2+ entry studies using transient transfections of SOAR-dimers were cultured in media containing 0.5 μM EGTA (Sigma-Aldrich) with low Ca2+ in order to protect cells from cytotoxicity. Fluorescence ratiometric imaging was measured utilizing a Leica DMI 6000B fluorescence microscope and Hamamatsu camera ORCA-Flash 4 controlled by the Slidebook 6.0 software (Intelligent Imaging Innovations) as described previously (Zhou et al., 2018a). At 2 s intervals, excitation at 340 nm (F340) and 380 nm (F380) was performed and emission fluorescence was collected at 505 nm. Intracellular Ca2+ levels are shown as F340/F380 ratios that are representative of typically 10-50 single cells per coverslip. Expression levels for CFP-Orai3 (WT, L282D, L285D, L282/L285D), and either YFP-SOAR dimer constructs (WT and F394H), mCherry-STIM1, STIM1-YFP constructs (WT and F394H), and YFP vector were in a narrow fluorescence range. For studies evaluating the effects of the LK-CFP-OraiM4x on store-operated calcium entry and oscillations (Figures 5, S5, and S6), LK-CFP-Orai-M4x (O1M4x, O3M4x WT, O3M4x-L285D, and LK-CFP control) expression levels were kept in a narrow range. All Ca2+ imaging experiments were performed at room temperature and representative traces of at least three independent repeats are shown. Scatter dot bar graphs shown for all cells recorded are with means ± SEM.
NFAT nuclear translocation assays
For NFAT1-GFP nuclear translocation assays in HEK293 WT (Figure 5), cells were transiently transfected with 4 μg NFAT1-GFP as expressed above. After baseline readings, carbachol (Cch) was added (after 4 min). In experiments dealing with HeLa-GFP-NFAT1 (Figure S6), a single baseline picture was taken before addition of thapsigargin (TG). A ratio of nuclear GFP-labeled NFAT1 to cytosolic levels were measured on a cell-to-cell basis.
Electrophysiological measurements
Whole-cell patch-clamp recording was performed on HEK-Orai1/2/3TKO cells that were transiently transfected with CFP-Orai3 or mutant constructs thereof, with or without STIM1-YFP. ER Ca2+ stores were passively depleted prior to Ca2+ functionality experiments on constructs with the pipette solution containing: 135 mM Cs-aspartate, 10 mM HEPES, 8 mM MgCl2 and 10 mM BAPTA (pH 7.2 with CsOH). The bath solution contained: 130 mM NaCl, 4.5 mM KCl, 5 mM HEPES, 10 mM Dextrose, 10 mM TEA-Cl and 20 mM CaCl2 (pH 7.2 with NaOH). For 2-APB response experiments with CFP-Orai3 constructs, pipette solution contained (in mM):135 Cs-Aspartate, 10 HEPES, 4 MgCl2, 10 EGTA and 3.6 CaCl2 (pH 7.2 with CsOH). Quantities of EGTA and CaCl2 were calculated using WEBMAXCLITE (http://web.stanford.edu/Bcpatton/webmaxc2) to maintain cytosolic Ca2+ at ~90 nM throughout experiments. The bath solution contained 50 μM 2-APB in addition to other contents listed above. Currents were recorded in the standard whole-cell configuration using the EPC-10 amplifier (HEKA). Glass electrodes with a typical resistance of 2-4 MΩ were pulled using a P-97 pipette puller (Sutter Instruments). A 50-ms step to −100 mV from a holding potential of 0 mV, followed by a 50-ms ramp from −100 to 100 mV, was delivered every 2 s. Currents were filtered at 3.0 kHz and sampled at 20 kHz. A +10 mV junction potential compensation was applied to correct the liquid junction potential between the bath and pipette solutions. The current measure at −100 mV was used in I/V curves. All data were acquired with Patch Master and analyzed using FitMaster and Graph Pad Prism 6.
