

Abstract
PURPOSE
Adrenocortical carcinoma (ACC) is a rare aggressive pediatric malignancy with distinct biology. Its treatment follows the principles developed for adults; pediatric-specific studies are scarce.
PATIENTS AND METHODS
Prospective single-arm risk-stratified interventional study. Study objectives were (1) to describe the outcome of patients with stage I ACC treated with adrenalectomy alone; (2) to describe the outcome of stage II patients (completely resected > 200 cc or > 100 g) treated with adrenalectomy and retroperitoneal lymph node dissection; and (3) to describe the outcome of patients with stage III or IV treated with mitotane and chemotherapy.
RESULTS
Between September 2006 and May 2013, 78 patients (77 eligible, 51 females) were enrolled. The 5-year event-free survival estimates for stages I (24 patients), II (15 patients), III (24 patients), and IV (14 patients) were 86.2%, 53.3%, 81%, and 7.1%, respectively. The corresponding 5-year overall survival estimates were 95.2%, 78.8%, 94.7%, and 15.6%, respectively. On univariate analysis, age, stage, presence of virilization, Cushing syndrome, or hypertension, germline TP53 status, and presence of a somatic ATRX mutation were associated with outcome. On multivariable analysis, only stage and age were significantly associated with outcome. The probabilities of mitotane and chemotherapy feasibility events were 10.5% and 31.6%, respectively.
CONCLUSION
Outcome for children with stage I ACC is excellent with surgery. Outcome for patients with stage II disease is inferior despite retroperitoneal lymph node dissection. Patients with stage III ACC have an excellent outcome combining surgery and chemotherapy. Patients with stage IV ACC are older and have a poor outcome; new treatments should be explored for this high-risk group. The combination of mitotane and chemotherapy as prescribed in ARAR0332 resulted in significant toxicity; one third of patients with advanced disease could not complete the scheduled treatment.
INTRODUCTION
Childhood adrenocortical carcinoma (ACC) is rare; only 25 cases are expected to occur annually in the United States for an estimated annual incidence of 0.2-0.3 cases per million children and adolescents.1 Internationally, however, its incidence varies; it is 10-15 times higher in southern Brazil.2,3 Germline TP53 mutations have been implicated in 50%-65% of cases generally, and in 95% of cases in Brazil, where the TP53p.R337H variant is prevalent.4
CONTEXT
Key Objective
To investigate the role of extended surgery with retroperitoneal lymph node dissection (RPLND) for large localized tumors, and the tolerance and efficacy of a mitotane and cisplatin-based regimen for advanced childhood adrenocortical carcinoma (ACC).
Knowledge Generated
Two thirds of children with ACC carry a germline TP53 mutation. Although patients with small localized tumors can be cured with surgery, RPLND failed to improve outcomes for patients with completely resected large tumors. Patients with unresectable disease and those with tumor spillage benefit from chemotherapy; however, the mitotane- and cisplatin-based regimen results in high toxicity rates. The outcome of metastatic patients is dismal.
Relevance
Risk-adapted therapies can be developed for childhood ACC. However, the role of RPLND for patients with localized disease is not clear. Although chemotherapy and mitotane play a role for a subset of patients with locally advanced disease, current regimens are toxic and should be modified to maximize risk-benefit.
Data from the International Pediatric Adrenocortical Tumor Registry3,5 and population-based studies6,7 have characterized its biology, clinical features, and prognostic factors. Children with ACC present typically in the first 5 years of life, with a strong female predominance, and almost universally with virilization. Biologically, childhood ACC has distinctive features; its genomic landscape is characterized by copy-neutral loss of heterozygosity of chromosomes 11 and 17 associated with germline TP53 pathogenic variants, universal insulin-like growth factor-2 overexpression, and somatic mutations in ATRX and CTNNB1.8
The principles of therapy have been adapted from those established for adults.9,10 Surgery is the mainstay of therapy, and for children with advanced disease, chemotherapy and mitotane have been proposed. Low stage and complete resection are the most important prognostic factors11,12,2; more than 90% of patients with small localized tumors are long-term survivors, compared with 10% of those with metastatic disease.3,12 Despite complete tumor resection, disease recurs in 50% of patients with large localized tumors.11,3 The reason for this increased risk of recurrence is not well understood; tumor spillage is common, and studies in adults have suggested that retroperitoneal lymph node involvement may play a role.13
Cooperative efforts have been pivotal in the advancement of pediatric oncology. Rare pediatric tumors, however, have remained research orphans. Building on the evidence generated by the International Pediatric Adrenocortical Tumor Registry, the Children's Oncology Group developed the ARAR0332 study, a risk-based trial that sought to further the knowledge on childhood ACC.
PATIENTS AND METHODS
Study Objectives
The study objectives of ARAR0332 were (1) to describe the outcome of stage I with surgery only; (2) to describe the outcome of stage II (completely resected > 200 cc or > 100 g) with adrenalectomy and retroperitoneal lymph node dissection (RPLND); and (3) to describe the outcome of stage III or IV with mitotane and chemotherapy.