Western blot analysis
HEK cells were washed with cold PBS solution and lysed using RIPA lysis buffer (Sigma-Aldrich with 1x protease inhibitor mixture) for 30 min, followed by centrifugation at 14,000 X g for 10 min at 4°C. Supernatants were collected and protein isolates were quantified using Bio-Rad DC kits. Protein was then loaded onto 4%–12% NuPAGE BisTris precast gels and transferred to Bio-Rad Immuno-Blot PVDF membrane. Membranes were blocked in 5% dry milk PBS-T solution for 1 hr at room temperature, then incubated with primary antibody (1:1000 Anti-Orai1) at 4°C overnight. The membrane underwent three washes in PBS-T and subsequently incubated in secondary antibody for 1 hr. at room temperature. An additional three washes with PBS-T were then performed. Horseradish peroxidase activity was examined with the Pierce ECL Plus Western Blot blotting substrate (ThermoFisher Scientific) and fluorescence was collected using the FluorChem M imager (ProteinSimple). Expression of LK-CFP-Orai1-M4x and CFP-Orai1 WT was undertaken using Orai1 antibody (Sigma Chemicals, O8264) against the C-terminal 288-301 segment of the M4x peptide, and recognizing the Orai1-M4x peptide construct as essentially a single band (Figure S7). Corresponding antibodies for Orai2 and Orai3 peptides are not available. Accurate quantification of expression of Orai1, Orai2 and Orai3 peptide constructs was determined by CFP intensity measurement.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data generated was collected in Excel and analyzed with the use of Graphpad Prism (Ver. 8 and 9). The mean ± SEM is graphed, with P values < 0.05 (*), < 0.01 (**), < 0.001 (***), and < 0.0001 (****) considered significant. One-way ANOVA and Dunnett’s test were used for multiple group comparisons, and Welch’s unpaired t test used in the case of two-group comparisons.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal to Orai1 | Sigma-Aldrich | Cat#O8264; RRID:AB_1078883 |
| Chemicals, peptides, and recombinant proteins | ||
| 2-aminoethoxydiphenyl borate | Millipore-Sigma | Cat#8.41636 |
| Fura 2-acetoxymethyl ester | Sigma-Aldrich | Cat#F0888 |
| Ionomycin | Sigma-Aldrich | Cat#I9657 |
| EGTA | Sigma-Aldrich | Cat#324626 |
| YFP-SOAR concatemers | Zhou et al., 2015 (Mutagenix) | N/A |
| NFAT1-GFP | Yoast et al., 2020 | N/A |
| Poly-L-Lysine | Sigma-Aldrich | Cat#P4832 |
| Thapsigargin | Sigma-Aldrich | Cat#T9033 |
| Carbachol | Sigma-Aldrich | Cat#212385-M |
| Critical commercial assays | ||
| QuikChange Lightning Site-Directed Mutagenesis Kit | Agilent | Cat#210518 |
| Experimental models: Cell lines | ||
| HEK293 cells | ATCC | Cat#CRL-1573; RRID:CVCL_0045 |
| HEK293 Orai1-3 TKO cells | Yoast et al., 2020 | N/A |
| HEK293 STIM1/2 DKO cells | Zhou et al., 2018a | N/A |
| HeLa-GFP-NFAT1 cells | Zhou et al., 2016 | N/A |
| Jurkat T cells | ATCC | Cat#TIB-152; RRID:CVCL_0367 |
| Oligonucleotides | ||
| O1-M4x Forward: GGCATGGACGAGCTGTACAAGAAGCTTGCTGACCGACAGTTCCAGGAGCTCA | Zhou et al., 2015 | N/A |
| O1-M4x Reverse: ACAGCTCATCCTTAAGTCGACTCGAGATGCGCTAGGCAsTAGTGGCTGCCGGG | Zhou et al., 2015 | N/A |
| O2-M4x Forward: GGCATGGACGAGCTGTACAAGAAGCTTGCTGAGCGCCACAACCGCGAGATCG | This paper | N/A |
| O2-M4x Reverse: GGCCCGCGGTACCGTCGACTGCAGAATTCGCCTACAAGACCTGCAGGCTGCG | This paper | N/A |
| O3-M4x Forward: GGCATGGACGAGCTGTACAAGAAGCTTGCTGACCGCTACAAGCAGGAACTAG | This paper | N/A |
| O3-M4x Reverse: GGCCCGCGGTACCGTCGACTGCAGAATTCGCTCACACAGCCTGCAGCTCCCC | This paper | N/A |
| O1-O3 M4x mutant primers | This paper | See Table S1 |
| Software and algorithms | ||
| SlideBook Software Ver. 6.0 | Intelligent Imaging Innovations | https://www.intelligent-imaging.com |
| GraphPad Prism 6, 8, and 9 | GraphPad Software, Inc. | https://www.graphpad.com |
| Excel | Microsoft | https://www.microsoft.com |
| WebMaxCLITE | Stanford | http://web.stanford.edu/Bcpatton/webmaxc2 |
| SnapGene Viewer | SnapGene | https://www.snapgene.com |
| DNASTAR Lasergene Software | DNASTAR | https://www.dnastar.com |
| Other | ||
| FluorChem M Imaging System | Protein-simple | N/A |
Highlights.
PM-tethered Orai channel M4x peptides undergo Leu-specific binding to SOAR dimers
The Leu specificity of M4x peptides mimics that of Orai channel activation by STIM1
Orai3 M4x peptides block Ca2+ oscillations and NFAT translocation driven by SOCE
M4x helices precisely define the coupling interface between STIM and Orai proteins
ACKNOWLEDGMENTS
This work was supported by NIH R01 grants GM120783 and GM109279 and NIH R35 grant GM131916 to D.L.G., F31 predoctoral fellowship GM125376 to R.M.N., F31 predoctoral fellowship HL152619 to J.H.B., and Penn State University JFDP grant to Y.Z.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109322.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Amcheslavsky A, Safrina O, and Cahalan MD (2013). Orai3 TM3 point mutation G158C alters kinetics of 2-APB-induced gating by disulfide bridge formation with TM2 C101. J. Gen. Physiol 142, 405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amcheslavsky A, Safrina O, and Cahalan MD (2014). State-dependent block of Orai3 TM1 and TM3 cysteine mutants: insights into 2-APB activation. J. Gen. Physiol 143, 621–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryksin AV, and Matsumura I (2010). Overlap extension PCR cloning: a simple and reliable way to create recombinant plasmids. Biotechniques 48, 463–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Zhou Y, Nwokonko RM, Loktionova NA, Wang X, Xin P,Trebak M, Wang Y, and Gill DL (2016). The Orai1 store-operated calcium channel functions as a hexamer. J. Biol. Chem 291, 25764–25775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Nwokonko RM, Loktionova NA, Abdulqadir R, Baraniak JH Jr., Wang Y, Trebak M, Zhou Y, and Gill DL (2018). Pore properties of Orai1 calcium channel dimers and their activation by the STIM1 ER calcium sensor. J. Biol. Chem 293, 12962–12974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeHaven WI, Smyth JT, Boyles RR, Bird GS, and Putney JW Jr. (2008). Complex actions of 2-aminoethyldiphenyl borate on store-operated calcium entry. J. Biol. Chem 283, 19265–19273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derler I, Plenk P, Fahrner M, Muik M, Jardin I, Schindl R, Gruber HJ, Groschner K, and Romanin C (2013). The extended transmembrane Orai1 N-terminal (ETON) region combines binding interface and gate for Orai1 activation by STIM1. J. Biol. Chem 288, 29025–29034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derler I, Jardin I, and Romanin C (2016). Molecular mechanisms of STIM/Orai communication. Am. J. Physiol. Cell Physiol 310, C643–C662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derler I, Butorac C, Krizova A, Stadlbauer M, Muik M, Fahrner M, Frischauf I, and Romanin C (2018). Authentic CRAC channel activity requires STIM1 and the conserved portion of the Orai N terminus. J. Biol. Chem 293, 1259–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahrner M, Muik M, Schindl R, Butorac C, Stathopulos P, Zheng L, Jardin I, Ikura M, and Romanin C (2014). A coiled-coil clamp controls both conformation and clustering of stromal interaction molecule 1 (STIM1). J. Biol. Chem 289, 33231–33244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S (2019). CRAC channels and disease - From human CRAC channelopathies and animal models to novel drugs. Cell Calcium 80, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorio Pla A, Kondratska K, and Prevarskaya N (2016). STIM and ORAI proteins: crucial roles in hallmarks of cancer. Am. J. Physiol. Cell Physiol 310, C509–C519. [DOI] [PubMed] [Google Scholar]