Eligibility
Patients < 22 years of age who were not pregnant or nursing with newly diagnosed, previously untreated ACC, and adequate performance and organ function, were eligible. Central pathology review was required for eligibility.14,15 The trial was approved by the Pediatric Central Institutional Review Board of the National Cancer Institute, and by the institutional review boards of participating institutions. It was registered at ClinicalTrials.gov (identifier: NCT00304070) and opened in two institutions in Southern Brazil. Informed consent from the patient, parent, or guardian was obtained before enrollment.
Treatment
Staging system was modified from Sandrini et al16 (Table 1).2,3 The protocol included three strata: stratum 1 consisted of patients with stage I tumors, stratum 2 consisted of patients with stage II tumors, and stratum 3 consisted of patients with stage III and IV tumors. Treatment for stage I was tumor resection. Treatment for stage II included resection and RPLND, which could be done either upfront or as a second procedure. Treatment for stages III and IV was eight cycles of chemotherapy, and mitotane for 8 months, with surgery of primary tumor and metastases as clinically indicated at the discretion of the treating team. Surgical guidelines are included in the Data Supplement (online only). Each cycle of chemotherapy consisted of cisplatin 50 mg/m2/dose (days 1-2), etoposide 100 mg/m2/dose (days 1-3), and doxorubicin 25 mg/m2/dose (days 4-5). Filgrastim 5 mcg/kg/dose was started on day 6 and given daily until neutrophil recovery. Mitotane was given daily and adjusted to plasma concentrations of 14-20 mcg/mL. Toxicity was assessed by National Cancer Institute Common Terminology Criteria for Adverse Events (version 4) for patients who received chemotherapy. The proportion of patients experiencing toxicity was tabulated separately for cycles 1-4 and 5-8. The maximum grade of each toxicity for each period was recorded. The number and percent of patients with each toxicity type whose maximum grade was 3 or greater was tabulated.
TABLE 1.
Stage and Treatment Administered in ARAR0332

Mutation Analysis
Germline TP53 status and functional activity were determined as previously reported.4 Based on prior studies performed in a cohort that included a subset of ARAR0332 patients demonstrating a pattern of recurring somatic alterations in CTNNB1, TP53, and ATRX, the mutational status of those genes was determined as previously reported.8
Statistical Methods
Event-free survival (EFS) was defined as time from enrollment to the earliest of disease progression, diagnosis of second malignancy, death, or last follow-up. Overall survival (OS) was defined as the time from enrollment to the earliest of death or last follow-up. Patients who experienced disease progression, second malignancy, or death were considered to have experienced an event; otherwise, patients were censored at last follow-up. EFS and OS as functions of time since enrollment were estimated using the method of Kaplan and Meier.17 Median follow-up for OS was calculated by the reverse-Kaplan-Meier methods as suggested by Schemper and Smith.18
Study design.
Accrual and follow-up provided sufficient precision to address the three primary aims. We planned for 7 years of enrollment with 2 years of follow-up after the last enrolled patient, at which time each of the stratum-specific tests of hypothesis were to be done. With the target number of patients in each stratum, the asymptotic distribution of the Kaplan-Meier estimate of the 2-year EFS was compared with (1) stage I—24 patients; 90%; (2) stage II—15 patients; 50%; and (3) stages 3 and 4—40 patients; 15%. We conducted a post hoc analysis comparing the results of stage III and stage IV patients separately with the target value of 15%. Detailed statistical properties and interim monitoring are described in the Data Supplement.
Feasibility of therapy delivery.
All patients enrolled on stratum 3 and who received at least one cycle of therapy were considered in the analysis for tolerability. We planned to evaluate 40 patients for this secondary aim. Any patients who had mitotane stopped because of toxicity were considered to have experienced a mitotane feasibility-event (MFE). If six or more patients experienced an MFE, the study treatment was to be identified as associated with excessive MFE rate. If the true MFE rate was 10%, the regimen would be considered tolerable with probability 90%; if the true MFE rate was 25%, the regimen would be considered intolerable with probability 90%. Patients who had at least one agent of the combination stopped were considered to have experienced a chemotherapy feasibility event (CFE). Patients who completed therapy or were removed from protocol therapy for disease progression, second malignancy, or death unrelated to protocol therapy were considered to have successfully tolerated treatment. If 11 or more patients experienced a CFE, the study treatment was to be identified as associated with excessive chemotherapy toxicity. If the true CFE rate was 20%, the regimen would be considered tolerable with probability 91%; if the true CFE rate was 40%, the regimen would be considered intolerable with probability 92%.
Exploratory analyses.
Analyses relating patient characteristics to risk of EFS event were conducted using a relative risk regression model.17 Analyses assessing the association between patient characteristics measured at the time of study enrollment used contingency table methods. P values for the associations considered were calculated using the exact conditional test of proportions.19 The association between patient stage and patient age and tumor volume distribution was assessed using the Kruskal-Wallis test.20 For all exploratory testing, a P value ≤ .05 was considered as evidence of a statistically significant association.