- Frischauf I, Muik M, Derler I, Bergsmann J, Fahrner M, Schindl R, Groschner K, and Romanin C (2009). Molecular determinants of the coupling between STIM1 and Orai channels: differential activation of Orai1-3 channels by a STIM1 coiled-coil mutant. J. Biol. Chem 284, 21696–21706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover PJ, and Lewis RS (2011). Stoichiometric requirements for trapping and gating of Ca2+ release-activated Ca2+ (CRAC) channels by stromal interaction molecule 1 (STIM1). Proc. Natl. Acad. Sci. USA 108, 13299–13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou X, Pedi L, Diver MM, and Long SB (2012). Crystal structure of the calcium release-activated calcium channel Orai. Science 338, 1308–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou X, Burstein SR, and Long SB (2018). Structures reveal opening of the store-operated calcium channel Orai. eLife 7, e36758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Heo WD, Grimley JS, Wandless TJ, and Meyer T (2005). An inducible translocation strategy to rapidly activate and inhibit small GTPase signaling pathways. Nat. Methods 2, 415–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki T, Lange I, and Feske S (2009). A minimal regulatory domain in the C terminus of STIM1 binds to and activates ORAI1 CRAC channels. Biochem. Biophys. Res. Commun 385, 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Lu J, Xu P, Xie X, Chen L, and Xu T (2007). Mapping the interacting domains of STIM1 and Orai1 in Ca2+ release-activated Ca2+ channel activetion. J. Biol. Chem 282, 29448–20456. [DOI] [PubMed] [Google Scholar]
- Li Z, Liu L, Deng Y, Ji W, Du W, Xu P, Chen L, and Xu T (2011). Graded activation of CRAC channel by binding of different numbers of STIM1 to Orai1 subunits. Cell Res 21, 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou J, Fivaz M, Inoue T, and Meyer T (2007). Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc. Natl. Acad. Sci. USA 104, 9301–9306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lis A, Peinelt C, Beck A, Parvez S, Monteilh-Zoller M, Fleig A, and Penner R (2007). CRACM1, CRACM2, and CRACM3 are store-operated Ca2+ channels with distinct functional properties. Curr. Biol 17, 794–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wu G, Yu Y, Chen X, Ji R, Lu J, Li X, Zhang X, Yang X, and Shen Y (2019). Molecular understanding of calcium permeation through the open Orai channel. PLoS Biol 17, e3000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maus M, Jairaman A, Stathopulos PB, Muik M, Fahrner M, Weidinger C, Benson M, Fuchs S, Ehl S, Romanin C, et al. (2015). Missense mutation in immunodeficient patients shows the multifunctional roles of coiled-coil domain 3 (CC3) in STIM1 activation. Proc. Natl. Acad. Sci. USA 112, 6206–6211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally BA, Somasundaram A, Jairaman A, Yamashita M, and Prakriya M (2013). The C- and N-terminal STIM1 binding sites on Orai1 are required for both trapping and gating CRAC channels. J. Physiol 591, 2833–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muik M, Frischauf I, Derler I, Fahrner M, Bergsmann J, Eder P, Schindl R, Hesch C, Polzinger B, Fritsch R, et al. (2008). Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J. Biol. Chem 283, 8014–8022. [DOI] [PubMed] [Google Scholar]
- Niu L, Wu F, Li K, Li J, Zhang SL, Hu J, and Wang Q (2020). STIM1 interacts with termini of Orai channels in a sequential manner. J. Cell Sci 133, jcs239491. [DOI] [PubMed] [Google Scholar]
- Palty R, and Isacoff EY (2016). Cooperative Binding of Stromal Interaction Molecule 1 (STIM1) to the N and C Termini of Calcium Release-activated Calcium Modulator 1 (Orai1). J. Biol. Chem 291, 334–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, Walz T, Garcia KC, Dolmetsch RE, and Lewis RS (2009). STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 136, 876–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinelt C, Lis A, Beck A, Fleig A, and Penner R (2008). 2-Aminoethoxydiphenyl borate directly facilitates and indirectly inhibits STIM1-dependent gating of CRAC channels. J. Physiol 586, 3061–3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakriya M, and Lewis RS (2015). Store-Operated Calcium Channels. Physiol. Rev 95, 1383–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothberg BS, Wang Y, and Gill DL (2013). Orai channel pore properties and gating by STIM: implications from the Orai crystal structure. Sci. Signal 6, pe9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindl R, Bergsmann J, Frischauf I, Derler I, Fahrner M, Muik M, Fritsch R, Groschner K, and Romanin C (2008). 2-aminoethoxydiphenyl borate alters selectivity of Orai3 channels by increasing their pore size. J. Biol. Chem 283, 20261–20267. [DOI] [PubMed] [Google Scholar]
- Scrimgeour N, Litjens T, Ma L, Barritt GJ, and Rychkov GY (2009). Properties of Orai1 mediated store-operated current depend on the expression levels of STIM1 and Orai1 proteins. J. Physiol 587, 2903–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soboloff J, Rothberg BS, Madesh M, and Gill DL (2012). STIM proteins: dynamic calcium signal transducers. Nat. Rev. Mol. Cell Biol 13, 549–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stathopulos PB, Schindl R, Fahrner M, Zheng L, Gasmi-Seabrook GM, Muik M, Romanin C, and Ikura M (2013). STIM1/Orai1 coiled-coil interplay in the regulation of store-operated calcium entry. Nat. Commun 4, 2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirado-Lee L, Yamashita M, and Prakriya M (2015). Conformational Changes in the Orai1 C-Terminus Evoked by STIM1 Binding. PLoS One 10, e0128622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trebak M, and Kinet JP (2019). Calcium signalling in T cells. Nat. Rev. Immunol 19, 154–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Várnai P, Tóth B, Tóth DJ, Hunyady L, and Balla T (2007). Visualization and manipulation of plasma membrane-endoplasmic reticulum contact sites indicates the presence of additional molecular components within the STIM1-Orai1 Complex. J. Biol. Chem 282, 29678–29690. [DOI] [PubMed] [Google Scholar]
- Wang Y, Deng X, Zhou Y, Hendron E, Mancarella S, Ritchie MF, Tang XD, Baba Y, Kurosaki T, Mori Y, et al. (2009). STIM protein coupling in the activation of Orai channels. Proc. Natl. Acad. Sci. USA 106, 7391–7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Deng X, Mancarella S, Hendron E, Eguchi S, Soboloff J, Tang XD, and Gill DL (2010). The calcium store sensor, STIM1, reciprocally controls Orai and Cav1.2 channels. Science 330, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang Y, Zhou Y, Hendron E, Mancarella S, Andrake MD, Rothberg BS, Soboloff J, and Gill DL (2014). Distinct Orai-coupling domains in STIM1 and STIM2 define the Orai-activating site. Nat. Commun 5, 3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen M, and Lewis RS (2018). Physiological CRAC channel activation and pore