RESULTS
The study opened in September 2006 and closed in May 2013. Seventy-eight patients were enrolled of whom 77 were eligible (one patient was enrolled in error after death) and were included in the outcome analyses; data were current as of March 2019 (Fig 1). Patient characteristics are summarized in Table 2, and grade 3 or higher adverse events in Appendix Table A1 (online only). There was a significant association between age and stage (P = .001); patients with stage IV were older (median 13 years) when compared to those with stage I (1.5 years), II (2 years), and III (3 years). There was a higher proportion of stage I and lower proportion of stage IV patients in the Brazilian versus North American institutions, and more patients in the Brazilian sites had a germline TP53 pathogenic variant compared with North American institutions (95% v 50%, respectively).
FIG 1.

Patient flow diagram of ARAR0332.
TABLE 2.
Patient Characteristics of 77 Eligible Patients Enrolled on ARAR0332

There was a trend toward increased tumor volume with increasing stage; median volume was 37.3 cm3, 296.4 cm3, 351.5 cm3, and 706 cm3 for stages I, II, III, and IV, respectively. Ninety percent of patients presented with evidence of hormonal hypersecretion. There was an association between the endocrine phenotype and stage. Virilization was present at diagnosis in 91.3%, 78.6%, 69.6%, and 50% of stage I, II, III, and IV cases, respectively (P = .05); Cushing syndrome was present in 20.8%, 13.3%, 16.7%, and 61.5% of stage I, II, III, and IV cases, respectively (P = .02); and hypertension was diagnosed within 3 months of ACC in 12.5%, 20%, 29.2%, and 64.3% of stage I, II, III, and IV cases, respectively (P = .009).
Among the 15 stage II patients, 13 were documented to have undergone an RPLND (one patient refused, and the reason in the second case was unknown) and only one patient had nodal disease; this patient had seven positive lymph nodes, did not receive adjuvant chemotherapy, and remains in remission. The operative notes to assess the adequacy of the RPLND were available in 11 of those patients; the median number of lymph nodes resected was 4 (range, 1-30). Among the 24 patients with stage III, five were because of unresectable tumor or macroscopic residual, and 19 because of microscopic disease or spillage. Of the 17 stage III patients who underwent an RPLND or sampling, one patient was found to have nodal disease. Metastatic sites in the 14 stage IV patients were liver (3), lung (4), combined liver and lung (3), and multiple sites including lung (4).
Primary Analysis
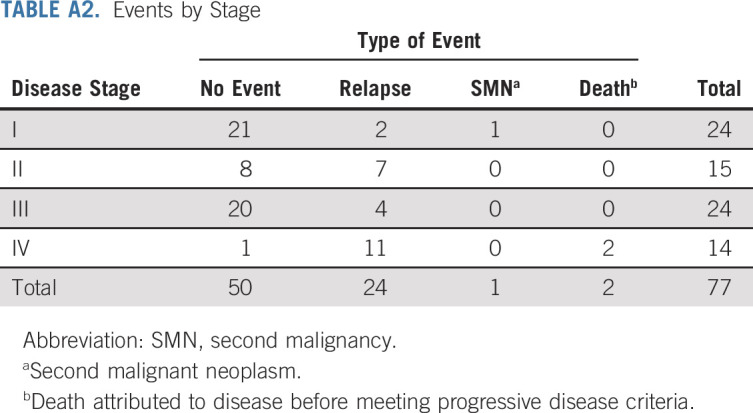
Twenty-seven events were observed (Appendix Table A2, online only) 24 patients had tumor relapse, two died of disease as first event, and one patient with stage I developed a precursor B-cell lymphoblastic leukemia. Seven stage II patients had tumor relapse (two locoregional, two combined local or lung, one combined local or liver, one lung, and one unknown). With a median follow-up for OS of 60 months, the 5-year EFS and OS estimates were 62.9% (95% CI, 50.6 to 73.0) and 76.7% (95% CI, 64.7 to 85.1), respectively. The 5-year EFS estimates for stages I, II, III, and IV were 86.2% (95% CI, 62.9 to 95.4), 53.3% (95% CI, 26.3 to 74.4), 81% (95% CI, 56.9 to 92.5), and 7.1% (95% CI, 0.5 to 27.5), respectively (Fig 2A). The corresponding 5-year OS estimates were 95.2% (95% CI, 70.7 to 99.3), 78.8% (95% CI, 47.3 to 92.7), 94.7% (95% CI, 68.1 to 99.2), and 15.6% (95% CI, 2.5 to 39.2), respectively (Fig 2B). There were no differences in outcome by stage between Brazilian and North American patients. Based on the study design, we concluded that the strategy of surgery and observation for stage I and chemotherapy for stage III warrant adoption. However, the conclusion for stage II and stage IV was that the strategies of RPLND and systemic chemotherapy, respectively, do not provide sufficient improvement for those groups to warrant adoption (Table 3).
FIG 2.
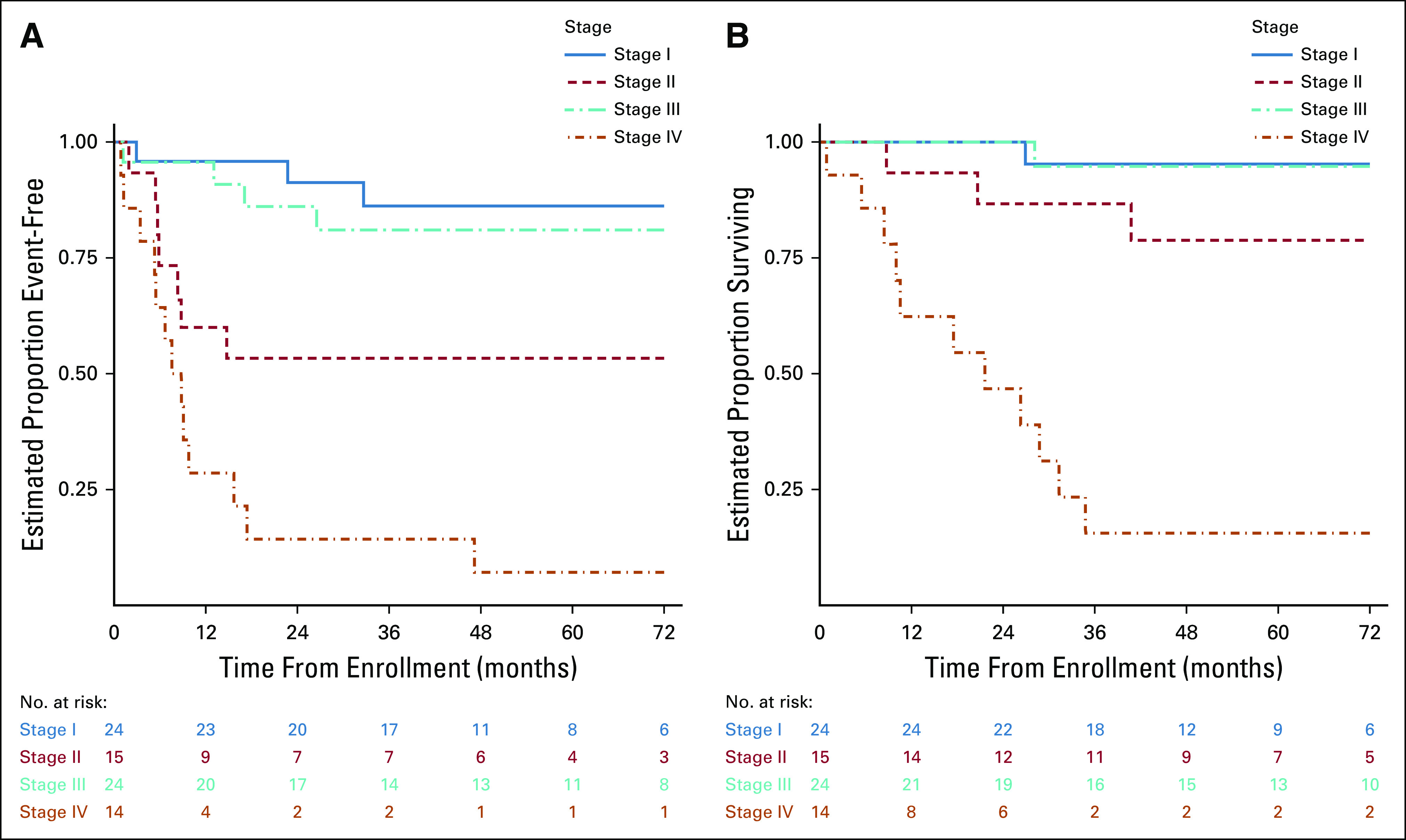
(A) EFS and (B) OS probabilities for 78 patients enrolled on ARAR0332. EFS, event-free survival; OS, overall survival.
TABLE 3.
Primary Analysis Results

Secondary Analyses
Sixty-two patients consented for germline TP53 testing. For one case, sequencing could not be performed; of the remaining 61 cases, 20 (32.8%) had wild-type TP53 sequence and 41 (67.2%) had a pathogenic variant (20 of them p.R337H) (Table 2). Among the mutant cases, p53 protein activity was low (0%-15%) in 17 variants, medium (34%-35%) in three, medium-high (69%) in one (p.R337H), and it could not be performed in one variant. When comparing outcomes by germline p53 function, presence of normal function was significantly associated with higher disease stage (P = .006) and worse outcome (Table 4).
TABLE 4.
Outcomes According to Patient Characteristics

The results of somatic mutation analysis for TP53, CTNNB1, and ATRX are depicted in Table 2. Disease stage was not associated to the presence of somatic TP53 or ATRX mutations; however, mutated CTNNB1 was more frequent in stage IV patients; four of the seven patients with mutated CTNNB1 had metastatic disease (P = .015). Age at diagnosis was not correlated with the presence of somatic TP53 or CTNNB1 mutations. There was a significant association between the presence of a somatic ATRX mutation (which was always in the presence of a TP53 mutation) and older age; six of the seven patients with somatic ATRX mutation were older than the median age of the cohort (P = .046).