properties require STIM1 binding to all six Orai1 subunits. J. Gen. Physiol 150,1373–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen M, Lokteva LA, and Lewis RS (2016). Functional Analysis of Orai1 Concatemers Supports a Hexameric Stoichiometry for the CRAC Channel. Biophys. J 111, 1897–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung PS, Yamashita M, Ing CE, Pomès R, Freymann DM, and Prakriya M (2018). Mapping the functional anatomy of Orai1 transmembrane domains for CRAC channel gating. Proc. Natl. Acad. Sci. USA 115, E5193–E5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung PS, Yamashita M, and Prakriya M (2020). Molecular basis of allosteric Orai1 channel activation by STIM1. J. Physiol 598, 1707–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoast RE, Emrich SM, Zhang X, Xin P, Johnson MT, Fike AJ, Walter V, Hempel N, Yule DI, Sneyd J, et al. (2020). The native ORAI channel trio underlies the diversity of Ca2+ signaling events. Nat. Commun 11, 2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan JP, Zeng W, Dorwart MR, Choi YJ, Worley PF, and Muallem S (2009). SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol 11, 337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias DA, Violin JD, Newton AC, and Tsien RY (2002). Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 296, 913–916. [DOI] [PubMed] [Google Scholar]
- Zal T, and Gascoigne NR (2004). Photobleaching-corrected FRET efficiency imaging of live cells. Biophys. J 86, 3923–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Kozak JA, Jiang W, Yeromin AV, Chen J, Yu Y, Penna A, Shen W, Chi V, and Cahalan MD (2008). Store-dependent and -independent modes regulating Ca2+ release-activated Ca2+ channel activity of human Orai1 and Orai3. J. Biol. Chem 283, 17662–17671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Zhou MH, Hu C, Kuo E, Peng X, Hu J, Kuo L, and Zhang SL (2013). Differential roles of the C and N termini of Orai1 protein in interacting with stromal interaction molecule 1 (STIM1) for Ca2+ release-activated Ca2+ (CRAC) channel activation. J. Biol. Chem 288, 11263–11272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wang X, Wang X, Loktionova NA, Cai X, Nwokonko RM, Vrana E, Wang Y, Rothberg BS, and Gill DL (2015). STIM1 dimers undergo unimolecular coupling to activate Orai1 channels. Nat. Commun 6, 8395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Cai X, Loktionova NA, Wang X, Nwokonko RM, Wang X, Wang Y, Rothberg BS, Trebak M, and Gill DL (2016). The STIM1-binding site nexus remotely controls Orai1 channel gating. Nat. Commun 7, 13725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Cai X, Nwokonko RM, Loktionova NA, Wang Y, and Gill DL (2017). The STIM-Orai coupling interface and gating of the Orai1 channel. Cell Calcium 63, 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Nwokonko RM, Cai X, Loktionova NA, Abdulqadir R, Xin P, Niemeyer BA, Wang Y, Trebak M, and Gill DL (2018a). Cross-linking of Orai1 channels by STIM proteins. Proc. Natl. Acad. Sci. USA 115, E3398–E3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wang Y, and Gill DL (2018b). Assessing the Molecular Nature of the STIM1/Orai1 Coupling Interface Using FRET Approaches. In Calcium Entry Channels in Non-Excitable Cells, Kozak JA and Putney JW Jr., eds. (CRC Press; ), pp. 127–144. [PubMed] [Google Scholar]
- Zhou Y, Nwokonko RM, Baraniak JH Jr., Trebak M, Lee KPK, and Gill DL (2019). The remote allosteric control of Orai channel gating. PLoS Biol 17, e3000413. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting the current study are available from the corresponding authors upon request.