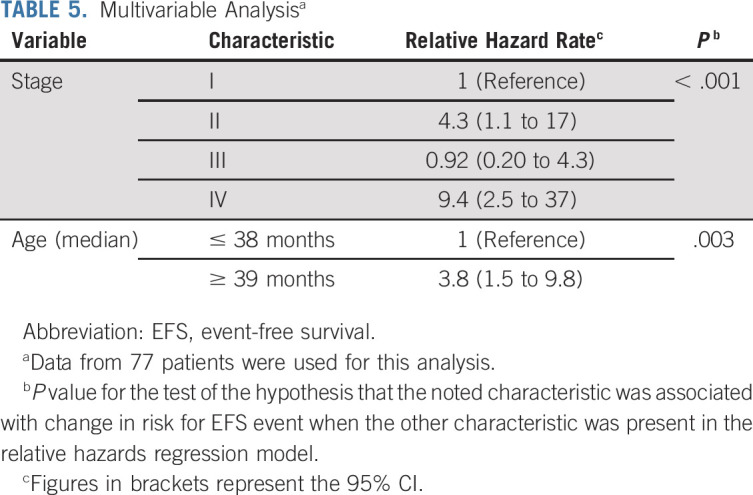
On univariate analysis, age, stage, presence of virilization, Cushing syndrome, or hypertension, predicted p53 function, and the combination of somatic TP53 and ATRX mutations were associated with outcome (Table 4). On multivariable analysis, only stage and age were significantly associated with outcome (Table 5).
TABLE 5.
Multivariable Analysisa
Toxicity or Feasibility Analysis
Among 38 evaluable patients, four had an MFE and 12 had a CFE, for MFE and CFE probabilities of 10.5% (2.9%-24.8%) and 31.6% (17.5%-48.7%), respectively. Based on the study design, we concluded that the chemotherapeutic regimen was not feasible and that further modifications would be required to improve tolerance.
DISCUSSION
Herein, we have reported the results of ARAR0332, a prospective risk-based study for children with ACC. As shown, pediatric ACC has distinct features that separate it from its adult counterpart. First, it is strongly associated with germline TP53 mutations, which were present in 53% of the cases analyzed, compared with < 10% in adults.21 Second, childhood ACC presents at a very early age, and the age continuum defines clinical presentation and prognosis.
Patients with stage I had an excellent outcome, confirming that surgery alone can be curative.3 An RPLND was planned for patients with larger tumors (stage II), based on the high recurrence rate in this group of patients,12,3 and the premise that residual tumor in lymph nodes may contribute to relapse.11,3 Studies performed in adults have shown lymphatic spread at recurrence,22 supporting the rationale for nodal dissection. However, the inclusion of an RPLND was not associated with improved EFS, with only 53% being event-free at 5 years. Although our intervention failed to improve outcomes as hypothesized, it is possible that surgery was not completed as prescribed since the median number of resected lymph nodes was low. In a multivariable analysis performed in a cohort of 283 adult patients with ACC, patients undergoing RPLND (defined as ≥ 5 nodes resected) had a significantly reduced recurrence risk and disease-related death than those not having nodal dissection.23 In this same series, 25.5% of patients undergoing RPLND were found to have nodal metastases, compared with 5.5% of patients who had < 5 nodes resected. In our series, only two patients (6.6%) undergoing an RPLND were found to have positive nodes; whether this is a true proportion or an underestimate because of inadequate RPLND is not clear. RPLND is not a commonly performed procedure in children, which may explain the low compliance with surgical guidelines. A similarly low compliance was reported for children and adolescents with paratesticular rhabdomyosarcoma, a disease in which RPLND affects outcome.24 Further research will be required to improve outcomes for stage II patients; however, the high salvage rates suggest that adjuvant chemotherapy may play a role.
Compared with the suboptimal results for stage II, patients with stage III had excellent outcomes, supporting the use of adjuvant chemotherapy. Unfortunately, the chemotherapy regimen was poorly tolerated. Future studies should explore the optimal regimen for stage II and III patients, define the role of combined therapy versus single agent mitotane, and the optimal duration of mitotane treatment. Several retrospective studies performed in adults have shown a favorable impact of mitotane on relapse-free survival.25-27 Based on these retrospective data, international panels recommend adjuvant mitotane for adults with high recurrence risk (European Network for the Study of Adrenal Tumor stage III, R1 resection, or Ki67 > 10%).28 For patients with lower relapse risk, a randomized clinical trial is testing the efficacy of adjuvant mitotane.29
Little information is available about the use of mitotane in chilren, although response rates appear to be similar to those in adults.30 Compliance with mitotane administration is a limitation, and monitoring for neurotoxicity is particularly important as mitotane has been associated with motor and speech developmental delays.31
Similar to adults, the outcomes of patients with metastatic disease was very poor, highlighting the need for new approaches. In a pan-genomic characterization of adult ACC, at least one alteration of potential driver genes was found in 69% of tumors, with 51 potentially actionable alterations.21 Tumor-infiltrating lymphocytes have been correlated with improved outcomes in adult ACC,32 and checkpoint inhibitors have shown potential, with response rates ranging from 6% to 23%.33-36 We have previously reported the association of major histocompatibility complex class expression with outcome, suggesting that immune responses modulate tumorigenesis and may help identify those who could benefit from checkpoint inhibitors.37 Responses to pembrolizumab have been reported in children.38
We previously reported the genomic landscape of pediatric ACC.8 Mutations in TP53 were the most common, followed by ATRX mutations (which are concomitant with TP53 mutations) and activating mutations of CTNNB1 (which are mutually exclusive of TP53 mutations), and more than 90% of the tumors showed copy-number loss of heterozygosity at 11p15 and insulin-like growth factor-2 overexpression.8 In the current analysis, we sought to further investigate the impact of these three broad genomic groups, as defined by mutations in CTNNB1, TP53, and TP53 and ATRX combined. The small sample size limits the depth of the analysis; however, our data confirm the adverse outcome associated with mutations in ATRX. The presence of TP53 germline pathogenic variants was associated with lower stage and better outcomes, consistent with recent methylation studies.5 It is possible that awareness and screening influenced early diagnosis and outcomes in patients with TP53 germline mutations. Further biological characterization of pediatric ACC, including genomic and methylation studies will be required for further risk-adaptation.
Although the rarity of the disease conditioned the small size of the cohort and the interpretation of some of the findings, ARAR0332 has shown the potential of developing prospective studies in rare cancers. The success of this initiative, together with other coordinated efforts in Europe,12 could provide a platform for international studies.
In summary, treatment of pediatric ACC can follow a risk-adapted approach, with surgery alone for patients with small tumors. RPLND as conducted in this study failed to improve outcome for patients with larger tumors, and thus its role as a standalone treatment strategy is uncertain. Patients with stage III demonstrate an excellent outcome combining surgery and chemotherapy, whereas the outcome for patients with metastatic disease remains poor.
Appendix
TABLE A1.

TABLE A2.
Events by Stage
Mark D. Krailo
Consulting or Advisory Role: Merck Sharp & Dohme
Travel, Accommodations, Expenses: Merck Sharp & Dohme
Emilia M. Pinto
Patents, Royalties, Other Intellectual Property: Genotyping assays to identify mutations in XAF1 pending to St Jude Children's Research Hospital
Farzana Pashankar
Consulting or Advisory Role: Novartis
David Malkin
Consulting or Advisory Role: Bayer
Jonathan D. Wasserman
Consulting or Advisory Role: Bayer
Gerard Zambetti
Research Funding: Johnson & Johnson
Patents, Royalties, Other Intellectual Property: MCL1 antibody license (Rockland Labs) to St Jude Children's Research Hospital. I receive small royalty on an annual basis, Patent pending for Genotyping assays to identify mutations in XAF1 Provisional application #62/659,427; Foreign filing April 18, 2019
Alberto S. Pappo
Honoraria: Bayer, Roche
Consulting or Advisory Role: Merck, Loxo/Bayer, EUSA Pharma, Debbio
No other potential conflicts of interest were reported.
SUPPORT
Supported by Chair's Grant U10 CA98543, NCTN Operations Center Grant U10CA180886, Statistics and Data Center Grant U10 CA98413, and NCTN Statistics and Data Center Grant U10CA180899 of the Children's Oncology Group; by Imaging and Radiation Oncology Core Grant U10 CA29511 from the National Cancer Institute, National Institutes of Health; the Canadian Institutes for Health Research; Grant P30CA21765 of the National Institutes of Health; the American Lebanese Syrian Associated Charities, and by the St Baldrick's Foundation. A complete listing of grant support for research conducted by Children's Cancer Study Group and Pediatric Oncology Group before initiation of the Children's Oncology Group grant in 2003 is available online (http://www.childrensoncologygroup.org/admin/grantinfo.htm).
AUTHOR CONTRIBUTIONS
Conception and design: Carlos Rodriguez-Galindo, Mark D. Krailo, Christopher B. Weldon, John Hicks, David Malkin, Antonio G. de Oliveira Filho, Michael P. LaQuaglia, Deborah A. Ward, Gerard Zambetti, Alberto S. Pappo, Raul C. Ribeiro
Provision of study materials or patients: Eliana M. Caran, Maria J. Mastellaro
Collection and assembly of data: Carlos Rodriguez-Galindo, Mark D. Krailo, Emilia M. Pinto, Eliana M. Caran, John Hicks, Jonathan D. Wasserman, Antonio G. de Oliveira Filho, Deborah A. Ward, Gerard Zambetti, Maria J. Mastellaro, Alberto S. Pappo, Raul C. Ribeiro
Data analysis and interpretation: Carlos Rodriguez-Galindo, Mark D. Krailo, Emilia M. Pinto, Farzana Pashankar, Christopher B. Weldon, Li Huang, John Hicks, M. Beth McCarville, David Malkin, Michael P. LaQuaglia, Gerard Zambetti, Alberto S. Pappo, Raul C. Ribeiro
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Treatment of Pediatric Adrenocortical Carcinoma With Surgery, Retroperitoneal Lymph Node Dissection, and Chemotherapy: The Children's Oncology Group ARAR0332 Protocol
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Mark D. Krailo
Consulting or Advisory Role: Merck Sharp & Dohme
Travel, Accommodations, Expenses: Merck Sharp & Dohme
Emilia M. Pinto
Patents, Royalties, Other Intellectual Property: Genotyping assays to identify mutations in XAF1 pending to St Jude Children's Research Hospital
Farzana Pashankar
Consulting or Advisory Role: Novartis
David Malkin
Consulting or Advisory Role: Bayer
Jonathan D. Wasserman
Consulting or Advisory Role: Bayer
Gerard Zambetti
Research Funding: Johnson & Johnson
Patents, Royalties, Other Intellectual Property: MCL1 antibody license (Rockland Labs) to St Jude Children's Research Hospital. I receive small royalty on an annual basis, Patent pending for Genotyping assays to identify mutations in XAF1 Provisional application #62/659,427; Foreign filing April 18, 2019
Alberto S. Pappo
Honoraria: Bayer, Roche
Consulting or Advisory Role: Merck, Loxo/Bayer, EUSA Pharma, Debbio
No other potential conflicts of interest were reported.
REFERENCES
- 1.Siegel DA, King J, Tai E, et al. Cancer incidence rates and trends among children and adolescents in the United States, 2001–2009 Pediatrics 134e945–e9552014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Michalkiewicz E, Sandrini R, Figueiredo B, et al. Clinical and outcome characteristics of children with adrenocortical tumors. An analysis of 254 cases from the International Pediatric Adrenocortical Tumor Registry J Clin Oncol 22838–8452004 [DOI] [PubMed] [Google Scholar]
- 3.Ribeiro RC, Pinto EM, Zambetti GP, et al. The International Pediatric Adrenocortical Tumor Registry initiative: Contributions to clinical, biological, and treatment advances in pediatric adrenocortical tumors Mol Cell Endocrinol 35137–432012 [DOI] [PubMed] [Google Scholar]
- 4.Wasserman JD, Novokmet A, Eichler-Jonsson C, et al. Prevalence and functional consequence of TP53 mutations in pediatric adrenocortical carcinoma: A Children's Oncology Group study J Clin Oncol 33602–6092015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clay MR, Pinto EM, Cline C, et al. DNA methylation profiling reveals prognostically significant groups in pediatric adrenocortical tumors: A report from the International Pediatric Adrenocortical Tumor Registry. JCO Precis Oncol. 2019 doi: 10.1200/PO.19.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McAteer JP, Huaco JA, Gow KW.Predictors of survival in pediatric adrenocortical carcinoma: A Surveillance, Epidemiology, and End Results (SEER) program study J Pediatr Surg 481025–10312013 [DOI] [PubMed] [Google Scholar]
- 7.Gulack BC, Rialon KL, Englum BR, et al. Factors associated with survival in pediatric adrenocortical carcinoma: An analysis of the National Cancer Data Base (NCDB) J Pediatr Surg 51172–1772016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pinto EM, Chen X, Easton J, et al. Genomic landscape of paediatric adrenocortical tumours. Nat Commun. 2015;6:6302. doi: 10.1038/ncomms7302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berruti A, Terzolo M, Sperone P, et al. Etoposide, doxorubicin and cisplatin plus mitotane in the treatment of advanced adrenocortical carcinoma: A large prospective phase II trial Endocr Relat Cancer 12657–6662005 [DOI] [PubMed] [Google Scholar]
- 10.Fassnacht M, Terzolo M, Allolio B, et al. Combination chemotherapy in advanced adrenocortical carcinoma N Engl J Med 3662189–21972012 [DOI] [PubMed] [Google Scholar]
- 11.Rodriguez-Galindo C, Figueiredo BC, Zambetti GP, et al. Biology, clinical characteristics, and management of adrenocortical tumors in children Pediatr Blood Cancer 45265–2732005 [DOI] [PubMed] [Google Scholar]
- 12.Cecchetto G, Ganarin A, Bien E, et al. Outcome and prognostic factors in high-risk childhood adrenocortical carcinomas: A report from the European Cooperative Study Group on Pediatric Rare Tumors (EXPeRT) Pediatr Blood Cancer. 2017;64:e26368. doi: 10.1002/pbc.26368. [DOI] [PubMed] [Google Scholar]
- 13.Crucitti F, Bellantone R, Ferrante A, et al. The Italian Registry for Adrenal Cortical Carcinoma: Analysis of a multiinstitutional series of 129 patients. The ACC Italian Registry Study Group Surgery 119161–1701996 [DOI] [PubMed] [Google Scholar]
- 14.Weiss LM, Medeiros LJ, Vickery AL.Pathologic features of prognostic significance in adrenocortical carcinoma Am J Surg Pathol 13202–2061989 [DOI] [PubMed] [Google Scholar]
- 15.Wieneke JA, Thompson LDR, Heffess CS.Adrenal cortical neoplasms in the pediatric population: A clinicopathologic and immunophenotypic analysis of 83 patients Am J Surg Pathol 27867–8812003 [DOI] [PubMed] [Google Scholar]
- 16.Sandrini R, Ribeiro RC, DeLacerda L.Childhood adrenocortical tumors J Clin Endocrinol Metab 822027–20311997 [DOI] [PubMed] [Google Scholar]
- 17.Kaplan E, Meier P.Nonparametric estimation from incomplete observations J Am Stat Assoc 53457–4811958 [Google Scholar]
- 18.Schemper M, Smith T.A note on quantifying follow-up studies of failure time Control Clin Trials 17343–3461996 [DOI] [PubMed] [Google Scholar]
- 19.Bishop Y, Feiberg S, Holland P. Discrete Multivariate Analysis. Cambridge, MA: MIT Press; 1975. [Google Scholar]
- 20.Kruskal W, Wallis W.Use of ranks in one-criterion analysis of variance J Am Stat Assoc 47583–6211952 [Google Scholar]
- 21.Zheng S, Cherniack Andrew D, Dewal N, et al. Comprehensive pan-genomic characterization of adrenocortical carcinoma Cancer Cell 29723–7362016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reibetanz J, Rinn B, Kunz AS, et al. Patterns of lymph node recurrence in adrenocortical carcinoma: Possible implications for primary surgical treatment Ann Surg Oncol 26531–5382019 [DOI] [PubMed] [Google Scholar]
- 23.Reibetanz J, Jurowich C, Erdogan I, et al. Impact of lymphadenectomy on the oncologic outcome of patients with adrenocortical carcinoma Ann Surg 255363–3692012 [DOI] [PubMed] [Google Scholar]
- 24.Hamilton EC, Miller CC, Joseph M, et al. Retroperitoneal lymph node staging in paratesticular rhabdomyosarcoma—Are we meeting expectations? J Surg Res 22444–492018 [DOI] [PubMed] [Google Scholar]
- 25.Else T, Williams AR, Sabolch A, et al. Adjuvant therapies and patient and tumor characteristics associated with survival of adult patients with adrenocortical carcinoma J Clin Endocrinol Metab 99455–4612014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Terzolo M, Angeli A, Fassnacht M, et al. Adjuvant mitotane treatment for adrenocortical carcinoma N Engl J Med 3562372–23802007 [DOI] [PubMed] [Google Scholar]
- 27.Berruti A, Grisanti S, Pulzer A, et al. Long-term outcomes of adjuvant mitotane therapy in patients with radically resected adrenocortical carcinoma J Clin Endocrinol Metab 1021358–13652017 [DOI] [PubMed] [Google Scholar]
- 28.Fassnacht M, Dekkers OM, Else T, et al. European Society of Endocrinology Clinical Practice Guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors Eur J Endocrinol 179G1–G462018 [DOI] [PubMed] [Google Scholar]
- 29.Adiuvo: Efficacy of adjuvant mitotane treatment in prolonging recurrence-free survival in patients with adrenocortical carcinoma at low-intermediate risk of recurrence submitted to radical resection. http://www.epiclin.it/adiuvo
- 30.Zancanella P, Pianovski MAD, Oliveira BH, et al. Mitotane associated with cisplatin, etoposide, and doxorubicin in advanced childhood adrenocortical carcinoma. Mitotane monitoring and tumor regression J Pediatr Hematol Oncol 28513–5242006 [DOI] [PubMed] [Google Scholar]
- 31.De Leon DD, Lange BJ, Walterhouse D, et al. Long-term (15 years) outcome in an infant with metastatic adrenocortical carcinoma J Clin Endocrinol Metab 874452–44562002 [DOI] [PubMed] [Google Scholar]
- 32.Landwehr LS, Altieri B, Schreiner J, et al. Interplay between glucocorticoids and tumor-infiltrating lymphocytes on the prognosis of adrenocortical carcinoma. J Immunother Cancer. 2020;8:e000469. doi: 10.1136/jitc-2019-000469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Le Tourneau C, Hoimes C, Zarwan C, et al. Avelumab in patients with previously treated metastatic adrenocortical carcinoma: Phase 1b results from the JAVELIN solid tumor trial. J Immunother Cancer. 2018;6:111. doi: 10.1186/s40425-018-0424-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carneiro BA, Konda B, Costa RB, et al. Nivolumab in metastatic adrenocortical carcinoma: Results of a phase 2 trial J Clin Endocrinol Metab 1046193–62002019 [DOI] [PubMed] [Google Scholar]
- 35.Habra MA, Stephen B, Campbell M, et al. Phase II clinical trial of pembrolizumab efficacy and safety in advanced adrenocortical carcinoma. J Immunother Cancer. 2019;7:253. doi: 10.1186/s40425-019-0722-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raj N, Zheng Y, Kelly V, et al. PD-1 blockade in advanced adrenocortical carcinoma J Clin Oncol 3871–802020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pinto EM, Rodriguez-Galindo C, Choi JK, et al. Prognostic significance of major histocompatibility complex class II expression in pediatric adrenocortical tumors: A St Jude and Children's Oncology Group Study Clin Cancer Res 226247–62552016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geoerger B, Kang HJ, Yalon-Oren M, et al. Pembrolizumab in paediatric patients with advanced melanoma or a PD-L1-positive, advanced, relapsed, or refractory solid tumour or lymphoma (KEYNOTE-051): Interim analysis of an open-label, single-arm, phase 1–2 trial Lancet Oncol 21121–1332020 [DOI] [PubMed] [Google Scholar]